[level-membership-for-pathology-category]38
Embryonal neuroepithelial neoplasms of the CNS
EMBRYONAL NEUROEPITHELIAL NEOPLASMS

 a tendency to disseminate through CSF pathways
a tendency to disseminate through CSF pathways
 a dominant population of small undifferentiated cells
a dominant population of small undifferentiated cells



In the WHO classification (2007), listed embryonal neuroepithelial neoplasms are:
 Medulloblastoma and its variants:
Medulloblastoma and its variants:
 CNS primitive neuroectodermal tumor (PNET) and its variants:
CNS primitive neuroectodermal tumor (PNET) and its variants:

 EMBRYONAL NEUROEPITHELIAL NEOPLASMS
EMBRYONAL NEUROEPITHELIAL NEOPLASMS
 Represents about 5% of intracranial primary neoplasms.
Represents about 5% of intracranial primary neoplasms.
 Represents about 20% of childhood CNS neoplasms.
Represents about 20% of childhood CNS neoplasms.
 Represents over 90% of childhood CNS PNETs.
Represents over 90% of childhood CNS PNETs.
 Can present at any age, but 60% occur in patients less than 10 years of age.
Can present at any age, but 60% occur in patients less than 10 years of age.
 Generally midline, filling the fourth ventricle.
Generally midline, filling the fourth ventricle.
 Belongs to the SHH molecular subgroup, when in the lateral cerebellar cortex.
Belongs to the SHH molecular subgroup, when in the lateral cerebellar cortex.

 Regarded as high-risk if a large cell or anaplastic pathological variant.
Regarded as high-risk if a large cell or anaplastic pathological variant.
 Has a 5-year survival of 80% in standard-risk patients.
Has a 5-year survival of 80% in standard-risk patients.
 Has a 5-year survival of 25% in high-risk patients.
Has a 5-year survival of 25% in high-risk patients.
 Therapies are associated with significant adverse effects, e.g. cognitive impairment.
Therapies are associated with significant adverse effects, e.g. cognitive impairment.










MACROSCOPIC APPEARANCES
Many embryonal neoplasms are circumscribed, pink or gray neoplasms, which may contain areas of hemorrhage, necrosis, or calcification (Fig. 38.1). CNS neuroblastomas and medulloepitheliomas sometimes contain cysts. All embryonal neoplasms have the capacity to invade the brain and spinal cord, and this will often be evident microscopically, if not macroscopically. However, infiltration occurs to a variable degree, and is rarely as diffuse as demonstrated by some astrocytic tumors.
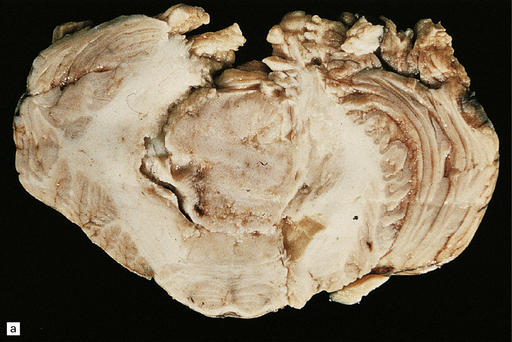
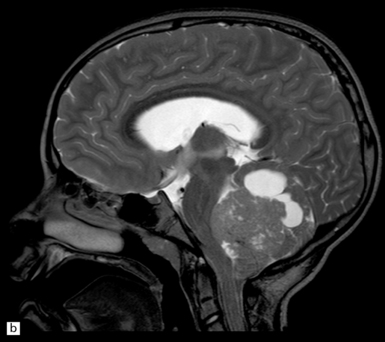
38.1 Medulloblastoma.
(a) A soft homogeneous mass destroys and occupies the fourth ventricle. (b) A sagittal midline MR image through the brain showing a medulloblastoma in the posterior fossa between the cerebellum and brain stem. (Dr N Sabin, St Jude Children’s Research Hospital.)
The texture of embryonal neoplasms varies. Some are soft, but some medulloblastomas in the lateral cerebellar cortex and some cerebral neuroblastomas tend to be firm because they contain areas of desmoplasia. Neoplastic cells occasionally metastasize through the CSF pathways (Fig. 38.2).
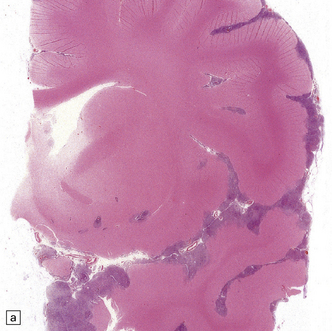
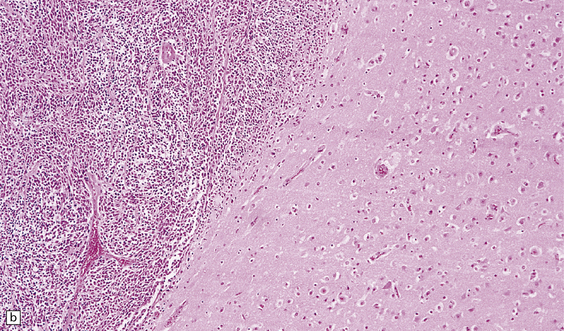
38.2 Supratentorial PNET in the subarachnoid space.
(a) There is an extensive infiltration of the subarachnoid space by basophilic cells from a PNET. Several distinct parenchymal deposits are also present. (b) From the same case, a mass of small cells fills the subarachnoid space and has begun to invade the pial surface of the cerebrum.
 FAMILIAL CANCER SYNDROMES FEATURING MEDULLOBLASTOMAS
FAMILIAL CANCER SYNDROMES FEATURING MEDULLOBLASTOMAS














 GENETIC ASPECTS OF MEDULLOBLASTOMA
GENETIC ASPECTS OF MEDULLOBLASTOMA
 Common chromosomal abnormalities include:
Common chromosomal abnormalities include:
• Chromosome 17 abnormalities (~60%)
• Isodicentric chromosome 17q (~35%)
 Gene-specific abnormalities include:
Gene-specific abnormalities include:
 Molecular subgroups derived from gene expression profiling have been classified into four main groups (see Table 38.1):
Molecular subgroups derived from gene expression profiling have been classified into four main groups (see Table 38.1):


MICROSCOPIC APPEARANCES
Medulloblastoma
The classic medulloblastoma is composed of isomorphic cells with a high nuclear:cytoplasmic ratio (Fig. 38.3). Sheets of hyperchromatic round or oval nuclei set against a neuropil-like matrix give a monotonous appearance, with scattered mitotic figures and apoptotic bodies in the background. Necrosis is variably present, but angiogenesis with endothelial hyperplasia is a rare feature in these neoplasms. Though frequently forming a mass in the fourth ventricle, the medulloblastoma is an invasive neoplasm. Its cells have a tendency to spread along the pial surface of the cerebellum, invading the underlying cortex in swathes. Infiltration of the leptomeninges can produce a striking desmoplasia (Fig. 38.3).
