Discovery and Characterization of Cancer Genetic Susceptibility Alleles
Stephen J. Chanock and Elaine A. Ostrander
• The discovery of cancer susceptibility regions across the genome provides opportunities to understand defining events in tumor development and identify cellular pathways that contribute to the complex development of cancer.
• Regions of the genome that harbor susceptibility alleles can be determined with use of association studies in families or populations and linkage studies within families.
• New technologies, together with the annotation of genetic variation across the human genome, are accelerating the pace of discovery and characterization of cancer susceptibility alleles. The conclusive identification of a gene or a regulatory region contributes to an understanding of defining events in tumor development.
• The spectrum of cancer susceptibility alleles includes mutations in genes that are highly penetrant, which indicates that persons born with a mutant allele have a high probability of developing cancer and common variants that impart a small additional risk for cancer.
• Association studies and linkage-based studies both require collection of accurate clinical and family history data by clinicians, and both offer hope for precision medicine. Precision medicine is based on a molecular understanding of cancer and specifically uses biomarkers, such as susceptibility alleles, to inform clinical and public health decisions.
Introduction
For generations, investigators have pursued the heritable contribution to cancer. Seminal studies in families with several members affected with breast cancer, colorectal cancer, melanoma, or a constellation of cancers (e.g., Li-Fraumeni syndrome) provided evidence for rare mutations with strong effects.1 Family-based and twin studies indicate an excess familial cancer aggregation for nearly all types of cancers, although the estimates vary greatly across cancer types. These observations suggested that it would be possible to map cancer genes and thus estimate the genetic contribution to each molecular type of cancer, even in unrelated populations. Until the past decade, progress has been slow. However, the pace at which new genetic regions harboring cancer susceptibility alleles have been discovered has accelerated substantially as a result of three converging factors: first, a high-quality draft sequence of the human genome was produced2,3; second, its subsequent annotation has resulted in the appreciation of a wide spectrum of variation across the genome4; and third, the development of technical platforms that enable interrogation of genetic variation across the genome has changed both the economics and speed with which genetic studies can be performed. The scope of studies has thus changed dramatically, expanding from family-based studies to larger population-based studies of unrelated individuals. These studies have been fueled by the precipitous drop in price for interrogating single nucleotide polymorphisms (SNPs), the most common type of variant in the genome, or massive parallel sequence analysis of entire or partial genomes. To keep pace with the new streams of large data sets, investigators have forged new collaborations and developed computational tools for analyzing larger data sets in search of new cancer susceptibility alleles.
Cancer susceptibility alleles have been discovered with the use of a variety of approaches, yielding a range of inherited genetic variants, from rare mutations with strong effects (e.g., highly penetrant) to common genetic polymorphisms, each of which confers a small risk for cancer.1 Susceptibility alleles can increase a person’s risk of developing cancer either within families or across populations. It is notable that not all susceptibility alleles have equal estimated effects. Consequently, the observed spectrum of established susceptibility alleles reflects an inverse relationship between the effect size and the frequency of the genetic variation (Fig. 22-1).5,6 Highly penetrant mutations are rare and have a strong predictive value for developing one or more cancers. These highly penetrant mutations are generally discovered in family studies using linkage analysis and, more recently, next-generation sequencing analysis in and across pedigrees in which several family members are affected with the same or a constellation of cancers. More frequent susceptibility alleles have smaller effect sizes and are discovered using association studies in which the genomes of a set of affected cases are compared with that of unaffected control subjects.7
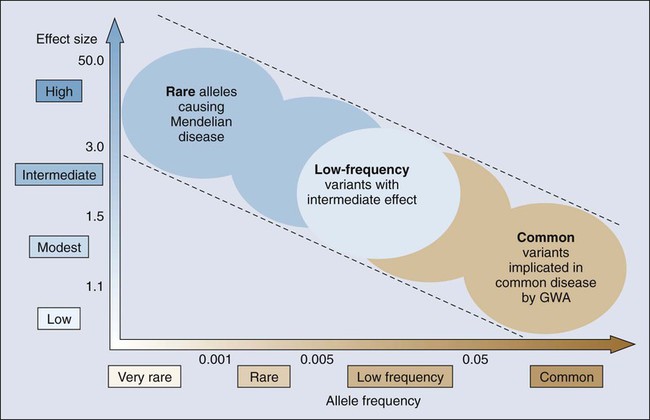
Genetic mapping of cancer susceptibility genes can identify regions of the genome harboring genes that play a role in cancer susceptibility but also nongenic regions that can regulate genes and pathways of interacting genes. Although the direct public health impact associated with conclusively establishing a specific cancer susceptibility allele may not be immediately apparent, its contribution to understanding tumor development and metastasis is invaluable, expanding possible pathways and putative targets for intervention downstream.8 Moreover, the possible clinical value of known susceptibility alleles will continue to increase as more comprehensive maps of susceptibility alleles emerge for specific cancers. Thus far, there are more distinct susceptibility alleles per cancer than there are susceptibility alleles that contribute to the risk for multiple cancers. To define the genetic architecture (Fig. 22-1), namely, the constellation of susceptibility alleles that contributes to a specific cancer, further efforts are required to define comprehensive sets of variants, which in turn should emerge as vital tools in both public health and individual (known as precision medicine) assessments of cancer risk.9
Fundamental Science
Genetic Variation in the Human Genome
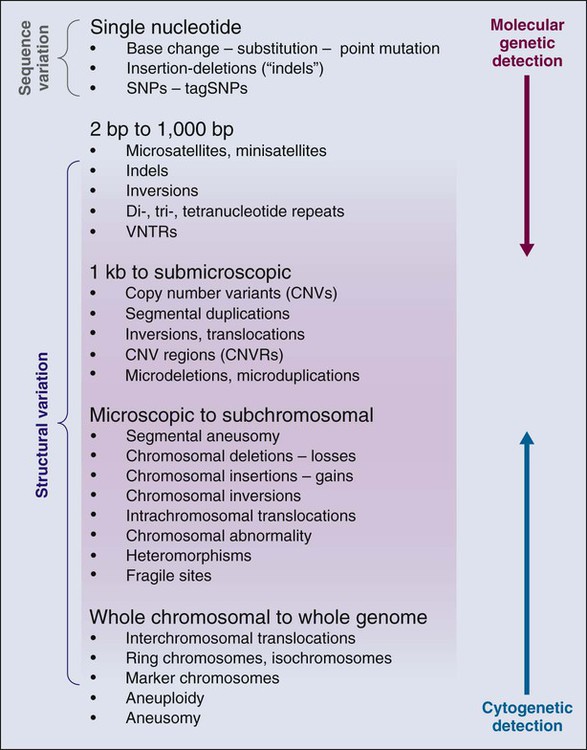
The annotation of genetic variation in the genome has provided important clues to elucidation of the genetic history of distinct populations, possible interactions between environmental or pathogen challenges, and the heterogeneous distribution of human cancers. The differences in the spectrum of allele frequencies and the types of genetic variation, from SNPs to large copy number variants, have become indispensable tools for geneticists to map diseases (Fig. 22-2).4,10–12 The basic principle has been to observe distinct patterns of genetic variation between affected and unaffected individuals, whether in families or population studies.
