Childhood Lymphoma
John T. Sandlund and Mihaela Onciu
• Malignant lymphoma, which comprises both Hodgkin lymphoma and non-Hodgkin lymphoma (NHL), is the third most common malignancy in childhood.
• Among children younger than age 15 years, there is a slight predominance of NHL, whereas HL is more frequent among children up to age 18 years.
• There are approximately 500 newly diagnosed cases of pediatric NHL in the United States each year.
• NHL is more common in boys than girls and in white children than black.
• There are geographic differences with respect to the frequency of histologic subtypes of NHL. Burkitt lymphoma is the predominant subtype in equatorial Africa and northeast Brazil, where it is associated with Epstein-Barr virus in the majority of cases, in contrast to the infrequent association observed in the United States and Western Europe.
• Children with immunodeficiency conditions are at increased risk of developing NHL. These include those with ataxia-telangiectasia (A-T), Wiskott-Aldrich syndrome, and X-linked lymphoproliferative syndrome (XLP). Children with acquired immunodeficiency disorders, including the acquired immunodeficiency syndrome (AIDS), and those receiving immunosuppressive therapy following bone marrow or organ transplantation are also at increased risk.
• The most common subtypes of NHL in children are Burkitt lymphoma, lymphoblastic lymphoma, anaplastic large-cell lymphoma, and diffuse large B-cell lymphoma (including the mediastinal subtype).
• Burkitt lymphoma is a mature B-cell lymphoma of germinal center origin, characterized by a very high proliferation rate, resulting from the activation of the c-myc oncogene as a result of juxtaposition to one of the immunoglobulin genes, through one of three characteristic balanced chromosomal translocations, that is, t(8;14), t(2;8), and t(8;22).
• Lymphoblastic lymphoma is typically of precursor T-cell immunophenotype, and may be associated with reciprocal translocations involving a T-cell receptor gene. Rare cases may be of precursor B-cell lineage.
• Anaplastic large-cell lymphoma is a peripheral (postthymic) T-cell lymphoma, characterized by large anaplastic (“hallmark”) cells expressing CD30 and, in the vast majority of pediatric cases, anaplastic lymphoma kinase (ALK), as a result of a balanced translocation involving the ALK gene, for example, t(2;5).
• Diffuse large B-cell lymphomas are a biologically heterogeneous category of mature B-cell lymphomas of germinal center or postgerminal center origin, composed predominantly of large cells, with a diffuse growth pattern.
• The clinical features at diagnosis are determined by primary sites of disease, which vary according to histologic subtype.
• Children with Burkitt lymphoma usually present with an abdominal mass and associated gastrointestinal symptoms, whereas those with advanced-stage lymphoblastic lymphoma typically present with a mediastinal mass associated with a spectrum of respiratory symptoms.
• Children with large-cell lymphoma or HL may present with disease in either the abdomen or mediastinum.
Diagnosis and Differential Diagnosis
• Infectious processes, such as bacterial adenitis, histoplasmosis, tuberculosis, and Epstein-Barr virus infection, may simulate lymphoma.
• A comprehensive characterization of the biological features of tissue will help distinguish NHL from other small round blue cell tumors, including Ewing sarcoma, neuroblastoma, and rhabdomyosarcoma.
• The workup should include a history and physical examination, complete blood count, chemistry panel (including electrolytes, blood urea nitrogen, creatinine, uric acid, phosphorus, calcium, and lactate dehydrogenase [LDH]), diagnostic imaging studies (computed tomographic scan of chest, abdomen and pelvis, nuclear imaging with a PET scan), and HIV screen.
• For children with NHL, stage is usually assigned according to the St. Jude system, whereas children with HL are staged using the Ann Arbor system.
• The treatment plan is determined on the basis of histology, stage, immunophenotype, and in some cases, clinical symptoms such as fever, weight loss, and night sweats.
• Children with advanced-stage Burkitt lymphoma are generally treated with intensive cyclophosphamide-based regimens given over a relatively short period of time, whereas children with lymphoblastic lymphoma are generally treated with regimens derived from strategies for children with acute lymphoblastic leukemia (ALL).
• Among children with large-cell lymphoma, the treatment plan varies with respect to tumor cell immunophenotype.
• Involved-field radiation therapy has a role in certain cases of HL but is rarely indicated for children with NHL.
• Children with refractory or recurrent disease are generally considered to have a poor prognosis and are therefore candidates for intensive or novel salvage treatment regimens.
• Those with chemosensitive disease are potential candidates for an intensification phase with hematopoietic stem cell rescue.
• The most concerning late effects of therapy include cardiac toxicity, infertility, and the development of a second malignancy.
• The risks for these complications are determined in part by the components of initial therapy.
• The most important predictors of treatment outcome for children with NHL are treatment protocol and tumor burden, as reflected by stage and serum LDH.
Introduction
Substantial clinical and laboratory advances have improved our understanding of both the pathogenesis and treatment of the malignant lymphomas of childhood and adolescence, which comprise Hodgkin lymphoma and the non-Hodgkin lymphomas (NHLs).1–5 These are the third most common type of cancer in children in the United States, comprising approximately 15% of newly diagnosed cases in this age group each year.6–11 Among children less than 15 years of age, the NHLs account for approximately 60% of cases. However, when children up to age 18 are included, there is a slight predominance of Hodgkin lymphoma.10,11
The NHLs of childhood are markedly different from those of adulthood.4,12 Diffuse high-grade extranodal subtypes account for the majority of pediatric cases, whereas low- and intermediate-grade lymphomas are predominant in adults. These differences are probably due in part to age-related maturational changes in the immune system and consequently in the types of cells susceptible to malignant transformation.5 The differences between adults and children in histologic subtype underlie the differing clinical features, staging, and treatment strategies in these age groups.4
The clinical presentation, staging, histologic subtypes, and treatment strategies in children and adults with HL are less dissimilar; however, some differences are worth noting.13 Epidemiological studies have suggested three distinct forms that depend on age: the childhood form in patients 14 years of age or younger, the young adult form (ages 15 to 34 years), and an older adult form in individuals 55 to 74 years of age.14 Among the four histologic subtypes of HL described in the Rye classification system,15 the nodular sclerosing subtype is most common in children, occurring in 40% of younger children and 70% of adolescents.16 Mixed-cellularity HL occurs in approximately 30% of cases and is more common in children with HIV infection and in those less than 10 years of age—it is frequently associated with an advanced stage (with extranodal extension) at presentation.16 Lymphocyte-predominant HL accounts for approximately 10% to 15% of pediatric cases, is usually associated with localized disease at presentation, and occurs more commonly in younger patients and in boys. The fourth subtype of Hodgkin lymphoma, lymphocyte depletion, is very rare in children.13
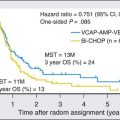
Despite excellent event-free survival rates in HL, there are well-recognized challenges related to the sequelae of therapy, which include endocrine dysfunction, chemotherapy-induced sterility, radiation-induced abnormalities in bone growth, chemotherapy- and radiation-related second cancers, and late cardiac deaths.13 Most current studies are exploring strategies that maintain an excellent treatment result while reducing late effects. For example, a combined-modality approach with low-dose radiation and combination chemotherapy may reduce the bone growth abnormalities associated with high-dose extended-field radiation therapy.13 However, as therapy is modified in an attempt to reduce the risk of late effects, the rates of event-free survival may be compromised.19–19 For example, attempts at reducing alkylating agent–related late effects in patients with advanced-stage disease by substituting other chemotherapeutic agents have lowered event-free survival rates.17,20 There have been some recent clinical trials that have also attempted to reduce toxicity without compromising outcome. Metzger et al. reported that among children with favorable-risk HL and a complete early response to vincristine, doxorubicin, methylprednisolone (VAMP) chemotherapy, a high rate of 2-year event-free survival could be achieved with limited use of radiotherapy.21 The results of the CCG 5942 study, which examined the benefit of adding involved field radiation therapy (IFRT) for patients who achieved a complete response to chemotherapy, were recently published.22 They reported a statistically significant improvement in EFS for those who received IFRT; however, there was no associated improvement in overall survival. The refinement of a risk-adapted treatment approach to the management of pediatric HL remains a challenge.23 These and other issues regarding the management of children with HL will be discussed in Chapter 105. The remainder of this chapter will focus on the NHLs of childhood.
Epidemiology and Pathogenesis
NHLs may occur at any age in childhood, but are unusual in children younger than age 3 years; the median age at diagnosis is approximately 10 years.4,24 There are approximately 500 new cases of pediatric NHL in the United States each year.7,9,10,25 In contrast to Hodgkin lymphoma, which has a bimodal age distribution with peaks in early and late adulthood, the incidence of NHLs increases steadily with age.10 NHL occurs almost twice as commonly in whites as in blacks, and two to three times more often in boys than in girls; the explanation for these differences has yet to be elucidated.5
There are specific populations at increased risk for the development of NHL.5,26–29 These include individuals with primary immunodeficiency syndromes,5,28,29 including ataxia-telangiectasia (A-T), Wiskott-Aldrich syndrome, X-linked lymphoproliferative syndrome (XLP), common variable immunodeficiency, Nijmegen syndrome, autoimmune lymphoproliferative syndrome (ALPS). It is important that these syndromes be recognized so that appropriate therapy can be designed. For example, in the management of children with A-T who develop a malignancy, involved field irradiation and radiomimetics such as bleomycin should be avoided. Children with A-T are also at increased risk for the development of late-onset hemorrhagic cystitis following exposure to cyclophosphamide. Boys with XLP are at increased risk for developing fatal infectious mononucleosis and/or B-cell lymphomas. XLP should be considered in any boy with a high-grade B-cell lymphoma whose brother has had either fatal infectious mononucleosis or B-cell lymphoma, or in any boy who has had two primary B-cell lymphomas. Children who have received immunosuppressive therapy (e.g., recipients of bone marrow or organ transplants) and those with the acquired immunodeficiency syndrome (AIDS) are also at a higher risk for developing NHL.27 The overall prevalence of lymphomas among children with HIV infection is approximately 1.6%.27 Among pediatric HIV-positive hemophiliacs, NHL is 36 times more frequent than in HIV-negative children with factor 8 deficiency. The majority of the HIV-associated NHLs have a B-cell immunophenotype with either aggressive B-cell/Burkitt or large-cell morphology. Proliferative lesions of mucosa-associated lymphoid tissue (MALT), which may be either benign or malignant, have also been described in children with HIV infection.27 Although deficient T-cell function has been implicated in these congenital and acquired immunodeficiency states, further study is required to fully clarify the mechanisms of pathogenesis.
There are well-recognized geographic differences in both the incidence and the distribution of histologic subtypes of NHL.5,6,30 For example, NHLs are very rare in Japan but are very common in equatorial Africa. More specifically, Burkitt lymphoma accounts for approximately one-half of all childhood malignancies in equatorial Africa and is the predominant NHL subtype in Northeastern Brazil and areas of the Middle East.30 In contrast, lymphoblastic lymphomas are the predominant histologic subtype in southern India.31 In some parts of the world, the distribution of histologic subtypes of childhood NHL has yet to be firmly established.
