Cutaneous T-Cell Lymphoma and Cutaneous B-Cell Lymphoma
Christiane Querfeld and Steven T. Rosen
• Cutaneous lymphomas represent 3.9% of non-Hodgkin lymphomas.
• The annual incidence of cutaneous T-cell lymphoma (CTCL) in the United States is approximately 9.6 cases per 1 million population with a median age of 60 years at initial presentation.
• CTCL accounts for up to 71% and cutaneous B-cell lymphoma (CBCL) accounts for up to 29% of all cutaneous lymphomas.
• CTCL may be indolent as in mycosis fungoides (MF) or aggressive as in Sézary syndrome (SS).
• The malignant T cells in MF/SS are characterized by a CD4+ T-helper (Th)/memory cell phenotype with frequent loss of CD7 and/or CD26 and the expression of skin homing markers such as CLA, CCR4, and CCR10. MF/SS demonstrate an altered immune biology and the accumulation of cytogenetic abnormalities during disease progression with a predominant Th2 cytokine profile in advanced stages.
• Primary CBCLs such as cutaneous follicle center lymphoma have failed to show similar alterations compared with their nodal counterpart. The prognosis of both primary cutaneous follicle center lymphoma and cutaneous marginal zone lymphoma is usually excellent.
• The gene expression profile of primary cutaneous large B-cell lymphoma, leg type, is similar to that of nodal diffuse large B-cell lymphoma (DLBCL) arising from germinal center or post–germinal center–activated B cells with constitutive nuclear factor–κB (NF-κB) pathway activation and strong expression of the IRF4/MUM1 transcription factor and carries a worse prognosis.
• All patients with cutaneous lymphoma should initially be seen and co-managed by a multidisciplinary cutaneous lymphoma team consisting of members from dermatology and oncology with the support of other health care professionals such as radiation oncologists, pathologists, and clinical psychologists.
• Routine evaluation should include complete physical examination, complete blood cell count with differential, and chemistry panel with lactate dehydrogenase (LDH) level, skin biopsy for histology, immunophenotyping and gene rearrangement studies, and lymph node biopsies in cases with enlarged nodes at presentation.
• Imaging studies and flow cytometry panel of peripheral blood should be reserved for patients with clinical and laboratory findings suggestive of systemic disease and/or prominent lymphadenopathy. Bone marrow biopsy is a consideration in advanced-stage disease. Histopathological and molecular results should be correlated with clinical findings and patients classified according to the World Health Organization/European Organization of Research and Treatment of Cancer (WHO/EORTC) consensus classification.
• Early-stage MF usually responds well to skin-directed therapies such as phototherapy, radiation, topical nitrogen mustard, retinoid, or corticosteroid skin creams.
• Treatment for patients with advanced stages of MF or SS generally requires systemic therapies, including biological or immune therapies, histone deacetylase inhibitors, conventional chemotherapeutic agents, or combinations of these agents.
Introduction
A variety of non-Hodgkin lymphomas can involve the skin, either primarily or secondarily. Primary cutaneous lymphomas present in the skin with no evidence of extracutaneous disease at the time of diagnosis. They comprise a heterogeneous group of cutaneous T-cell lymphomas (CTCLs), cutaneous B-cell lymphomas (CBCLs), natural killer (NK) cell, and precursor hematopoietic neoplasms with distinct variability in clinical presentation, histopathology, immunophenotyping, gene rearrangement, and prognosis. The diversity of clinical and pathological manifestations among subsets of cutaneous lymphomas has led to much controversy over its diagnosis and classification and to the proposition of new variants and the establishment of consensus guidelines by a joint effort of the World Health Organization and European Organization for Research and Treatment of Cancer (WHO/EORTC) in 2005.1 These guidelines were incorporated into the revised fourth WHO classification of tumors of hematopoietic and lymphoid tissues in 2008 using the existing framework for nodal lymphomas; however, differences remain, particularly to the subclassification of CBCLs.2
Based on the WHO/EORTC classification, cutaneous lymphomas are classified as indolent, intermediate, or aggressive.1,3 The two most commonly recognized types of CTCL are mycosis fungoides (MF), which is generally indolent, and Sézary syndrome (SS), which is an aggressive and leukemic form of disease (Table 107-1). MF and SS together comprise 53% of all cutaneous lymphomas and are often synonymously named as CTCL. Notably, cutaneous manifestations of human T-cell lymphotrophic virus type I (HTLV-I)–associated adult T-cell lymphoma/leukemia (ATLL) occur in about 50% of cases and can mimic a slowly progressing course of MF.4,5 The second common CTCL type encompasses the spectrum of primary cutaneous CD30+ lymphoproliferative disorders, including lymphomatoid papulosis and cutaneous anaplastic large cell lymphoma; this CTCL type comprises 20% of all cutaneous lymphomas. Other, rarer types include the subcutaneous panniculitis–like T-cell lymphoma, which is generally indolent, and the group of primary cutaneous peripheral T-cell lymphomas (PTCLs) that includes the provisional entities cutaneous aggressive epidermotropic CD8+ T-cell lymphoma, cutaneous γ/δ T-cell lymphoma, and cutaneous CD4+ small/medium-sized pleomorphic T-cell lymphoma, with the last generally recognized as more indolent than the others.
Table 107-1
The WHO/EORTC Consensus Classification for Primary Cutaneous Lymphomas with Relative Frequency and 5-Year Survival Rate
| WHO-EORTC | Frequency (%) | 5-Year Survival (%) |
| CUTANEOUS T-CELL AND NK-CELL LYMPHOMA | ||
| INDOLENT | ||
| Mycosis fungoides | 44 | 88 |
| Follicular mycosis fungoides | 4 | 80 |
| Pagetoid reticulosis | <1 | 100 |
| Granulomatous slack skin | <1 | 100 |
| CD30+ LYMPHOPROLIFERATIVE DISORDERS | ||
| Anaplastic large cell lymphoma | 8 | 95 |
| Lymphomatoid papulosis | 12 | 100 |
| Subcutaneous panniculitis–like T-cell lymphoma | 1 | 82 |
| CD4+ small/medium pleomorphic T-cell lymphoma | 2 | 72 |
| AGGRESSIVE | ||
| Sézary syndrome | 3 | 24 |
| Cutaneous peripheral T-cell lymphoma, unspecified | 2 | 16 |
| Cutaneous aggressive CD8+ T-cell lymphoma | <1 | 18 |
| Cutaneous γ/δ T-cell lymphoma | <1 | |
| Cutaneous NK/T-cell lymphoma, nasal-type | <1 | |
| CUTANEOUS B-CELL LYMPHOMA | ||
| Indolent | ||
| Follicle center cell lymphoma | 11 | 95 |
| Marginal zone lymphoma | 7 | 99 |
| Intermediate Clinical Behavior | ||
| Large B-cell lymphoma of the leg | 4 | 55 |
| Cutaneous diffuse large B-cell lymphoma, other | <1 | 50 |
| Intravascular large B-cell lymphoma | <1 | 65 |
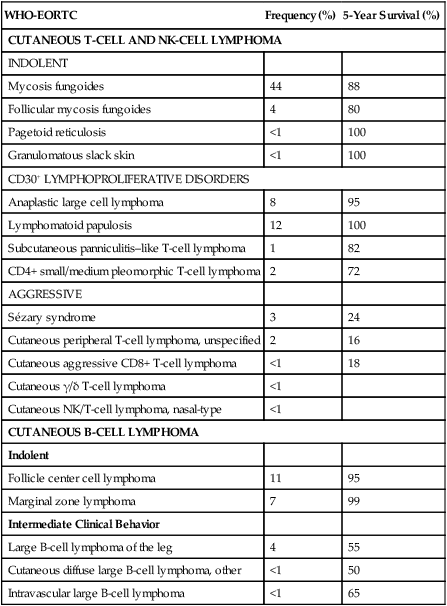
Modified from Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood 2005;105:3768–85.
Epidemiology
Cutaneous lymphomas represent about 3.9% of all non-Hodgkin lymphomas. Frequency and survival data used in the WHO/EORTC classification are based on Dutch and Austrian registries.1 Cutaneous lymphoma incidence patterns were studied using data from 3884 patients from 16 Surveillance Epidemiology and End Results (SEER) program registries in the United States during 1980 through 2005.6 Data revealed that CTCLs account for 71% and CBCLs account for 29% of all lymphomas. MF accounted for 38% of all CTCLs, followed by cutaneous PTCL (20%) and CD30+ lymphoproliferative disorders (10%). The age-adjusted annual incidence of CTCLs in the United States with most cases classified as MF has increased from 2.8 per million (1973-1977) to 9.6 per million (1998-2002) according to data from Criscione and Weinstock, and from 5.0 per million to 12.7 per million, according to Bradford and associates.6,7 MF is the most common type of CTCL, with a predominance of male patients of approximately 2 : 1 and a predominance of African-American patients of 1.6 : 1. The median age at presentation is between 50 and 70 years with only a small number of cases occurring in children and young adults. CBCL incidence rates are highest in non-Hispanic whites with a male predominance and an exponential increase with age. The most common primary CBCL subtypes are cutaneous DLBCL, follicle center cell lymphoma, and marginal zone lymphoma, comprising 40%, 30%, and 25% of all cases of CBCL, respectively.