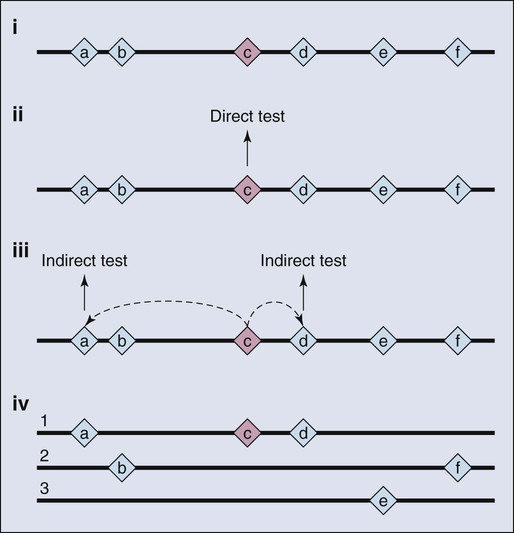
As a consequence of the enormous scope of human genetic variation, the search for susceptibility alleles has broadened and for most study designs has focused on conclusively discovering “markers” that highlight the region of the genome where a disease susceptibility alleles resides.13 Sets of markers to be tested are drawn from dense maps of human genetic variation that are publicly available. The approach is not predicated on testing the actual casual variant, at least not initially, but instead identifying one or more surrogates that are highly correlated with the variant actually underlying the susceptibility allele. Although embracing this “indirect” approach has had great value (Fig. 22-3), it comes at a price, namely, additional steps to sort through the correlated variants and then conduct the functional studies needed to illuminate the underpinnings of the susceptibility allele.13 In other words, further work is required to characterize the mutations directly responsible for contribution to disease susceptibility (also known as causal mutations).14
Until it was possible to envision a whole genome sequence, genetics had created and modified maps of relative coordinates based on incomplete constructs. Sets of markers can be thought of as molecular street signs, which allowed one to knowingly navigate his or her way up or down a chromosome. Early on, “genetic maps” provided a stable reference for mapping highly penetrant mutations, primarily in families.15 These maps were based on empirical evidence of recombination hot spots. The long-standing value of functional elements, herein recombination frequencies, served adequately for the mapping of disease and traits before the draft sequences of genomes began to appear. The emergence of a physical map (currently tractable for more than 92% of the genome) has accelerated the mapping of traits and diseases because the field has closed in on absolute coordinates for the genome. That is, we generally know the nucleotide location of a given marker or gene in millions of base pairs from the end or terminus of the chromosome. Investigators still use the principles uncovered in studying genetic maps to pinpoint alleles on the physical map.
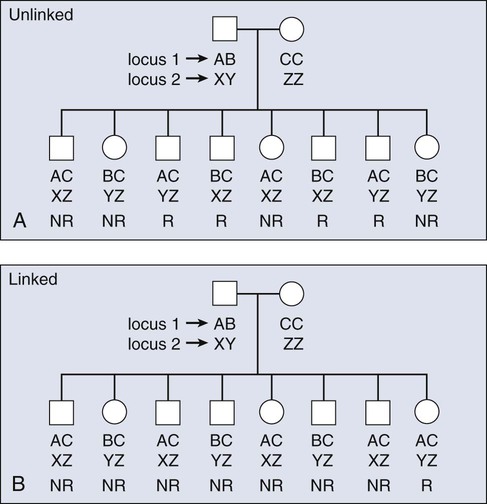
The principles of meiotic recombination are key to understanding the relationship between genetic loci, here defined as genetic variants that map to unique coordinates on the physical map. The correlation between genetic markers is fundamental to both association and linkage analysis. In meiosis, the cell division leading to gamete formation and homologous chromosomes are paired. Each chromosome consists of two identical strands (chromatids), with each chromosome pairing composed of four strands. Homologous chromosomes separate from each other during the process of meiosis except at one or two zones of contact in a process that leads to genetic recombination (Fig. 22-4). Mendel’s second law, independent assortment, states that alleles of genes at unlinked loci segregate or assort independently of one another. Deviations from independent assortment occur when genes are located close to one another, in which case alleles assort together more than 50% of the time. In this scenario, the associated loci are “linked.” Distributed throughout the genome are recombination hot spots, which “divide” the genome. These hot spots can vary by population genetic history, providing an opportunity to compare groups and use the differences to pinpoint possible susceptibility alleles, especially if substantive differences exist between populations with respect to cancer incidence.
