DNA Damage Response Pathways and Cancer
James M. Ford and Michael B. Kastan
• DNA repair and the cellular response to DNA damage are critical for maintaining genomic stability.
• Defects in DNA repair or the response to DNA damage encountered from endogenous or external sources results in an increased rate of genetic mutations, often leading to the development of cancer.
• Inherited mutations in DNA damage response pathway genes often result in cancer susceptibility.
• The major active pathways for DNA repair in humans are nucleotide excision repair, base excision repair, mismatch DNA repair, translesional DNA synthesis, and homologous recombination or nonhomologous end joining processes for double-strand break repair.
• Inherited defects in nucleotide excision repair lead to the skin cancer–prone syndrome xeroderma pigmentosum, as well as Cockayne syndrome and trichothiodystrophy.
• Inherited defects in base excision repair can result in colorectal cancer susceptibility.
• Inherited defects in mismatch repair result in Lynch syndrome (hereditary nonpolyposis colorectal cancer syndrome), leading to increased incidence of gastrointestinal cancers, endometrial cancer, and other malignancies.
• Inherited defects in DNA double-strand break repair and response pathways underlie a number of cancer prone disorders, including ataxia-telangiectasia, Nijmegen breakage syndrome, Bloom syndrome, Werner syndrome, Rothmund-Thomson syndrome, and Fanconi anemia.
• Persons with Li-Fraumeni syndrome, who are highly prone to cancer because of inherited p53 mutations, and persons with breast-ovarian cancer syndrome, who are highly prone to cancer because of inherited mutations of the BRCA1 and BRCA2 genes, exhibit defects in multiple DNA repair and DNA damage response pathways.
• Because most cancer therapeutic agents that are currently used damage DNA, understanding how normal cells and tumor cells respond to and repair DNA damage is an important aspect of understanding cancer therapeutic responses and toxicities of treatment.
• The presence of mutations in DNA damage response and repair pathways in tumors present opportunities for developing therapeutics that target alternative damage response pathways and can be selectively lethal to the mutated tumor cells, an approach called “synthetic lethality.”
Introduction
Cancer is a disease caused in large part by the accumulation over time of changes to the normal DNA sequence, resulting in alterations, loss, or amplification of genes important for normal cellular functions and growth properties, including many proto-oncogenes and tumor suppressor genes. Nearly all cancers are clonal in origin; that is, they originate from a single progenitor cell rather than from a group of cells. The development of cancer in a particular cell type or tissue is caused by a series of specific mutations, each of which could be caused by DNA replication errors or unrepaired endogenous or exogenous DNA damage, or which could be the result of inherited mutations. In persons with the most common cancers, multiple genetic events occur in many different genes during the process of carcinogenesis, suggesting that an early and perhaps necessary event in the cancer process is an underlying defect in mechanisms to maintain genomic stability.3–3 In fact, alterations in the specific genes required for recognizing, processing, and responding to DNA damage may result in an enhanced rate of accumulation of additional mutations, recombinational events, chromosomal abnormalities, and gene amplification.4 Whether such a “mutator phenotype” is a required event for human tumorigenesis remains a controversial issue,5,6 but clear examples of such a process are evident and will be considered here.
Many converging lines of experimental evidence reveal the complexity of the cellular responses to DNA damage and their role in malignant transformation.7 A number of interrelated biochemical pathways exist that influence the following actions: (1) the metabolism of potentially mutagenic or carcinogenic agents, (2) the efficiency and manner by which damaged DNA is recognized and repaired, (3) cell cycle progression and the coordination of DNA replication and cell division relative to the repair of lesions, and (4) the decision point determining survival or the active induction of programmed death of cells carrying different types and amounts of DNA damage. Many cellular pathways have evolved that require hundreds of gene products for the repair of DNA damage and involve excision of damaged DNA bases, joining of broken DNA strands, or replication bypass of DNA lesions (Table 10-1). The central role of DNA damage responses in neoplastic transformation has been highlighted by the discovery that mutations in numerous classes of genes required for DNA repair and the maintenance of genomic integrity result in a predisposition to the development of many malignancies.8 In fact, a number of rare, inherited disorders have been described that appear to be caused by defects in the repair of DNA lesions (Table 10-2), and many of these disorders are associated with an increased risk of the development of cancers.9
Table 10-1
| DNA Repair Pathway | Type of DNA Damage | Approximate No. of Genes |
| Nucleotide excision repair | Bulky or helix-distorting DNA adducts, for example, ultraviolet photoproducts and carcinogen adducts | 37 |
| Base excision repair | Oxidative DNA damage; spontaneous depurination | 40 |
| Mismatch repair | Mispaired nucleotides | |
| 1-15 nucleotide insertion-deletion loops | 26 | |
| Homologous recombination | Double-strand DNA breaks, DNA cross-links | 20 |
| Nonhomologous end joining | Double-strand DNA breaks | 10 |
Table 10-2
Human Genetic Diseases Involving Defects in DNA Damage Response Pathways