Etiology
Despite rising incidence rates of CTCL, its etiology remains elusive. It has been suggested that the condition results from persistent antigen stimulation; however, its antigen-driving process is not known. Microbiological, environmental, occupational, and lifestyle factors have been suggested in the etiology of CTCL, but none has yet been verified.10–10 Unlike ATLL, which is etiologically associated with HTLV-I, most patients with CTCL are serologically negative for HTLV-I.11,12 Other investigators have found serologic evidence for Epstein-Barr virus and cytomegalovirus, but these findings have not been substantiated.15–15 Immunosuppression and/or immunosuppressive therapy might be a risk factor for the development of CTCL, as documented in patients after treatment with tumor necrosis factor-α (TNF-α) antagonists, treatment of organ transplant or of Hodgkin disease, and in human immunodeficiency virus (HIV)-positive patients.16–19
Cutaneous T-Cell Lymphoma
Mycosis Fungoides and Sézary Syndrome
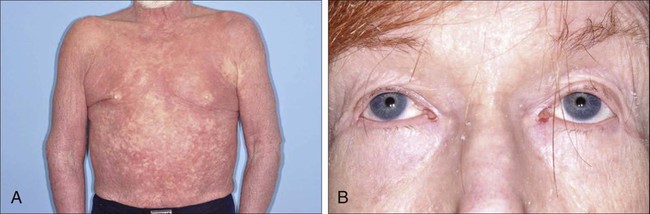
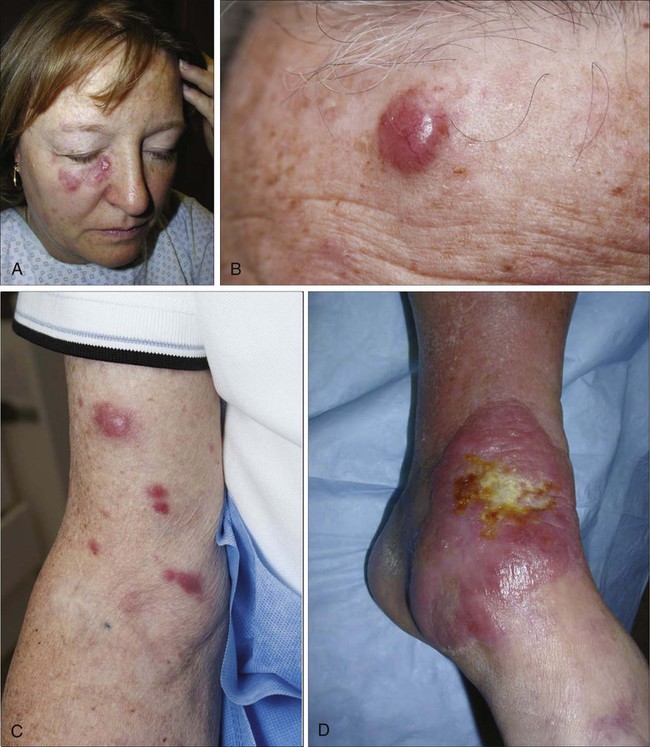
MF is the prototype of CTCL and the best-studied entity. MF classically presents as an indolent course, with slow progression over years or sometimes decades. The disease may evolve from erythematous patches to infiltrated plaques and eventually to tumors (Fig. 107-1). However, about 30% of patients have skin tumors (T3) or erythroderma (T4) only at initial presentation.20 Patients usually have a prolonged history of a rash in sun-protected areas, such as the lower abdomen, upper thighs, buttocks, inner upper arms, and breasts in women. Pruritus may or may not have be associated. The majority of patients remain in clinical stages limited to the skin; however, 20% of patients progress into more aggressive and advanced disease with either cutaneous or extracutaneous tumor manifestations, with an estimated 5-year survival rate of 25% to 40%. MF has numerous clinical and histologic variants. Besides the conventional type of MF, three distinct variants have been recognized in the 2005 WHO/EORTC classification, including folliculotropic MF, pagetoid reticulosis, formerly known as Worringer-Kolopp disease, and granulomatous slack skin, characterized by the development of lax skin folds (Fig. 107-2).
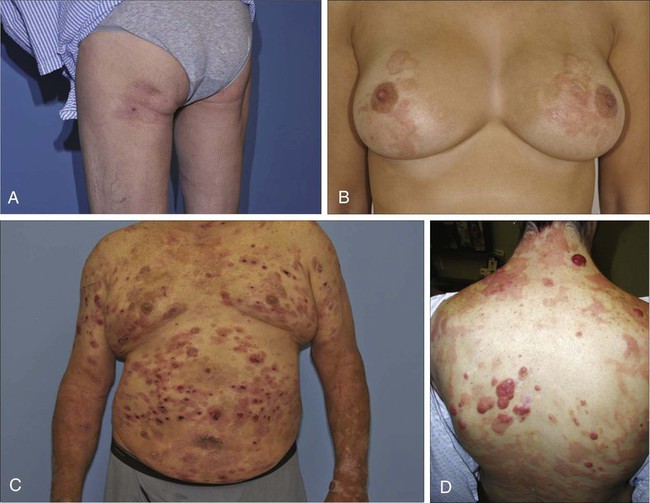
Patients with folliculotropic MF have erythematous papules, plaques, and nodules with follicular involvement with or without alopecia; comedonal, acneiform, and cystic lesions; and associated intractable pruritus.21 Folliculotropic MF is frequently seen on the head and neck area in contrast to conventional MF. The clinical course of folliculotropic MF, particularly in early stages, is more aggressive; these patients respond poorly to topical therapy alone and require systemic therapy to achieve clinically meaningful responses. The 10-year progression-free survival is 45% compared with 91% in patients with conventional MF. Pagetoid reticulosis is a rare variant of MF with an indolent course and is predominantly seen in middle-aged men. Patients have a solitary psoriasiform plaque most often located on the lower extremity or foot. In contrast to classic MF, the epidermotropic atypical lymphocytes show a CD8+ phenotype by immunohistochemistry. Granulomatous slack skin is extremely rare and clinically characterized by indurated plaques resembling lax skin folds often in axillary and/or inguinal areas. On histology, there is a diffuse dermal infiltrate of giant cells and atypical lymphocytes with destruction of elastic fibers.22,23
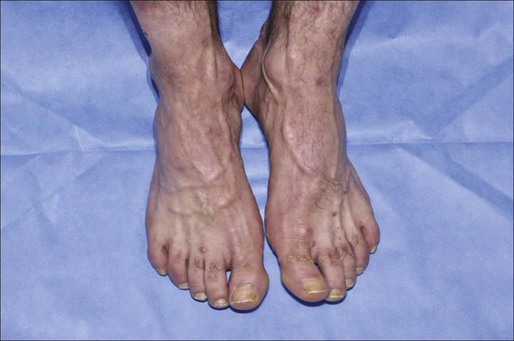
Sézary syndrome (SS) is part of the spectrum of erythrodermic CTCLs as proposed by the International Society for Cutaneous Lymphomas (ISCL) (Table 107-2).24 Erythrodermic CTCLs can either develop de novo (as with SS) or as a progression of preexisting MF (as with erythrodermic MF). Patients who have erythrodermic MF may also have coexisting patches, plaques, or tumors. Three percent to 5 percent of all newly reported cases of CTCL are SS. SS is characterized by circulating, atypical, malignant T lymphocytes with cerebriform nuclei (Sézary cells), by the presence of erythroderma, and often by lymphadenopathy. Severe, disabling pruritus, ectropion, lymphoma-associated alopecia, palmoplantar keratoderma with fissures, and dystrophic nails are other common associated features (Fig. 107-3). Concomitant infections with dermatophytes or bacteria are common. Patients with SS are immunocompromised because of defective T-cell function and therefore predisposed for opportunistic skin infections such as colonization with Staphylococcus aureus.25
Table 107-2
Proposed Classification for Erythrodermic Cutaneous T-Cell Lymphoma (CTCL) and Relative Hematologic Criteria Devised by the International Society for Cutaneous Lymphoma (ISCL)
| Erythrodermic CTCL | Preexisting MF | Blood Findings | TNMB Classification |
| Sézary syndrome | Rarely | Leukemic | T4, N0–3, M0–1, B2* |
| Erythrodermic MF | Always | Absent or minimal | T4, N0–3, M0–1, B0–1* |
| Erythrodermic CTCL, NOS | Absent | Absent or minimal | T4, N0–3, M0–1, B0–1* |

MF, mycosis fungoides; NOS, not otherwise specified; TNMB, tumor, node, metastasis, blood.
*B0, <5% circulating Sézary cells; B1, Sézary cell count of <1000 cells/m3 or <20% atypical T cells on peripheral smear; B2, Sézary cell count of >1000 cells/m3 or >20% atypical T cells on peripheral smear.
From Vonderheid EC, Bernengo MG, Burg G, et al. Update on erythrodermic cutaneous T-cell lymphoma: report of the International Society for Cutaneous Lymphomas. J Am Acad Dermatol 2002;46:95–106.
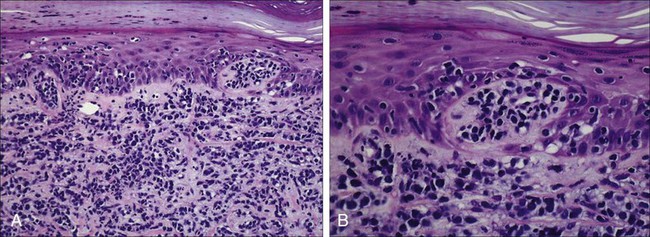
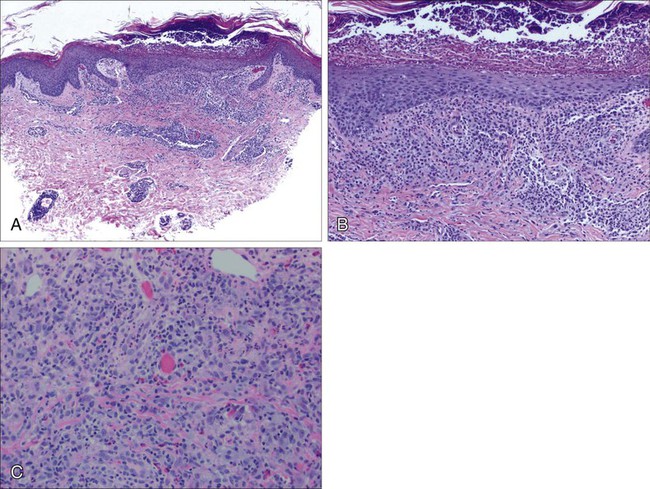
A histopathological diagnosis of MF has proved to be difficult, because in initial and erythrodermic stages of disease inflammatory cells often predominate and may resemble benign dermatoses.26 It is not uncommon that patients may undergo multiple biopsies before a diagnosis can be rendered. A diagnostic algorithm for early MF was proposed by the ISCL and the Cutaneous Lymphoma Task Force of the EORTC that takes the clinical and histopathological findings into account (Table 107-3).27 Characteristic diagnostic features of classic MF/SS are a papillary dermal band–like infiltrate with atypical lymphocytes with hyperchromatic, hyperconvoluted nuclei, variable findings of inflammatory cells, and epidermotropism with infrequently seen Pautrier microabscesses (Fig. 107-4).28 Most MF/SS patients have the phenotype of the CD4+ T-helper (Th)/memory lymphocyte. Only a small number of cases, such as pagetoid reticulosis or hypopigmented variant of MF, are of the cytotoxic/suppressor T-cell subset expressing a CD8+ phenotype. SS patients also show commonly nonspecific histopathological findings, and a definitive diagnosis may be made by flow cytometry.29,30 It is important to differentiate tumor stage MF from non-MF subtypes of CTCL. In conventional MF, tumor lesions usually develop in the setting of patch/plaque disease and not de novo with the latter previously classified as tumor d’emblée type MF. It is now recognized that many of these cases represent non-MF cases and may fit best within the group of primary cutaneous PTCL, not otherwise specified.
