Control of the Cell Cycle
• Most cells in postnatal tissues are quiescent. Exceptions include abundant cells of the hematopoietic system, skin, and gastrointestinal mucosa, as well as other minor progenitor populations in other tissues.
• Many quiescent cells can reenter into the cell cycle with the appropriate stimuli, and the control of this process is essential for tissue homeostasis.
• The key challenges for proliferating cells are to make an accurate copy of the 3 billion bases of DNA (S phase) and to segregate the duplicated chromosomes equally into daughter cells (mitosis).
• Progression through the cell cycle is dependent on both intrinsic and extrinsic factors, such as growth factor or cytokine exposure, cell-to-cell contact, and basement membrane attachments.
• The internal cell cycle machinery is controlled largely by oscillating levels of cyclin proteins and by modulation of cyclin-dependent kinase (Cdk) activity. One way in which growth factors regulate cell cycle progression is by affecting the levels of the D-type cyclins, Cdk activity, and the function of the retinoblastoma protein.
• Cell cycle checkpoints are surveillance mechanisms that link the rate of cell cycle transitions to the timely and accurate completion of prior dependent events. p53 is a checkpoint protein that induces cell cycle arrest, senescence, or death in response to cellular stress.
• Checkpoints minimize replication and segregation of damaged DNA or the abnormal segregation of chromosomes to daughter cells, thus protecting cells against genome instability.
• Disruption of cell cycle controls is a hallmark of all malignant cells. Frequent tumor-associated alterations include aberrations in growth factor signaling pathways, dysregulation of the core cell cycle machinery, and/or disruption of cell cycle checkpoint controls.
• Because cell cycle control is disrupted in virtually all tumor types, the cell cycle machinery provides multiple therapeutic opportunities.
Introduction
Most cells in the adult body are quiescent—that is, they are biochemically and functionally active but do not divide to generate daughter cells. However, specific populations retain the ability to proliferate throughout the adult life span, which is essential for proper tissue homeostasis. For example, cells of the hematopoietic compartment and the gut have a high rate of turnover, and active proliferation is therefore essential for the maintenance of these tissues. On average, about 2 trillion cell divisions occur in an adult human every 24 hours (about 25 million per second). The decision about whether to proliferate is tightly regulated.1,2 It is influenced by a variety of exogenous signals, including nutrients and growth factors, as well inhibitory factors and the interaction of the cell with its neighbors and with the underlying extracellular matrix. Each of these factors stimulates intracellular signaling pathways that can either promote or suppress proliferation. The cell integrates all of these signals, and if the balance is favorable, the cell will initiate the proliferation process.
During the past three decades, extensive effort has been placed on unraveling the basic molecular events that control this process. Studies in a variety of organisms have identified evolutionarily conserved machinery that controls eukaryotic cell cycle transitions through the action of key enzymes, including cyclin-dependent kinases (Cdks) and other kinases.3 It is essential that proliferating cells copy their genomes and segregate them to the daughter cells with high fidelity. Eukaryotic cells therefore have evolved a series of surveillance pathways, termed “cell cycle checkpoints,” that monitor for potential problems during the cell cycle process.4 Human cells are continuously exposed to external agents (e.g., reactive chemicals and ultraviolet light) and to internal agents (e.g., byproducts of normal intracellular metabolism, such as reactive oxygen intermediates) that can induce DNA damage. The cell cycle checkpoints detect DNA damage and activate cell cycle arrest and DNA repair mechanisms, thereby maintaining genomic integrity.
Anything that disrupts proper cell cycle progression can lead to either the reduction or expansion of a particular cell population. It is now clear that such changes are a hallmark of tumor cells, which carry mutations that impair signaling pathways; that is, they suppress proliferation and/or activate pathways that promote proliferation. In addition, most (if not all) human tumor cells have mutations within key components of both the cell cycle machinery and checkpoint pathways.1,5,6 This characteristic has important clinical implications, because the presence of these defects can modulate cellular sensitivity to chemotherapeutic regimens that induce DNA damage or mitotic catastrophe. This chapter focuses on the mechanics of the cell cycle and checkpoint-signaling pathways and discusses how this knowledge can lead to the efficient use of current anticancer therapies and to the development of novel agents.
The Cell Division Cycle
Overview of the Cell Cycle Machinery
Cell division proceeds through a well-defined series of stages (Fig. 4-1). First, the cell moves from the quiescent (also known as G0) state into the first gap phase, or G1, in which the cell essentially is readying itself for the cell division process. This process involves a dramatic upregulation of both transcriptional and translational programs not only to yield the proteins required to regulate cell division but also to essentially double the complement of macromolecules so that one cell can give rise to two cells without a loss of cell size. Not surprisingly, this process takes a significant amount of time (from 8 to 30 hours) and energy. Studies with cultured cells show that mitogenic growth factors are essential for continued passage through the G1 phase. Specifically, if growth factors are withdrawn at any point during this phase, the cell will not divide. However, as it nears the end of the G1 phase, the cell passes through a key transition point, called the restriction point, whereupon it becomes growth factor independent and is fully committed to undergoing cell division.1 Within an hour or two, the cell enters the synthesis phase, or S phase, in which each of the chromosomes is replicated once and only once. The cell then enters a second gap phase, called G2, which lasts 3 to 5 hours, and then initiates mitosis, or the M phase, a rapid phase (lasting about 1 hour) in which the chromosomes are segregated. Upon completion of mitosis, the daughter cells can enter quiescence or initiate a second round of cell division, depending on the milieu.
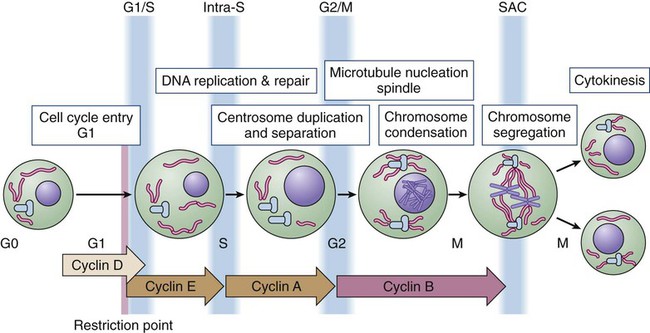
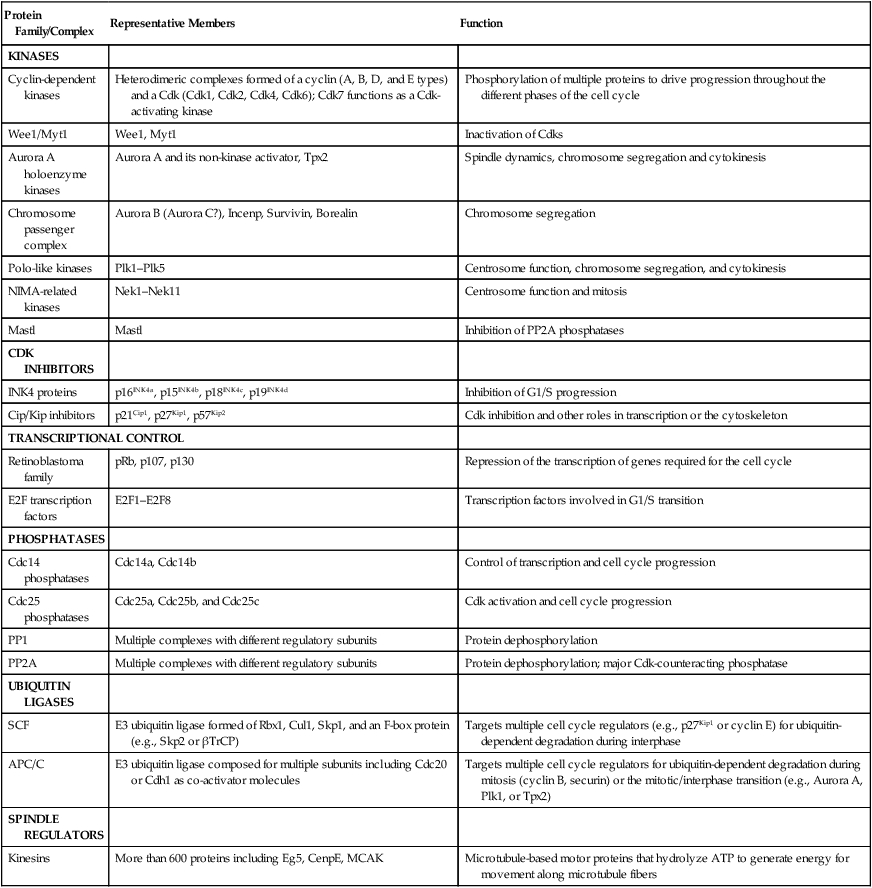
Progression throughout the different phases of the cell cycle depends on the activity of key molecules that drive transcription, translation, or the structural changes required for cell division (Table 4-1). A large number of these changes are modulated by protein phosphorylation and dephosphorylation, but other molecular processes such as SUMOylation, acetylation, or ubiquitin-dependent protein degradation are crucial for ordered cell cycle progression. Many of these cell cycle regulators have been involved in tumor development or may be attractive targets for cancer therapy and will be introduced in the following sections.