Consequently, if two loci are located on different chromosomes or far apart on the same chromosome, their alleles will assort randomly, transmitting to the same gamete 50% of the time. Such loci are “unlinked.” For a chromosomal segment, the probability of a genetic recombination event occurring between a pair of markers is proportional to the distance between them. This probability is expressed as a recombination frequency (q), where θ = number of recombinant offspring/number of total offspring. The closer a marker and disease gene were located on a chromosome, the lower the probability they would be dissociated during recombination events. Conversely, the farther apart they were, the higher the probability they would appear “unlinked” in multiple generations of a family. The recombination frequency values range from 0 for markers that are so closely linked that crossover events essentially never occur to 0.5 for genes that assort randomly—for instance, those on different chromosomes or chromosome arms. Within small intervals, when the probability of multiple crossovers is negligible the relationship between the recombination fraction (θ) and the distance between two genes (x), is simply x = θ.16 After a mathematical adjustment for the small possibility of double recombinants, recombination fractions are expressed in units called centimorgans (cMs).17 One percent recombination (θ = 0.01) is equal to 1 cM for the genetic map, which, in the human genome, corresponds to about one million base pairs.
The spectrum of human genetic variation varies by the frequency of polymorphisms, which often is substantial between populations, as well as the length of the variant. The most common sequence variation is the substitution of a single base, known as an SNP, which, by definition, is observed in at least 1% in one or more populations. The minor allele frequency (MAF) refers to the lower allele frequency, and it can vary by population. The number of SNPs increases across the genome as the frequency decreases.18 A substantially larger fraction of genetic variation exists for single base substitutions below 1%, and many of these are population private, reflecting the population genetics history.18 The majority of SNPs with an MAF greater than 10% are common to all human populations, but the actual frequencies can vary greatly. Reported SNPs are cataloged in the dbSNP database (http://www.ncbi.nlm.nih.gov/snp), which is an important reference that points to emerging data sets and is useful in interpreting variants identified through DNA sequencing.
A small subset of SNPs are located in exons, of which a fraction change the predicted amino acid. SNPs that can alter the coding sequence are known as nonsynonymous SNPs, whereas those that are silent are termed synonymous. Although great interest has been expressed in coding SNPs, partly because they appear to be more interpretable, very few of the known associations between a disease and a common SNP marker (MAF >10%) are for coding SNPs. On the other hand, rare highly penetrant mutations mainly map to coding changes or preterminal stop codons. Many of the reported disease mutations are cataloged in a public database, the Online Mendelian Inheritance in Man (http://www.ncbi.nlm.nih.gov/sites/entrez?db=OMIM/).
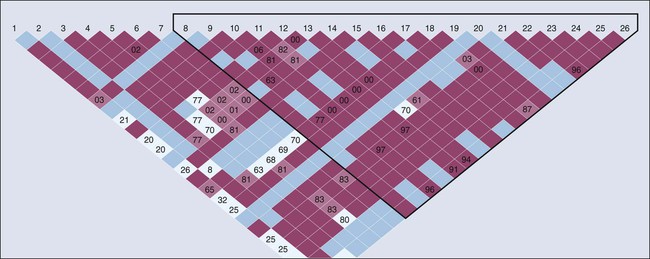
SNPs become fixed in populations over multiple generations and are generally not inherited independent of the adjacent variants. Recombination hot spots can separate sets of highly correlated variants, resulting in “blocks of haplotypes” (Fig. 22-5).19 These segments of a chromosome, which usually are quite small, are transmitted as a unit from one generation to the next. The correlation between SNPs is an estimate of linkage disequilibrium (LD), which is classically defined as the nonrandom association of alleles at different loci. Individual SNPs that always track together are said to be in strong LD. This correlation can be eroded over time by recombination (exchange of genetic material) during meiosis, and SNPs can be defined as being in weak LD20—that is, a correlation exists, but it is not strong. We measure the degree of LD with use of either D′ or r2 coefficients; both give similar information, but the latter are more highly dependent on the MAFs of the adjacent SNPs and are generally more favored by geneticists.
The concept of LD is important because it enables investigators to evaluate sets of SNPs and determine proxies for other, untested SNPs, which is useful for “indirect” mapping. Thus if a group of SNPs are in strong LD and are always inherited together, one can test for the alleles of just one reference SNP and immediately have information regarding which alleles are segregating to a given individual for all the adjacent SNPs. By extension, estimates of LD are useful to construct haplotypes in unrelated subjects. With new reference data sets (e.g., the 1000 Genome Project), it is possible to impute untested variants against the backbone of stable data sets.18 The computational efficiencies enable estimation of the correlation between sets of markers and the construction of haplotypes.21 Still, the most reliable approach is to resolve the phase of haplotypes in multigeneration pedigrees, in which haplotypes can be traced; alternatively, one can infer the relationship of alleles in unrelated subjects with computational tools.22 Phase refers to the parental (and grandparental) chromosome of origin for a set of alleles.23 This specific information regarding a set of markers in LD can, in turn, be useful for determining where a disease allele originates.
The annotation of the human genome has revealed a wide spectrum of structural variations, which may be either cytologically visible or detected by either microarray chips or actual sequence analysis (Fig. 22-2). For instance, short tandem repeats are a class of polymorphisms in which a small number of base pairs are reiterated, such as “CACACA.” Polymerase chain reaction primers are used to define the physical location of one short tandem repeat from the remaining 50,000 that litter the genome. Also known as microsatellites, they have been effectively used for linkage analysis and forensic investigation. Structural variants of all sizes can include deletions, insertions, and duplications collectively known as copy number variations (CNVs).12–12 In addition, infrequent inversions and translocations of pieces of DNA are present that vary in size. Some of these inversions and translocations are quite common; for example, chromosome 17 harbors an inversion of 3.5 million base pairs in approximately 20% of the European population.24 CNVs have been shown to influence gene dosage and therefore can contribute to risk for cancer, as demonstrated for a chromosome 1 CNV and the risk for childhood neuroblastoma.25 Accurately determining CNVs from SNP microarrays continues to be a formidable technical challenge, but with new resources and sequencing technologies, termed “next generation sequencing,” it is anticipated that precision will continue to improve, which, in turn, should lead to improved detection of CNVs associated with disease outcomes.