Table 107-3
Key Diagnostic Criteria for Mycosis Fungoides/Sézary Syndrome
| Criteria | Major (2 points) | Minor (1 point) |
| CLINICAL | ||
| Persistent and/or progressive patches/plaques plus: | Any 2 | Any 1 |
| Sun-protected areas | ||
| Size/shape variation | ||
| Poikiloderma | ||
| HISTOPATHOLOGICAL | ||
| Superficial lymphoid infiltrate plus: | ||
| Epidermotropism without spongiosis | Both | Either |
| Lymphoid atypia | ||
| Molecular | ||
| Clonal TCR rearrangement | Present | |
| Immunophenotypic | ||
| CD2, CD3, CD5 < 50% of T cells | Any 1 | |
| CD7 < 10% T cells | ||
| Epidermal discordance from CD2, CD3, CD5, or CD7 phenotype of dermal T cells |
From Olsen E, Vonderheid E, Pimpinelli N, et al. Revisions to the staging and classification of mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the Cutaneous Lymphoma Task Force of the European Organization of Research and Treatment of Cancer (EORTC). Blood 2007;110:1713–22.
Immunophenotyping by immunohistochemistry or by flow cytometry and molecular studies have an important role in the evaluation of CTCL and aid into the diagnosis. There is no unique marker for the malignant lymphocyte, but an increased CD4 to CD8 ratio with an aberrant loss of T-cell lineage markers such as CD5, CD7, and/or CD26 is a common finding.29 A clonal gene rearrangement of the T-cell receptor can be detected in many cases and support the diagnosis using standard polymerase chain reaction (PCR) methods, but this has been complicated by the dilemma of clonality detected in benign skin disorders.* Furthermore, data on microsatellite DNA studies suggest that tumor cells may arise from multiple subclones and show a multiple-lineage progression.31
Staging and Prognosis
Accurate staging of patients with MF/SS is essential both for its prognostic value and for treatment decisions. The widely used and recommended staging system for CTCL relies on the TNMB (tumor, node, metastasis, blood) classification adopted by Bunn and Lamberg in 1979, which was revised by the ISCL and EORTC (Table 107-4).27,32 It considers the extent of skin involvement (T), presence of lymph node (N) and visceral disease (M), and detection of Sézary cells in the peripheral blood (B). Patients with patch/plaque disease are classified as either stage IA (T1, N0, M0, B0) with less than 10% of body surface area (BSA) involved or stage IB (T2, N0, M0, B0) with more than 10% BSA involved. By rule-of-thumb, the palm and digit of one hand represent 1% of BSA. Stage IIA disease (T1–2, N1, M0, B0) includes patients with skin findings of stage IA/IB disease with architectural preservation of any clinically abnormal lymph nodes. Currently, two histopathological grading systems, the Dutch and the National Cancer Institute/Veterans Administration (NCI-VA) classification system for lymph node evaluation, are used. Whereas the Dutch system is based on the presence of large cerebriform nuclei (>7.5 µm) and the degree of architectural effacement, the NCI-VA system uses the relative numbers of such cells in the paracortex of lymph nodes and nodal architecture to determine the extent of disease. Nodal involvement is characterized by partial (N2) or complete (N3) architectural effacement. Stage IIB (T3, N0–2, M0, B0) is associated with the development of skin tumors with or without associated lymph node involvement. The ISCL/EORTC has recommended definitions based on the degree of blood involvement for subsets of erythrodermic CTCL to address the differences among erythrodermic CTCL. A B1 rating is defined as a Sézary cell count of less than 1000 cells/m3 or less than 20% Sézary cells on peripheral smear. A B2 rating would be more than 1000 cells/m3 or greater than 20% Sézary cells on peripheral smear. Patients with erythroderma without significant lymph node involvement are stratified into stage IIIA (T4, N0–2, M0, B0) and stage IIIB (T4, N0–2, M0, B1) based on the presence of low blood tumor burden (B1) that allows for tracking of its prognostic significance in erythrodermic CTCL. High blood tumor burden (B2) is an independent prognostic variable as shown by others, and the B2 rating is now considered to be comparable to nodal involvement (N3).32a Stage IVA is therefore any skin stage with either blood (B2) or nodal disease (N3). Stage IVB is defined by any stage with visceral involvement (M1). The ISCL/EORTC considers splenomegaly even without biopsy confirmation as visceral disease. Suspected liver or bone marrow disease should be confirmed by biopsy.
Table 107-4
TNMB Classification and Staging for Patients with Mycosis Fungoides/Sézary Syndrome
| T (SKIN) | |
| T1 | Limited patch/plaque (<10% of BSA) |
| T2 | Generalized patch/plaque (>10% of BSA) T2a = patch only |
| T3 | One or more tumors (>1 cm) |
| T4 | Generalized erythroderma (>80% of BSA) |
| N (NODES) | |
| N0 | No clinically abnormal peripheral lymph nodes |
| N1 | Clinically abnormal peripheral lymph nodes Dutch grade 1, NCI grade LN0–2 |
| N1a | Clone negative |
| N1b | Clone positive |
| N2 | Clinically abnormal peripheral lymph nodes Dutch grade 2, NCI grade LN3 |
| N2a | Clone negative |
| N2b | Clone positive |
| N3 | Clinically abnormal peripheral lymph nodes Dutch grade 3, NCI grade LN4, clone positive or negative |
| Nx | Clinically abnormal peripheral lymph nodes No histologic confirmation |
| M (VISCERA) | |
| M0 | No visceral organ involvement |
| M1 | Visceral organ involvement (pathology confirmation required) |
| B (BLOOD) | |
| B0 | Atypical circulating cells not present (<5%) |
| B0a | Clone negative |
| B0b | Clone positive |
| B1 | Atypical circulating cells present (>5%) |
| B1a | Clone negative |
| B1b | Clone positive |
| B2 | ≥1000/µL Sézary cells, clone positive |
| Stage | T | N | M | B |
| IA | 1 | 0 | 0 | 0,1 |
| IB | 2 | 0 | 0 | 0,1 |
| IIA | 1,2 | 1,2 | 0 | 0,1 |
| IIB | 3 | 0-2 | 0 | 0,1 |
| IIIA | 4 | 0-2 | 0 | 0 |
| IIIB | 4 | 0-2 | 0 | 1 |
| IVA1 | 1-4 | 0-2 | 0 | 2 |
| IVA2 | 1-4 | 3 | 0 | 0-2 |
| IVB | 1-4 | 0-3 | 1 | 0-2 |

BSA, body surface area; TNMB, tumor, node, metastasis, blood.
Histologic classification of lymph node (LN) involvement is also used for prognostic value and for treatment decisions in patients diagnosed with MF/SS.33,34 It was found that the LN rating correlates with disease progression and survival.33,34 Moreover, detection of a monoclonal T-cell population within the lymph nodes is associated with an inferior survival and outcome regardless of the LN class.35 LN1 rating defines reactive changes, LN2 and LN3 nodes describe small or large clusters of atypical cells in paracortical T-cell regions, whereas LN4 nodes define frank effacement.
The most important prognostic factors for survival remain the T stage (skin tumor burden), extracutaneous manifestation, and patient age.36 The Stanford experience with MF/SS patients demonstrates the impact of stage on overall survival (OS) rate (see Fig. 107-4).36 Although the OS rate of all patients with MF is 68% at 5 years and 17% at 30 years, the specific survival of patients ranges widely depending on T classification and stage at initial presentation. Patients with SS have an estimated 5-year survival rate of 24%. The largest study, consisting of 525 patients, showed an OS of 97% in patients with T1 (less the 10% body surface involvement) at 5 years, compared with 40% and 41% in T3 and T4 disease, respectively. In addition, several independent adverse prognostic factors have been identified, including large cell transformation, follicular mucinosis, thickness of tumor infiltrate, and increased LDH and β2-microglobulin levels.39–39 Patients with large circulating Sézary cells were also found to have a worse prognosis.40 A high Sézary cell count, loss of T-cell subset markers such as CD5 and CD7, and chromosomal abnormalities in T cells are also independently associated with a poor outcome.41 The existence of a blood clonal T-cell population, detected by PCR, was of poor predictive survival value, independent of the T stage and lymph node involvement.42 Recent multivariant analyses in survival outcomes and prognostic factors of MF and SS patients using the ISCL/EORTC revised staging proposal confirmed that the presence of a T-cell clone in blood (identical to the cutaneous T-cell clone) in the absence of morphologic evidence of blood involvement (B0b) was associated with a significantly worse OS rate and disease-specific survival (DSS) rate compared with those patients with no peripheral blood T-cell clone (B0a).43 In contrast, the presence of a high number of cytotoxic CD8+ T lymphocytes in the cutaneous infiltrate, as well as the density of epidermal Langerhans cells greater than 90 cells/mm2, is associated with a better prognosis.44,45
Transformed Mycosis Fungoides/Sézary Syndrome
Large cell transformation is generally associated with a poor outcome. A recent study on prognostic factors in 100 patients with large cell transformed MF showed a median survival rate of 24 months (range, 1 to 235 months) with a 5-year DSS and OS rate of 38% and 33%, respectively.39 These results are similar to those from previous studies with median survival rates ranging from 12 to 36 months and 5-year OS rate from 11% to 32%.38,46–48 The most important prognostic factors included advanced stage at transformation, CD30 negativity, folliculotropic MF, and increased extent of skin lesions. There is no difference in outcome in patients with clinical stage IIB with or without large cell transformation. In contrast, another recent study of 1502 patients with MF/SS, including 70 patients with transformed MF at the time of first diagnosis, suggested a better prognosis of patients with LCT compared with all previous studies; the median survival was 8.3 years, and the 5-year OS and DSS rates were 63% and 65%, respectively.43 Current guidelines recommend a more aggressive approach to patients with large cell transformation49; however, the recent studies highlight that not all patients with large cell transformation may do worse.