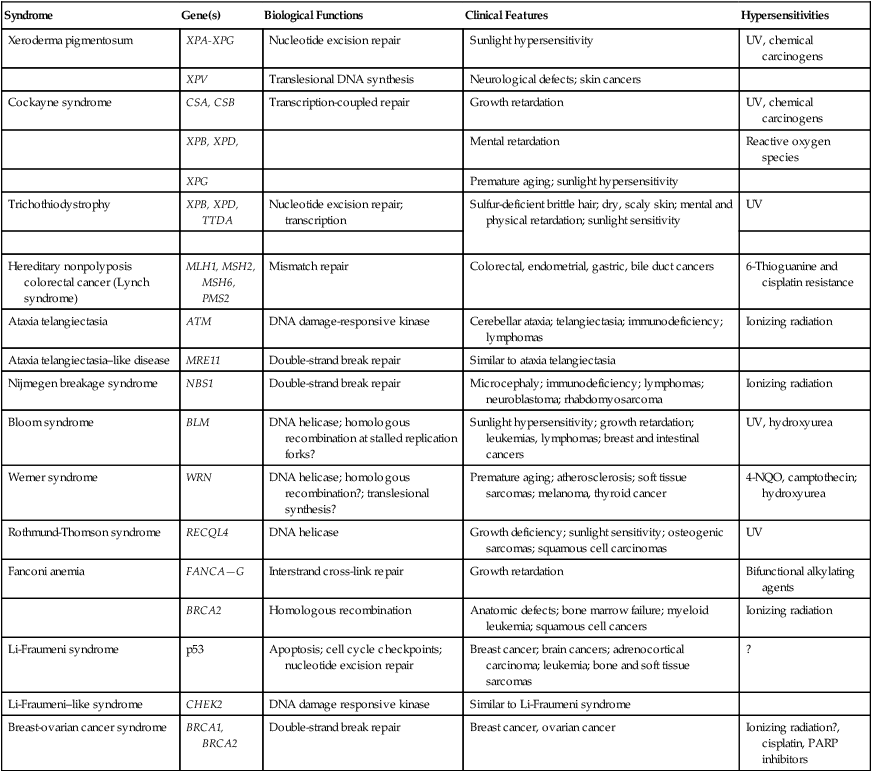
| Syndrome | Gene(s) | Biological Functions | Clinical Features | Hypersensitivities |
| Xeroderma pigmentosum | XPA-XPG | Nucleotide excision repair | Sunlight hypersensitivity | UV, chemical carcinogens |
| XPV | Translesional DNA synthesis | Neurological defects; skin cancers | ||
| Cockayne syndrome | CSA, CSB | Transcription-coupled repair | Growth retardation | UV, chemical carcinogens |
| XPB, XPD, | Mental retardation | Reactive oxygen species | ||
| XPG | Premature aging; sunlight hypersensitivity | |||
| Trichothiodystrophy | XPB, XPD, TTDA | Nucleotide excision repair; transcription | Sulfur-deficient brittle hair; dry, scaly skin; mental and physical retardation; sunlight sensitivity | UV |
| Hereditary nonpolyposis colorectal cancer (Lynch syndrome) | MLH1, MSH2, MSH6, PMS2 | Mismatch repair | Colorectal, endometrial, gastric, bile duct cancers | 6-Thioguanine and cisplatin resistance |
| Ataxia telangiectasia | ATM | DNA damage-responsive kinase | Cerebellar ataxia; telangiectasia; immunodeficiency; lymphomas | Ionizing radiation |
| Ataxia telangiectasia–like disease | MRE11 | Double-strand break repair | Similar to ataxia telangiectasia | |
| Nijmegen breakage syndrome | NBS1 | Double-strand break repair | Microcephaly; immunodeficiency; lymphomas; neuroblastoma; rhabdomyosarcoma | Ionizing radiation |
| Bloom syndrome | BLM | DNA helicase; homologous recombination at stalled replication forks? | Sunlight hypersensitivity; growth retardation; leukemias, lymphomas; breast and intestinal cancers | UV, hydroxyurea |
| Werner syndrome | WRN | DNA helicase; homologous recombination?; translesional synthesis? | Premature aging; atherosclerosis; soft tissue sarcomas; melanoma, thyroid cancer | 4-NQO, camptothecin; hydroxyurea |
| Rothmund-Thomson syndrome | RECQL4 | DNA helicase | Growth deficiency; sunlight sensitivity; osteogenic sarcomas; squamous cell carcinomas | UV |
| Fanconi anemia | FANCA—G | Interstrand cross-link repair | Growth retardation | Bifunctional alkylating agents |
| BRCA2 | Homologous recombination | Anatomic defects; bone marrow failure; myeloid leukemia; squamous cell cancers | Ionizing radiation | |
| Li-Fraumeni syndrome | p53 | Apoptosis; cell cycle checkpoints; nucleotide excision repair | Breast cancer; brain cancers; adrenocortical carcinoma; leukemia; bone and soft tissue sarcomas | ? |
| Li-Fraumeni–like syndrome | CHEK2 | DNA damage responsive kinase | Similar to Li-Fraumeni syndrome | |
| Breast-ovarian cancer syndrome | BRCA1, BRCA2 | Double-strand break repair | Breast cancer, ovarian cancer | Ionizing radiation?, cisplatin, PARP inhibitors |
Types of DNA Damage
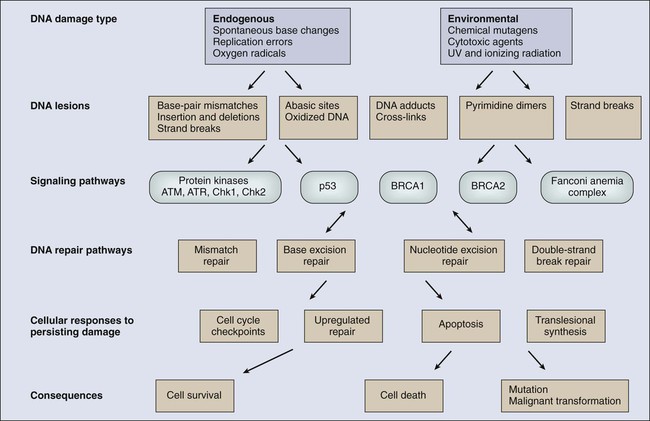
DNA undergoes many types of spontaneous modifications, and it also can react with physical and chemical agents, some of which are endogenous products of normal cellular metabolism (e.g., reactive oxygen species), whereas others, including ionizing radiation and ultraviolet (UV) light, are threats from the external environment (Fig. 10-1). One pronounced example is exposure to genotoxic compounds in cigarette smoke, which currently are responsible for the most common cancers in Western countries. Most active chemotherapeutic agents function by damaging DNA through alkylation, cross-linking, and other means, and mechanisms to repair these lesions determine the sensitivity and resultant resistance of a tumor to such treatments. Damage to DNA can cause genetic mutations, and these mutations can lead to the development of cancer. DNA damage also may result in cell death that can have serious consequences for the organism of which the cell is a part—for example, loss of irreplaceable neurons in the brain. Accumulation of damaged DNA is thought to contribute to some of the features of aging.8 Therefore it is not surprising that a complex set of cellular surveillance and repair mechanisms has evolved to reverse or limit potentially deleterious DNA damage. Some of these DNA repair systems are so important that life cannot be sustained without them. An increasing number of human hereditary diseases that are characterized by severe developmental problems or a predisposition to cancer have been linked to deficiencies in DNA repair (Table 10-2).
Consequences of DNA Damage
The results of DNA damage are diverse and usually adverse. Acute effects arise from disturbed DNA metabolism, triggering cell-cycle arrest or cell death. Long-term effects result from irreversible mutations contributing to oncogenesis and inherited genetic disorders. Many lesions block transcription, which has elicited the development of a dedicated repair system, transcription-coupled repair (TCR), which displaces or removes the stalled RNA polymerase and ensures preferential repair of lesions within the transcribed strand of expressed genes.12–12 Transcriptional stress due to DNA lesions that block RNA polymerase and DNA strand breaks caused by DNA damage or stalled replication forks constitute two major signals for DNA damage–inducible responses, including apoptosis,15–15 through both p53-dependent and independent mechanisms.16
Lesions also may interfere with DNA replication. A class of more than 10 specialized DNA polymerases were discovered that appear devoted to overcoming damage-induced replication stress.19–19 These special polymerases take over temporarily from the stalled replicative DNA polymerases. Although translesion polymerases protect the genome, this solution to replication blocks comes at the expense of a higher replicative error rate, and mutations in some of these polymerases cause cancer susceptiblity.9 Therefore detection of DNA lesions may occur by blocked transcription, replication, or specialized sensors.