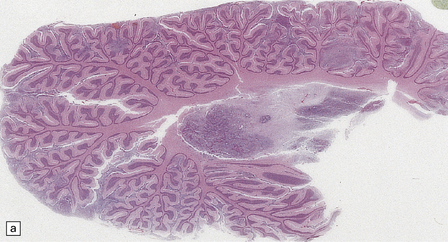
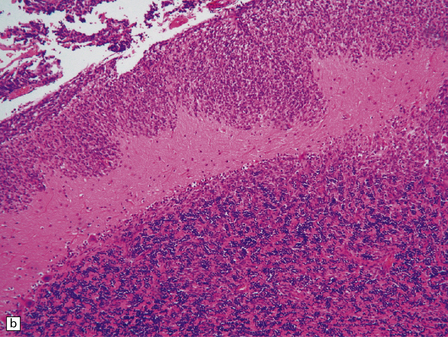
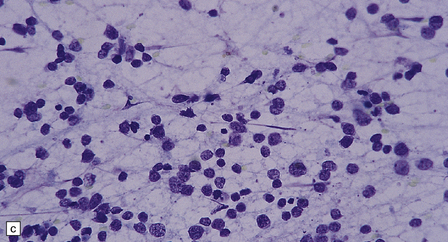
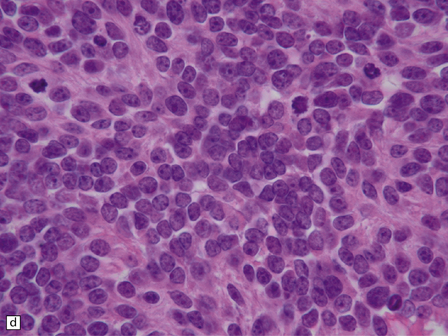
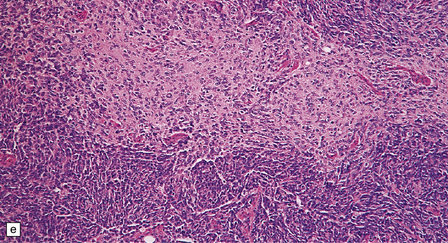
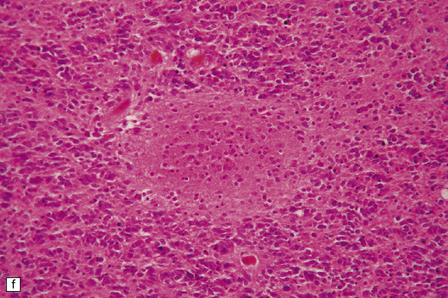
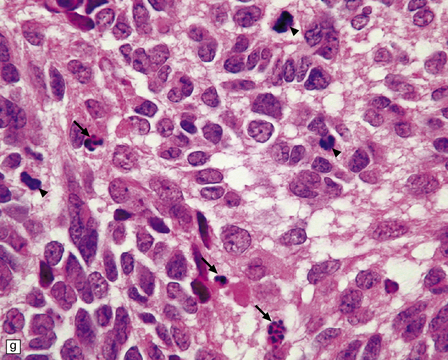
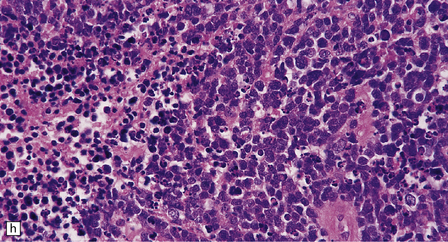
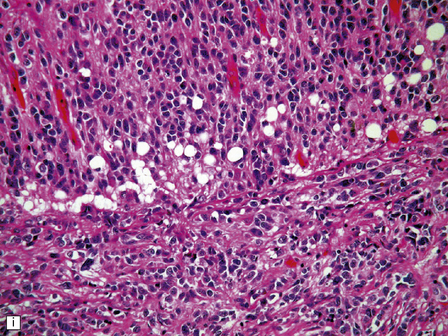
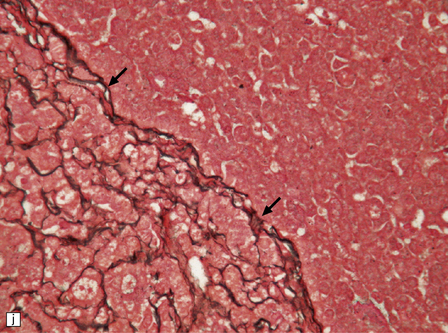
38.3 Medulloblastoma.
(a) Multiple tumor deposits and leptomeningeal spread of medulloblastoma cells are evident in this section of cerebellum. (b) Centripetal spread through the cerebellar cortex is a frequent finding when there is leptomeningeal disease. (c) This smear preparation shows scattered bare hyperchromatic nuclei. (d) Uniform small cells with round nuclei and a high density characterize the classic medulloblastoma. (e) A monotonous sheet of densely packed small cells is interrupted by an area with a reduced cell density. (f) Approximately 7% of classic medulloblastomas contain hypocellular nodules that contain neurocytic cells with a low growth fraction, but critically there is no internodular desmoplasia. (g) Abundant mitoses (arrowheads) and apoptotic bodies (arrows) are typical. (h) Micronecrosis. (i) Desmoplasia results when medulloblastoma cells spill out into the leptomeninges (bottom half of image). (j) The leptomeningeal desmoplasia (arrows) is demonstrated well by a reticulin preparation.
As with other CNS PNETs, the medulloblastoma has the capacity for divergent neuroepithelial differentiation (Fig. 38.4). Neuronal differentiation may take the form of rosettes that lack a central canal or capillary (Homer–Wright rosettes). A more obvious neuronal morphophenotype occurs in about 7% of classic medulloblastomas with the formation of nodules of neurocytic cells with a reduced growth fraction or regions containing both neurocytic and ganglion cells (ganglioneuroblastoma phenotype). These (biphasic) neoplasms do not show any desmoplasia. Groups of tumor cells without obvious neuronal morphology may show immunoreactivity for synaptophysin or neurofilament proteins. Ultrastructural examination reveals secretory granules in some cells. Medulloblastoma cells with a clear-cut astrocytic morphophenotype are very rare, but the cytoplasm of some neoplastic cells may be immunoreactive for GFAP. In searching for focal neuroepithelial differentiation, care should be taken to distinguish reactive astrocytes and entrapped native neurons from neoplastic cells (Fig. 38.4).
