Chronic Myeloid Leukemia
Hagop Kantarjian and Jorge Cortes
• About 5000 cases of chronic myeloid leukemia (CML) occur per year in the United States; CML represents 15% of all leukemias.
• Median age is 55 to 60 years at diagnosis.
• Common findings include fatigue, anemia, abdominal discomfort, splenomegaly, and leukocytosis.
• At diagnosis 40% to 50% of patients are asymptomatic.
• White blood cell count is usually greater than 50 × 109 cells/L, with a left-shifted differential, basophilia, and thrombocytosis.
• Chronic myelomonocytic leukemia
• Other myeloproliferative disorders
• History and physical examination, complete blood cell count with differential and platelet count, and chemistries
• Bone marrow aspiration and biopsy
• Testing for the presence of the Philadelphia (Ph) chromosome by cytogenetic analysis; testing for the presence of the BCR-ABL fusion gene by fluorescent in situ hybridization (FISH) and by quantitative polymerase chain reaction (qPCR) analysis
• Hydroxyurea or a BCR-ABL tyrosine kinase inhibitor (TKI; e.g., imatinib mesylate, nilotinib, dasatinib) is given initially to control leukocytosis and thrombocytosis.
• Imatinib induces a complete hematologic and cytogenetic response in most patients; estimated 8- to 10-year survival rate is 85% (93% if only CML-related deaths considered).
• Randomized studies of frontline therapy with second-generation TKIs (e.g., nilotinib, dasatinib) show better results than those of imatinib.
• New TKIs (e.g., dasatinib, nilotinib, bosutinib, ponatinib) are active after imatinib failure.
• Allogeneic stem cell transplantation may be curative but is associated with considerable morbidity and mortality.
Introduction
Chronic myeloid (or myelogenous) leukemia (CML) is a clonal hematopoietic stem cell disorder. It is characterized by overproduction of myeloid cells, a result of excessive proliferation, and reduced apoptosis. Clinical findings include fatigue, splenomegaly, leukocytosis, and anemia. Basophilia and thrombocytosis are common.3–3 CML is defined by the presence of a characteristic cytogenetic abnormality, the Philadelphia (Ph) chromosome, a reciprocal balanced translocation between the long arms of chromosome 9 and 22, t(9;22)(q34;q11.2). This results in a BCR-ABL fusion gene, which is causally related to the disease pathophysiology.6–6
Incidence, Epidemiology, and Etiology
CML accounts for 15% of cases of leukemia in the United States. There is a slight male preponderance (male-to-female ratio, 1.6 : 1). Its annual incidence is about 1.5 cases per 100,000 individuals. About 5,000 cases of CML are diagnosed annually. This incidence has not changed during the past few decades, and it increases with age. The median age at diagnosis is 55 to 65 years; it is uncommon in children and adolescents; only 2.7% of patients with CML are younger than 20 years.7
Before imatinib therapy, the prevalence of CML was about 25,000 cases in the United States. Now that the annual mortality after imatinib therapy has been reduced to 2% or less, the prevalence of CML will continue to rise, reaching a plateau (in the next 20 years) at about 180,000 cases (when the annual incidence will equal the annual mortality: based on a hazard risk of annual mortality of 1.5 for patients with CML compared with age-matched healthy individuals).8 This increase will change CML from an uncommon disorder to a prevalent one.
There are no known familial associations in CML. Its risk is not increased in monozygotic twins or in relatives of patients with CML. There are no known common etiologic agents identified in CML. Ionizing radiation (exposure to nuclear bombs or accidents; radiation treatment of ankylosing spondylitis and cervical cancer) has increased the risk of CML. Its peak incidence is 5 to 10 years after exposure and is dose related. The risk of CML is not increased in individuals working in the nuclear industry. Radiologists working without adequate protection (before 1940) had an increased risk of developing CML, but no such risk has been found in recent studies. Benzene exposure increases the risk of acute myelogenous leukemia but not of CML. CML is not a frequent secondary leukemia after treatment of other cancers with radiation and/or alkylating agents.9,10
Pathogenesis
Molecular Pathogenesis
The Ph chromosome abnormality is present in more than 90% of patients with typical CML (Fig. 101-1). It results from a balanced reciprocal translocation between the long arms of chromosomes 9 and 22, t(9;22)(q34;q11.2). It is found in hematopoietic cells but not in other human cells. Its origin is close to the pluripotent stem cell, and it is present in erythroid, myeloid, monocytic, and megakaryocytic cells, less commonly in B lymphocytes, rarely in T lymphocytes, but not in marrow fibroblasts. The breakpoint of chromosome 9 results in translocation of the cellular oncogene ABL1 to a region on chromosome 22 coding for the major breakpoint cluster region (BCR). ABL1 is a homolog of V-ABL, the Abelson virus that causes leukemia in mice. This juxtaposes a 5′ portion of BCR to a 3′ position of ABL1 and produces a new hybrid oncogene, BCR-ABL. This codes for a novel BCR-ABL oncoprotein of molecular weight 210 kDa (p210BCR-ABL). The p210BCR-ABL oncoprotein results in uncontrolled kinase activity, which causes excessive proliferation and reduced apoptosis of CML cells,13–13 leads to a growth advantage of CML cells to normal cells, and suppresses normal hematopoiesis. The normal stem cells, although suppressed, persist and reemerge after effective therapy of CML.
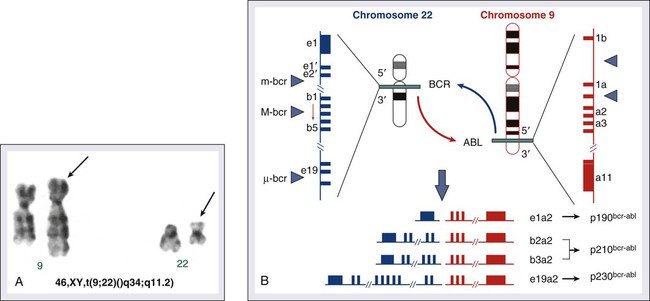
In Ph-positive acute lymphocytic leukemia, the breakpoint in BCR often occurs in a more centromeric region called the minor BCR region (mBCR). This produces a smaller BCR gene opposing ABL1, and therefore a fusion gene, messenger RNA, and BCR-ABL oncoprotein (p190BCR-ABL) of smaller sizes. A third rare breakpoint distal to the major BCR region, called µ-BCR, produces a p230BCR-ABL hybrid oncoprotein associated with a very indolent course of CML (see Fig. 101-1).14
What causes the BCR-ABL molecular rearrangement is unknown. Molecular techniques that amplify detection of BCR-ABL to 1 in 108 detect it in marrow cells of 25% to 30% of normal adults, in 5% of infants, but not in cord blood.15 Because CML develops in only 1.5 of 100,000 individuals (i.e., 1 to 2 per 25,000 to 30,000 individuals who express BCR-ABL in their bone marrow), additional molecular events or lack of immune recognition of the clonal cells may contribute to the development of CML.