Biological Properties
Immunopathogenesis
Most malignant T cells in CTCL are clonally derived from CD4+ CD45RO+ Th cells.50 In early stages of MF, the T-cell infiltrate consists of both malignant CD4+ and reactive CD8+ T cells with a dominance of Th1 cytokines such as interferon-γ (IFN-γ), interleukin (IL)-12, and IL-2.51 In later stages, there is a gradual increase of malignant CD4+ T cells, decrease in nonmalignant CD8+ T cells, and a shift to Th2 cytokine dominance (IL-4, IL-5, IL-10, and IL-13).52 These changes correlate with disease progression, host immunosuppression, and susceptibility to infection.53 Biological immune modifiers such as IFN-α, IFN-γ, and IL-12 are therapeutically effective in CTCL by stimulating Th1 cytokines and boosting host immune responses. An IL-17–producing T-cell (Th17) population in cutaneous lesions of MF/SS patients was also identified. Interestingly, the in vitro stimulation of MF/SS cells with IL-17 did not enhance their cell proliferation.54 In another study, IL-17 protein was found to be mediated by IL-2/IL-15 through the Janus-activated kinase (JAK3)/signal transducer and activator of transcription (STAT3) pathway.55 Whether distinct CTCL subtypes derive from Th1, Th2, or Th17 cells remains to be shown.
Controversial results were found for programmed cell death (PD)-1 expression. Increased PD-1 expression has been shown on circulating CD4+ cells in SS patients when compared with MF patients, which could imply a role for an aberrant increase in PD-1 expression in the progression of tumors.56 Follicular Th cells display a distinct gene expression profile positive for PD-1, CXCL13, and BCL-6. Recent data showed increased PD-1 expression in pseudolymphoma and cutaneous CD4+ small/medium-sized pleomorphic T-cell lymphoma, suggesting a follicular Th cell phenotype.57,58 Further research is needed in this area to determine whether the increase in PD-1 expression protects the tumor cells from elimination or if the increased PD-1 expression is a response of immunocompetent cells that are simply chronically stimulated by tumor antigens.
The malignant T cells in MF express skin-homing markers of a skin-resident effector memory T cell, whereas circulating T cells in SS/erythrodermic CTCL express markers of a central memory T cell.59 These differences may explain their different clinical behavior and response to treatment. The malignant skin-resident T cells express the chemokine receptors (CCR)4 and CCR10 among others that are required for migration and homing into the skin. Malignant circulating T cells express CCR7 and L-selectin required for lymph node and peripheral blood. The chemokine ligand (CCL)17, a CCR4 ligand expressed on epidermal keratinocytes, endothelial cells, and dendritic cells, facilitates extravasation and migration of CTCL cells into the skin and epidermis. CCL27, a CCR10 ligand expressed on keratinocytes, has been implicated in both skin and nodal homing of CTCL cells.60 Anti-CCR4 is being evaluated in clinical trials including CTCL patients.
Persistent activation of the neoplastic T cells is demonstrated by the constitutive phosphorylation of intracellular signaling protein STAT3.61 These cells may express the activation markers CD45RO and the IL-2α receptor (CD25) that has provided a target for biological therapy with denileukin diftitox.62 Naturally occurring regulatory T cells (Tregs; CD4+, CD25+, FOXP3+) also express the CD25 molecule. These cells suppress the activity of other immune cells, thus maintaining immunologic tolerance. Tregs appear to be dysregulated in CTCL.63 Early cutaneous MF lesions contain numerous FOXP3+ infiltrating Tregs that decrease in number in advanced lesions. The high frequency of FOXP3+ infiltrating Tregs may suppress tumor proliferation and has been correlated with improved survival.63 SS patients have very low levels of Tregs but high levels of malignant T cells expressing a Treg-phenotype (FOXP3+ CD25−). These malignant FOXP3+ Tregs express cytotoxic T-lymphocyte antigen (CTLA-4), IL-10, and transforming growth factor–β (TGF-β), which suppress immunity and diminish the antitumor response.53
CTLA-4 is a co-inhibitory molecule expressed on T cells that inhibits T-cell activation and proliferation and confers resistance against activation-induced cell death.64,65 Tregs also constitutively express CTLA-4, which is necessary for their functioning to maintain peripheral tolerance and to prevent autoimmunity.66,67 High CTLA-4 expression was found in peripheral blood mononuclear cells from patients with MF, and higher expression levels correlate with increased tumor burden. Th1-derived cytokines such as IL-2 and IFN-γ upregulate expression of CTLA-4.68 Whether increased CTLA-4 expression relates to Treg-like properties that CTCL cells acquire during disease progression is not clear.
Malignant CD4+ T cells from cutaneous lesions and peripheral blood samples in MF and SS have decreased and/or defective Fas expression and decreased Fas expression has been correlated with more aggressive disease as well as resistance to Fas-mediated apoptosis.69–72 Downregulation of Fas in CTCL occurs through multiple mechanisms: mutations in the Fas gene, production of nonfunctioning splice variants, and promoter hypermethylation.70,73 In this context, malignant T cells in CTCL may acquire resistance to FasL signaling through the increased expression of cFLIP, an intracellular apoptosis inhibitor.69 T cells generally express BCL-2, which inhibits apoptosis and is widely and stably expressed in all stages of MF.74 STAT3 directly regulates BCL-2/BAX genes involved in apoptosis.75 Data suggested that inhibition of STAT3 signaling in CTCL cells through the JAK inhibitor Ag490 induced apoptosis through decreased expression of antiapoptotic BCL-2 and increased expression of pro-apoptotic BAX protein.76
Molecular Pathogenesis
Improvements in microarray technology and computational analysis of genomic data have led to discoveries of underlying chromosomal mutations in tumor suppressor and oncogenes involved in CTCL.79–79 Chromosomal regions with significant gains include 8q (including the MYC oncogene), 17q, and 10p13 (including GATA3, a transcription factor that promotes Th2 cytokine production).79 Additionally, a recent study suggested that amplifications on 4q12 (including KIT), 7p11.2 (including EGFR), and 17q25.1 may be highly associated with patients refractory to treatment.79 Patients with large cell transformation show chromosomal clonal evolution with changes in ploidy levels and structural aberrations.80 Specific oncogenes have been examined for defining new prognostic factors in CTCL. Deletions have been found on chromosomes 17p (including TP53), 10p and 10q (including PTEN and FAS), 13q including RB1, and 9p21.3 (including CDKN2A).79 Expression profiles showed upregulation of genes involved in the TNF signaling pathway and decreased expression of tumor suppressor genes such as TGF-β receptor II whereas EPHA4 and TWIST were overexpressed.81,82 Aberrant expression of STAT4, GATA3, T-plastin, CD1D, TRAIL, CDO1, and DNM3 was also found and may serve as potential targets for novel treatment strategies.83,84 Evidence of epigenetic silencing of hypermethylation of individual genes or gene-specific promoters associated with tumor progression have been observed in more advanced stages of CTCL.31,85 In CTCL, promoter hypermethylation leads to dysregulation of cell cycle (CDKN2B, CDKN2A, TP73), apoptosis (TMS1, TP73), DNA repair (MGMT), chromosomal instability (CHFR), and microsatellite instability (MLH1) genes and proteins.85–89 Promoter hypermethylation of CDKN2B, CDKN2A, and MLH1 were found in both early and advanced stages of MF and SS, suggesting that early epigenetic alterations were responsible for the inactivation of these genes.89–89 The multilineage progression of CTCL is also supported by microsatellite instability, as determined by microdissected analyses of tumor infiltrates.31 Several studies have found microRNA (miRNA) signatures in CTCL, but data were inconsistent and distinct genetic markers for diagnosis and prognostication were not identified.90,91 More recently, a study has shown that miRNA classifier can distinguish CTCL from benign skin disorders.92 A microarray screen found that five miRNAs (miR-203, miR-205, miR-326, miR-663b, and miR-711) distinguish CTCL from benign skin diseases with accuracy greater than 90%.93 Interestingly, these five miRNA classifiers were not affected by drug treatment. In tumor stage MF, miR-93, miR-92A, and miR-155 were upregulated in comparison with benign inflammatory skin diseases.94 In SS, most miRNAs were downregulated but miR-21, miR-486, and miR-214 are upregulated and involved in apoptotic resistance.95
CD30+ Lymphoproliferative Disorders
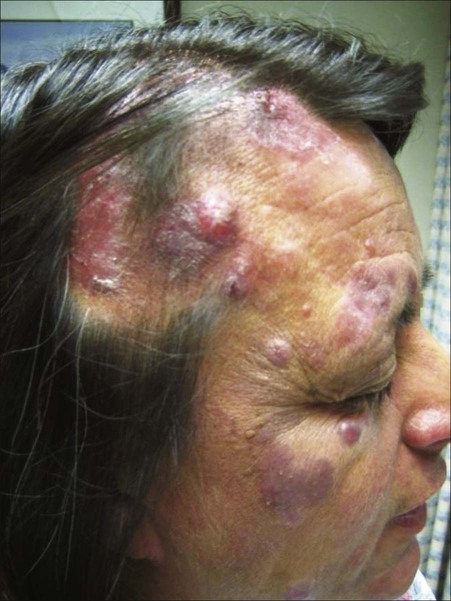
Lymphomatoid papulosis (LyP) and primary cutaneous anaplastic large cell lymphoma (ALCL) represent the spectrum of CD30+ lymphoproliferative disorders. The use of the term CD30+ lymphoproliferative disorders in the initial clinical and pathological assessment is preferable over LyP or ALCL because of overlapping clinical, histologic, and immunophenotypic characteristics. LyP is a chronic self-healing and recurrent condition composed of erythematous papules and nodules on the trunk and extremities that often occur in clusters or as many disseminated lesions (Fig. 107-5). The number of lesions may vary from patient to patient, and they tend to become hemorrhagic and necrotic with eventual spontaneous regression. The association with a second malignant lymphoma varies depending on the series but is probably between 10% and 20% of cases. The most common associated lymphomas are MF, ALCL, and Hodgkin disease and, rarely, other systemic lymphomas.96 The prognosis for patients with LyP is otherwise excellent, with a 100% 5-year survival rate.97 Three histologic types have been identified, characterized as type A, B, and C. Type A and type C consist of large lymphocytes resembling Reed-Sternberg cells. Type A cells are embedded in a dense inflammatory background, whereas type C cells form large sheets imitating CD30+ large T-cell lymphoma. Type B simulates classic MF features with epidermotropism and a dermal band–like infiltrate composed of small to medium-sized cells.98
