[level-membership-for-hematology-oncology-and-palliative-medicine-category]72
Cancer of the Lung
Non–Small Cell Lung Cancer and Small Cell Lung Cancer
Leora Horn, Rosana Eisenberg, David Gius, Katherine N. Kimmelshue, Pierre P. Massion, Joe Bill Putnam, Clifford G. Robinson and David P. Carbone
• Constitutes 80% to 85% of new cases of lung cancer in North America.
• Most frequent histologies: adenocarcinoma, squamous cell carcinoma, and large cell carcinoma.
• Leading cause of cancer-related death in the United States for both men and women; represents one of the most preventable forms of cancer death.
• Tobacco use causes 80% of lung cancer deaths.
• Low-dose computed tomography (CT) scan in high-risk individuals is associated with high sensitivity but low specificity for detection of early stage disease.
• Screening remains controversial.
• History and physical examination, routine hematologic and biochemical testing.
 CT scan (with contrast) evaluating lungs, mediastinum, liver, and adrenals
CT scan (with contrast) evaluating lungs, mediastinum, liver, and adrenals
 Positron emission tomography (PET) scan
Positron emission tomography (PET) scan
 If patient has locally advanced or metastatic disease, add magnetic resonance imaging (MRI) of brain
If patient has locally advanced or metastatic disease, add magnetic resonance imaging (MRI) of brain
 If PET is not available, add bone scan
If PET is not available, add bone scan
• Bronchoscopy and/or CT-guided and/or ultrasound-guided biopsy.
• If disease is clinically more advanced, use the least invasive biopsy procedure and sample to confirm advanced disease.
• Mediastinal node evaluation:
 If disease is in early stage (I or II) and PET of mediastinum is negative, proceed directly to surgery. If PET is positive, proceed to biopsy via endobronchial ultrasound (EBUS), or endoscopic ultrasound (EUS), or mediastinoscopy, depending on node location.
If disease is in early stage (I or II) and PET of mediastinum is negative, proceed directly to surgery. If PET is positive, proceed to biopsy via endobronchial ultrasound (EBUS), or endoscopic ultrasound (EUS), or mediastinoscopy, depending on node location.


 Stereotactic body radiotherapy in selected cases.
Stereotactic body radiotherapy in selected cases.

 Postoperative chemotherapy prolongs survival in patients with a good performance status.
Postoperative chemotherapy prolongs survival in patients with a good performance status.
 Stereotactic body radiotherapy in selected cases.
Stereotactic body radiotherapy in selected cases.
 Postoperative chemotherapy useful in resected IIIA in patients with a good performance status.
Postoperative chemotherapy useful in resected IIIA in patients with a good performance status.

 Concurrent chemotherapy combined with radiation therapy in patients with a good performance status.
Concurrent chemotherapy combined with radiation therapy in patients with a good performance status.
 Concurrent chemotherapy combined with radiation is superior to radiation alone; increased toxicity limits this approach in frail patients.
Concurrent chemotherapy combined with radiation is superior to radiation alone; increased toxicity limits this approach in frail patients.
 Clinical trials of new approaches should be a high priority.
Clinical trials of new approaches should be a high priority.
 Primary chemotherapy with platinum-based doublet improves the quality and quantity of life.
Primary chemotherapy with platinum-based doublet improves the quality and quantity of life.
 All tumors should be sent for molecular testing.
All tumors should be sent for molecular testing.

 No advantage for triplet therapy.
No advantage for triplet therapy.
 Non–platinum-based therapy is appropriate in selected patients.
Non–platinum-based therapy is appropriate in selected patients.
 Single-agent therapy reserved for select populations.
Single-agent therapy reserved for select populations.
 There is a clear need for more clinical trials at this stage.
There is a clear need for more clinical trials at this stage.
• Second- or third-line therapy
 Further treatment with any of several agents is appropriate with progression after chemotherapy if patient sustains a good performance status; docetaxel, pemetrexed, erlotinib, and crizotinib are all U.S. Food and Drug Administration (FDA)-approved.