There are also geographic differences in the clinical and biological features of some NHLs.5,31,32 For example, Burkitt lymphoma in equatorial Africa (endemic Burkitt lymphoma) frequently involves the jaw, abdomen, orbit, paraspinal area, and central nervous system, whereas the common sites of involvement associated with Burkitt lymphoma in the United States and Western Europe (sporadic Burkitt lymphoma) include the abdomen, bone marrow, and nasopharynx.5 The predominant chromosome 8 breakpoints, as well as the breakpoints within the immunoglobulin heavy-chain gene (on 14q32) differ between sporadic and endemic cases.33 In addition, sporadic cases have more complex karyotypic abnormalities than endemic cases in addition to the classic translocations, suggesting distinct mechanisms of malignant transformation.34 The endemic cases are associated with IgM expression, whereas the sporadic cases generally are not.5 The sporadic and endemic cases also differ with respect to Epstein-Barr virus association.32 The overlap of the lymphoma belt in equatorial Africa with the malaria belt prompted speculation that an infectious agent might be involved in lymphomagenesis. This led to the discovery of the Epstein-Barr virus (EBV) and its association with African Burkitt lymphoma.5 Although a direct role in pathogenesis has not been demonstrated, the circumstantial evidence for its involvement is compelling. It has been suggested that as a B-cell mitogen, EBV increases the target pool of cells potentially susceptible to malignant transformation.32 Supporting this hypothesis is evidence that Rag gene expression can be induced by EBV, theoretically increasing the likelihood of a translocation occurring in immature B cells that are about to rearrange their immunoglobulin genes.35 The potential role of Epstein-Barr nuclear antigen (EBNA)-1 in pathogenesis has been suggested by experiments demonstrating that lymphomas develop in mice transgenic for EBNA-1.36 Moreover, an identified EBNA-1 variant has been shown to be associated with the majority of Burkitt lymphoma cases studied, prompting investigators to speculate that this tumor-associated mutation alters EBNA-1 function in a way that directly or indirectly provides a growth advantage for the lymphoma cell.37 A more direct role for EBV in lymphomagenesis is suggested by studies of the EBV-positive Burkitt lymphoma cell line, Akata, which loses its malignant phenotype with spontaneous loss of EBV; however, the malignant phenotype is regained with EBV reinfection.38 EBV association has been reported in approximately 90% of the endemic (i.e., African) Burkitt tumors and in approximately 15% of sporadic cases (United States and Western Europe).5 Aberrant and disrupted expression of the EBV genome has recently been reported in cases of sporadic Burkitt lymphoma that were EBV-negative by conventional EBNA screening.39 This observation, coupled with the 50% rate of EBV association in Burkitt tumors in other parts of the world (including Brazil, Russia, Argentina, and Chile), suggest a widespread role for this virus in lymphomagenesis.
Pathology and Biology
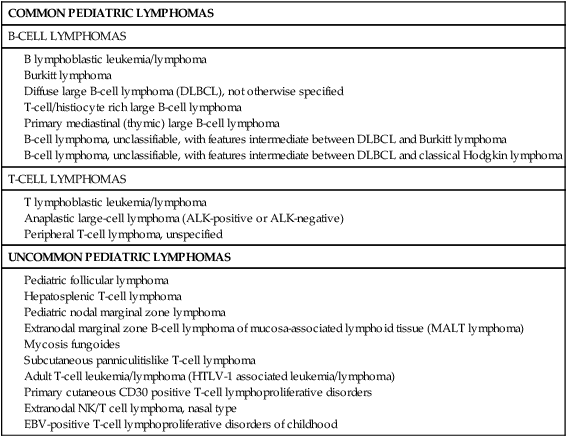
The classification systems applied to pediatric NHL are similar to those used for lymphomas occurring in adults. In the National Cancer Institute (NCI) Working Formulation for clinical use, published in 1982,12 which subclassified NHLs based on their morphologic appearance and clinical aggressiveness into three grades (low, intermediate, and high), most of the childhood NHLs would be designated as high-grade lymphomas (Figure 97-1). The Revised European-American Lymphoma (REAL) Classification introduced in the mid-1990s40 and the World Health Organization (WHO) Classification initially published in 2001 and updated in 200841,42 define the different categories of NHL as distinct clinicopathological entities, with well-defined morphologic, immunophenotypic, and genetic features, named, where feasible, according to their postulated counterparts in the normal lymphoid system. This has led to the definition of several lymphoma subtypes within the high-grade lymphoma category previously assigned to most pediatric lymphomas. These include Burkitt lymphoma, diffuse large B-cell lymphoma (DLBCL, including a mediastinal or thymic subtype), B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and Burkitt lymphoma,42 precursor B or T lymphoblastic lymphomas, and anaplastic large-cell lymphoma (ALCL). Some low-grade lymphoma subtypes, including follicular center cell lymphoma and marginal zone lymphoma may also occur in children where they appear to have distinct clinicopathological features, albeit with less frequency. The WHO classification of pediatric NHLs is summarized in Table 97-1.
Table 97-1
Pediatric Non-Hodgkin Lymphoma According the World Health Organization (WHO) Classification 2008
Pediatric nodal marginal zone lymphoma
Extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma)
Subcutaneous panniculitislike T-cell lymphoma
Adult T-cell leukemia/lymphoma (HTLV-1 associated leukemia/lymphoma)
Primary cutaneous CD30 positive T-cell lymphoproliferative disorders
Extranodal NK/T cell lymphoma, nasal type
EBV-positive T-cell lymphoproliferative disorders of childhood
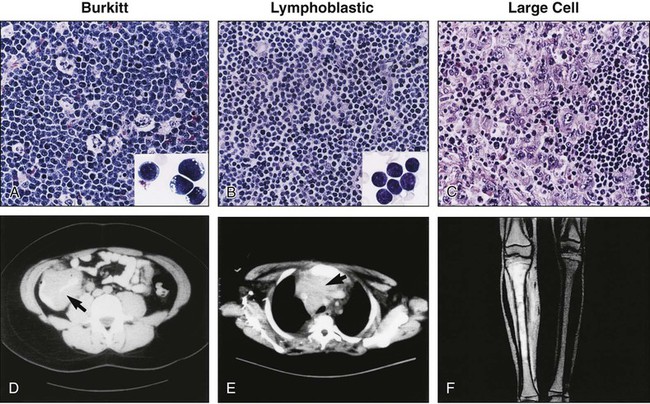
Burkitt Lymphoma
Modern classification systems41 define Burkitt lymphoma as a mature B-cell neoplasm composed of monomorphic medium-sized cells (“small noncleaved cell”), with an extremely short doubling time, resulting from c-myc gene translocations. Burkitt lymphoma and leukemia (classically known as acute lymphoblastic leukemia (ALL) L3 in the French American British classification) form a biological continuum. B-cell lymphomas with a similar proliferation rate and genetic features, previously termed “small noncleaved cell lymphoma, Burkitt-like,” have also been incorporated in this clinicopathological entity as one of its morphologic variants (see later).
Histologically, Burkitt lymphoma classical variant, present in endemic cases, as well as in many of the sporadic cases, is characterized by a diffuse growth pattern, uniform medium-sized cells (typically equal in size to the nuclei of adjacent histiocytes), with coarsely clumped nuclear chromatin and one to three nucleoli. When observed in Wright-Giemsa-stained smears, these cells are large, with moderate amounts of deeply basophilic cytoplasm and prominent cytoplasmic vacuoles. These tumors typically contain numerous mitotic figures, and, matching the high cell turnover rate, they have numerous “tingible-body” macrophages with pale cytoplasm that contain ingested apoptotic cellular debris and impart a characteristic “starry-sky appearance” on low magnification. Burkitt lymphomas that are otherwise typical, but show a greater degree of pleomorphism in nuclear size and shape, and one to two large nucleoli in the neoplastic cells, were classified as the atypical Burkitt/Burkittlike variant in the previous WHO Classification (2001). These lymphomas represent a subset of the tumors previously designated as “small noncleaved cell lymphoma, Burkitt-like.” This morphologic variant can be seen in sporadic cases and in immunodeficiency-associated Burkitt lymphoma. Finally, Burkitt lymphomas with plasmacytoid differentiation (eccentric nuclei with a single central nucleolus, basophilic cytoplasm that contains abundant monotypic immunoglobulin on immunohistochemical staining) represents a third morphologic variant. This latter variant can also be seen with immunodeficiency-associated lymphoma. The most recent WHO Classification (2008)42 no longer defines morphologic variants of Burkitt lymphoma, but rather recognizes the wide morphologic spectrum of a single disease.
The immunophenotype of Burkitt lymphoma, regardless of its morphologic variants, is that of a mature B cell of germinal center or postgerminal center type.41 Burkitt lymphoma cells strongly express CD19, CD20, CD22, CD24, as well as CD10 and BCL6, and are negative for BCL2, CD5, CD23, CD34, and TdT. The neoplastic cells also express surface immunoglobulin (typically IgM, less commonly IgA or IgG) with light-chain (kappa or lambda) restriction. Staining for proliferation markers (Ki-67) highlights a growth fraction of nearly 100% in all the histologic variants. Recent studies have demonstrated that MYC expression detected by immunohistochemical staining highly correlates with MYC gene rearrangements when it has nuclear localization and is present in a high percentage (>70%) of the lymphoma cells.43,44 Demonstration of this high proliferation rate is required for the diagnosis of Burkitt lymphoma. Rare cases may exhibit a precursor B-cell immunophenotype associated with the characteristic Burkitt chromosomal translocations.45,46 These cases show morphologic and immunophenotypic features intermediate between a classical Burkitt lymphoma and a precursor B cell. The latter includes expression of CD34 and TdT and lack of surface immunoglobulin expression. Recognition of these cases as Burkitt lymphoma/leukemia typically requires integration with the cytogenetic findings.
