[level-membership-for-anesthesiology-category]
Embolic Disorders
Paloma Toledo MD, MPH, Andrew M. Malinow MD
Chapter Outline
Embolic disease during pregnancy includes amniotic fluid embolism, venous thromboembolism, and venous air embolism. Each of these entities varies in its incidence, clinical course, and consequences. Embolic events account for almost one fifth of all maternal deaths in the United States.1 Early recognition, diagnosis, and treatment are necessary to reduce associated morbidity and to avoid mortality.
Amniotic Fluid Embolism
Death attributable to an amniotic fluid embolism (AFE) was first reported by Meyer in 1926.2 Early reports described a syndrome of fatal peripartum shock attributed to emboli of amniotic fluid mechanically obstructing the maternal pulmonary circulation.3 Although the pathophysiology of this disease remains poorly understood, current evidence suggests that emboli alone are insufficient to precipitate this infrequent, idiosyncratic, and devastating condition. Rather, fetal material in the maternal circulation has the potential to trigger a massive cascade of inflammatory and hemostatic reactions that culminate in cardiopulmonary collapse and disseminated intravascular coagulation.
Epidemiology
The incidence of amniotic fluid embolism is difficult to establish because (1) AFE is a diagnosis of exclusion, (2) there is no universally accepted definition for identifying cases of AFE, and (3) differing ascertainment methods yield divergent rates of AFE. Registry data from the United Kingdom suggest an event rate between 0.8 and 2 per 100,000 deliveries.4,5 Cross-sectional analyses of administrative data in Australia and the United States suggest higher rates—3.3 and 7.7 per 100,000 deliveries, respectively.6,7 A systematic review published in 2009 estimated that the pooled incidence of AFE in North America was approximately 1 : 15,200 (95% confidence interval [CI], 1 : 13,900 to 1 : 16,700), whereas the incidence in Europe was three times lower at 1 : 53,800 (95% CI, 1 : 48,800 to 1 : 59,900).8 This heterogeneity likely reflects variations in ascertainment procedures, rather than true differences by continent of origin. One limitation of analyses of secondary databases is that the diagnosis of AFE may not have been validated. For example, one regional surveillance system in Australia has developed the capacity to systematically review records for all cases identified from administrative data. By only counting those women who experienced one of the cardinal symptoms of AFE, with no other potential explanation, the reported incidence decreased from 6.3 to 3.3 cases per 100,000 pregnancies.9
Risk Factors
Maternal demographic factors such as older age and race or ethnicity have been associated with AFE in population-based studies.5,7 Other obstetric factors such as abnormal placentation, placental abruption, eclampsia, multiple gestation, induction of labor, artificial or spontaneous rupture of membranes, and operative delivery have also been associated with AFE.5,7,10–12 Because nonreassuring fetal heart rate (FHR) tracings can complicate AFE in labor, cesarean delivery may be a consequence, rather than cause, of intrapartum AFE. Nonetheless, a strong association between cesarean birth and postpartum AFE persists among women in the United Kingdom Obstetric Surveillance System (UKOSS) dataset (adjusted odds ratio, 8.8).5 The proportion of excess postpartum AFE events associated with cesarean delivery (the population-proportional attributable risk for cesarean delivery) from the UKOSS dataset was estimated to be 62%.5
Pathophysiology
In 1941, Drs. Steiner and Lushbaugh, two pathologists from the University of Chicago, described a case series of eight autopsies after fatal intrapartum shock.3 Examination of lung tissue from these cases revealed embolic material of squamous cells, mucin, meconium, and amorphous eosinophilic material.3 Because all of these patients were described as having tumultuous labors with stronger than usual uterine contractions, it was presumed that the forceful contractions loosened or tore the placenta and forced the emboli into the maternal circulation.3 Yet, periods of uterine tachysystole are the least likely times for maternoplacental exchange of embolic material to occur as uterine blood flow ceases.13 The tachysystole is probably a result of endogenous norepinephrine release and, therefore, is likely temporally related to, but not causative for, embolic material transfer.13
Large intravenous boluses of human meconium suspended in human amniotic fluid can precipitate cardiovascular collapse in rabbits and dogs,3 but injection of autologous amniotic fluid fails to reproduce the AFE syndrome in many animal models.14 The passage of fetal squames, lanugo hair, and mucin into the maternal pelvic vasculature appears to be a common event at term,15 and fetal material has been identified in pulmonary arterial samples aspirated from critically ill women who did not have AFE.16–18
The exact trigger for the reaction in women with AFE is not known but may be a rare pathologic fetal antigen or a common antigen presented in an unusual way—in amount, timing, or frequency of entry into the maternal circulation.19 AFE appears to be a systemic inflammatory response associated with the inappropriate release of endogenous inflammatory mediators.20 Whatever the trigger, several maternal endogenous mediators appear to play an important role in the initial reaction, including arachidonic acid metabolites (i.e., thromboxane, prostaglandins, leukotrienes, endothelins).20
Approximately 40% of women in a United States national AFE registry had a history of allergy or atopy, leading some authors to suggest an anaphylactoid mechanism.12 In support of this theory, the symptoms of AFE could be blocked by the administration of a leukotriene inhibitor in a rabbit model.21 However, a series of case reports now suggests that levels of tryptase and histamine are not dramatically or universally elevated among women experiencing the AFE syndrome.22,23 Although mast cell degranulation may contribute to the pathophysiology of AFE, this effect may be a secondary product of the inflammatory cascade, rather than causal, and thus the term anaphylactoid syndrome of pregnancy may be a misnomer.23
Other immune-mediated mechanisms have also been implicated in AFE. A case-control study demonstrated low levels of the complement components C3 and C4 among women diagnosed with AFE compared with controls, suggesting that complement activation may also contribute to the inflammatory cascade.22 Whether complement plays a primary19 or secondary role24 in the AFE syndrome is unknown. A heat-stable pressor agent in meconium has been suggested as another possible mediator of AFE; in a goat model this agent caused a circulatory response similar to that seen in human AFE.25
Alternatively, the hemodynamic consequences of AFE could derive from activation of the coagulation system, mediated through platelet activation. Fetal squamous cells and syncytiotrophoblasts display high concentrations of tissue factor and phosphatidylserine.26–28 Tissue factor irreversibly aggregates platelets,29 leading to platelet degranulation, which releases thromboxane, serotonin, and additional mediators that amplify the immunologic systems. Serotonin is a potent pulmonary vasoconstrictor that produces vasodilation in the systemic vasculature and may lead to early right-sided heart failure in the AFE syndrome.27,30,31
Coagulopathy develops in the majority of women who survive the initial cardiovascular collapse.12,32 One proposed mechanism involves tissue factor. As pregnancy progresses, increasing amounts of tissue factor, a potent procoagulant, accumulates in the amniotic fluid.27,28 Tissue factor binds factor VII, thus activating the extrinsic pathway and triggering clotting by activating factor X, with the subsequent development of a consumptive coagulopathy.27 A second possible mechanism is that amniotic fluid has a thromboplastin-like effect, which induces platelet aggregation, releases platelet factor III, and activates the clotting cascade.33 Other components of amniotic fluid, the amniochorion, and the placenta have also been implicated in contributing to the coagulopathy. Uterine atony, whether due to a specific myometrial depressant or from uterine hypoperfusion, may exacerbate hemorrhage and consumptive coagulopathy in AFE.34
There is conflicting evidence regarding whether the bleeding seen in AFE is due primarily to a consumptive coagulopathy versus massive fibrinolysis. In one in vitro study, investigators added 10 to 60 µL of clear autologous amniotic fluid to 330 µL of blood from volunteers undergoing uneventful cesarean delivery.35 Thromboelastographic analysis of the specimens revealed accelerated clot formation but no evidence of fibrinolysis. However, because of the rapid and severe hypofibrinogenemia observed during AFE, some investigators have suggested that severe hyperfibrinolysis is also present.36 Future work is necessary to further refine our understanding of the coagulopathy that accompanies AFE.
Clinical Presentation
Whereas the classic presentation of AFE includes acute respiratory distress, cardiovascular collapse, and coagulopathy near the time of delivery, a broad range of AFE syndromes have been described in situations in which other diagnoses were excluded (Table 39-1).12,20,37
TABLE 39-1
Features of Amniotic Fluid Embolism at Presentation
| Percent Exhibiting Feature (n = 60)* |
Percent Exhibiting Feature as First Symptom or Sign (n = 60)* |
|
| Maternal hemorrhage† | 65% | 2% |
| Hypotension† | 63% | 8% |
| Shortness of breath† | 62% | 20% |
| Coagulopathy† | 62% | 0% |
| Premonitory symptoms† (i.e., restlessness, agitation, numbness, tingling) | 47% | 30% |
| Acute fetal compromise | 43% | 20% |
| Cardiac arrest | 40% | 8% |
| Dysrhythmias | 27% | 5% |
| Seizure | 15% | 7% |
* Some women had multiple features; therefore, the totals are greater than 100%.
† Cardinal feature of amniotic fluid embolism. Twenty-seven percent of women experienced at least four of the five cardinal features.
Data from Knight M, Tuffnell D, Brocklehurst P, et al. Incidence and risk factors for amniotic-fluid embolism. Obstet Gynecol 2010; 115:910-7.
Currently, the UKOSS provides the most comprehensive prospective surveillance for amniotic fluid embolism in the world. All hospitals in the United Kingdom with a consultant-led maternity unit report all suspected cases of AFE, along with monthly delivery volume data, for central review and reporting.5,38 Cases of AFE are defined using either clinical criteria that include acute hypotension, cardiac arrest, acute hypoxemia, and/or coagulopathy in the absence of any other potential explanation for the symptoms and signs observed, or pathologic evidence indicating the presence of fetal squames or hair in the maternal lungs. All women in the UKOSS registry demonstrated at least one cardinal feature of AFE, including shortness of breath, hypotension, coagulopathy, maternal hemorrhage, or premonitory symptoms (see Table 39-1).5
A national registry in the United States accepted voluntary submissions of medical records for patients with suspected AFE beginning in 1988. The diagnosis of AFE was confirmed based on meeting all entry criteria outlined in Box 39-1. Data for 46 confirmed cases of AFE were last published in 1995.12
AFE most often occurs during labor; intrapartum events comprise 56% of cases in the UKOSS registry5 and 70% in the U.S. registry.12 In the U.S. registry, seizure and dyspnea were the two most common presenting symptoms in women who collapsed before delivery.12 Maternal symptoms may precede FHR changes, as shown in Figure 39-1. Fetal bradycardia, or the abrupt onset of variable decelerations that progress to fetal bradycardia, may also herald AFE in labor.12
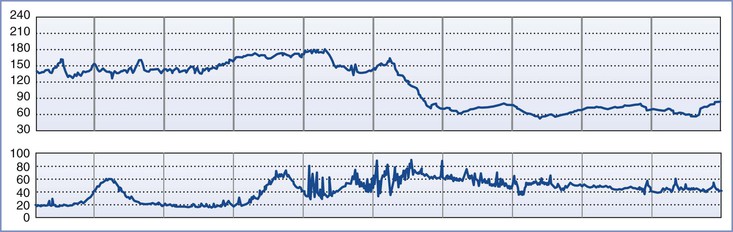
FIGURE 39-1 Fetal heart rate tracing in a patient with amniotic fluid embolism. Maternal symptoms began just before the onset of spontaneous uterine hypertonus and fetal bradycardia. (Modified from Clark SL, Hankins GDV, Dudley DA, et al. Amniotic fluid embolism: analysis of the national registry. Am J Obstet Gynecol 1995; 172:1158-67.)
AFE can present after abdominal trauma,39 after first-trimester abortion,40 in the second trimester,41 at the time of delivery,12 and in the postpartum period.42 Although acknowledging that AFE has been reported up to 48 hours postpartum,34 both the U.S. and U.K. registries require the diagnosis of AFE to be made within 30 minutes of delivery.4,12
Close inspection of hemodynamic data reveals a biphasic cardiovascular response during AFE. During the initial phase, acute pulmonary hypertension results in right ventricular dilation, a decrease in cardiac output, and ventilation-perfusion (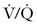 ) mismatch resulting in oxygen desaturation. Early arterial blood gas analysis may demonstrate evidence of profound shunt.12 In the U.S. registry, 11 of 17 patients with a blood gas sample drawn within 30 minutes of the acute event demonstrated an initial PaO2 less than 30 mm Hg while breathing an FIO2 of 100%.12 Release of endogenous catecholamines may produce a brief period of systemic hypertension and uterine tachysystole that precedes hypotension or cardiac arrest.12 Electrocardiographic findings are nonspecific and vary from ST-wave and T-wave abnormalities to arrhythmias or asystole. A chest radiograph may show diffuse bilateral heterogeneous or homogenous areas of opacity.
) mismatch resulting in oxygen desaturation. Early arterial blood gas analysis may demonstrate evidence of profound shunt.12 In the U.S. registry, 11 of 17 patients with a blood gas sample drawn within 30 minutes of the acute event demonstrated an initial PaO2 less than 30 mm Hg while breathing an FIO2 of 100%.12 Release of endogenous catecholamines may produce a brief period of systemic hypertension and uterine tachysystole that precedes hypotension or cardiac arrest.12 Electrocardiographic findings are nonspecific and vary from ST-wave and T-wave abnormalities to arrhythmias or asystole. A chest radiograph may show diffuse bilateral heterogeneous or homogenous areas of opacity.