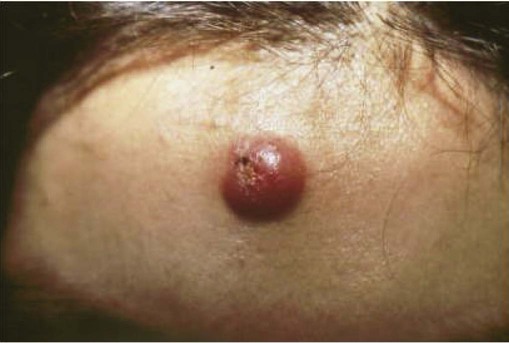
Primary cutaneous ALCL typically arises in solitary or localized nodules (Fig. 107-6), and typically affects older patients.101–101 Regional lymph node involvement is seen in 25% of patients at presentation. This tumor has an excellent prognosis as confirmed in several studies, in contrast to the transformation of MF to a CD30+ large cell variant. It shows histologic and immunophenotypic overlap with LyP. Therefore, clinical features are important in distinguishing ALCL from LyP. Cytomorphology shows a diffuse, nonepidermotropic infiltrate with cohesive sheets of large CD30+ lymphocytes (Fig. 107-7). In most cases tumor cells show anaplastic features; less commonly there is a pleomorphic or immunoblastic appearance. However, there is no difference in the prognosis and survival rate. Primary cutaneous ALCL rarely carries the t(2;5) translocation and is usually negative for anaplastic large cell lymphoma kinase (ALK) protein. These lesions may undergo spontaneous regression, as do the lesions of LyP. The mechanism of tumor regression remains unknown. CD30 ligand–mediated cytotoxicity may participate in the pathophysiology of clinical regression.102 A few cases have been reported showing that CD30/CD56 co-expression is associated with disease progression, as well as increased fascin levels.103 In cases with progression, point mutations and deletions on TGF-β receptor genes I and II have been found, leading to the loss of its tumor-suppressive properties.104 MUM1 expression is upregulated in CD30+ lymphoproliferative disorders, but its use for distinction for LyP from ALCL is controversial.105,106 The diagnosis of CD30+ lymphoproliferative disorder requires the exclusion of other cutaneous infiltrates characterized by CD30 expression.98 These conditions include reactive and neoplastic diseases such as arthropod bites, scabies, pityriasis lichenoides, Langerhans cell histiocytosis, CBCLs with immunoblastic or large cell features, CD30+ large cell transformation of mycosis fungoides, CD30+ cutaneous NK/T-cell lymphoma, and secondary cutaneous involvement of systemic ALCL. Therefore, clinical exclusion of these conditions is essential to establish a diagnosis of CD30+ lymphoproliferative disorder and to avoid unnecessary aggressive therapy.
Treatment of LyP is not known to alter the natural history of the disease or the risk for developing associated malignancies.97 There is no role for multiagent systemic chemotherapies. Many patients do not require treatment if their disease is asymptomatic, especially given the possibility of spontaneous resolution of lesions. Observation in patients with few lesions is recommended, whereas in patients with more disseminated disease, low-dose methotrexate or ultraviolet light therapy is effective in clearing disease. In contrast, localized radiation therapy for solitary or localized lesions is the preferred treatment for cutaneous ALCL, with methotrexate or systemic chemotherapy reserved for cases with high tumor burden and/or extracutaneous involvement. CD30 is an attractive molecule for targeted therapy; the antibody conjugate brentuximab vedotin (SGN-35) is currently emerging for primary cutaneous ALCL and CD30+ transformed MF.
Therapy for Cutaneous T-Cell Lymphoma
The development of treatment strategies for patients with CTCL relies on the extent of disease (TNMB stage) and its impact on a patient’s quality of life. No treatment is considered to be curative, and survival has not been shown to benefit from early intervention even with multiagent chemotherapies combined with electron-beam radiation but was associated with significant toxicities.107 Early-stage (IA to IIA) MF has a favorable prognosis. The disease is limited to the upper dermis and epidermis of the skin, which is ideal to apply skin-directed therapies. Current topical therapies include corticosteroids, nitrogen mustard (mechlorethamine), retinoids/rexinoids (e.g., bexarotene), carmustine (BCNU), phototherapy with psoralens and ultraviolet light A (PUVA), UVB or narrowband (NB)-UVB, and total-skin electron-beam therapy (TSEBT).108
Topical Therapy
Of the available UV light therapies, PUVA therapy has been widely used with established benefit in early-stage MF for more than 25 years. Treatment is given three times per week. A dose of 0.6 mg/kg of 8-methoxypsoralen is taken 1 to 2 hours before UV light exposure. Patients are required to use protective eye glasses for 12 to 24 hours after a PUVA treatment to prevent the formation of cataracts. Success rates with PUVA are 90% for stage IA, 76% for stage IB, 78% for stage IIA, 59% for stage IIB%, and 61% for stage III CTCL.111–111 The most common reported acute side effects were erythema, pruritus, and nausea. Long-term PUVA exposure was associated with an increased risk for chronic photodamage and nonmelanoma skin cancer.111 Recent consensus from the EORTC Cutaneous Lymphoma Task Force indicates that patients with patches and thin plaques should be given NB-UVB treatment, whereas PUVA should be reserved for patients with folliculotropic MF, failure of NB-UVB treatment, or dark complexion, because of the treatment’s carcinogenic effects and a shortage of available treatment centers.112 Patients with refractory disease in early stages benefit from combination therapies such as PUVA or NB-UVB with low-dose systemic bexarotene or INF-α.113
Topical corticosteroids are frequently prescribed to induce clearing of skin lesions in patients with limited patches (stage IA) and commonly used in combination with UV light therapy. High-potency topical corticosteroids have also been shown to have an overall response rate greater than 90% in patch-stage MF with some side effects of irritant dermatitis and cutaneous atrophy with no signs of adrenal insufficiency detected.114,115 Nitrogen mustard (mechlorethamine) belongs to a class of agents known as alkylators.116 The topical formulation has been widely used as a first-line treatment of early stage MF since 1959. It may be applied locally or to the entire skin once daily. Nitrogen mustard–induced DNA damage has been the primary mechanism responsible for its systemic anticancer effects, but the mechanism of action of the topical formulation remains elusive. Topical nitrogen mustard has a complete response rate of 76% to 80% for stage IA and 35% to 68% complete response rate for stage IB disease.116 Common side effects are contact hypersensitivity reactions.
Bexarotene gel is a retinoid X receptor agonist approved for treatment of early-stage MF.117,118 In clinical trials, the complete response rate was 21%, with a 63% overall response rate and a 75% response rate in treatment-naive stage IA/IIA MF patients. The median duration of remission was 24 months.119,120 A dose-response effect with greater efficacy at higher concentrations and frequencies of application was observed. Patients without prior therapies achieved a higher response rate of 75%. The majority of the patients tolerated the topical 1.0% formulation twice daily. A total of 35 patients (52%) applied the gel for more than 1 year. Bexarotene gel commonly causes skin irritation. Tazarotene gel, a retinoic acid receptor agonist, is another topical retinoid approved for use in psoriasis and acne and has been shown to improve skin lesions in refractory MF and may be useful as an adjuvant topical treatment.121
Total skin electron-beam therapy (TSEBT) is a procedure that involves administering ionizing radiation to the entire skin surface.110 TSEBT has been shown to be an effective therapy for palliation of the cutaneous symptoms of MF and SS.110 Because the electron-beam radiation in TSEBT has greater energy and depth of penetration than other skin-directed therapies, it may be an option for treatment of stage T2 or T3 MF.110 Options may be limited for white patients, and TSEBT toxicity can be cosmetically disfiguring and is associated with an increased risk for developing nonmelanoma skin cancer. A recent study from Stanford University followed 180 patients with T2 or T3 stage MF with TSEBT treatment and found that all patients had over 50% improvement in skin involvement, with 63% achieving complete clinical response; the rates were 75% for patients with stage T2 MF and 47% for patients with stage T3 MF, with a median duration of response of 29 months in patients with stage T2 and 9 months in patients with stage T3.110 These results confirm previous studies that show conventional-dose TSEBT is significantly more efficacious in stage T2 than T3 MF. Despite the standard dose of 30 to 36 Gy, relapses are common and repeat treatment courses are limited owing to cumulative toxicities. A clinical phase II trial with low-dose TSEBT (10 Gy) showed an overall response rate of 90% with a complete response or very good partial response (<1% skin affected with patches/plaques) of 70%.122 The median response duration was 5 months, ranging from 83 to 469 days for complete response and very good partial response, respectively.