Genetically, Burkitt lymphoma is characterized by several translocations that rearrange the c-myc gene on chromosome 8q24, bringing it under the controlling sequences (enhancers) for immunoglobulin heavy- or light-chain genes and leading to overexpression of this gene in the neoplastic B cells.47–52 These translocations include the t(8;14)(q24;q32) seen in 85% to 90% of the cases, that involves the c-myc and IgH gene loci, and the less common variants, t(2;8)(p11;q24) and t(8;22)(q24;q11.2), that juxtapose c-myc to the immunoglobulin light-chain genes, Ig kappa (on 2p11) and Ig lambda (on 22q11), respectively. The breakpoints involving c-myc and IgH are highly variable, and cluster in regions different between endemic and sporadic Burkitt lymphoma. The IgH breakpoints most often involve the VDJ region in endemic Burkitt lymphoma, and the switch region Sµ in the sporadic cases, suggesting that the neoplastic transformation occurs at different stages of B-cell development in these two disease subtypes.33 Similarly, the c-myc breakpoints appear to cluster in two different regions that correlate with the location of the IgH breakpoints.53 This high variability in breakpoints leads to difficulty in generating polymerase chain reaction (PCR) assays that can detect this translocation. Long-range PCR combined with nested PCR strategies have allowed detection of the t(8;14) and of the variant translocations.54,55 Approximately 70% to 80% of the sporadic cases have additional chromosomal abnormalities, the most common of which involve chromosomes 1 (1q), 6 (6q), 13 (13q), 17, and 22.56 Some of these alterations (such as those of chromosome 13q), appear to have a negative impact on prognosis in pediatric Burkitt lymphoma.56
The biology of Burkitt lymphoma has been extensively characterized. MYC, a widely studied oncogene, initially discovered because of its involvement in the t(8;14), appears to play a central role in malignant transformation and the biological behavior of Burkitt lymphoma.50,51 MYC overexpression has been described in up to 50% of all human cancers,57 occurring through both epigenetic and genetic mechanisms (that include chromosomal translocations and genomic amplifications). In Burkitt lymphoma, MYC is deregulated primarily by mutation/translocation and by overexpression.58 Its oncogenic properties have been demonstrated both in vitro and in vivo, by using a variety of animal models.52,57
MYC is a nuclear transcription factor. It is the founding member of a family of transcription factors of the basic helix-loop-helix-leucine zipper (BHLH-LZ) classes. MYC operates as a heterodimeric complex with a cofactor named MAX to bind specific DNA sequences, thereby transcriptionally regulating hundreds to thousands of target genes.59 These genes are involved in diverse programs, including cell cycle, cell growth, apoptosis, protein translation, cell adhesion, various metabolic pathways, angiogenesis, and DNA repair. MYC can promote cell proliferation through cyclins D, B1 and A, and CDK4, that lead to G1 to S progression. It also inhibits differentiation, increases protein synthesis, and suppresses genes that encode cytoskeletal and cell adhesion molecules, thus contributing to neoplastic transformation. An interesting aspect of MYC biology, with major implications in neoplasia, is its role in apoptosis. MYC sensitizes cells to apoptosis and, therefore it appears that mechanisms that would block this downstream effect are necessary in the process of malignant transformation, allowing for cell proliferation to take precedence over cell death.60 Apoptosis can be inhibited by a variety of proteins, such as BCL2, BCL-XL, cFLIP, mTOR, Mdm2/Hdm2, Twist, Bmi1, and Cul7. Apoptosis promoters include a variety of proteins typically regarded as tumor suppressors, such as P53, ARF, PUMA, ATM, BAX, BID, BIM, Fas/CD95, and 4EBP1. Extensive studies of tumor cell lines and tumors arising in animal models,57,61 as well as of primary human tumor samples, have shown that in most cases, MYC overexpression is accompanied by either the overexpression of one of the apoptosis suppressors (e.g., BCL2) or by inactivation of one of the pro-apoptotic factors (e.g., P53 or ARF).62 In animal models, alterations in P53-related pathways seem to play a major role in the biology of Burkitt lymphoma. The relevance of these findings to human Burkitt lymphoma is currently under study. Limited studies performed in pediatric sporadic Burkitt lymphoma suggest that these findings may be applicable to primary tumor samples as well.63
Lymphoblastic Lymphoma
Modern classifications designate these neoplasms as B and T lymphoblastic leukemia/lymphoma, in an attempt to reflect the morphologic, immunophenotypic, genetic, and possibly biological continuum between the lymphomatous and the leukemic presentation in these blastic tumors.41 Indeed, at the present time, the separation between these two presentations is largely arbitrary, with lymphoblastic neoplasms involving less than 25% of the marrow cellularity being treated as lymphoma with marrow involvement, with those exceeding this cut-off being considered ALLs.10,64,65
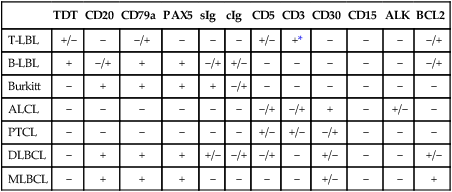
Immunophenotypically, the majority of lymphoblastic neoplasms designated as lymphomas (~90%) are of T lineage, whereas the remaining show B lineage differentiation (Table 97-2).68–68 Very rare cases of NK cell origin have been described.67,69,70 The antigen expression profile of lymphoblastic lymphomas is largely similar to that of their leukemic (ALL) counterparts. Most of the lymphoblastic neoplasms express markers of early lymphoid differentiation, including terminal deoxynucleotidyl transferase (TdT), CD34, and CD10. TdT and CD34 expression may be absent in a subset of cases, requiring careful correlation with morphology and other immunophenotypic features for the differential diagnosis with mature (peripheral) T-cell and B-cell lymphomas. T-lymphoblastic lymphomas additionally express T-lineage antigens, including CD1a, CD2, CD3, CD4, CD5, CD7, and CD8. By analogy with the stages of intrathymic T-cell differentiation, several subtypes of T-lymphoblastic neoplasms have been defined, including an early double-negative stage (CD1a–, cytoplasmic CD3+, CD7+, variably positive for other pan–T cell antigens such as CD2 and CD5), early cortical thymic stage (CD1a+, CD2+, cytoplasmic CD3+, often CD4+, CD8+, CD5+, CD7+, CD21+), and late cortical thymic stage (CD1a–, CD2+, surface CD3+, CD5+, CD4+ or CD8+). The majority of T-cell lymphoblastic lymphomas correspond to a late stage of intrathymic maturation.71,72 Gene expression profiling studies73 have shown that different genetic lesions correlate with malignant transformation at different stages of intrathymic T-cell maturation in T-cell ALL (see later). T lymphoblastic lymphomas are characterized by a more frequent expression of T-cell receptor αβ than γδ, as compared with T-ALL.74 The immunophenotypic profile of true precursor B lymphoblastic lymphoma (without peripheral blood or bone marrow involvement) has been less extensively characterized, because of availability of paraffin-embedded tissue only, in many of the cases.77–77 By immunohistochemistry, these neoplasms are positive for TdT, CD34, CD10, pan-B cell antigens (CD79a, CD22, PAX-5) and often negative for CD20, a mature B-cell antigen, as well as for immunoglobulin kappa and lambda light chains. These features, along with the morphology and clinical presentation, are helpful in the differential diagnosis with Burkitt lymphoma and DLBCL, as both mature B-cell neoplasms that are typically negative for TdT and CD34, and strongly positive for CD20.78–81
Table 97-2
Immunophenotypic Features of the Most Common Pediatric Non-Hodgkin Lymphomas
| TDT | CD20 | CD79a | PAX5 | sIg | cIg | CD5 | CD3 | CD30 | CD15 | ALK | BCL2 | |
| T-LBL | +/− | − | −/+ | − | − | − | +/− | +* | − | − | − | −/+ |
| B-LBL | + | −/+ | + | + | −/+ | +/− | − | − | − | − | − | −/+ |
| Burkitt | − | + | + | + | + | −/+ | − | − | − | − | − | − |
| ALCL | − | − | − | − | − | − | −/+ | −/+ | + | − | +/− | − |
| PTCL | − | − | − | − | − | − | +/− | +/− | −/+ | − | − | − |
| DLBCL | − | + | + | + | +/− | −/+ | −/+ | − | +/− | − | − | +/− |
| MLBCL | − | + | + | + | − | − | − | − | +/− | − | − | + |
Limited information is available regarding the cytogenetic abnormalities encountered in T-lymphoblastic lymphoma.68 It is generally assumed that they resemble those of T-lineage ALL. Recurrent chromosomal abnormalities often include reciprocal translocations that disrupt developmentally important transcription factor genes and lead to their overexpression, as a result of rearrangements to loci for T-cell receptor (TCR) genes, most commonly TCRA (14q11.2) and TCRB (7q35). For example, the TAL1/SCL gene (1p32) is involved in the t(1;14)(p32;q11) present in approximately 3% of T-ALLs.72 The HOX11 transcription factor gene (10q24) is rearranged as part of the t(10;14)(q24;q11.2) and the t(7;10)(q35;q24).82,83 The LMO1 (11p15) and LMO2 (11p13) oncogenes are disrupted by the t(11;14) and t(7;11), respectively.84 Other less common translocations do not involve the TCR genes. For example, the t(9;17) translocation is more commonly detected in T-lineage lymphoblastic lymphoma than precursor T-ALL,85 and is often associated with a mediastinal mass and aggressive disease course. The t(10;11)(p12;q14), leading to the AF10-CALM fusion gene, has been rarely reported in pediatric lymphoblastic lymphoma cases,86,87 and limited studies suggest a poor prognostic significance.88 The t(8;13)(p11;q11-14) has been reported in rare cases of T-cell lymphoblastic lymphoma that present with eosinophilia and myeloid hyperplasia.91–91
Much progress has been accomplished in the recent years in the understanding of the biology of pediatric T-cell lymphoblastic neoplasms. Molecular studies combined with gene expression profiling have uncovered several oncogenes that appear to play important roles in malignant transformation and correlate with disease subgroups with potential prognostic and therapeutic implications. TAL1/SCL shows activating mutations in up to 50% of all T-ALL,73,92 independent of detectable translocations, and correlates with leukemias arrested at the late cortical stage of thymocyte maturation expressing αβ TCR, and with an inferior overall survival.73 HOX11(TLX1) mutations are present in ~30% of all T-ALL, more commonly in adults than in children92 and in early cortical T-ALL expressing αβ TCR, and correlate with a superior survival in limited studies.73 LYL1 is mutated in up to 22% of pediatric T-ALL, correlating with the double-negative early thymocyte stage of differentiation and an inferior survival.73 The MLL gene is mutated in 4% to 8% of T-ALL cases, associated with maturation arrest at early thymocytes stages, expression of γδ TCR, and no impact on prognosis.92 Certain molecular alterations may provide opportunities for therapeutic targeting. Cryptic deletions of the INK4/ARF locus at 9p21, leading to alterations of the p14/p16 loci are present in up to 75% of all T-ALL,92 leading to defects in cell cycle control. Because these alterations likely lead to neoplasia through the retinoblastoma (Rb1) and P53 oncogenes, targeting the latter genes may prove effective in a significant proportion of T-ALL. In addition, 10% to 20% of T-ALL harbor constitutively activated tyrosine kinases (LCK, FLT3, ABL1), which may also provide targets for tyrosine kinase inhibitor drugs.92 Finally, NOTCH1-activating mutations are present in more than 50% of T-ALL.93 This transcription factor activates a wide variety of cellular pathways related to cell growth, using c-MYC as an essential mediator. Inhibition of these signaling pathways has been shown to inhibit cell growth in vitro, in T-ALL cell lines.94
Anaplastic Large-Cell Lymphoma
ALCL is a lymphoma of mature (peripheral) T cells, characterized by strong expression of CD30 (Ki-1)95 and, morphologically, by the presence of large, “hallmark” cells.41 These lymphomas may present as systemic disease or as primary cutaneous neoplasms. A subset of the systemic lymphomas expresses the anaplastic lymphoma kinase (ALK) protein as a result of ALK gene rearrangements and have been designated by the most recent WHO classification42 as ALCL, ALK positive. These constitute the most common subtype of ALCL encountered in children. Only rare cases of primary cutaneous ALK + ALCL have been reported in both pediatric and adult patients.96 Cases of ALK-negative primary cutaneous lymphoma have also been reported in children.97
ALCL was initially described as a lymphoma composed of large anaplastic cells with pleomorphic nuclei (hence the designation), and sinusoidal lymph node involvement, that expressed CD30 (Ki-1).95,98 This histologic subtype of ALCL is now designated as the common (classic) subtype. The availability of an immunohistochemical assay for ALK expression has allowed the recognition of lymphomas with very heterogeneous morphology as ALCL. Consequently, the term ALK lymphoma or ALKoma was proposed for this category of tumors, to better accommodate morphologic variants that lack obvious anaplastic morphology.99 ALCL typically involves the lymph node sinuses and interfollicular area, with subtotal effacement of the lymph node architecture. Regardless of their morphologic subtype, ALK-positive ALCLs consist of a mixture in variable proportions of large anaplastic “hallmark” cells with kidney- or horseshoe-shaped nuclei, and small atypical lymphoid cells with irregular nuclear outlines, as well as inflammatory cells that may include histiocytes and plasma cells. In the common (classical) type the hallmark cells predominate, sometimes forming cohesive clusters and sheets, whereas in the small cell type, these cells are sparse and the neoplastic infiltrate consists predominantly of small lymphoid cells.100 In the lymphohistiocytic type, the neoplastic cells may be difficult to identify within an exuberant polymorphous inflammatory background that includes histiocytes and plasma cells, is usually poor in neutrophils and eosinophils, and sometimes shows associated fibroblastic proliferation, vascular proliferation, and fibrosis.101 Occasionally, the fibrotic component may impart a partial nodularity to the tumor. Within this infiltrate, immunohistochemical staining for CD30 or ALK will highlight large tumor cells that typically aggregate around blood vessels. In the monomorphic type, the hallmark cells may be difficult to find, with the tumor consisting predominantly of a monotonous, highly mitotic population of large immunoblastic cells. This latter variant may be associated with a “starry-sky” appearance because of frequent tingible-body macrophages. Rarely, ALCL may manifest with prominent (leukemic) peripheral blood involvement (at diagnosis or at relapse).102–106 In such cases, the lymphoma cells seen on the peripheral blood smear include a mixture in variable proportions of small lymphoid cells with marked nuclear membrane irregularity (“cerebriform”) and large immunoblastic cells with deeply basophilic and occasionally vacuolated cytoplasm, and coarsely clumped nuclear chromatin. In most cases, there is a predominance of small cells, very similar to the cellular composition of the small cell variant. Notably, in such cases a significant proportion of the peripheral blood leukocytosis usually consists of granulocytes that may show prominent left shift and toxic changes. These findings, combined with the clinical picture of fever, malaise, and respiratory distress may distract from the atypical lymphoid population present. In the sarcomatoid type,107 the tumor cells are sparse and present within an edematous stroma that contains atypical proliferating fibroblasts. In these cases, the tumor cells may have the classical morphology or a spindle cell appearance and may cluster around blood vessels. Rare histologic subtypes of ALCL include giant-cell rich,98 signet-ring cell,108 neutrophil-rich,111–111 and eosinophil-rich.112 The latter three have only been reported in ALK-negative ALCL.