The fusion BCR-ABL gene and the p210 oncoprotein can be found in some patients with typical morphologic CML, in whom the t(9;22)(q34;q11.2) is not identified. These patients have a response to therapy and a survival rate similar to those with Ph-positive cases. Patients with Ph-negative and BCR-ABL-negative CML (see later discussion) constitute a different entity (atypical CML) and have a worse prognosis.16,17 The molecular pathophysiology of CML transformation is poorly understood. Several molecular events have been associated with CML transformation, including point mutations or deletions in the TP53 tumor suppression gene, MYC amplification, deletions in the CDKN2A tumor suppressor gene, alteration of the RB retinoblastoma gene, and others. The causal relation of these events to transformation is not clear.
Animal Models of Chronic Myeloid Leukemia
Experimental models have established a causal relationship between the BCR-ABL molecular events and the development of CML. Transgenic mice that express Bcr-Abl developed acute leukemia. A Bcr-Abl–expressing retrovirus used to infect murine bone marrow cells that later repopulate irradiated mice resulted in myeloproliferative disorders, including a CML-like syndrome.20–20 Bcr-Abl, under the control of a tetracycline-repressible promoter, expressed in mice, resulted in a lymphoid leukemia that reversed in the presence of tetracycline,21 attesting to the leukemic potential of Bcr-Abl as a sole oncogenic abnormality. The recapitulation of CML-like disorders in animal models mediated by the Bcr-Abl molecular events established them as a root cause for CML and a legitimate molecular target for therapeutic interventions with TKIs.
Clinical Presentation
Chronic Phase
Common signs and symptoms of CML, when present, result from anemia and splenomegaly. These include fatigue, weight loss, malaise, early satiety, and left upper quadrant fullness or pain (Table 101-1). Rare manifestations include bleeding (associated with a low platelet count and/or platelet dysfunction), thrombosis (associated with thrombocytosis and/or marked leukocytosis), gouty arthritis (from elevated uric acid levels), priapism (usually with marked leukocytosis or thrombocytosis), retinal hemorrhages, and upper gastrointestinal ulceration and bleeding (from elevated histamine levels due to basophilia). Leukostatic symptoms (dyspnea, drowsiness, loss of coordination, confusion) caused by sludging in the pulmonary or cerebral vessels, are uncommon in chronic-phase CML despite white blood cell (WBC) counts exceeding 100 × 109 cells/L.
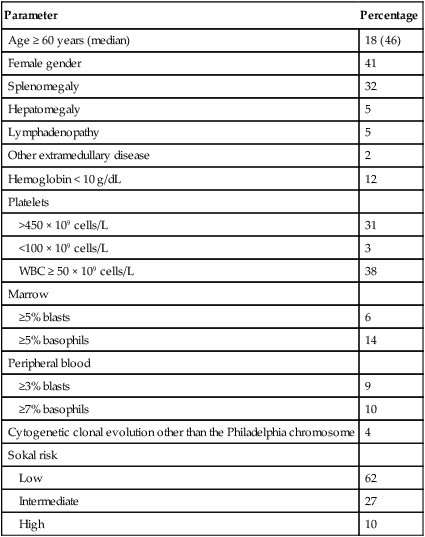
Table 101-1
Features of Patients with Newly Diagnosed Philadelphia Chromosome–Positive Chronic Myelogenous Leukemia in Chronic Phase Referred to MD Anderson Cancer Center (1990–present; N = 1469)
| Parameter | Percentage |
| Age ≥ 60 years (median) | 18 (46) |
| Female gender | 41 |
| Splenomegaly | 32 |
| Hepatomegaly | 5 |
| Lymphadenopathy | 5 |
| Other extramedullary disease | 2 |
| Hemoglobin < 10 g/dL | 12 |
| Platelets | |
| >450 × 109 cells/L | 31 |
| <100 × 109 cells/L | 3 |
| WBC ≥ 50 × 109 cells/L | 38 |
| Marrow | |
| ≥5% blasts | 6 |
| ≥5% basophils | 14 |
| Peripheral blood | |
| ≥3% blasts | 9 |
| ≥7% basophils | 10 |
| Cytogenetic clonal evolution other than the Philadelphia chromosome | 4 |
| Sokal risk | |
| Low | 62 |
| Intermediate | 27 |
| High | 10 |
The bone marrow is hypercellular with marked myeloid hyperplasia. The myeloid-to-erythroid ratio is usually 15 : 1 to 20 : 1. About 15% of patients have 5% or more blast cells in the peripheral blood or bone marrow at diagnosis. Increased reticulin fibrosis is common (30% to 40% grade 3/4 reticulin fibrosis by silver staining), but collagen fibrosis is rare at presentation.22 Interestingly, whereas the “spent phase” of myelofibrotic CML was commonly reported in the past (with busulfan therapy) as an end-stage CML event, it has become uncommon in the eras of IFN-α and imatinib therapy. Imatinib effectively reduces myelofibrosis in CML.23,24
Accelerated and Blastic Phases
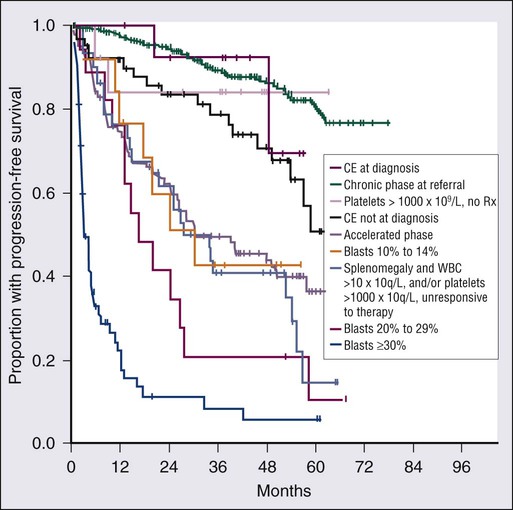
The definitions of accelerated and blastic phases of CML are shown in Table 101-2. In most patients the transformation from chronic to advanced phase is insidious, with the disease becoming more difficult to control. This phase is referred to as the accelerated phase. The criteria of accelerated phase are variable. In one study, features that correlated with a median survival of 18 months or less included a blast percentage equal or greater than 15%, blasts plus promyelocytes equal or greater than 30%, basophils equal or greater than 20%, a platelet count less than 100 × 109 cells/L unrelated to therapy, and cytogenetic clonal evolution.25 Features of accelerated-phase CML should be revisited because some (e.g., clonal evolution more favorable, blasts >15% less favorable) have different prognostic implications from recent studies (Fig. 101-2).26 Five to 10 percent of patients are seen in the accelerated phase; these patients have an estimated 8-year survival rate of 75% when treated frontline with TKIs.27 The accelerated phase is also associated with worsening anemia and splenomegaly, organ infiltration (liver, lymph nodes, skin, bones, or other tissues), and constitutional symptoms (aches, fever, malaise, weight loss).