Systemic Therapy
Biological Therapies
Extracorporeal photopheresis, approved in 1988 by the U.S. Food and Drug Administration (FDA) for the palliative treatment of patients with CTCL, is best suited for patients with SS who are within 2 years of disease onset and have near-normal counts of CD8+ T cells and NK cells and modest tumor burden.123 Overall response rates have ranged from 31% to 73% when CTCL patients have been treated with extracorporeal photopheresis as monotherapy, but efficacy has been shown to be greater when extracorporeal photopheresis is used in various combinations with IFN-α, IFN-γ, granulocyte-macrophage colony-stimulating factor, and bexarotene, owing to enhancement of antitumor immunity.124,125 The novel continuous-flow separation system (THERAKOS, CELLEX) has been developed based on the current UVAR XTS photopheresis device and is designed to reduce treatment times and extracorporeal volumes.
IFN-α is one of the most widely used first-line treatments and probably the most effective single agent in the treatment of CTCL. It has shown a wide range of antiviral, antiproliferative, and immunomodulatory effects. The exact mechanism with which interferons exert their antitumor effects remains unknown. Th1 cytokines support cytotoxic T-cell–mediated immunity, and it has been speculated that IFN-α maintains or enhances a Th1-cell population balance for an effective cell-mediated response to malignant T lymphocytes. A response rate of 73% in stage IA/IIA and a 60% response rate in stage IIB/IVA disease has been seen with IFN-α monotherapy.126 When IFN-α is used in combination with PUVA, both overall response rates and response duration show improvement, with studies demonstrating overall response rates and complete response rates of 98% and 84%, respectively.127,128 Although the optimal dose and duration has not been established yet in CTCL, current experience suggests therapy should be given at a starting dose of 1 to 3 million units five times weekly with gradual escalation to 6 to 9 million units daily or as tolerated.
Denileukin Diftitox.
Denileukin diftitox (Ontak) is an IL-2–diphtheria toxin fusion protein targeted against malignant T cells expressing CD25, the high-affinity IL-2 receptor. Denileukin diftitox was approved by the FDA in 1999 for the treatment of patients with CTCL refractory to standard treatment options. In general, response rates in patients with relapsed and refractory MF/SS range from 30% to 37%.129 A recent randomized phase III trial in CD25+ CTCL demonstrated an overall response rate of 44% for patients treated with denileukin diftitox versus 15.9% for patients on placebo. CTCL patients were randomly assigned to receive denileukin diftitox 9 µg/kg/d (n = 45), denileukin diftitox 18 µg/kg/d (n = 55), or placebo infusions (n = 44). In addition, patients treated with both doses of denileukin diftitox had a significantly higher progression-free survival rate than patients on placebo.130 The incidence of grade 3 and 4 capillary leak syndrome was seen in two to three patients (3.6%) at doses of 18 µg/kg/d. A key remaining question of whether response to denileukin diftitox depends on expression of CD25 was explored in a retrospective study of complete responders in previous phase II and phase III trials. This study found no difference in response between patients with CD25+ and CD25− disease.131
Histone Deacetylase Inhibitors.
HDACIs were initially developed to modulate chromatin condensation by acetylation of histones affecting gene expression. More recently, their effects on posttranslational modification of many intracellular proteins have been recognized.132 Vorinostat (suberoylanilide hydroxamic acid; Zolinza), an orally administered HDACI, was FDA approved in 2006 for treatment of relapsed/refractory CTCL. A phase IIB trial was conducted in 74 patients with stage IB/IVA CTCL, including 82% with stage IIB or higher disease. The overall response rate was 29.7%. Oral vorinostat was administered at 400 mg daily. The median time to response was 2 months, and median duration of response was not reached but was estimated to be greater than 6.1 months. In addition, 43.4% of patients with severe pruritus experienced relief. The most common adverse effects included diarrhea, fatigue, and nausea, but most were of grade 2 or lower. Other significant grade 3 side effects included pulmonary embolism (5%) and thrombocytopenia (5%). In this study, QTc interval prolongation was observed in 3 patients with no reported clinical sequelae, none of which were grade 3. There were no cases of infection.133 Another phase II trial of vorinostat conducted in 33 patients with advanced or refractory CTCL at multiple doses demonstrated an overall response rate of 24% with a median time to response of 3 months and a median duration of response of 3.7 months. Forty-five percent of patients experienced relief from pruritus.134 In addition, analysis of lesion biopsy specimens in responding patients demonstrated a shift in localization of phosphorylated STAT3 from nuclear to cytoplasmic, suggesting that vorinostat may inhibit proliferation of CTCL cells by inactivating STAT3.134 Further in vitro work has shown that CTCL patients with high nuclear levels of STAT1 and pSTAT3 are resistant to vorinostat.135 Combination therapy has also been investigated, and phosphatidylinositol-3-kinase (PI3K) inhibitors have been found to synergize with vorinostat in reducing cell viability.136
Romidepsin (depsipeptide, Istodax) is another HDACI approved by the FDA for patients with relapsed/refractory CTCL. Two phase II trials were conducted in a total of 167 patients suffering from relapsed, refractory, or advanced CTCL.137,138 Romidepsin was administered intravenously at 14 mg/m2 on days 1, 8, and 15 of a 28-day cycle. In both trials, the overall response rate was 34% and the complete response rate was 6%. In both trials, the median time to response was 2 months and the median duration of response was longer than 12 months. Most common adverse effects were fatigue, nausea, vomiting, and anorexia. Severe adverse effects included leukopenia, lymphopenia, granulocytopenia, thrombocytopenia, and anemia. Earlier studies have shown that histone deacetylase inhibitors may be associated with electrocardiographic abnormalities such as QTc interval prolongation.139 In one trial, 9% of patients had QTc prolongation and 80% had asymptomatic T-wave flattening or ST-segment depression.137 The other trial had no patients with QTcF values greater than 480 msec or an increase of more than 60 msec over baseline. It was also found that antiemetics might contribute to QTcF prolongation.138
Panobinostat (LBH589) is another HDACI that was shown in a phase I trial to induce clinical responses in CTCL patients.140 Preliminary results of a phase II trial of oral panobinostat have been reported. Panobinostat was administered at a dose of 20 mg on days 1, 3, and 5 weekly in bexarotene-treated patients and bexarotene-naïve patients. In 62 bexarotene-treated patients, 17.7% achieved a response. In 35 bexarotene-naïve patients, 12.1% achieved a response. Thrombocytopenia, neutropenia, pruritus, diarrhea, and hypophosphatemia were the most common grade 3 or 4 toxicities. Two patients had a QTcF greater than 480 ms and 4 had an increase in QTcF greater than 60 msec from baseline.141
Monoclonal Antibodies.
Monoclonal antibodies (mAb) target tumor cells via cell surface markers upregulated on malignant T cells such as CD4, CD52, and CCR4. Zanolimumab (Hu-Max CD4) is a humanized anti-CD4 mAb that has been shown in vitro to mediate antibody-dependent cellular cytotoxicity (ADCC) primarily in CD4+CD45RO+ T cells.142 Zanolimumab also blocks T-cell activation by macrophages in Pautrier microabscesses via induction of inhibitory signaling pathways involving SHIP-1 and DOK-1.143 In two phase II studies done in 47 patients with refractory early- and advanced-stage CTCL, zanolimumab was given intravenously at a weekly dose of 280 mg and 560 mg for patients with early-stage disease and at 280 mg and 980 mg for those with late-stage disease. The overall response rate was 56% in patients with MF treated with a high dose and 15% at a lower dose. In patients with SS, the overall response rate was 20% in patients treated with a high dose and 25% at a lower dose. Adverse events included skin inflammation (24%) and infections of the skin and upper respiratory tract (49%).144
Alemtuzumab (Campath), a humanized mAb against CD52 surface antigen expressed on most malignant T cells, has been shown to mediate ADCC,145,146 complement-mediated cell lysis,147 and apoptosis.148 A phase II study was conducted in 22 patients with advanced CTCL at a dose of 30 mg intravenously three times a week. The overall response rate was 55% (32% complete response; 23% partial response). A greater effect was observed in patients with erythrodermic CTCL (overall response rate, 69%) than on plaque or tumor CTCL (40%).149 To investigate this preferential effect on erythrodermic CTCL, another phase II study conducted in 19 patients with advanced refractory erythrodermic CTCL found an overall response rate of 84%.150 Serious adverse events in these studies included infections and hematologic toxicity. Infections, occurring primarily in patients who had received three or more treatments, included cytomegalovirus reactivation, fever of unknown origin, herpes simplex virus reactivation, pulmonary aspergillosis, and Mycobacterium pneumonia. Hematologic toxicities included anemia, neutropenia, and thrombocytopenia. One study found adverse effects of congestive heart failure and arrhythmias after alemtuzumab treatment.151 However, several studies since have found no correlation with cardiac toxicity.150,152
Chemotherapy.
Conventional systemic treatments include chemotherapeutic agents and biological immunomodulatory therapies. Gemcitabine (Gemzar) and pegylated doxorubicin (Doxil) are being used as newer initial single-agent chemotherapeutic choices.153,154 A phase II trial of gemcitabine reported a 68% overall response rate in 25 patients with refractory advanced CTCL.155 In advanced, untreated CTCL, gemcitabine was shown to result in a 75% response rate in 32 patients.153 Another study showed a response rate of 88% for pegylated liposomal doxorubicin.154
Targeted Antifolate Therapy.
Methotrexate is the traditional antifolate used in therapy for lymphomas. It inhibits dihydrofolate reductase that converts dihydrofolate to tetrahydrofolate, which is required for synthesis of thymidylate and purine nucleotides involved in DNA and RNA synthesis. Pralatrexate (Folotyn) belongs to a class of novel folate analogs, the 10-deazaaminopterins, designed with greater affinity than methotrexate for receptor-reduced folate carrier (RFC), leading to improved drug internalization through membrane transport. It is also a better substrate for polyglutamylation than methotrexate, leading to greater intracellular retention158–158 and tenfold greater cytotoxicity than methotrexate in lymphoma cell lines.159 Pralatrexate has been FDA-approved for relapsed or refractory PTCL.