Further treatment with any of several agents is appropriate with progression after chemotherapy if patient sustains a good performance status; docetaxel, pemetrexed, erlotinib, and crizotinib are all U.S. Food and Drug Administration (FDA)-approved.

• Accounts for approximately 15% of new cases of lung cancer each year in United States; incidence is decreasing.
• Subtypes include small cell carcinoma and combined small cell carcinoma (most often combined with squamous cell carcinoma, adenocarcinoma or large cell carcinoma).
• History and physical examination, routine histologic and biochemical testing.
 CT scan (with contrast) evaluating lungs, mediastinum, liver, and adrenals.
CT scan (with contrast) evaluating lungs, mediastinum, liver, and adrenals.
 PET useful with clinically early stage disease.
PET useful with clinically early stage disease.
 Brain evaluation (MRI preferred).
Brain evaluation (MRI preferred).
 If there are signs or symptoms of bone involvement, a bone scan or PET scan is recommended.
If there are signs or symptoms of bone involvement, a bone scan or PET scan is recommended.
 Mediastinal node evaluation is not required unless as a convenient site for biopsy.
Mediastinal node evaluation is not required unless as a convenient site for biopsy.

 Concurrent etoposide plus cisplatin and radiation to the intrathoracic disease.
Concurrent etoposide plus cisplatin and radiation to the intrathoracic disease.
 Prophylactic cranial irradiation in complete (or near complete) responders.
Prophylactic cranial irradiation in complete (or near complete) responders.
 Etoposide (or irinotecan) plus cisplatin or carboplatin.
Etoposide (or irinotecan) plus cisplatin or carboplatin.
 No evidence a third agent or dose intensification improves outcome.
No evidence a third agent or dose intensification improves outcome.
 Research studies are a clear priority given the lack of progress in this arena.
Research studies are a clear priority given the lack of progress in this arena.
 Prophylactic cranial irradiation in complete (or near complete) responders.
Prophylactic cranial irradiation in complete (or near complete) responders.
 Consider consolidative radiotherapy (RT) to primary in complete responders.
Consider consolidative radiotherapy (RT) to primary in complete responders.
 Second-line therapy useful mainly in patents who have experienced a longer treatment-free interval and a good performance status.
Second-line therapy useful mainly in patents who have experienced a longer treatment-free interval and a good performance status.
 Topotecan FDA-approved; however, any of several chemotherapy agents offer short-term benefit.
Topotecan FDA-approved; however, any of several chemotherapy agents offer short-term benefit.
 Research protocols are a preferred choice for patients with disease recurrence.
Research protocols are a preferred choice for patients with disease recurrence.
Introduction
Although reports of pulmonary malignancies date to antiquity, lung cancer is largely a disease of modern man. Prior to 1900, lung cancers were viewed as “matters of medical curiosity not known to be in any degree influenced by medicine and too rare to be of much practical importance.”1 By the mid-20th century, following the introduction of cheap, mass-produced cigarettes, however, lung cancer had become epidemic and firmly established as the leading cause of cancer-related death in North America and Europe.2 It should not be forgotten that lung cancer is potentially one of the most preventable of all of the major malignancies afflicting man. Its primary cause is tobacco smoke.3 King James I was among the first to chronicle the adverse health effects of tobacco smoke,4 but it was Raymond Pearl’s landmark 1938 report that conclusively established the devastating impact smoking has on longevity.5 It would be another decade before tobacco smoking was firmly established as a causative agent of lung cancer6,7 and nearly 30 more years before the emergence of the U.S. Surgeon General’s initial report of the ill effects of tobacco smoking. Regrettably it would be yet another 3 decades before the tobacco industry publicly acknowledged this obvious truth, but only after a long, drawn out battle of misinformation and deception,8 ironically helped along by the unwitting (perhaps) complicity of physicians.9 With the belated recognition of the etiologic role of tobacco smoke the incidence of lung cancer has started to decline in North America and parts of Europe. For the most part the decline is seen most clearly in men. Only recently has this decline become apparent in women in the United States, following a similar decline among men 15 to 20 years prior.10 In short, the story of lung cancer is replete with controversy,11 politics,12 pessimism,13 and, more recently, guarded optimism.14 This chapter focuses on our perceptions of modern-day management of lung cancer with an emphasis on advances made over the past 5 years.