Table 101-2
Definitions of the Accelerated and Blastic Phases of Chronic Myelogenous Leukemia
| Criteria | MD Anderson Cancer Center* | International Bone Marrow Transplant Registry | World Health Organization |
| ACCELERATED PHASE | |||
| Percent blasts | 15-29 | 10-29 | 10-19 |
| Percent blasts + promyelocytes | ≥30 | ≥20 | NA |
| Percent basophils | ≥20 | ≥20% (basophils + eosinophils) | ≥20 |
| Platelets (×109/L) | <100 | Unresponsive ↑, persistent ↓ | <100 or > 1000 unresponsive |
| Cytogenetics | CE | CE | CE not at diagnosis |
| WBC | NA | Difficult to control, or doubling in < 5 days | NA |
| Anemia | NA | Unresponsive | NA |
| Splenomegaly | NA | Increasing | NA |
| Other | Chloromas, myelofibrosis | Megakaryocyte proliferation, fibrosis | |
| BLASTIC PHASE | |||
| 30% or more blasts; or extramedullary blastic disease, except for the World Health Organization classification, which requires 20% or more. | |||
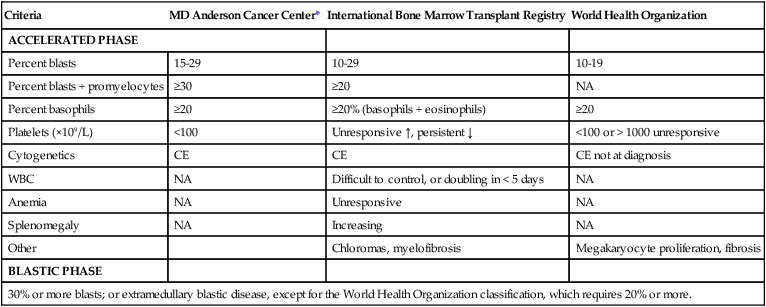
CE, clonal evolution; NA, not applicable; WBC, white blood cells.
*These criteria were also used in all the IFN-α and tyrosine kinase inhibitors studies.
Diagnosis
Reverse-transcriptase polymerase chain reaction (RT-PCR) amplifies the region around the junction between BCR and ABL1. It is highly sensitive for the detection of minimal residual disease. PCR testing can either be qualitative, providing information about the presence of the BCR-ABL transcript, or quantitative, assessing the amount of BCR-ABL transcripts. Qualitative PCR may be useful for diagnosis of CML; quantitative PCR (qPCR) is ideal for monitoring response to therapy and is usually performed by real-time PCR. Simultaneous peripheral blood and marrow PCR studies show a high level of concordance. False-positive and false-negative results can happen with PCR. False-negative results may be from poor-quality RNA or failure of the reaction or from atypical transcripts (e.g., b3a3); false-positive results can be due to contamination. A 0.5- to 1-log coefficient of variability in some samples can occur depending on testing procedures, sample handling, and laboratory experience.28,29
Diagnostic and Monitoring Procedures
The improved rates of complete cytogenetic response and of molecular response with imatinib now require new techniques that measure these responses more accurately (rather than relying on evaluation of only 20 metaphases by routine cytogenetic studies), with less painful procedures (peripheral blood rather than marrow samples), and with methods that measure minimal disease below the level of detection by routine karyotypic analysis (molecular studies). FISH studies can assess rapidly the disease status in 200 cells; this assessment may be done in cells in interphase and can be performed on blood specimens. Real-time PCR studies usually measure the ratio of the abnormal message, BCR-ABL, to a control gene (e.g., ABL). Because this is variable by laboratory and with time, efforts are ongoing to standardize the measurement of BCR-ABL transcripts in relation to an international standard (IS).30 Recent monitoring procedures in CML have emphasized replacing bone marrow studies (cytogenetics) with peripheral blood studies (FISH, qPCR) as more reliable and measuring deeper levels of response to TKIs. FISH studies are reliable in patients in complete cytogenetic response with a strong concordance between negative FISH studies and complete cytogenetic response. However, the concordance between the percent of Ph positivity by FISH and cytogenetic studies is not perfect. Recently, landmark studies have emphasized the importance of BCR-ABL transcripts of 10% or less as early indicators for response and long-term prognosis and that the finding of BCR-ABL transcripts of 1% or less (IS) (grossly equivalent to a complete cytogenetic response) is an important response criterion at 1 year or later. In general, BCR-ABL transcripts less than 10% (IS) are equivalent to a partial cytogenetic response with Ph-positive metaphases less than or equal to 35%; and BCR-ABL transcripts less than or equal to 1% (IS) are equivalent to a complete cytogenetic response.31–34 A BCR-ABL transcript of less than or equal to 0.1% (IS) (approximately a 3-log reduction of disease from a standardized baseline) has been associated with a low risk of relapse and with favorable PFS. A negative qPCR analysis (undetectable BCR-ABL transcripts; 4.5-log reduction or more) may be technique dependent and is referred to as a complete molecular response.