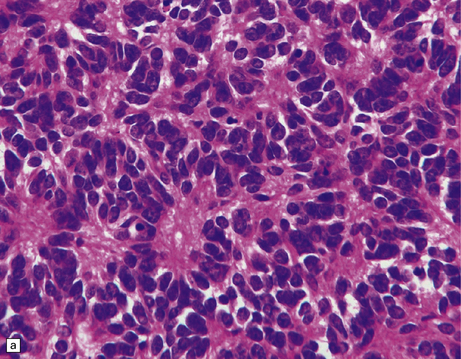
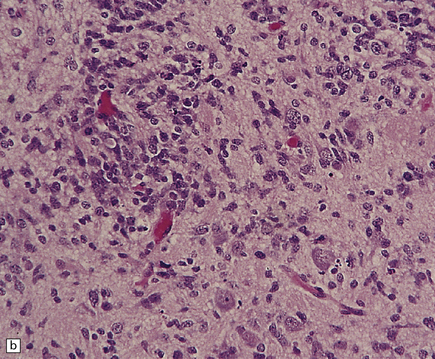
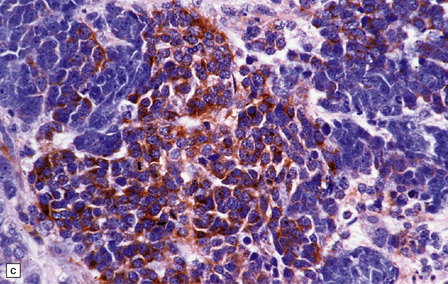
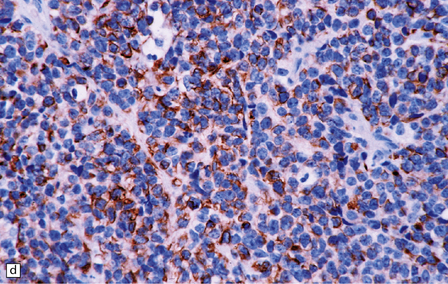
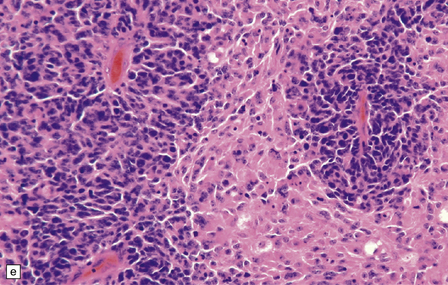
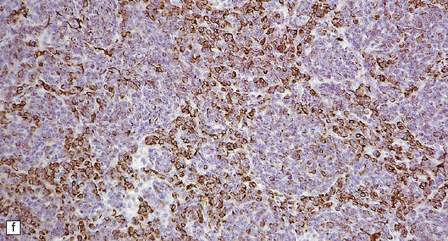
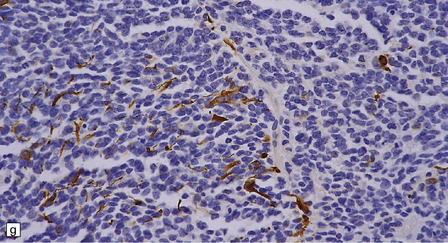
38.4 Neuroepithelial differentiation in a medulloblastoma.
(a) Rosette (Homer–Wright) formation is found mainly in classic medulloblastomas, but can sometimes coexist with an anaplastic phenotype. (b) Ganglion or neurocytic cells occur in about 5% of medulloblastomas.(c) A focal or widespread neuronal immunophenotype may be revealed with antibodies to synaptophysin or (rarely) (d) neurofilament proteins. (e) An astrocytic morphophenotype among groups of cells is a rare finding. (f) More frequent is a scattered population of GFAP-positive cells. (g) The GFAP-positive processes of intermingling reactive astrocytes are commonly found in embryonal tumors and should not be mistaken for glial differentiation.
The range of nuclear size in medulloblastomas is more extensive than might initially be apparent. Nuclei in a classic medulloblastoma have less than half the area of nuclei in the large cell variant, which is characterized by groups of cells with round nuclei and a prominent single nucleolus. In the large cell variant, neoplastic cells that show considerably more pleomorphism than those in classic medulloblastomas are interspersed between the defining groups of large cells, assuming a polyhedral form as they flatten themselves into a pavement-like arrangement (Fig. 38.5). The term anaplastic medulloblastoma has been attached to medulloblastomas with both this phenotype and features of increased cell turnover, such as a high mitotic count and abundant apoptosis. The anaplastic medulloblastoma forms a continuum with the large cell variant (Fig. 38.6), and both have a more aggressive biologic behavior than the classic medulloblastoma.
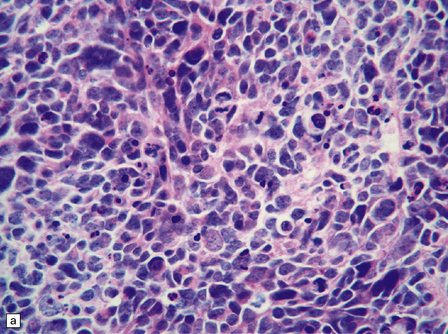
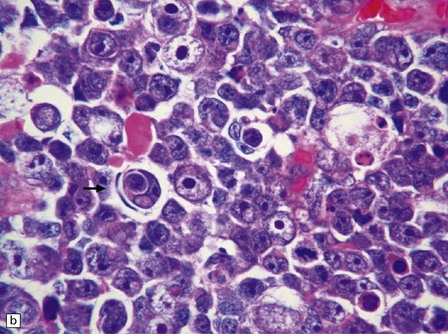
38.5 Anaplastic medulloblastoma.
(a) The anaplastic medulloblastoma is characterized by large cells, nuclear pleomorphism, and a high mitotic count. (b) Cell wrapping (arrow) is another characteristic feature. Note the scattered large cells with round nuclei and single nucleolus, demonstrating overlap with the large cell phenotype.
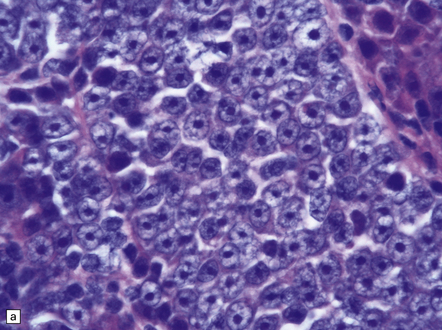
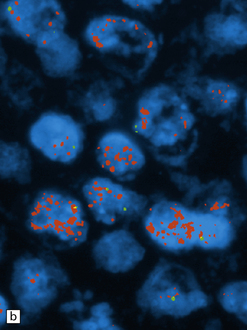
38.6 Large cell medulloblastoma.
(a) This variant combines areas with the anaplastic phenotype and groups of cells with large round nuclei and a single round nucleolus. (b)MYC amplification, as demonstrated here by interphase fluorescence in situ hybridization (red signals, MYC; green signals, chromosome 8p control probe), is often seen in large cell or anaplastic medulloblastomas that present with metastatic disease. MYC amplification is an indicator of poor outcome.
