[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Endocrine Complications
Manpreet K. Chadha and Donald L. Trump
• Endocrine dysfunction may occur as a direct result of cancer or may be a consequence of cancer therapy (e.g., surgery, radiation, chemotherapy, biological agents, and hormone therapy). Endocrine dysfunction may be an intentional consequence or an adverse effect of antineoplastic therapy.
• Hypopituitarism with clinically significant deficiencies of growth hormone, thyrotropin, gonadotropin, and corticotrophin may result from radiation (cranial or total body irradiation), surgery, or chemotherapy.
• Thyroid dysfunction from neck irradiation, immune therapy (interleukin-2), and small molecule tyrosine kinase inhibitors such as sunitinib may result in either hyperthyroidism or hypothyroidism.
• Gonadal dysfunction after surgery, radiotherapy, or chemotherapy results in disruption of puberty, infertility, and premature menopause.
• Adrenal dysfunction from agents such as ketoconazole or aminoglutethimide may result in glucocorticoid or mineralocorticoid deficiency.
• Pancreatitis and, occasionally, pancreatic exocrine or endocrine deficiencies may result from chemotherapy (l-asparaginase and streptozotocin).
• A detailed history along with a complete physical examination is critical for diagnosis. Locations of primary and metastatic tumors along with past and current therapies are necessary elements of evaluation.
• Signs and symptoms such as delayed or precocious puberty, fatigue, weight loss or gain, amenorrhea, orthostatic hypotension, hyperpigmentation, or electrolyte abnormalities should prompt consideration of unrecognized endocrine dysfunction.
• When one hormonal deficiency is identified, others should be sought.
• Basal serum hormone concentrations are usually sufficient; however, dynamic testing might be required to diagnose partial deficiencies.
• Patients often have multiple, concurrent hormone deficiencies. Replacement therapy should be started as soon as possible.
• Primary hypothyroidism is characterized by a low free thyroxine (T4) level and an elevated thyroid-stimulating hormone (TSH) level, whereas central hypothyroidism is associated with a low free T4 level and inappropriately normal or low TSH levels. Replacement with levothyroxine is indicated and is highly effective.
• Hyperthyroidism is caused by increased T4 and/or T3 (triiodothyronine) levels with a low serum TSH level. Treatment options include surgery, radioiodine ablation, or antithyroid medications (e.g., propylthiouracil). Rarely, hyperthyroidism may be associated with production of TSH-like substances by germ cell or choriocarcinoma. Management in these instances involves thyroid suppression and treatment of the primary tumor.
• Low- or high-dose corticotrophin testing can distinguish between central and primary causes of adrenal insufficiency. Acute adrenal insufficiency is a medical emergency and should be treated by immediate parenteral glucocorticoid replacement and supportive care. Chronic insufficiency is treated by oral glucocorticoid supplement with or without mineralocorticoid.
Syndrome of Inappropriate Antidiuretic Hormone Secretion
• Hyponatremia is classically associated with high-dose cyclophosphamide and vinca alkaloid administration. Measurement of serum and urine osmolality, renal function tests, and assessment of volume status of a patient are the key to diagnosis. Treatment involves fluid restriction and increased salt intake. Patients with refractory cases might need loop diuretics, doxycycline, or newer vasopressin receptor blockers.
Introduction
Endocrine dysfunction is an increasing cause of morbidity in patients with cancer. In the Childhood Cancer Survivor Study, one or more endocrine conditions were reported in 43% of childhood brain tumor survivors.1 Timely recognition and management of endocrine dysfunction are essential to prevent further morbidity and impairment of quality of life in patients with cancer. Box 61-1 outlines the causes of endocrine dysfunction among patients with cancer. Appropriate evaluation and treatment of common endocrinopathies are discussed in the latter sections of this chapter. A special section is included on surveillance of childhood cancer survivors for detection of late endocrine complications of various cancer therapies. A brief section is also included on endocrine adverse effects from newer therapeutic agents. Tumors of endocrine origin and neuroendocrine tumors are discussed in relevant sections of this textbook.
Role of Surgical Therapy
Historically, surgery has been used as a means of disrupting normal endocrine function and was the first effective therapy for advanced breast or prostate cancers.2 Response rates of 15% to 30% were reported after hypophysectomy or adrenalectomy in patients with advanced breast cancer.3 However, these procedures resulted in significant morbidity, including hypoadrenalism and hypopituitarism, requiring lifelong replacement therapy. For premenopausal women, ovarian ablation by surgical oophorectomy remains a therapeutic option in metastatic and adjuvant settings. These surgical procedures have been largely supplanted by pharmacologic agents such as luteinizing hormone-releasing agonists along with aromatase inhibitors (that inhibit adrenal steroidogenesis) to attain functional castration.4 Orchiectomy, whether surgical or medical, is a critical therapeutic option for men with advanced prostate cancer.5
Similarly, resection of a tumor involving other endocrine glands may result in deficiencies of hormones secreted from these glands: thyroid (hypothyroidism), parathyroids (hypoparathyroidism), pancreas (diabetes mellitus), ovaries (hypogonadism), testes (hypogonadism), or adrenals (hypoadrenalism). Unilateral gland resection rarely results in noticeable hormone deficiencies. Extensive neck surgery and irradiation for advanced head and neck cancers may result in parathyroid hormone deficiency, which can be due to interference with the vascular supply of the parathyroids. Permanent hypoparathyroidism can result inadvertently from total thyroidectomy; the reported incidence is as high as 40%.6 Subtotal removal of parathyroid glands as a part of therapy for parathyroid hyperplasia can also cause hypoparathyroidism. It is often possible to preserve parathyroid function by careful surgical technique and/or by autotransplanting the parathyroid tissue to another part of the body.