Echocardiography typically demonstrates a dilated, akinetic right ventricle, pulmonary hypertension, and a normally-contracting left ventricle with a nearly obliterated cavity.43,44 Initially, right ventricular failure leads to right ventricular dilation, which compresses the left ventricle and impedes left ventricular filling and cardiac output.43,44 A second phase commences when right ventricular function improves,44 typically 15 to 30 minutes after the initial event. At this point, left ventricular failure may persist due to ischemic injury to the left ventricle,45 or direct myocardial depression,34 and is accompanied by decreased systemic ventricular resistance, decreased left ventricular stroke index, and pulmonary edema.46,47 Women who survive to the second phase may also experience hemorrhage and disseminated intravascular coagulopathy. Laboratory analysis may reveal anemia, thrombocytopenia, prolonged prothrombin time or activated partial thromboplastin time (aPTT) or both, and decreased fibrinogen levels.12,34 In a minority of cases there is massive hemorrhage and disseminated intravascular coagulopathy without preceding cardiopulmonary collapse.37
Because many of the signs and symptoms are nonspecific, the differential diagnosis for AFE is extensive and should include nonobstetric, obstetric, and anesthetic causes (Box 39-2). Even though the time course and clinical presentation of many of these competing diagnoses are similar, only amniotic fluid embolism and placental abruption result in a relatively sudden onset of consumptive coagulopathy after maternal collapse. However, partial AFE syndromes have been described in the literature.20 Therefore, the absence of coagulopathy should not exclude the possibility of AFE.
Confirmatory Tests
To date, there is no definitive test to confirm the diagnosis of AFE, although the UKOSS considers the finding of fetal material in the maternal pulmonary vasculature at autopsy to be pathognomonic for AFE.5 However, fetal squamous cells and trophoblasts are commonly found in the maternal circulation of healthy parturients. Furthermore, differentiating between maternal and fetal cells histologically is challenging.18 Clinicians should therefore not place invasive monitors solely for the purpose of aspirating cells of fetal origin.
Several biochemical markers have been suggested. Although some of these may be promising based on preliminary data and theoretical understanding of AFE, studies of test performance are limited by delayed sample acquisition and small sample size owing to the rarity and unpredictability of the AFE syndrome.
Several markers suggest an anaphylactic/anaphylactoid mechanism. Mast cells release tryptase and histamine during degranulation; tryptase has been used as a marker for anaphylaxis because its half-life is longer than that of histamine. Elevations in serum tryptase have been reported in some parturients with AFE.23 However, a case series found normal tryptase and urinary histamine levels in nine women with presumed AFE.22 Pulmonary mast cell counts have also been suggested; however, this measurement can only be obtained using immunohistochemistry at autopsy and therefore is of limited applicability in the clinical setting. In one observational study, the mean pulmonary mast cell count per fixed area for parturients who died of AFE was similar to that of parturients who died of anaphylactic shock but was higher than the mean counts in both pregnant and nonpregnant control patients.48 These data, together with the finding of minimal to no elevation in serum tryptase, suggest that pulmonary mast cell degranulation may be a secondary process in AFE.23
Complement activation may cause mast cell degranulation, and there is some evidence supporting widespread complement activation, and depressed C3 and C4 levels, in AFE.22 However, because complement is activated during acute respiratory distress syndrome and other inflammatory states, results are not specific for AFE. Zinc coproporphyrin49 and sialyl Tn antigen22 are two biomarkers that are components of meconium and have been associated with the AFE syndrome. Sialyl Tn is a mucinous glycoprotein that originates in the fetal gastrointestinal tract and is also associated with mucinous gastrointestinal tumors. Most recently, insulin-like growth factor–binding protein-1 (IGFBP-1) was identified as a sensitive and specific biomarker for AFE in a case-control study conducted in 13 delivery centers in France.50 IGFBP-1 levels exceeded 104.5 µg/L in 23 of 25 women with AFE and remained below 95 µg/L in all patients with postpartum hemorrhage due to atony, thrombotic pulmonary embolism, or uncomplicated labor. A single false-positive result was found in a woman with acute fatty liver of pregnancy. Based on the median concentration of IGFBP-1 in the amniotic fluid and blood samples, the investigators estimated that 6 to 92 mL of amniotic fluid passed into the maternal circulation in women who experienced the AFE syndrome.50
Management
Although no single intervention has been shown to reliably reverse the AFE syndrome, prompt recognition and aggressive resuscitation may improve maternal and fetal outcomes. Maternal resuscitation should focus on three priorities: (1) maintenance of oxygenation; (2) hemodynamic support; and (3) correction of coagulopathy (Box 39-3).
On diagnosis of AFE, 100% oxygen should be delivered to the patient. Given the risk for coagulopathy and hemorrhage, large-bore intravenous access is warranted. An arterial line and central venous pressure catheter may facilitate hemodynamic monitoring, blood sampling, and vasopressor administration. Transesophageal echocardiography may be useful to guide volume resuscitation and selection of appropriate vasopressor therapy.
In addition to standard resuscitative measures, other management strategies have been reported for AFE. The use of cardiopulmonary bypass, extracorporeal membrane oxygenation, continuous hemofiltration, and exchange transfusions have all been described in the literature.5,43,51 It is speculated that these technologies may filter amniotic fluid or vasoactive mediators from the systemic circulation. Strategies for management of the early right-sided heart failure seen in AFE include inhaled nitric oxide, prostacyclin, right ventricular assist devices, and vasopressors such as vasopressin, dobutamine, and milrinone.52,53 The use of extracorporal membrane oxygenation and intra-aortic balloon counterpulsation has also been reported for management of left-sided heart failure.54
In AFE, intact neonatal survival is related to the time interval from the onset of maternal compromise to delivery.12 In the event of maternal cardiopulmonary arrest, the American Heart Association recommends that delivery of the fetus should occur within 5 minutes to increase the probability of good outcomes for both the mother and her neonate.55 Although the operating room may provide a more favorable environment for surgery and resuscitation, simulation studies have suggested that resuscitation quality and the arrest-to-delivery interval both suffer with maternal transport56,57; therefore, strong consideration should be given to performing a bedside perimortem cesarean delivery.
Because coagulopathy will likely ensue for the majority of AFE survivors, the obstetric and anesthesia providers should activate a massive transfusion protocol as soon as AFE is suspected. Blood and component therapy should be guided by the clinical presentation (see Chapter 38). Close communication with the blood bank is paramount because large quantities of blood products may be needed. Analysis of the U.K. registry demonstrated that patients who developed a coagulopathy received between 12 and 106 units of blood products.4 Potential pharmacologic therapies for the coagulopathy associated with AFE include antifibrinolytic agents (e.g., tranexamic acid, aprotinin), recombinant factor VIIa (rVIIa), prothrombin complex concentrate, and fibrinogen concentrate.36,58,59
The use of rVIIa to treat intractable hemorrhage in the AFE syndrome is controversial. The use is off-label in obstetric hemorrhage (see Chapter 38). Recombinant factor VIIa binds to tissue factor and initiates clotting via the extrinsic pathway. Although individual case reports have described improvement in hemostasis, rVIIa has been associated with thrombotic complications. A meta-analysis of 25 trials involving 3849 nonobstetric bleeding patients without hemophilia found a significant increase in arterial thromboembolic events among patients who received rVIIa (relative risk [RR], 1.45; 95% CI, 1.02 to 2.05).60
A case-controlled study based on a systematic review of case reports of women with the AFE syndrome identified a twofold increase in risk for death or permanent disability among 16 women treated with rVIIa compared with 28 control patients who underwent surgery to control bleeding but did not receive rVIIa.58 Among survivors, treatment with rVIIa was associated with more permanent disability (risk ratio, 4.0; 95% CI, 1.5 to 10.4), largely attributed to thrombosis in major organs.58 An unmeasured increase in severity of disease in the treatment group compared with the control group could explain these dismal results because the population of patients who did not survive to the time of operation was excluded from the analysis. In this case-controlled study of case reports,58 50% of parturients treated with rVIIa survived, whereas in the 2010 UKOSS dataset,5 93% of the parturients treated with rVIIa survived (n = 14). However, the UKOSS data did not report neurologic outcomes or thromboembolic complications.5 The differences in outcomes between these two studies may be explained in part by differing definitions of AFE and differences in case ascertainment. Only five cases in the case-controlled study58 were peer-reviewed publications; the remainder were obtained from abstracts presented at national meetings and from registry data.
Maternal and Perinatal Outcomes
The maternal mortality ratio associated with AFE has been reported between 0.5 and 1.7 deaths per 100,000 live births.8 AFE accounted for 7.5% of pregnancy-related deaths in the United States between 1998 and 2005.1 The case-fatality rate from the UKOSS data was 20% (95% CI, 11 to 32),5 significantly lower than the reported fatality rate of 61% in the 1988 to 1994 analysis of the U.S. AFE registry.12 In the UKOSS dataset, patients who died of AFE were more likely to be from ethnic minority groups than were survivors, even after adjusting for confounders such as age, socioeconomic status, obesity, and parity (adjusted odds ratio [aOR], 11.8; 95% CI, 1.4 to 99.5).5 Similar racial/ethnic disparities were seen in the United States.1 Improvements in the management of critically ill parturients may have contributed to the decline in the case-fatality rate observed in the two studies, but the disparity in outcomes by race or ethnicity suggests further systems improvements are necessary.
Cardiac arrest complicated 87% of AFE cases reported to the U.S. registry but only 40% of cases identified by UKOSS.5,12 This difference may reflect more comprehensive ascertainment in the United Kingdom that captures less severe cases; alternatively, improvements in early recognition and management of AFE over the past 15 years may explain improved outcomes recently reported from the United Kingdom compared with those previously published from the U.S. registry.
Neurologic outcomes for survivors are poor. The 1995 analysis of the U.S. registry reported a 15% overall rate of intact neurologic survival, with only 8% neurologically intact after cardiac arrest.5,12 Neonates have a high overall survival rate, with approximately 80% of infants surviving,5,12 but also have low rates of intact neurologic survival, with only 39% of survivors neurologically intact.12 Intact neurologic survival for infants is related to the cardiac arrest-to-delivery interval; delays of greater than 15 minutes are associated with worse outcomes.12
Recurrent AFE has not been reported.
Thromboembolic Disorders
Incidence
Venous thromboembolic events (VTE) in pregnancy refer to deep vein thrombosis (DVT) and pulmonary thromboembolism (PTE). The incidence of pregnancy-related thromboembolic events is 1.0 to 1.7 events per 1,000 pregnancies.61–63 The range of reported incidences may reflect differing study designs and diagnostic criteria for thromboembolism, in addition to biologic differences among populations. Analysis of administrative data from a nationally representative sample of United States hospital admissions that included 9,058,162 pregnancy admissions and 73,834 postpartum admissions demonstrated 1.36 cases of DVT and 0.36 cases of PTE per 1,000 deliveries.61 Fifteen to 24 percent of pregnant women with an untreated DVT develop a PTE.61,64,65
There is a fivefold greater odds of thromboembolic events during pregnancy (odds ratio [OR], 4.6; 95% CI, 2.7 to 7.8), and a 60-times greater odds in the postpartum period (95% CI, 26.5 to 135.9) than in nonpregnant patients.66 Study results conflict as to whether there is an increased risk by trimester. A meta-analysis of 12 studies that evaluated the period of risk for DVT in pregnancy found that 21.9% of antepartum DVTs develop in the first trimester (95% CI, 17.4 to 27.3), 33.7% in the second trimester (95% CI, 28.1 to 39.8), and 47.6% in the third trimester (95% CI, 39.2% to 56.2%).67 The highest risk for thromboembolic events occurs postpartum. Analysis of data from a 30-year population-based cohort (n = 50,080 births) revealed that the risk for both DVT and PTE was highest in the first week postpartum (incidence rate, 3573 per 100,000 woman-years [95% CI, 2475 to 4993 per 100,000]), with a progressive decline thereafter.68
Overall, the incidence of DVT appears to be stable or decreasing while the incidence of PTE appears to be increasing.63,68 Despite the increased incidence of PTE, mortality from venous thromboembolism is decreasing.1,62,69,70 From 1985 to 2005, the leading cause of direct maternal deaths, as reported by the Confidential Enquiries into Maternal Deaths in the United Kingdom (CMACE), was PTE.69 In the most recent report (2006-2008), the overall rate of maternal death decreased compared with the previous triennium, largely as a result of the decreased rate of deaths due to PTE; PTE was no longer the leading cause of direct maternal deaths.69 This change is likely the result of early recognition of at-risk patients, as well as the increasing use of protocols for peripartum thromboprophylaxis. Ten percent of all maternal deaths in the Centers for Disease Control and Prevention (CDC) Pregnancy Mortality Surveillance System from 1998 to 2005, and 15% of all direct deaths in the 2006 to 2008 CMACE report, were attributable to PTE.1,69
Risk Factors
The two most important risk factors for thromboembolic events in pregnancy and the postpartum period are a previous history of thromboembolism and a diagnosis of thrombophilia.61,63,70 Essentially all known thrombophilias increase risk for VTE in pregnancy, with the greatest risk increase noted in women homozygous for the factor V Leiden mutation.71 Antenatal immobilization and obesity are the most important modifiable risk factors for VTE,72 and their combination has a multiplicative effect.73 In the most recent CMACE report, 81% of the 16 patients who died of PTE were either overweight or obese.69 Compared with vaginal birth, cesarean delivery essentially doubles the risk for postpartum venous thromboembolism, with greater increases noted for unplanned, as opposed to elective, cesarean delivery.61–63 The American College of Chest Physicians has defined major and minor risk factors for postcesarean VTE (Box 39-4).74
Pathophysiology
Virchow’s triad describes three factors that contribute to an increased risk for thromboembolism: (1) venous stasis, (2) vascular damage, and (3) hypercoagulability. The incidence of each factor is increased during pregnancy or in the postpartum period. Venous stasis occurs due to venocaval compression and possibly decreased mobility later in pregnancy. Separation of the placenta from the uterine wall traumatizes the endometrium, which accelerates the coagulation cascade. Finally, pregnancy is a relatively hypercoagulable state, associated with enhanced platelet turnover, coagulation, and fibrinolysis.75,76 Thrombin generation and the concentration of clotting factors increase during pregnancy, including factors I (fibrinogen), V, VII, VIII, IX, X, and XII.75 Platelet count typically remains unchanged or is decreased during pregnancy. Fibrinolytic activity decreases during the 48 hours after delivery and enhances clot stability in the early postpartum period.75
The prognosis of PTE depends on the following factors: (1) the size and number of emboli; (2) concurrent cardiopulmonary function; (3) the rate of clot fragmentation and lysis; (4) the presence or absence of a source for recurrent emboli; and (5) the location of the embolism (proximal or main pulmonary artery embolism is more symptomatic than segmental embolization).77 Massive PTE can occlude the pulmonary vasculature and precipitate cardiopulmonary arrest; smaller emboli may also lead to cardiopulmonary failure by triggering pulmonary arterial vasospasm78 and secondary pulmonary edema.79 Local platelets embedded in the clot release serotonin, adenosine diphosphate, and thrombin, factors that promote both vasoconstriction and bronchoconstriction.80 Redistribution in pulmonary blood flow leads to “hyperperfusion” of otherwise low 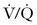 zones in unaffected areas of the lung; the resulting intrapulmonary shunts can generate hypoxemia disproportionate to the cross-sectional area occluded by clot.81 At the same time, regional hypoxic pulmonary vasoconstriction exacerbates pulmonary hypertension initiated by mechanical and humoral factors. Intracardiac shunting may develop when elevated right ventricular pressure forces blood across a probe-patent foramen ovale. Elevated pulmonary venous pressure and aggressive intravenous volume replacement may disrupt capillary integrity and exacerbate pulmonary edema.79
zones in unaffected areas of the lung; the resulting intrapulmonary shunts can generate hypoxemia disproportionate to the cross-sectional area occluded by clot.81 At the same time, regional hypoxic pulmonary vasoconstriction exacerbates pulmonary hypertension initiated by mechanical and humoral factors. Intracardiac shunting may develop when elevated right ventricular pressure forces blood across a probe-patent foramen ovale. Elevated pulmonary venous pressure and aggressive intravenous volume replacement may disrupt capillary integrity and exacerbate pulmonary edema.79
The increase in right ventricular pressure leads to right ventricular dilation, with increased wall tension and oxygen demand, and a leftward shift of the interventricular septum.78 Compression of the left ventricle combined with a decrease in preload impairs left ventricular function, cardiac output, and coronary arterial perfusion, with eventual myocardial ischemia and cardiopulmonary failure.