On immunohistochemical staining, ALCL cells are characteristically positive for CD30 and ALK (in the ALK-positive cases) and usually positive for CD45 (leukocyte common antigen). In the small cell variant (including the forms with leukemic presentation), the small cells may be CD30 negative and usually show a much lower level of expression of ALK than the large “hallmark cells” also manifest within the same tumors. The majority of ALK-positive ALCLs are also EMA positive (and cytokeratin negative).113 Many tumors express CD43. Although the majority of these tumors are of T-lineage when examined at the molecular level, many of them fail to express several pan T-cell antigens as detectable by immunohistochemistry, hence the apparent “null-cell phenotype.” More than 50% of these lymphomas fail to express CD3, CD5, CD7, and CD45RO.114 The majority of ALCL will express CD2 and CD4, whereas they are only exceptionally CD8 positive. Also, regardless of the CD4/CD8 expression status, the tumor cells are very often positive for cytotoxic T-cell antigens (e.g., TIA-1, perforin, granzyme B). Rare ALCLs also express CD56, which has been proposed as a factor of poor prognosis. Flow cytometric analysis usually shows a similar immunophenotype. In addition, 40% to 50% of the analyzed cases have been found to express antigens typically associated with myeloid differentiation, such as CD11b, CD13, CD15, and CD33.117–117
All ALK-positive ALCLs show aberrant expression of ALK, a membrane-bound receptor tyrosine kinase that is not expressed by any normal lymphoid elements. Across normal tissues, ALK expression can only be found within the brain, in scattered neurons.118 The aberrant ALK expression seen in ALCL is the result of various chromosomal translocations that juxtapose the ALK locus (chromosome 2p23) to other partner genes, which in turn lead to the activation and determine the subcellular localization of the ALK (as seen by immunohistochemistry). The most common translocation manifest in up to 75% of ALCL is t(2;5)(p23;q35).119–123 The result of this translocation is a fusion protein (p80) that includes the portion of the ALK protein with associated tyrosine kinase activity and the oligomerization domain of nucleophosmin (NPM) encoded in 5q35.118,124,125 Normal NPM molecules dimerize and shuttle between the cytoplasm and the nucleus (nucleolus). This is why, in most ALCL that contain the NPM-ALK fusion, ALK expression can be detected immunohistochemically in the nucleus and the cytoplasm. Several other translocations have been described in ALK-positive ALCL (frequency 2% to 5%). They include t(1;2)(q21;p23) (fusion partner tropomyosin 3 gene, TPM3),122,126 t(2;3)(p23;q21) (fusion partner tropomyosin receptor kinase-fused gene, TFG), t(2;22)(p23;q11) (fusion partner clathrin heavy-chain gene, CLTCL),127 and inv(2)(p23;q35) (fusion partner Pur H gene, ATIC).128 In all these cases, the fusion partners for ALK are proteins that dimerize (aspect that appears to be essential for activating the kinase function of ALK) but typically localize to the cytoplasm. Therefore in ALCL containing these translocations, ALK expression can be detected only in the cytoplasm. The presence of NPM-ALK can be explored for diagnostic purposes using PCR, RT-PCR, or fluorescence in situ hybridization (FISH). Aberrant ALK expression appears to play an essential role in tumorigenesis in ALCL.129,130 ALK inhibitors are currently being studied as potential therapeutic agents for this type of lymphoma.
Numerous studies have uncovered signaling pathways that are important in the biology of the ALK-positive ALCL, and especially in tumors that harbor the NPM-ALK fusion product. These pathways appear to be important in NPM-ALK-induced tumorigenesis and are attractive therapeutic targets. They include the Jak/STAT (specifically the Jak3 and STAT3 family members) and the PI3Kinase/Akt pathways. Inhibition of these pathways has been shown to induce apoptosis and inhibit tumor cell growth in studies performed on lymphoma cell lines. Inhibition of the Src kinase pp60src and pharmacological blockade of Hsp90 have likewise been shown to induce lymphoma cell apoptosis in vitro.131
Diffuse Large B-cell Lymphoma
The WHO classification defines DLBCL as a mature B-cell neoplasm with diffuse growth pattern, composed of large lymphoma cells (i.e., larger than the normal macrophage nuclei).41 Several morphologic variants are accepted (all of which may be encountered in children).132 These variants include centroblastic, immunoblastic, T-cell/histiocyte rich, and anaplastic. DLBCL may manifest as a de novo lymphoma or as progression of a preexisting low-grade lymphoma. In children, de novo DLBCL is the more common occurrence. In a small percentage of cases, a preexisting follicular lymphoma component may be identified.
DLBCL is a tumor with diffuse growth pattern composed, in variable proportions, of several types of large lymphoma cells that include centroblasts, immunoblasts, and anaplastic cells. The predominance of one or another cell type leads to the various morphologic subtypes. Centroblasts are medium-sized to large cells with scanty cytoplasm, irregular nuclear outlines, vesicular chromatin, and several peripherally located medium-sized nucleoli. Immunoblasts are large cells with abundant basophilic cytoplasm; vesicular or clumped chromatin; eccentric nuclei; and large, single, centrally located nucleoli. Anaplastic cells are similar to those seen in ALCL and include giant cells with pleomorphic nuclei or Reed-Sternberg-like morphology. In the centroblastic variant (>80% of the pediatric DLBCL),133 the tumor consists of a polymorphous mixture of centroblasts and immunoblasts, with <90% immunoblasts. In the immunoblastic variant (<10% of the pediatric DLBCL),133 >90% of the lymphoma cells are immunoblasts. In the rare anaplastic variant, frequent anaplastic cells are admixed with centroblasts and immunoblasts. Occasionally, a cohesive growth pattern may be present in the latter subtype. In T-cell/histiocyte-rich B-cell lymphoma, the bulk of the tumor consists of inflammatory cells, whereas the neoplastic cells (centroblasts, immunoblasts, Reed-Sternberg-like) represent <10% of cellularity. Rare cases of DLBCL with plasmablastic morphology, immunohistochemical expression of ALK, and the presence of ALK fusion transcripts have been reported.134,135
In all subtypes of DLBCL, the lymphoma cells strongly express CD45 and CD20, as well as other B-cell antigens, such as CD19, CD22, and CD24. A subset of DLBCL may express CD10, BCL-6, and BCL-2 (resembling follicle center cell lymphoma). Occasional DLBCLs are positive for CD5. CD30 expression is typically present in the anaplastic subtype and may be seen in the T cell–rich subtype. Light-chain restricted immunoglobulin expression may be demonstrated in many cases, although a significant proportion of these lymphomas may lack immunophenotypic evidence of immunoglobulin expression. Such cases show clonal rearrangements of the immunoglobulin genes when analyzed by molecular means.136 Some of the DLBCL may show high proliferation indices when analyzed by Ki-67 immunohistochemistry, but they typically do not exceed 90%, a feature helpful for the distinction from Burkitt lymphoma. The ALK-positive plasmablastic B-cell lymphomas have unique immunophenotypic features, which reflect their distinctive morphologic differentiation. They lack expression of mature B-cell antigens such as CD20, and express weak CD79a, CD138, and abundant cytoplasmic immunoglobulin, typically IgA. They are CD30 negative in most cases.134
When cytogenetic studies are performed, these tumors typically show complex chromosomal abnormalities. Immunophenotypic and genetic studies including gene expression profiling by several methodologies have demonstrated that DLBCL is a biologically heterogeneous group of neoplasms that includes at least two major subgroups: a germinal center (GC)-like group and an activated peripheral blood B-cell (ABC)-like group.139–139 The GC-like group is characterized by expression of CD10 and BCL-6, while negative for multiple myeloma oncogene (MUM1); contains ongoing Ig gene mutations; may show the t(14;18), 12q12 gains, amplification of c-rel gene (chromosome 2p); low levels of Blimp-1 gene mRNA expression; and a more favorable outcome. A significant number of the GC-like tumors occurring in adults also harbor the t(14;18), IGH/BCL2 translocation. The ABC-like group lacks expression of CD10 and BCL-6 as well as ongoing Ig gene mutations; may show trisomy 3 or gains on chromosomes 3q or 18q21-22 and losses at 6q21-22; has constitutive activation of nuclear factor kappa B (NF-κB), high levels of Blimp-1 mRNA expression, with lack of protein expression; aberrant IL-4-induced STAT6 signaling; and is associated with a poor prognosis.140
Recent studies have shown that in pediatric DLBCL, the distribution and characteristics of these subgroups are different, with children showing a much higher frequency of the centroblastic variant (83% vs. 75%, respectively) and of the GC-like subtype (83% vs. 40% to 48%, respectively), when compared with the adult tumors, and with a much lower incidence of the ABC-like subtype in this age group.133 In addition, the t(14;18) appears to be virtually absent in the pediatric GC-like DLBCL.133 Furthermore, these disease subgroups do not appear to have an impact on prognosis in children.133 All these insights offer interesting opportunities for differential, age-appropriate therapeutic targeting in the various disease subtypes in DLBCL. The most recent WHO Classification (2008)42 has introduced an additional category of aggressive B-cell lymphoma, designated as “B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and Burkitt lymphoma. This is a heterogeneous category of mature B-cell lymphomas, which includes cases of DLBCL with a gene expression profile similar to Burkitt lymphoma, many of which harbor dual translocations involving MYC and BCL2 (“double-hit lymphomas”), correlating with a particularly poor outcome. It is not yet clear if cases of DLBCL with high proliferation rate and immunophenotype resembling Burkitt lymphoma, but lacking a detectable MYC translocation, should also be included in the latter category.