DNA Damage Response Pathways
DNA damage checkpoints initially were defined as regulatory pathways that control the ability of cells to arrest the cell cycle in response to DNA damage, allowing time for repair.20 However, in addition to controlling cell cycle arrest, proteins involved in these pathways have been shown to control the activation of DNA repair pathways,21–25 the movement of DNA repair proteins to sites of DNA damage,26–31 and activation of transcriptional responses.34–34 When damage is too significant, a cell may opt for the ultimate mode of rescue by initiating apoptosis to benefit the organism at the expense of losing a cell.37–37 Because the DNA damage response pathway has been better defined at a molecular level, it has been seen as a network of interacting pathways that together execute the response.38 Initial recognition of DNA damage occurs by a variety of damage-specific DNA binding proteins; these proteins, either by themselves or together with complexes of associated proteins that are not directly involved in DNA repair, may signal the DNA damage response.39 Transduction and amplification of the DNA damage signal often is carried out by an overlapping set of conserved protein kinases, including the phosphoinositide-3-kinase-related proteins, which include ataxia telangiectasia mutated (ATM) and ATM- and Rad3-related (ATR) proteins, the checkpoint kinases Chk1 and Chk2, and others.7,40–45 Many of these protein kinases are themselves targets for phosphorylation and activation; they then further target downstream genes critical to oncogenesis, such as p53 and BRCA1.40,46–48 The ultimate targets of this highly regulated DNA damage response include mechanisms for DNA repair, and although much of DNA repair is constitutive, a number of regulatory connections between the DNA damage response pathway and DNA repair have emerged.7 In mammals, a large number of genes involved in DNA repair are transcriptionally induced in response to DNA damage, suggesting that many facets of repair are inducible, similar to the RecA-dependent SOS response in bacteria that enhances DNA repair and mutagenesis after DNA damage.25,49 In fact, the p53 tumor suppressor gene is a central mediator of the DNA damage–inducible transcriptional response in humans, and p53 mutant mammalian cells are deficient in several aspects of DNA repair.21–25,34,50 Therefore the mammalian DNA damage–inducible response pathway is highly regulated and fine-tuned to determine if a particular cell type proceeds to a cell cycle checkpoint and DNA repair, or cell death, after an insult with significant damage. Defects at any level of these pathways can alter repair and result in carcinogenesis (Fig. 10-1).
Types of DNA Repair and Their Contribution to Cancer
DNA repair may be defined as those cellular responses associated with the restoration of the normal nucleotide sequence after events that damage or alter the genome.51 Given the wide variety of DNA damage a cell encounters, it is not surprising that an equally large number of repair systems are available to handle these insults. Indeed, many of the repair systems are broadly overlapping and interacting, with several sharing certain strategies and even specific gene products. Much of what is known regarding the basic mechanisms of many types of DNA repair comes from the study of lower organisms, such as bacteria and yeast, because these pathways have been highly conserved through evolution. Inherited defects in any of the major DNA repair pathways in humans, in general, predisposes to malignancy, and several of these syndromes will be discussed in detail. In humans, a great deal has been learned regarding DNA repair from the often rare, autosomal-recessive hereditary syndromes associated with defects in DNA repair genes.9
Nucleotide Excision Repair
The most versatile and ubiquitous mechanisms for DNA repair are those in which the damaged or incorrect part of a DNA strand is excised and then the resulting gap is filled by repair replication using the complementary strand as a template. The redundancy of genetic information provided by the duplex DNA structure is essential to the maintenance of the genome by this “cut and patch” mode known as excision repair. Each DNA strand can serve as a template for replication-based repair of the other strand. Excision repair was discovered in the early 1960s through basic studies on the effects of UV irradiation on DNA synthesis and repair replication in bacteria.54–54 Nucleotide excision repair (NER) functions to remove many types of lesions, including bulky base adducts of chemical carcinogens, intrastrand cross-links, and UV-induced cyclobutane pyrimidine dimers and 6-4 photoproducts. Such lesions may serve as structural blocks to transcription and replication as a result of distortion of the helical conformation of DNA, and they also may result in mutations if translesional replication occurs or if they are not repaired correctly. The sequential steps for NER are (1) recognition of the damaged site, (2) incision of the damaged DNA strand near the site of the defect, (3) removal of a stretch of the affected strand containing the lesion, (4) repair replication to replace the excised region with a corresponding stretch of normal nucleotides using the complementary strand as a template, and (5) ligation to join the repair patch at its 3′ end to the contiguous parental DNA strand.55,56 This excision repair pathway can remove DNA damage from sites throughout the genome and is termed global genomic repair (GGR). Most human NER genes have been identified and cloned, and many have been shown to be mutated in persons with hereditary NER-deficient, cancer-prone diseases (Fig. 10-2).25,50,57
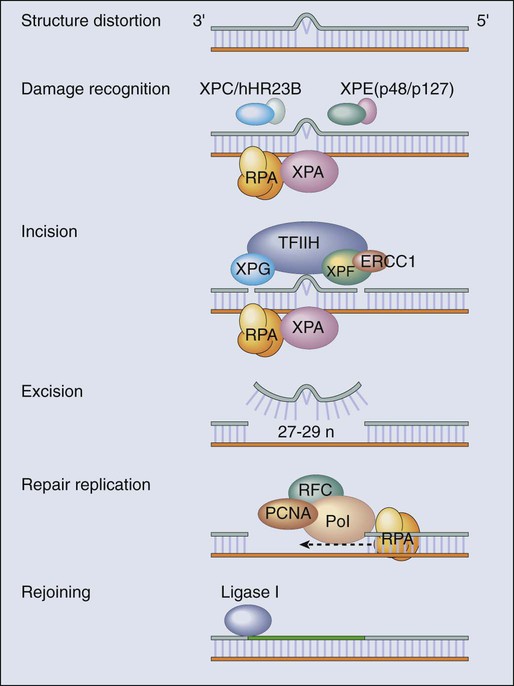
A unique problem arises if a bulky lesion is encountered by a translocating RNA polymerase-making messenger RNA before repair enzymes have removed the damage and restored intact DNA. The polymerase may be arrested at the site of the lesion and prevent access to the damage by repair enzymes. Furthermore, the arrest of transcription in human cells is a strong signal for p53 activation and can trigger apoptosis.16 In this situation, a dedicated excision repair pathway known as transcription-coupled repair (TCR) comes to the rescue to displace the RNA polymerase and then efficiently repairs the blocking lesion so that transcription may resume and the cell may survive.49 The role of defects in TCR, if any, in cancer development remain unclear. Recent evidence suggests that cells from patients with Cockayne syndrome (CS), trichothiodystrophy (TTD), and xeroderma pigmentosum (XP)/CS share a defect in repair of oxidative DNA damage, which may explain the overlapping progeroid features of these syndromes.58
Recently it has become apparent that the GGR subpathway of NER is damage inducible and highly regulated by both transcriptional and posttranslational mechanisms after DNA damage, in concert with damage-inducible cell cycle checkpoints and apoptosis.7,50 In fact, the p53 gene, which is central to maintaining genomic stability in human cells, is required for efficient GGR of UV light–induced and carcinogen-induced DNA damage and functions as a DNA damage–activated transcription factor that directly regulates the expression of several NER damage recognition genes.23–25,34 Similarly, several other important cancer-related genes have been shown to transcriptionally regulate the DNA damage recognition NER genes XPC and DDB2, including BRCA1 and E2F1.61–61 Therefore the GGR pathway of NER appears relevant to suppressing DNA damage–induced malignancy and is highly regulated by genes involved in tumor suppression.