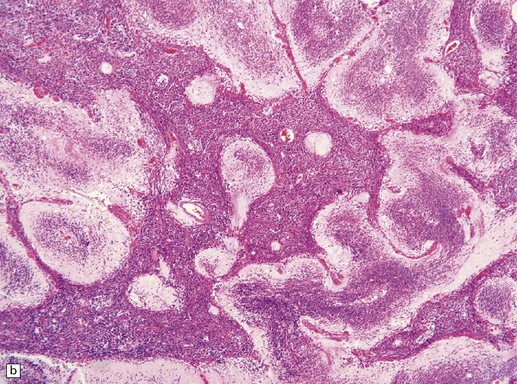
Focal desmoplasia is common in embryonal neuroepithelial neoplasms, but the desmoplastic variant of medulloblastoma is also characterized by distinctive reticulin-free nodules of neurocytic cells among the reticulin-rich internodular areas, in which the cells appear more pleomorphic (Fig. 38.7). Nodules are distinctive tumor microenvironments. Cells in nodules often have a lower nuclear:cytoplasmic ratio than surrounding cells, and their uniform appearance matches that of the central neurocytoma. Relative to internodular cells, those in nodules have a reduced growth fraction and a greater apoptotic rate. The histology of the desmoplastic variant merges with that of the paucinodular desmoplastic medulloblastoma, and with a distinctive subtype of desmoplastic tumor, the medulloblastoma with extensive nodularity (MBEN). Nodules are not only frequent in this tumor, but are larger than in the standard desmoplastic medulloblastoma, having oval or reniform shapes (Fig. 38.8). A neuronal immunophenotype is typical of nodules in the MBEN. The MBEN usually presents in infancy, and is associated with a good outcome.
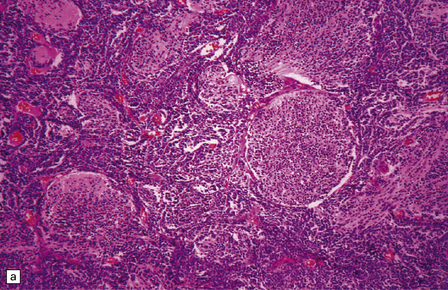
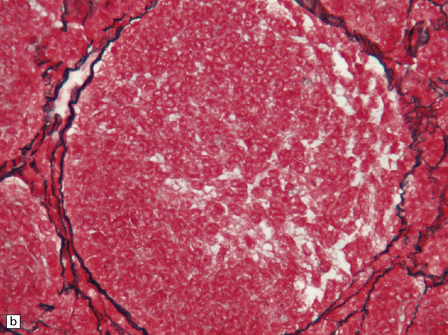
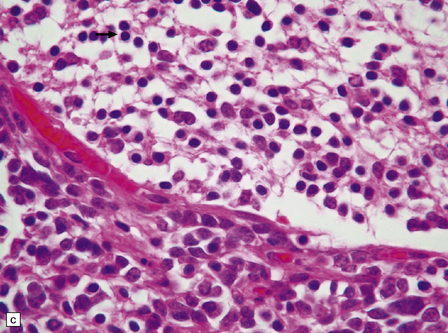
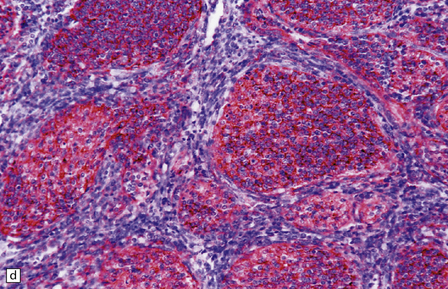
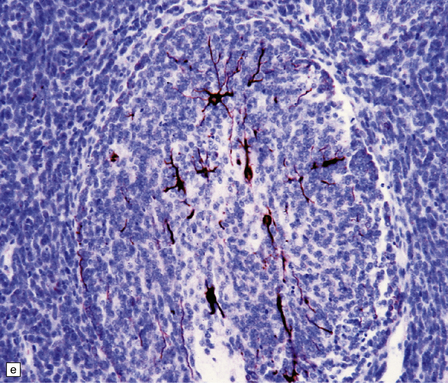
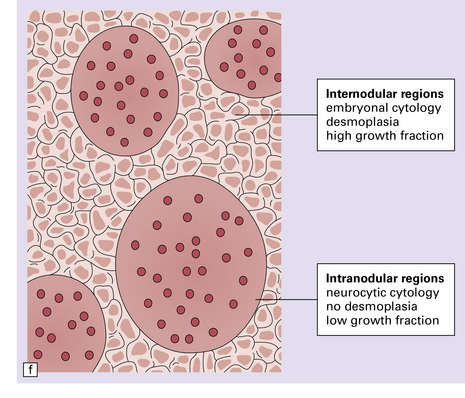
38.7 Desmoplastic medulloblastoma.
(a) All but very rare desmoplastic medulloblastomas have a nodular architecture, the desmoplasia occurring in the internodular regions (b). The desmoplasia around nodules stands out in a reticulin preparation. (c) Extranodular cells among strands of collagen show more nuclear pleomorphism than the intranodular cells with their round nuclei and neurocytic features (arrow). (d) An intranodular neurocytic morphology is usually accompanied by a neuronal immunophenotype, as shown here by immunopositivity for synaptophysin. (e) Reactive astrocytes are frequently centered on nodules. (f) Graphic showing differences in morphology between internodular and intranodular regions in the D/N medulloblastoma.
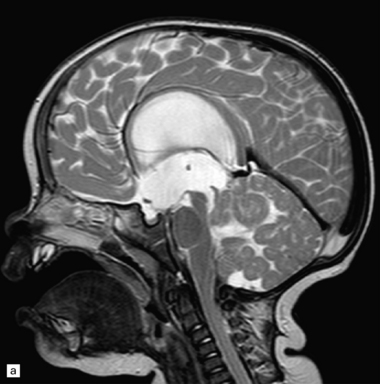
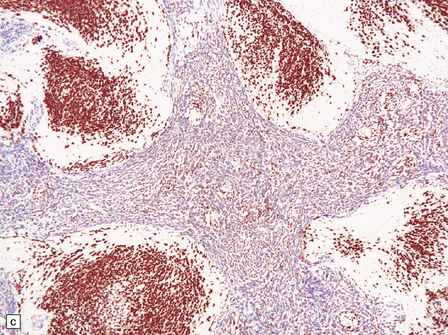
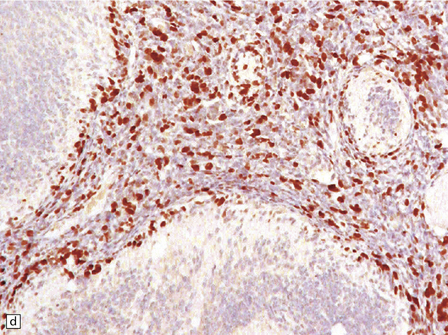
38.8 Medulloblastoma with extensive nodularity.
(a) Sagittal MRI image of the nodular architecture often apparent with this tumor. (Courtesy of Dr Noah Sabin, St Jude Children’s Research Hospital.) (b) This variant represents the ultimate expression of the nodular phenotype. (c) Intranodular cells have neurocytic features, including immunoreactivity for NEU-N. (d) The growth fraction of intranodular cells, as indicated by Ki-67 nuclear immunoreactivity, is very low compared with that of internodular cells.