Primary Mediastinal (Thymic) Large B-Cell Lymphoma (Med-DLBCL)
Med-DLBCL is a clinicopathologically and biologically distinct subtype of DLBCL that accounts for less than 10% of all large-cell lymphomas in children.40,141 It has also been designated as mediastinal clear-cell lymphoma of B-cell type and mediastinal diffuse large-cell lymphoma with sclerosis. It is currently thought to originate in the CD19+ CD21– B cells and asteroid B cells, that reside in the thymic medulla.142
Histologically, Med-DLBCLs can present as any of the DLBCL variants. Approximately half of the cases can have prominent associated stromal fibrosis, adding to the difficulty in obtaining suitable diagnostic biopsy samples in many of these cases. Cytologically, the neoplastic cells may have the appearance of centroblasts or immunoblasts, or may have abundant pale cytoplasm and markedly lobated, “flowerlike” nuclear membrane outlines (so-called “clear-cell” appearance).41,143–148
Immunophenotypically, Med-DLBCL shows a mature B-cell immunophenotype, with expression of CD45 and of pan-B-cell antigens, including CD19, CD20, CD79a, and PAX-5. There is typically partial and variable coexpression of CD30.149 Some cases may express CD23. Other antigens expressed include BCL-2 and BCL-6.150 The neoplastic cells are typically negative for CD10, CD21, immunoglobulin, and HLA class I and II molecules.151
Little conventional cytogenetic data is available largely because of the small tumor samples typically available in these patients. Alternative approaches have shown consistent gains of material on chromosome 2p (seen in ~25% of the cases), with amplification of c-REL, a gene encoding for a transcription factor important in B-cell development, as well as gains of material on 9p (50% to 75% of the cases) with amplification of the JAK2 gene, whose product is important in the JAK/STAT signaling pathway. Other alterations that have been described involve the chromosomes 6p (MHC class I locus), Xq, 12q, and the P53 gene.142,152,153
Biologically, Med-DLBCL has the profile of activated germinal center or postgerminal center cells. The lymphoma cells contain clonal Ig gene rearrangements with somatic mutations,136 BCL-6 gene mutations, and express MUM1/IRF4, BCL6, and the B cell–specific transcription factors OCT-2, BOB.1, and PU.1. Overexpression of an IL-4-inducible gene, FIG1 (IL-4I 1), suggests the possible oncogenic activation of signaling pathways such as IL-4/IL-13 or Janus kinase 2 (JAK2). Gene expression profiling experiments have demonstrated a close resemblance of this lymphoma with that of classical HL, suggesting that at least in the mediastinal location, these neoplasms may share a common origin from a thymic B cell. Notably, both tumors show overexpression of genes involved in cytokine signaling pathways, such as IL-13, tumor necrosis factor (TNF), and NF-κB. These findings suggest that targeting of some of these overexpressed genes or of activated NF-κB could represent therapeutic possibilities in these tumors.142
Uncommon Pediatric Lymphomas
Lymphomas that occur infrequently in children and adolescents include follicle center cell lymphoma,154 hepatosplenic lymphoma,155 panniculitislike T-cell lymphoma,156 mycosis fungoides,157–160 T/NK lymphoma, nasal-type natural killer,161 marginal zone lymphoma (including the extranodal MALT type),162 and HTLV-1-associated leukemia/lymphoma.163,164 The clinical and biological features of these lymphomas in children are generally similar to those observed in adults; however, this conclusion is drawn from the very few reported pediatric cases. Pediatric follicular lymphomas and pediatric marginal zone lymphomas appear to have a spectrum of clinicopathological features distinct from the tumors seen in adults and will be detailed below.
Pediatric Follicular Lymphoma42
Because of the relatively small numbers of cases reported, little is known about the genetics and biology of the follicular lymphomas occurring in children. It appears that in addition to rare cases of classic follicular lymphomas, resembling phenotypically and genetically those seen in adults, children may present with a second subtype, which is in fact more common in this age group.154,165–170 These follicular lymphomas, typically limited to one site (most commonly lymph nodes in the head and neck area or testis/epididymis) are characterized by effacement of the normal architecture by neoplastic follicles composed of large, expansive, irregular germinal centers with geographic appearance, and thin or absent mantle zones. Cytologically, the germinal centers are typically composed of monotonous predominantly large centroblasts, with occasional cases showing numerous mitotic figures and a “starry-sky” appearance because of numerous tingible-body macrophages. Most of these cases have morphologically the appearance of grade 2 or grade 3 (WHO) follicular lymphoma, albeit without the prognostic significance that this grading would have in classical adult follicular lymphoma.
Pediatric Nodal Marginal Zone Lymphoma42
These cases appear to have distinctive clinical and morphologic characteristics, based on very limited studies addressing this entity.162 These lymphomas present predominantly in males with typically asymptomatic disease, mostly stage I, involving most often lymph nodes in the head and neck area. Histologically, these lymphomas are largely similar to those seen in adults, with the distinctive feature of lymphoma cells often colonizing follicles with progressively transformed germinal centers.
Clinical Presentation
The primary sites of involvement and the extent of disease spread determine the clinical features of NHL at presentation (see Figure 97-1).4,5,25 Although adults usually present with nodal disease, children with NHL usually present with extranodal disease, most frequently involving the abdomen (31% percent of cases), the mediastinum (26% of cases), or head and neck region (29% of cases).4 These are rapidly growing tumors associated with hematogenous disease spread, and the majority of children with NHL present with locally invasive or advanced-stage disease. Involvement of the central nervous system is characterized by the presence of cranial nerve palsies and/or cerebrospinal fluid pleocytosis. When the bone marrow is involved, the distinction between NHL and leukemia is somewhat arbitrary: if more than 25% of the marrow is replaced by lymphoblasts, the patient is considered to have leukemia; if less than 25%, the patient is considered to have advanced-stage NHL with marrow involvement.
There is a striking relationship between the histologic subtype and the presenting site of disease in the lymphoblastic and Burkitt lymphomas.4 Among the lymphoblastic lymphomas, the typical primary site of involvement is the mediastinum and/or head and neck region, but rarely the abdomen, whereas the Burkitt lymphomas typically present in the abdomen or head and neck region, but rarely the mediastinum. In contrast to patients with lymphoblastic and Burkitt lymphomas, children with large-cell lymphomas may present with disease at almost any location. Involvement of the central nervous system at diagnosis is associated with both the Burkitt and lymphoblastic histiotypes, but rarely with large-cell histology.171 In contrast, spread to the bone marrow may occur in any of these three histologic subtypes.4,172
Primary involvement of the abdomen, as is typical of Burkitt lymphoma, may be associated with nausea, vomiting, or abdominal pain at presentation. These tumors usually arise from the distal ileum and result in obstruction of the bowel by either intussusception or direct compression of the lumen. Other primary sites of involvement in the abdomen include the appendix and/or large bowel. Abdominal tumors may be associated with malignant ascites; involvement of kidney, liver, or lymph node; and invasion of adjacent structures, including the abdominal wall.4,5 Involvement of the pelvis may include ureteral compression with associated hydronephrosis.
Primary involvement of the mediastinum, as is typical of advanced-stage lymphoblastic lymphoma and mediastinal large B-cell lymphoma, may be associated with respiratory symptoms ranging from mild cough to severe respiratory distress caused by direct tumor compression of the airway, requiring emergency attention (see Emergency Situations).4,5 Associated pleural effusions may further complicate the respiratory status. Compression of the superior vena cava by the tumor may obstruct venous blood return, resulting in swelling of the neck, shoulder, and face (“superior vena cava syndrome”); this condition may predispose the patient to the development of deep venous thrombosis. In rare cases, children with mediastinal masses may also have cardiac irregularities or tamponade.5
Involvement of the central nervous system may cause symptoms resulting from increased intracranial pressure, including nausea, vomiting, headache, and vision changes; there may also be neurological abnormalities on physical examination resulting from palsies of cranial nerves innervating the face or extraocular muscles. Involvement of the bone marrow may be associated with bone pain, pallor, neutropenia, and/or thrombocytopenia with associated bruising and bleeding. Involvement of the skin occurs in approximately 4% of children with newly diagnosed NHL. When present, it is usually associated with CD30+ anaplastic large-cell histology (ALCL)175–175; however, lymphoblastic lymphomas (often non-T-cell immunophenotype) may also involve the skin.176,177
Diagnosis and Differential Diagnosis
The NHLs of childhood grow very rapidly; therefore an expeditious diagnostic workup in consultation with a pediatric oncologist is indicated if NHL is suspected (Figure 97-2). The diagnosis of NHL is most readily established by examination of tissue obtained by open biopsy of the involved site. A comprehensive characterization of the biological features of the tissue, including histologic, immunophenotypic, cytogenetic, and molecular studies, will help to distinguish NHL from the small round blue cell tumors, including Ewing sarcoma, neuroblastoma, and rhabdomyosarcoma. When patients (such as those with large anterior mediastinal masses and associated airway compression) are too unstable to undergo anesthesia for open biopsy, the diagnosis may be established by parasternal fine-needle aspiration or biopsy with local anesthesia.178 Among children who present with an associated pleural effusion, thoracentesis with cytologic examination of pleural fluid is usually diagnostic. For children with large abdominal Burkitt tumors, either percutaneous aspiration of the mass or paracentesis to obtain ascitic fluid often yields diagnostic cytologic and cytogenetic findings. A bone marrow and cerebrospinal fluid examination should be performed early in the workup of a child with suspected NHL, because these studies may be diagnostic and may preclude the need for more invasive procedures.
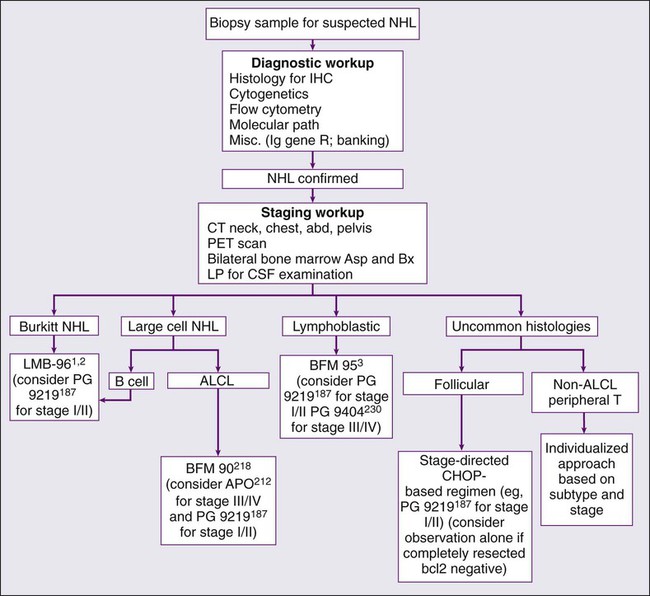
Initial Evaluation and Staging Workup
A prompt and meticulous staging workup is imperative, because treatment is determined in part by primary site and degree of disease spread. A complete history and physical examination, including documentation of the presence or absence of B symptoms by history, should be completed. Computed tomographic imaging of the chest, abdomen, and pelvis, as well as bone scans should be performed on all patients. Nuclear imaging studies such as gallium and bone scans have historically been used by many in the staging workup of children with newly diagnosed NHL.4,5,25 Currently, positron emission tomography (PET) is commonly used in the evaluation of adults with Hodgkin or non-Hodgkin lymphoma179; this modality has largely replaced the use of gallium scanning as part of the staging workup of children with NHL. Of note, not all cooperative group studies incorporate PET scanning in their protocols. Moreover, there remains some controversy regarding the interpretation and use of PET imaging in the staging workup and response evaluation of children with NHL.180–192 Bilateral posterior iliac crest aspirations and biopsies increase the chance of identifying overt marrow involvement, thus reducing the possibility of underestimating the stage of disease.193 The bone marrow samples should be submitted for cell count and differential, flow cytometric, cytogenetic, and molecular pathological analyses. Magnetic resonance imaging of the bone marrow has been shown to be effective in detecting occult disease is some studies194; however, it is not a standard component of the currently accepted pediatric NHL workup. A lumbar puncture should be performed for cytologic evaluation of the cerebrospinal fluid.
The stage of disease is usually assigned according to the St. Jude Staging System described by Murphy (Table 97-3),65 which was developed to accommodate the noncontiguous nature of disease spread, predominant extranodal involvement, and involvement of the bone marrow and central nervous system that characterize the pediatric NHLs. Stages I and II are considered to be limited-stage disease, whereas stages III and IV are advanced-stage disease. The initial laboratory evaluation should include a complete blood count with differential and a chemistry panel comprising electrolytes, blood urea nitrogen, creatinine, lactate dehydrogenase (LDH), calcium, phosphorus, and uric acid. An HIV screen should be performed on all patients newly diagnosed with lymphoma. Those who test positive may be at increased risk for therapy-related toxicity, including life-threatening infections. Serologic tests for EBV infection may be helpful when lymphoproliferative disease is highly suspected in the differential diagnosis; however, positive serologic results do not rule out a malignancy.
A single tumor (extranodal) with regional node involvement.
Two or more nodal areas on the same side of the diaphragm.
Two single (extranodal) tumors, with or without regional node involvement on the same side of the diaphragm.
A primary gastrointestinal tract tumor (usually in the ileocecal area), with or without involvement of associated mesenteric nodes, that is completely resectable.
Two single tumors (extranodal) on opposite sides of the diaphragm.
Two or more nodal areas above and below the diaphragm.
Any primary intrathoracic tumor (mediastinal, pleural, or thymic).
Extensive primary intraabdominal disease.
Any paraspinal or epidural tumor, whether or not other sites are involved.