Deep Vein Thrombosis
Clinical Presentation
The signs and symptoms of DVT are nonspecific and often mimic normal symptoms of pregnancy, specifically lower leg edema and pain. A systematic review of the anatomic distribution of DVT in symptomatic pregnant patients (six studies, pooled n = 124) identified left leg thrombus in 88% of pregnant women in whom the side of the DVT was reported.82 Most thrombi were proximally located in the iliac or femoral veins or both. This distribution is different from the anatomic distribution seen in nonpregnant patients, who are more likely to have thrombi in the distal calf vessels.82 A prospective observational study of serial ultrasonographic examinations in pregnant women (n = 24) found an increase in vessel diameter and a decrease in flow velocity in the proximal deep leg veins with increasing gestation; this finding was most notable in the common femoral vein.83 The flow velocity was slower in the left leg than in the right leg, presumably owing to uterine compression of the left iliac vein where it crosses the right iliac artery.83
Diagnosis
For patients with new-onset signs or symptoms suggestive of DVT, the American College of Obstetricians and Gynecologists (ACOG) recommends compression ultrasonography of proximal veins as the initial diagnostic test.84 If the test is negative, and involvement of the iliac vessels is not suspected, no further action other than routine surveillance is necessary. A positive result warrants treatment (see later discussion). If the results are negative or equivocal, and iliac vein thrombosis is suspected, clinicians may opt for magnetic resonance imaging or presumptive anticoagulation.84
The D-dimer test is useful in nonpregnant patients because it has a high sensitivity and a high negative predictive value. Unfortunately, D-dimer levels are increased in pregnancy, making interpretation of elevated levels difficult in pregnant women. In one prospective, longitudinal study, serial D-dimer levels were evaluated in 89 healthy pregnant women; values exceeded the normal nonpregnant reference range in all but one woman in the third trimester.85 Therefore, the D-dimer test is not currently recommended for diagnosis of DVT in pregnancy84; however, future work may delineate pregnancy-specific thresholds.
Pulmonary Thromboembolism
Clinical Presentation
Clinical suspicion for PTE is critical to ensure timely diagnosis and treatment (Table 39-2). Physical signs and symptoms may be subtle and limited to symptoms (e.g., shortness of breath) that mimic normal pregnancy. Palpitations, anxiety, chest pain that may be pleuritic, cyanosis, diaphoresis, and cough with or without hemoptysis may all indicate PTE. Physical examination of the patient commonly reveals tachypnea, crackles, decreased breath sounds (more common than rhonchi or wheezing), and tachycardia. Signs of right ventricular failure, including an accentuated or split second heart sound, jugular venous distention, a parasternal heave, and hepatic enlargement, may be apparent. The electrocardiogram may show signs of right ventricular strain, including a right-axis shift, P pulmonale, ST-segment abnormalities, and T-wave inversion, as well as supraventricular arrhythmias. One or more of the signs of DVT (calf or thigh edema, erythema, tenderness, palpable cord) generally accompanies the pulmonary or cardiovascular findings.64,77 Despite the observation that pulmonary shunt is a common feature of PTE, as many as 30% of all patients with a pulmonary embolus have an arterial PaO2 greater than 80 mm Hg, and the diagnosis of PTE cannot be excluded on the basis of an apparently normal PaO2.77
TABLE 39-2
Physical Findings in Pulmonary Embolism
| Finding | Patients Affected (%) |
| Tachypnea | 85 |
| Tachycardia | 40 |
| Fever | 45 |
| Accentuated second heart sound | 50 |
| Localized rales | 60 |
| Thrombophlebitis | 40 |
| Supraventricular dysrhythmia | 15 |
Modified from Spence TH. Pulmonary embolization syndrome. In Civetta JM, Taylor RW, Kirby RR, editors. Critical Care. Philadelphia, JB Lippincott, 1988:1091-102.
Invasive hemodynamic monitoring typically demonstrates (1) normal to low (< 15 mm Hg) pulmonary artery occlusion pressure, (2) increased mean pulmonary artery pressure (although typically < 35 mm Hg), and (3) increased (> 8 mm Hg) central venous pressure.79 Calculated pulmonary vascular resistance typically is more than 2.5 times normal resistance; right ventricular failure occurs when the mean pulmonary artery pressure exceeds 35 to 45 mm Hg.79 Left ventricular failure may occur secondary to poor left ventricular filling and arterial hypoxemia.
Diagnosis
There are no validated risk criteria, such as the Wells or Geneva criteria, for pregnant patients, which makes diagnosis of PTE in pregnancy challenging. If the pregnant patient has signs or symptoms suggestive of DVT in addition to the signs or symptoms of PTE, compression ultrasonography should be done. If the results are positive, treatment should ensue; however, if results are negative, further imaging is necessary. In the absence of symptoms of DVT, or if compression ultrasonography of the legs is negative, the choice of the next diagnostic test to be performed is controversial. Factors such as the likelihood of a nondiscriminatory test and the radiation exposure to both mother and fetus must be considered in decision-making.
The American Thoracic Society developed an evidence-based guideline for the evaluation of suspected PTE, which has been endorsed by the ACOG.86 A diagnostic algorithm for suspected PTE is shown in Figure 39-2. If there are no signs or symptoms of DVT, a chest radiograph should be performed, both to exclude alternative diagnoses, and to guide decision-making for the next most appropriate test. If the chest radiograph is normal, 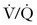 scanning should be performed.86 A retrospective study of 304 women who underwent computed tomographic angiography (CTA) or
scanning should be performed.86 A retrospective study of 304 women who underwent computed tomographic angiography (CTA) or 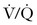 scanning at a single institution between 2001 and 2006 demonstrated that pregnant women with a normal chest radiograph have a fivefold higher rate of a nondiagnostic result from a CTA compared with a
scanning at a single institution between 2001 and 2006 demonstrated that pregnant women with a normal chest radiograph have a fivefold higher rate of a nondiagnostic result from a CTA compared with a 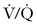 scan (RR, 5.3; 95% CI, 2.1 to 13.8).87 The use of chest radiography as a screening procedure may also minimize the amount of radiation to which a pregnant woman and her fetus are exposed if the pregnant patient is a candidate for
scan (RR, 5.3; 95% CI, 2.1 to 13.8).87 The use of chest radiography as a screening procedure may also minimize the amount of radiation to which a pregnant woman and her fetus are exposed if the pregnant patient is a candidate for 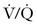 scanning.86 If the chest radiograph is abnormal, CTA is the next appropriate test because the proportion of nondiagnostic
scanning.86 If the chest radiograph is abnormal, CTA is the next appropriate test because the proportion of nondiagnostic 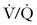 scans in the presence of an abnormal chest radiograph has been reported to be as high as 48%.88 If either the
scans in the presence of an abnormal chest radiograph has been reported to be as high as 48%.88 If either the  scan or CTA is positive, anticoagulation should ensue. The use of magnetic resonance pulmonary angiography has not been validated in the pregnant population. A new diagnosis modality,
scan or CTA is positive, anticoagulation should ensue. The use of magnetic resonance pulmonary angiography has not been validated in the pregnant population. A new diagnosis modality,  single-photon emission computed tomography (SPECT), has recently been introduced89,90; however, to date, the
single-photon emission computed tomography (SPECT), has recently been introduced89,90; however, to date, the  SPECT has not been evaluated in pregnant women.
SPECT has not been evaluated in pregnant women.
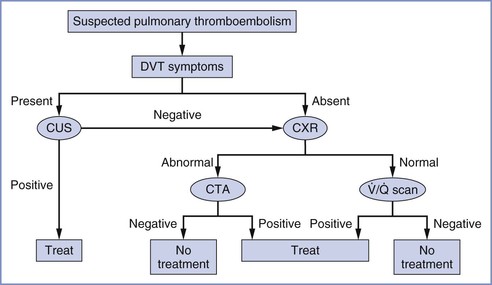
FIGURE 39-2 Diagnostic algorithm for workup of suspected pulmonary thromboembolism during pregnancy. CTA, computed tomography pulmonary angiography; CUS, compression ultrasonography; CXR, chest radiograph; DVT, deep vein thrombosis; 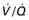 scan, ventilation-perfusion scan.
scan, ventilation-perfusion scan.
Perhaps the greatest concern with diagnostic imaging for PTE is maternal and fetal exposure to ionizing radiation. The teratogenic effects of ionizing radiation are discussed in Chapter 17).91 Both 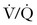 scanning and CTA are associated with low-doses of fetal radiation exposure (< 1 mGy).
scanning and CTA are associated with low-doses of fetal radiation exposure (< 1 mGy). 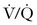 scanning delivers a higher fetal dose of radiation than CTA; however, the maternal radiation exposure is higher with CTA, particularly radiation to the breast tissue. A patient’s lifetime breast cancer risk may increase as much as 14% after CTA.92
scanning delivers a higher fetal dose of radiation than CTA; however, the maternal radiation exposure is higher with CTA, particularly radiation to the breast tissue. A patient’s lifetime breast cancer risk may increase as much as 14% after CTA.92
Management of Thromboembolic Disorders
Anticoagulation
All women with a new-onset thromboembolic event in pregnancy should be therapeutically anticoagulated.84 Patients with a previous history of thrombosis, certain high-risk populations, such as patients with acquired or inherited thrombophilias, or patients with a mechanical heart valve should be anticoagulated during pregnancy as well as the postpartum period. The ACOG practice bulletin on thromboembolism in pregnancy outlines which patients should receive prophylactic or therapeutic anticoagulation.84
Although the exact dose and regimen for anticoagulation remain controversial, two classes of drugs are typically used to initiate anticoagulation: low-molecular-weight heparin (LMWH) and unfractionated heparin (UFH) (see Chapter 44). Table 39-3 lists anticoagulation regimens commonly used in pregnancy. The ACOG does not make a distinction between UFH and LMWH for initiation of anticoagulation84; however, the American College of Chest Physicians recommends LMWH for prophylactic and therapeutic anticoagulation for pregnant women instead of UFH.74
TABLE 39-3
Commonly Used Anticoagulation Regimens during Pregnancy

aPTT, activated partial thromboplastin time; INR, international normalized ratio; LMWH, low-molecular weight heparin; SC, subcutaneous; UFH, unfractionated heparin.
Modified from American College of Obstetricians and Gynecologists. Thromboembolism in pregnancy. ACOG Practice Bulletin No. 123. Washington, DC. (Obstet Gynecol 2011; 118:718-28.)