Focal melanin production and evidence of striated muscle differentiation are exceptional features in medulloblastomas, giving rise to two distinctive subtypes of the classic neoplasm: medulloblastoma with melanotic differentiation and medulloblastoma with myogenic differentiation (Figs 38.9, 38.10).
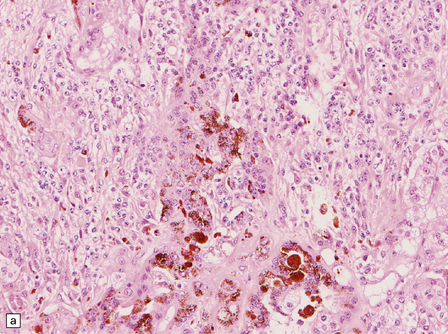
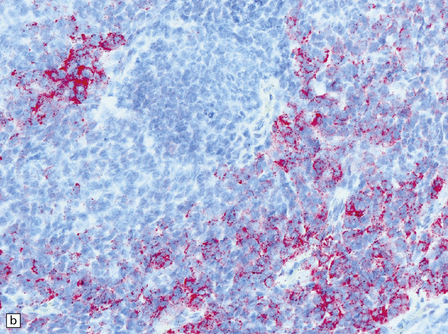
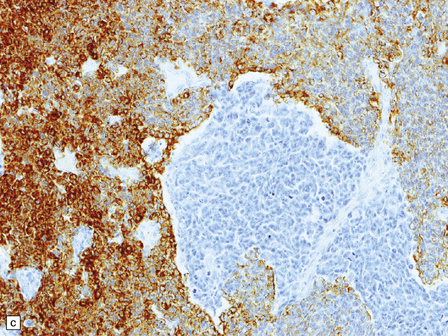
38.9 Medulloblastoma with melanotic differentiation.
(a) Cells that contain melanin and have an epithelioid morphology occur as a rare phenomenon in medulloblastomas that otherwise have a classic or anaplastic morphology. Sometimes, these cells can be hard to find, but are highlighted by immunoreactivities for HMB-45 (b) or cytokeratins (c).
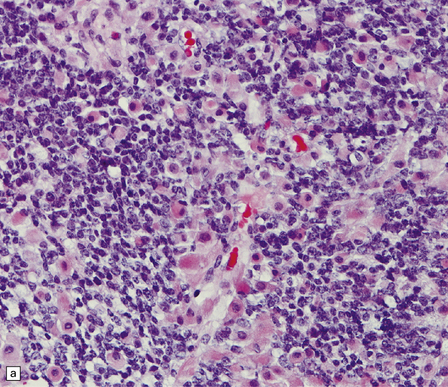
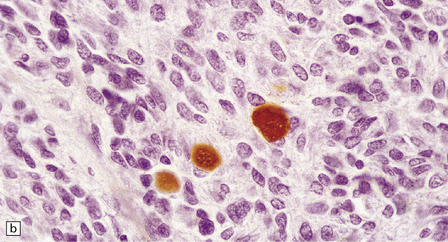
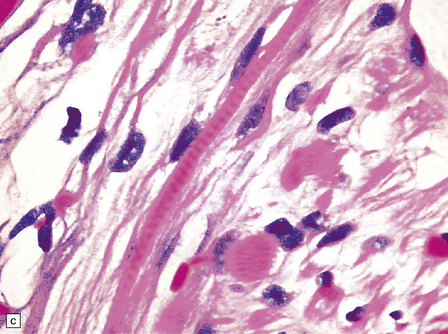
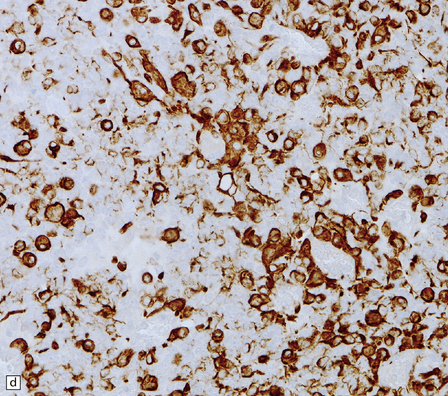
38.10 Medulloblastoma with myogenic differentiation.
(a) Scattered large cells have abundant eosinophilic cytoplasm. (b) These cells typically show immunoreactivity for myoglobin. (c) Muscle fibrils with cross-striations may occasionally manifest in this tumor. (d) Myogenic differentiation as demonstrated by immunoreactivity for desmin.
CNS (extracerebellar) PNET
The extracerebellar PNET and classic medulloblastoma have similar histologic appearances; both contain sheets of small cells with sparse cytoplasm, hyperchromatic nuclei, and a high mitotic count (Fig. 38.11). Dystrophic calcification is often found in supratentorial PNETs. The PNET may exhibit focal neuronal differentiation in the form of Homer Wright rosettes or scattered immunoreactivities for neurofilament proteins, class III β-tubulin, and synaptophysin. Immunohistochemistry may reveal coexisting GFAP-positive cells, providing evidence of divergent neuroepithelial differentiation. Desmoplasia may be found in PNETs, particularly if there is invasion of the leptomeninges.
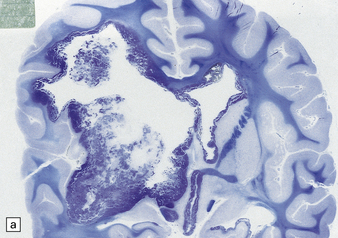
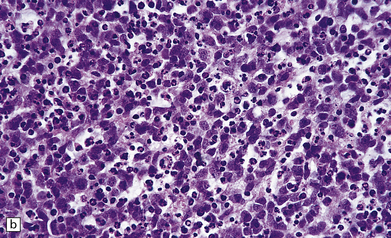
38.11 Extracerebellar PNET.
(a) This supratentorial PNET has destroyed a large part of one cerebral hemisphere and has spread beneath the ependyma. (b) A sheet of uniform cells with hyperchromatic nuclei contains abundant apoptotic bodies.
A cerebral embryonal neoplasm that shows focal neurocytic differentiation is sometimes referred to as a central neuroblastoma, rather than PNET. When this differentiation amounts to the production of scattered neoplastic cells with the cytologic features of small or large neurons, then the term ganglioneuroblastoma is used (Fig. 38.12).