Role of Radiation Therapy
Endocrine organs may be intentionally or unavoidably exposed to ionizing radiation during treatment for malignancy, and high-dose radiation may result in endocrine dysfunction. Box 61-2 lists factors that are known to be associated with a high risk of endocrine dysfunction after radiation. Assessment of late effects of radiation may be difficult and subjective. Various groups have attempted to develop a scoring system to standardize toxicity reporting and description. The most accurate scale for grading radiation-induced effects on normal tissues, including to the hypothalamic-pituitary axis and thyroid, is LENT-SOMA (Late Effects on Normal Tissue—Subjective, Objective, Management and Analytic).7 These scales grade the radiation-induced adverse effects on organs exposed to irradiation by using criteria similar to the common toxicity criteria grading of adverse effects developed by the National Cancer Institute (http://www.eortc.be/services/doc/ctc/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf).
Hypothalamic-Pituitary Axis
Anterior pituitary damage can result from irradiation of extracranial or primary brain tumors, especially those involving the pituitary. Total body irradiation as part of a bone marrow transplant preparative regimen and prophylactic cranial radiation in patients with acute lymphoblastic leukemia can also cause hypopituitarism.10–10 After curative irradiation for nasopharyngeal carcinoma, approximately 19% of patients have a deficiency in one or more anterior pituitary hormones as early as 2 years after therapy.10
Somatotrophs (cells that secrete growth hormone) are the most vulnerable to radiation damage; hence growth hormone deficiency (GHD) is the endocrine abnormality most commonly seen after cranial irradiation. GHD may occur in isolation after irradiation of the hypothalamic-pituitary axis even with doses less than 30 Gy. The clinical manifestations of GHD of reduction in growth velocity and short stature are most evident in the growing child.10 Although poor linear growth is very common in children with GHD, it is not universal or immediately apparent. Several studies suggest that the slowing of growth might not occur for the first year or two after the onset of GHD. In postpuberal individuals, GHD is associated with a decrease in muscle mass along with an increase in adiposity.
The hypothalamic neurons secrete gonadotropin-releasing hormone (GnRH) in pulses that are necessary for normal secretion of gonadotropins from the pituitary. This GnRH pulse generation is affected differentially by the dose of radiation that is received. Higher dose irradiation (>30 Gy) is associated with delayed sexual maturation because of gonadotropin deficiency from damage to GnRH secretory neurons.11 Lower doses (<30 Gy) can result in precocious puberty equally in both sexes with a radiation dose of 30 to 50 Gy. Radiation-induced precocious puberty might be caused by damage to inhibitory GABAergic neurons, leading to disinhibition and premature activation of GnRH neurons.11
Deficiency in other pituitary hormones is less common. Littley and colleagues12 described 251 patients who had been treated for pituitary disease with external radiotherapy. Five years after completion of treatment, these investigators noted a 9% dose-related incidence of thyroid-stimulating hormone (TSH) deficiency at 20 Gy, which increased to 52% at 42 to 45 Gy. A similar trend in frequency of adrenocorticotrophic hormone (ACTH) deficiency was seen. Hyperprolactinemia related to damage to inhibitory neurons that control prolactin secretion can be seen after high-dose radiotherapy (>40 Gy); it has been described in both sexes and all age groups but is most common in young women. Hyperprolactinemia occurs with a described frequency ranging from 20% to 50% of patients with nasopharyngeal and brain irradiation.10 Hyperprolactinemia can cause delayed puberty in children, galactorrhea or amenorrhea in adult women, and decreased libido and impotence in men.
Thyroid
Irradiation of the thyroid may produce hypothyroidism, Graves disease, silent thyroiditis, benign nodules, and thyroid cancers.13 Hancock and colleagues described a series of patients with thyroid dysfunction among patients treated with irradiation with or without chemotherapy for Hodgkin disease at Stanford University.14 Of 1787 patients, 1677 received irradiation to the thyroid. At 26 years of follow-up, the actuarial risk of thyroid disease was 67%. Hypothyroidism developed in the majority of the patients (47%). The risk of Graves disease was 7 to 20 times higher than that for healthy subjects. The risk of thyroid cancer was 15.6 times the expected risk for healthy subjects. These data remind clinicians to monitor thyroid function closely in patients who have been treated with upper mantle or cervical irradiation. Similar results were noted in the Childhood Cancer Survivor Study with an evaluable cohort of 1791 Hodgkin disease survivors (including 959 males). Among patients with Hodgkin disease, a 50% risk of hypothyroidism was found 20 years from the time of diagnosis in persons treated with 45 Gy or more.15 The total dose of irradiation received has been shown to correlate with the incidence of hypothyroidism in many studies.15–15 However, controversy exists regarding the effect of age and gender at the time of irradiation.15–15
Radiation-induced thyroid dysfunction is thought to be caused by damage to small thyroid vessels and to the glandular capsule. Histomorphologic features that are described in such patients include focal and irregular follicular hyperplasia, hyalinization, and fibrosis beneath the vascular endothelium; lymphocytic infiltration; single and multiple adenomas; and thyroid carcinomas.13,14
A rare complication of neck irradiation is acute radiation thyroiditis, which is more commonly associated with therapeutic doses of radioiodine for thyroid diseases. Patients typically have symptoms of fever, pain in the anterior cervical region, and transient hyperthyroidism. Hyperthyroidism with a clinical picture that resembles Graves disease may be seen after neck irradiation for Hodgkin disease.14 The incidence is uncertain because of the small number of cases reported. The clinical picture is characterized by diffuse thyroid enlargement, suppressed TSH, high levels of thyroid hormones, and development of thyroid autoantibodies. Ophthalmopathy, with or without overt hyperthyroidism, may be seen and is thought to be related to autoantibodies, similar to that seen in persons with classic Graves disease.
Parathyroid Glands
Several studies link prior head and neck irradiation and hyperparathyroidism. Cohen and colleagues16 followed a cohort of patients who were treated with radiation to the tonsils before the age of 16 years. Among the 2923 patients, 32 were found to have clinical hyperparathyroidism—a 2.5- to 2.9-fold increase compared with the general population in the same age group. A long latency period (>25 years) occurs between exposure and the onset of hyperparathyroidism. Parathyroid adenomas were found in most of the patients in whom this complication develops.17 Clinical presentations vary from asymptomatic increases in serum parathormone levels and hypercalcemia to disabling metabolic bone disease or nephrolithiasis. Persons with a history of head and neck radiation should be monitored with calcium levels periodically (every 1 to 2 years) and indefinitely.