Principles of Linkage Mapping
Many epidemiological studies indicate the presence of a familial contribution, such as the observation that family history of a specific cancer within first-degree relatives is associated with a doubling or more of risk among relatives, particularly in twin registries.26,27 In the case of prostate cancer, for instance, studies of selected hospital-based patient populations, population-based case-control studies, and cohort studies all demonstrate that a family history of disease is correlated with an increase in an individual’s risk. If the affected family members are first-degree relatives (e.g., brothers or fathers and sons), the risk increases from 1.7-fold to 3.7-fold. Younger ages at diagnosis and multiple affected relatives with the disease tend to be associated with even higher relative risk.28–31 For example, men with three or more first-degree relatives with prostate cancer have an almost elevenfold increased risk of the disease compared with men who have no known family history of the disease.29 For this reason, families ascertained for linkage analysis studies tend to be large, have multiple affected individuals, and feature people who were diagnosed with the disease at a comparatively young age.
Familial aggregation describes the occurrence of multiple cases of cancer within a family (Fig. 22-6). Clustering of familial cases may be due to shared environment, shared alleles of particular genes, or simply chance if the tumor is very common in the population. In mapping of cancer susceptibility genes for many cancers, particularly for breast and colon cancer, the most promising pedigrees for hereditary cancer are families with three or more first-degree relatives with a given cancer, three successive generations with cancer, or at least two siblings with the same cancer detected at a relatively young age. First-degree relatives are parents, offspring, or siblings.
Obtaining good clinical information for all persons in a family mapping study gives geneticists the power to stratify the data into more homogenous subsets, which increases statistical power for finding genes associated with any one particular aspect of a phenotype. For example, if a subset of individuals in the family in Figure 22-7 all had tumors of similar stage and grade, the data from this homogenous subset of individuals could be considered in isolation from the remainder of the affected cases, thus reducing heterogeneity and increasing power. Recall that for many common diseases, many susceptibility genes are likely to be present in the population.14 The ability to stratify families on the basis of clinical features of disease, family history, age at onset, and presence or absence of other cancers are approaches to develop homogenous subsets and improve success.
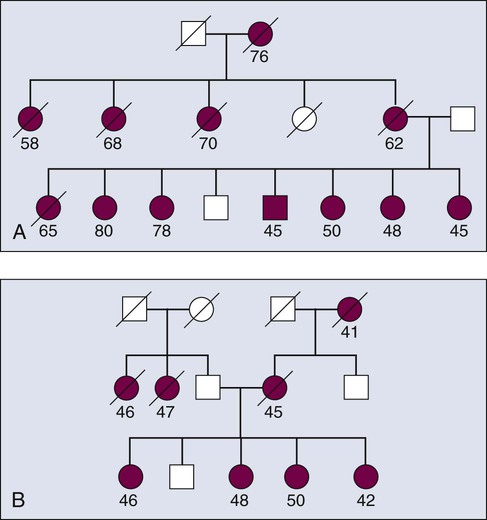
Theoretically, a given set of affected individuals within a family would all have cancer for the same reason—that is, each member would have inherited a mutated copy of the same gene. Because distinct mutations exist within a gene, each of which can confer high penetrance, the approach is predicated on finding a gene and not the specific mutation within a gene. For example, a number of mutations across the BRCA1 gene can confer an increased risk for breast and/or ovarian cancer, with measurable differences in penetrance.32 This latter point suggests that there are differential effects of disturbances of key biological pathways. Moreover, recent genome-wide association studies (GWAS) have begun to identify secondary genetic modifiers that further modulate the penetrance of BRCA1 mutations.33,34
Figure 22-7 demonstrates two types of seemingly useful families for linkage mapping studies. Both include a significant number of affected members. The first family has a large number of affected individuals (Fig. 22-5). However, some persons were affected very early in life, whereas others were diagnosed at later ages. It is likely that some persons have the disease because they inherited mutated copies of a particular gene, whereas others have the disease for sporadic reasons unrelated to the disease allele segregating in the family. Age at onset provides some guidance as to which persons are more likely to have hereditary versus sporadic forms of the disease, but age at onset is not absolute, and in the case of a disease with age-dependent penetrance, some people will be affected late in life, even though they carry a mutant allele, and others will be affected early in life for sporadic reasons. The second family shown in Figure 22-5 appears to be more informative for linkage mapping studies because the family includes several affected individuals and all were affected at a relatively early age. However, the presence of disease segregating on both sides of the family should be noted. The affected persons in the youngest generation could have cancer because they inherited mutant alleles from one or both sides of their family and one or multiple genes could be involved. Thus the family is of limited usefulness for mapping studies.
Finding Cancer Susceptibility Genes
Linkage analysis has been successful in identifying highly penetrant mutations in multiply affected families for both common and uncommon cancers. A combination of linkage and candidate gene analyses revealed mutations in CDKN2A or CDK4 in roughly 50% of cases of familial melanoma, although there appears to be heterogeneity in exposure to a strong carcinogen for melanoma—that is, ultraviolet sun rays.35,36 For a rare familial cancer, chordoma, a gene duplication of the T (brachyury) gene confers susceptibility.37 With next-generation sequencing, investigators are expected to return to families in whom the problem is not solved with linkage analysis and search of sets of susceptibility alleles that can explain an oligogenic risk model.
The breast cancer susceptibility genes BRCA1 and BRCA2 were among the first to be mapped because large and well-characterized families had been meticulously ascertained.38,39 The presence of ovarian cancer in some families and not in others and the presence of breast cancer in some male carriers allowed for creation of data sets enriched for the BRCA1 and BRCA2 genes, respectively. In turn, the initial identification of the BRCA1 gene and subsequent removal of BRCA1-linked families from remaining data sets provided further useful enrichment for BRCA2-linked families.39,40
For the breast cancer susceptibility genes BRCA1 and BRCA2, several founder mutations have been identified in different populations.41–44 For instance, a single BRCA2 mutation, 999del5, was initially found in 16 of 21 Icelandic families with breast cancer.45 All 16 of these families share a haplotype or pattern of alleles within the BRCA2 gene, suggesting a common ancestral origin. This pattern has since been replicated several times. Studies of breast cancer in Ashkenazi Jewish families have also demonstrated this point, contributing enormously to our knowledge of founder mutations for both BRCA1 and BRCA2.46,47 The three common founder mutations in this population, BRCA1-185delAG, 5382insC, and BRCA2-6174delT, have a combined population prevalence of 2% to 2.5%. With these observations in mind, investigators have frequently sought families for genetic mapping studies from regions of the world where marriage between related individuals is not discouraged and where geographic barriers have restricted gene flow.