LMWH has an enhanced ratio of antithrombotic (anti–factor Xa) to anticoagulant (anti–factor IIa) activity when compared with UFH and does not affect aPTT measurement.93 A 2005 systematic review (64 studies, pooled n = 2777) evaluated the efficacy and safety of LMWH in pregnancy. The rate of VTE was 0.9% (95% CI, 0.6% to 1.3%), the rate of significant maternal bleeding was 2.0% (95% CI, 1.5% to 2.6%), and there were no reported cases of heparin-induced thrombocytopenia when LMWH was used for any indication in pregnancy.93 An older systematic review (1999) did not find any significant increase in osteoporosis in pregnant women treated with LMWH compared with nonpregnant controls.94
The pharmacokinetics of LMWH are altered during pregnancy95; however, dose adjustment is not routinely required for prophylactic therapy.84 When LMWH is used for therapeutic anticoagulation, dosing can be adjusted based on anti–factor Xa activity; the desired peak level is 0.6 to 1.0 U/mL measured 4 hours after injection.84 Thromboelastography provides an alternative method to assess LMWH activity. Carroll et al.96 demonstrated that the delta reaction time (ΔR) measured by thromboelastography correlates with anti–factor Xa levels in the pregnant woman and allows LMWH doses to be adjusted to ensure anticoagulation or, conversely, confirms the absence of anticoagulation. LMWH is cleared by the kidney; therefore, dose reduction may be required in patients with renal failure.
UFH therapy may be used to initiate anticoagulation or to maintain therapy as the patient nears delivery (see later discussion). UFH exerts its anticoagulant activity by binding to antithrombin III and potentiates inactivation of other coagulation factors, including thrombin (IIa), factors IXa, Xa, XIa, and XIIa, and kallikrein. Typically, UFH is administered as a subcutaneous injection for both prophylactic and therapeutic therapy. Intravenous therapy becomes necessary when PTE causes hemodynamic instability; it is initiated with a bolus dose and maintained by infusion titrated to a therapeutic aPTT value. The aPTT measured 6 hours after an injection or dose adjustment should be maintained at 1.5 to 2.5 times the normal range.84 In pregnancy, the bioavailability of UFH decreases owing to an increase in heparin-binding proteins, increased plasma volume, increased renal clearance, and increased degradation by plasma heparinases.97 Based on pharmacodynamic studies, 5000 units of subcutaneous heparin are often inadequate to achieve prophylactic serum anti–factor Xa levels in the second half of pregnancy.98 Thus, for therapeutic anticoagulation, the ACOG currently recommends UFH 10,000 units subcutaneous twice daily, with aPTT monitoring and dose adjustment.84
In patients who develop heparin-induced thrombocytopenia, or severe cutaneous reactions to heparin, fondaparinux is the preferred anticoagulant owing to minimal cross-reactivity with UFH.84 Anticoagulation with other classes of drugs such as vitamin K antagonists, thienopyridines, direct factor Xa inhibitors, and direct thrombin inhibitors is possible in pregnancy, but the use of these medications is less common. Additionally, animal studies with rivaroxaban, a direct factor Xa inhibitor, and dabigatran, a direct thrombin inhibitor, have found teratogenic effects, reduced fetal viability, hemorrhagic changes, and placental abnormalities; thus, their use in pregnancy is not recommended.99
Antithrombotic Therapy and Anesthetic Implications
The greatest concern with neuraxial procedures in anticoagulated patients is spinal or epidural hematoma. A meta-analysis of the incidence of epidural hematoma in obstetric patients (8 studies, pooled n = 1.1 million) found an incidence of 1 : 183,000.100 Notwithstanding the very low incidence, the consequences of a spinal-epidural hematoma can be devastating. The American Society of Regional Anesthesia and Pain Medicine (ASRA) guidelines include a summary of 16 cases of spinal hematoma after neuraxial anesthesia in obstetric patients; approximately half of the cases had some form of permanent motor or sensory dysfunction.101
A case report and literature review published in 2005 described six published cases of spontaneous epidural hematoma in pregnancy.102 One proposed cause is that a pressure differential exists between the low-pressure epidural space and central venous pressure and that an increase in central venous pressure relative to the pressure in the epidural space leads to spontaneous rupture of the venous wall.102 Case reports of spontaneous epidural hematoma in anticoagulated pregnant women, including patients with preeclampsia, have been reported.103,104
In 2010, ASRA published updated guidelines for regional anesthesia in patients receiving antithrombotic or thrombolytic therapy.101 Among other topics, the updated guidelines address antithrombotic therapy during pregnancy. Owing to the relative paucity of outcome data in pregnant women, the ASRA suggests following the guidelines for surgical patients when developing clinical policy for obstetric patients with regard to initiation of neuraxial procedures and postpartum thromboprophylaxis (Table 39-4).101 The ASRA guidelines do not make recommendations for patients receiving doses of more than 10,000 units of UFH daily or for patients receiving UFH more than twice daily. These guidelines differ somewhat from those of the European Society of Anaesthesiology.101,105
TABLE 39-4
Time Interval for Administration of Neuraxial Anesthesia after Anticoagulation
| Therapy | American Society of Regional Anesthesia and Pain Medicine101 | European Society of Anaesthesiology105 |
| SC UFH, prophylactic | No delay* | 4-6 h†‡ |
| SC UFH, therapeutic | No recommendation*‡ | 8-12 h‡§ |
| LMWH, prophylactic | 10-12 h | 12 h |
| LMWH, therapeutic | 24 h | 24 h |
* No contraindication to neuraxial procedures with twice-daily dosing and total daily dose < 10,000 units.
† UFH ≤ 15,000 units/day.
‡ Check platelet count if UFH therapy for more than 4 days (American Society of Regional Anesthesia and Pain Medicine) or more than 5 days (European Society of Anaesthesiology).
§ Check activated partial thromboplastin time (aPTT) or activated clotting time (ACT) before initiation of neuraxial procedure.
LMWH, low-molecular weight heparin; SC, subcutaneous; UFH, unfractionated heparin.
For patients being treated with UFH for more than 4 days, a platelet count should be assessed before initiation of neuraxial anesthesia procedures.101 Measurement of aPTT is not necessary in patients receiving prophylactic UFH (≤ 5000 units twice a day) but may be indicated in patients receiving a large dose. Protamine reversal of UFH to facilitate more rapid administration of neuraxial anesthesia is not recommended. Further, protamine reversal of LMWH also is not recommended because it is unpredictable in reversing LMWH-induced anti–factor Xa activity.101 If anticoagulant dosing is provided outside the ranges specifically addressed by the ASRA guidelines, individualized risk-benefit assessment is necessary to ascertain the best timing for initiation of neuraxial procedures. In some cases, confirmation of coagulation parameters (e.g., aPTT, anti–factor Xa level) within the normal range before initiation of neuraxial anesthesia may help to clarify the relative risk/benefit ratio for an individual patient.101
Other anticoagulant medications sometimes administered to pregnant women include aspirin, warfarin, and newer anticoagulant medications. In patients receiving warfarin therapy, the ASRA recommends discontinuing warfarin therapy for 4 to 5 days and waiting for normalization of the international normalized ratio (INR). Neuraxial catheters are not recommended in patients receiving fondaparinux, and neuraxial techniques are not recommended in patients anticoagulated with direct thrombin inhibitors. Treatment with thrombolytics is an absolute contraindication to neuraxial anesthesia.101 By contrast, there is no contraindication to neuraxial anesthesia in patients who have received nonsteroidal anti-inflammatory drugs or aspirin if these drugs are used alone.101
If a patient is not a candidate for neuraxial anesthesia, noninvasive analgesic methods (e.g., intravenous opioids) should be offered for labor, and general anesthesia should be performed for operative procedures, including cesarean delivery.
The following anticoagulation strategies will help to facilitate safe and timely neuraxial analgesia/anesthesia for parturients101:
1. Women taking oral anticoagulants should transition to LMWH or UFH no later than 36 weeks’ gestation.
3. Intravenous UFH should be discontinued 4 to 6 hours before planned delivery.
Postpartum, prophylactic anticoagulation with LMWH or UFH can begin 12 hours after vaginal delivery or 2 hours* after neuraxial catheter removal, whichever occurs later. A 24-hour delay is required for the first dose of UFH or LMWH if (1) the patient underwent a cesarean delivery, (2) therapeutic dosing is required (regardless of the mode of delivery), or (3) blood was present during needle or catheter placement.101 The ASRA postpartum dosing recommendations differ from the guidelines of the ACOG and other international guidelines, including those of the Royal College of Obstetricians and Gynaecologists.84,106 Therefore, a multidisciplinary institutional review of anticoagulation policies is advisable.
Because of the risk for epidural hematoma—even in the absence of neuraxial procedures—anesthesiologists, obstetricians, and nursing staff must remain vigilant for signs and symptoms of epidural hematoma. These include (1) severe, unremitting backache; (2) neurologic deficit, including bowel or bladder dysfunction or radiculopathy; (3) tenderness over the spinous or paraspinous area; and (4) unexplained fever.107 Suspicion of epidural hematoma should lead to immediate diagnostic imaging of the spinal cord and neurosurgical consultation for possible spinal cord decompression. Neurologic recovery is a function of the severity of preoperative deficits, the duration of maximum deficit, and the interval between symptom onset and surgery; better outcomes are associated with a shorter symptom onset-to-surgery interval.108 A high index of suspicion is necessary because neurologic dysfunction may mimic local anesthetic-induced effects.109
Risks of general anesthesia in the anticoagulated patient include airway bleeding. Laryngoscopy and tracheal intubation should be as atraumatic as possible. The anesthesia provider should be aware that placement of nasopharyngeal and oropharyngeal airways, gastric tubes, and other devices (e.g., temperature probes, stethoscopes) carries the tangible risk for traumatic hemorrhage. Emergency surgery may necessitate the administration of protamine or the transfusion of blood products (e.g., plasma, platelets) to reverse anticoagulation and reduce the risk for hemorrhage during and after surgery.
Prevention of Thromboembolic Events
Given the morbidity and mortality associated with thromboembolic events, there is great interest in identifying and implementing strategies to reduce their occurrence. Several risk-stratification strategies have been proposed to prevent thromboembolic events, although these are based on expert opinion.74,106,110 A meta-analysis published in 2010 found that there was insufficient evidence on which to base recommendations for thromboprophylaxis in pregnancy and the postpartum period.111 The American College of Chest Physicians recommends the following for postcesarean delivery thromboprophylaxis based on the risk factors outlined in Box 39-474:
One decision-analysis study modeled the safety and efficacy of thromboprophylaxis with intermittent pneumatic compression stockings at cesarean delivery compared with universal subcutaneous heparin prophylaxis. The use of the pneumatic compression stockings was the preferred strategy because universal heparin prophylaxis would be associated with 13 cases of heparin-induced thrombocytopenia or hemorrhage for each VTE prevented.112 The reduction in VTE risk was similar between pneumatic compression stockings and universal heparin use.112 Casele and Grobman113 found intermittent pneumatic compression stockings to be cost-effective when compared with no thromboprophylaxis after cesarean delivery. Regardless of whether thromboprophylaxis is initiated in the hospital or not, women are at the highest risk for thrombotic events in the first week postpartum.68 Therefore, careful postdischarge planning and evaluation of VTE risk are equally important to inpatient management.
Venous Air Embolism
Venous air embolism (VAE) is a recognized complication of many surgical procedures.114 The first study of VAE during cesarean delivery was published in 1987.115 Subsequent research has revealed that VAE is a common occurrence during cesarean delivery. Most air emboli are small, but volumes greater than 200 to 300 mL, or 3 to 5 mL/kg, may be lethal.114 Early recognition and appropriate management are necessary for avoiding adverse outcomes.
Incidence
The reported incidence of VAE during cesarean delivery varies depending on the method used to ascertain the presence of air (Table 39-5). The most sensitive monitors detect volumes of air as low as 0.02 mL/kg.114 Studies that have used precordial Doppler monitoring in patients undergoing cesarean delivery with neuraxial anesthesia have found incidence rates ranging from 10% to 65%.115–119 One study used precordial Doppler monitoring to detect VAE and correlated the Doppler findings with transthoracic echocardiographic evidence of intracardiac air. Of the 42 patients who underwent cesarean delivery with neuraxial anesthesia, 11 (26%) had evidence of VAE, with perfect agreement between the Doppler and the echocardiographic monitoring (kappa = 1.0).117 One study of healthy parturients undergoing elective cesarean delivery under general anesthesia found that 29 of 30 parturients had evidence of intraoperative VAE.120 The authors defined VAE as a 0.1% increase from the baseline of expired nitrogen concentration (equivalent to 0.25 to 1.0 mL/kg venous air).120 Although the volume of entrained air may be lower with general anesthesia using positive-pressure ventilation than spontaneous ventilation,121 the use of prophylactic positive end-expiratory pressure has not been shown to decrease the incidence of VAE in a neurosurgical population.122
TABLE 39-5
Methods to Detect Venous Air Embolism
| Method of Detection | Sensitivity | Volume of Air Detected (mL/kg) |
| Transesophageal echocardiography | High | 0.02 |
| Precordial Doppler | High | 0.05 |
| Pulmonary artery catheter | High | 0.25 |
| Expired CO2 | Moderate | 0.5 |
| Expired nitrogen | Moderate | 0.5 |
Modified from Mirski MA, Lele AV, Fitzsimmons L, Toung TJK. Diagnosis and treatment of vascular air embolism. Anesthesiology 2007; 106:164-77.