Preliminary results of a multicenter dose-escalation phase II study in 54 patients with relapsed or refractory CTCL have been reported. The starting dose and schedule was 30 mg/m2 intravenously once per week for 3 of 4 weeks. An optimal dose of 15 mg/m2 for 3 of 4 weeks was defined, at which the overall response rate was 43%. At doses greater than 15 mg/m2, the rate was 50%. Most common grade 1 to grade 2 adverse effects included fatigue, mucositis, nausea, edema, epistaxis, pyrexia, constipation, and vomiting. Grade 3 adverse effects included thrombocytopenia, neutropenia, leukopenia, and anemia.160 In another report of 12 patients with MF with large cell transformation, who were part of the multicenter PROPEL trial for patients with PTCL, the overall response rate was 58% via investigator assessment and 25% via independent central review.161,162 Combination therapy with bortezomib is under exploration.163
Hematopoietic Stem Cell Transplantation.
The concept of high-dose combined chemotherapy followed by autologous bone marrow transplant or peripheral blood stem cell support has curative potential in various non-Hodgkin lymphomas, but experience in CTCL is limited. Autologous stem cell transplants have yielded disappointing results. Despite reported effective responses with complete response in most patients treated, relapses are frequent and may occur rapidly.164,165 Allogeneic hematopoietic stem cell transplantation (HSCT) with myeloablative conditioning regimens provides a graft-versus-tumor effect and avoids reinfusion of tumor cells—both features lacking in autologous HSCT. However, myeloablation places the patient at high risk for infections and graft-versus-host disease (GVHD), rendering it difficult to use in the elderly or those with multiple co-morbidities. With the broadened use of nonmyeloablative reduced-intensity conditioning regimens, allogeneic HSCT may now be better suited for patients with CTCL.166 A retrospective of 60 patients with advanced CTCL who received allogeneic HSCT and either reduced-intensity conditioning or myeloablative conditioning regimens had a complete response rate of 60.5% and an overall survival of 54% at 3 years. Overall survival rate at 3 years in patients who received reduced-intensity conditioning regimens was 63% compared with 29% in patients who received myeloablative conditioning regimens. The median age of patients was 46.5 years, indicating that allogeneic HSCT may be an effective treatment in younger as well as older patients.167 In another study of allogeneic HSCT with reduced-intensity conditioning regimens and pretreatment TSEBT for tumor debulking, 58% had a complete response and overall survival at 2 years was 79%. Causes of mortality included sepsis, metastatic non–small cell lung cancer, and disease progression.168 Cord blood transplantation has also been attempted with some success in Japan in cases of failure or inability to attempt allogeneic HSCT.169,170
Investigational Therapies
Lenalidomide.
Lenalidomide (Revlimid), an analog of thalidomide, is currently FDA approved for treatment of myelodysplastic syndrome171 and refractory/relapsed multiple myeloma.172 The immunomodulatory properties such as NK- and T-cell activation with induction of Th1 cytokine production and cytotoxic activity along with alteration of the tumor cell microenvironment through antiangiogenic, antiproliferative, and proapoptotic properties provided the rationale to use this agent in patients with CTCL.173 A phase II trial in 35 patients with advanced/refractory CTCL showed an overall response rate of 32%, a mean time to response of 3 months, and mean duration of response of 4 months.174 The most common side effects were fatigue, lower leg edema, gastrointestinal symptoms, leukopenia, and neutropenia. Temporary tumor flares, characterized by increase in size and number of skin lesions, tender swelling of lymph nodes, or increase in Sézary cell count were noted in 25% of patients after initial treatment. Data from this study also suggest that immunomodulatory effects of lenalidomide might be associated with decreased Treg and CD4+ T-cell numbers.
Oligonucleotides (Nuclear Acid Therapeutics).
Toll-like receptor (TLR) agonists represent a novel approach to stimulate an effective antitumor immune response in patients with CTCL through augmentation of either dendritic cells or T-cell effects. PF-3512676 (CPG-7909, ProMune) is a TLR-9–activating oligodeoxynucleotide and potent plasmacytoid dendritic cell stimulator175 that was shown in a dose-escalating phase I trial to induce an overall response rate in 32% of patients (3 complete responses; 6 partial responses) with treatment-refractory stage IB to IVA CTCL.176 Twenty-eight patients received subcutaneous doses (0.08, 0.16, 0.24, 0.28, 0.32, or 0.36 mg/kg) once weekly for 24 weeks.
Proteasome Inhibitors.
NF-κB is an oncogenic transcription factor normally sequestered in an inactive state by the inhibitory I-κB molecule. Various oncogenic signals activate NF-κB via phosphorylation of I-κB, leading to its degradation via the 26S proteasome. Downstream targets of NF-κB include BIRC2, BIRC3, and BCL-2.177 Bortezomib inhibits the 26S proteasome and therefore prevents degradation of I-κB and the activation of NF-κB.178 It has been shown to induce apoptosis in CTCL through the suppression of NF-κB–dependent antiapoptotic genes Birc2 (cIAP1) and Birc3 (cIAP2), but not BCL-2.177,179 A phase II trial was conducted in 15 patients with relapsed/refractory CTCL with a dose of 1.3 mg/m2 on days 1, 4, 8, and 11, every 21 days for a total of six cycles. The overall response rate was 67% (17% complete response, 50% partial response). Common adverse effects were neutropenia, thrombocytopenia, and sensory neuropathy.180 Currently, a phase I trial is being conducted in patients with refractory T-cell lymphoma using a combination of bortezomib and 5-azacytidine, a DNA methyltransferase inhibitor.
CCR4 Antibody.
KW-0761, a novel defucosylated humanized mAb against CCR4, enhances ADCC against malignant CTCL cells.181 In vitro studies of KW-0761 using MF/SS cell lines, primary MF/SS cells, and MF/SS mouse models showed not only significant ADCC-mediated antitumor activity, but also synergistic effect with IL-12, IFN-α2b, and IFN-γ. Phase I studies in ATLL and PTCL have demonstrated an overall response rate of 31% with minimal adverse effects consisting mainly of hematologic toxicities. 182 Phase II studies are currently ongoing in patients with PTCL, CTCL, and ATLL.
Cutaneous B-Cell Lymphomas
Primary CBCLs are B-cell lymphomas that present in the skin at the time of diagnosis without evidence of extracutaneous disease.1 They account for 20% to 29% of all primary cutaneous lymphomas with variable incidence in Europe and the United States.1,6 Primary CBCLs are clinically but not histologically distinct from their nodal counterparts.2 However, it is critical to distinguish primary CBCLs from systemic B-cell lymphomas with secondary skin involvement because their clinical behavior, prognosis, and management may differ considerably. The revised WHO/EORTC classification and the EORTC/ISCL consensus recommendations on CBCLs recognize three major subtypes (see Table 107-1).1,183 Primary cutaneous follicle center lymphoma and cutaneous marginal zone lymphoma are characterized by an indolent clinical behavior, whereas primary cutaneous large B-cell lymphoma of the leg presents as a more aggressive clinical behavior. Rare variants are intravascular large B-cell lymphoma and plasmacytoma. Because of the small number of these cases reported, conclusions on prognosis and outcome are not possible.
An accurate classification for CBCLs still remains unsatisfying, particularly for cases presenting as large cell morphology that were classified into primary cutaneous follicle center lymphoma. Recent published studies on gene expression profiles of CBCL have shown that gene microarray technology can be used to characterize the nature of distinct subgroups of CBCLs and thus be a helpful tool in lymphoma classification.184,185 Further molecular support was provided by a study that compared gene expression profiles of primary cutaneous follicle center lymphoma with large cell morphology with primary cutaneous large B-cell lymphoma, leg type.185 The expression profile of cutaneous large B-cell lymphoma, leg type, is similar to that of nodal DLBCL arising from germinal center or post germinal center–activated B cell with constitutive NF-κB pathway activation and strong expression of the IRF4/MUM1 transcription factor that carries a worse prognosis, whereas primary cutaneous follicle center lymphoma including cases with large cell morphology belongs to the germinal center–like phenotype with a favorable prognosis. Different micro-RNA profiles were found in indolent primary cutaneous marginal zone lymphoma and cutaneous follicle center lymphoma with significantly higher expression of miR-150 and miR-155 in cutaneous marginal zone lymphoma.186 When compared with outcome, low expression of miR-150 and miR-155 was associated with increased risk for progression in patients with cutaneous marginal zone lymphoma.
CBCLs are characterized by proliferation of B lymphocytes within the skin. To date, there are scant data available regarding the process of homing and expansion of clonal B cells. CBCLs have been proposed to be skin-associated lymphoid tissue (SALT)–related B-cell lymphomas induced by possible chronic antigen stimulation.187 Analysis of B-cell clonality in skin biopsy specimens showed molecular evidence for a trafficking B-cell component of SALT with a reliable detection for clonality in CBCLs using the standardized BIOMED-2 method that distinguished cutaneous malignant from benign B-cell–rich infiltrates.188,189 An etiologic role of HTLV-I, herpes simplex virus, Epstein-Barr virus, and hepatitis C virus was suggested in rare cases192–192 but remains largely unknown. An association between CBCLs and Borrelia burgdorferi infection was shown in Europe but not in the United States.195–195 Recent findings indicate that B-cell attractant chemokine 1 (BCA-1 [CXCL13]) and its BCA-1 receptor (CXCR5), which function as homing chemokines in normal lymphoid tissue, are constantly expressed in cutaneous lymphoproliferative B-cell disorders but not in normal skin, suggesting a possible role in lymphomagenesis.196
Clinically, CBCLs present as solitary or infrequently multiple and/or clustered, indolent, erythematous to violaceous papules, plaques, or nodules (Fig. 107-8). Microscopically, in contrast to CTCLs, the infiltrate typically involves the dermis with an uninvolved subepidermal zone, referred to as the grenz zone. Epidermotropism is reported but extremely rare. The B cells express CD19, CD20, CD22, and CD79a by immunohistochemistry. The distinction between CBCLs and B-cell pseudolymphoma is often difficult; additional information can be provided by immunophenotyping for light-chain restriction or by monoclonal rearrangement of immunoglobulin light-chain genes. Staging procedures include positron emission tomography/computed tomography (PET/CT), laboratory investigations such as complete blood cell count, complete metabolic panel, LDH, flow cytometry panel, and/or bone marrow biopsy. A recent consensus proposal of a TNM classification system applicable for all primary cutaneous lymphomas other than MF/SS has been introduced in an attempt to improve patient care, develop guidelines, and predict prognosis and outcome,197 but it currently relies only on anatomic information without available prognostic information.