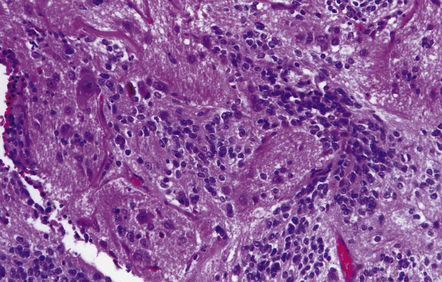
Ependymoblastoma
The ependymoblastic rosette is the key diagnostic feature of ependymoblastomas, which like other embryonal neuroepithelial neoplasms consist mainly of undifferentiated small cells (Fig. 38.13). Ependymoblastic rosettes are composed of multiple layers of small cells with hyperchromatic nuclei that surround a circular or elongated lumen. The lumen has a prominent internal limiting membrane, and mitotic figures are readily found in the rosettes. Ependymoblastic rosettes are not specific for PNETs; they occur rarely in atypical teratoid/rhabdoid tumors and in a tumor termed ‘embryonal tumor with abundant neuropil and true (ependymoblastic) rosettes’. The latter is now the most common setting for ependymoblastic rosettes, but this diagnosis has yet to be incorporated in the WHO classification of nervous system tumors. In time, this tumor is likely to supplant the term ‘ependymoblastoma’.
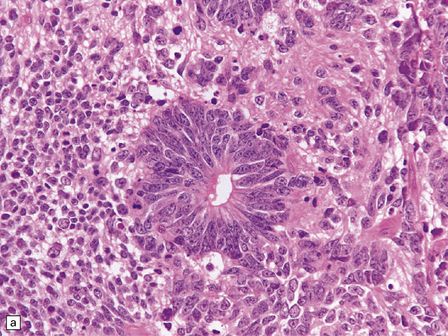
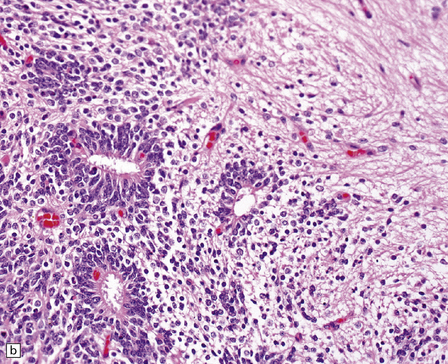
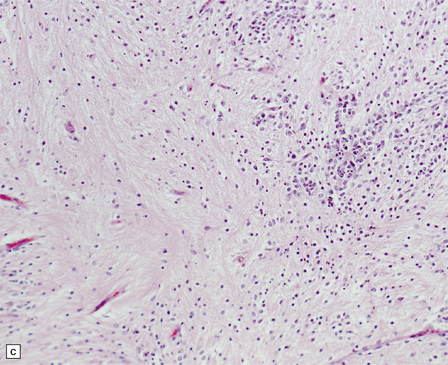
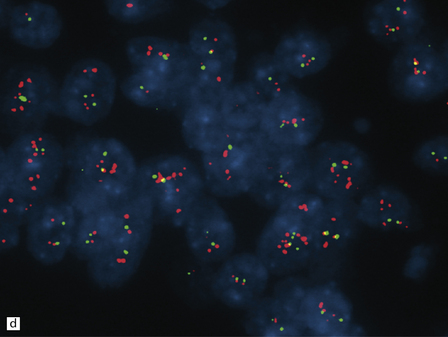
38.13 Embryonal tumor with abundant neuropil and true (ependymoblastic) rosettes (ETANER).
(a) Undifferentiated PNET-like embryonal cells surround an ependymoblastic rosette. This is the classic ‘ependymoblastoma’ phenotype. (b) Several rosettes are arranged against a neuropil-like matrix in this ETANER. (c) ETANERs are characterized by large regions of neuropil-like matrix, in which just a few neurocytic or ganglion cells are embedded. (d) ETANERs demonstrate focal amplification at the C19MC miRNA cluster on 19q13 (iFISH – red signal probe 19q13; green signal probe 19p).
Medulloepithelioma
The defining histologic attribute of the medulloepithelioma is an arrangement of tightly-packed multilayered cells into tubules that resemble embryonic neural tube. Convoluted ribbons and papillary structures may also be seen (Fig. 38.14). The tubules have a periodic acid–Schiff (PAS)-positive external basement membrane, and, in some cases, PAS-positive material lines their luminal aspect. The neoplastic cells have hyperchromatic nuclei and very scanty cytoplasm, and the mitotic count is high.
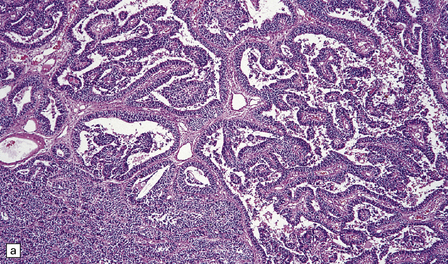
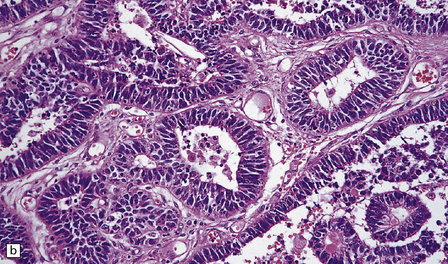
38.14 Medulloepithelioma.
(a) A papillary architecture is sometime seen in medulloepitheliomas, bringing choroid plexus carcinoma into the differential diagnosis. (b) Tubules of cells with hyperchromatic nuclei mimic primitive neural tube, and are characteristic of medulloepithelioma.
 DIFFERENTIAL DIAGNOSIS OF CNS EMBRYONAL NEOPLASMS
DIFFERENTIAL DIAGNOSIS OF CNS EMBRYONAL NEOPLASMS





 GENETIC ASPECTS OF AT/RT
GENETIC ASPECTS OF AT/RT AT/RT and other malignant rhabdoid tumors are associated with mutations of the SMARCB1 tumor suppressor gene on chromosome 22q, which codes for a subunit (BAF47, also known as integrase interactor 1 protein/INI1) of the SWI/SNF chromatin remodeling protein complex.
AT/RT and other malignant rhabdoid tumors are associated with mutations of the SMARCB1 tumor suppressor gene on chromosome 22q, which codes for a subunit (BAF47, also known as integrase interactor 1 protein/INI1) of the SWI/SNF chromatin remodeling protein complex.



• hemizygous deletion and mutation (including intragenic deletion) of SMARCB1
• homozygous deletion of SMARCB1
• loss of heterozygosity at the SMARCB1 locus plus SMARCB1 mutation
 The following tumors, which could involve the CNS, may also show loss of BAF47/INI1 expression:
The following tumors, which could involve the CNS, may also show loss of BAF47/INI1 expression:
Atypical teratoid/rhabdoid tumor (ATRT)
Cytologic polymorphism in an embryonal neoplasm often alerts the pathologist to the diagnostic possibility of an ATRT (Fig. 38.15). Small undifferentiated neuroepithelial cells with a high nuclear:cytoplasmic ratio occupy some areas of the ATRT, but cells elsewhere often have more cytoplasm and show more nuclear pleomorphism than expected in a typical PNET. Groups of classic rhabdoid cells with abundant eosinophilic cytoplasm and eccentrically placed nuclei can sometimes be found in the ATRT, and there may be large cells showing considerable nuclear pleomorphism. The mitotic count is high, and necrosis is common. Microvascular proliferation in an ATRT very rarely shows the endothelial proliferation characteristic of some gliomas.