The incidence of VAE does not appear to vary with maternal position. The Trendelenburg position could produce a pressure gradient between the right side of the heart and open venous sinuses in the surgical field. A meta-analysis of two randomized controlled trials, which compared 5 to 10 degrees reverse Trendelenburg position with supine positioning for cesarean delivery (pooled n = 130) found no difference in the incidence of air embolism (RR, 0.95; 95% CI, 0.65 to 1.26).123
The majority of VAE episodes are subclinical. The true incidence of fatal VAE in the obstetric population is unknown. One estimate using data on maternal deaths from the National Center for Health Statistics from 1974 to 1978 found that 25 of 2475 deaths were attributable to air embolism.124 More recent published estimates in the United States have not reported VAE as a cause of maternal death.1 In the most CMACE report from the United Kingdom, only 1 of 261 deaths was attributed to VAE.69 This death occurred during intercourse 2 weeks postpartum.69 This case highlights that VAE may occur in the nonoperative setting. Venous air embolism has been reported during pregnancy during vaginal examinations and during orogenital sex.125,126
Pathophysiology
A pressure gradient as small as −5 cm H2O between the surgical field and the heart allows a significant amount of air to be entrained into the venous circulation. Immediately after placental separation, the raw endometrial surface appears to be a location of significant air entrainment; almost all episodes of VAE during cesarean delivery are noted after the delivery of the placenta.118 In the noncesarean delivery setting, pressurized air in the vagina is believed to traverse the cervical canal, dissect around fetal membranes (if present), and enter the maternal circulation via subplacental sinuses.127
Small volumes of air can precipitate pulmonary vasospasm via a pathophysiology that mimics that of other embolic phenomena. Vasoactive mediators or mechanical obstruction of small vessels appear to induce pulmonary vasoconstriction that leads to 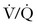 mismatch, hypoxemia, right-sided heart failure, arrhythmias, and hypotension. Fluid resuscitation and increased hydrostatic pressure may provoke interstitial pulmonary edema. A paradoxical air embolus into the arterial circulation (by means of a patent foramen ovale) can lead to cardiovascular and neurologic sequelae and morbidity. Large volumes of air (> 3 mL/kg) can generate cardiovascular collapse by creating an “air lock” that causes right ventricular outflow tract obstruction.
mismatch, hypoxemia, right-sided heart failure, arrhythmias, and hypotension. Fluid resuscitation and increased hydrostatic pressure may provoke interstitial pulmonary edema. A paradoxical air embolus into the arterial circulation (by means of a patent foramen ovale) can lead to cardiovascular and neurologic sequelae and morbidity. Large volumes of air (> 3 mL/kg) can generate cardiovascular collapse by creating an “air lock” that causes right ventricular outflow tract obstruction.
Clinical Presentation
Most air emboli are subclinical with no sequelae; however, massive VAE can manifest as a sudden, dramatic, and devastating event with hypotension, hypoxemia, and even cardiac arrest.128 In awake patients receiving neuraxial anesthesia, transient episodes of hypoxemia, dyspnea, or chest pain during uterine repair suggest VAE.115,116,118,119 In the patient receiving general anesthesia, evidence of VAE may be limited to hypoxemia and a slight decrease in end-tidal CO2.114 VAE may be more frequent when the uterus is exteriorized than when it is repaired within the abdomen, either owing to vertical elevation relative to the heart or traction on the uterus that opens venous sinuses.118,119 Clinically significant air emboli may be associated with hypotension, heart rate or rhythm changes, evidence of right-sided heart strain on the electrocardiogram, an increase in central venous pressure, and/or an increase in pulmonary artery pressure. Electrocardiographic changes consistent with myocardial ischemia (e.g., ST-segment depression) are relatively common during cesarean delivery,129,130 but the clinical significance of ST-segment depression and its relationship to VAE remain unclear.
A precordial stethoscope may detect a pathognomonic “millwheel murmur.” Transesophageal echocardiography may detect air in the right atria or the pulmonary artery. However, several small case series failed to identify VAE using echocardiography at the time of ST-segment depression during cesarean delivery.129 More recent evidence suggests that ST-segment depression in the immediate postdelivery period may be associated with rapid oxytocin administration.131,132
Management
The clinician should consider VAE in women who complain of intraoperative chest pain or dyspnea or experience sudden hypoxemia, hypotension, or arrhythmia during cesarean delivery. Although continuous precordial Doppler monitoring has been recommended by some experts,114 the rarity of hemodynamically significant VAE suggests that targeted use in high-risk patients (e.g., women with a known intracardiac shunt) may be more appropriate than routine application. Transthoracic or transesophageal echocardiography may help to confirm a diagnosis of VAE, to exclude alternative causes of hemodynamic instability, and to guide appropriate clinical management. Regardless, clinical suspicion of VAE should prompt appropriate management (Box 39-5).
Currently, there are no data to support central line insertion for air aspiration from the right side of the heart, but central line placement may be indicated to deliver potent vasopressors.114 Position changes to limit air entrainment are recommended. However, in a canine study of resuscitation positioning after a 2.5 mL/kg VAE,133 repositioning animals to direct air to the dependent portion of the right side of the heart did not improve cardiac function or survival; thus, this maneuver is not recommended. The use of the Valsalva maneuver and positive-end expiratory pressure should be avoided because an increase in right atrial pressure may result in a paradoxical embolism. In patients with delayed emergence from anesthesia, computed tomography or magnetic resonance imaging may be considered to evaluate for intracerebral air. Hyperbaric oxygen may improve neurologic outcomes if instituted within 6 hours of intracerebral air embolism.134
References
1. Berg CJ, Callaghan WM, Syverson C, Henderson Z. Pregnancy-related mortality in the United States, 1998 to 2005. Obstet Gynecol. 2010;116:1302–1309.
2. Meyer J. Embolia pulmonar amniocaseosa. Bras Med. 1926;2:301–303.
3. Steiner PE, Lushbaugh CC. Maternal pulmonary embolism by amniotic fluid as a cause of obstetric shock and unexpected deaths in obstetrics. JAMA. 1941;117:1245–1254.
4. Tuffnell DJ. United Kingdom amniotic fluid embolism register. BJOG. 2005;112:1625–1629.
5. Knight M, Tuffnell D, Brocklehurst P, et al. Incidence and risk factors for amniotic-fluid embolism. Obstet Gynecol. 2010;115:910–917.
6. Roberts CL, Algert CS, Knight M, Morris JM. Amniotic fluid embolism in an Australian population-based cohort. BJOG. 2010;117:1417–1421.
7. Abenhaim HA, Azoulay L, Kramer MS, Leduc L. Incidence and risk factors of amniotic fluid embolisms: a population-based study on 3 million births in the United States. Am J Obstet Gynecol. 2008;199:49 e1–8.
8. Conde-Agudelo A, Romero R. Amniotic fluid embolism: an evidence-based review. Am J Obstet Gynecol. 2009;201:445 e1–13.
9. Knight M, Berg C, Brocklehurst P, et al. Amniotic fluid embolism incidence, risk factors and outcomes: a review and recommendations. BMC Pregnancy Childbirth. 2012;12:7.
10. Kramer MS, Rouleau J, Liu S, et al. Amniotic fluid embolism: incidence, risk factors, and impact on perinatal outcome. BJOG. 2012;119:874–879.
11. Kramer MS, Rouleau J, Baskett TF, Joseph KS. Amniotic-fluid embolism and medical induction of labour: a retrospective, population-based cohort study. Lancet. 2006;368:1444–1448.
12. Clark SL, Hankins GD, Dudley DA, et al. Amniotic fluid embolism: analysis of the national registry. Am J Obstet Gynecol. 1995;172:1158–1167.
13. Paul RH, Koh KS, Bernstein SG. Changes in fetal heart rate-uterine contraction patterns associated with eclampsia. Am J Obstet Gynecol. 1978;130:165–169.
14. Rannou B, Rivard GE, Gains MJ, Bedard C. Intravenous injection of autologous amniotic fluid induces transient thrombocytopenia in a gravid rabbit model of amniotic fluid embolism. Vet Clin Pathol. 2011;40:524–529.
15. Leong AS, Norman JE, Smith R. Vascular and myometrial changes in the human uterus at term. Reprod Sci. 2008;15:59–65.
16. Kuhlman K, Hidvegi D, Tamura RK, Depp R. Is amniotic fluid material in the central circulation of peripartum patients pathologic? Am J Perinatol. 1985;2:295–299.
17. Clark SL, Pavlova Z, Greenspoon J, et al. Squamous cells in the maternal pulmonary circulation. Am J Obstet Gynecol. 1986;154:104–106.
18. Lee W, Ginsburg KA, Cotton DB, Kaufman RH. Squamous and trophoblastic cells in the maternal pulmonary circulation identified by invasive hemodynamic monitoring during the peripartum period. Am J Obstet Gynecol. 1986;155:999–1001.
19. Benson MD. A hypothesis regarding complement activation and amniotic fluid embolism. Med Hypotheses. 2007;68:1019–1025.
20. Clark SL. Amniotic fluid embolism. Clin Obstet Gynecol. 2010;53:322–328.
21. Azegami M, Mori N. Amniotic fluid embolism and leukotrienes. Am J Obstet Gynecol. 1986;155:1119–1124.
22. Benson MD, Kobayashi H, Silver RK, et al. Immunologic studies in presumed amniotic fluid embolism. Obstet Gynecol. 2001;97:510–514.
23. Benson MD. Current concepts of immunology and diagnosis in amniotic fluid embolism. Clin Dev Immunol. 2012;2012:946576.
24. Oikonomopoulou K, Ricklin D, Ward PA, Lambris JD. Interactions between coagulation and complement—their role in inflammation. Semin Immunopathol. 2012;34:151–165.
25. Hankins GD, Snyder RR, Clark SL, et al. Acute hemodynamic and respiratory effects of amniotic fluid embolism in the pregnant goat model. Am J Obstet Gynecol. 1993;168:1113–1129.
26. Aharon A, Brenner B, Katz T, et al. Tissue factor and tissue factor pathway inhibitor levels in trophoblast cells: implications for placental hemostasis. Thromb Haemost. 2004;92:776–786.
27. Lockwood CJ, Bach R, Guha A, et al. Amniotic fluid contains tissue factor, a potent initiator of coagulation. Am J Obstet Gynecol. 1991;165:1335–1341.
28. Zhou J, Liu S, Ma M, et al. Procoagulant activity and phosphatidylserine of amniotic fluid cells. Thromb Haemost. 2009;101:845–851.
29. Salem HH, Walters WA, Perkin JL, et al. Aggregation of human platelets by amniotic fluid. Br J Obstet Gynaecol. 1982;89:733–737.
30. Ulrich S, Huber LC, Fischler M, et al. Platelet serotonin content and transpulmonary platelet serotonin gradient in patients with pulmonary hypertension. Respiration. 2011;81:211–216.
31. Maclean MR, Dempsie Y. The serotonin hypothesis of pulmonary hypertension revisited. Adv Exp Med Biol. 2010;661:309–322.
32. Gilbert WM, Danielsen B. Amniotic fluid embolism: decreased mortality in a population-based study. Obstet Gynecol. 1999;93:973–977.
33. Courtney LD, Allington M. Effect of amniotic fluid on blood coagulation. Br J Haematol. 1972;22:353–355.
35. Harnett MJ, Hepner DL, Datta S, Kodali BS. Effect of amniotic fluid on coagulation and platelet function in pregnancy: an evaluation using thromboelastography. Anaesthesia. 2005;60:1068–1072.
36. Annecke T, Geisenberger T, Kurzl R, et al. Algorithm-based coagulation management of catastrophic amniotic fluid embolism. Blood Coagul Fibrinolysis. 2010;21:95–100.
37. Davies S. Amniotic fluid embolism and isolated disseminated intravascular coagulation. Can J Anaesth. 1999;46:456–459.
38. Knight M, Kurinczuk JJ, Tuffnell D, Brocklehurst P. The UK Obstetric Surveillance System for rare disorders of pregnancy. BJOG. 2005;112:263–265.
39. Ct Olcott, Robinson AJ, Maxwell TM, Griffin HA. Amniotic fluid embolism and disseminated intravascular coagulation after blunt abdominal trauma. J Trauma. 1973;13:737–740.
40. Cromey MG, Taylor PJ, Cumming DC. Probable amniotic fluid embolism after first-trimester pregnancy termination: a case report. J Reprod Med. 1983;28:209–211.
41. Kelly MC, Bailie K, McCourt KC. A case of amniotic fluid embolism in a twin pregnancy in the second trimester. Int J Obstet Anesth. 1995;4:175–177.
42. Margarson MP. Delayed amniotic fluid embolism following caesarean section under spinal anaesthesia. Anaesthesia. 1995;50:804–806.
43. Stanten RD, Iverson LI, Daugharty TM, et al. Amniotic fluid embolism causing catastrophic pulmonary vasoconstriction: diagnosis by transesophageal echocardiogram and treatment by cardiopulmonary bypass. Obstet Gynecol. 2003;102:496–498.
44. Shechtman M, Ziser A, Markovits R, Rozenberg B. Amniotic fluid embolism: early findings of transesophageal echocardiography. Anesth Analg. 1999;89:1456–1458.
45. Richards DS, Carter LS, Corke B, et al. The effect of human amniotic fluid on the isolated perfused rat heart. Am J Obstet Gynecol. 1988;158:210–214.
46. Clark SL, Cotton DB, Gonik B, et al. Central hemodynamic alterations in amniotic fluid embolism. Am J Obstet Gynecol. 1988;158:1124–1126.
47. Clark SL, Montz FJ, Phelan JP. Hemodynamic alterations associated with amniotic fluid embolism: a reappraisal. Am J Obstet Gynecol. 1985;151:617–621.