Role of Systemic Therapy
Effects of systemic chemotherapy on ovarian and testicular function are discussed in Chapter 60.
Hypothalamic-Pituitary Axis
In children, chemotherapeutic agents without any cranial irradiation may disrupt growth hormone (GH) secretion. Roman and colleagues18 studied growth and GH secretion in 60 children who were in complete remission after treatment with chemotherapy and surgery for solid tumors. They observed GH deficiency in 45% of those studied and found that the GHD group had received significantly higher doses of actinomycin D compared with the non-GHD group (P < .05). These investigators found no correlation with duration of treatment, length of follow-up, tumor type, sex, or age. Depending on the intensity of chemotherapy, significant height loss was detected in 40% to 70% of patients at 6-year follow-up. Adjuvant chemotherapy can also aggravate growth failure in children with brain tumors who receive craniospinal radiation.19 Rose and colleagues20 reported hypothalamic dysfunction in patients with non–central nervous system tumors who received chemotherapy but did not receive cranial irradiation and had no history of traumatic brain injury. Of 31 identified patients, GHD was identified in 15 (48%), central hypothyroidism was identified in 16 (52%), and pubertal abnormalities was identified in 10 (32%). GHD and hypothyroidism were coexistent in eight patients (26%). Overall, 81% had GHD, hypothyroidism, precocious puberty, or gonadotropin deficiency.
The syndrome of inappropriate antidiuretic hormone (SIADH) secretion is associated with various malignant tumors including certain primary brain tumors, hematologic malignancies, intrathoracic nonpulmonary cancers, skin tumors, gastrointestinal cancers, gynecologic cancer, breast and prostatic cancer, and sarcomas.21 SIADH may result from the effects of many chemotherapeutic agents, either by potentiation of antidiuretic hormone (ADH) effect or by increased ADH secretion. The most commonly implicated agents are vinca alkaloids and cyclophosphamide. The vinca alkaloids are reported to stimulate the central release of ADH from the neurohypophyseal system,22 whereas alkylating agents enhance renal tubular sensitivity to ADH.23 Regardless of the mechanism, the result is an increase in water reabsorption by the distal tubules of the kidney, leading to volume expansion and dilutional hyponatremia. Case reports also implicate platinum agents,24 vinorelbine,25 taxanes,26 and methotrexate.27 Clinically significant hyponatremia may occur with administration of these agents, and management requires fluid restriction and, at times, salt replacement.
Thyroid
Clinically evident thyroid dysfunction has rarely been associated with the use of standard chemotherapy agents. However, a growing body of literature points to the increased prevalence of endocrine dysfunction after bone marrow transplantation, following use of preparation regimens that do not include radiation. Thyroid dysfunction has been reported in as many as 50% of allogeneic bone marrow transplant recipients treated with busulfan and cyclophosphamide.28 Thyroid dysfunction may appear as low T3 syndrome (i.e., normal levels of free T4 and TSH and a below-normal level of free T3), chronic thyroiditis, and transient subclinical hyperthyroidism or hypothyroidism. Chemotherapy may potentiate radiation-induced damage to normal tissue.
Newer biological agents, tyrosine kinase inhibitors, and immune-based cancer treatments (e.g., ipilimumab, interferon-α, and interleukin-2) have been associated with various types of thyroid dysfunction.29, 30 Box 61-3 summarizes the well-described adverse effects of cancer therapies on thyroid function.
Hypothyroidism is a recognized complication of treatment with tyrosine kinase inhibitors. Sunitinib maleate is an oral tyrosine kinase inhibitor that was recently approved for the treatment of gastrointestinal stromal tumors and renal cell carcinoma. Desai and colleagues31 reported hypothyroidism in patients undergoing sunitinib therapy. One potential mechanism may be sunitinib-induced destructive thyroiditis through follicular cell apoptosis. However, there is no explanation for the apparent sensitivity of thyroid tissue compared with other endocrine organs. Sunitinib inhibits the RET tyrosine kinase that is involved in pathogenesis of medullary thyroid cancer; however, the relationship between specific tyrosine kinase inhibition and the development of hypothyroidism is unclear.
Imatinib, an inhibitor of the bcr-abl tyrosine kinase, interacts with thyroid hormone replacement and results in increased TSH levels in persons with hypothyroidism who are being treated with levothyroxine. However, it does not appear to have a direct effect on the thyroid but alters the levels of thyroxine-binding protein.32
Aminoglutethimide, which now is infrequently used to disrupt adrenal and peripheral steroid hormone synthesis, inhibits cholesterol conversion to pregnenolone. It causes thyroid dysfunction after long-term use as a result of the blockade of iodination of tyrosine.33 Chemotherapeutic agents can also interfere with circulating thyroid hormone, thereby altering free blood levels. The drug 5-fluorouracil increases total T3 and T4 levels, but free T4 index and TSH remain normal, indicating increased levels of thyroxine-binding globulin or enhanced binding capacity. l-asparaginase causes transient thyroxine-binding globulin deficiency by diminishing hepatic synthesis and also inhibits TSH secretion from the pituitary, resulting in decreased total T4 and free T4 levels. Transient hyperthyroidism after l-asparaginase therapy for acute lymphoblastic leukemia has also been observed. These thyroid abnormalities are mild and short-lived and generally do not require specific therapy.30
In postmenopausal women, tamoxifen therapy is associated with changes in thyroid hormone concentrations, although patients may remain clinically euthyroid. Mamby and colleagues34 evaluated the effect of tamoxifen, 10 mg orally, twice daily on the thyroid in a placebo-controlled trial. Thyroid function was examined before and 3 months after initiation of therapy. Serum thyroid-binding globulin increased, as did T4 uptake and T4 levels in the tamoxifen-treated group compared with the group that received a placebo. TSH levels and the free thyroxine index remained unchanged; patients were clinically euthyroid and did not require treatment.