Prognostic Factors
Tumor burden at diagnosis, reflected by both the disease stage and serum LDH, is an important predictor of outcome for children.4 In one large, single-institution study of childhood NHL, the treatment era, disease stage, and serum LDH level all emerged as independent and significant prognostic indicators.4 Serum IL-2 receptor levels, which reflect tumor burden, have also been shown to predict outcome.195,196 Of interest, stage was not found to be predictive of outcome among children with ALCL treated on the recently completed ALCL study.
A poor early response to therapy is considered by some to confer a poorer prognosis, as has been demonstrated in children with ALL.197 In the LMB-89 protocol, poor early response is a criterion for placement in the most intensive arm of therapy.198 The current COG study for children with advanced-stage lymphoblastic lymphoma is examining the impact of early response as determined both by diagnostic imaging and flow cytometric MRD technology. The use of flow cytometric MRD technology in children with T-lymphoblastic lymphoma demonstrated a poorer treatment outcome for those with a higher level of minimal disseminated disease (MDD) in the bone marrow at the time of diagnosis.199 Specifically, those with greater than 1% level of lymphoblasts had a poorer event-free survival compared to those with a lower level of detectable disease. Of interest, this study also demonstrated a comparable level of MDD in the bone marrow and peripheral blood—a similar observation as made in patients with T-ALL.200
Prognostic factors have also been identified with respect to specific histologic subtypes. For example, among adults with ALCL, ALK protein expression is a favorable prognostic factor.203–203 It is unclear if ALK status has prognostic significance in children, although one study suggested that it did not influence outcome.204 Additional data are needed on this subtype to be certain of its prognostic significance. In a multivariate analysis of clinical features in childhood ALCL, mediastinal, visceral (lung, liver, spleen), and skin involvement were associated with adverse risk.205 There are certain pathological features that have been shown to have prognostic significance in pediatric ALCL.206 For example, in the ALCL99 study, those with lymphohistiocytic–small cell variant components had a significantly poorer event-free survival compared to those lacking this feature. Another study suggested that those children with B-lineage ALCL may have a poorer prognosis.134 A poorer event-free survival has also been associated with a leukemic presentation.102 Among children with ALCL, the presence of circulating tumor cells in the peripheral blood or bone marrow as detected by either quantitative or qualitative PCR has been reported to be associated with a poorer outcome.207
Primary Treatment
Treatment of the NHLs of childhood has advanced dramatically over the past 30 years through the incremental development and clinical testing of effective multiagent chemotherapeutic regimens.4,5,25,171,198,205,208–246 In a randomized trial comparing two of the first successful regimens for treating childhood NHL (the cyclophosphamide-based COMP regimen and the multiagent LSA2L2 regimen designed for ALL), the Children’s Cancer Group clearly demonstrated that children with limited-stage disease fared well regardless of histology or treatment arm assignment, whereas treatment outcomes for children with advanced-stage disease varied with histology and treatment arm.208 Children with Burkitt lymphoma had a better outcome with the COMP regimen,208 whereas those with lymphoblastic NHL fared better with the LSA2L2 regimen.210–210 However, among those who presented with advanced-stage large-cell disease, neither treatment arm emerged as superior. In contrast to the stage- and histology-directed therapeutic approach that has been historically used in the United States, a stage- and immunophenotype-directed approach is predominant in Europe. Recent studies in both the United States and Europe suggest that the immunophenotype-directed approach has advantages for treating children with advanced-stage large-cell NHL, a family of lymphomas whose prognoses vary with immunophenotype.211
Surgery and radiation therapy have minimal roles in the management of childhood NHLs.4,5 Surgery is indicated only for diagnostic purposes and in cases where an ileocecal mass, associated with mesenteric nodes only, can be completely resected (resulting in downstaging from III to II, which requires less intensive systemic therapy; see Staging).247 Otherwise, aggressive debulking procedures should not be undertaken. Prospective randomized trials have demonstrated that the incorporation of IFRT into a multiagent chemotherapy regimen does not improve outcome.214,244 The use of radiation therapy in the management of relapse, central nervous system disease, and certain emergency situations will be discussed in other sections.
Initial Management
The serum chemistry values should be reviewed before chemotherapy is started. Many patients with bulky Burkitt or lymphoblastic lymphomas present with hyperuricemia, hyperphosphatemia, and renal dysfunction because of the rapid turnover of lymphoblasts. These metabolic abnormalities are exacerbated by chemotherapy, which rapidly lyses tumor cells. Tumor lysis releases purines, potassium, and phosphorus into the bloodstream, resulting in the deposition of uric acid, xanthines, and phosphates in the renal tubules, which causes further renal dysfunction—a clinical condition termed “tumor lysis syndrome.”5,248 This problem can be precluded or reduced if patients with advanced-stage disease are vigorously hydrated (3 to 4 L/m2/day) prior to receiving chemotherapy. Alkalization is usually necessary to maintain a urine pH of approximately 7.0. The urinary excretion of uric acid is reduced at an acidic pH, whereas the excretion of phosphorus is impaired by overalkalinization. Allopurinol, a xanthine oxidase inhibitor, has historically been helpful in the management and prevention of hyperuricemia by blocking ongoing production of uric acid. Urate oxidase, a uricolytic agent used for many years in Europe, has advantages that make it a preferable alternative.249 It results in a precipitous drop in uric acid by converting it to allantoin, precludes the need for vigorous alkalinization, and is associated with preservation of normal renal function and avoidance of dialysis in most cases. Rasburicase, a recombinant form of this drug, is also effective in reducing uric acid, has a much lower risk of associated allergic reactions, and has recently become a standard part of initial management.250 In the rare case in which urine output is unacceptable, addition of mannitol followed by furosemide (Lasix®) may be necessary.
Limited-Stage Disease
The excellent treatment results (survival rates of 85% to 95% at 5 years) achieved in children with limited-stage NHL have prompted an emphasis on new treatment strategies that reduce both morbidity and the risk of late sequelae (e.g., anthracycline-induced cardiomyopathy, sterility, and second malignancies) while maintaining or improving the treatment outcome.198,213,214,229,232,251 Various approaches to reduction of treatment intensity have been investigated. The Pediatric Oncology Group (POG) performed two sequential randomized trials that examined the need for IFRT and maintenance chemotherapy, respectively.214 In the first study, patients were randomly assigned to receive either chemotherapy alone (three courses of cyclophosphamide, doxorubicin, vincristine, and prednisone [CHOP] followed by a 24-week continuation phase of 6-mercaptopurine and low-dose methotrexate) or chemotherapy plus IFRT. In the subsequent study, patients were randomly assigned to receive the three courses of CHOP with or without the 24-week continuation phase.214 These studies demonstrated that both IFRT and the continuation phase of therapy could be safely eliminated without compromising outcome, with one exception. Children with limited-stage lymphoblastic lymphoma had lower event-free survival rates than did those with nonlymphoblastic histology. Even with the 24-week continuation phase, one-third of the patients with lymphoblastic lymphoma developed recurrent disease. With salvage therapy, there was no reported difference in overall survival between those with lymphoblastic and nonlymphoblastic histology. The optimal management of limited-stage lymphoblastic lymphoma remains controversial. The French Society of Pediatric Oncology (SFOP) uses a more aggressive initial approach to avoid the need for retreatment.228 Children with limited-stage lymphoblastic lymphoma are entered on the same protocol used for advanced-stage lymphoblastic lymphoma, and children with limited-stage nonlymphoblastic lymphoma are candidates for less-intensive regimens only if there is complete resection of disease.198 The current COG trial for lymphoblastic lymphoma is piloting an arm of ALL-derived therapy for those children presenting with limited-stage disease.
Advanced-Stage Disease
Attempts to improve the treatment result in children with advanced-stage NHL have relied on further intensification of therapy. Historically, the general approach in the United States has been histology-directed, whereas in Europe an immunophenotype-directed approach has predominated. For example, for many years French investigators have entered patients with Burkitt lymphoma and B-cell large-cell lymphoma on the same protocol.2,198 This approach is now widely used in the United States.
Burkitt Lymphoma
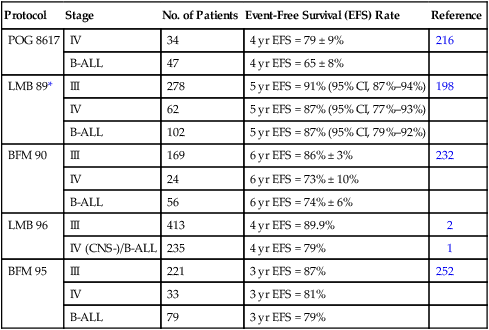
The dramatic improvements achieved in the treatment of advanced-stage Burkitt lymphoma and B-cell ALL represent one of pediatric oncology’s undisputed success stories. With current therapy, which is generally cyclophosphamide based, very intensive, and given over a relatively short period of time (4 to 8 months), approximately 80% to 90% of patients are long-term event-free survivors. Results achieved with the COMP regimen were first improved on by the incorporation of high-dose methotrexate and/or high-dose cytarabine.212,215–218 Two studies during the same period showed that the duration of therapy for stage III patients could be shortened to 2 to 4 months without compromising patient outcome.217,221 Further improvements in treatment outcome were achieved with further intensification of therapy (escalation of cyclophosphamide, methotrexate, and cytarabine dosages) and by the addition of new active agents, including etoposide and ifosfamide.198,232,234 The LMB-89 regimen designed by the SFOP, for example, has produced one of the best results to date.198 In this protocol, children receive high-dose methotrexate (3 g/m2), fractionated CHOP and low-dose cytarabine given over approximately 5 months if marrow blast cells at diagnosis are <70%. For children with >70% marrow blast cells or with central nervous system (CNS) involvement at diagnosis, therapy is intensified (methotrexate dose is escalated to 8 g/m2, high-dose cytarabine and etoposide are incorporated, and the total duration of therapy is extended to approximately 8 months). A subsequent international trial (LMB-96) examined the safety of intensity reduction in the Group B and C arms of the LMB-89 study.1,2 The intensity of Group B therapy was safely reduced (i.e., no escalation of cyclophosphamide in the second COPADM course, and deletion of the maintenance sequence) without compromising the excellent outcome; however, intensity of therapy of the Group C arm could not be safely reduced.