48. Fineschi V, Gambassi R, Gherardi M, Turillazzi E. The diagnosis of amniotic fluid embolism: an immunohistochemical study for the quantification of pulmonary mast cell tryptase. Int J Legal Med. 1998;111:238–243.
49. Kanayama N, Yamazaki T, Naruse H, et al. Determining zinc coproporphyrin in maternal plasma—a new method for diagnosing amniotic fluid embolism. Clin Chem. 1992;38:526–529.
50. Legrand M, Rossignol M, Dreux S, et al. Diagnostic accuracy of insulin-like growth factor binding protein-1 for amniotic fluid embolism. Crit Care Med. 2012;40:2059–2063.
51. Kaneko Y, Ogihara T, Tajima H, Mochimaru F. Continuous hemodiafiltration for disseminated intravascular coagulation and shock due to amniotic fluid embolism: report of a dramatic response. Intern Med. 2001;40:945–947.
52. McDonnell NJ, Chan BO, Frengley RW. Rapid reversal of critical haemodynamic compromise with nitric oxide in a parturient with amniotic fluid embolism. Int J Obstet Anesth. 2007;16:269–273.
53. Nagarsheth NP, Pinney S, Bassily-Marcus A, et al. Successful placement of a right ventricular assist device for treatment of a presumed amniotic fluid embolism. Anesth Analg. 2008;107:962–964.
54. Hsieh YY, Chang CC, Li PC, et al. Successful application of extracorporeal membrane oxygenation and intra-aortic balloon counterpulsation as lifesaving therapy for a patient with amniotic fluid embolism. Am J Obstet Gynecol. 2000;183:496–497.
55. Vanden Hoek TL, Morrison LJ, Shuster M, et al. Part 12: cardiac arrest in special situations: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2010;122:S829–S861.
56. Lipman S, Daniels K, Cohen SE, Carvalho B. Labor room setting compared with the operating room for simulated perimortem cesarean delivery: a randomized controlled trial. Obstet Gynecol. 2011;118:1090–1094.
57. Lipman SS, Wong JY, Arafeh J, et al. Transport decreases the quality of cardiopulmonary resuscitation during simulated maternal cardiac arrest. Anesth Analg. 2013;116:162–167.
58. Leighton BL, Wall MH, Lockhart EM, et al. Use of recombinant factor VIIa in patients with amniotic fluid embolism: a systematic review of case reports. Anesthesiology. 2011;115:1201–1208.
59. Stroup J, Haraway D, Beal JM. Aprotinin in the management of coagulopathy associated with amniotic fluid embolus. Pharmacotherapy. 2006;26:689–693.
60. Simpson E, Lin Y, Stanworth S, et al. Recombinant factor VIIa for the prevention and treatment of bleeding in patients without haemophilia. Cochrane Database Syst Rev. 2012;(3).
61. James AH, Jamison MG, Brancazio LR, Myers ER. Venous thromboembolism during pregnancy and the postpartum period: incidence, risk factors, and mortality. Am J Obstet Gynecol. 2006;194:1311–1315.
62. Jacobsen AF, Skjeldestad FE, Sandset PM. Incidence and risk patterns of venous thromboembolism in pregnancy and puerperium—a register-based case-control study. Am J Obstet Gynecol. 2008;198:233 e1–7.
63. Liu S, Rouleau J, Joseph KS, et al. Epidemiology of pregnancy-associated venous thromboembolism: a population-based study in Canada. J Obstet Gynaecol Can. 2009;31:611–620.
64. Sipes SL, Weiner CP. Venous thromboembolic disease in pregnancy. Semin Perinatol. 1990;14:103–118.
65. Rutherford SE, Phelan JP. Deep venous thrombosis and pulmonary embolism in pregnancy. Obstet Gynecol Clin North Am. 1991;18:345–370.
66. Pomp ER, Lenselink AM, Rosendaal FR, Doggen CJ. Pregnancy, the postpartum period and prothrombotic defects: risk of venous thrombosis in the MEGA study. J Thromb Haemost. 2008;6:632–637.
67. Ray JG, Chan WS. Deep vein thrombosis during pregnancy and the puerperium: a meta-analysis of the period of risk and the leg of presentation. Obstet Gynecol Surv. 1999;54:265–271.
68. Heit JA, Kobbervig CE, James AH, et al. Trends in the incidence of venous thromboembolism during pregnancy or postpartum: a 30-year population-based study. Ann Intern Med. 2005;143:697–706.
69. Centre for Maternal and Child Enquiries (CMACE). Saving Mothers’ Lives: reviewing maternal deaths to make motherhood safer: 2006–08. The Eighth Report on Confidential Enquiries into Maternal Deaths in the United Kingdom. BJOG. 2011;118(Suppl 1):1–203.
70. James AH, Tapson VF, Goldhaber SZ. Thrombosis during pregnancy and the postpartum period. Am J Obstet Gynecol. 2005;193:216–219.
71. Robertson L, Wu O, Langhorne P, et al. Thrombophilia in pregnancy: a systematic review. Br J Haematol. 2006;132:171–196.
72. Knight M. Antenatal pulmonary embolism: risk factors, management and outcomes. BJOG. 2008;115:453–461.
73. Jacobsen AF, Skjeldestad FE, Sandset PM. Ante- and postnatal risk factors of venous thrombosis: a hospital-based case-control study. J Thromb Haemost. 2008;6:905–912.
74. Bates SM, Greer IA, Middeldorp S, et al. VTE, thrombophilia, antithrombotic therapy, and pregnancy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e691S–736S.
75. Gerbasi FR, Bottoms S, Farag A, Mammen E. Increased intravascular coagulation associated with pregnancy. Obstet Gynecol. 1990;75:385–389.
76. Gerbasi FR, Bottoms S, Farag A, Mammen EF. Changes in hemostasis activity during delivery and the immediate postpartum period. Am J Obstet Gynecol. 1990;162:1158–1163.
77. Stein PD, Beemath A, Matta F, et al. Clinical characteristics of patients with acute pulmonary embolism: data from PIOPED II. Am J Med. 2007;120:871–879.
78. Wood KE. Major pulmonary embolism. Crit Care Clin. 2011;27:885–906.
79. Hollingsworth H, Pratter M, Irwin R. Acute respiratory failure in pregnancy. J Intensive Care Med. 1989;4:11–34.
80. Stratmann G, Gregory GA. Neurogenic and humoral vasoconstriction in acute pulmonary thromboembolism. Anesth Analg. 2003;97:341–354.
82. Chan WS, Spencer FA, Ginsberg JS. Anatomic distribution of deep vein thrombosis in pregnancy. CMAJ. 2010;182:657–660.
83. Macklon NS, Greer IA, Bowman AW. An ultrasound study of gestational and postural changes in the deep venous system of the leg in pregnancy. Br J Obstet Gynaecol. 1997;104:191–197.
84. American College of Obstetricians and Gynecologists. Thromboembolism in pregnancy. [ACOG Practice Bulletin No. 123. Washington, DC] Obstet Gynecol. 2011;118:718–728.
85. Kovac M, Mikovic Z, Rakicevic L, et al. The use of D-dimer with new cutoff can be useful in diagnosis of venous thromboembolism in pregnancy. Eur J Obstet Gynecol Reprod Biol. 2010;148:27–30.
86. Leung AN, Bull TM, Jaeschke R, et al. An official American Thoracic Society/Society of Thoracic Radiology clinical practice guideline: evaluation of suspected pulmonary embolism in pregnancy. Am J Respir Crit Care Med. 2011;184:1200–1208.
87. Cahill AG, Stout MJ, Macones GA, Bhalla S. Diagnosing pulmonary embolism in pregnancy using computed-tomographic angiography or ventilation-perfusion. Obstet Gynecol. 2009;114:124–129.
88. Forbes KP, Reid JH, Murchison JT. Do preliminary chest X-ray findings define the optimum role of pulmonary scintigraphy in suspected pulmonary embolism? Clin Radiol. 2001;56:397–400.
89. Gutte H, Mortensen J, Jensen CV, et al. Detection of pulmonary embolism with combined ventilation-perfusion SPECT and low-dose CT: head-to-head comparison with multidetector CT angiography. J Nucl Med. 2009;50:1987–1992.
90. Sostman HD, Miniati M, Gottschalk A, et al. Sensitivity and specificity of perfusion scintigraphy combined with chest radiography for acute pulmonary embolism in PIOPED II. J Nucl Med. 2008;49:1741–1748.
91. Baysinger CL. Imaging during pregnancy. Anesth Analg. 2010;110:863–867.
92. Bhalla S, Lopez-Costa I. MDCT of acute thrombotic and nonthrombotic pulmonary emboli. Eur J Radiol. 2007;64:54–64.
93. Greer IA, Nelson-Piercy C. Low-molecular-weight heparins for thromboprophylaxis and treatment of venous thromboembolism in pregnancy: a systematic review of safety and efficacy. Blood. 2005;106:401–407.
94. Sanson BJ, Lensing AW, Prins MH, et al. Safety of low-molecular-weight heparin in pregnancy: a systematic review. Thromb Haemost. 1999;81:668–672.
95. Sephton V, Farquharson RG, Topping J, et al. A longitudinal study of maternal dose response to low molecular weight heparin in pregnancy. Obstet Gynecol. 2003;101:1307–1311.
96. Carroll RC, Craft RM, Whitaker GL, et al. Thrombelastography monitoring of resistance to enoxaparin anticoagulation in thrombophilic pregnancy patients. Thromb Res. 2007;120:367–370.
97. Barbour LA, Smith JM, Marlar RA. Heparin levels to guide thromboembolism prophylaxis during pregnancy. Am J Obstet Gynecol. 1995;173:1869–1873.
98. Brancazio LR, Roperti KA, Stierer R, Laifer SA. Pharmacokinetics and pharmacodynamics of subcutaneous heparin during the early third trimester of pregnancy. Am J Obstet Gynecol. 1995;173:1240–1245.
99. Cutts BA, Dasgupta D, Hunt BJ. New directions in the diagnosis and treatment of pulmonary embolism in pregnancy. Am J Obstet Gynecol. 2013;208:102–108.
100. Ruppen W, Derry S, McQuay H, Moore RA. Incidence of epidural hematoma, infection, and neurologic injury in obstetric patients with epidural analgesia/anesthesia. Anesthesiology. 2006;105:394–399.
101. Horlocker TT, Wedel DJ, Rowlingson JC, et al. Regional anesthesia in the patient receiving antithrombotic or thrombolytic therapy: American Society of Regional Anesthesia and Pain Medicine Evidence-Based Guidelines (Third Edition). Reg Anesth Pain Med. 2010;35:64–101.
102. Jea A, Moza K, Levi AD, Vanni S. Spontaneous spinal epidural hematoma during pregnancy: case report and literature review. Neurosurgery. 2005;56:E1156.
103. Forsnes E, Occhino A, Acosta R. Spontaneous spinal epidural hematoma in pregnancy associated with using low molecular weight heparin. Obstet Gynecol. 2009;113:532–533.
104. Case AS, Ramsey PS. Spontaneous epidural hematoma of the spine in pregnancy. Am J Obstet Gynecol. 2005;193:875–877.
105. Gogarten W, Vandermeulen E, Van Aken H, et al. Regional anaesthesia and antithrombotic agents: recommendations of the European Society of Anaesthesiology. Eur J Anaesthesiol. 2010;27:999–1015.
106. Royal College of Obstetricians and Gynaecologists. Green-top Guideline no. 37b. The acute management of thrombosis and embolism during pregnancy and the puerperium. Royal College of Obstetricians and Gynecologists: London; 2010 http://www.rcog.org.uk/files/rcog-corp/GTG37b_230611.pdf.
107. Dickman CA, Shedd SA, Spetzler RF, et al. Spinal epidural hematoma associated with epidural anesthesia: complications of systemic heparinization in patients receiving peripheral vascular thrombolytic therapy. Anesthesiology. 1990;72:947–950.
108. Lawton MT, Porter RW, Heiserman JE, et al. Surgical management of spinal epidural hematoma: relationship between surgical timing and neurological outcome. J Neurosurg. 1995;83:1–7.
109. Cheney FW, Domino KB, Caplan RA, Posner KL. Nerve injury associated with anesthesia: a closed claims analysis. Anesthesiology. 1999;90:1062–1069.
110. Duff P. A simple checklist for preventing major complications associated with cesarean delivery. Obstet Gynecol. 2010;116:1393–1396.
111. Tooher R, Gates S, Dowswell T, Davis LJ. Prophylaxis for venous thromboembolic disease in pregnancy and the early postnatal period. Cochrane Database Syst Rev. 2010;(5).
112. Quinones JN, James DN, Stamilio DM, et al. Thromboprophylaxis after cesarean delivery: a decision analysis. Obstet Gynecol. 2005;106:733–740.
113. Casele H, Grobman WA. Cost-effectiveness of thromboprophylaxis with intermittent pneumatic compression at cesarean delivery. Obstet Gynecol. 2006;108:535–540.
114. Mirski MA, Lele AV, Fitzsimmons L, Toung TJ. Diagnosis and treatment of vascular air embolism. Anesthesiology. 2007;106:164–177.
115. Malinow AM, Naulty JS, Hunt CO, et al. Precordial ultrasonic monitoring during cesarean delivery. Anesthesiology. 1987;66:816–819.
116. Karuparthy VR, Downing JW, Husain FJ, et al. Incidence of venous air embolism during cesarean section is unchanged by the use of a 5 to 10 degree head-up tilt. Anesth Analg. 1989;69:620–623.
117. Fong J, Gadalla F, Pierri MK, Druzin M. Are Doppler-detected venous emboli during cesarean section air emboli? Anesth Analg. 1990;71:254–257.