Adrenal
Medical ablation of adrenal function has been extensively evaluated. Aminoglutethimide and ketoconazole both suppress adrenal function. These drugs appear to have their effect through the inhibition of important cytochrome P-450 isozymes, which are necessary for adrenal steroid synthesis.33,35 Aminoglutethimide, at doses of 1000 to 1500 mg/day, and ketoconazole, at doses of 800 to 1200 mg/day, produce adrenal insufficiency in 30% to 40% of patients. Although glucocorticoid replacement is generally required in these patients during treatment, mineralocorticoid replacement is usually not required. The antiadrenal effects of ketoconazole and aminoglutethimide are reversible with treatment discontinuation. Full recovery within 1 to 2 weeks is usual.
Mitotane (1-dichloro-2-[o-chlorophenyl]-2-[p-chlorophenyl]-ethane) is an oral chemotherapeutic agent that is used to treat adrenal carcinoma and has potent antiadrenal effects. It is used primarily to treat adrenal hyperfunction associated with adrenal carcinoma or ectopic production of corticotrophin.36 Although its mechanism of action is incompletely understood, adrenal necrosis and permanent adrenal insufficiency can result, necessitating lifelong glucocorticoid administration.
Abiraterone acetate, a pregnenolone analog, is a small molecule that irreversibly inhibits CYP17, a rate-limiting enzyme in androgen biosynthesis. It is approved for treatment of metastatic, castration-resistant prostate cancer. Abiraterone administration decreases the production of cortisol by blocking the activity of 17α hydroxylase. This process leads to increased production of ACTH after diminished negative feedback, which, in turn, stimulates production of steroid precursors above the 17α hydroxylase level. One of these precursors is corticosterone, which is only a weak inhibitor of ACTH. The increased production of corticosterone leads to fluid retention, hypokalemia, and hypertension. These effects can be prevented by concomitant administration of low doses of prednisone, corticosterone, or dexamethasone.37
Pancreas
Pancreatic exocrine or endocrine insufficiency attributable to chemotherapy is uncommon. Although rare, cases of diabetes mellitus have been associated with l-asparaginase therapy.38 Hyperglycemia is usually transient and responds to intravenous fluids and drug discontinuation. A plausible although unproven mechanism underlying this phenomenon may be inhibition of protein synthesis by l-asparaginase, leading to interference with insulin production. High triglyceride levels have also been associated with l-asparaginase use, although these changes are not clearly associated with an increased incidence of pancreatitis in such patients. The occurrence of pancreatitis seems to be independent of the individual or cumulative asparaginase dose.
High-dose cytarabine therapy can result in pancreatitis. Siemers and colleagues39 described two patients with evidence of pancreatitis among 30 patients treated with high-dose cytarabine.39 Streptozotocin is a nitrosourea that is used primarily for the treatment of pancreatic endocrine tumors. In preclinical models in many species, streptozotocin causes β-cell necrosis and insulin-dependent diabetes.40 Mild glucose intolerance has been described in patients receiving this agent; however, frank diabetes requiring specific treatment is rare.
Androgen ablation therapy may also be associated with diabetes. Keating and colleagues41 reported an increased incidence of diabetes in patients with prostate cancer who were treated with GnRH agonists. A potential mechanism is the increase in body fat mass associated with hypogonadism, which results in insulin resistance.
Phosphatidyl inositol 3 kinase (PI3K) inhibitors, which are presently in clinical trials with promising results in persons with breast cancer, have been recognized to cause hyperglycemia. This effect is related to decreased glucose uptake by cells rather than any effect on insulin or glucagon secretion. Mammalian target of rapamycin (mTOR) inhibitors also produce hyperglycemia.42
Role of Biological Agents
Immune therapies may cause thyroid dysfunction. Interleukin-2 therapy is associated with both hypothyroidism and hyperthyroidism, although the former is more common. In the original report by Atkins and colleagues,43 seven patients (21%) had laboratory evidence of hypothyroidism, a decline in the serum thyroxine concentration and serum free thyroxine index, and an increase in the serum TSH concentration 6 to 11 weeks after treatment. All five symptomatic patients had borderline or elevated serum antimicrosomal antibody titers after treatment; two had serum antibodies to thyroglobulin. The proposed mechanism is autoimmune with development of antithyroid antibodies.
Ipilimumab (anti–CTLA-4 antibody) has been shown to induce hypophysitis and result in immune-mediated hypopituitarism. In a recent phase 3 trial, nine patients treated with ipilimumab (1.8%) experienced severe or life-threatening hypopituitarism. Several other endocrine adverse effects including hypothyroidism, adrenal insufficiency, or hypogonadism were also noted. Primary thyroid disease may present as hyperthyroidism with Graves disease or as hypothyroidism as a result of destructive thyroiditis. Secondary hypothyroidism may result from decreased production of TSH as a result of hypophysitis and panhypopituitarism. Immune-mediated pancreatitis was also noted in the study. The median time to onset of endocrine symptoms was 11 weeks; delayed symptoms were observed in some patients even after completion of the intended four courses. Some of the endocrine dysfunction is thought to be reversible with cessation of therapy with ipilimumab. Limited data are available on the long-term sequelae of ipilimumab therapy.29 Patients treated with ipilimumab should be monitored for clinical signs or symptoms associated with pituitary, thyroid, or adrenal disease. Baseline monitoring of TSH, repeated with every cycle of treatment, is recommended to detect hypothyroidism. Evaluation and testing for other endocrinopathies should be guided by clinical symptoms. Treatment includes hormone replacement therapy, corticosteroids, and cessation of ipilimumab based on the severity of the complication.44
Interferon-α therapy may increase ACTH, prolactin, growth hormone, and cortisol levels.45 An assessment of interferon-α–induced endocrine stimulation in patients with myeloproliferative disorders demonstrated that on day 1 of therapy, a significant stimulation of the hypothalamic-pituitary axis was apparent, an effect that disappeared by the third week of therapy. The acute stimulatory effect of interferon-α on cortisol release appears to be mediated by the release of hypothalamic corticotropin-releasing hormone. Alterations in the levels of sex hormones during interferon therapy have been reported, and male sexual dysfunction has been noted.45
Evaluation and Treatment of Common Endocrine Dysfunction
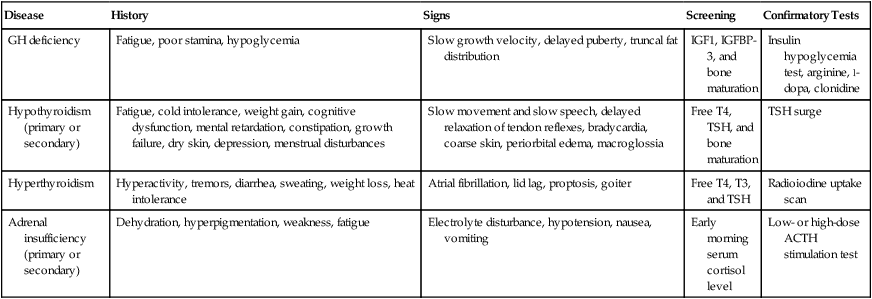
A detailed history, including treatment history and physical examination, should be performed for any patient who is suspected of having endocrine dysfunction. Evaluation should be directed by this information and the location and type of tumor. The initial approach to the diagnostic workup is outlined in the following sections. Endocrinology consultation should be sought for more complex and multisystem involvement. Table 61-1 provides a brief outline of the evaluation of common endocrine disorders.