Table 4-1
Molecules Involved in Cell Cycle Regulation
| Protein Family/Complex | Representative Members | Function |
| KINASES | ||
| Cyclin-dependent kinases | Heterodimeric complexes formed of a cyclin (A, B, D, and E types) and a Cdk (Cdk1, Cdk2, Cdk4, Cdk6); Cdk7 functions as a Cdk-activating kinase | Phosphorylation of multiple proteins to drive progression throughout the different phases of the cell cycle |
| Wee1/Myt1 | Wee1, Myt1 | Inactivation of Cdks |
| Aurora A holoenzyme kinases | Aurora A and its non-kinase activator, Tpx2 | Spindle dynamics, chromosome segregation and cytokinesis |
| Chromosome passenger complex | Aurora B (Aurora C?), Incenp, Survivin, Borealin | Chromosome segregation |
| Polo-like kinases | Plk1–Plk5 | Centrosome function, chromosome segregation, and cytokinesis |
| NIMA-related kinases | Nek1–Nek11 | Centrosome function and mitosis |
| Mastl | Mastl | Inhibition of PP2A phosphatases |
| CDK INHIBITORS | ||
| INK4 proteins | p16INK4a, p15INK4b, p18INK4c, p19INK4d | Inhibition of G1/S progression |
| Cip/Kip inhibitors | p21Cip1, p27Kip1, p57Kip2 | Cdk inhibition and other roles in transcription or the cytoskeleton |
| TRANSCRIPTIONAL CONTROL | ||
| Retinoblastoma family | pRb, p107, p130 | Repression of the transcription of genes required for the cell cycle |
| E2F transcription factors | E2F1–E2F8 | Transcription factors involved in G1/S transition |
| PHOSPHATASES | ||
| Cdc14 phosphatases | Cdc14a, Cdc14b | Control of transcription and cell cycle progression |
| Cdc25 phosphatases | Cdc25a, Cdc25b, and Cdc25c | Cdk activation and cell cycle progression |
| PP1 | Multiple complexes with different regulatory subunits | Protein dephosphorylation |
| PP2A | Multiple complexes with different regulatory subunits | Protein dephosphorylation; major Cdk-counteracting phosphatase |
| UBIQUITIN LIGASES | ||
| SCF | E3 ubiquitin ligase formed of Rbx1, Cul1, Skp1, and an F-box protein (e.g., Skp2 or βTrCP) | Targets multiple cell cycle regulators (e.g., p27Kip1 or cyclin E) for ubiquitin-dependent degradation during interphase |
| APC/C | E3 ubiquitin ligase composed for multiple subunits including Cdc20 or Cdh1 as co-activator molecules | Targets multiple cell cycle regulators for ubiquitin-dependent degradation during mitosis (cyclin B, securin) or the mitotic/interphase transition (e.g., Aurora A, Plk1, or Tpx2) |
| SPINDLE REGULATORS | ||
| Kinesins | More than 600 proteins including Eg5, CenpE, MCAK | Microtubule-based motor proteins that hydrolyze ATP to generate energy for movement along microtubule fibers |
Cyclin-Dependent Kinases and Their Regulators
The Cdks constitute a large subfamily of highly conserved Ser/Thr kinases that are defined by their dependence on a regulatory subunit, called a cyclin.9–9 The first identified human Cdk, called Cdk1 (originally cdc2), was cloned by virtue of its ability to complement a mutant cdc2 yeast strain.10 Subsequent studies identified additional human Cdks and determined that they regulate distinct cell cycle stages; for example, Cdk4 and Cdk6 regulate cell cycle entry, whereas Cdk2 may have specific roles during the G1-to-S transition and S phase. Cdk1 is essential in the control of G2 and mitosis and also may play additional roles in earlier stages. The human genome encodes about 15 additional Cdks, although the functional relevance of many of them is still unknown.3,8,9
The activity of these kinases is controlled by multiple regulatory mechanisms. Cdks act in association with a cyclin subunit that binds to the conserved PSTAIRE helix within the kinase.11 Cyclin binding causes a reorientation of residues within the active sites that is essential for kinase activity.7,11 The associated cyclin also determines the substrate specificity of the resulting cyclin-Cdk complex. The cyclins are quite divergent, especially in their N-terminal sequences, but they all share a highly conserved 100-amino acid sequence, called the cyclin box, that mediates Cdk binding and activation.8 As their name implies, cyclins originally were identified as proteins whose expression was restricted to a particular stage of the cell cycle12 because of cell cycle–dependent regulation of both cyclin gene transcription and protein degradation. The human genome encodes more than 25 cyclin-like proteins, yet only four distinct subclasses—D-, E-, A-, and B-type cyclins—are involved in cell cycle regulation (see Fig. 4-1).8 Each of these classes has a few paralogs (e.g., cyclin D1, D2, and D3; cyclin E1 and E2; cyclin A1 and A2; and cyclin B1, B2, and B3). The relative roles of these paralogs are not completely clear in most cases. Although some functional redundancy may exist, published evidence suggests differences in regulation, expression pattern, and substrate specificity.8,13
The activation of cyclin-Cdk complexes requires considerable posttranslational regulation.8,14 First, kinase activation is dependent on phosphorylation of a threonine residue that is adjacent to the active site (Thr160 in Cdk2). This phosphorylation is catalyzed by a kinase, called Cdk-activating kinase (CAK).15,16 In mammalian cells, phosphorylation occurs after cyclin binding. Although it appears that at least two mammalian CAKs exist, the major CAK is a trimolecular complex composed of Cdk7, cyclin H, and Mat1. The Cdk7-cyclinH-Mat1 complex also is required for the control of basal transcription via regulation of RNA polymerase II function.16 Second, when it is first formed, the cyclin-Cdk complex frequently is subject to inhibitory phosphorylation of Thr14 and Tyr15 residues within the Cdk’s active site by the Wee1 (Tyr15) and Myt1 (Thr14 and Tyr15) kinases.8,17 Activation of the cyclin/Cdk complex is then dependent on the action of a dual-specificity phosphatase called Cdc25. Mammalian cells have three different Cdc25 proteins, called Cdc25a, Cdc25b, and Cdc25c, which show some specificity for different cyclin-Cdk complexes.18
Cdks are modulated by a series of CdK inhibitors (CKIs) that play a key role in establishing the activity of the cyclin-Cdk complexes in response to either external signals or internal stresses.19 The CKIs can be divided into two distinct families based on their biological properties. The first CKI family is named INK4, based on their roles as INhibitors of CDK4. The INK4 family has four members called p16INK4a, p15INK4b, p18INK4c, and p19INK4d (encoded by the CDKN2A-D genes in humans). These INK4 proteins specifically prevent the binding of cyclins to monomeric Cdk4 and Cdk6 but do not inhibit other Cdks.11,19 The second CKI family is named Cip/Kip and includes three members: p21Cip1 (also called p21Waf1), p27Kip1, and p57Kip2 (encoded by the CDKN1A-C genes in humans).19 These Cip/Kip proteins have two major activities. First, they do not bind to monomeric Cdks but associate with and inhibit the activity of cyclin-Cdk complexes already formed. Second, Cip/Kip proteins may promote the assembly of cyclin D-Cdk4/6 complexes without dramatically perturbing its kinase activity.19,20 This activity is somehow modulated by phosphorylation of Cip/Kip proteins by Src, Jak2, and Akt kinases,21–24 directly linking Cdk regulation with the activity of these upstream mitogenic pathways. In addition to regulating the cell cycle, Cip/Kip proteins play important roles in apoptosis, transcriptional regulation, cell fate determination, cell migration, and cytoskeletal dynamics.25,26
Retinoblastoma Proteins and E2F Transcription Factors
The retinoblastoma protein (pRb) originally was identified by virtue of its association with hereditary retinoblastoma.27 It behaves as a classic tumor suppressor: Affected persons inherit a germline mutation within one RB1 allele, and loss of heterozygosity is seen in all of the tumors. Subsequent studies showed that the transforming ability of small DNA tumor viruses, including human papilloma virus, adenovirus, and simian virus, was dependent on the ability of virally encoded oncoproteins (E7, E1A, and SV40, respectively) to bind and inhibit pRb.28,29 Moreover, the RB1 gene is inactivated in approximately one third of all sporadic human tumors.
pRb and the pRb-related proteins p107 and p130, collectively known as the pocket proteins, are transcriptional repressors whose major function is to inhibit the expression of cell-cycle related proteins (Table 4-1).30 This suppressive activity is largely dependent on the ability to prevent cell cycle entry through inhibition of the E2F transcription factors.30,31 The E2F proteins regulate the cell cycle–dependent transcription of numerous targets, including core components of the cell cycle control (e.g., cyclin E and cyclin A) and DNA replication (e.g., Cdc6, Cdt1, and the Mcm proteins) machineries.31,32 pRb regulates E2F through two distinct mechanisms. First, its association with E2F is sufficient to block the transcriptional activity of E2F.33,34 Second, the pRb-E2F complex can recruit histone deacetylases to the promoters of E2F-responsive genes and thereby actively repress their transcription.35,36 Cell cycle entry requires the phosphorylation of pRb by cyclin-Cdk complexes and the consequent dissociation of pRb from E2F.1
Studies to date have identified eight E2F genes that encode nine different E2F proteins.31,32 Pocket proteins can regulate a subset of these factors: E2F1, E2F2, E2F3a, E2F3b, E2F4, and E2F5. These E2F proteins associate with a dimerization partner, called DP, and the resulting complexes function primarily as either activators (E2F1, E2F2, and E2F3a) or repressors (E2F4 and E2F5) of transcription under the direction of the pocket proteins. Recent observations suggest that several of these factors may act either as positive or negative regulators of transcription depending on the cell type or the differentiation state.31,37–39 Most classic E2F target genes are regulated by the coordinated action of these repressor and activator E2Fs.