118. Vartikar JV, Johnson MD, Datta S. Precordial Doppler monitoring and pulse oximetry during cesarean delivery: detection of venous air embolism. Reg Anesth. 1989;14:145–148.
119. Handler JS, Bromage PR. Venous air embolism during cesarean delivery. Reg Anesth. 1990;15:170–173.
120. Lew TW, Tay DH, Thomas E. Venous air embolism during cesarean section: more common than previously thought. Anesth Analg. 1993;77:448–452.
121. Adornato DC, Gildenberg PL, Ferrario CM, et al. Pathophysiology of intravenous air embolism in dogs. Anesthesiology. 1978;49:120–127.
122. Giebler R, Kollenberg B, Pohlen G, Peters J. Effect of positive end-expiratory pressure on the incidence of venous air embolism and on the cardiovascular response to the sitting position during neurosurgery. Br J Anaesth. 1998;80:30–35.
123. Cluver C, Novikova N, Hofmeyr GJ, Hall DR. Maternal position during caesarean section for preventing maternal and neonatal complications. Cochrane Database Syst Rev. 2010;(6).
124. Kaunitz AM, Hughes JM, Grimes DA, et al. Causes of maternal mortality in the United States. Obstet Gynecol. 1985;65:605–612.
125. Hoppe KK, Fialkow MF, Dighe M, Cheng E. Intrauterine air embolism associated with a rectovaginal fistula in a pregnant woman. Obstet Gynecol. 2011;118:481–484.
126. Hill BF, Jones JS. Venous air embolism following orogenital sex during pregnancy. Am J Emerg Med. 1993;11:155–157.
127. Nelson PK. Pulmonary gas embolism in pregnancy and the puerperium. Obstet Gynecol Surv. 1960;15:449–481.
129. Mathew JP, Fleisher LA, Rinehouse JA, et al. ST segment depression during labor and delivery. Anesthesiology. 1992;77:635–641.
130. Palmer CM, Norris MC, Giudici MC, et al. Incidence of electrocardiographic changes during cesarean delivery under regional anesthesia. Anesth Analg. 1990;70:36–43.
131. Svanstrom MC, Biber B, Hanes M, et al. Signs of myocardial ischaemia after injection of oxytocin: a randomized double-blind comparison of oxytocin and methylergometrine during Caesarean section. Br J Anaesth. 2008;100:683–689.
132. Jonsson M, Hanson U, Lidell C, Norden-Lindeberg S. ST depression at caesarean section and the relation to oxytocin dose: a randomised controlled trial. BJOG. 2010;117:76–83.
133. Geissler HJ, Allen SJ, Mehlhorn U, et al. Effect of body repositioning after venous air embolism: an echocardiographic study. Anesthesiology. 1997;86:710–717.
134. Blanc P, Boussuges A, Henriette K, et al. Iatrogenic cerebral air embolism: importance of an early hyperbaric oxygenation. Intensive Care Med. 2002;28:559–563.
* The U.S. Food and Drug Administration recommends waiting at least 4 hours after removal of a neuraxial catheter before administering a postprocedure dose of LMWH. (U.S. Food and Drug Administration. Low molecular weight heparins: drug safety communication—recommendations to decrease risk of spinal column bleeding and paralysis. November 6, 2013. Available at http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm373918.htm. Accessed November 2013.)
[/level-membership-for-anesthesiology-category][not-level-membership-for-anesthesiology-category]
Embolic Disorders
Paloma Toledo MD, MPH, Andrew M. Malinow MD
Chapter Outline
Embolic disease during pregnancy includes amniotic fluid embolism, venous thromboembolism, and venous air embolism. Each of these entities varies in its incidence, clinical course, and consequences. Embolic events account for almost one fifth of all maternal deaths in the United States.1 Early recognition, diagnosis, and treatment are necessary to reduce associated morbidity and to avoid mortality.
Amniotic Fluid Embolism
Death attributable to an amniotic fluid embolism (AFE) was first reported by Meyer in 1926.2 Early reports described a syndrome of fatal peripartum shock attributed to emboli of amniotic fluid mechanically obstructing the maternal pulmonary circulation.3 Although the pathophysiology of this disease remains poorly understood, current evidence suggests that emboli alone are insufficient to precipitate this infrequent, idiosyncratic, and devastating condition. Rather, fetal material in the maternal circulation has the potential to trigger a massive cascade of inflammatory and hemostatic reactions that culminate in cardiopulmonary collapse and disseminated intravascular coagulation.
Epidemiology
The incidence of amniotic fluid embolism is difficult to establish because (1) AFE is a diagnosis of exclusion, (2) there is no universally accepted definition for identifying cases of AFE, and (3) differing ascertainment methods yield divergent rates of AFE. Registry data from the United Kingdom suggest an event rate between 0.8 and 2 per 100,000 deliveries.4,5 Cross-sectional analyses of administrative data in Australia and the United States suggest higher rates—3.3 and 7.7 per 100,000 deliveries, respectively.6,7 A systematic review published in 2009 estimated that the pooled incidence of AFE in North America was approximately 1 : 15,200 (95% confidence interval [CI], 1 : 13,900 to 1 : 16,700), whereas the incidence in Europe was three times lower at 1 : 53,800 (95% CI, 1 : 48,800 to 1 : 59,900).8 This heterogeneity likely reflects variations in ascertainment procedures, rather than true differences by continent of origin. One limitation of analyses of secondary databases is that the diagnosis of AFE may not have been validated. For example, one regional surveillance system in Australia has developed the capacity to systematically review records for all cases identified from administrative data. By only counting those women who experienced one of the cardinal symptoms of AFE, with no other potential explanation, the reported incidence decreased from 6.3 to 3.3 cases per 100,000 pregnancies.9
Risk Factors
Maternal demographic factors such as older age and race or ethnicity have been associated with AFE in population-based studies.5,7 Other obstetric factors such as abnormal placentation, placental abruption, eclampsia, multiple gestation, induction of labor, artificial or spontaneous rupture of membranes, and operative delivery have also been associated with AFE.5,7,10–12 Because nonreassuring fetal heart rate (FHR) tracings can complicate AFE in labor, cesarean delivery may be a consequence, rather than cause, of intrapartum AFE. Nonetheless, a strong association between cesarean birth and postpartum AFE persists among women in the United Kingdom Obstetric Surveillance System (UKOSS) dataset (adjusted odds ratio, 8.8).5 The proportion of excess postpartum AFE events associated with cesarean delivery (the population-proportional attributable risk for cesarean delivery) from the UKOSS dataset was estimated to be 62%.5
Pathophysiology
In 1941, Drs. Steiner and Lushbaugh, two pathologists from the University of Chicago, described a case series of eight autopsies after fatal intrapartum shock.3 Examination of lung tissue from these cases revealed embolic material of squamous cells, mucin, meconium, and amorphous eosinophilic material.3 Because all of these patients were described as having tumultuous labors with stronger than usual uterine contractions, it was presumed that the forceful contractions loosened or tore the placenta and forced the emboli into the maternal circulation.3 Yet, periods of uterine tachysystole are the least likely times for maternoplacental exchange of embolic material to occur as uterine blood flow ceases.13 The tachysystole is probably a result of endogenous norepinephrine release and, therefore, is likely temporally related to, but not causative for, embolic material transfer.13
Large intravenous boluses of human meconium suspended in human amniotic fluid can precipitate cardiovascular collapse in rabbits and dogs,3 but injection of autologous amniotic fluid fails to reproduce the AFE syndrome in many animal models.14 The passage of fetal squames, lanugo hair, and mucin into the maternal pelvic vasculature appears to be a common event at term,15 and fetal material has been identified in pulmonary arterial samples aspirated from critically ill women who did not have AFE.16–18
The exact trigger for the reaction in women with AFE is not known but may be a rare pathologic fetal antigen or a common antigen presented in an unusual way—in amount, timing, or frequency of entry into the maternal circulation.19 AFE appears to be a systemic inflammatory response associated with the inappropriate release of endogenous inflammatory mediators.20 Whatever the trigger, several maternal endogenous mediators appear to play an important role in the initial reaction, including arachidonic acid metabolites (i.e., thromboxane, prostaglandins, leukotrienes, endothelins).20
Approximately 40% of women in a United States national AFE registry had a history of allergy or atopy, leading some authors to suggest an anaphylactoid mechanism.12 In support of this theory, the symptoms of AFE could be blocked by the administration of a leukotriene inhibitor in a rabbit model.21 However, a series of case reports now suggests that levels of tryptase and histamine are not dramatically or universally elevated among women experiencing the AFE syndrome.22,23 Although mast cell degranulation may contribute to the pathophysiology of AFE, this effect may be a secondary product of the inflammatory cascade, rather than causal, and thus the term anaphylactoid syndrome of pregnancy may be a misnomer.23
Other immune-mediated mechanisms have also been implicated in AFE. A case-control study demonstrated low levels of the complement components C3 and C4 among women diagnosed with AFE compared with controls, suggesting that complement activation may also contribute to the inflammatory cascade.22 Whether complement plays a primary19 or secondary role24 in the AFE syndrome is unknown. A heat-stable pressor agent in meconium has been suggested as another possible mediator of AFE; in a goat model this agent caused a circulatory response similar to that seen in human AFE.25
Alternatively, the hemodynamic consequences of AFE could derive from activation of the coagulation system, mediated through platelet activation. Fetal squamous cells and syncytiotrophoblasts display high concentrations of tissue factor and phosphatidylserine.26–28 Tissue factor irreversibly aggregates platelets,29 leading to platelet degranulation, which releases thromboxane, serotonin, and additional mediators that amplify the immunologic systems. Serotonin is a potent pulmonary vasoconstrictor that produces vasodilation in the systemic vasculature and may lead to early right-sided heart failure in the AFE syndrome.27,30,31
Coagulopathy develops in the majority of women who survive the initial cardiovascular collapse.12,32 One proposed mechanism involves tissue factor. As pregnancy progresses, increasing amounts of tissue factor, a potent procoagulant, accumulates in the amniotic fluid.27,28 Tissue factor binds factor VII, thus activating the extrinsic pathway and triggering clotting by activating factor X, with the subsequent development of a consumptive coagulopathy.27 A second possible mechanism is that amniotic fluid has a thromboplastin-like effect, which induces platelet aggregation, releases platelet factor III, and activates the clotting cascade.33 Other components of amniotic fluid, the amniochorion, and the placenta have also been implicated in contributing to the coagulopathy. Uterine atony, whether due to a specific myometrial depressant or from uterine hypoperfusion, may exacerbate hemorrhage and consumptive coagulopathy in AFE.34
There is conflicting evidence regarding whether the bleeding seen in AFE is due primarily to a consumptive coagulopathy versus massive fibrinolysis. In one in vitro study, investigators added 10 to 60 µL of clear autologous amniotic fluid to 330 µL of blood from volunteers undergoing uneventful cesarean delivery.35 Thromboelastographic analysis of the specimens revealed accelerated clot formation but no evidence of fibrinolysis. However, because of the rapid and severe hypofibrinogenemia observed during AFE, some investigators have suggested that severe hyperfibrinolysis is also present.36 Future work is necessary to further refine our understanding of the coagulopathy that accompanies AFE.
Clinical Presentation
Whereas the classic presentation of AFE includes acute respiratory distress, cardiovascular collapse, and coagulopathy near the time of delivery, a broad range of AFE syndromes have been described in situations in which other diagnoses were excluded (Table 39-1).12,20,37
TABLE 39-1
Features of Amniotic Fluid Embolism at Presentation
| Percent Exhibiting Feature (n = 60)* |
Percent Exhibiting Feature as First Symptom or Sign (n = 60)* |
|
| Maternal hemorrhage† | 65% | 2% |
| Hypotension† | 63% | 8% |
| Shortness of breath† | 62% | 20% |
| Coagulopathy† | 62% | 0% |
| Premonitory symptoms† (i.e., restlessness, agitation, numbness, tingling) | 47% | 30% |
| Acute fetal compromise | 43% | 20% |
| Cardiac arrest | 40% | 8% |
| Dysrhythmias | 27% | 5% |
| Seizure | 15% | 7% |
* Some women had multiple features; therefore, the totals are greater than 100%.
† Cardinal feature of amniotic fluid embolism. Twenty-seven percent of women experienced at least four of the five cardinal features.
Data from Knight M, Tuffnell D, Brocklehurst P, et al. Incidence and risk factors for amniotic-fluid embolism. Obstet Gynecol 2010; 115:910-7.
Currently, the UKOSS provides the most comprehensive prospective surveillance for amniotic fluid embolism in the world. All hospitals in the United Kingdom with a consultant-led maternity unit report all suspected cases of AFE, along with monthly delivery volume data, for central review and reporting.5,38 Cases of AFE are defined using either clinical criteria that include acute hypotension, cardiac arrest, acute hypoxemia, and/or coagulopathy in the absence of any other potential explanation for the symptoms and signs observed, or pathologic evidence indicating the presence of fetal squames or hair in the maternal lungs. All women in the UKOSS registry demonstrated at least one cardinal feature of AFE, including shortness of breath, hypotension, coagulopathy, maternal hemorrhage, or premonitory symptoms (see Table 39-1).5
A national registry in the United States accepted voluntary submissions of medical records for patients with suspected AFE beginning in 1988. The diagnosis of AFE was confirmed based on meeting all entry criteria outlined in Box 39-1. Data for 46 confirmed cases of AFE were last published in 1995.12
AFE most often occurs during labor; intrapartum events comprise 56% of cases in the UKOSS registry5 and 70% in the U.S. registry.12 In the U.S. registry, seizure and dyspnea were the two most common presenting symptoms in women who collapsed before delivery.12 Maternal symptoms may precede FHR changes, as shown in Figure 39-1. Fetal bradycardia, or the abrupt onset of variable decelerations that progress to fetal bradycardia, may also herald AFE in labor.12
FIGURE 39-1 Fetal heart rate tracing in a patient with amniotic fluid embolism. Maternal symptoms began just before the onset of spontaneous uterine hypertonus and fetal bradycardia. (Modified from Clark SL, Hankins GDV, Dudley DA, et al. Amniotic fluid embolism: analysis of the national registry. Am J Obstet Gynecol 1995; 172:1158-67.)