Epidemiology
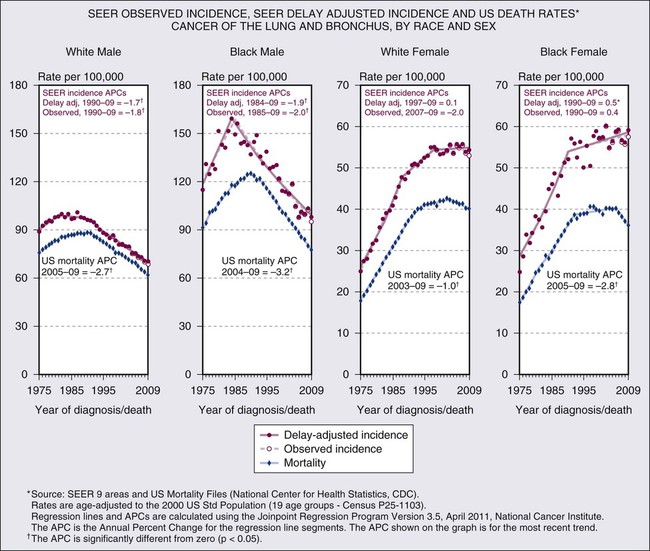
Lung cancer was an uncommon disease in the early part of the 20th century but then began an epidemic rise to far surpass all other cancers in numbers and rates of death.15 Lung cancer is the leading cause of cancer-related mortality with an estimated 244,180 new cases and 164,770 deaths anticipated in 2012.16 Lung cancer is relatively rare in persons younger than age 40 years, but rates rise steadily until age 80 years and then taper off; the projected lifetime probability of developing lung cancer is estimated to be approximately 8% among males and approximately 6% among females.17 The incidence of lung cancer varies by racial and ethnic group, with the highest age-adjusted incidence rates among African American men (Fig. 72-1). The excess in age-adjusted rates among African Americans occurs only among men, but examinations of recent age-specific rates show that before age 50 years, mortality from lung cancer is more than 25% higher among African American women compared with white women.17 Incidence and mortality rates among Hispanic, Native, and Asian Americans are only 40% to 50% those of whites.
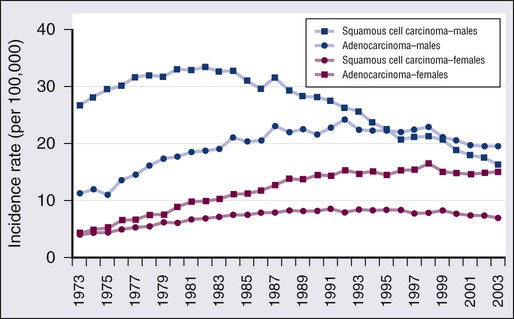
The trends in overall lung cancer rates mask differences in temporal patterns according to lung cancer cell type. Figure 72-2 shows changes in age-adjusted incidence rates for squamous cell carcinoma and for adenocarcinoma during 1973 to 2003 based on data from nine continuing cancer registries in the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) program of cancer registration. Among men, squamous cell cancers predominated in the first two thirds of this period. The decline in incidence of lung cancer among men was first apparent for squamous cell tumors. Decreases began in the early 1980s so that by the mid-1990s rates of squamous cell carcinoma among men had dropped below those for adenocarcinoma, which did not peak until over a decade later. Among women, adenocarcinomas have been more common than squamous cell carcinomas during the past 3 decades. The adenocarcinoma excess among women has become more pronounced over time, as rates of squamous cell cancers increased steadily through the 1980s before beginning to decline, whereas adenocarcinoma did not plateau until about a decade later. Although not shown, rates of small cell carcinoma, the third most frequent cell type, tended to parallel those for squamous cell cancer among both sexes. Trends for other cell types of lung cancer, including large cell carcinomas, tend to be intermediate between those of squamous cell carcinomas and adenocarcinomas. As discussed later in the chapter, although 5-year relative survival rates for lung cancer have improved over time, the survival rates are low, currently approximately 15% overall.17 Some variation exists by sex, race, cell type, and molecular status, with slightly higher survival among whites than blacks and females than males, but for no group does the overall 5-year relative survival exceed 20%.