Cell Cycle Phosphatases
Cdc14 phosphatases are the major Cdk-counteracting proteins in yeast. Although two family members exist in mammals, their relevance in the cell cycle is not well understood.40,41 In eukaryotes, two major complexes, PP1 and PP2A, account for more than 90% of protein phosphatase activity. In fact, these enzymes correspond to hundreds of phosphatase complexes assembled from a few catalytic subunits (PP1α, PP1β/δ, and PP1γ1/2 for PP1, and the Cα and Cβ isoforms for PP2A) and a diverse array of regulatory subunits.42 Recent evidence suggests that these protein families cooperate in the dephosphorylation of most cell cycle kinase targets, including the retinoblastoma family or mitotic phosphoproteins.40,43,44 PP1 and PP2A are major phosphatases responsible for pRb dephosphorylation during mitotic exit, although the relative roles of these complexes or the particular holoenzymes involved are not clear.43,44 Similarly, both PP1 and PP2A are required for dephosphorylation of hundreds of mitotic proteins that are phosphorylated by Cdk1, as well as the other mitotic kinases. Thus it has been suggested that the cell cycle ultimately is regulated by the dynamic equilibrium between Cdks (and partially by the other mitotic kinases) and PP1/PP2A activity. In the absence of Cdk activity, the balance tilts in favor of the phosphatases. When Cdks are activated, phosphatase activity is overtaken. Cdk1 is able to directly inhibit PP1 by direct phosphorylation of the catalytic subunit. The Cdk-dependent inhibition of PP2A, on the other hand, is not direct; rather, it is mediated by a new kinase known as Greatwall in flies and Xenopus or Mastl in mammals.47–47 Cdk1 phosphorylates and activates Mastl, which in turn phosphorylates Arpp-19 and Ensa, two highly related proteins that function as inhibitors of a particular PP2A holoenzyme encompassing a regulatory subunit of the B55 family.45,46,48,49 Reactivation of PP1 and PP2A phosphatases is a mandatory step for the exit from mitosis and the transition to interphase.52–52
Ubiquitin-Dependent Protein Degradation
The original observation that cyclin levels are tightly regulated during the cell cycle implies that these proteins are not only regulated at the transcriptional level but also at the protein level. It is now evident that ubiquitin-mediated protein degradation is a major regulatory mechanism to ensure ordered transition through the different phases of the cell division cycle. Ubiquitylation depends on an enzymatic cascade, in which ubiquitin ligases recruit specific substrates for modification. About 600 ubiquitin ligases are encoded by the human genome. Among them, the Skp1–Cullin1–F-box (SCF) and the anaphase-promoting complex/cyclosome (APC/C) are known for driving the degradation of cell cycle regulators to accomplish irreversible cell cycle transitions.55–55 SCF has three core components: a RING finger protein, called Rbx1, which recruits the E2-ubiquitin conjugate; a cullin (Cul1); and Skp1 (Fig. 4-2).54 Skp1 acts to recruit a family of proteins, called F-box proteins, that determines the target specificity of the SCF complex. Once SCF binds its substrate, it transfers a ubiquitin molecule to lysine residues within the target protein to create a polyubiquitin chain, which targets the substrate to the proteasome for degradation.56
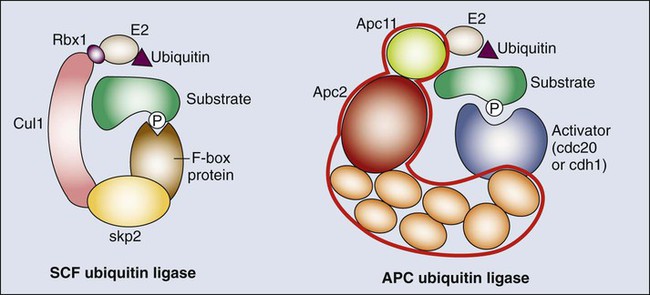
The APC/C is a much larger complex, but it also contains a RING finger protein, called Apc11, to recruit the E2-ubiquitin conjugate, and a core cullin subunit (Apc2). In addition, APC/C is activated by a cofactor that, in a manner comparable with that of the F-box proteins of SCF, establishes substrate specificity (see Fig. 4-2).57 Cdc20 is the mitotic cofactor of APC/C, and it targets several cell cycle regulators (A- and B-type cyclins, Nek2, and securin) during mitotic entry and the metaphase-to-anaphase transition. Cdh1 (also known as FZR1 in mammals) replaces Cdc20 during mitotic exit and is the cofactor responsible for the elimination of many cell cycle regulators in the G0 or G1 phase to prevent unscheduled DNA replication.57,58 Recent studies have provided new insights into the intricate relationship between ubiquitylation and the cell division apparatus, including new roles for atypical ubiquitin chains, new mechanisms of regulation, and extensive cross talk between ubiquitylation enzymes.56
The Mitotic Spindle and Mitotic Kinases and Kinesins
In addition to the central role of Cdks in the cell cycle, many other kinases play critical roles in cell cycle progression.3 Aurora and Polo kinases were first identified in genetic studies in flies as a result of their essential role in mitotic progression.61–61 Each of these families of kinases is represented by a single member in yeast, whereas three Aurora kinases (Aurora-A, Aurora-B, and Aurora-C) and 5 Polo-like kinases (Plk1 through Plk5) exist in humans (Table 4-1).60,62 Aurora A participates in several processes required for building a bipolar spindle, including centrosome separation and microtubule dynamics. Aurora A is activated by Tpx2, and the Aurora A–Tpx2 holoenzyme may have critical implications in tumor development.63 Aurora B, on the other hand, is part of the chromosome passenger complex (CPC) that localizes to the kinetochores from prophase to metaphase and to the central spindle and midbody in cytokinesis. Other components of the CPC include Incenp, survivin, and borealin, and this complex regulates proper microtubule-kinetochore attachment and promotes biorientation during mitosis.61 Aurora C may play similar roles to Aurora B, although it is mostly expressed in germ cells and may play a specific role in meiosis and during the first embryonic cycles.66–66 Plk1 functions as a major regulator of centrosome maturation, mitotic entry, and cytokinesis, whereas Plk4 is a critical regulator of centriole duplication.59,67–69 The other members of the family, Plk2, Plk3, and Plk5, are mostly involved in stress responses during interphase or in neuron biology.3,62
A different group of additional cell cycle kinases with potential interest in tumor biology is the never in mitosis, gene A (NIMA)-related kinase family (Nek1 through Nek11). Four of these proteins, Nek2, Nek6, Nek7, and Nek9, are involved in cell cycle progression, whereas all other family members are likely to play critical roles in cilia and centrioles.3,70,71 Nek2, the closest relative to the Aspergillus NIMA kinase, localizes to the centrosome and plays a role in establishing the bipolar spindle by initiating the separation of centrosomes and contributing to microtubule organization at the G2/M transition by phosphorylating several centrosomal substrates. Nek2 also may play additional roles in chromosome condensation and the mitotic checkpoint. Nek9, Nek6, and Nek7 function in a kinase cascade that participates in centrosome separation and the formation and/or maintenance of the mitotic spindle.70,72 The activity of all these kinases is functionally linked to many other microtubule-associated proteins such as kinesin motor proteins, which determine the dynamic behavior of the mitotic spindle required for chromosome movement during mitosis.73,74
Entry into the Cell Cycle
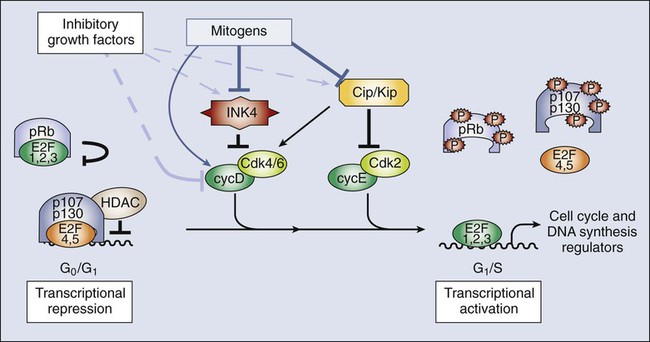
Because most adult cells are quiescent, the mechanisms that determine their quiescent state and their reentry into the cell cycle upon the appropriate stimuli are key determinants of tissue homeostasis. In quiescent cells, several DP-E2F complexes associate with the promoters of E2F-responsive genes and recruit pocket proteins, along with their associated histone deacetylases, to actively repress their transcription.75,76 This repression machinery therefore prevents the expression of proteins required for DNA synthesis and chromosome segregation. In addition, CKIs normally are expressed in quiescent cells, preventing the activation of Cdks.19 D-type cyclins are present at very low levels in most quiescent cells, in large part because they are phosphorylated by an abundant kinase called Gsk3β and then exported to the cytoplasm for degradation.77
In response to mitogenic signaling, the activity of Gsk3β is inhibited, preventing the degradation of D-type cyclins. In addition, mitogens directly induce the transcription of this class of cyclins. Notably, individual mitogenic signaling pathways not only cooperate in inducing a single D-type cyclin but also may induce different D-type cyclins.78,79 Transcriptional induction of D-type cyclins promotes cell cycle entry through two distinct mechanisms. First, D-type cyclins associate with Cdk4 and Cdk6, and the resulting complexes phosphorylate pRb, partially inactivating its transcriptional suppressor function (Fig. 4-3).80–83 Second, the D-type cyclins titrate CKIs away from other Cdks and thereby promote their activation.19,84 Cdk activity increases, and the phosphorylation of the pocket proteins causes them to release their associated DP-E2Fs. Repressive E2Fs such as E2F4 and E2F5 dissociate from the DNA, and the free E2F complexes—DP-E2F1, DP-E2F2, and DP-E2F3—now occupy the promoters and activate their transcription. Cyclin E is itself an E2F-responsive gene, and this regulation creates a strong feed-forward loop. Cyclin E binds to Cdk2, and this complex may promote further pRb inactivation.85,86 Cyclin E–Cdk2 also phosphorylates the Cdk inhibitor p27Kip1 on Thr187.87,88 This action creates a high-affinity binding site for the SCF ubiquitin ligase bound to the F-box protein Skp2, leading to p27Kip1 degradation during the G1/S transition.89–92 Finally, cyclin E-Cdk2 phosphorylates itself on multiple sites, creating a recognition site for SCF-Fbw7/Cdc4 and thereby ensuring its own destruction.93,94 The fact that lack of Cdk2 in the mouse does not result in defective mitotic cycles suggests that the activity of this protein overlaps with other Cdks, with Cdk1 being the best candidate.97–97 Indeed, Cdk1 is able to bind interphase cyclins such as cyclin D and cyclin E, and it is sufficient for G1/S transition, at least in the absence of other interphase Cdks.98,99 On the other hand, cyclin E–Cdk2 may have specific functions in the duplication of centrosomes required for formation of the mitotic spindle,102–102 and it has an essential role in meiosis, although the molecular basis for this requirement is not fully understood.96,103–105
DNA Replication
The DNA replication machinery is optimized to ensure that the genome is copied once—and only once—in each cell cycle.106 This optimization is achieved through a two-step process that first establishes a prereplication complex (pre-RC) at each origin of replication, a process that is frequently referred to as origin licensing, and subsequently transforms pre-RCs into the preinitiation complex (pre-IC) that activates DNA replication (Fig. 4-4). These two steps occur at distinct stages of the cell cycle to ensure that origins are only licensed once per cell cycle and rereplication cannot occur. Pre-RC formation takes place during G1. The first step in this process is the recruitment of the multiprotein complex called the origin recognition complex to the origin DNA.107 The origin recognition complex recruits additional proteins including Cdc6, Cdt1, and finally the mini chromosome maintenance (MCM) complex, a helicase that is required to unwind the DNA strands to form the pre-RC. Once cells enter S phase, the transformation of the pre-RC to the pre-IC requires the activity of two kinases: a Cdk and the Ddf4-dependent kinase, which is composed of the Dbf4 regulatory subunit and the Cdc7 kinase.108,109 The action of these kinases allows numerous additional proteins to associate with the pre-RC and form the pre-IC.110 Assembly of the pre-IC is thought to trigger DNA unwinding by the MCM complex, recruitment of the DNA polymerases, and initiation of the replication process, frequently called “origin firing.”