Monitoring response to imatinib-based therapy may use different approaches depending on the investigators’ experience, whether changes of residual disease affect subsequent therapy, availability of methodologies, and other factors. In general, patients with newly diagnosed CML require an initial bone marrow analysis (to evaluate the percentage of blasts and basophils and whether clonal evolution is present) and then once a year (to detect cytogenetic abnormalities in both the Ph-positive and Ph-negative cell). Some experts recommend bone marrow studies every 3 to 6 months in the first year because of the prognostic significance of the Ph-positive status from routine marrow cytogenetic studies. Practically, peripheral blood FISH studies every 3 to 4 months provide a reasonable estimate of the response profile in the first year. Once Ph-positive cells are 0% by FISH, the complete cytogenetic response can be confirmed by a bone marrow analysis with routine cytogenetics and subsequent monitoring of minimal residual disease performed by peripheral FISH and qPCR studies. If BCR-ABL transcripts are less than 0.1% (IS), qPCR may be used as the single monitoring procedure. The simultaneous use of FISH and qPCR allows for verification of concordance of results in patients who are in complete cytogenetic response but not in major molecular response. In a patient in stable complete cytogenetic response, FISH and qPCR studies may be performed every 6 months. If concerns arise regarding changes in qPCR values, the study can be repeated more frequently (e.g., every 3 months). Some investigators suggest a twofold or 0.5-log increase in transcript levels may correlate with the development of mutations or with relapse.30,31 However, these analyses stem from individual laboratories with significant expertise and may not apply to routine practice. In general, in a patient in complete cytogenetic response, an increase of BCR-ABL transcripts should be confirmed before a change of therapy is considered, if at all. Significant fluctuations in BCR-ABL transcripts may also suggest poor treatment compliance and require discussions with the patient regarding compliance with TKI therapy.
Important Landmarks for Response or Failure to TKI Therapy
Among patients receiving frontline imatinib therapy, an optimal response requires achievement at least a partial cytogenetic response (Ph-positive metaphases ≤ 35%) by 6 months of therapy and a complete cytogenetic response by 1 year of therapy. Recent studies have suggested that, with qPCR, an optimal response would be a reduction of the BCR-ABL transcripts to less than or equal to 10% (IS) at 3 to 6 months and to less than or equal to 1% (IS) after 1 year of therapy. Failure to achieve BCR-ABL transcripts of less than or equal to 10% (IS) or a partial cytogenetic response at 3 to 6 months, and failure to achieve and maintain a complete cytogenetic response beyond 1 year of imatinib therapy, indicate a worse prognosis. It has not been established yet whether a change of therapy at the early time points (3 to 6 months) improves outcome.34–34 When second-generation TKIs (e.g., nilotinib, dasatinib) are used as frontline therapy, earlier responses are expected.35 An optimal response is considered to be achievement of complete cytogenetic response after 3 to 6 months of therapy. Patients not in complete cytogenetic response by that time have a worse event-free survival, but their survival is still excellent (estimated 3-year survival > 90%),35 therefore precluding the need to consider allogeneic SCT in this minority of patients (5% to 10% of total population) but advising closer observation to identify early patients who may require a change of therapy (e.g., to newer-generation TKIs such as ponatinib or allogeneic SCT).
Prognosis
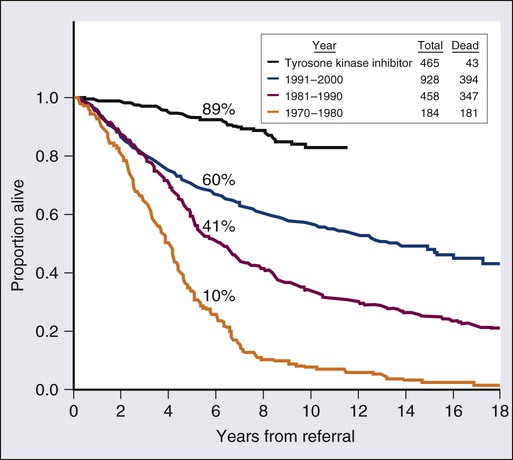
Prognosis in CML has changed drastically over the past 10 years. Before the availability of TKIs, the median survival of patients with CML in chronic phase (85% to 90% of newly diagnosed patients) was 6 to 7 years with IFN-α with or without cytarabine. For candidates for allogeneic SCT, the expected 20-year survival rate was 40% to 50% in younger patients with a matched related donor. With TKI therapy the estimated 10-year survival rate in chronic phase is more than 80%.27,36 With an annual mortality of 1% to 2% in the first 10 years, the estimated median survival may exceed 25 years (Fig. 101-3).27,36 Thus, chronic-phase CML may have now changed to an indolent disorder in which most patients may survive normally with continued oral TKI therapy. Therapy with TKIs has also changed the prognosis in accelerated phase.27 Patients with clonal evolution as the only sign of accelerated disease have an estimated 5-year survival rate of 80%.37 Survival in blastic phase is also better with imatinib therapy, but only modestly so (median survival improved from 3 to 6 months to 12 to 15 months).27 Prognosis of lymphoid blastic-phase disease is slightly more favorable, with a response rate to anti–acute lymphocytic leukemia chemotherapy with TKIs of 60% and a median survival of 12 to 18 months.
Before imatinib, several prognostic models (Sokal, Hasford) divided patients in chronic phase into low-, intermediate-, and high-risk groups based on pretreatment adverse prognostic factors, including older age, splenomegaly, thrombocytosis, higher percentage of blasts and basophils, and presence of cytogenetic clonal evolution.38,39 Therapy with imatinib and other TKIs has now altered the prognostic significance of several of these factors, including deletion of derivative chromosome 9, age, and myelofibrosis.23,24,40 Myelofibrosis, in fact, resolves quite effectively with imatinib therapy.24 Currently, the most relevant prognostic factor of imatinib therapy is the degree of cytogenetic and molecular response to initial imatinib therapy (in the first 3 to 12 months).32–35 Thus, the introduction of TKI therapy in CML will require new definitions of the accelerated phase and understanding of the adverse factors and risk models in CML. Recent prognostic factor analyses of patients on TKI therapy suggest that the three persistent independent adverse factors include severe basophilia, high peripheral blast percent, and massive splenomegaly. A recently proposed European risk score (EUTOS score) categorizes patients into two prognostic groups based on the percent of peripheral basophils and spleen size (in centimeters below the costal margin). This model has not been consistently reproduced.41