Risk Factors
The cause of the large majority (80%) of lung cancers is cigarette smoking.18,19 There is a massive compilation of scientific evidence from epidemiological studies conducted around the world since the 1950s demonstrating the link between smoking and lung cancer. Epidemiological research has also revealed that several other factors have been implicated as causes of lung cancer, although none to the extent of tobacco.20,21 Cigarette smokers have been shown to have large increases in the risk of lung cancer. A recent deep-sequencing study suggested that one genetic mutation is induced for every 15 cigarettes smoked. Numerous investigations typically show 10 fold or greater increases in risk of this cancer among smokers compared with those who have never smoked.18,19 One of the largest studies is the American Cancer Society’s prospective cohort study of more than 1 million Americans in which a more than 20 fold increase of lung cancer has been observed among men who were current smokers at the start of the followup in the early 1980s.18,19 In contrast, even the most prolonged and intense exposures to asbestos, perhaps the most prominent occupational cause of lung cancer, are associated with no more than about fivefold increase in risk of lung cancer.22
Risks of lung cancer are lower among persons who quit smoking than among those who continue smoking.18,19 The reductions in risk indicate that quitting smoking is beneficial (and conversely that continuing to smoke is harmful). Risk among former smokers on average is less than one-half that of those who continue to smoke. In the American Cancer Society cohort study cited above, former smokers had a ninefold increase in lung cancer compared with men who had never smoked versus the 20 fold excess in those who continued to smoke.18,19 Such relative reductions have been consistently seen, with the size of the reduction in risk increasing the longer the person has quit smoking, although generally even long-term former smokers have higher risks of lung cancer than those who never smoked.18,19
Cigarette smoking has been shown to increase risk of all the major lung cancer cell types.18–21 The magnitude of the increase varies by histologic type, however, with highest risks for squamous cell, small cell, and large cell carcinomas of the lung. Some early studies tended to show only small increases in risk of adenocarcinoma among smokers, but more recent studies indicate that the excess of lung adenocarcinoma is substantial.18,19,21
Cigarette smoke has also been implicated in increasing risk of lung cancer among nonsmokers. A 2006 update of the Surgeon General’s report declared that there is sufficient evidence to list passive smoking as an established cause of lung cancer and called for further control of environmental tobacco smoke (ETS) exposures.23 The risk from ETS is far less than from active smoking, with approximately a 20% to 30% increase in lung cancer observed among nonsmokers married for many years to smokers, in comparison to the 2000% increase among continuing active smokers. Nevertheless, cancer-control activities based on the knowledge that ETS exposure may convey an increased risk of lung cancer have helped reduce exposures in public places and have also provided additional incentive for smokers to quit the habit.