Locus heterogeneity can be reduced by studying families from isolated or inbred populations. Fewer disease alleles are predicted to segregate with a particular phenotype in a population derived from a limited number of founders. Studies of colon cancer in Finland and studies of breast cancer in Iceland and in Ashkenazi Jewish populations illustrate these points very well. In Finland, two variants in the DNA mismatch repair gene MLH1, termed mutations one and two, account for 51% of all Finnish families with verified or putative cases of hereditary nonpolyposis colorectal cancer.48 Nineteen families with mutation one and six families with mutation two underwent further investigation by haplotype analysis with use of 15 microsatellite markers surrounding the MLH1 locus. The presence of two distinct, large, conserved disease haplotypes, one in families with mutation one and the other in families with mutation two, indicated that these families are likely to descend from two common ancestors born in the sixteenth century and the eighteenth century, respectively.
Principles of Association Testing
Although genetic linkage analysis has been the workhorse for discovery of mutations underlying Mendelian disorders, geneticists have also considered strategies to map complex diseases, namely, those in which multiple distinct genetic regions plus environmental factors contribute to risk for disease. Linkage analysis did not fare well when applied to complex diseases, primarily because of insufficient power to detect association of smaller effect sizes for multiple susceptibility alleles and complexities in phenotype assignment. Risch and Merikangas49 pointed out the shortcomings of linkage for complex disease mapping and made the case for association analyses in populations of unrelated subjects. Their projections have been born out in the age of GWAS.50
In response to new platforms that can simultaneously test large numbers of genetic variants and the perceived opportunity to more efficiently search for genetic susceptibility to complex, common diseases, such as most cancers, the testing strategy for association studies shifted from candidate gene studies to GWAS. Before the advent of GWAS, investigators chose specific variants based on prior hypotheses and analyzed underpowered studies, yielding a sea of false-positive reports. Of the thousands of candidate gene association studies performed prior to the GWAS era, fewer than 10 have been robustly replicated in cancer studies. The most notable examples include common variants in NAT2 and GSTM1 in persons with bladder cancer and alcohol dehydrogenase genes (ADH1B and ADH7) in persons with aerodigestive cancers.51–54 As the annotation of the human genome emerged with first the International HapMap and then the 1000 Genomes Project, the approach shifted toward utilizing surrogate markers across the genome designed to capture the majority of common genetic variation in reference continental populations from Africa, Asia, and Europe with a minor allele frequency of greater than approximately 1%.55,56
GWAS have been successful across a spectrum of diseases and traits, yielding more than 2000 regions of the genome that harbor common susceptibility variants associated with more than 150 diseases/traits.50 The approach is based on an initial scan across the genome followed by independent replication.57 The results of scanning hundreds of thousands of SNPs are analyzed using an “agnostic” statistical approach utilizing logistic regression analysis, often—but not always—adjusted for critical covariates (e.g., age, study, and measures of subtle differences in underlying population genetics history). In other words, GWAS are pursued free of prior hypotheses, unlike standard linkage analysis. In this regard, one region is not favored over another.
The GWAS strategy is based on testing “indirectly” for the actual genetic variant responsible for the association using surrogate markers in cases and control subjects to point to regions that harbor susceptibility alleles.13 Thus the actual functional marker directly responsible for the effect does not have to be actually tested in the scan. Instead, its surrogate, which is in LD as measured by a high correlation (r2 > 0.8), can be replicated in subsequent studies, pointing to the susceptibility allele(s). Hence substantial effort is required to “fine-map” the region—that is, finding all of the correlated variants before choosing which ones to examine in follow-up studies. In many regions, variants with lower minor allele frequencies are also highly correlated with the GWAS marker and, on occasion, a less common allele with a stronger effect has been identified, yielding a so-called “synthetic association.”58 However, to date, the majority of susceptibility alleles cannot be explained by less common variants with stronger effects.59
Patterns of LD vary between populations, both in specific regions and across the genome.19 This pattern variation results in major differences in the minor allele frequencies between populations on a per-SNP basis, as well as the intervals of LD, defined by recombination hot spots. In rare circumstances, the differences between incidence in disease, such as prostate cancer between men of European and African ancestry, can be explored using admixture linkage analysis. Notably, one of the first GWAS signals for prostate cancer on 8q24 was also found by admixture analysis.60,61
The basic principle behind an association study is that a statistically significant difference exists in the distribution of one or more alleles between cases and control subjects, indicating the location of a susceptibility allele that contributes to cancer risk. However, in association studies, the findings can uncover mutations that are highly penetrant and strongly correlated with risk for development of cancer, as seen in the family linkage studies previously discussed. This situation is reflected in the fact that the estimated odds ratios are substantially smaller in GWAS, almost always conferring a ratio of less than 1.4.62 Thus low-effect susceptibility alleles are neither necessary nor sufficient for development of a cancer. Hence for most forms of cancer, we expect that many susceptibility alleles exist, each of which contributes a small effect to the disease.
Investigators use one of several commercial SNP microarray chips to scan the genome and compute rank p values for prioritization of promising genetic markers for replication studies in one or more independent data sets of cases and control subjects. Because of the daunting challenge of false-positive results in testing so many markers, a community-wide standard has emerged that protects against false-positive findings; this protection is achieved when studies report markers that surpass the threshold of genomewide significance, now generally defined as a trend association test with a p value of 5 × 10−8.57 These data can be reported in the primary scan, if it is large enough, or in a combined analysis of the scan and follow-up replication studies or large metaanalyses, which combine data from several independently collected data sets. Because GWAS discover loci that are highly associated with specific markers, surpassing this threshold ensures that a very small probability of a false-positive result exists, which is particularly important because extensive follow-up analyses are required to map and investigate the biological underpinning of the susceptibility allele.