Further evidence for the importance of exquisite regulation of DNA damage recognition and repair activity to carcinogenesis comes from a new appreciation for the role of the ubiquitin-proteasome system in maintenance of genomic stability.62 Many complex intracellular signaling processes, including DNA repair, are controlled not only by the regulated expression of proteins but also by their assembly and targeted degradation through posttranslational ubiquitination. With regard to NER, the same DNA damage recognition proteins found to be transcriptionally induced after DNA damage (XPC and DDB2) are also rapidly ubiquitylated after DNA damage by an E3 ubiquitin ligase activity that contains the DDB2 binding partner DDB1, resulting in a higher order of regulation.50 The ubiquitin-proteasome system has been found to regulate other repair pathways as well, including translesional synthesis and the Fanconi anemia–associated homologous recombination.62 Therefore mammalian cells have evolved a proteolytic pathway to limit their repair capability through restricting certain types of DNA repair activities. Considering that most DNA damage recognition complexes identify a variety of DNA damages or metabolic conditions rather than binding to specific DNA sequences, it is plausible that a “checkpoint” mechanism is required to limit DNA damage binding from interfering with other cellular processes that involve unconventional DNA structures, and that after inducible expression of DNA repair genes, levels are actively reduced to avoid gratuitous DNA repair and associated mutagenesis mediated by error-prone polymerases.
Despite the plethora of mechanisms for DNA repair described herein, it is likely that the cellular replication machinery will nevertheless encounter lesions in the DNA template strand that block replication during each cell cycle. The solution adapted by cells is DNA damage tolerance, in which DNA is synthesized past the damaged bases and subsequently excised after the replication fork has passed—a process known as translesional synthesis and carried out by a newly identified class of specialized error-prone DNA polymerases, termed ζ (zeta) to σ (sigma).19–19 These Y-family DNA polymerases take over temporarily from the stalled replicative DNA polymerases (δ [delta] and ε [epsilon]). They have more flexible base-pairing properties, thus permitting translesion DNA synthesis, with each polymerase probably designed for a specific category of injury. Although translesion polymerases protect the genome, this solution to replication blocks comes at the expense of a higher error rate. For instance, inherited defects in polymerase eta (pol η), encoded for by the XPV/POLH/RAD30 gene, which specializes in relatively error-free bypassing of UV-induced cyclobutane pyrimidine dimers, causes a variant form of the skin cancer–prone disorder XP.63,64
Human Nucleotide Excision Repair Deficient Syndromes and Cancer
A direct correlation between unrepaired DNA damage and carcinogenesis in humans was first established when James Cleaver found that the cancer-prone hereditary disease XP involved a defect in the repair of DNA lesions produced by UV light.65 Since then, at least four syndromes have been attributed to inborn errors in NER: XP, CS, TTD, and UV sensitivity syndrome (UVSS), all of which are characterized by exquisite sun sensitivity.
XP is a rare, autosomal-recessive disease in which homozygous individuals display several characteristics: (1) extreme sensitivity of the skin to sun exposure, which is evident by 1 year of age; (2) pigmentation abnormalities and premalignant lesions in sun-exposed skin; (3) increases up to 4000-fold in the incidence of skin cancers (predominantly squamous and basal cell carcinomas, but also melanomas) and ocular neoplasms, occurring three to five decades earlier than in the general population; and (4) a ten- to twentyfold increased incidence of internal cancers in non—sun-exposed sites.9,66,67 Overall, life span is reduced by approximately 30 years among persons with XP, and many die as a result of malignancies.68 Approximately 20% of patients with XP also display progressive neurological degeneration, characterized by peripheral neuropathy, sensorineural deafness, progressive mental retardation, and cerebellar and pyramidal tract involvement.69 XP occurs worldwide in all ethnic groups and with a frequency varying from 1 to 10 patients per million.
The biochemical defect in cells in most persons with XP is in NER,65 although in a small number of cases (termed XP variants), excision repair appears normal and a defect exists in bypass replication at unrepaired lesions as a result of a mutation in the pol η translesional synthesis gene (XPV).70 Complementation analysis via fusion of cells from different patients has demonstrated genetic heterogeneity within XP and provided evidence for the existence of at least seven excision-deficient complementation groups, termed XP-A to XP-G, in addition to XP variant.9
CS is an autosomal-recessive disease that is associated with defective TCR of UV-damaged and oxidative-damaged DNA.73–73 It is characterized by cutaneous photosensitivity, cachectic dwarfism, skeletal abnormalities, retinal degeneration, cataracts, severe mental retardation, and neurological degeneration characterized by primary demyelination.69,74 In contrast to patients with XP, those with CS are not at increased risk for the development of skin cancers. The average life span of persons with CS is only 12 years, with most patients succumbing to infectious or renal complications, rather than cancer.75 CS is characterized by the existence of at least three complementation groups. Several patients in XP groups B, D, and G have been found to share the DNA repair defects and clinical features of CS together with the cutaneous manifestations of XP.76,77 One model for the severe developmental problems in persons with CS is that endogenous oxidative damage in metabolically active cells, including neurons, is blocking transcription. The lack of TCR could then lead to apoptosis of these essential cells. Alternatively, CS could be a “transcription disease” caused by defects in TFIIH-associated gene products and resulting in inadequate gene expression.