Management
Chronic-Phase CML
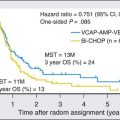
In the 1980s to 1990s, the two frontline therapies in CML were IFN-α–based regimens and allogeneic SCT.42 The positive results with imatinib therapy led to its establishment as standard frontline therapy. Allogeneic SCT is now delayed as a second- or third-line strategy after failure of imatinib and/or other TKIs. Two recent randomized trials of standard imatinib (400 mg orally daily) versus second-generation TKIs using nilotinib in two different dose schedules (300 or 400 mg orally twice daily) or dasatinib (100 mg in a single dose daily) showed nilotinib and dasatinib to be associated with better rates of early surrogate end points for long-term survival, including higher rates of complete cytogenetic response, major molecular response, and complete molecular response. The newer TKIs were associated with lower rates of transformation to accelerated or blastic phases and with lower rates of events and progression. With the current follow-up, the survival rates with imatinib versus second-generation TKIs are not different. Based on these results, the U.S. Food and Drug Administration (FDA) granted approval in 2009 for nilotinib and dasatinib to be used as frontline therapies in newly diagnosed chronic CML.43,44
Imatinib, Nilotinib, and Dasatinib
The role of imatinib therapy in CML was established in a series of pivotal trials of imatinib after IFN-α failure in chronic, accelerated, and blastic phases of CML and later in a frontline international randomized study of interferon plus cytarabine versus STI571 (the IRIS trial).45,46
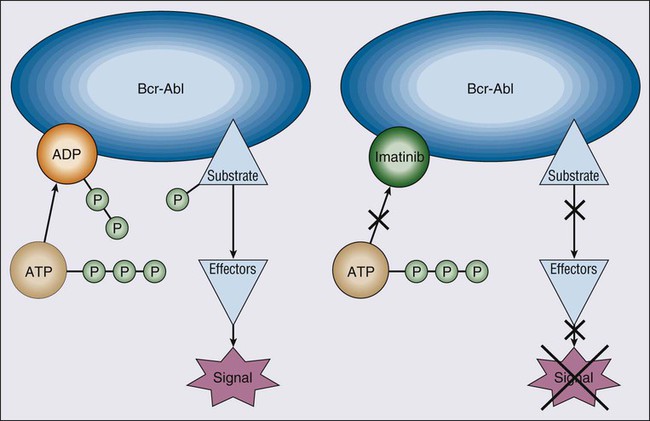
Imatinib mesylate, a 2-phenylaminopyrimidine derivative, binds to the canonical adenosine triphosphate (ATP)-binding site of the ABL kinase domain. It blocks phosphorylation of tyrosine residues on substrate protein. Blocking ATP binding inactivates the ABL kinase, because it cannot transfer phosphate to its substrate. Inhibiting phosphorylation prevents activation of signal transduction pathways that cause CML (Fig. 101-4). Imatinib inhibits several tyrosine kinases, including p210BCR-ABL, p190BCR-ABL, v-ABL, c-ABL, c-Kit, and platelet-derived growth factor receptor (PDGF-R).
In the IRIS study, 1106 patients with newly diagnosed CML were randomized to imatinib 400 mg orally daily versus IFN-α plus cytarabine. Imatinib was associated with significantly higher rates of major (Ph-positive metaphases ≤ 35%; 85% vs. 22%) and complete cytogenetic responses (74% vs. 8%), and lower rates of progression (3% vs. 20%) and transformation (1.5% vs. 7%) after 12 months of therapy.45 An update of the 8-year follow-up in the 553 patients treated with imatinib continued to show excellent results: projected complete cytogenetic response rate (single-time response) of 83%; annual rate of progression to accelerated-blastic phase of 4% in the first 2 to 3 years and 1% or less later on imatinib therapy; and annual mortality rate of 1% to 2%. At the 8-year follow-up, 304 patients (55%) were still on imatinib. Rates of transformation and mortality seem to have been reduced in years 4 through 8 (≤2%) compared with the first 3 years (5% to 8%). The estimated 8-year survival rate was 85%. The estimated 8-year survival rate excluding non-CML–related deaths was 93%.47 Survival was also better with imatinib versus IFN-α plus cytarabine (survival rates 89% vs. 86%), but the difference was not drastic, because of the crossover design of the IRIS study and the commercial availability of imatinib, both resulting in a change from IFN-α plus cytarabine to imatinib in about 90% of patients within a median of 9 months from start of therapy. Comparison of imatinib results to historical experiences demonstrates more significant survival differences.27,36
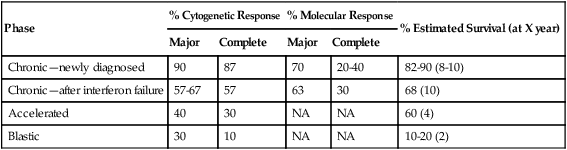
Among patients in late chronic-phase CML after IFN-α failure receiving imatinib, the long-term results show a complete cytogenetic response rate of 41% and estimated 6- and 10-year survival rates of 76% and 68%, respectively.48,49 In accelerated-phase CML the complete cytogenetic response rate was 40% and the estimated 4-year survival rate was 60%.50 In blastic-phase CML, response rates were lower and more transient and the median survival duration only 6 to 12 months (Table 101-3).51
Table 101-3
Results of Imatinib Therapy in Chronic Myelogenous Leukemia
| Phase | % Cytogenetic Response | % Molecular Response | % Estimated Survival (at X year) | ||
| Major | Complete | Major | Complete | ||
| Chronic—newly diagnosed | 90 | 87 | 70 | 20-40 | 82-90 (8-10) |
| Chronic—after interferon failure | 57-67 | 57 | 63 | 30 | 68 (10) |
| Accelerated | 40 | 30 | NA | NA | 60 (4) |
| Blastic | 30 | 10 | NA | NA | 10-20 (2) |
In the IRIS study, the cumulative incidence of major molecular response was 70% and of disappearance of BCR-ABL transcripts 30% to 50%.47 Achieving a major molecular response in complete cytogenetic response at 12 months was associated with estimated 5-year rates of transformation-free survival and survival rates of 100%.47 Disappearance of BCR-ABL transcripts was initially observed in a minority of patients (5%) but is now reported with longer-term follow-ups in 30% to 50% of patients.52 This rate depends on the sensitivity of the qPCR. In general, a reduction of BCR-ABL transcripts by 4.5 log or more (or BCR-ABL transcripts ≤ 0.0032 [IS]) may reach levels below detection in most molecular laboratories.