Because GWAS can be effectively scaled to accelerate discovery, the major challenge ahead is to determine how to establish the critical connection between the genetic markers of susceptibility alleles and carcinogenesis. The rapid pace of discovery using the GWAS approach has not been matched by research to interpret and understand the functional significance of different alleles that are correlated with cancer phenotypes. The gap between the number of new independent markers and a biological understanding of the loci continues to widen at an accelerated pace because of the differences in scientific approaches. The GWAS approach is scalable using surrogate markers across the genome so that with larger sample sets, further discovery is possible. However, rarely is the marker also the functional variant. Thus follow-up analyses are generally required to characterize each susceptibility allele. The patterns of genetic variation vary greatly across regions, thus requiring a detailed fine mapping of each region before choosing individual genetic variants for laboratory study (Fig. 22-7).
Study Design and Association Studies
Until the age of GWAS, a vigorous debate ensued among persons conducting candidate gene studies regarding the impact of possible differences in underlying population substructure. The testing of thousands of uncorrelated SNP markers in GWAS has provided investigators with the opportunity to sift out persons who differ substantively across the genome. Several different analytical algorithms can distinguish the degree of admixture across chromosomes, based on referential continental population sets (e.g., International HapMap or 1000 Genome Projects).63,64
Association Studies in Cancer
To date, more than 350 genomic regions have been conclusively established (i.e., achieving the threshold of genomewide significance, which protects against the likelihood of a false-positive discovery) for more than two dozen distinct types of cancers.6 The majority of susceptibility alleles are specific to one cancer, but at least nine regions harbor alleles that contribute to two or more cancers. Nearly all cancer susceptibility alleles discovered by GWAS have an MAF greater than 10%, with a handful in the 5% to 10% range as reported in large studies. Overall, the per-allele estimated effect size has been small, with odds ratio between 1.1 and 1.4. Several of the alleles reported in pediatric cancers have risks of 1.6 to 1.8, but for the adult cancers the norm is low.65 Among the most significant is testicular cancer, which is known to have a high heritability in families and twin studies; GWAS identified a susceptibility allele with a per-allele effect estimate of greater than 2.5 on chromosome 12 (KITLG).66,67
Thus far, a small fraction of susceptibility alleles have been associated with more than one distinct cancer type; these alleles are quite informative and reveal possible common carcinogenic mechanisms underlying distinct cancers. Although it is not unexpected to detect the human leukocyte antigen (HLA) regions for cancers of the immune system or those driven by viral infections, the mapping of the HLA alleles require detailed analyses. Commercial arrays and imputation provide inadequate discrimination of the region, particularly because of its complex structure. Other technologies will be required to comprehensively explore this region in cancer susceptibility. The region flanking the MYC oncogene on 8q24 harbors at least five independent loci associated with prostate cancer, as well as loci associated with cancers of the breast, colon, and bladder and chronic lymphocytic leukemia.60,68–76 The nearby MYC oncogene is a plausible candidate gene, and recent work suggests that allelic differences in enhancers could directly or indirectly interact with MYC, although little evidence exists to suggest that coding changes are important.79–79
A region on 5p15.33 harbors a range of susceptibility alleles for many cancers, including rare and common SNP alleles. Thus far, 10 distinct cancers have been observed in GWAS, and at least five independent alleles in this region contain the telomerase gene (TERT).80–89 Rare mutations in TERT track with congenital dyskeratosis (an inherited bone marrow failure syndrome), idiopathic pulmonary fibrosis, acute myelogenous leukemia, and chronic lymphocytic leukemia.92–92 The pleiotropy in this region hints at complex gene-gene or gene-environment interactions. For instance, a protective allele for one cancer appears to be a susceptibility allele for another cancer; it is remarkable that the same allele has inverse effects for two skin cancers, basal cell carcinoma and melanoma.
The discovery of new susceptibility alleles is now being driven by the use of imputation. Imputation is based on several computational algorithms that infer untested and highly correlated SNPs based on reference data sets (e.g., International HapMap or 1000 Genome Projects)93; it is predicated on the basis of the observed LD between SNPs in reference populations drawn from distinct regions. A notable exception is the recent identification of a rare SNP on 8q24, conferring susceptibility to prostate cancer, which was discovered and characterized in the isolated population of Iceland.72 However, the usefulness of imputation for SNPs below 1% is limited in the general or admixed populations, mainly because of the enormous number of population-private rare variants.
The fine mapping of susceptibility alleles has led to the discovery of independent signals nearby, indicating that more than one allele contributes to cancer risk. The first prostate cancer susceptibility allele on 8q24 has now blossomed to five separate alleles, at least one of which is apparent only in African Americans, because of its rarity in European and Asian populations. An intergenic region of 11q13 harbors several independent prostate cancer susceptibility alleles, but also nearby are distinct alleles for renal and breast cancer, each of which works through separate mechanisms.94,95
The formation of numerous international consortia promise to maintain the pace of discovery of common susceptibility variants, not only in populations of European ancestry but those of other ancestry, including studies from Asia and Africa. To date, the majority of reported GWAS studies have been in subjects of European ancestry, but progressively more studies have been conducted in subjects of Asian and African ancestry. In a few instances, alleles have been identified in specific populations with substantially higher frequency. For instance, a region of chromosome 17q21 harbors a prostate cancer susceptibility allele in African Americans, for which the best marker has an MAF of 5%, whereas in persons of European background it is below 1%.96 Additional alleles in 8q24 have been reported in men of African American background, but they do not fully explain the difference in incidence.