TTD is an autosomal-recessive condition sharing many of the signs and symptoms of CS, with the additional hallmark of brittle, sulfur-deficient hair and nails due to reduced cysteine content in the component proteins. As with CS, several of the responsible genes implicated are XPB and XPD, but there is a third complementation group, TTD-A, in which mutations of a small 8 kDa TFIIH stabilizing factor GRF2H5 have been identified.58 The favored model for TTD is that of a transcription deficiency with respect to the genes relevant to the phenotype, including sulfur-containing proteins.78 It is also conceivable that TTD and CS could be diseases of “premature cell death” in which the transcription deficiency and deficiency in TCR could cause the apoptosis of certain classes of metabolically active cells that sustain significant endogenous oxidative damage (e.g., neurons).
UVSS is a rare disease characterized by severe photosensitivity but without cancer susceptibility. As with CS, cells from the three identified complementation groups lack TCR. Through use of exome sequencing and genetic and proteomic approaches, authors of three recent studies have identified mutations in the UV-stimulated scaffold protein A (UVSSA) gene as being responsible for UVSS group A. These findings suggest a new mechanistic model involving UVSSA and the ubiquitin-specific protease 7 (USP7) in the fate of stalled RNA polymerase II during TCR.79
Analysis of the specific abnormalities in NER displayed by the various genetic complementation groups of XP, CS, TTD, and UVSS allow correlations to be drawn with their heterogeneous clinical features. Specifically, only the subgroups of patients who display a defect in GGR are at significantly increased risk for developing UV-induced malignancies. In contrast, the neurological symptoms and developmental abnormalities associated with other complementation groups of XP and CS are found only in the groups that are defective in TCR. The fact that the TFIIH complex, containing the XP-B and XP-D proteins, is common to both core NER and transcriptional initiation, supports the suggestion that the clinical phenotype of patients with defects in TCR may actually be due to abnormalities in transcription, rather than in repair itself.78
Although these observations may explain the molecular basis for many of the clinical characteristics of XP and CS, they present an apparent paradox with regard to the cancer risk of these patients. Many currently recognized oncogenes and tumor suppressor genes are known to possess important cellular functions and to be actively expressed in normal cells. Because CS cells are defective in the repair of actively expressed genes, it would be reasonable to expect that patients with CS would acquire mutations in genes leading to transformation more readily than normal patients. However, this expectation is not supported by the clinical picture. It has been demonstrated that defects in TCR specifically activate DNA damage–induced apoptosis,13,14 which may eliminate potentially mutagenic premalignant cells.
Base Excision Repair
A major source of DNA damage to cellular genomes arises from normal metabolism in the cytoplasmic environment through hydrolysis and exposure to reactive metabolites that cause oxidation and alkylation of DNA. The repair system primarily involved in identifying and removing such lesions, as well as for dealing with the spontaneous loss of purines from DNA, is the base excision repair (BER) pathway.80,81 The essential nature of BER for viability is highlighted by the fact that although a number of BER proteins have been discovered, only recently has a single human hereditary disease been identified that appears to result from a mutation in a gene unique to this pathway.82–85 The enormous task required for BER is exemplified by the fact that in 1 hour, humans spontaneously lose on the order of a trillion guanines from their DNA, each of which must be replaced. Similarly, a large number of cytosines become deaminated spontaneously and the resulting product, uracil, must be removed and replaced with cytosine to restore the correct nucleotide sequence. In most cases, BER is initiated by one of a set of lesion-specific glycosylases that recognize the altered or inappropriate base and cleaves it from its sugar moiety in the DNA (Fig. 10-3). Different DNA glycosylases remove different types of damage, conferring specificity to the process. Once the base is removed, the apurinic/apyrimidinic (AP) site is removed by an AP endonuclease or an AP lyase, which cleaves the DNA strand 5′ or 3′ to the AP site, respectively. The remaining deoxyribose phosphate residue is excised by a phosphodiesterase, with the resulting gap filled by a DNA polymerase, and the strand is sealed by DNA ligase. The major oxidized purine lesion is 8-oxo-7,8-dihydroguanine (8-oxoG), which is abundant and has strong mutagenic properties. Oxidized pyrimidines include thymine glycol, 5-hydroxycytosine, and formamidopyrimidines. Oxidized bases, including both 8-oxoG and thymine glycol, share the property of blocking DNA replication and transcription and must be repaired efficiently to maintain genomic stability.86,87
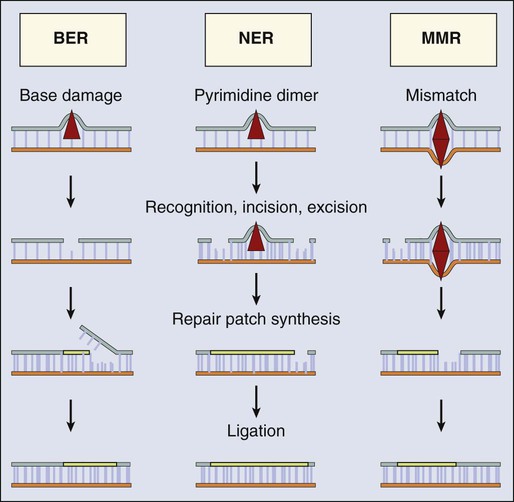
In mammalian cells, the gene functions responsible for the strand incision steps of BER include the glycosylases hNTH1, which removes oxidized pyrimidines, hOGG1, which targets oxidized purines, and MYH, which removes adenines mispaired with an 8-oxoG, together with AP endonuclease 1.88 Attempts to engineer mice that are deficient in the core enzymes required for BER typically have resulted in early embryonic death, whereas knockout of individual glycosylases produce mice with no overt phenotype at all.89 This phenomenon attests to the importance of the repair of DNA lesions from endogenous causes during embryonic development, as well as the likely redundancy between individual glycosylases. It also is consistent with the near absence of known human hereditary diseases characterized by defects in BER genes.
However, given the mutagenic and cytotoxic potential of the classes of DNA damage that are BER substrates, it seems likely that altered activity in these pathways would result in enhanced cancer risk. The most direct evidence for a role for BER in cancer comes from the discovery that germline mutations in the MYH gene, which are involved in processing 8-oxoG lesions, are associated with recessive inheritance of a predisposition to develop multiple colorectal adenomas (polyposis) and colon cancers.84–84 Tumors from affected persons exhibit excess transversions of a guanine-cytosine pair to a thymine-adenine pair in the APC gene, which itself is associated with colon carcinogenesis and is causative of familial adenomatous polyposis. Therefore biallelic inherited mutations in MYH result in a polyposis-like syndrome termed MYH-associated polyposis. In persons with MYH-associated polyposis, tens to hundreds of polyps tend to develop by the age of 40 years, and nearly 50% of such persons present with colon cancer.85,90
A great deal of effort has gone into attempts to identify functional polymorphic genetic variation in DNA repair genes that modulate activity and affect individual cancer risk. To date, genetic epidemiologic studies of single-nucleotide polymorphisms (SNPs), particularly in BER genes, have been inconsistent.91 However, metaanalyses have identified modest associated increased lung cancer risk with OGG1 and XRCC1 SNPs.92 Continued advances in SNP maps and high-throughput genotyping methods will facilitate the analysis of multiple polymorphisms within multiple genes using haplotypes, and with larger sample sizes, perhaps future studies will reveal more clear risk associations. The genetic variation of DNA repair genes is discussed in Box 10-1.