Higher doses of imatinib of 600 to 800 mg daily may overcome resistance to standard-dose imatinib in some patients.52 In patients with accelerated-phase CML, high-dose imatinib was associated with higher response rates and longer survival.50
Imatinib is associated with mild-to-moderate side effects, including nausea, vomiting, diarrhea, rashes, muscle cramps, bone aches, periorbital or leg edema, and weight gain. Serious but uncommon side effects (1% to 2%) include hepatic, renal, or cardiopulmonary dysfunction. These are manageable with dose reductions or treatment interruptions.53 Drug-related myelosuppression occurs in 10% to 30% of patients in newly diagnosed CML. This is managed with brief treatment interruptions and/or dose modifications or with growth factors (e.g., erythropoietin for anemia, filgrastim for neutropenia). Chromosomal abnormalities may appear in the Ph-negative cells in 5% to 10% of responding patients. These include abnormalities observed in myelodysplastic syndrome and acute myeloid leukemia, such as chromosome 5 or 7 abnormalities, trisomy 8, 20q−, and others. These chromosomal abnormalities are probably the result of unmasking of a fragile stem cell prone to development of CML or to genetic instability. Such changes disappear spontaneously in up to 70% of cases. Evolution to a Ph-negative myelodysplastic syndrome or acute myeloid leukemia is rare and probably part of the new natural course of CML.54
A phase III frontline study in newly diagnosed CML (Evaluating Nilotinib Efficacy and Safety in Clinical Trials–Newly Diagnosed Patients [ENESTnd]) compared the standard dose of imatinib of 400 mg orally daily with two schedules of nilotinib 400 mg orally twice daily (original approved dose schedule) and a lower-dose schedule of 300 mg orally twice daily. This study demonstrated a significant improvement in the primary end point of the study, achievement of major molecular response by 12 months of therapy, favoring nilotinib, resulting in the FDA approval of nilotinib 300 mg orally twice daily as a standard of care for patients with newly diagnosed chronic-phase CML.44 With a minimum follow-up of 3 years, the updated results continue to show better results with nilotinib in relation to achievement of major molecular response (70% to 73% with nilotinib vs. 53% with imatinib; P < .0001), deeper molecular responses, defined as MR4.0 (BCR-ABL transcript levels < 0.01%) (IS) and MR4.5 (BCR-ABL transcript levels < 0.0032%) (IS). Importantly, the incidence of disease transformation to accelerated or blastic phase was lower with nilotinib (3.2% with nilotinib 300 mg twice a day, 2.1% with nilotinib 400 mg twice a day) compared with imatinib (6.7%). Survival rates were not significantly different with nilotinib versus imatinib. A similar phase III frontline study comparing imatinib 400 mg orally daily to dasatinib 100 mg orally daily (DASISION trial) also showed a significant difference in the primary study end point, confirmed complete cytogenetic response by 12 month of therapy, favoring dasatinib, and thus resulting in the FDA approval of dasatinib 100 mg daily as a standard of care for newly diagnosed CML. The longer follow-up in the study continues to confirm the benefit of dasatinib in relation to early surrogate end points of long-term outcome, including rates of major molecular response and transformation to accelerated- and blastic-phase disease.43
Therapy with imatinib 400 mg daily, nilotinib 300 mg orally twice daily (on an empty stomach), or dasatinib 100 mg single daily dose is started as soon as the diagnosis of CML is established. Allopurinol is recommended until the WBC count is less than 10 × 109 cells/L. Tumor lysis syndrome is rare, except in occasional patients in advanced-phase disease who require hydration and closer monitoring. With TKIs, the WBC and platelet counts decrease in the first 2 to 4 weeks and normalize within 1 to 2 months. Complete blood cell counts are recommended weekly during the first month of therapy and then every 1 to 2 months. For patients with accelerated- or blastic-phase CML, complete blood cell counts should be performed more frequently and as indicated clinically. Therapy with TKIs should be continued indefinitely. Discontinuation of imatinib treatment even among patients with durable complete molecular responses (>2 years) has been associated with molecular relapse in about half.55 High-dose imatinib (i.e., 600 to 800 mg daily) may be considered in CML transformation or Ph-positive acute lymphocytic leukemia (in combination with chemotherapy) or in patients with cytogenetic relapse while undergoing therapy with imatinib.
Allogeneic Stem Cell Transplantation
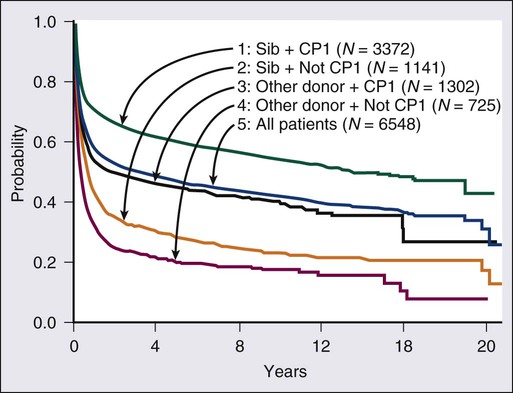
Allogeneic SCT is curative in selected patients with CML. It is most effective during the chronic phase. Among patients in first chronic phase who receive a transplant from a matched sibling, the estimated 20-year survival rate is 40% to 50% (Fig. 101-5). The report from the International Bone Marrow Transplant Registry (IBMTR), compiling data from more than 6000 patients undergoing transplants, showed a 5-year survival rate of 60% for first chronic-phase/sibling donor patients but a 20-year survival rate of 40% to 45%.56 Most of the 10% to 15% additional deaths between years 5 and 20 were due to transplant-associated complications rather than CML relapse. Chronic morbidities for SCT include graft-versus-host disease (GVHD), infertility, cataracts, hip necrosis, second cancers, and GVHD-associated organ damage (pulmonary, hepatic) or immune-mediated complications. Transplant-related mortality ranges from 5% to 50%, depending on several factors, including patient age, whether the donor is related or unrelated, the degree of matching, and other factors, such as positivity for cytomegalovirus (CMV), preparative and post-transplant regimens, and institutional expertise.56–61 Disease-free survival rates with related allogeneic SCT are 40% to 80% in chronic-phase CML. The disease-free survival rates are 60% to 80% among patients younger than 30 to 40 years of age and 30% to 40% in patients older than age 50 years. Disease-free survival rates range from 30% to 60% in accelerated-phase CML and from 5% to 30% in blastic-phase CML. Patients with clonal evolution as the only accelerated-phase criterion have disease-free survival rates of 60%. Patients receiving a transplant in second chronic-phase CML have favorable disease-free survival rates of about 40%.
Improving Results
Allogeneic SCT is safest and results are best in younger patients with an HLA-identical sibling donor. Improvement of SCT results can be achieved through (1) better molecular characterization of the HLA phenotype and genotype and matching of unrelated donors, (2) improved management of complications, including viral infections (e.g., CMV), (3) performing SCT before CML evolution to accelerated or blastic phases, (4) treatment of relapse after SCT before overt disease occurs (i.e., at time of molecular relapse), and (5) safer and better conditioning regimens.56–61
Monitoring Outcome and Treatment of Relapse
Relapse can be managed effectively with withdrawal of immune suppressive therapy, imatinib or another TKI, donor lymphocyte infusions, IFN-α, or a second SCT. Imatinib results in remission rates of 40% to 60% if the relapse is molecular or cytogenetic in chronic phase.62 Imatinib or other TKIs are currently preferred as a first salvage option after allogeneic SCT relapse to donor lymphocyte infusions, because these infusions may exacerbate GVHD. TKIs are less effective if CML relapse is hematologic or in transformation. Donor lymphocyte infusions induce durable remission rates of 60% with molecular or cytogenetic relapse but may exacerbate GVHD (which can be fatal) and result in intense myelosuppression in 20% of patients.