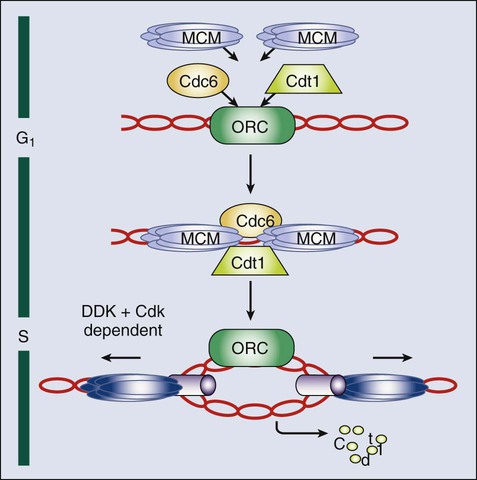
The transformation of the pre-RC to the pre-IC can occur at different time points in S phase, depending on whether the origin fires early or late.106 The system can tolerate this heterogeneity because the pre-RC is disassembled after firing and cannot re-form until the subsequent cell cycle. This process occurs through several mechanisms. The MCM complex travels with the replication fork in its role as the DNA helicase. Some evidence also indicates that phosphorylation of Orc1 reduces its ability to bind to origins. Finally, and most important, Cdt1 is prevented from participating in pre-RC formation outside of the G1 phase in two distinct ways. First, Cdt1 is marked for destruction by ubiquitination.111 This process is mediated by SCF-Skp2 and particularly by an E4 ubiquitin ligase that includes Rbx1 (to recruit the E2-ubiquitin), a cullin (Cul4), Ddb1, and Dtl/Cdt2 (the substrate specificity factor).114–114 Importantly, this Cul4-Ddb1-Dtl/Cdt2 complex functions independently of Cdt1 phosphorylation. Instead, Cdt1 is targeted only when proliferative cell nuclear antigen is present on the DNA, which occurs primarily as a consequence of the initiation of DNA replication.115 Second, cells possess a protein called geminin that sequesters Cdt1 and prevents it from participating in pre-RC formation. Geminin is present specifically in S, G2, and early M phase cells. However, the APC/C ubiquitinates geminin and thereby triggers its destruction. This action creates a window between anaphase of mitosis and the late G1 phase (when APC/C-Cdh1 is inactivated) in which geminin is absent and therefore Cdt1 is free to participate in pre-RC formation.116
The A-type cyclins are first transcribed late during the G1 phase under the control of the E2F transcription factors in a similar manner to that of cyclin E. However, in contrast to cyclin E, cyclin A associates with both Cdk2 and Cdk1 and acts at two distinct cell cycle stages.117,118 First, cyclin A is required for S phase progression, which was established by showing that injection of either cyclin A antisense constructs or antibodies was sufficient to block S phase progression.117,119 Notably, cyclin A–Cdk2 enters the nucleus at the start of S phase and is specifically localized at nuclear replication foci, and thus it is thought to be actively involved in the firing of replication origins.120 As was described previously, cyclin A–Cdk2 also is required to phosphorylate E2F-1 and mediate its degradation, which is required to prevent E2F1 from triggering apoptosis.121,122
Mitosis
There are two A-type cyclins and three B-type cyclins with different expression patterns. Cyclin A1 and cyclin B2 are mostly expressed in meiotic cells, although these proteins also may be present in small quantities in mitotic cells.8 The analysis of mutant mouse models shows that cyclin A1 or B2 loss has no detectable effect on development, whereas cyclin A2 and B1 are absolutely required for embryogenesis.8 Cyclin B3 is an early meiotic cyclin that is expressed in the leptotene and zygotene phases of spermatogenesis.123,124 Interestingly, a new oncogenic fusion has been observed between BCOR (encoding the BCL6 co-repressor) and CCNB3 (encoding the testis-specific cyclin B3) on the X chromosome, indicating a biologically distinct class of sarcomas.125 Most of the information discussed in this chapter therefore relates to cyclin A2 and B1.
Cyclin A2 is active during the G2 phase and at the beginning of mitosis.117 Here it is thought to play a key role in initiating the condensation of chromatin and also might participate in the activation of the cyclin B–Cdk1 complexes. Cyclin A2 is destroyed upon nuclear envelop breakdown in an APC/C-Cdc20–dependent manner.126 A-type cyclins are then substituted by B-type cyclins. Cyclin B1 protein accumulates steadily through the G2 phase and associates specifically with Cdk1, although it also may associate with Cdk2.118 However, the resulting cyclin B1–Cdk1 complex is mostly sequestered in the cytoplasm, and it is retained in an inactive form throughout the G2 phase via the inhibitory phosphorylation of Thr14 and Tyr15 in Cdk1’s active site by the Myt1 and, to a lesser extent, Wee1 kinases.129–129 Activation of cyclin B1–Cdk1 occurs in a highly synchronous manner during G2-prophase transition (see Fig. 4-1).130 This activation is mediated by two changes. First, the activities of Myt1 and Wee1 are downregulated at the transition between the G2 and M phases. Second, a dramatic increase in the activity of the Cdc25 phosphatases occurs that relieves the inhibitory phosphorylation of Thr14 and Tyr15. These activity changes are triggered by the phosphorylation of Myt1, Wee1, Cdc25a, and Cdc25c. Three different kinases are thought to contribute to this phosphorylation: Polo-like kinase (Plk1), cyclin A–Cdk1, and cyclin B1–Cdk1 itself. The involvement of cyclin B1–Cdk1 creates a powerful feed-forward loop; once a small amount of cyclin B1–Cdk1 is activated, it simultaneously inactivates its own inhibitors and activates its activators, enabling a rapid transformation of the entire cyclin B1–Cdk1 pool from the inactive state to the active state. Once active, cyclin B1–Cdk1 phosphorylates components of the centrosomes and initiates a process called centrosome separation, in which the centrosomes move to opposing poles of the nascent spindle, an event essential for formation of the mitotic spindle.131 Cyclin B1–Cdk1 then translocates across the nuclear membrane (which is still intact at this point in mitosis) to orchestrate mitotic events. Cdk1 phosphorylates nearly a hundred proteins and participates in such activities as chromosome condensation, nuclear envelop breakdown, modifications in the Golgi apparatus, and formation and dynamics of the mitotic spindle.8 Cdk1-deficient mouse embryos arrest at the two-cell state, indicating that this kinase is absolutely required for mitosis and cannot be compensated by other mammalian Cdks.99
The mitotic machinery is optimized to ensure that the replicated chromosomes are faithfully segregated to the daughter cells. This segregation is achieved through the use of a specialized microtubule-based structure, the mitotic spindle, on which the original chromosomes and their newly replicated copies, called sister chromatids, align and then are partitioned to opposite poles of the cell. The mitotic spindle is a highly dynamic structure that is maintained by many protein families, including motor molecules and other microtubule-associated proteins.132,133 The appropriate side-by-side alignment of the sister chromatids, termed biorientation, is facilitated by the physical tethering of the sister chromatids to one another. This process, called cohesion, actually occurs in S phase in a manner that is coordinated with the replication process.136–136 Cohesin is mediated by four proteins that together make up the cohesin complex. Two of these proteins, Smc1 and Smc3, have a long coiled-coil structure with a dimerization domain at one end that allows them to heterodimerize to form a V-like structure. Importantly, the remaining ends of Smc1 and Smc3 can associate with each another to form a functional adenosine triphosphate (ATP) domain. This domain acts in an ATP-dependent manner to recruit two additional proteins, Scc1 and Scc3, which form a closed-ring structure that most likely encircles the chromosomes.137,138 Cohesin loading onto chromosomes, catalyzed by a separate complex called kollerin, is thought to be mediated by the entry of DNA into cohesin rings, whereas dissociation, catalyzed by Wapl and several other cohesin subunits, is mediated by the subsequent exit of DNA.139
Largely on the basis of morphologic features, mitosis is divided into five different stages—prophase, prometaphase, metaphase, anaphase, and telophase (Fig. 4-5)—before separation of the daughter cells or cytokinesis. Prophase is essentially a preparative stage. One of the major events is modification of the DNA. In a process called resolution, the sister chromatids are untangled via the action of topoisomerase II.137,138 Resolution requires removal of the chromosome arm cohesin through phosphorylation of Scc3 by Plk1 and histone H3 by the aurora B kinase. Importantly, the cohesin complex at the centromere is somehow protected from this modification by a protein called shugosin (Sgo).142–142 In addition to resolution, the sisters undergo condensation—essentially, packaging into a more compact chromatin structure. This process involves two multimeric complexes, condensin I and II, which also contribute to sister chromatid resolution and requires phosphorylation by mitotic Cdks.137 During prophase, the nuclear envelope is still intact; consequently, differences in subcellular localization of the condensin and Cdk complexes allow only condensin II and cyclin A-Cdk1 (nuclear), and not condensin I and cyclin B-Cdk1 (cytoplasmic), to initiate condensation. The second major event in prophase is the translocation of the cytoplasmic cyclin B1-Cdk1 to the nucleus, where it phosphorylates components of the nuclear envelope and triggers its breakdown,143 which defines the transition from prophase to prometaphase.
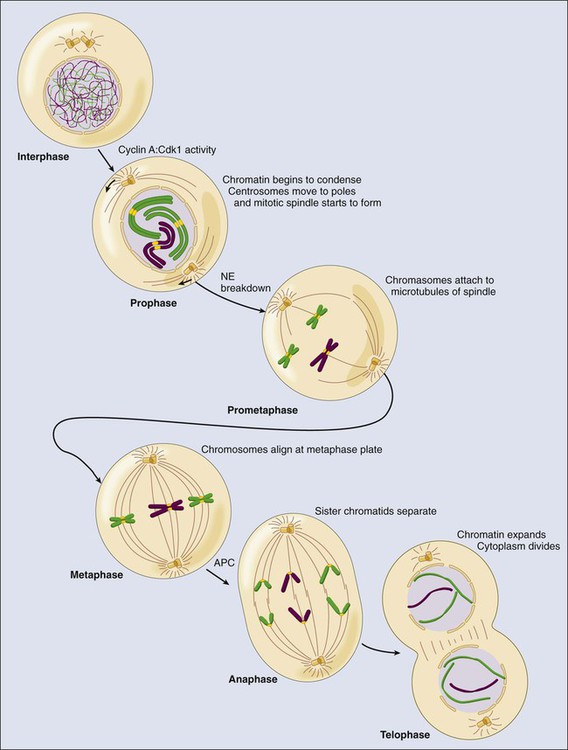
During prometaphase, the condensation process is accelerated because condensin I and cyclin B-Cdk1 now have access to the DNA. The sister chromatids become attached to spindle microtubules through a structure called the kinetochore, which is assembled onto centromeric DNA.146–146 Microtubules nucleated from the centrosomes attach to the kinetochore through a process called search and capture, in which individual microtubules grow and shrink until they contact and bind the kinteochore.145,147 Typically, one sister chromatid of the pair attaches first, and this attachment is further stabilized through the recruitment of additional microtubules from the same pole of the mitotic spindle to create a kinetochore fiber—that is, highly bundled microtubules bound to the kinetochore. The sister chromatids oscillate in the cell until the second sister chromatid is captured by microtubules emanating from the other pole. These oscillations continue until all of the chromosomes are properly aligned on the metaphase plate.
Metaphase is defined as the point at which all of the chromosome pairs are fully condensed, attached to the mitotic spindle, and aligned at the center—termed the “metaphase plate.” The pulling of the kinetochore fibers toward the poles creates tension through the cohesin complex at the kinetochores, which indicates that the sister chromatids have achieved appropriate biorientation. The cell constantly monitors the attachments of microtubules to the chromosomes, and the tension that is generated by microtubules on the kinetochores ensures that the sister chromatids are properly aligned at the metaphase plate.148,149 This process is the basis of one of several cell cycle checkpoints, called the mitotic spindle assembly checkpoint (SAC), that will be described in more detail in the following sections.