Mismatch Repair
Mismatch repair (MMR) is another example of an excision repair mechanism that utilizes a similar strategy for genomic maintenance (Fig. 10-3). MMR is a process that corrects mismatched nucleotides in the otherwise complementary paired DNA strands; the mismatched nucleotides arise from DNA replication errors and recombination, as well as from some types of base modifications.104–104 This repair mode also can deal with small loops of single-stranded DNA at sites of insertions or deletions in the duplex DNA structure. The importance of this repair mechanism in maintaining genetic stability is illustrated by the observation that its absence results in a large increase in the frequency of spontaneously occurring mutations, particularly in microsatellite sequences of highly repetitive DNA.105 Some of these spontaneous mutations arise from mistakes introduced during DNA replication, in spite of the operation of a “proofreading” system that also helps to ensure the high fidelity of replication. In humans, genetic defects in several MMR genes have been linked to Lynch syndrome (hereditary nonpolyposis colorectal cancer), as well as to sporadic cancers, that exhibit instability in regions of DNA containing short repetitive sequences of nucleotides, a feature known as microsatellite instability (MSI).
As with other modes of excision repair, four principal steps are required for MMR: (1) mismatch recognition, (2) recruitment of additional MMR factors, (3) identification of the newly synthesized DNA strand containing the mismatched nucleotides, followed by their excision, and (4) resynthesis of the excised tract and ligation. The biochemical workings of this pathway are best understood in bacteria, but a similar set of events occurs in human cells. Based on functional homologies to their bacterial counterparts, and sequence homology to corresponding yeast genes, a number of human genes have been cloned that participate in MMR, including those homologous to the bacterial MutS mismatch recognition protein (hMSH2, hMSH3, and hMSH6) and to the bacterial MutL gene (hMLH1 and hPMS2).108–108 In humans, heterodimers of the MSH2/6 proteins recognize single base-pair mismatches and short insertion-deletion loops, whereas MSH2/3 dimers recognize longer loops. Heterodimeric complexes of MLH1/PMS2 and MLH1/PMS1 interact with the MSH complexes and replication factors for strand discrimination and DNA excision. Similar to NER and BER, additional proteins are then recruited for repair replication based on the original DNA template.
The MMR system also may interact with DNA damage as a result of certain alkylators and intercalating agents that assume similar structural alterations in DNA as mismatches and actually result in erroneous or futile MMR cycles, ultimately resulting in apoptosis. Thus intact MMR may confer chemosensitivity to these chemotherapeutic agents, and MMR defective tumors may exhibit resistance to certain drugs.109
Human MMR Deficiency and Cancer
Lynch syndrome is the most common hereditary colorectal cancer predisposition syndrome.110,111 Lynch syndrome is an autosomal-dominant inherited condition with an incidence of 1 : 1000 in the general population. It accounts for ~5% of all cases of colorectal cancer. Persons with Lynch syndrome also are at elevated risk for cancers of the endometrium, ovary, stomach, small bowel, and other sites. Persons with Lynch syndrome have an 80% lifetime risk for colorectal cancer and a 50% lifetime risk for endometrial cancer. The discovery of MSI (i.e., the frequent alteration in the tract lengths of certain short repetitive nucleotide sequences) in some hereditary colorectal cancers provided the first indication that the etiology of these cancers might involve a problem in the MMR system.112 The finding of germline MMR gene defects in persons with Lynch syndrome established that these defects are the cause of the enhanced incidence of cancer.108–108 Germline mutations in the MLH1 and MSH2 genes together account for more than half of all cases of Lynch syndrome. Defects in the MSH6 gene cause a late-onset Lynch syndrome phenotype, and rare mutations in the PMS2 gene also can result in Lynch syndrome. No strong genotype-phenotype correlations have been observed to date, but mutations in the MSH2 gene do appear to be associated with more extracolonic manifestations than are mutations in the MLH1 gene.
MSI has been identified as a source of the genomic instability driving tumorigenesis in a number of sporadic tumor types, in addition to those that arise in the context of inherited germline mutations of MMR genes.112,113 For example, up to 20% of sporadic colon cancers exhibit MSI, particularly when manifesting in the ascending colon and in persons younger than 50 years old, with most due to epigenetic silencing of MLH1 gene expression by promoter hypermethylation.114,115 Whether through genetic or epigenetic inactivation, loss of MMR results in the elevated rate of mutations, particularly at microsatellite sequences, several of which occur in the coding sequences of other genes that often are found mutated in cancers, including transforming growth factor-β type II receptor, BAX, and the mismatch repair genes MSH3 and MSH6, themselves. Therefore clear genetic evidence demonstrates that the phenotype of genetic instability associated with defects in MMR results in the genotype of tumors that arise as a result of these defects.116 Intriguingly, the survival of patients with MSI-associated colorectal cancer is better than that of persons with more typical tumors exhibiting chromosomal instability.119–119 These tumors also demonstrate different sources of genomic instability, resulting in distinct biological and pathological characteristics.120 Whether their more favorable outcome reflects differences in clinical behavior, responsiveness to therapy, or both remains to be fully determined. Given the readily available phenotypic assays for Lynch syndrome (MSI and immunohistochemistry for the four causative gene products), “universal” screening is now performed on all colon and endometrial cancers at many centers, leading to cost-effective identification of families with Lynch syndrome and, it is hoped, improved clinical outcomes.121,122
Double-Strand Break Repair