Table 61-1
Diagnostic Evaluation of Common Endocrine Disorders
| Disease | History | Signs | Screening | Confirmatory Tests |
| GH deficiency | Fatigue, poor stamina, hypoglycemia | Slow growth velocity, delayed puberty, truncal fat distribution | IGF1, IGFBP-3, and bone maturation | Insulin hypoglycemia test, arginine, l-dopa, clonidine |
| Hypothyroidism (primary or secondary) | Fatigue, cold intolerance, weight gain, cognitive dysfunction, mental retardation, constipation, growth failure, dry skin, depression, menstrual disturbances | Slow movement and slow speech, delayed relaxation of tendon reflexes, bradycardia, coarse skin, periorbital edema, macroglossia | Free T4, TSH, and bone maturation | TSH surge |
| Hyperthyroidism | Hyperactivity, tremors, diarrhea, sweating, weight loss, heat intolerance | Atrial fibrillation, lid lag, proptosis, goiter | Free T4, T3, and TSH | Radioiodine uptake scan |
| Adrenal insufficiency (primary or secondary) | Dehydration, hyperpigmentation, weakness, fatigue | Electrolyte disturbance, hypotension, nausea, vomiting | Early morning serum cortisol level | Low- or high-dose ACTH stimulation test |
Hypothalamic-Pituitary Axis Disorders
Growth Hormone Deficiency
Evaluation
The assessment of pituitary GH production is difficult because GH secretion is pulsatile and serum GH levels are often low between the pulses. Therefore measurement of a single serum GH level is of limited use in diagnosing GHD. Serum insulin-like growth factor (IGF1) and IGF binding protein–3 (IGFBP-3) concentrations may be measured as a surrogate marker for GH production, and further evaluation is indicated if these results are below the mean for normal children of the same age. An IGF1 level below 84 ng/mL with the Esoterix assay reportedly is highly indicative of GHD.46 Confirmation can be obtained with GH secretion provocative tests.
According to the consensus guidelines for the diagnosis and treatment of adults with GH deficiency, the insulin hypoglycemia test is the gold-standard GH provocative test. According to the Food and Drug Administration (FDA), GHD is diagnosed if the maximum stimulated serum GH concentration is less than 5.1 µg/L (polyclonal radioimmunoassay) or less than 2.5 µg/mL (immunochemiluminescent assay). Although it is reliable, the insulin-induced hypoglycemia test requires careful and strict monitoring. It is contraindicated in debilitated patients, those with cardiovascular or cerebrovascular disease, and those with a history of seizure, abnormal electroencephalogram, or a history of brain surgery.46 In these patients, the combined arginine/growth hormone releasing hormone (GHRH) stimulation test may be used. The arginine stimulation test involves intravenous infusion of 0.5 g/kg body weight (to a maximum of 30 g) of arginine given during a period of 30 minutes and measuring serum growth hormone level at 0, 30, 60, 90, and 120 minutes. Although the insulin hypoglycemia test was considered the gold standard for GHD, the arginine-GHRH test is much safer, is 95% sensitive and 91% specific (at a cutoff of 4.1 µg/L), and has largely replaced the insulin hypoglycemia test.
The diagnosis of GHD in childhood is complex and requires clinical and growth assessment combined with biochemical tests and radiologic evaluation. GHD may be an isolated finding or a component of multiple pituitary hormone deficiency. In a child with clinical criteria for GHD, a peak GH concentration less than 10 mg/L has traditionally been used to support the diagnosis after a provocative GH test.46 Supportive evidence includes very short height (more than 2.5 standard deviations below the mean height for healthy children of the same age), delayed bone age, poor growth velocity (less than the 25th percentile), and a predicted adult height substantially below the mean parental height. IGF-I/IGFBP-3 levels and GH provocation tests should be performed after hypothyroidism has been excluded as a cause of slow growth. Great care should be taken in using insulin or glucagon provocative tests in a young child, and testing should be monitored by an experienced team.
Treatment
GH therapy in adults is approved by the FDA only if evidence of hypothalamic or pituitary disease exists and a subnormal serum GH response to a provocative test occurs. The goal of the therapy is to improve muscle and cardiac function, restore normal body composition, and improve serum lipid levels. The usual starting dose for adults between 30 to 60 years of age is 300 µg/day; the dose is increased every 1 to 2 months by 100 to 200 µg/day, guided by clinical response and measurement of serum GH levels.46
Hyperprolactinemia
Evaluation
The presenting symptoms of hyperprolactinemia in adults include amenorrhea, galactorrhea, impotence, and infertility. The diagnosis of hyperprolactinemia is made by a random serum prolactin level measurement. Dynamic testing of the lactotrophin reserve with thyrotropin-releasing hormone is not useful because it does not differentiate between the different causes of hyperprolactinemia.47 Hypothyroidism must also be ruled out as a cause of clinical findings, and a careful review of medications should be performed to rule out drug-induced hyperprolactinemia.