Primary cutaneous follicle center lymphoma is the most common subtype, comprising 40% of CBCLs. Primary cutaneous follicle center lymphoma shows predilection for head, neck, and trunk in elderly patients with a median age of 60 and male predominance of approximately 1.5 : 1. The clinical course is usually indolent, with an excellent overall survival up to 97%.1 However, relapses occur frequently. The large round cell morphology might be associated with higher progression and poorer prognosis. Primary cutaneous follicle center lymphoma is defined as a proliferation of centrocytes (small to large cleaved cells) and centroblasts (large round cells with prominent nuclei) showing a nodular or diffuse infiltrate in the majority of cases and presenting only rarely a true follicular pattern (Fig. 107-9).198 In contrast to their nodal counterpart, BCL-2 is usually not expressed by neoplastic cells, but low expression levels have been reported, and the t(14 : 18) translocation is rarely detected.199 In addition to CD10+ and BCL-6+ expression, primary cutaneous follicle center lymphoma has also an aberrant expression of CD45RA and CD43, and thus provides a helpful clue to distinguish it from pseudolymphomas.200,201
Primary cutaneous marginal zone lymphoma represents the second most common subtype of CBCLs. It predominantly occurs on the upper and lower extremities. The median age at onset is 55 years, and more females are affected than males. The reported survival rates are 97% to 100%, although relapses occur commonly. Histologically, primary cutaneous marginal zone lymphoma has features of mucosa-associated lymphoid tissue lymphomas and show a nodular or diffuse dermal infiltrate with a heterogeneous cellular infiltrate of small lymphocytes, plasmacytoid cells, plasma cells, and reactive germinal centers that may be infiltrated by neoplastic cells.1 Eosinophilic intranuclear (Dutcher body) or intracytoplasmic (Russell body) inclusions of immunoglobulin are common. Diagnosis can be difficult, because of the variable composition of the infiltrate that may be interpreted as a reactive process or as primary cutaneous follicle center lymphoma. In contrast to primary cutaneous follicle center lymphoma, marginal zone lymphoma is negative for BCL-6 and CD10, but positive for BCL-2. In 50% of the cases, CD43 is markedly expressed. Large cell transformation and head and neck presentation may be associated with a worse prognosis.
Primary CBCL, leg type, is a more aggressive type confined to elderly patients with a median age of 76 at diagnosis and a female predominance.183 Most cases have a germinal center–activated B-cell phenotype. Histopathology shows a diffuse dermal infiltrate with predominance of large B cells with multilobulated nuclei, composed of centroblasts and immunoblasts, with the presence of small cleaved cells and a minor admixed infiltrate component. Malignant cells are positive for BCL-2, MUM1, and FoxP1 with variable expression of BCL-6. CD10 is negative. The prognosis is less favorable than for other CBCLs. The 5-year overall survival rate for patients with a solitary tumor is 70%, compared with 27% and 0% for patients presenting with localized or multilocalized disease, respectively.
Therapy
CBCLs are mostly characterized by a favorable prognosis with a low risk for systemic spread. Therapeutic approaches range from simple observation to local radiotherapy and/or surgical excision, with multiagent chemotherapy and immune-based modalities as additional options, especially for more aggressive CBCLs. Given its possible association with B. burgdorferi, there is also the use of antibiotics for the treatment of primary cutaneous marginal zone lymphoma. Although most of the data on treatment approaches is limited to small retrospective studies, the EORTC/ISCL provided consensus recommendations for the treatment of the three main types of CBCLs (Table 107-5).183 In addition, the National Comprehensive Cancer Network continues to provide updated treatment guidelines on CBCLs. Radiation is often the preferred therapy for solitary or localized grouped lesions. Reported complete remission rates range from 92% to 100%, with 5-year survival rates ranging from 73% to 89%.183,202,203 However, cutaneous recurrences are fairly common and were observed in up to 46% of patients. Surgical excision can be considered for small lesions. These options may not be best in young patients with facial lesions at risk for disfigurement or in patients who have multifocal skin disease or sites involved that are ill suited for radiotherapy (i.e., scalp) in addition to the high recurrence rates. Alternative therapies currently pursued, and with promising outcomes, are immune-based modalities, such as rituximab, IFN-α, and imiquimod. Intralesional IFN can be employed as an alternative when lesions are few.204,205 Topical imiquimod therapy in one study seemed to be less potent for CBCL than for CTCL with two of three patients with solitary or localized CBCL achieving a partial response.206
Table 107-5
Treatment Recommendations for Primary Cutaneous B-Cell Lymphomas
| EORTC/ISCL Consensus Recommendations183 | NCCN Guidelines | |||
| Type and Extent | No. Patients | First-Line Therapy | Alternative Therapy | First Line |
| Marginal zone lymphoma | 288 | |||
| Solitary/localized | ||||
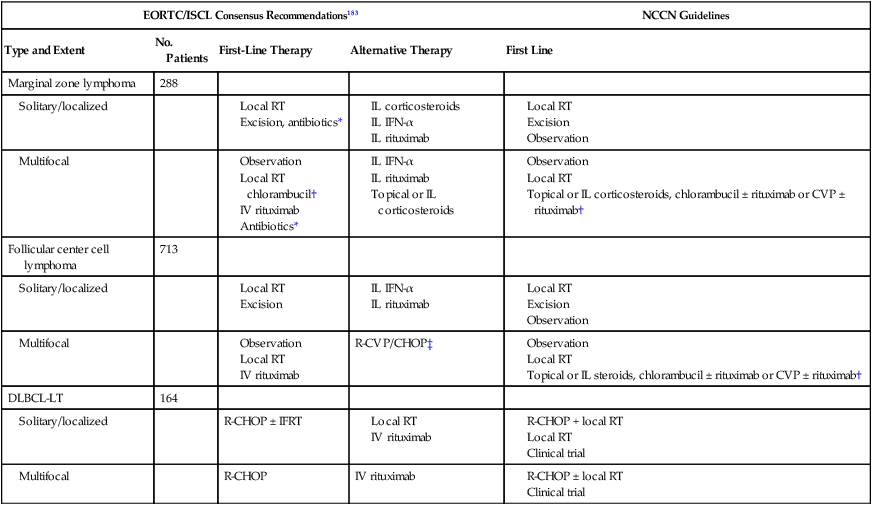
*In cases of evidence for Borrelia burgdorferi infection.
†Or other single or combination regimens appropriate for low-grade B-cell lymphomas.
‡In exceptional cases or for patients developing extracutaneous disease.
Adapted from EORTC/ISCL Consensus Recommendations183 and National Comprehensive Cancer Network Guidelines.
Treatment with rituximab, a chimeric antibody against CD20 surface marker, has been reported to produce overall response rates of 48% to 85% depending on the lymphoma type.183 The treatment is usually well tolerated without adverse reactions, except for mild transient cytopenias or mild infusion reactions. More recently, complete remissions have been reported in cases treated with low-dose intralesional rituximab.207,208 In patients with distinct subtypes such as DLBCL or disseminated cutaneous or extracutaneous spread, the cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) regimen or CHOP-like chemotherapy seems to be the treatment of choice.209 However, successful outcomes with rituximab as monotherapy have also been reported.210 Moreover, the addition of rituximab to the CHOP regimen (R-CHOP) increased the complete response rate and prolonged overall survival rate in elderly patients with DLBCL, without a clinically significant increase in toxicity.211 R-CHOP is the standard therapy for systemic DLBCL and, when feasible, is the preferred first-line treatment for primary cutaneous large B-cell lymphoma, leg type.212 Grange and colleagues compared different treatment cycles in 70 patients with primary cutaneous large B-cell lymphoma and demonstrated a higher benefit when patients were treated with rituximab and anthracycline-based multiagent chemotherapy compared with other therapies, including radiation therapy, single-agent chemotherapy, or multiagent chemotherapy with rituximab.213 In this study, however, there was no statistical difference in overall 2-year survival rate. Rituximab alone induced an objective response rate of 75% in eight cases of primary cutaneous large B-cell lymphoma, leg type, with a 5.25-month median disease-free survival rate.214 Because this disease often occurs in the elderly, radiation therapy alone or with rituximab is often chosen for those with localized disease in whom the toxicities of combination chemotherapy are best avoided.
General Health Care
Many patients are disabled by their pruritus and skin appearance. Emollients should be recommended in patients with dryness and scaling. In addition, topical treatment with mid-potent corticosteroids such as triamcinolone applied once or twice daily can minimize skin irritation and pruritus, particularly in patients with SS. A short-term course with systemic corticosteroids often gives immediate relief of more diffuse symptoms. Oral antihistamines, gabapentin, aprepitant, and/or mirtazapine may be also of benefit for pruritus. Patients with more widespread cutaneous disease or generalized erythroderma should be screened for secondary infections such as staphylococcal or streptococcal species and/or dermatophytes and treated with systemic antibiotics and antifungal agents. Bleach baths, adopted from treatment strategies for children with severe atopic dermatitis, can minimize colonization of S. aureus.215 Patients with advanced stages are at risk for infections and sepsis due to their immunosuppressive stage. Treatment-associated toxicities can be pronounced, particularly in elderly patients with comorbid medical conditions, poor performance status, and/or severe skin disease or in patients whose life expectancy is short. Dose adjustments in those patients are often required to preserve quality of life because treatment is always regarded to be palliative and greater disease elimination does not outweigh the increased risk for toxicities.