Double-strand breaks (DSBs) in DNA induced by ionizing radiation, chemicals, and replication across single-strand breaks and during repair of interstrand DNA cross-links usually are dealt with through the recombination machinery. An unrepaired DSB is a highly lethal event, and even a single occurrence in the entire genome is thought to be sufficient to signal cell cycle checkpoints that prevent attempted DNA synthesis or cell division until repair has been completed, or apoptosis, if it is improperly repaired. DSBs also pose problems during mitosis, because intact chromosomes are a prerequisite for proper chromosome segregation during cell division. Thus these lesions often induce various sorts of chromosomal aberrations, including aneuploidy, deletions, and chromosomal translocations—all of which are associated with carcinogenesis. Genetic recombination is the principal mechanism for dealing with DSBs that involve homologous stretches of nucleotides at the ends to be joined. If no such homology is present, however, another system exists for nonhomologous end joining, which is more prone to error (Fig. 10-4). Recent studies have identified a cascade of protein kinases involved in signaling cellular processes in response to DSBs. Many of these protein kinases have been found defective in cancer-prone disorders exhibiting genomic instability, such as the ATM protein45 and the Chk2 protein kinase associated with a Li-Fraumeni–like cancer susceptibility syndrome.125–125 A major target for these kinase activities is the p53 tumor suppressor gene product. This protein, when phosphorylated, is activated and involved in G1 arrest and apoptosis after ionizing radiation,40,41,126–129 and when found mutated in the germline, it results in the Li-Fraumeni cancer susceptibility syndrome. Many other enzymes involved in the actual DNA transactions necessary for DSB repair have been found in cancer-prone disorders, including MRE11 (AT-like disorder), NBS1 (Nijmegen breakage syndrome), BRCA1 and BRCA2 (breast-ovarian cancer syndrome), and the RecQ-like helicases (Werner, Bloom, and Rothmund-Thomson syndromes).130
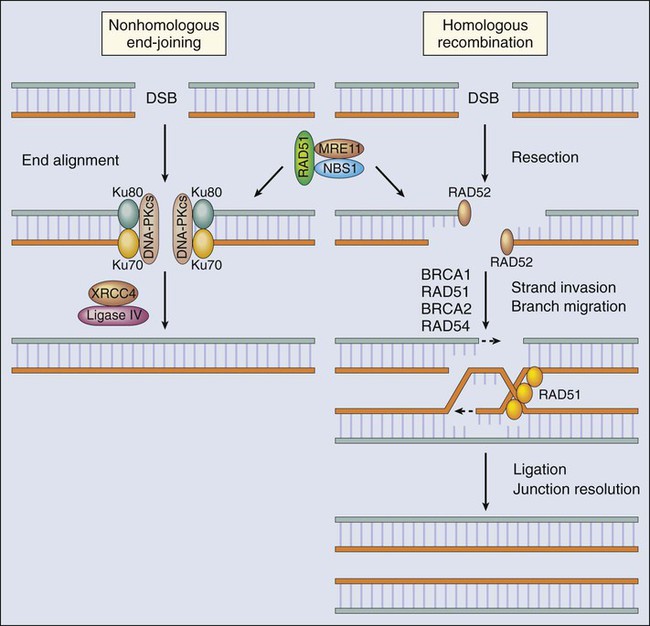
Ataxia Telangiectasia
Ataxia telangiectasia (AT) originally was identified in humans with a severe sensitivity to ionizing radiation. Later it was learned that AT also affects the immune system and the cerebellum. The neurological defect causes progressive loss of motor control leading to lack of coordination and balance. Respiratory infections develop in children with this disease as a consequence of deficiencies in the immune and nervous systems. A single gene, ATM, is responsible for the multiple and surprisingly diverse symptoms of this disease, including a predisposition to lymphoma and leukemia. Cancer develops in more than 10% of patients with AT at an early age. AT is an autosomal-recessive disease with an incidence of nearly 1 in 100,000 live births. Persons who are heterozygous for AT mutations, about 1% of the general population, may have an increased predisposition to cancer, in particular breast cancers, especially in persons expressing an ATM with missense mutations, resulting in a dominant-negative effect on their wild-type ATM gene.133–133 The ATM gene product is a central signaling protein in the DNA damage response, and cells lacking ATM fail to execute many critical cellular responses to DNA damage. For example, one hallmark of AT is what has been termed “x-ray resistant DNA synthesis.” It is now known that the ATM gene product is a key element in delaying the initiation of DNA replication after DNA damage, resulting in strand breaks. In addition, ATM directly phosphorylates the NBS1 protein, resulting in its association with the MRE11 and RAD50 gene products, which together are required for both nonhomologous end-joining and homologous recombination of DSBs.134 Inherited germline mutations of the NBS1 and MRE11 genes, themselves, result in clinical variants of AT, termed Nijmegen breakage syndrome and AT-like disorder, respectively.134,135 Therefore ATM, NBS1, and MRE11 are central to the DNA damage response pathways that are critical for regulating recombination and repair after DSBs. Defects in many of the component proteins in the pathway result in genomic instability and a predisposition to cancer. Interestingly, ATM also recently has been implicated in cellular processes other than DNA damage responses, including insulin signaling and mitochondrial function.136,137 Thus, although only a single gene is mutated in AT, loss of ATM may result in a pleiotropic disease phenotype in patients because of its multifaceted functions.
Other Cancer-Prone Disorders Associated with Genomic Instability
Diseases Involving Homologues of RecQ
In at least three cancer-prone diseases in humans, the defect is in a homologue of the RecQ gene originally discovered in bacteria.138 The product of RecQ is a helicase, which in Escherichia coli is involved in processing the nascent DNA at arrested replication forks. Helicases are enzymes that separate the complementary strands of nuclei-acid duplexes using energy derived from adenosine triphosphate hydrolysis. In humans, RecQ helicases are thought to function at the interface between DNA replication and recombination in dealing with damaged replication forks and interact with many other nuclear proteins that are required for DNA metabolism.139 The biological and clinical effects of the homozygous deficiency of these genes can be quite dramatic and profound. Thus Bloom syndrome is characterized by an extremely high frequency of genetic exchanges (so-called sister chromatid exchanges) that cause genomic instability and lymphoma, leukemias, and solid tumors of the gastrointestinal tract and breast.140,141 Of interest, in recent studies it has been found that persons who are heterozygous for a BLM mutation have an increased susceptibility to develop colorectal cancer, potentially as a result of haploinsufficiency.142,143 In persons with Werner syndrome, the deficiency in another RecQ homologue results in remarkable features of premature aging, as well as predisposition to sarcomas, melanoma, and cancer of the thyroid.146–146 Yet another RecQ homologue defect, Rothmund-Thomson syndrome, is characterized by growth deficiency and cancer predisposition, in particular to osteogenic sarcomas.147,148
p53 Gene and Li-Fraumeni Syndrome
The discovery of the p53 tumor suppressor gene more than 30 years ago inspired widespread investigations with the aim of understanding the basic biology behind its role in maintaining genomic stability and the cellular response to DNA damage. The p53 gene is one of the most commonly mutated genes in human cancers,149 and its product is a multifunctional protein that regulates many physiological processes, including cell cycle checkpoints, apoptosis, and DNA repair.25,127,150–152 The primary role of p53 in tumor suppression has been attributed to its function as a transcription factor, regulating expression of several hundred different cellular genes,153 although it clearly exhibits transcription-independent activities as well. Indeed, p53 appears to act as a central “node” that lies at the intersection of upstream signaling cascades induced by DNA damage and cellular stress responses and downstream DNA repair and DNA damage response pathways. In response to a variety of genotoxic stimuli, p53 protein is stabilized through a series of posttranslational modifications153 and activated for sequence-specific DNA binding, and it transcriptionally regulates downstream target genes that contain a consensus p53 response element in their promoter or intronic segments. These p53 target genes include those important for cell cycle checkpoints, such as p21,154 apoptosis, such as BAX and PERP,155 and DNA repair, such as the DDB2 and XPC genes required for NER.23,24,34 In addition, p53 protein may act in a transcription-independent manner to modulate BER through interactions with DNA polymerase-β and OGG1 and homologous recombination in conjunction with RecQ proteins.152