Thyroid Disorders
Evaluation
Primary hypothyroidism is characterized by a high serum TSH concentration (normal range: 0.5 to 5 mU/L) and a low serum free T4 concentration (normal range: 0.8 to 1.8 ng/dL). Secondary hypothyroidism is characterized by a low serum T4 concentration and a serum TSH concentration that is not appropriately elevated. To distinguish between pituitary and hypothalamic causes of hypothyroidism, a thyrotropin-releasing hormone stimulation test and/or imaging studies of the sellar and suprasellar region should be performed. Patients who are diagnosed with central hypothyroidism should also be evaluated for coexistent adrenocortical, gonadal, and posterior pituitary function, and, in children, growth hormone function. As noted previously, patients with cranial irradiation or traumatic brain injury are at high risk for panhypopituitarism.48
Primary hyperthyroidism is associated with a low TSH level and a high free T4 level. Graves disease is associated with uniformly high 24-hour radioiodine uptake, whereas toxic adenoma is associated with focal high uptake. If free T4 and T3 levels are high with a normal or high TSH level in a clinically hyperthyroid patient, pituitary magnetic resonance imaging should be performed to look for a pituitary mass (TSH-secreting adenoma).48
Adrenal Disorders
Treatment
Adrenal insufficiency requires glucocorticoid supplementation and at times mineralocorticoid supplementation. Pituitary or isolated ACTH deficiency is not characterized by mineralocorticoid deficiency. Patients with symptomatic adrenal insufficiency should be treated with hydrocortisone or cortisone in the early morning and afternoon. The usual initial oral dose is 25 mg of hydrocortisone (15 mg in the morning and 10 mg in the afternoon). This dose may be decreased over time, with the goal of using the minimal effective dose to prevent weight gain and osteoporosis.49 Patients with primary adrenal insufficiency require mineralocorticoid replacement with fludrocortisone (0.05 to 2 mg orally each day). During periods of stress, patients with adrenal insufficiency require higher than usual doses of hydrocortisone, and in severe illness, they might require intravenous high-dose hydrocortisone therapy as a result of acute adrenal crisis. Box 61-4 outlines a treatment algorithm for acute adrenal crisis, which should be treated as an oncologic emergency.
Surveillance of Childhood Cancer Survivors
Improved therapy for most childhood cancers has led to an increasing population of childhood cancer survivors. These persons are at risk for long-term endocrine complications related to the tumor and/or the treatment they received. The risk of a particular endocrinopathy depends on the tumor location and the dose and duration of radiotherapy and chemotherapy received. Box 61-5 provides a summary of recommended yearly surveillance in childhood cancer survivors for endocrine complications.50 Close follow-up should be performed every 4 to 6 months if the initial tests are normal but the child remains symptomatic.
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Endocrine Complications
Manpreet K. Chadha and Donald L. Trump
• Endocrine dysfunction may occur as a direct result of cancer or may be a consequence of cancer therapy (e.g., surgery, radiation, chemotherapy, biological agents, and hormone therapy). Endocrine dysfunction may be an intentional consequence or an adverse effect of antineoplastic therapy.
• Hypopituitarism with clinically significant deficiencies of growth hormone, thyrotropin, gonadotropin, and corticotrophin may result from radiation (cranial or total body irradiation), surgery, or chemotherapy.
• Thyroid dysfunction from neck irradiation, immune therapy (interleukin-2), and small molecule tyrosine kinase inhibitors such as sunitinib may result in either hyperthyroidism or hypothyroidism.
• Gonadal dysfunction after surgery, radiotherapy, or chemotherapy results in disruption of puberty, infertility, and premature menopause.
• Adrenal dysfunction from agents such as ketoconazole or aminoglutethimide may result in glucocorticoid or mineralocorticoid deficiency.
• Pancreatitis and, occasionally, pancreatic exocrine or endocrine deficiencies may result from chemotherapy (l-asparaginase and streptozotocin).
• A detailed history along with a complete physical examination is critical for diagnosis. Locations of primary and metastatic tumors along with past and current therapies are necessary elements of evaluation.
• Signs and symptoms such as delayed or precocious puberty, fatigue, weight loss or gain, amenorrhea, orthostatic hypotension, hyperpigmentation, or electrolyte abnormalities should prompt consideration of unrecognized endocrine dysfunction.
• When one hormonal deficiency is identified, others should be sought.
• Basal serum hormone concentrations are usually sufficient; however, dynamic testing might be required to diagnose partial deficiencies.
• Patients often have multiple, concurrent hormone deficiencies. Replacement therapy should be started as soon as possible.
• Primary hypothyroidism is characterized by a low free thyroxine (T4) level and an elevated thyroid-stimulating hormone (TSH) level, whereas central hypothyroidism is associated with a low free T4 level and inappropriately normal or low TSH levels. Replacement with levothyroxine is indicated and is highly effective.
• Hyperthyroidism is caused by increased T4 and/or T3 (triiodothyronine) levels with a low serum TSH level. Treatment options include surgery, radioiodine ablation, or antithyroid medications (e.g., propylthiouracil). Rarely, hyperthyroidism may be associated with production of TSH-like substances by germ cell or choriocarcinoma. Management in these instances involves thyroid suppression and treatment of the primary tumor.
• Low- or high-dose corticotrophin testing can distinguish between central and primary causes of adrenal insufficiency. Acute adrenal insufficiency is a medical emergency and should be treated by immediate parenteral glucocorticoid replacement and supportive care. Chronic insufficiency is treated by oral glucocorticoid supplement with or without mineralocorticoid.
Syndrome of Inappropriate Antidiuretic Hormone Secretion
• Hyponatremia is classically associated with high-dose cyclophosphamide and vinca alkaloid administration. Measurement of serum and urine osmolality, renal function tests, and assessment of volume status of a patient are the key to diagnosis. Treatment involves fluid restriction and increased salt intake. Patients with refractory cases might need loop diuretics, doxycycline, or newer vasopressin receptor blockers.