Other successful treatment strategies for advanced-stage Burkitt lymphoma and B-cell ALL have been reported (Table 97-4). Excellent results have been achieved by the BFM (Berlin-Frankfurt-Munster) cooperative group using a regimen that incorporates high-dose methotrexate (5 g/m2), ifosfamide, etoposide, doxorubicin, and steroids.232 BFM trials have also examined the impact of methotrexate infusion time; they found that a 4-hour infusion time was noninferior to a 24-hour infusion for children with limited-stage disease but not for those with advanced-stage disease.252 The current BFM trial incorporates a rituximab window that precedes conventional therapy.253 Sequential NCI studies have shown that adding etoposide, high-dose cytarabine, and ifosfamide to a regimen of cyclophosphamide, doxorubicin, vincristine, and high-dose methotrexate improved the treatment result.234 Importantly, these more aggressive approaches have shown encouraging preliminary results in adults with the same malignancies.254
Table 97-4
Treatment Outcome for Advanced-Stage Burkitt Lymphoma
| Protocol | Stage | No. of Patients | Event-Free Survival (EFS) Rate | Reference |
| POG 8617 | IV | 34 | 4 yr EFS = 79 ± 9% | 216 |
| B-ALL | 47 | 4 yr EFS = 65 ± 8% | ||
| LMB 89* | III | 278 | 5 yr EFS = 91% (95% CI, 87%–94%) | 198 |
| IV | 62 | 5 yr EFS = 87% (95% CI, 77%–93%) | ||
| B-ALL | 102 | 5 yr EFS = 87% (95% CI, 79%–92%) | ||
| BFM 90 | III | 169 | 6 yr EFS = 86% ± 3% | 232 |
| IV | 24 | 6 yr EFS = 73% ± 10% | ||
| B-ALL | 56 | 6 yr EFS = 74% ± 6% | ||
| LMB 96 | III | 413 | 4 yr EFS = 89.9% | 2 |
| IV (CNS-)/B-ALL | 235 | 4 yr EFS = 79% | 1 | |
| BFM 95 | III | 221 | 3 yr EFS = 87% | 252 |
| IV | 33 | 3 yr EFS = 81% | ||
| B-ALL | 79 | 3 yr EFS = 79% |
Lymphoblastic Lymphoma
Most of the successful treatment plans for children with advanced-stage lymphoblastic lymphoma are derived from therapies designed to treat children with high-risk ALL (Table 97-5). These regimens, which generally use multiple chemotherapy agents (up to 10 different agents) and are given over a 15- to 32-month period, result in an approximate 65% to 90% event-free survival rate at 4 to 5 years.208–210,222,223,228,231,235 The multiagent nature of these regimens makes it difficult to determine the relative merit of individual components. For example, the need for high-dose methotrexate is now in dispute. The French (SFOP) achieved an excellent result by incorporating courses of high-dose methotrexate into the previously studied LSA2L2 regimen.228 Moreover, the BFM group has recently reported very encouraging results (3-year event-free survival of approximately 90%) of a regimen that features an intensive high-dose methotrexate (5 g/m2) consolidation course.235 In a subsequent study, it was demonstrated that prophylactic cranial irradiation could be safely eliminated without compromising the excellent outcome.3 Although definitive data are lacking, this result may reflect the higher levels of intracellular methotrexate polyglutamates produced in T-cell lymphoblasts by high-dose methotrexate as compared to low-dose methotrexate.255 In contrast, data from a recently published POG study suggested that high-dose methotrexate may not be needed for advanced-stage lymphoblastic lymphoma in the context of an anthracycline and l-asparaginase rich backbone.256,257 Moreover, preliminary data from a recently completed COG study also suggests that high-dose methotrexate can be safely eliminated—if replaced by extended intrathecal therapy.258 l-Asparaginase is also commonly used in many successful regimens. In this regard, a POG study demonstrated a survival advantage to those randomized to receive additional l-asparaginase.241 Efforts to improve the treatment result have also included the addition of new active agents (e.g., epipodophyllotoxins)222,259 and the incorporation of a reinduction235 or late intensification phase.
Table 97-5
Treatment Outcome for Advanced-Stage Lymphoblastic Non-Hodgkin Lymphoma
| Protocol | Stage | No. of Patients | Event-Free Survival Rate | Reference |
| LSA2L2 (modified) CCG-551 |
III and IV | 124 | 5-y EFS = 64% | 208 |
| BFM 90 | III | 82 | 5-y EFS = 90% ± 3% | 235 |
| IV | 19 | 5-y EFS = 95% ± 5% | ||
| X-H SJCRH | III and IV | 22 | 4-y DFS = 73% | 222 |
| APO (Dana Farber) | III and IV | 21 | 3-y DFS = 58% ± 23% | 246 |
| A-COP + (POG) | III | 33 | 3-y DFS = 54% ± 9% | 223 |
| SFOP LMT81 | III | 33 | 57 mo EFS = 79% (SE, 4%) | 228 |
| IV and ALL | 43 | 57 mo EFS = 72% (SE, 4%) | ||
| CCG: LSA2L2 (modified) | I–IV | 243 | 5-y EFS = 74% | 231 |
| vs. | ||||
| ADCOMP | I–IV | 138 | 5-y EFS = 64% | |
| POG8704: no extra Asp | III and IV | 83 | 4-y CCR = 64% (SE, 6%) | 241 |
| vs. | ||||
| extra Asp | III and IV | 84 | 4-y CCR = 78% (SE, 5%) | |
| BFM 95 | III and IV(CNS–) | 156 | 5-y DFS = 88% | 3 |
| NHL 13 | III and IV | 41 | 5-y EFS = 82.% | 259 |
| POG 9404 | III and IV | 137 | 5-y EFS = 84.6% | 256 |
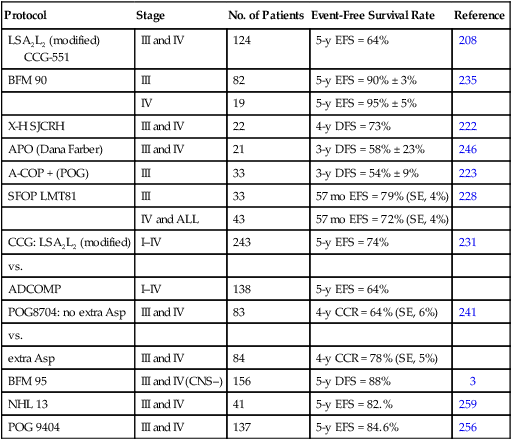
Large-Cell Lymphoma
Historically, the biological heterogeneity of advanced-stage large-cell lymphoma coupled with quite varied treatment strategies reported made it difficult to identify an optimal treatment approach (Table 97-6).171 Histology-directed therapies in the United States, primarily CHOP-based, resulted in a 50% to 70% event-free survival rate at 3 years.171,208,211,219,224–226 Although some studies have examined the feasibility of eliminating agents associated with significant late effects,219,239 some have examined the benefit of incorporating additional active agents (e.g., intermediate- and high-dose methotrexate, intermediate- and high-dose cytarabine, ifosfamide, and carboplatin).240,260 POG investigators reported that children with large-cell lymphoma of the B-cell immunophenotype had a better outcome than did those with a non-B-cell immunophenotype—a finding which suggested that immunophenotype-directed therapies for pediatric large-cell lymphomas should be further pursued.211 In this regard, European trials for children with large-cell lymphoma have historically assigned treatment on the basis of immunophenotype (e.g., B cell, T cell, CD30+). Currently, trials in both the United States and Europe for large-cell lymphoma are directed toward specific immunophenotypically defined categories as designated in the most recent WHO classification system.42 The two most common large-cell types encountered in children include DLBCL and ALCL.
Table 97-6
Treatment Outcome for Advanced Stage Large-Cell Non-Hodgkin Lymphoma
| Protocol | Stage | No. of Patients | Event-Free Survival Rate | Reference |
| CHOP | III and IV | 21 | 3-y EFS = 62% ± 11% | 171 |
| MACOP-B | III and IV | 11 | 3-y EFS = 55% ± 16% | 226 |
| COMP | III and IV | 42 | 5-y EFS = 52% | |
| vs. | 208 | |||
| LSA2L2 | III and IV | 18 | 5-y EFS = 43% | |
| APO | III and IV | 62 | 3-y EFS = 72 ± 6% | |
| vs. | 239 | |||
| ACOP+ | III and IV | 58 | 4-y EFS = 62 ± 7% |
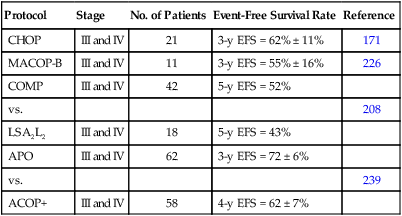
Diffuse Large B-Cell Lymphoma.
The SFOP reported equally excellent outcomes for children with either large B-cell or Burkitt lymphoma who were treated on the same B-cell regimen (i.e., LMB-89).198 This observation was confirmed in the recently completed international collaborative LMB-96 trial, which resulted in a 4-year event-free survival rate of 92.5% for patients with DLBCL (excluding primary mediastinal disease) compared to 93.5% for those with Burkitt lymphoma. In this trial, those with primary mediastinal disease had a somewhat inferior outcome (4-year event-free survival of 71.5%).2 Other studies have also suggested that children with mediastinal B large cell lymphomas (MBCLC) have a slightly worse outcome compared to those with DLBCL.141,144 The anthracycline-based APO regimen was also shown to be active in children with DLBCL (i.e., 4-year event-free survival of 63.8%); however, the result is somewhat inferior to that of the LMB-96 approach.239,240
There is a recognized need to identify novel strategies for the mediastinal large B-cell subtype of pediatric NHL. The addition of an immunotherapeutic competent is a consideration. In this regard, the activity of the DA-EPOCH-R regimen261 that incorporates rituximab (anti-CD20) into a backbone of more conventional agents delivered in a prolonged infusional fashion is currently being studied in children.
Anaplastic Large-Cell Lymphoma.
The therapeutic approaches for children with CD30+ ALCL are quite varied.238,240,245 An immunophenotype-directed strategy has been used in Europe for many years. The German BFM (Berlin-Frankfurt-Munster) group has treated children with CD30+ ALCL with a B-cell approach (Burkitt lymphoma-like), achieving one of the best published outcomes to date (5-year event-free survival of approximately 76% for all patients).227,245 The activity of a B-cell approach (NHL 9002), based on the very successful French LMB-89 B-cell protocol was studied in The United Kingdom Children’s Cancer Study Group. In the NHL 9002 study, those with stage II, III, and non-CNS+ stage IV ALCL were treated with the Group B arm of the LMB-89 regimen (5-year event-free survival rate of 55%).205 It has yet to be determined why this approach appeared to be inferior to the B-cell approach used in the BFM trials; however, it has been suggested that it may be a result of the more frequent use of alkylators in the BFM regimen. In the French (SFOP) trials, children with CD30+ ALCL have been238 enrolled on the HM 89 and HM 91 protocols, which both feature an initial treatment phase (one course of COP followed by two courses of COPADM) based on their very successful B-cell protocol, LMB-89. The maintenance phases differed from LMB-89: the HM 89 protocol featured alternating courses of VEM (etoposide, methotrexate, and cyclophosphamide) and VAD (vincristine and doxorubicin) for a total of eight courses and the HM 91 regimen featured alternating courses of VEBBP (vinblastine, etoposide, bleomycin, and prednisone) and Sequence 1 (vincristine, methotrexate, cyclophosphamide, and doxorubicin) for a total of eight courses. A 60% (54% to 76%) 3-year event-free survival for all stages treated with HM89 and HM91; however, the 3-year event-free survival was only 55% for stage III and IV patients.
A histology-directed approach has historically been used in the United States to treat children with large-cell lymphoma. One of the most widely used regimens is APO, which features an induction phase (doxorubicin, vincristine, prednisone, and intrathecal methotrexate) followed by sequential maintenance phases (doxorubicin, methotrexate, vincristine, prednisone, 6-mercaptopurine, and intrathecal methotrexate).224,239,240 A randomized trial performed by the POG examined the potential benefit of adding intermediate-dose methotrexate and high-dose cytarabine to an APO backbone and found no difference in outcome regardless of immunophenotype.240 A 4-year event-free survival and overall survival of 71.8% and 88.1%, respectively, was reported for the 86 patients with advanced-stage disease enrolled on the study.
Trials in Europe and the United States were designed to build on the successful results achieved with both the BFM B-cell and APO strategies. Both trials examined the benefit of adding vinblastine, an agent that has activity as a single agent in multiply-relapsed ALCL patients.242,262 The multinational European ALCL99 trial included a randomization for vinblastine with a BFM backbone for high-risk patients. Although there initially appeared to be an advantage in the arm with an extended vinblastine maintenance component, after completion of the vinblastine, there was no significant difference in long-term event-free survival between the two arms (i.e., vinblastine vs. no vinblastine).263 Another important observation in this trial was that the treatment outcome for children randomized to receive high-dose methotrexate (3 g/m2) given over 3 hours (without intrathecal methotrexate) was not significantly different than that achieved when methotrexate was given at 1 g/m2 over 24 hours with intrathecal methotrexate; moreover, there was less toxicity associated with the shorter infusion time.264 Preliminary results from the APO trial, which also featured a randomization of vinblastine for patients with advanced-stage ALCL, do not reveal any improvement in treatment outcome associated with vinblastine, although the final report is pending.265
Rare Histologic Subtypes
The most common low-grade B-cell malignancies encountered in children include follicular lymphomas and marginal zone B-cell lymphomas. One study suggests that those with Bcl-2-negative tumors are more likely to be limited stage and less likely to relapse.154 Children with localized follicular lymphoma have been successfully treated with CHOP-based limited-stage therapeutic approaches; however, there have been reports of children who have remained disease free following complete resection without adjuvant chemotherapy.154,266,267 In this regard, conservative treatment measures have also been successful in the management of children with marginal zone B-cell lymphomas.162
The prognosis for those children who present with non-ALCL mature (“peripheral”) T-cell lymphomas is poorer than that for those with either follicular or marginal zone B-cell lymphomas.268,269 For example, children with hepatosplenic lymphomas have an aggressive disease course, and often have recurrent disease even after an initial good response to aggressive therapy.270–274 There is clearly a need for organized national trials to improve our treatment approaches to these and other infrequently encountered histologic subtypes.