Persons with the rare autosomal-dominant Li-Fraumeni syndrome (LFS) are at increased risk for the development of a number of common tumors at an early age because of an inherited germline defect in one allele of the p53 gene, including soft tissue and osteosarcomas, breast cancer, brain tumors, lymphomas, leukemia, and adrenocortical carcinomas. Mutations in the p53 tumor suppressor gene account for 70% to 85% of classic LFS cases.158–158 Although the heterozygote carriers of a defective p53 allele do not appear to have clinical problems or somatic DNA repair defects, when the second allele has been mutated or lost, the absence of functional p53 results in severe problems for the cell and substantial cancer susceptibility for the host. First of all, the p53 controlled pathway of apoptosis is disengaged, and thus severely damaged cells will survive and be at risk for carcinogenic transformation because of their genomic instability. That genomic instability derives from the fact that p53 is also an important regulator of cell cycle checkpoints. Thus as with the situation in AT, the cells continue to progress through their growth cycle rather than pausing to allow time for DNA lesions to be repaired. Finally, p53 serves an important regulatory function in NER and perhaps in BER and recombination, and in its absence, some important mutagenic lesions are simply not repaired. That nonrepair, of course, is a major contributor to the genomic instability and the consequent development of tumors.
BRCA1, BRCA2, and Breast-Ovarian Cancer Susceptibility
Hereditary breast cancer includes a broad group of hereditary predisposition conditions in which breast cancer is a component tumor; these conditions account for approximately 5% to 10% of all breast cancer cases. Hereditary syndromes of breast and ovarian cancer susceptibility have been particularly associated with germline mutations of two genes, BRCA1 and BRCA2, as well as rare cases due to mutations in the p53 gene in persons with LFS, the PTEN gene in persons with Cowden disease, and perhaps ATR, CHEK2, and others. Recent experimental data suggest that both the BRCA genes may be involved in multiple DNA repair activities.59,159–163
The BRCA1/2 genes are large and complex. Many hundreds of different germline mutations have been detected in each gene, but only rare sporadic breast or ovarian cancers have been found to harbor BRCA1/2 mutations. Increasing evidence suggests that BRCA1/2 genes are centrally involved in various aspects of DNA repair and DNA damage response pathways. For example, BRCA1 is phosphorylated after exposure to DNA-damaging agents by ATM, ATR, and CHEK2, associates with a number of DNA repair proteins including MSH2, MSH6, ATM, RAD51, and the RAD50-MRE11-NBS1 protein complex after DNA damage, and localizes to nuclear foci with these proteins after treatment ionizing radiation and UV radiation.28,164,165 The association of BRCA1 with RAD51, an enzyme involved in the coordination of recombination, suggests its involvement in DSB repair, and strong data implicate BRCA1 in homologous recombination.166,167 Other studies suggest that BRCA1 may regulate cellular processes through transcriptional co-activation. BRCA1 has been shown to transcriptionally regulate the NER genes XPC and DDB2 and affect GGR of UV- and cisplatin-induced DNA damage,59,168,169 as well as transcriptionally regulate BER genes and affect BER of oxidative DNA damage.163,169,170 Chromosomal instability also is characteristic of breast tumors that harbor BRCA2 mutations, probably as a result of defective recombination-mediated DSB repair. BRCA2 has been shown to bind to the RAD51 protein, an enzyme involved in the coordination of recombination, and together co-localize to nuclear sites containing DNA strand breaks caused by ionizing radiation. Structural studies of BRCA2 DNA binding domains suggest that they may facilitate interactions of RAD51 with single-stranded DNA during recombination.171,172 The genomic instability associated with mutations of BRCA1/2, therefore, may in part be a result of the intact but error-prone nonhomologous end-joining repair pathway.160 See Box 10-2 for a discussion of DNA repair and cancer treatment.
Fanconi Anemia, Cancer, and Interstrand Cross-link Repair
The centrality and interwoven nature of DNA repair pathways for genomic stability have been highlighted by findings regarding Fanconi anemia, a rare, autosomal-recessive disease that confers an increased risk of acute myeloid leukemia, squamous-cell carcinomas of the head and neck and esophagus, gynecologic carcinomas, and liver tumors at a young age.184,185 At least 13 subtypes of Fanconi anemia have been determined by complementation analyses, and germline mutations in genes have been identified for most of the subtypes.186,187 The proteins encoded by these genes all cooperate in a common DNA repair pathway involved in interstrand cross-link repair and processing of arrested replication forks.188,189 Germline homozygous inactivating mutations of the BRCA2 gene result in the D1 group.190 A “core complex” of eight of these proteins are subunits of a nuclear E3 ligase, which is required for the monoubiquitination of the downstream FANCD2 protein, which itself links Fanconi anemia proteins to BRCA1 in the response to DNA damage by co-localizing to nuclear sites occupied by RAD51 and BRCA2.191 This process also is regulated by the ATM protein kinase, which phosphorylates FANCD2 in response to DNA damage.192 It has long been appreciated that cells from persons with Fanconi anemia are hypersensitive to DNA cross-linking agents, such as mitomycin C and cisplatin, in addition to being modestly sensitive to ionizing radiation. Therefore the Fanconi anemia proteins appear to function at an interface between several DNA repair and DNA damage response pathways.
Conclusions and Future Directions
Recent molecular biology and genetic research has provided ample evidence to support the long-standing prediction that genomic instability is a major factor driving the onset and progression of carcinogenesis.38 Overlapping and interacting mechanisms for DNA repair and the cellular response to DNA damage are critical components for the maintenance of genomic stability. Alterations in these pathways often are early events in the multistep acquisition of genetic mutations leading to cancer development. Continued exploration of the DNA damage response will prove important for our improved understanding of cancer etiology, prevention, genetic susceptibility, diagnosis, and treatment.