AFE can present after abdominal trauma,39 after first-trimester abortion,40 in the second trimester,41 at the time of delivery,12 and in the postpartum period.42 Although acknowledging that AFE has been reported up to 48 hours postpartum,34 both the U.S. and U.K. registries require the diagnosis of AFE to be made within 30 minutes of delivery.4,12
Close inspection of hemodynamic data reveals a biphasic cardiovascular response during AFE. During the initial phase, acute pulmonary hypertension results in right ventricular dilation, a decrease in cardiac output, and ventilation-perfusion () mismatch resulting in oxygen desaturation. Early arterial blood gas analysis may demonstrate evidence of profound shunt.12 In the U.S. registry, 11 of 17 patients with a blood gas sample drawn within 30 minutes of the acute event demonstrated an initial PaO2 less than 30 mm Hg while breathing an FIO2 of 100%.12 Release of endogenous catecholamines may produce a brief period of systemic hypertension and uterine tachysystole that precedes hypotension or cardiac arrest.12 Electrocardiographic findings are nonspecific and vary from ST-wave and T-wave abnormalities to arrhythmias or asystole. A chest radiograph may show diffuse bilateral heterogeneous or homogenous areas of opacity.
Echocardiography typically demonstrates a dilated, akinetic right ventricle, pulmonary hypertension, and a normally-contracting left ventricle with a nearly obliterated cavity.43,44 Initially, right ventricular failure leads to right ventricular dilation, which compresses the left ventricle and impedes left ventricular filling and cardiac output.43,44 A second phase commences when right ventricular function improves,44 typically 15 to 30 minutes after the initial event. At this point, left ventricular failure may persist due to ischemic injury to the left ventricle,45 or direct myocardial depression,34 and is accompanied by decreased systemic ventricular resistance, decreased left ventricular stroke index, and pulmonary edema.46,47 Women who survive to the second phase may also experience hemorrhage and disseminated intravascular coagulopathy. Laboratory analysis may reveal anemia, thrombocytopenia, prolonged prothrombin time or activated partial thromboplastin time (aPTT) or both, and decreased fibrinogen levels.12,34 In a minority of cases there is massive hemorrhage and disseminated intravascular coagulopathy without preceding cardiopulmonary collapse.37
Because many of the signs and symptoms are nonspecific, the differential diagnosis for AFE is extensive and should include nonobstetric, obstetric, and anesthetic causes (Box 39-2). Even though the time course and clinical presentation of many of these competing diagnoses are similar, only amniotic fluid embolism and placental abruption result in a relatively sudden onset of consumptive coagulopathy after maternal collapse. However, partial AFE syndromes have been described in the literature.20 Therefore, the absence of coagulopathy should not exclude the possibility of AFE.
Confirmatory Tests
To date, there is no definitive test to confirm the diagnosis of AFE, although the UKOSS considers the finding of fetal material in the maternal pulmonary vasculature at autopsy to be pathognomonic for AFE.5 However, fetal squamous cells and trophoblasts are commonly found in the maternal circulation of healthy parturients. Furthermore, differentiating between maternal and fetal cells histologically is challenging.18 Clinicians should therefore not place invasive monitors solely for the purpose of aspirating cells of fetal origin.
Several biochemical markers have been suggested. Although some of these may be promising based on preliminary data and theoretical understanding of AFE, studies of test performance are limited by delayed sample acquisition and small sample size owing to the rarity and unpredictability of the AFE syndrome.
Several markers suggest an anaphylactic/anaphylactoid mechanism. Mast cells release tryptase and histamine during degranulation; tryptase has been used as a marker for anaphylaxis because its half-life is longer than that of histamine. Elevations in serum tryptase have been reported in some parturients with AFE.23 However, a case series found normal tryptase and urinary histamine levels in nine women with presumed AFE.22 Pulmonary mast cell counts have also been suggested; however, this measurement can only be obtained using immunohistochemistry at autopsy and therefore is of limited applicability in the clinical setting. In one observational study, the mean pulmonary mast cell count per fixed area for parturients who died of AFE was similar to that of parturients who died of anaphylactic shock but was higher than the mean counts in both pregnant and nonpregnant control patients.48 These data, together with the finding of minimal to no elevation in serum tryptase, suggest that pulmonary mast cell degranulation may be a secondary process in AFE.23
Complement activation may cause mast cell degranulation, and there is some evidence supporting widespread complement activation, and depressed C3 and C4 levels, in AFE.22 However, because complement is activated during acute respiratory distress syndrome and other inflammatory states, results are not specific for AFE. Zinc coproporphyrin49 and sialyl Tn antigen22 are two biomarkers that are components of meconium and have been associated with the AFE syndrome. Sialyl Tn is a mucinous glycoprotein that originates in the fetal gastrointestinal tract and is also associated with mucinous gastrointestinal tumors. Most recently, insulin-like growth factor–binding protein-1 (IGFBP-1) was identified as a sensitive and specific biomarker for AFE in a case-control study conducted in 13 delivery centers in France.50 IGFBP-1 levels exceeded 104.5 µg/L in 23 of 25 women with AFE and remained below 95 µg/L in all patients with postpartum hemorrhage due to atony, thrombotic pulmonary embolism, or uncomplicated labor. A single false-positive result was found in a woman with acute fatty liver of pregnancy. Based on the median concentration of IGFBP-1 in the amniotic fluid and blood samples, the investigators estimated that 6 to 92 mL of amniotic fluid passed into the maternal circulation in women who experienced the AFE syndrome.50
Management
Although no single intervention has been shown to reliably reverse the AFE syndrome, prompt recognition and aggressive resuscitation may improve maternal and fetal outcomes. Maternal resuscitation should focus on three priorities: (1) maintenance of oxygenation; (2) hemodynamic support; and (3) correction of coagulopathy (Box 39-3).
On diagnosis of AFE, 100% oxygen should be delivered to the patient. Given the risk for coagulopathy and hemorrhage, large-bore intravenous access is warranted. An arterial line and central venous pressure catheter may facilitate hemodynamic monitoring, blood sampling, and vasopressor administration. Transesophageal echocardiography may be useful to guide volume resuscitation and selection of appropriate vasopressor therapy.
In addition to standard resuscitative measures, other management strategies have been reported for AFE. The use of cardiopulmonary bypass, extracorporeal membrane oxygenation, continuous hemofiltration, and exchange transfusions have all been described in the literature.5,43,51 It is speculated that these technologies may filter amniotic fluid or vasoactive mediators from the systemic circulation. Strategies for management of the early right-sided heart failure seen in AFE include inhaled nitric oxide, prostacyclin, right ventricular assist devices, and vasopressors such as vasopressin, dobutamine, and milrinone.52,53 The use of extracorporal membrane oxygenation and intra-aortic balloon counterpulsation has also been reported for management of left-sided heart failure.54
In AFE, intact neonatal survival is related to the time interval from the onset of maternal compromise to delivery.12 In the event of maternal cardiopulmonary arrest, the American Heart Association recommends that delivery of the fetus should occur within 5 minutes to increase the probability of good outcomes for both the mother and her neonate.55 Although the operating room may provide a more favorable environment for surgery and resuscitation, simulation studies have suggested that resuscitation quality and the arrest-to-delivery interval both suffer with maternal transport56,57; therefore, strong consideration should be given to performing a bedside perimortem cesarean delivery.
Because coagulopathy will likely ensue for the majority of AFE survivors, the obstetric and anesthesia providers should activate a massive transfusion protocol as soon as AFE is suspected. Blood and component therapy should be guided by the clinical presentation (see Chapter 38). Close communication with the blood bank is paramount because large quantities of blood products may be needed. Analysis of the U.K. registry demonstrated that patients who developed a coagulopathy received between 12 and 106 units of blood products.4 Potential pharmacologic therapies for the coagulopathy associated with AFE include antifibrinolytic agents (e.g., tranexamic acid, aprotinin), recombinant factor VIIa (rVIIa), prothrombin complex concentrate, and fibrinogen concentrate.36,58,59
The use of rVIIa to treat intractable hemorrhage in the AFE syndrome is controversial. The use is off-label in obstetric hemorrhage (see Chapter 38). Recombinant factor VIIa binds to tissue factor and initiates clotting via the extrinsic pathway. Although individual case reports have described improvement in hemostasis, rVIIa has been associated with thrombotic complications. A meta-analysis of 25 trials involving 3849 nonobstetric bleeding patients without hemophilia found a significant increase in arterial thromboembolic events among patients who received rVIIa (relative risk [RR], 1.45; 95% CI, 1.02 to 2.05).60
A case-controlled study based on a systematic review of case reports of women with the AFE syndrome identified a twofold increase in risk for death or permanent disability among 16 women treated with rVIIa compared with 28 control patients who underwent surgery to control bleeding but did not receive rVIIa.58 Among survivors, treatment with rVIIa was associated with more permanent disability (risk ratio, 4.0; 95% CI, 1.5 to 10.4), largely attributed to thrombosis in major organs.58 An unmeasured increase in severity of disease in the treatment group compared with the control group could explain these dismal results because the population of patients who did not survive to the time of operation was excluded from the analysis. In this case-controlled study of case reports,58 50% of parturients treated with rVIIa survived, whereas in the 2010 UKOSS dataset,5 93% of the parturients treated with rVIIa survived (n = 14). However, the UKOSS data did not report neurologic outcomes or thromboembolic complications.5 The differences in outcomes between these two studies may be explained in part by differing definitions of AFE and differences in case ascertainment. Only five cases in the case-controlled study58 were peer-reviewed publications; the remainder were obtained from abstracts presented at national meetings and from registry data.
Maternal and Perinatal Outcomes
The maternal mortality ratio associated with AFE has been reported between 0.5 and 1.7 deaths per 100,000 live births.8 AFE accounted for 7.5% of pregnancy-related deaths in the United States between 1998 and 2005.1 The case-fatality rate from the UKOSS data was 20% (95% CI, 11 to 32),5 significantly lower than the reported fatality rate of 61% in the 1988 to 1994 analysis of the U.S. AFE registry.12 In the UKOSS dataset, patients who died of AFE were more likely to be from ethnic minority groups than were survivors, even after adjusting for confounders such as age, socioeconomic status, obesity, and parity (adjusted odds ratio [aOR], 11.8; 95% CI, 1.4 to 99.5).5 Similar racial/ethnic disparities were seen in the United States.1 Improvements in the management of critically ill parturients may have contributed to the decline in the case-fatality rate observed in the two studies, but the disparity in outcomes by race or ethnicity suggests further systems improvements are necessary.
Cardiac arrest complicated 87% of AFE cases reported to the U.S. registry but only 40% of cases identified by UKOSS.5,12 This difference may reflect more comprehensive ascertainment in the United Kingdom that captures less severe cases; alternatively, improvements in early recognition and management of AFE over the past 15 years may explain improved outcomes recently reported from the United Kingdom compared with those previously published from the U.S. registry.
Neurologic outcomes for survivors are poor. The 1995 analysis of the U.S. registry reported a 15% overall rate of intact neurologic survival, with only 8% neurologically intact after cardiac arrest.5,12 Neonates have a high overall survival rate, with approximately 80% of infants surviving,5,12 but also have low rates of intact neurologic survival, with only 39% of survivors neurologically intact.12 Intact neurologic survival for infants is related to the cardiac arrest-to-delivery interval; delays of greater than 15 minutes are associated with worse outcomes.12
Recurrent AFE has not been reported.
Thromboembolic Disorders
Incidence
Venous thromboembolic events (VTE) in pregnancy refer to deep vein thrombosis (DVT) and pulmonary thromboembolism (PTE). The incidence of pregnancy-related thromboembolic events is 1.0 to 1.7 events per 1,000 pregnancies.61–63 The range of reported incidences may reflect differing study designs and diagnostic criteria for thromboembolism, in addition to biologic differences among populations. Analysis of administrative data from a nationally representative sample of United States hospital admissions that included 9,058,162 pregnancy admissions and 73,834 postpartum admissions demonstrated 1.36 cases of DVT and 0.36 cases of PTE per 1,000 deliveries.61 Fifteen to 24 percent of pregnant women with an untreated DVT develop a PTE.61,64,65
There is a fivefold greater odds of thromboembolic events during pregnancy (odds ratio [OR], 4.6; 95% CI, 2.7 to 7.8), and a 60-times greater odds in the postpartum period (95% CI, 26.5 to 135.9) than in nonpregnant patients.66 Study results conflict as to whether there is an increased risk by trimester. A meta-analysis of 12 studies that evaluated the period of risk for DVT in pregnancy found that 21.9% of antepartum DVTs develop in the first trimester (95% CI, 17.4 to 27.3), 33.7% in the second trimester (95% CI, 28.1 to 39.8), and 47.6% in the third trimester (95% CI, 39.2% to 56.2%).67
[/not-level-membership-for-anesthesiology-category]