Anaphase is characterized by the segregation of the chromosomes.150 This event is controlled by the mitotic ubiquitin ligase APC/C-Cdc20. APC/C-Cdc20 ubiquitinates, and thereby triggers the degradation of, cyclin B1 and a protein called securin.53 Both securin and cyclin B1–Cdk1 complexes are able to bind and inhibit a protease called separase.53,150,151 APC/C-Cdc20 activity results in the degradation of cyclin B and securin and the subsequent separase activation. Once released, separase cleaves the Scc1 component of the cohesin complex, which opens the cohesin ring, unlinking the sister chromatids and allowing them to be pulled to opposite poles (see Fig. 4-5). The spindle poles then move farther apart to ensure that the chromosomes are fully segregated. The separase-dependent cleavage of Scc1 also is essential to link segregation of chromatids with the separation of centrioles during mitotic exit.152
During telophase, the mitotic spindle disassembles, leaving a single centrosome and a single set of chromosomes with each nascent daughter cell. Cyclin B1 degradation and the subsequent inactivation of Cdk1 results in DNA decondensation, and the nuclear envelope reforms around the segregated chromosomes to create two nuclei. These events are not only dependent on the loss of Cdk kinase activity, but the activation of mitotic exit phosphatases such as PP1 and PP2A is essential for the dephosphorylation of Cdk substrates.52–52 During anaphase, Cdh1, which is inhibited by Cdk-dependent phosphorylation during mitosis, is dephosphorylated and replaces Cdc20 as the main APC/C activator.53 APC/C-Cdh1 is responsible for the degradation of multiple cell cycle regulators, including Cdc20.53,58 APC/C-Cdh1 also activates the ubiquitination and degradation of geminin, allowing accumulation of Cdt1 for origin relicensing in the subsequent G1 phase, and the mitotic cyclins, allowing loss of Cdk kinase activity. Loss of Cdh1 does not result in major abnormalities during mitotic exit but results in earlier entry into the following S phase because of increased Cdk activity and DNA damage.153,154 Cdh1 therefore is required to prevent unscheduled entry into S phase and genomic instability.58
Finally, the cell undergoes cytokinesis, or cytoplasmic division. This process involves formation of a structure containing actin and myosin, called the contractile ring, on the inner face of the cell membrane. The position of the contractile ring is carefully controlled. For most mammalian cells, the ring begins to form in anaphase, and its position is established by the position of the metaphase plate. As the membrane grows, the contractile ring contracts steadily to form a constriction, termed the “cleavage furrow,” which ultimately separates the two nuclei and forms the two daughter cells.132,155,156 This process partially depends on several mitotic kinases such as Aurora B, a component of the CPC, and Plk1, which are located at the midbody. Plk1 controls the activity of RhoA guanosine triphosphatases, which are major regulators of the actomyosin ring required for abscission.67,157 Aurora B also ensures that abscission does not occur in the presence of DNA bridges to avoid DNA damage in a process known as the abscission checkpoint.156,158
Cell Cycle Checkpoints
At key transitions during eukaryotic cell cycle progression, signaling pathways monitor the successful completion of events in one phase of the cell cycle before proceeding to the next phase. These regulatory pathways are commonly referred to as cell cycle checkpoints.4 In a broader context, cell cycle checkpoints are signal transduction pathways that link the rate of cell cycle phase transitions to the timely and accurate completion of prior dependent events. Checkpoint surveillance functions are not confined to monitoring normal cell cycle progression; they also are activated by both external and internal stress signals. To minimize the possibility of errors, checkpoints exist at the four different phases of the cell cycle: G1 (to prevent entry into S phase), intra S phase, G2 (to prevent mitosis), and M (to avoid mitotic exit with abnormal segregation of chromosomes) (see Fig. 4-1).
G1/S Checkpoint
The molecular pathway that determines cell cycle entry upon the appropriate mitogenic stimuli (previously described) is not considered a cell cycle checkpoint sensu estricto. However, several of their components also are used by a checkpoint that monitors DNA alterations before replication. In G1 cells, double-stranded DNA breaks (DSBs) are the most common and most deleterious type of DNA damage. The central components of the DNA damage response (DDR) are two members of the phosphoinositide 3-kinase-related kinase family: ataxia telangiectasia mutated (ATM) and ATM- and rad3-related (ATR).159 ATM originally was identified by virtue of its mutation in a hereditary syndrome, ataxia-telangiectasia, which is associated with radiation hypersensitivity and cancer predisposition.160 ATR also is associated with a hereditary syndrome called Seckel syndrome.161 Early studies suggested that ATM and ATR played distinct roles in the response to DSBs (ATM) versus replicative defects and single-stranded breaks (ATR). However, we now know that the regulation is more complex; considerable cross-talk occurs between ATM and ATR, and they share many mediators and effectors, but the precise composition and role of the DDR complexes vary depending on both the type of damage and the stage of the cell cycle (Fig. 4-6).162
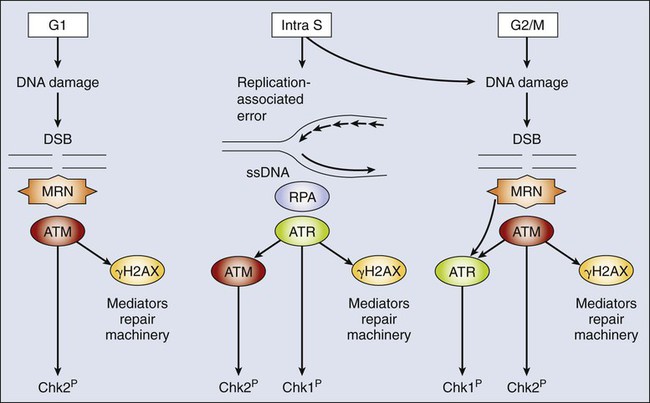
These DSBs are recognized by the multifunctional Mre11-Rad50-Nbs1 (MRN) complex.159,163 This complex recruits ATM to the site of damage. The active ATM then recruits proteins to modify the chromatin at the region of the break and activate repair and signaling. As a first step in this process, ATM phosphorylates histone H2AX to form γH2AX, which helps hold the damaged ends together and acts as a binding platform for additional factors, including Mdc1, 53BP1, and Brca1, as well as more MRN and ATM. In contrast to the S and G2 response, no recruitment of ATR to DSB in G1 cells occurs, and thus ATM is solely responsible for checkpoint activation. The recruitment of additional ATM amplifies the signal, and ATM acts via phosphorylation and activation of the effector kinase Chk2.164,165
Chk2 influences the G1 cell cycle arrest via two mechanisms (Fig. 4-7). First, it phosphorylates all three members of the Cdc25 family. Phospho-Cdc25a is ubiquitinated by SCF-TrCPβ and degraded, whereas phospho-Cdc25b and phospho-Cdc25c are bound and sequestered by a cytoplasmic protein called 14–3-3.18,166 This process is a rapid response that can take effect within minutes after DNA damage, and it has a widespread effect on cell cycle progression by preventing activation of Cdks. Second, Chk2 phosphorylates p53, a critical regulator of cell cycle checkpoints.167 In normal, nonstressed cells, p53 protein is maintained at low steady-state levels because it has a very short half-life. This half-life is a result of rapid ubiquitination of p53 by Hdm2 (the human ortholog of murine Mdm2 protein) and its consequent degradation.168,169 The importance of Mdm2 for maintenance of appropriate p53 levels in vivo is highlighted by the fact that absence of Mdm2 in knockout mice results in early embryonic lethality that is rescued by a dual knockout of Mdm2 and p53.170,171 Phosphorylation of p53 by Chk2 is sufficient to prevent its association with Hdm2/Mdm2,170 which leads to an accumulation of p53, which functions as a transcriptional activator. p53 induces expression of many genes involved in cell cycle arrest, including the Cdk inhibitor p21Cip1.172,173 This p53-mediated arrest takes longer to develop than does the Cdc25 response (because it requires transcription and protein synthesis), but it appears to be much more robust. Moreover, in addition to inducing cell cycle arrest, p53 has the capacity to induce apoptosis through the transcriptional activation of proapoptotic regulators (e.g., the BH3-only proteins Puma and Noxa).174
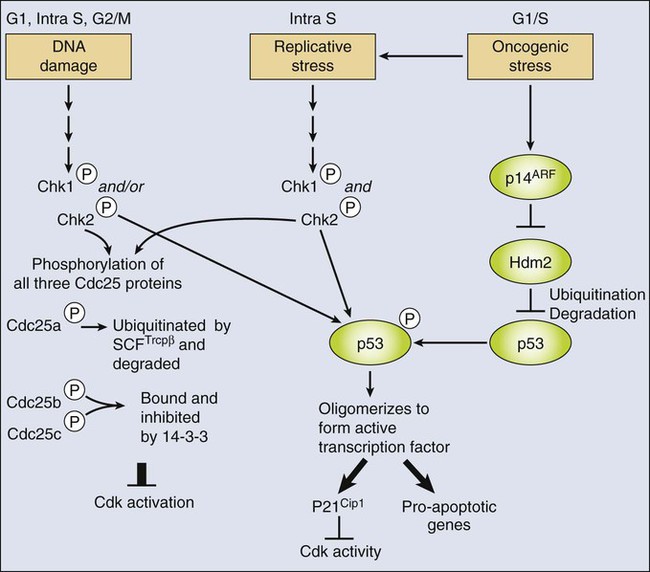
Importantly, p53 also is activated by other stress signals (see Fig. 4-7). In particular, it is now well established that numerous oncogenes trigger a stress response (called oncogene-induced stress) that leads to the activation of p53.175,176 The emerging view is that this process occurs through two distinct mechanisms. First, oncogene activation is thought to yield replicative stress that activates p53 via activation of Chk kinases and phosphorylation of Hdm2/Mdm2 as just described.177,178 Second, many oncogenes activate transcription of cell cycle inhibitors such as p16INK4a, p15INK4a, p21CIP1, or p19ARF in a p53-independent manner.175,179–182 p19ARF is a protein encoded by the INK4A/ARF (CDKN2A) locus, and it actually shares two coding exons, which are read in alternate reading frames (hence the name ARF) with the p16INKa tumor suppressor.181 The ARF protein product, called p14ARF in humans and p19ARF in mice, binds to Hdm2/Mdm2 and prevents it from regulating p53.183–186 As with the DDR, this mechanism frees p53 to activate the transcription or proarrest or proapoptotic targets.
As an additional DNA damage response in G1 cells, genotoxic agents also inhibit origin licensing by way of an ATM/ATR-independent process that is achieved through regulation of Cdt1.187 As described previously, Cdt1 is required for pre-RC formation. In an undamaged cell, Cdt1 is available during the G1 phase but is inhibited after origin firing by degradation (mediated by the SCF-Skp2 and Cul4-Ddb1-Dtl/Cdt2 ubiquitin ligases) and geminin binding. As a key feature of this regulatory system, Cdt1 is completely resistant to Cul4-Ddb1-Dtl/Cdt2 in the G1 phase. However, DNA damage allows the Cul4-Ddb1-Dtl/Cdt2 complex to ubiquitinate Cdt1 and induce its degradation. Importantly, the degradation of Cdt1 is extremely rapid, occurring within minutes of the DNA damage. Both Cdt1 and Cdt2 are phosphorylated in response to DNA damage, which results in the ubiquitination of Cdt1. This process depends on the activity of the p97 AAA+-ATPase and its cofactor Ufd1, a complex required for the extraction of ubiquinated proteins from the chromatin.113,188,189 As a result, origin licensing is completely blocked until the damage is repaired and Cdt1 is resynthesized.