Differences in study design can lead to conflicting conclusions, including the biological interpretation of the association. Initially, two distinct GWAS in prostate cancer reported contrary results for alleles on chromosome 19q13.33, which harbors the gene responsible for the prostate serum antigen (PSA).71,97 In a GWAS using cohort studies, the effect appears to be related to PSA levels, whereas in a study using advanced cases and control subjects with low PSA levels, the effect points toward carcinogenesis. Follow-up studies, including fine mapping of the KLK3 gene (which encodes PSA) and further replication, have revealed evidence that the locus could be associated with both prostate cancer susceptibility and PSA levels.100–100
Genetic Architecture Underlying Cancer Susceptibility
The underlying genetic architecture can differ by cancer sites with respect to the relative contribution of common and rare variants, with the latter detected as highly or weakly penetrant mutations. As indicated in Figure 22-1, the emerging picture suggests that variants of low effect size are commonly seen in the population and contribute a fraction of the heritable risk for the cancer. Although ongoing studies are expected to generate a more comprehensive catalog of variants in each class (e.g., common and rare), early modeling of empirical data suggest differences between cancers.103–103
Because a number of groups are investigating pedigrees that were not solved by linkage mapping, it is likely that some proportion of their disease may be due to sets of common SNPs or may be oligogenic in nature, such that a few moderately or weakly penetrant mutations contribute to their cancer risk. Studies of families with breast cancer aggregation have led to the discovery of susceptibility alleles, each with moderate effects of estimated relative risk between two and four. Interestingly, the majority of these susceptibility alleles map to genes in pathways related to BRCA1 activity. Furthermore, the genes harboring alleles with frequencies in the range of 1% include the ATM, BRIP1, CHEK2, ERCC2, PALB2, RAD51C, and RAD51D. These susceptibility alleles were discovered by sequencing persons from high-risk pedigrees and on occasion looking at the effect of the rare allele in the general population. These studies require large sample sizes, which are available for breast cancer. However, for other cancers, novel findings exist for both common cancers such as prostate cancer (e.g., 8q24 and HOXB13), as well as less common cancers, such as testicular germ cell cancer.72,104,105 Corroborative laboratory work can supplement analyses in families and population-based studies. For instance, a rare variant in the MITF gene that has an allele frequency of approximately 1% increases the risk for melanoma, a dangerous skin cancer.106,107 After familial testing showed incomplete penetrance and association studies suggested an effect in unrelated populations, laboratory investigation revealed that the mutation resulted in impaired SUMOylation and differentially regulated MITF targets.
Unraveling the Cancer Biology of Cancer Susceptibility Alleles
To unravel the biological underpinnings of cancer susceptibility regions, investigators are turning to new resources and approaches. Consortium guidelines have been developed to accelerate the pace of characterization of cancer susceptibility alleles.108 For instance, progress in understanding regions identified by GWAS has recently received a major boost by the publication of the ENCODE (ENCyclopedia Of DNA Elements) Project, a far-reaching project to map the functional genome.111–111 The ENCODE papers have begun to shed light on the biology of the regulation of the genome, specifically cataloging signposts and markers of biological activity. The ENCODE Project has sharpened our vision of the inner workings of how the genome functions. With use of ENCODE, it is possible to trace the errors that contribute to cancer susceptibility. A greater than expected fraction of cancer susceptibility alleles map to regions of high probability for a functional effect on gene regulation, using multiple types of ENCODE data. Although this type of study did not pinpoint a specific variant, it suggests that a subset of regulatory SNPs are promising for follow-up work. Moreover, it is now possible to look at patterns of regulatory variation in persons and across populations, integrating experimental data with computational tools, using several in silico programs that should enable assessment of known susceptibility regions and prioritize variants for further study, with a higher priority for functional value.114–114 Already, new insights into breast cancer susceptibility loci identified by GWAS have been generated with use of ENCODE insights.115
One of the first prostate cancer susceptibility alleles identified by GWAS was on chromosome 10q11, which localizes to the beta-microseminoprotein gene (MSMB).116 Its gene product has been the subject of research in early detection studies as a possible biomarker. A number of groups have shown that the risk allele, a T in the promoter region, decreases transcriptional activity and also tracks with lower expression of the gene product in prostate cancer tissue.117,118 When prostate cancer progresses from early to late stages, the expression of MSMB progressively decreases, and loss of MSMB expression is associated with disease recurrence after radical prostatectomy. On the other side of the MSMB gene is a second gene, NCOA4, which is upregulated by this same promoter SNP, providing evidence for a more complex biological effect.119 Chimeric transcripts between MSMB and NCOA4 have also been observed.120 Ongoing studies are examining the relationship between the MSMB allele and urinary levels of MSP.
The search for functional variants underlying GWAS signals has begun to uncover new insights, some with potential clinical potential. For example, investigation of the bladder cancer GWAS signal on 8q24.2, followed by RNA sequencing and genetic and functional analysis, identified a variant that is strongly associated with increased messenger RNA expression of the prostate stem cell antigen (PSCA) gene.121–124 Furthermore, this variant creates an alternative translation start site and leads to increased expression of PSCA on the cell surface, where it can be subjected to immunotherapy with anti-PSCA antibody, an emerging therapy for several cancers. Because the genotype of the GWAS variant is predictive of PSCA protein expression, a genetic test could be used to identify patients with bladder cancer who could benefit from the anti-PSCA therapy.
A few of the GWAS susceptibility alleles have been conclusively shown to interact with environmental exposures. An SNP in NAT2 is important for bladder cancer susceptibility only in people who have “ever smoked,” whereas in persons who have never smoked, no effect occurs.124 The GWAS of lung cancer, which is strongly driven by tobacco exposure, have revealed that select alleles are operative only in smokers, whereas others are observed only in nonsmokers.127–127 The strongest signal seen in smoking-related lung cancer GWAS on chromosome 15q25 is not evident in nonsmoking women in Asia, suggesting that the contribution of 15q25 is not associated with lung cancer, independent of smoking.128
Initially, many investigators attempted to elucidate the functional genetic variant and a mechanism underpinning the genetic association between clearance of hepatitis C virus (HCV), a risk factor for liver cancer, and genetic variants upstream of IFNL3, previously called IL28B on chromosome 19, discovered in several GWAS.131–131 However, RNA-sequencing analysis in primary human hepatocytes has uncovered a new gene, IFNL4, which is generated by a complex dinucleotide insertion/deletion variant in LD with the GWAS markers.132 The deletion allele of this variant causes a frame shift, which leads to a novel protein that induces an interferon-type response. The genetic analysis showed that the IFNL4 genetic variant has a strong effect on HCV clearance, especially in persons of African ancestry. The strength and the effect size of the genetic association in IFNL4 leading to spontaneous and treatment-induced clearance of HCV are large enough to pursue in clinical studies.