Introduction
Endocrine dysfunction is an increasing cause of morbidity in patients with cancer. In the Childhood Cancer Survivor Study, one or more endocrine conditions were reported in 43% of childhood brain tumor survivors.1 Timely recognition and management of endocrine dysfunction are essential to prevent further morbidity and impairment of quality of life in patients with cancer. Box 61-1 outlines the causes of endocrine dysfunction among patients with cancer. Appropriate evaluation and treatment of common endocrinopathies are discussed in the latter sections of this chapter. A special section is included on surveillance of childhood cancer survivors for detection of late endocrine complications of various cancer therapies. A brief section is also included on endocrine adverse effects from newer therapeutic agents. Tumors of endocrine origin and neuroendocrine tumors are discussed in relevant sections of this textbook.
Role of Surgical Therapy
Historically, surgery has been used as a means of disrupting normal endocrine function and was the first effective therapy for advanced breast or prostate cancers.2 Response rates of 15% to 30% were reported after hypophysectomy or adrenalectomy in patients with advanced breast cancer.3 However, these procedures resulted in significant morbidity, including hypoadrenalism and hypopituitarism, requiring lifelong replacement therapy. For premenopausal women, ovarian ablation by surgical oophorectomy remains a therapeutic option in metastatic and adjuvant settings. These surgical procedures have been largely supplanted by pharmacologic agents such as luteinizing hormone-releasing agonists along with aromatase inhibitors (that inhibit adrenal steroidogenesis) to attain functional castration.4 Orchiectomy, whether surgical or medical, is a critical therapeutic option for men with advanced prostate cancer.5
Similarly, resection of a tumor involving other endocrine glands may result in deficiencies of hormones secreted from these glands: thyroid (hypothyroidism), parathyroids (hypoparathyroidism), pancreas (diabetes mellitus), ovaries (hypogonadism), testes (hypogonadism), or adrenals (hypoadrenalism). Unilateral gland resection rarely results in noticeable hormone deficiencies. Extensive neck surgery and irradiation for advanced head and neck cancers may result in parathyroid hormone deficiency, which can be due to interference with the vascular supply of the parathyroids. Permanent hypoparathyroidism can result inadvertently from total thyroidectomy; the reported incidence is as high as 40%.6 Subtotal removal of parathyroid glands as a part of therapy for parathyroid hyperplasia can also cause hypoparathyroidism. It is often possible to preserve parathyroid function by careful surgical technique and/or by autotransplanting the parathyroid tissue to another part of the body.
Role of Radiation Therapy
Endocrine organs may be intentionally or unavoidably exposed to ionizing radiation during treatment for malignancy, and high-dose radiation may result in endocrine dysfunction. Box 61-2 lists factors that are known to be associated with a high risk of endocrine dysfunction after radiation. Assessment of late effects of radiation may be difficult and subjective. Various groups have attempted to develop a scoring system to standardize toxicity reporting and description. The most accurate scale for grading radiation-induced effects on normal tissues, including to the hypothalamic-pituitary axis and thyroid, is LENT-SOMA (Late Effects on Normal Tissue—Subjective, Objective, Management and Analytic).7 These scales grade the radiation-induced adverse effects on organs exposed to irradiation by using criteria similar to the common toxicity criteria grading of adverse effects developed by the National Cancer Institute (http://www.eortc.be/services/doc/ctc/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf).
Hypothalamic-Pituitary Axis
Anterior pituitary damage can result from irradiation of extracranial or primary brain tumors, especially those involving the pituitary. Total body irradiation as part of a bone marrow transplant preparative regimen and prophylactic cranial radiation in patients with acute lymphoblastic leukemia can also cause hypopituitarism.10–10 After curative irradiation for nasopharyngeal carcinoma, approximately 19% of patients have a deficiency in one or more anterior pituitary hormones as early as 2 years after therapy.10
Somatotrophs (cells that secrete growth hormone) are the most vulnerable to radiation damage; hence growth hormone deficiency (GHD) is the endocrine abnormality most commonly seen after cranial irradiation. GHD may occur in isolation after irradiation of the hypothalamic-pituitary axis even with doses less than 30 Gy. The clinical manifestations of GHD of reduction in growth velocity and short stature are most evident in the growing child.10 Although poor linear growth is very common in children with GHD, it is not universal or immediately apparent. Several studies suggest that the slowing of growth might not occur for the first year or two after the onset of GHD. In postpuberal individuals, GHD is associated with a decrease in muscle mass along with an increase in adiposity.
The hypothalamic neurons secrete gonadotropin-releasing hormone (GnRH) in pulses that are necessary for normal secretion of gonadotropins from the pituitary. This GnRH pulse generation is affected differentially by the dose of radiation that is received. Higher dose irradiation (>30 Gy) is associated with delayed sexual maturation because of gonadotropin deficiency from damage to GnRH secretory neurons.11 Lower doses (<30 Gy) can result in precocious puberty equally in both sexes with a radiation dose of 30 to 50 Gy. Radiation-induced precocious puberty might be caused by damage to inhibitory GABAergic neurons, leading to disinhibition and premature activation of GnRH neurons.11
Deficiency in other pituitary hormones is less common. Littley and colleagues12 described 251 patients who had been treated for pituitary disease with external radiotherapy. Five years after completion of treatment, these investigators noted a 9% dose-related incidence of thyroid-stimulating hormone (TSH) deficiency at 20 Gy, which increased to 52% at 42 to 45 Gy. A similar trend in frequency of adrenocorticotrophic hormone (ACTH) deficiency was seen. Hyperprolactinemia related to damage to inhibitory neurons that control prolactin secretion can be seen after high-dose radiotherapy (>40 Gy); it has been described in both sexes and all age groups but is most common in young women. Hyperprolactinemia occurs with a described frequency ranging from 20% to 50% of patients with nasopharyngeal and brain irradiation.10 Hyperprolactinemia can cause delayed puberty in children, galactorrhea or amenorrhea in adult women, and decreased libido and impotence in men.