Intra-S Phase Checkpoint
One of the major goals of cell cycle checkpoints is to prevent the deleterious consequences of replicating damaged DNA. Therefore S phase cells must respond virtually instantaneously to DNA damage to halt initiation of new replication forks throughout the S phase.190 The most deleterious damage is DSBs. DSBs can occur through the action of DNA damaging agents (from either extrinsic or intrinsic sources) or as a consequence of the replication process itself—for example, if the replication fork passes through nicked DNA or replication stalls at sites of DNA damage. The cell senses the damage in different ways, depending on whether the lesion is associated with replication. Ultimately, both ATM and ATR are recruited to the site of damage, but the order of binding is different.162,190 Replication-linked DSBs are distinguished by the presence of single-stranded DNA (ssDNA), a hallmark of the replication process. The ssDNA is coated by replication protein A (RPA) and bound by ATR and its regulator subunit ATRIP, even during the normal replication process. In response to DNA damage, the ATR kinase is activated, and it then recruits a variety of complexes that mediate both repair and checkpoint activation, including ATM. In contrast, nonreplication-associated DSBs initially recruit and activate ATM through the MRN-dependent process described previously for the G1/S checkpoint. However, in S-phase cells, DSB resection causes the formation of ssDNA (through the action of the MRN endonuclease), which is then bound by RPA and ATR/ATRIP.162,190 Thus in S-phase cells, ATR and ATM jointly orchestrate the DDR. ATR contributes to the checkpoint response in a similar manner to ATM: it activates Chk1, which also can phosphorylate the Cdc25 proteins and p53.191–194
G2 Checkpoint
Whereas the G1/S and intra-S phase checkpoints prevent cells from unfaithful replication, the G2 checkpoint is required to prevent the passage of DNA lesions to the two daughter cells during mitosis.159,195 DSBs are detected exactly as described previously for the S-phase nonreplication-associated DSBs. Similarly, the ATR/Chk1 and ATM/Chk2 pathways enforce arrest through inhibition of G2 and mitotic Cdk complexes via the rapid removal of the Cdc25 phosphates and the p53-dependent induction of p21CIP1 to inhibit mitotic Cdk complexes (cyclin A/B in combination with Cdk1/2 kinases).194,196,197 Ubiquitin ligases also are involved in this process. SCF-βTrCP regulates the levels of Cdc25, Claspin, and Wee1, whereas APC/C-Cdh1 is critical for the elimination of Plk1, a kinase essential for checkpoint recovery.197,198
Spindle Assembly Checkpoint
The SAC acts to ensure that appropriate partitioning of the chromosomes occurs during mitosis.148 The concept that chromosome segregation is prevented until all condensed sister chromatid pairs are aligned at the metaphase plate with the appropriate biorientation has already been introduced. This process actually is controlled by a signaling network that constitutes the SAC (Fig. 4-8). The core components of this checkpoint—called Mad1, Mad2, BubR1, Bub1, and Bub3 in humans—originally were identified through screens in yeast for “mitotic arrest deficient” (MAD) and “budding uninhibited by benzimidazole” (BUB) mutants.148 Other components of the checkpoint include the Mps1 kinase and the three subunits of the Rod, Zwich, and ZW10 (RZZ) complex.199 During prometaphase, these proteins localize to the outer kinetochore and, in the absence of biorientation, prevent the Cdc20 activator from binding to the APC/C.
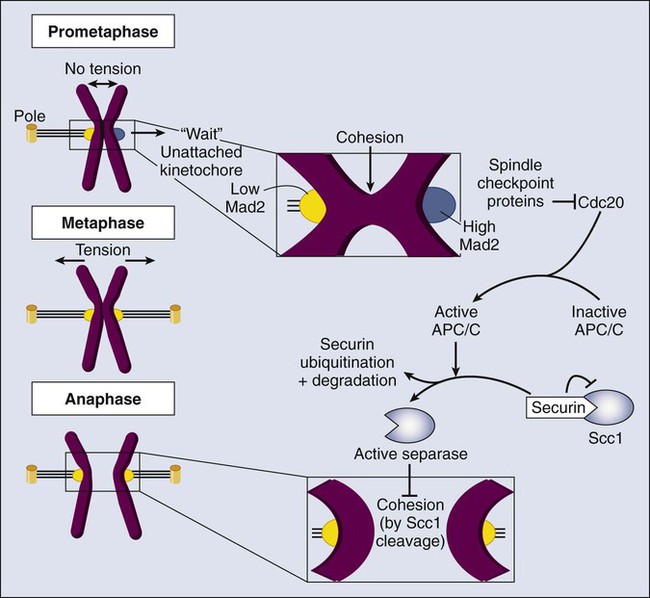
The kinetochore is made of approximately 30 scaffold proteins that link chromatin and the mitotic spindle.146,200–202 Additional regulatory elements include sensors of microtubule-kinetochore attachment and a complex signaling pathway that modulates APC/C-Cdc20 activity. Lack of tension or lack of attachment at the kinetochore results in stable Mad1-Mad2 complexes that convert an inactive open-Mad2 conformation into a closed-Mad2 conformation that is able to bind to Cdc20.148,203,204 The Mad2-Cdc20 association triggers the recruitment of BubR1-Bub3 into an APC/C-inhibitory complex (the mitotic checkpoint complex [MCC]). Because the closed-Mad2 signal is diffusible, a single unattached kinetochore is sufficient to form these complexes and inhibit APC/C-Cdc20 activity. As a result, separase is inhibited by the high levels of securin and cyclin B–Cdk1 complexes, being unable to cleave the centromeric cohesin (see Fig. 4-8). Once all chromosomes are bipolarly attached to the mitotic spindle, the SAC is satisfied and the Mad1 : Mad2 complex is removed from the kinetochores.199,205 How the checkpoint signaling is inhibited currently is unclear. Cdc20 is now released from the MCC complex and activates the APC/C, leading to the rapid ubiquitination and degradation of cyclin B and securin. Inactivation of these two proteins results in two major processes. First, lack of securin and cyclin B results in the activation of separase, a caspase-like protease that cleaves cohesion, the molecule that holds sister chromatids together at the centromere. This process results in the transition to anaphase when chromosomes separate toward the two poles of the spindle.138,150 Second, inhibition of Cdk1, due to the lack of cyclin B, leads to the activation of mitotic phosphatases such as PP1 and PP2A. How this process occurs is not clear at present, although it may involve the inactivation of phosphatase inhibitors such as Inh-1 or Mastl.50,52 APC/C-Cdc20–dependent inactivation of Cdk1 and activation of PP1 and PP2A results in dephosphorylation of mitotic substrates followed by disassembly of the spindle, decondensation of chromosomes, and reassembly of the nuclear envelop during telophase.
Cell Cycle Deregulation in Human Cancers
The central relevance of cell cycle regulation in cancer is underscored by the finding that virtually all human tumors carry mutations in (a) the basic cell cycle machinery that controls entry into the cell cycle and (b) checkpoint regulators such as the p53 pathway (Table 4-2) and by the observation that (c) most human tumors display aberrant chromosome numbers.1,5,6,181,206 Together, the pRb and p53 pathways are critical gatekeepers of cell cycle progression and stress response, and their function has crucial implications in the maintenance of genome integrity.207,208
Table 4-2
Alteration of Cell Cycle Regulators in Human Tumors*
| Protein (Gene) | Tumors Associated with Mutations or Altered Expression | Hereditary Syndromes Associated with Germline Mutations |
| ATM | Breast carcinomas, lymphomas, leukemias | Ataxia-telangiectasia |
| Aurora-A (AURKA) | Wide array of tumors | NR |
| Bub1 | Colon, lung, and pancreatic cancer | NR |
| BubR1 (BUB1B) | Wilms tumor, rhabdomyosarcoma, and acute leukemia | Mosaic variegated aneuploidy and premature chromatid separation syndrome |
| Brca1/2 | Breast and ovarian carcinoma | Familial breast and ovarian cancer |
| Cdc25a | Carcinomas of the breast, lung, head, and neck and lymphoma | NR |
| Cdc25b | Carcinomas of the breast, lung, head, and neck and lymphoma | NR |
| Cdk4 | Wide array of cancers | Familial melanoma |
| Cdk6 | Wide array of cancers | NR |
| Chk1 | Colorectal and endometrial carcinomas | NR |
| Chk2 | Carcinomas of the breast, lung, colon, urogenital tract, and testis | Li-Fraumeni syndrome |
| Cyclin D1 (CCND1) | Wide array of cancers | NR |
| Cyclin D2 (CCND2) | Lymphoma and carcinomas of the colon, testis, and ovary | NR |
| Cyclin D3 (CCND3) | Lymphoma, pancreatic carcinoma | NR |
| Cyclin E (CCNE1/2) | Wide array of cancers | NR |
| Mdm2 (HDM2) | Soft tissue tumors, osteosarcomas, esophageal carcinomas | NR |
| Mps1 (TTK) | Lung, gastric and bladder cancer | NR |
| Mre11 | Lymphoma | Ataxia-telangiectasia–like disorder |
| Nbs1 | Lymphomas, leukemias | Nijmegen breakage syndrome |
| p15INK4b (CDKN2B) | Wide array of cancers | NR |
| p16INK4a (CDKN2A) | Wide array of cancers | Familial melanoma |
| p27Kip1 (CDKN1B) | Wide array of cancers | NR |
| p53 (TP53) | Wide array of cancers | Li-Fraumeni syndrome |
| p57Kip2 (CDKN1C) | Bladder carcinomas | NR |
| p130 (RBL2) | Wide array of cancers | NR |
| pRb (RB1) | Wide array of cancers | Familial retinoblastoma |
| Tpx2 | Lung, bone, and pancreatic cancer | NR |
*Only genetic alterations or defects that are present in more than 10% of primary tumors are represented.