During the practice of applying stringent quality control metrics to GWAS data, a pattern of unexpected deviations emerged that have led to the investigation of the detection of genetic mosaicism, defined as two or more distinct karyotypes within an individual.133 Using SNP data, it is possible to estimate that approximately 1% of the adult population harbors one or more large somatic events.134,135 For hematopoietic malignancies, using cohort studies, it is possible to detect somatic events well in advance of the diagnosis of chronic lymphocytic leukemia. A more refined understanding of mosaicism in the aging genome promises new insights into genomic instability as it relates to carcinogenesis, especially in the hematopoietic system.
GWAS in large consortium have looked at intermediate exposures, which are strongly implicated in cancer risk, such as body mass and other quantitative traits (e.g., tobacco use and alcohol consumption), yielding new insights into biology.101,125,136–140 The study of height in hundreds of thousands of subjects for whom GWAS data are available has uncovered novel pathways and provided a more stable assessment of the contribution of common SNPs (MAF > 5%) to a trait such as height. Polygenic models for many SNPs, each with small effects not yet conclusively discovered by GWAS, define the fraction of heritability for important traits.141
Clinical Implications of Cancer Susceptibility Alleles
Of the more than 350 independent cancer susceptibility alleles for more than two dozen cancers, only a few are also associated with one or more cancer.6 This finding suggests that distinct alleles may influence the etiology of a cancer and a different set may influence clinical outcomes, such as progression or metastasis. For instance, none of the 75 reported regions for prostate cancer clearly separates men with aggressive cancer from those with indolent disease. In the childhood cancer neuroblastoma, regions have been identified that are associated with more aggressive disease.142,143 The discipline of pharmacogenomics has been accelerated by the same trends that have driven the discovery and characterization of germline genetic susceptibility alleles.144,145
Susceptibility alleles have been identified for risk for second cancers: these alleles include highly penetrant mutation of the retinoblastoma gene leading to osteogenic sarcoma and common SNPs on chromosome 6q21 after radiation therapy for pediatric Hodgkin disease.146 Distinct regions contribute to pharmacogenomics, defined as the contribution of germline genetic variation to response rates or toxicity associated with therapeutic modalities (e.g., medicine, surgery, or therapeutic radiation).147 The discipline of pharmacogenomics has been accelerated by the same trends that have driven the discovery and characterization of germline genetic susceptibility alleles.144,145
The effect of highly penetrant germline mutations on cancer outcomes and more specifically cancer risk has generated newfound interest in the contribution of the germline mutations to cancer outcomes across all cancers. In women with invasive ovarian cancer, patients with germline mutations in BRCA1 or BRCA2 carry an improved 5-year overall survival.148,149
Next-Generation Sequencing Analysis
With the availability of next-generation sequencing platforms, investigators are searching for less common and rare variants to explain a proportion of heritability of cancer susceptibility.150 Next-generation sequencing platforms perform massively parallel sequence analysis of major fractions of the genome with a high degree of redundancy, which is needed to minimize the error in calling sequence variants. These technologies are reshaping the scope of genetic studies in families and is starting to do so in population-based studies.
This transition will accelerate the identification of many possible variants, as the number of uncommon and rare variants increases. In large-scale sequencing of a large fraction of the exome (defined as the targetable exons of known genes), there are thousands of novel variants per individual, some reflecting rare and population-private variants. Consequently, the statistical challenge of parsing rare variants in unrelated population studies is daunting, especially as the minor allele frequency decreases, because the sample sizes required to detect alleles with low to moderate effect sizes becomes larger.151 Large databases and studies should be developed to conduct larger discovery analyses. For the foreseeable future, the discovery paradigm has shifted to include correlative laboratory confirmation of promising variants. Certainly, the ENCODE resource will be helpful in prioritizing variants for laboratory study.111
Clinical Relevance and Applications
Genetic Counseling and Testing
The identification of cancer susceptibility alleles has prompted rapid transition of common SNPs to individualized disease prevention and public health policy.152 To date, the data do not adequately support clinical usefulness, despite suggestions to the contrary by commercial direct-to-consumer groups.153,154 The identification of cancer susceptibility SNPs has triggered a debate regarding when and how to transition them into clinical care. Because the cumulative set of SNPs for any one disease still provides only a fraction of the population risk for a cancer, namely less than 10% overall, it is difficult to integrate single or small sets of SNPs into clinical paradigms without further study to augment the list to increase sensitivity and specificity.155 Although strong commercial pressure has been exerted, clinical studies have yet to provide conclusive evidence supporting transition into clinical practice. Eventually, it may be possible to reclassify high-risk versus low-risk persons in anticipation of deciding on a preventive or early detection program.153 It is also important to keep in mind that SNPs may not be informative for every cancer, because differences in the underlying genetic architectures may influence the putative utility of introducing sets of independent SNPs into clinical practice.
What the Future Holds
The sequence of the human genome has been referred to as an “instruction book for human biology,”156 and it has become clear that many dynamic factors interact in regulating and responding to the human genome, including environmental, behavioral, and lifestyle factors. Locked within the sequence of each person’s DNA is the information that can enable pursuit of a healthy lifestyle, but encoded as well are the sequence errors that could determine each person’s susceptibility to a spectrum of diseases and outcomes.
The results of the comprehensive sequence analyses of paired sets of genome (e.g., germline and somatically altered cancers) have uncovered a large spectrum of mutational events and epigenetic alterations.157–161 These newfound insights will provide opportunities to carefully unravel the interrelationship between the germline susceptibility alleles and cancer etiology plus progression. Already we can see somatic alterations that can explain exceptional responses to targeted therapies.162 A more complete understanding of the molecular pathways involved in cancer susceptibility will suggest avenues for the development of both methods of diagnosis and treatment. Identification of specific genes offers the promise of genetic testing to persons at risk, as well as the hope for targeted therapeutics. Finally, understanding the specific variation offers the promise of twenty-first century “precision medicine” in which lifestyle, diet, and preventative therapies come together to offer patients a full spectrum of choices for maintaining their personal health.