Imatinib Resistance
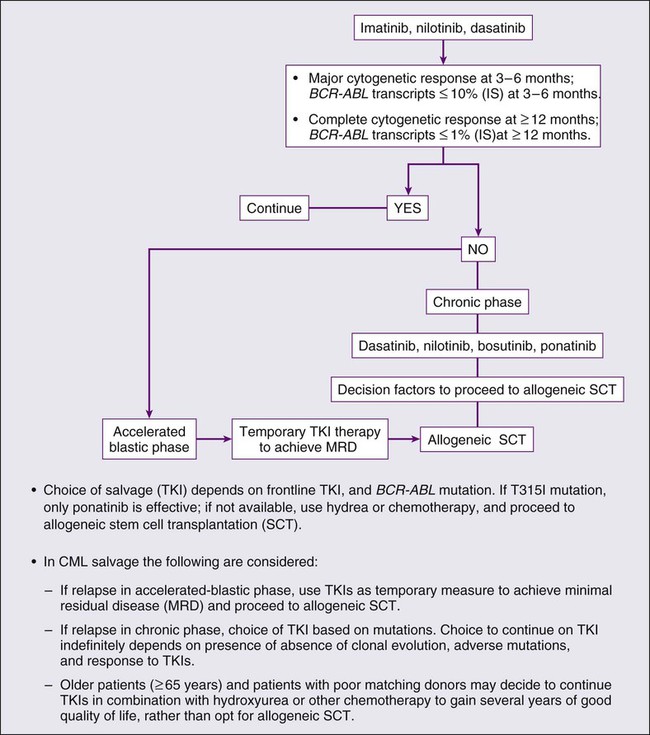
Patients who become resistant to imatinib have several treatment options, including allogeneic SCT and treatment with the new-generation TKIs and other agents. Imatinib resistance in chronic-phase CML is associated with an estimated 4-year survival rate of 60%. However, if progression is in accelerated or blastic phases, the prognosis is poor.63 For patients in accelerated or blastic phase after imatinib resistance, every attempt should be made to refer them to allogeneic SCT. If patients become resistant to imatinib in chronic phase, a trial of one of the new-generation TKIs is reasonable. Subsequent therapy (i.e., allogeneic SCT) may depend on the presence of clonal evolution, presence and type of mutation, early response to TKI therapy, patient age, and donor availability and degree matching. In all cases of imatinib resistance, mutational analysis is recommended to identify patients with T315I or other mutations that can be selectively nonresponsive to one of the new-generation TKIs. If a T315I mutation is identified, second-generation TKIs are not effective and the patient should be referred for allogeneic SCT as soon as possible and in the interim the disease should be controlled with hydroxyurea, cytarabine, combinations of standard older therapies, or investigational agents (ponatinib, omacetaxime) that might be effective against T315I mutations. A treatment algorithm for CML is proposed in Figure 101-6.
Dasatinib
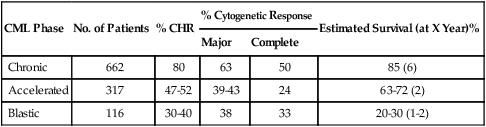
Dasatinib (Sprycel) is a potent, oral, multitargeted kinase inhibitor of critical oncogenic kinases including BCR-ABL, Src, C-kit, and PDGF-R. This dual Abl/Src inhibitor is 300 times more potent than imatinib in preclinical models and is effective against all imatinib-resistant kinase domain mutations except T315I. Pivotal phase II studies, referred to as START (Src/abl Tyrosine kinase inhibition Activity Research Trials of dasatinib), demonstrated high efficacy of dasatinib in all phases of CML after imatinib failure and in Ph-positive acute lymphocytic leukemia. This led to its approval for this indication in 2006. In 387 patients with chronic-phase CML after imatinib failure, dasatinib 70 mg orally twice daily produced a CHR rate of 90%, a major cytogenetic response rate of 52%, a complete cytogenetic response rate of 39%, and an estimated 10-month PFS rate of 92%.64 Response rates were higher in patients with imatinib intolerance versus resistance but were similar by whether mutations were present or absent. Results were similarly encouraging in accelerated blastic phases and in Ph-positive acute lymphocytic leukemia (Table 101-4).64,65 A randomized study of dasatinib 70 mg orally twice daily versus high-dose imatinib (400 mg orally twice daily) in patients with chronic-phase CML after failure on imatinib 400 to 600 mg daily showed dasatinib to be associated with higher rates of major cytogenetic response (53% vs. 33%; P = .017), complete cytogenetic response (44% vs. 18%, P = .025), and PFS (estimated 24-month rate 86% vs. 65%; P = .0012).66 Cytopenias were more common with dasatinib; pleural effusions occurred in 17% of patients on dasatinib. Pleural effusions were often exudates and were managed with treatment interruptions, diuretics and short courses of corticosteroids, and resumption of dasatinib at lower dose schedules. Myelosuppression was also managed with dose interruptions and reductions. A four-arm randomization study of different dasatinib dose schedules (50 mg twice daily, 100 mg single-dose daily, 70 mg twice daily, 140 mg single-dose daily) in 670 patients with chronic-phase CML after imatinib failure showed that a single daily dasatinib dose of 100 mg produced equivalent treatment results with fewer side effects.67 This dasatinib dose is now standard for chronic-phase CML. At the 6-year follow up, dasatinib 100 mg therapy resulted in a cumulative complete cytogenetic response rate of 50%, a major molecular response rate of 42%, and an estimated 6-year survival rate of 71%.68
Table 101-4
Results of Dasatinib Phase II Studies in Chronic Myeloid Leukemia (CML) After Imatinib Failure
| CML Phase | No. of Patients | % CHR | % Cytogenetic Response | Estimated Survival (at X Year)% | |
| Major | Complete | ||||
| Chronic | 662 | 80 | 63 | 50 | 85 (6) |
| Accelerated | 317 | 47-52 | 39-43 | 24 | 63-72 (2) |
| Blastic | 116 | 30-40 | 38 | 33 | 20-30 (1-2) |
Nilotinib
Nilotinib (Tasigna, AMN107) was rationally designed by replacement of the N-methylpiperazine binding group of imatinib to optimize the binding affinity and selectivity for the Abl kinase. Nilotinib, a selective BCR-ABL kinase inhibitor, is 30 times more potent than imatinib in preclinical models and was active against all imatinib-resistant mutations except T315I. The pivotal phase II studies with nilotinib 400 mg orally twice daily showed activity after imatinib failure. In the phase II pivotal trial of nilotinib in 321 patients in chronic phase after imatinib failure, the major cytogenetic response rate was 59%, the complete cytogenetic response rate was 44%, and the estimated 2-year survival rate was 87%. Side effects were modest, including grade 3/4 myelosuppression in 20% to 30%; no pleural effusions were observed.69
The activity of nilotinib in accelerated- and blastic-phase CML after imatinib failure was also encouraging, although response rates were lower and response durations shorter (Table 101-5).70,71 Nilotinib has been approved by the FDA (October 2007) for the treatment of CML in chronic or accelerated phases after imatinib failure.