Unscheduled Cell Cycle Entry in Cancer
The alterations in the cell cycle machinery that occur most frequently include loss or mutation of the pRb tumor suppressor; overexpression of cyclins, Cdks, and Cdc25 phosphatases; and loss of expression of CKIs.1 Mutations that affect the pRb pathway have been identified in most human cancers.1,27,208 The RB1 gene originally was identified by virtue of its mutation in both familial and sporadic retinoblastoma, but it is defective in many other tumor types, especially osteosarcoma and lung cancer. Indeed, more than 90% of small-cell lung cancers have mutant RB1, suggesting that disruption of the pRb pathway (through the genetic or epigenetic targeting of RB1 or upstream signaling components) is a requirement for the genesis of lung cancer.209 It is important to note that inactivation of the parallel and interconnecting p14ARF-p53 axis is also essential in functionally pRb-deficient lung cells to bypass efficient apoptosis.181
In tumors that lack RB1 mutations, alterations in other components of the signaling pathways that regulate pRb frequently are found, including cyclin overexpression or loss of CKIs. Nearly 50% of invasive breast cancers have elevated cyclin D expression compared with surrounding normal breast epithelium, and in transgenic mice with overexpression of human cyclin D1 or cyclin E in mammary gland cells, mammary adenocarcinomas develop.212–212 Similarly, Cdk4 and Cdk6 gene amplification occurs in breast cancers, sarcomas, gliomas, and melanomas, and specific translocations lead to cyclin D or Cdk6 overexpression in hematopoietic disorders.1,8,213 Modifications of CKIs that act upstream of pRb activity also commonly are found in human tumors. The CKI p27Kip1 often is aberrantly expressed in human breast cancer, and reduced p27Kip1 protein levels are correlated with more aggressive breast tumors.216–216 Likewise, decreased expression of the CKI p57Kip2 is found in human bladder cancers.217 Although p21Cip1 is not commonly mutated in human cancer, its expression is strongly reduced in multiple tumors as a consequence of defective p53 signaling.218 p16INK4a appears to be the CKI most frequently affected; it was identified as a tumor suppressor that is associated with familial melanoma, and it is inactivated by point mutation, deletion, and/or promoter methylation in approximately 30% of all human tumors.213,219,220 In contrast, point mutations in p15INK4b, p18INK4c, and p19INK4d are rare, but promoter methylation and reduced protein expression of some of these inhibitors have been seen in a variety of tumor types.1,213,221–223 Germline mutations in p16INK4a predispose individuals to melanoma, whereas deletion of p15INK4b and p16INK4a is linked to the pathogenesis of lymphomas, mesotheliomas, and pancreatic cancers.1,213,219 Although point mutations in Cdks are not common in human tumors, a specific mutation of Cdk4 (R24C) that prevents its inhibition by INK4 proteins has been found in persons with familial melanoma.224 Knockin mice harboring this mutation show not only an increased susceptibility to melanoma but also the development of multiple tumor types, indicating the relevance of this regulatory circuit in human tumors.225,226 Both Cdc25a and Cdc25b phosphatases also are overexpressed in more than 30% of primary breast tumors, 40% to 60% of non–small-cell lung cancers, 50% of head and neck tumors, and a significant fraction of non-Hodgkin’s lymphomas.18,227,228 All these events can result in increased activation of Cdks, defective pRb signaling, unscheduled cell proliferation, and override of checkpoint arrest.
Mutations in p53 and Checkpoint Regulators
The most frequently altered cell cycle checkpoint-signaling molecule is the p53 tumor suppressor. The importance of p53-dependent signaling in tumor suppression is underscored by the frequency of mutation (~50%) in sporadic tumors229 and the finding that germline mutations of p53 result in Li-Fraumeni syndrome, a highly penetrant familial cancer syndrome that is associated with significantly increased rates of brain tumors, breast cancers, and sarcomas.230 In human tumors that lack p53 gene mutation, p53 function may be disrupted by alterations in cellular proteins that modulate the levels, localization, and biochemical activity of p53. For example, in some tumors with wild-type p53 alleles, Mdm2 gene amplification occurs, resulting in Mdm2 protein overexpression and subsequent p53 inactivation.231 In human papillomavirus-induced cervical carcinoma, p53 typically is not mutated; however, the human papillomavirus E6 protein binds p53 and targets it for degradation, abrogating p53-dependent signaling.232
Proteins that reside upstream of p53 (including ATM and Chk2) also are targeted for mutation in human tumors, and their discovery and analysis have greatly deepened our insight into DNA damage response-signaling pathways. ATM mutations occur in ataxia-telangiectasia, a disorder in which patients have increased sensitivity to radiation and an elevated incidence of leukemias, lymphomas, and breast cancer.160,233 ATM-null mice exhibit growth retardation, neurological dysfunction, infertility, defective lymphocyte maturation, and sensitivity to ionizing radiation.234,235 The DNA double-strand break repair gene Mre11 also is mutated in persons with an ataxia-telangiectasia–like disorder.236
Mutations of Chk2 and Chk1 also arise in human cancers. Chk2 mutations have been reported in several cancers, including lung cancer, whereas Chk1 mutations have been observed in human colon and endometrial cancers.237,238 In addition, heterozygous alteration of Chk2 occurs in a subset of persons with Li-Fraumeni syndrome who lack p53 gene mutations.239 These findings support the theory that in human tumors in which p53 is intact, the function of this signaling pathway might be disrupted by alterations in cellular proteins that modulate the levels or activity of p53. In addition, the breast cancer susceptibility tumor suppressors Brca1 and Brca2 are known to participate in the DNA damage response and repair.240 Similarly, Fanconi’s anemia proteins, which originally were identified by virtue of their association with a recessive development disorder called Fanconi’s anemia, which is associated with increased cancer predisposition (particularly acute myeloid leukemia), also function in the DNA damage response.240 Mutations in the Nbs1 gene, a component of the MRN complex that sensors damage, are responsible for Nijmegen breakage syndrome, a rare autosomal-recessive disorder characterized by chromosome instability, hypersensitivity to ionizing radiation, and high susceptibility to the development of tumors.163,241
Aneuploidy and Chromosomal Instability
Abnormal chromosome numbers, called “aneuploidy,” is a frequent feature of cancer cells.6,206,242 In addition, many cancer cells also display chromosomal instability (CIN) or an aberrantly high change in the karyotype. CIN may lead to aneuploidy, but not all aneuploidy cells display CIN, because many cancer cells exhibit stable aneuploidy karyotypes. Aneuploidy has a high rate of frequency (more than 75%) in human tumors, and about one quarter of the genome is affected by whole-arm or whole-chromosome number alterations.243 Yet it is not clear whether aneuploidy is a cause or consequence of cancer.6,244
In vitro estimates suggest that normal cells missegregate a chromosome every hundred cell divisions.206 This rate is dramatically increased in cells that display CIN, which missegregate a chromosome in every one to three cell divisions in vitro.245 These defects occur for multiple reasons, including aberrant centrosome numbers, abnormal microtubule-kinetochore attachments, and imperfect SAC. In particular, there has been considerable speculation that disruption of the SAC could occur during tumor progression.246,247 Notably, inactivating mutations in BUB1 have been identified in human colon carcinoma cell lines, which are known to have a high degree of aneuploidy.248 These mutations facilitate the transformation of cells that lack the breast cancer susceptibility gene, Brca2.249 The gene encoding BubR1, BUB1B, is also mutated in persons with mosaic variegated aneuploidy and the premature chromatid separation syndrome. Both BUB1 and BUB1B also are silenced by promoter hypermethylation in specific tumors.250 Additional mutations and epigenetic alterations have been found in the SAC components Mad1 and Mad2.250 Most of these alterations are loss-of-function mutations, suggesting that the SAC is not functional in a variety of tumors. Moreover, haploinsufficiency of Mad2 has been shown to cause elevated rates of lung tumor development in mice.251 Interestingly, overexpression of Mad2 also results in increased tumor susceptibility and frequent relapses of chromosomally unstable tumors.252 In fact, Mad2 is also frequently overexpressed in human tumors. Several other mitotic regulators such as separase, securin, condensins, Cdc20, or Aurora kinases, as well as the SAC component Mps1, are included in the overexpression signature that marks chromosomally unstable cancers.250,253 All these data, together with the recent evidence gathered in mouse models, suggest that small aberrations, either overexpression or downregulation, in the levels of mitotic and SAC regulators may contribute to aneuploidy and/or CIN in human tumors.6,247 Because functional disruption of SAC proteins results in the abrogation of the checkpoint and a failure to arrest in mitosis in the presence of microtubule poisons such as taxol and nocodazole, these aberrations also may play a role in the response to mitotic poisons currently used in the clinic.254
Chromosome missegregation does not only result in whole-chromosome aneuploidy but also may generate chromosome breaks and rearrangements. A possible mechanism for DNA damage caused by chromosome missegregation is based on the defective DNA replication of the micronuclei present in cells affected by aneuploidy.255 These micronuclei undergo asynchronous DNA replication, resulting in DNA damage and pulverization of chromosomes. This process may provide, at least partially, an explanation for “chromothripsis,” a situation in which chromosomes undergo massive local DNA breakage and rearrangements.255,256 In addition, chromosomes that missegregate may be damaged during cytokinesis, resulting in DNA double-strand breaks that can lead to unbalanced translocations in the daughter cells.257 All these processes provide additional mechanisms by which defective chromosome segregation may induce genomic aberrations during tumor development.