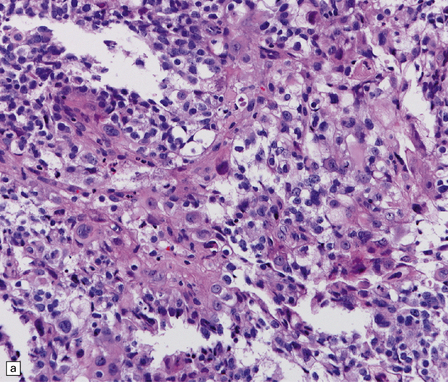
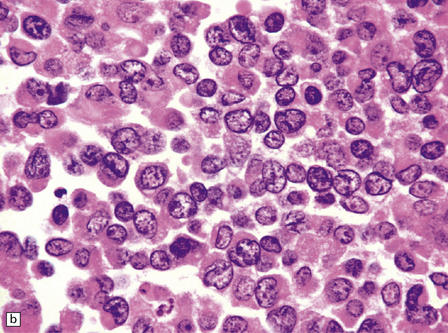
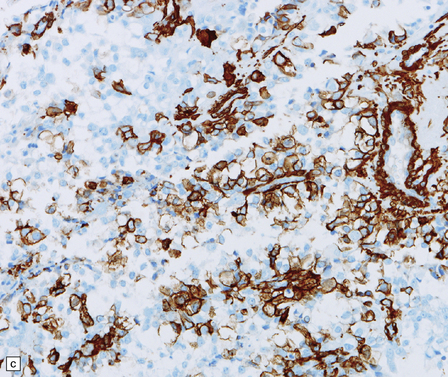
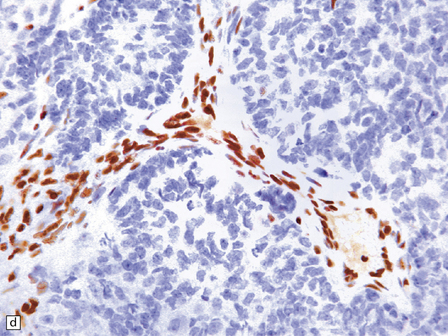
38.15 Atypical teratoid/rhabdoid tumor (AT/RT).
(a) This tumor’s cytology is more polymorphous than that of other embryonal tumors and often includes clusters of rhabdoid cells (b). By immunohistochemistry, AT/RTs express multiple epithelioid and mesenchymal markers, including smooth muscle actin (c), as well as neuroepithelial markers. (d) Tumor cells, but not normal cells in blood vessels, fail to express the SMARCB1 gene product (BAF47/INI1).
The diagnosis of ATRT is made using immunohistochemistry with an antibody to the SMARCB1 gene product (INI1), which is a subunit of the SWI/SNF chromatin remodeling complex. In ATRTs, immunoreactivity for INI1 is lost in tumor cells, but preserved in normal tissue elements, such as blood vessels, which act as an internal positive control (Fig. 38.15).
REFERENCES
Cho, Y.J., Tsherniak, A., Tamayo, P., et al. Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol.. 2011;29:1424–1430.
Eberhart, C.G., Kepner, J.L., Goldthwaite, P.T., et al. Histopathologic grading of medulloblastomas: a Pediatric Oncology Group study. Cancer.. 2002;94:552–560.
Ellison, D.W. Childhood medulloblastoma: novel approaches to the classification of a heterogeneous disease. Acta Neuropathol.. 2010;120:305–316.
Ellison, D.W., Dalton, J., Kocak, M., et al. Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol.. 2011;121:381–396.
Ellison, D.W., Kocak, M., Dalton, J., et al. Definition of disease-risk stratification groups in childhood medulloblastoma using combined clinical, pathologic, and molecular variables. J Clin Oncol.. 2011;29:1400–1407.
Ellison, D.W., Onilude, O.E., Lindsey, J.C., et al. beta-Catenin status predicts a favorable outcome in childhood medulloblastoma. J Clin Oncol.. 2005;23:7951–7957.
Fernandez-L, A., Squatrito, M., Northcott, P., et al. Oncogenic YAP promotes radioresistance and genomic instability in medulloblastoma through IGF2-mediated Akt activation. Oncogene. 2011. [[Epub ahead of print]].
Giangaspero, F., Perilongo, G., Fondelli, M.P., et al. Medulloblastoma with extensive nodularity: a variant with favorable prognosis. J Neurosurg.. 1999;91:971–977.
Giangaspero, F., Rigobello, L., Badiali, M., et al. Large-cell medulloblastomas. A distinct variant with highly aggressive behavior. Am J Surg Pathol.. 1992;16:687–693.
Gibson, P., Tong, Y., Robinson, G., et al. Subtypes of medulloblastoma have distinct developmental origins. Nature.. 2010;468:1095–1099.
Gilbertson, R.J., Ellison, D.W. The origins of medulloblastoma subtypes. Annu Rev Pathol.. 2008;3:341–365.
Han, Y.G., Kim, H.J., Dlugosz, A.A., et al. Dual and opposing roles of primary cilia in medulloblastoma development. Nat Med.. 2009;15:1062–1065.
McManamy, C.S., Pears, J., Weston, C.L., et al. Nodule formation and desmoplasia in medulloblastomas-defining the nodular/desmoplastic variant and its biological behavior. Brain Pathol.. 2007;17:151–164.
Northcott, P.A., Korshunov, A., Witt, H., et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol.. 2011;29:1408–1414.
Parsons, D.W., Li, M., Zhang, X., et al. The genetic landscape of the childhood cancer medulloblastoma. Science.. 2011;331:435–439.
Phillips, C.L., Miles, L., Jones, B.V., et al. Medulloblastoma with melanotic differentiation: case report and review of the literature. J Neurooncol.. 2011;103:759–764.
Remke, M., Hielscher, T., Northcott, P.A., et al. Adult medulloblastoma comprises three major molecular variants. J Clin Oncol.. 2011;29:2717–2723.
Rorke, L.B., Trojanowski, J.Q., Lee, V.M., et al. Primitive neuroectodermal tumors of the central nervous system. Brain Pathol.. 1997;7:765–784.
Roussel, M.F., Hatten, M.E. Cerebellum development and medulloblastoma. Curr Top Dev Biol.. 2011;94:235–282.
Rutkowski, S., von Hoff, K., Emser, A., et al. Survival and prognostic factors of early childhood medulloblastoma: an international meta-analysis. J Clin Oncol.. 2010;28:4961–4968.