Thyroid
Irradiation of the thyroid may produce hypothyroidism, Graves disease, silent thyroiditis, benign nodules, and thyroid cancers.13 Hancock and colleagues described a series of patients with thyroid dysfunction among patients treated with irradiation with or without chemotherapy for Hodgkin disease at Stanford University.14 Of 1787 patients, 1677 received irradiation to the thyroid. At 26 years of follow-up, the actuarial risk of thyroid disease was 67%. Hypothyroidism developed in the majority of the patients (47%). The risk of Graves disease was 7 to 20 times higher than that for healthy subjects. The risk of thyroid cancer was 15.6 times the expected risk for healthy subjects. These data remind clinicians to monitor thyroid function closely in patients who have been treated with upper mantle or cervical irradiation. Similar results were noted in the Childhood Cancer Survivor Study with an evaluable cohort of 1791 Hodgkin disease survivors (including 959 males). Among patients with Hodgkin disease, a 50% risk of hypothyroidism was found 20 years from the time of diagnosis in persons treated with 45 Gy or more.15 The total dose of irradiation received has been shown to correlate with the incidence of hypothyroidism in many studies.15–15 However, controversy exists regarding the effect of age and gender at the time of irradiation.15–15
Radiation-induced thyroid dysfunction is thought to be caused by damage to small thyroid vessels and to the glandular capsule. Histomorphologic features that are described in such patients include focal and irregular follicular hyperplasia, hyalinization, and fibrosis beneath the vascular endothelium; lymphocytic infiltration; single and multiple adenomas; and thyroid carcinomas.13,14
A rare complication of neck irradiation is acute radiation thyroiditis, which is more commonly associated with therapeutic doses of radioiodine for thyroid diseases. Patients typically have symptoms of fever, pain in the anterior cervical region, and transient hyperthyroidism. Hyperthyroidism with a clinical picture that resembles Graves disease may be seen after neck irradiation for Hodgkin disease.14 The incidence is uncertain because of the small number of cases reported. The clinical picture is characterized by diffuse thyroid enlargement, suppressed TSH, high levels of thyroid hormones, and development of thyroid autoantibodies. Ophthalmopathy, with or without overt hyperthyroidism, may be seen and is thought to be related to autoantibodies, similar to that seen in persons with classic Graves disease.
Parathyroid Glands
Several studies link prior head and neck irradiation and hyperparathyroidism. Cohen and colleagues16 followed a cohort of patients who were treated with radiation to the tonsils before the age of 16 years. Among the 2923 patients, 32 were found to have clinical hyperparathyroidism—a 2.5- to 2.9-fold increase compared with the general population in the same age group. A long latency period (>25 years) occurs between exposure and the onset of hyperparathyroidism. Parathyroid adenomas were found in most of the patients in whom this complication develops.17 Clinical presentations vary from asymptomatic increases in serum parathormone levels and hypercalcemia to disabling metabolic bone disease or nephrolithiasis. Persons with a history of head and neck radiation should be monitored with calcium levels periodically (every 1 to 2 years) and indefinitely.
Role of Systemic Therapy
Effects of systemic chemotherapy on ovarian and testicular function are discussed in Chapter 60.
Hypothalamic-Pituitary Axis
In children, chemotherapeutic agents without any cranial irradiation may disrupt growth hormone (GH) secretion. Roman and colleagues18 studied growth and GH secretion in 60 children who were in complete remission after treatment with chemotherapy and surgery for solid tumors. They observed GH deficiency in 45% of those studied and found that the GHD group had received significantly higher doses of actinomycin D compared with the non-GHD group (P < .05). These investigators found no correlation with duration of treatment, length of follow-up, tumor type, sex, or age. Depending on the intensity of chemotherapy, significant height loss was detected in 40% to 70% of patients at 6-year follow-up. Adjuvant chemotherapy can also aggravate growth failure in children with brain tumors who receive craniospinal radiation.19 Rose and colleagues20 reported hypothalamic dysfunction in patients with non–central nervous system tumors who received chemotherapy but did not receive cranial irradiation and had no history of traumatic brain injury. Of 31 identified patients, GHD was identified in 15 (48%), central hypothyroidism was identified in 16 (52%), and pubertal abnormalities was identified in 10 (32%). GHD and hypothyroidism were coexistent in eight patients (26%). Overall, 81% had GHD, hypothyroidism, precocious puberty, or gonadotropin deficiency.
The syndrome of inappropriate antidiuretic hormone (SIADH) secretion is associated with various malignant tumors including certain primary brain tumors, hematologic malignancies, intrathoracic nonpulmonary cancers, skin tumors, gastrointestinal cancers, gynecologic cancer, breast and prostatic cancer, and sarcomas.21 SIADH may result from the effects of many chemotherapeutic agents, either by potentiation of antidiuretic hormone (ADH) effect or by increased ADH secretion. The most commonly implicated agents are vinca alkaloids and cyclophosphamide. The vinca alkaloids are reported to stimulate the central release of ADH from the neurohypophyseal system,22 whereas alkylating agents enhance renal tubular sensitivity to ADH.23 Regardless of the mechanism, the result is an increase in water reabsorption by the distal tubules of the kidney, leading to volume expansion and dilutional hyponatremia. Case reports also implicate platinum agents,24 vinorelbine,25 taxanes,26 and methotrexate.27 Clinically significant hyponatremia may occur with administration of these agents, and management requires fluid restriction and, at times, salt replacement.
Thyroid
Clinically evident thyroid dysfunction has rarely been associated with the use of standard chemotherapy agents. However, a growing body of literature points to the increased prevalence of endocrine dysfunction after bone marrow transplantation, following use of preparation regimens that do not include radiation. Thyroid dysfunction has been reported in as many as 50% of allogeneic bone marrow transplant recipients treated with busulfan and cyclophosphamide.28 Thyroid dysfunction may appear as low T3 syndrome (i.e., normal levels of free T4 and TSH and a below-normal level of free T3), chronic thyroiditis, and transient subclinical hyperthyroidism or hypothyroidism. Chemotherapy may potentiate radiation-induced damage to normal tissue.
Newer biological agents, tyrosine kinase inhibitors, and immune-based cancer treatments (e.g., ipilimumab, interferon-α, and interleukin-2) have been associated with various types of thyroid dysfunction.29, 30 Box 61-3 summarizes the well-described adverse effects of cancer therapies on thyroid function.