Therapeutic Manipulation of Cell Cycle Controls
Targeting Cyclin-Dependent Kinase Activity
Considerable debate has ensued about whether inhibition of Cdk activity is a rational strategy for anticancer therapies.258 Cdk activity frequently is elevated in human tumors, but it also is required to maintain specific cell populations in adults (e.g., the hematopoietic compartment and gut) that are essential for viability.3 Thus the key issue is whether tumor cells may require different Cdks for proliferation or whether sufficient difference in the Cdk activity exists to create a therapeutic window. During the last few years, the analysis of Cdk and cyclin mouse models has yielded considerable insight into this question but also has raised additional questions.259 These mouse models show that the cell cycle machinery is extremely robust; it adapts easily to the loss of Cdks or cyclins by using other Cdks or cyclins to substitute for the missing activity. For example, cells are able to proliferate without specific interphase Cdks because novel cyclin-Cdk complexes can be formed that allow the cell cycle to progress.95,96,98,99 This ability raises the possibility that tumor cells will develop resistance to Cdk-inhibitory drugs rapidly simply by adapting their cell cycle machinery. On the positive side, studies in mouse models clearly show that tumors can be more dependent on Cdk activity, or at least a specific Cdk activity, than are normal tissues. For example, loss of D-type cyclin-dependent kinase has been shown to have little or no effect on the development and maintenance of many tissues but can greatly suppress the development of certain tumor types, depending on the tissue and the identity of the initiating oncogenic lesions.260–263 Mammary gland tumor proliferation dramatically depends on Cyclin D1-Cdk4 complexes, whereas the absence of these molecules does not alter normal mammary gland development.261,264,265 Interestingly, cyclin D1–Cdk4 is required for Her2 (ErbB2)- or HRas-driven tumors, whereas it is dispensable for Myc- or Wnt-induced neoplasias, suggesting that the efficacy of Cdk inhibitors may have a strong dependence on the genetic background of target tumors.261,266 Similarly, Cdk4 is required for KRAS-mutant lung tumors, but it seems dispensable for similar lung tumors with wild-type KRAS alleles.263 In light of these complexities, efforts have been placed on generating pharmacologic inhibitors of Cdks that either are Cdk-specific or have pan-Cdk activities.259 Numerous small molecule inhibitors have been developed, and many are in clinical trials (Table 4-3).259,267,268
Table 4-3
Cell Cycle Regulators with Therapeutic Interest
| Target | Representative Small-Molecule Inhibitors | Preclinical or Clinical Data |
| MITOTIC SPINDLE | ||
| Tubulin | Stabilizing: taxanes, eleutherobins, epothilones, laulimalide, sarcodictyins, and discodermolide; polymerization inhibitors: vinca-alkaloids, crypto-phycins, halichondrins, estramustine | In clinical use for solid and hematopoietic tumors |
| CenpE | GSK923295 | Clinical trials in advanced solid tumors |
| Eg5 (KSP) | ARRY-520, MK-0731, Ispinesib (SB-715992), SB-743921 | Clinical trials in advanced solid tumors, lymphoma, and taxane-resistant cancer |
| KINASES | ||
| ATR | ETP-46464, NVP-BEZ235, Schisandrin B | Preclinical studies: synthetic lethal with mutations that induce replicative stress |
| Aurora kinases | AT9283, CYC116, GSK1070916A, MLN8237, PF-03814735, VX-680 | Clinical trials in solid tumors and hematopoietic malignancies |
| Cdks (1, 2, 4, 6) | AG-024322, EM-1421, LEE-011, LY2835219, P276-00, PD-0332991 | Clinical trials in solid tumors and leukemias |
| Chk1 | LY2606368, UCN01 | Clinical trials in combination with DNA damaging agents |
| Mps1 | AZ3146, MPS1-IN-1, MPS1-IN-2, NMS-P715, reversine | Preclinical studies |
| PARP | ABT-888, AZD-2281, AG014699, BMN-673, CEP 9722, MK 4827 | Clinical trials in solid tumors and hematopoietic malignancies |
| Plk1 | BI2536, BI6727, GSK461364, NMS-1286937, TKM-080301 | Clinical trials in solid tumors and hematopoietic malignancies |
| PHOSPHATASES | ||
| Cdc25 | ARQ-501, IRC 083864 | Preclinical studies and clinical trials in advanced solid tumors |
| UBIQUITIN LIGASES | ||
| APC/C | TAME | Preclinical studies |
Targeting DNA Damage Response Proteins
In the past decade, there has been a growing appreciation that many tumors cells carry mutations that disrupt their DDR. This characteristic is a major factor in establishing the resistance of tumors to chemotherapeutic agents, many of which work by causing DNA damage and triggering apoptosis through induction of DNA damage pathways. Therefore considerable attention has focused on designing cancer treatments that would be effective in cells with an impaired DDR. Because it is difficult to restore the function of mutant or missing proteins, the prevailing strategy is to identify drugs that would synergize with the defective DDR to selectively kill the tumor cells and not the normal cells. For example, inhibitors of poly(adenosine diphosphate [ADP]-ribose) polymerase (PARP) selectively kill cells that lack either Brca1 or Brca2.269,270 The rationale for this action is that these proteins provide two alternative repair mechanisms in response to DNA damage: homologous recombination (Brca1 and Brca2) and base excision repair (PARP). Therefore loss of one but not both of these pathways can be tolerated. As a second example, inhibition of Chk1 sensitizes p53 mutant cells to DNA damage.271 Because p53 is mutated in approximately half of all human tumors and the absence of p53 is a major predictor of poor response to classic chemotherapeutic agents, considerable efforts are being made to develop small molecular inhibitors of Chk1. Although the initial model proposed that this effect was due to the simultaneous abrogation of the G2 (Chk1) and G1 (p53) checkpoints, new evidence suggests that the toxicity of Chk1 inhibitors may be due to the generation of replicative stress (RS) in cells, with the less restrictive S phase entry due to the lack of p53.272 Chk1 inhibitors therefore might synergize with other mutations that promote a promiscuous S phase entry. A similar rationale applies to other molecules that target the RS response, such as inhibitors of the Chk1-activating kinase ATR. Indeed, ATR inhibitors display a selective effect in Myc-driven tumors that display high levels of RS as a consequence of the overexpression of Myc.273
Targeting the Mitotic Spindle and the SAC
Microtubule poisons, such as taxol and vinblastine, kill cancer cells by exploiting their effects in their spindle checkpoint.274 Taxanes, such as docetaxel or paclitaxel, are microtubule-stabilizing drugs widely used to treat breast and ovarian tumors, non–small-cell lung cancer, and Kaposi sarcoma. Vinca alkaloids, such as vinblastine or vincristine, belong to microtubule destabilizing compounds and have shown clinical efficacy against a broad range of hematologic malignancies. Both classes of drugs bind tubulin and inhibit microtubule dynamics, impairing the formation of a functional spindle. The SAC senses lack of proper attachment of kinetochores and arrests cells in prometaphase in the presence of these compounds. Thus inhibiting spindle dynamics results in abnormal chromosome segregation that frequently results either in aneuploidy or cell death. About 30 microtubule poisons are currently in clinical development.
The success of these molecules in the clinic led to the search for additional compounds that target specific regulators of microtubule dynamics rather than tubulin itself. In a pioneer screening, monastrol was identified as an inhibitor of the kinesin Eg5, a protein required for centrosome separation and the formation of a bipolar spindle.275 New drugs have been characterized that result in similar defects by inhibiting CenpE, another kinesin with critical roles in microtubule-to-kinetochore attachment.278–278 Inhibition of Aurora A or Plk1 also may be considered to be antispindle strategies because these kinases, although involved in other mitotic processes, are essential for centrosome maturation and separation and the formation of a bipolar spindle.61,276 In general, the inhibition of all these molecules results in an SAC-dependent arrest that impairs proliferation or viability of targeted cells.276
Similarly to what was discussed for the DNA damage checkpoint, a different concept is provided by the use of SAC inhibitors (checkpoint abrogation) to increase instability in cancer cells. Checkpoint kinases such as Mps1 and Aurora B (not considered to be a “core” checkpoint protein but involved in an error-correction mechanism) are required for the proper bipolar spindle attachment of chromosomes. Inhibition of these kinases results in rapid exit from mitosis without chromosome segregation generating tetraploid or aneuploid cells. Importantly, inhibition of these molecules prevents the mitotic arrest in the presence of microtubule poisons and therefore generates aberrant cells.61,279 It has been shown that the reduction in these checkpoint proteins makes tumor cells more sensitive than untransformed cells to low doses of spindle poisons.280 The efficacy of several Aurora or Mps1 small-molecule inhibitors currently is being tested in preclinical or clinical assays (Table 4-3).61,276,279
Targeting Mitotic Entry and Exit
As indicated in the earlier sections of this chapter, multiple enzymatic activities are required for mitosis. Cdk1 is the major engine in this process, and its activity is essential for mitosis. However, it has not been considered as a major target because its inhibition may have strong undesired effects.259 Yet some evidence suggests that it may be an interesting target in specific situations. Cdk1 is specifically required in cells transformed with Myc but not in cells transformed by other oncogenes. Inhibition of Cdk1 rapidly downregulates survivin expression, a protein required to avoid apoptosis in the presence of an excess of Myc. Cdk1 inhibition therefore results in Myc-dependent apoptosis and regression of Myc-dependent lymphoma and hepatoblastoma tumors.281 Cdk1 also is implicated in DNA repair by homologous recombination, and its inhibition results in impaired Brca1 activity. Cdk1 inhibition synergizes with PARP inhibitors, and partial inhibition of Cdk1 therefore may sensitize BRCA-proficient cancer cells to inhibit PARP, suggesting specific applications of Cdk1-targeting compounds in cancer cells.282 Cdk1 activators such as Cdc25 phosphatases are additional targets whose inhibition results in impaired Cdk activity and cell cycle arrest.18 The therapeutic utility of inhibiting additional mitotic kinases such as Mastl, Nek proteins, or Haspin, among others, is currently undergoing preclinical evaluation.3,276,283
Inhibiting mitotic entry or progression or preventing spindle dynamics may result in different outcomes, including cell death or the exit from the cell cycle without chromosome segregation.276 In fact, a rapid exit from mitosis is a major resistance mechanism that generates viable cells in the presence of mitotic poisons. This finding led to the evaluation of mitotic exit pathways as new targets for therapy.284 In fact, genetic ablation of APC/C-Cdc20 completely prevents mitotic exit, and these mutant cells arrest in mitosis until they die.50 This action results in complete tumor repression in mouse models, and a first generation of APC/C inhibitors is currently available to test this strategy in preclinical studies (Table 4-3).50,285
Targeting Aneuploidy
Aneuploidy is a hallmark of cancer and is now considered as a highly attractive therapeutic target.206,286 Most tumor cells are aneuploid, whereas this abnormality is infrequent (although this has not been precisely established) in wild-type cells. This specificity is especially interesting given the problems that most therapeutic strategies that target mitosis have in specifically inhibiting tumor cells. Aneuploid cells display specific defects in cell cycle kinetics, growth rate, metabolism, and the response to specific stresses.287,288 These defects can be exploited by targeting specific cellular pathways that protect cells from the deleterious effects of aneuploidy. For instance, inhibiting the proteotoxic and metabolic stress pathways specifically reduces the viability of aneuploidy cells because of the unbalanced load of proteins in these cells.289 As previously described, targeting the mitotic checkpoint may increase the levels of aneuploidy in the tumor cells, which opens the opportunity to combine different types of antimitotic strategies to further induce CIN and aneuploidy and inhibit the pathways that cancer cells use to tolerate this abnormal state.286
Summary
During the past several decades, investigators have uncovered a wealth of information about the proteins that control the division of human cells. A key finding is that deregulation of the cell cycle machinery and/or its checkpoints is a universal alteration in human cancer.1,2,290 Because the basic machinery that controls the cell cycle is similar in all cell types, the hope is that common strategies will be developed against a wide variety of cancers. Even though several of the currently used anticancer therapies target nonselective and nonmechanism-based targets, their effectiveness, albeit limited in many cases, is likely due to the fact that they ultimately target cell cycle regulatory or DDR-signaling pathways, the status of which is different in normal cells versus tumor cells. Identifying all the components of the cellular machinery that control the cell cycle both positively and negatively is vital to the continued development of anticancer agents that can preferentially eliminate cancer cells and minimize the toxicity to normal tissues. The complexity of the human genome makes this task difficult because many members of the major protein families that regulate the cell cycle have not been studied thus far. In addition, our knowledge of the in vivo relevance of these proteins in different tissues is limited. It is obvious from the mouse models studied that the efficacy of inhibiting specific cell cycle targets depends on the tumor type and the oncogenic environment in each specific tumor. As our understanding of cell cycle regulation and checkpoint signaling improves, the goal is to use this knowledge in the design of mechanism-based therapeutics that will bring anticancer therapy to a new level. There can be little doubt of the value of targeting the cell cycle in drug discovery.