Table 101-5
Results of Nilotinib Phase II Studies in Chronic Myeloid Leukemia (CML) after Imatinib Failure
| CML Phase | No. of Patients | % CHR | % Cytogenetic Response | Estimated Survival (at X Year)% | |
| Major | Complete | ||||
| Chronic | 321 | 74 | 59 | 44 | 87 (2) |
| Accelerated | 137 | 31 | 32 | 21 | 70 (2) |
| Blastic | 136 | 21-24 | 38-52 | 30-32 | 27 (2) |

Bosutinib
Bosutinib is an orally available dual Src/Abl inhibitor that is 30 to 200 times more potent than imatinib. It has minimal inhibitory activity against c-Kit and PDGF-R and therefore is expected to produce less myelosuppression and fewer pleural effusions. In studies of 288 patients with chronic-phase CML after imatinib failure treated with bosutinib 500 mg orally daily, the major cytogenetic response rate was 53%, the complete cytogenetic response rate was 41%, and the estimated 2-year survival rate was 92%.72 Grade 3/4 toxicities included diarrhea (8%), rashes (9%), and thrombocytopenia (5% to 10%).73
Selection of Sequential Therapies
Old Traditional Standards of Care Revisited
IFN-α showed anti-CML activity in the 1980s and was a standard of care in CML until the discovery of imatinib. IFN-α induced CHR rates of 50% to 80% in untreated chronic-phase CML and cytogenetic response rates of 40% to 60% (major in 10% to 40%; complete in 5% to 30%).76 A meta-analysis of randomized studies of IFN-α versus hydroxyurea or busulfan showed that IFN-α therapy was associated with better survival than hydroxyurea or busulfan (5-year survival rates 57% vs. 42%, P < .00001).77 IFN-α has minimal activity in accelerated or blastic phases of CML. It is associated with significant side effects including flulike symptoms, fever, chills, myalgias, fatigue, depression, neuropathy, diarrhea, memory problems, immune-mediated complications, myelosuppression, and others.78 Achievement of a major or complete cytogenetic response with IFN-α is associated with significantly better long-term survival.76 Combinations of IFN-α and low-dose cytarabine may improve results over IFN-α alone.79 New formulations of IFN-α attached to polyethylene glycol (PEG) prolong its half-life, allow weekly administration, reduce toxicities, and may also improve results (at least with a particular pegylated IFN-α2a formulation, Pegasys).80
Accelerated- and Blastic-Phase CML
In accelerated phase, imatinib produces a complete hematologic response rate of 40%, a major cytogenetic response rate of 30%, and an estimated 4-year survival rate of 60%. In accelerated-phase CML after imatinib failure, dasatinib and nilotinib are associated with CHR rates of 30% to 40%, major cytogenetic response rate of 32% to 43% (complete cytogenetic, 21% to 24%), and an estimated 2-year survival rate of about 70%. The second-generation TKIs probably produce better results than imatinib, either alone or in combinations.81
The three TKIs have shown activity in blastic-phase CML, and two of them (imatinib and dasatinib) have been approved for the indication. The results are modest, and combination modalities should be pursued. These include acute myeloid leukemia regimens plus TKIs (e.g., idarubicin + cytarabine + imatinib) in myeloid-undifferentiated blastic phase and acute lymphoid leukemia regimens plus TKIs (e.g., hyper-CVAD + imatinib or dasatinib) in lymphoid blastic phase.82,83 In the latter, central nervous system prophylaxis should be included because about 30% of patients may develop central nervous system disease. In all such instances, patients should be referred for allogeneic SCT as soon as possible.
Allogeneic SCT in accelerated-phase CML results in 5-year disease-free survival rates of 15% to 30%. If SCT is done in a second chronic phase the results are better, with disease-free survival rates of 40% to 50%. In blastic phase, survival after allogeneic SCT is 5% to 15%.84
Special Considerations
Ph-Negative CML
Pregnancy
If CML is diagnosed in a pregnant woman, her disease can be managed with leukapheresis if indicated during the first trimester of pregnancy, with hydroxyurea therapy subsequently until delivery and then with more definitive therapy. Anecdotal reports of successful deliveries after IFN-α therapy during pregnancy have occurred; IFN-α is antiangiogenic and may have adverse effects on the fetus that are not evident because the reports have included only a few patients. If a woman becomes pregnant while on imatinib therapy, the drug should be discontinued. Among 125 infants delivered of women with CML on imatinib (with the drug being discontinued after pregnancy was evident), 12 had birth defects, including 3 with a syndrome of ocular, renal, and skeletal abnormalities.85 Partners of men on imatinib have been delivered of healthy infants. However, the numbers are too small to make definitive conclusions.
Future Directions
Targeted therapy with TKIs has significantly improved prognosis in CML, particularly for patients in chronic phase. Emergence of resistance in a minority of patients with newly diagnosed CML suggests the complex interactions in CML pathophysiology and the need to improve therapy. Long-term therapy with TKIs is expensive and presents a socioeconomic issue. Frontline therapy with new-generation TKIs may reduce or prevent emergence of resistance due to mutations or other mechanisms and increase the proportion of patients with durable disappearance of BCR-ABL transcripts (which may lend itself to possible treatment discontinuation and molecular cures in a fraction of patients). Combinations of TKIs with other active agents that may target the downstream events should also be explored. These include inhibitors of pathways involving Raf, farnesylation, mTOR, JAK/STAT, MEK/MAPK, PI3K/AKT, or others. Combinations of TKIs with classic anticancer agents such as hydroxyurea, cytarabine, omacetaxime, IFN-α, and others are in progress. Vaccine and stem cell–directed strategies in the setting of minimal residual disease are investigational.86