CNS Prophylaxis and Treatment
The prevention of disease spread to the CNS is an important component of most successful modern treatment protocols. This generally involves both the administration of chemotherapy directly into the cerebrospinal fluid (intrathecally by lumbar puncture or less commonly by the intraventricular route) and the administration of systemic high-dose chemotherapy (e.g., methotrexate, cytarabine). Prophylactic intrathecal therapy may be unnecessary in children who have limited-stage disease sparing the head and neck region214 or in some subtypes of advanced non–B cell large-cell lymphoma.275 There does not appear to be any benefit to the incorporation of cranial radiation as CNS prophylaxis for children with NHL. Although it had been used in a successful BFM study for children with advanced-stage lymphoblastic lymphoma, a subsequent BFM study demonstrated it could be safely eliminated without compromising outcome.3 A St. Jude study also demonstrated that prophylactic cranial irradiation could be safely eliminated in the management of pediatric advanced-stage lymphoblastic lymphoma.259
Emergency Situations
Various emergencies may arise in the management of children with NHL.5 First, children who present with large anterior mediastinal masses associated with severe respiratory distress require immediate attention. If there is significant airway compression, deep sedation should be avoided. Chemotherapy should be started as soon as possible if the diagnostic samples have been obtained. If they have not, local radiation therapy may be the best approach, because it spares more peripheral disease for subsequent biopsy and study. Steroids may be necessary in some cases, but this therapy may alter tumor histology and preclude an accurate tissue diagnosis.
A small percentage of children may present with epidural tumors causing cord compression and corresponding neurologic deficits. Delivery of appropriate chemotherapy is usually all that is required; however, if there is no prompt recovery of neurologic function, a radiation therapist should be consulted about the possible need for low-dose local irradiation.5
Treatment Complications
As the survival of children with malignant lymphomas has improved, there has been increased attention focused on treatment-related late effects.276 The most serious treatment sequelae in survivors of childhood NHL include anthracycline-related cardiomyopathy, cyclophosphamide-related sterility, second cancers, and transfusion-related hepatitis. Attempts to reduce or eliminate these complications have included the dose reduction or elimination of both specific chemotherapeutic agents as well as involved field irradiation. Improving the process of blood product screening has also been an important area of focused research.
The design of clinical trials for pediatric NHL has been strongly influenced by the desire to avoid anthracycline-induced cardiac toxicity. Although it has been demonstrated that adults may tolerate cumulative doses of doxorubicin of 550 mg/m2, much lower cumulative doses have been shown to have clinically significant effects on ventricular contractility in children.277 Factors that have been shown to be predictive of cardiac dysfunction include cumulative anthracycline dosage, higher anthracycline dose intensity, younger age at time of treatment, female sex, time interval since completion of therapy, and combined-modality therapy that includes mediastinal irradiation.278,279 Future trials that focus on the reduction of cumulative anthracycline dosage and the utility of cardioprotectant agents are indicated.219
The preservation of fertility is another important concern in the development of optimal therapeutic strategies for children with NHL. A dose-related depletion of germinal cells is associated with the use of alkylating agents such as cyclophosphamide and ifosfamide; these agents tend to be more gonadotoxic in males. Studies thus far have suggested that sterility is likely at cumulative doses of cyclophosphamide greater than 7.5 g/m2, whereas fertility is usually maintained at cumulative dosages less than 4 g/m2. In this regard, a number of pediatric NHL trials have attempted to eliminate or reduce the dosage of cyclophosphamide.239
Pediatric NHL trials have also focused on the elimination of involved field irradiation. In the first of two POG trials for limited-stage NHL, it was demonstrated that IFRT could be safely eliminated without compromising outcome.214 A similar observation was made for children with advanced-stage disease in a St. Jude study.244 Although involved field irradiation is not used in most current NHL trials, cranial irradiation is considered in the management of overt CNS involvement in children with lymphoblastic lymphoma.
Follow-Up
Management of Primary Treatment Failure
Children with refractory or recurrent disease, particularly after receiving modern intensive therapy, are generally considered to have a poor prognosis. Thus, aggressive or novel approaches are often used to treat relapse. Current approaches generally comprise multiagent salvage chemotherapy followed by an intensification phase that may include autologous or allogeneic hematopoietic stem cell transplantation (HSCT). Multiagent salvage regimens that have been studied include DHAP (dexamethasone, high-dose cytarabine, and platinum),226 which has been shown to be active in childhood and adult recurrent large-cell lymphoma, and VIPA (etoposide, ifosfamide, and high-dose cytarabine),280 which has been shown to be active in children with relapsed Burkitt lymphoma. Other active salvage regimens for children with refractory or recurrent malignant lymphoma include ICE (ifosfamide, carboplatin, and etoposide).281 The addition of rituximab to the ICE regimen has been shown to be active for patients with recurrent CD20+ high-grade mature B-cell lymphomas (e.g., Burkitt lymphoma, DLBCL). The MIED (high-dose methotrexate, ifosfamide, etoposide, and dexamethasone) has been shown to be active in children with recurrent nonlymphoblastic lymphoma.260 Children who are found to have chemosensitive recurrent disease are usually considered for an intensification phase of chemotherapy followed by an HSCT. In a recent report, however, the benefit of HSCT in this setting was questioned.243 Although published data on the use of HSCT in children with recurrent or refractory NHL is limited compared to that reported for adults, there are reported studies supporting its use.232,282–290 For example, European cooperative group trials demonstrated that some children with Burkitt lymphoma who had a poor early response to therapy could by successfully salvaged with high-dose chemotherapy followed by an autologous HSCT.232,285,286,288,291 Moreover, the Spanish Working Party for Bone Marrow Transplantation reported a 58% event-free survival rate following HSCT in children with either recurrent/refractory NHL or high-risk NHL in first complete remission.285 The SFOP reported that 8 of 24 children with NHL who failed initial therapy were long-term disease-free survivors following HSCT.286 In a review of 22 children at the St. Jude Children’s Research Hospital, approximately 45% were survivors following intensive chemotherapy and HSCT.290 It is difficult to make direct comparisons between the published studies of HSCT in children because they vary with respect to type of HSCT (autologous versus allogeneic), preparative regimen, numbers of patients, and histologic subtypes studied.
It appears, however, that histologic subtype should be considered in determining the type of HSCT (autologous vs. allogeneic) for children with refractory or recurrent NHL.292 Among children with Burkitt lymphoma, an autologous approach has been shown to be beneficial for those with a poor early response.232,282,286 For those with disseminated recurrent Burkitt lymphoma involving the marrow, many would favor an allogeneic approach if a suitable donor can be identified; however, following RICE, some patients with high-grade mature B-cell lymphomas were in long-term clinical complete remission following autologous HSCT if they responded well to RICE.293 An autologous HSCT has been shown to be effective strategy for some children with recurrent ALCL.284,290,292 The BFM study reported though that among those children who received an autologous HSCT for recurrent ALCL, those with either a CD3+ immunophenotype or an early relapse had a poorer prognosis.294 A recent study by the BFM identified an excellent outcome using an allogeneic HSCT strategy for those with high-risk ALCL relapses.295 Among children with recurrent lymphoblastic lymphoma, the results with autologous HSCT have been discouraging.292,243 Most would consider an allogeneic HSCT for children with recurrent lymphoblastic lymphoma if a suitable donor can be identified.
Various preparative regimens have been successfully used in the salvage of children with refractory or recurrent NHL.232,282–290 Two of the earliest reported regimens are BACT (carmustine, cytarabine, cyclophosphamide, and thioguanine) and BEAM (carmustine, etoposide, cytarabine, and melphalan).282,283,289 High-dose busulfan was reported as an important component of a successful French (SFOP) preparative regimen.286 Gordon et al. reported the successful salvage of children with recurrent peripheral T cell lymphoma using a preparative regimen that featured thiotepa284; however, it was associated with significant mucositis.
Currently, most would consider an autologous HSCT for children with chemosensitive recurrent ALCL, and in those with Burkitt lymphoma who have a poor early response or in some cases of recurrent disease. If a suitable donor is available, an allogenic HSCT approach would be considered for children with widely disseminated recurrent lymphoblastic lymphoma or Burkitt lymphoma involving the bone marrow. An allogeneic HSCT approach may also be considered for those with high-risk recurrent ALCL. It is clear that there is need for additional prospective clinical trials to evaluate the optimal preparative regimen and HSCT approach for children with recurrent or refractory NHL. In this regard, the European Lymphoma Bone Marrow Transplantation Registry has suggested that the potential graft versus lymphoma effect of allogeneic HSCT be studied.288
After Completion of Therapy Clinic
It is important that all children who have completed therapy for NHL be followed annually in an “after completion of therapy” or late effects clinic.296,297 The purposes of these visits are both to screen for therapy-related late effects (see section on complications of therapy) and to provide education and counseling about the medical and psychosocial issues that affect cancer survivors. These clinics also provide an opportunity for children and young adults to participate in important research initiatives that focus on cancer prevention and control, psychosocial problems, and treatment-related sequelae.
Future Directions
Despite dramatic improvements in the treatment of childhood NHL, approximately 20% to 30% of patients either do not achieve a complete remission or develop recurrent disease.4,5,25 Treatment-related late effects that place cancer survivors at risk are of additional concern.276 Therefore, the development of more effective and less toxic therapy remains an ongoing challenge. The identification of clinical and biological features that are predictive of treatment failure may help in the refinement of a risk-adapted treatment approach to children with NHL.
Improvement in treatment outcome may be achieved by the incorporation of new active agents, or the development of novel schedules for the delivery of currently used agents. Novel therapeutic approaches include the incorporation of immunotherapeutic approaches into multiagent chemotherapeutic regimens. In this regard, trials are underway for children with CD20+ B-cell lymphomas that incorporate the anti-CD20 antibody rituximab—an agent that has already been shown to be valuable in the treatment of some adults with CD20+ B-cell lymphomas.298,299 For example, the current BFM trial for high-grade mature B-cell lymphomas features a rituximab window.253 The results of this window were recently published and indicated that rituximab was active and generally well tolerated. Current studies in COG are piloting the incorporation of rituximab into both Group B and C arms of LMB-96-based therapy. A phase I/II study for children with CD30+ ALCL has been designed to examine the safety and efficacy of a newly developed anti-CD30 antibody.300 Antibody drug conjugates that target CD30 have been studied in adults with CD30+ lymphomas301; studies in children are pending. Other potential therapies include the use of protein-specific cytotoxic T-lymphocytes, a strategy that has been successful in preventing and treating EBV-related posttransplantation lymphoproliferative disease.302 Novel approaches may also include the incorporation of small molecule inhibitors, as well as antisense and anti-idiotype strategies. In this regard, the COG is currently studying a MET/ALK small molecule inhibitor in children with ALK-positive malignancies.303 Molecular characterization of the chromosomal abnormalities associated with the NHLs of childhood is providing us with tools that enhance diagnosis, disease classification, and monitoring of the response to therapy (detection of minimal residual disease). A clearer understanding of molecular pathogenesis may provide insights that permit the development of therapeutic strategies that target tumor-specific molecular lesions.