Sachdeva, M.U., Vankalakunti, M., Rangan, A., et al. The role of immunohistochemistry in medullomyoblastoma: a case series highlighting divergent differentiation. Diagn Pathol.. 2008;3:18.
Taylor, M.D., Mainprize, T.G., Rutka, J.T. Molecular insight into medulloblastoma and central nervous system primitive neuroectodermal tumor biology from hereditary syndromes: a review. Neurosurgery.. 2000;47:888–901.
Taylor, M.D., Northcott, P.A., Korshunov, A., et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2011;123:465–472.
Behdad, A., Perry, A. Central nervous system primitive neuroectodermal tumors: a clinicopathologic and genetic study of 33 cases. Brain Pathol.. 2010;20:441–450.
Biegel, J.A. Molecular genetics of atypical teratoid/rhabdoid tumor. Neurosurg Focus.. 2006;20:E11.
Burger, P.C., Yu, I.T., Tihan, T., et al. Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a Pediatric Oncology Group study. Am J Surg Pathol.. 1998;22:1083–1092.
Chacko, G., Chacko, A.G., Dunham, C.P., et al. Atypical teratoid/rhabdoid tumor arising in the setting of a pleomorphic xanthoastrocytoma. J Neurooncol.. 2007;84:217–222.
Cruz-Sanchez, F.F., Haustein, J., Rossi, M.L., et al. Ependymoblastoma: a histological, immunohistological and ultrastructural study of five cases. Histopathology.. 1988;12:17–27.
Dunham, C., Sugo, E., Tobias, V., et al. Embryonal tumor with abundant neuropil and true rosettes (ETANTR): report of a case with prominent neurocytic differentiation. J Neurooncol.. 2007;84:91–98.
Eaton, K.W., Tooke, L.S., Wainwright, L.M., et al. Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer.. 2011;56:7–15.
Gessi, M., Setty, P., Bisceglia, M., et al. Supratentorial primitive neuroectodermal tumors of the central nervous system in adults: molecular and histopathologic analysis of 12 cases. Am J Surg Pathol.. 2011;35:573–582.
Hasselblatt, M., Gesk, S., Oyen, F., et al. Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol.. 2011;35:933–935.
Judkins, A.R., Ellison, D.W. Ependymoblastoma: dear, damned, distracting diagnosis, farewell!*. Brain Pathol.. 2010;20:133–139.
Karch, H., Urich, S.B. Medulloepithelioma: definition of an entity. J Neuropathol Exp Neurol.. 1972;31:27–53.
Kleinschmidt-DeMasters, B.K., Birks, D.K., Aisner, D.L., et al. Atypical teratoid/rhabdoid tumor arising in a ganglioglioma: genetic characterization. Am J Surg Pathol.. 2011;35:1894–1901.
Li, M., Lee, K.F., Lu, Y., et al. Frequent amplification of a chr19q13.41 microRNA polycistron in aggressive primitive neuroectodermal brain tumors. Cancer Cell.. 2009;16:533–546.
Li, M.H., Bouffet, E., Hawkins, C.E., et al. Molecular genetics of supratentorial primitive neuroectodermal tumors and pineoblastoma. Neurosurg Focus.. 2005;19:E3.
Molloy, P.T., Yachnis, A.T., Rorke, L.B., et al. Central nervous system medulloepithelioma: a series of eight cases including two arising in the pons. J Neurosurg.. 1996;84:430–436.
Mork, S.J., Rubinstein, J.L. Ependymoblastoma. A reappraisal of a rare embryonal tumor. Cancer.. 1985;55:1536–1542.
Perry, A., Miller, C.R., Gujrati, M., et al. Malignant gliomas with primitive neuroectodermal tumor-like components: a clinicopathologic and genetic study of 53 cases. Brain Pathol.. 2009;19:81–90.
Rorke, L.B., Packer, R.J., Biegel, J.A. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg.. 1996;85:56–65.
Rubinstein, L.J. The definition of the ependymoblastoma. Arch Pathol.. 1970;90:35–45.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]38
Embryonal neuroepithelial neoplasms of the CNS
EMBRYONAL NEUROEPITHELIAL NEOPLASMS
a tendency to disseminate through CSF pathways
a dominant population of small undifferentiated cells
In the WHO classification (2007), listed embryonal neuroepithelial neoplasms are:
Medulloblastoma and its variants:
CNS primitive neuroectodermal tumor (PNET) and its variants:
EMBRYONAL NEUROEPITHELIAL NEOPLASMS
Represents about 5% of intracranial primary neoplasms.
Represents about 20% of childhood CNS neoplasms.
Represents over 90% of childhood CNS PNETs.
Can present at any age, but 60% occur in patients less than 10 years of age.
Generally midline, filling the fourth ventricle.
Belongs to the SHH molecular subgroup, when in the lateral cerebellar cortex.
Regarded as high-risk if a large cell or anaplastic pathological variant.
Has a 5-year survival of 80% in standard-risk patients.
Has a 5-year survival of 25% in high-risk patients.
Therapies are associated with significant adverse effects, e.g. cognitive impairment.
MACROSCOPIC APPEARANCES
Many embryonal neoplasms are circumscribed, pink or gray neoplasms, which may contain areas of hemorrhage, necrosis, or calcification (Fig. 38.1). CNS neuroblastomas and medulloepitheliomas sometimes contain cysts. All embryonal neoplasms have the capacity to invade the brain and spinal cord, and this will often be evident microscopically, if not macroscopically. However, infiltration occurs to a variable degree, and is rarely as diffuse as demonstrated by some astrocytic tumors.
38.1 Medulloblastoma.
(a) A soft homogeneous mass destroys and occupies the fourth ventricle. (b) A sagittal midline MR image through the brain showing a medulloblastoma in the posterior fossa between the cerebellum and brain stem. (Dr N Sabin, St Jude Children’s Research Hospital.)
The texture of embryonal neoplasms varies. Some are soft, but some medulloblastomas in the lateral cerebellar cortex and some cerebral neuroblastomas tend to be firm because they contain areas of desmoplasia. Neoplastic cells occasionally metastasize through the CSF pathways (Fig. 38.2).
38.2 Supratentorial PNET in the subarachnoid space.
(a) There is an extensive infiltration of the subarachnoid space by basophilic cells from a PNET. Several distinct parenchymal deposits are also present. (b) From the same case, a mass of small cells fills the subarachnoid space and has begun to invade the pial surface of the cerebrum.
FAMILIAL CANCER SYNDROMES FEATURING MEDULLOBLASTOMAS
GENETIC ASPECTS OF MEDULLOBLASTOMA
Common chromosomal abnormalities include:
• Chromosome 17 abnormalities (~60%)
• Isodicentric chromosome 17q (~35%)
Gene-specific abnormalities include:
Molecular subgroups derived from gene expression profiling have been classified into four main groups (see Table 38.1):