Although cigarette smoking is the dominant cause of lung cancer, several other risk factors for this cancer have been identified.20,21 These include occupational exposures to asbestos and other workplace agents, some of which have been evaluated for nearly as long as cigarette smoking. Among the occupational agents considered as known lung carcinogens are arsenic, bischloromethyl ether, hexavalent chromium, mustard gas, nickel (as found in certain nickel refining processes), and polycyclic aromatic hydrocarbons.20,21,24 Several other occupational exposures have been associated with increased rates of lung cancer, but the causal nature of the association is not clear. Epidemiological studies have attempted to assess the potentially synergistic interrelationship between certain workplace exposures and smoking. Risk of lung cancer among men exposed to asbestos who also smoked was originally thought to be exceptionally high (with early reports of 50 fold or greater excesses compared with unexposed nonsmokers), but recent modeling of larger data pools suggests that asbestos and tobacco combine to enhance lung cancer risk in a less than multiplicative manner.25 Occupational observations have also provided clues to the mechanisms of lung cancer induction. Risk of lung cancer among asbestos-exposed workers, for example, is increased primarily among those with underlying asbestosis, raising the possibility that the scarring and inflammation produced by this fibrotic nonmalignant lung disease may, in many cases (though likely not in all), be the trigger for asbestos-induced lung cancer.26
Increased risks of lung cancer have also been associated with other factors. Diet and nutrition are thought to be involved, as numerous investigations have shown somewhat higher risks of this cancer among those with low fruit and vegetable intake during adulthood.20,21 The early observational studies led to hypotheses that specific nutrients, in particular retinoids and carotenoids, might have chemopreventive effects for lung cancer. Randomized clinical trials were launched, but failed to validate this hypothesis when reports from interventions involving supplementation with beta-carotene in trials both in Finland and the United States found increased rather than decreased incidence of lung cancer among the supplemented.27,28 The current consensus regarding diet and lung cancer remains muddled, with a minor role for nutritional factors likely, but difficult to assess epidemiologically. Ionizing radiation has been established as a lung carcinogen, most convincingly demonstrated from studies showing modestly increased rates of this cancer among persons exposed to the atomic bombs of Hiroshima and Nagasaki and large excesses among workers exposed to α irradiation from radon in underground uranium mining.20,21 Extrapolations from the high exposures in mines to low-level radon exposures in homes, as well as direct observations from case-control studies assessing measured levels in homes, suggest that prolonged radon exposures above the recommended remedial levels might impart a risk of lung cancer equal to or greater than that of ETS.29 Prior lung diseases, such as asbestosis (mentioned above), chronic bronchitis, emphysema, and tuberculosis have also been linked to increased risks of lung cancer. Although smoking itself is a cause of the chronic obstructive pulmonary diseases (COPDs), the link between chronic bronchitis and emphysema and lung cancer persists after adjustment for smoking, with as much as double the smoking-adjusted cancer risk among those with COPD.20,21,30
Familial clustering of lung cancer has been observed, raising the possibility of inherited traits that may increase risk among some individuals, with risk about doubled in families with prior lung cancer.20,21 Individuals with inherited mutations in retinoblastoma and p53 (Li-Fraumeni syndrome) genes may develop lung cancer. Genome-wide association studies have recently identified four genetic loci for lung cancer risk, including two on 5p15 (TERT-CLPM1L), 15q25.1 (CHRNA5-CHRNA-3 nicotinic acetylcholine receptor subunits) and 6p21 (BAT3-MSH5).33–33 A rare germline mutation involving the epidermal growth factor receptor (EGFR) within exon 20 that results in an amino acid substitution at position 790 from threonine to methionine (T790M) has been linked to lung cancer susceptibility in never smokers.34,35 Smoking also clusters within families, so some of the familial aggregation of lung cancer may be smoking related. Nevertheless, there appear to be multiple genetic factors that help determine the way in which individuals metabolize, detoxify, repair, or otherwise respond to lung carcinogens, including the carcinogens in cigarette smoke, as discussed in more detail later in this chapter.
Smoking Cessation
Given the undeniable link between cigarette smoking and lung cancer,3 it is incumbent upon physicians to promote tobacco abstinence and help their patients who smoke to stop smoking.36 Smoking cessation, even well into middle age, can minimize an individual’s subsequent risk of lung cancer, and stopping before middle age avoids more than 90% of the risk attributable to tobacco.37,38 By contrast, there is little health benefit realized by simply “cutting back.”39 Among victims of lung cancer smoking cessation is associated with improved survival,40–43 fewer side effects from therapy,44 and an overall improvement in quality of life.45 Often forgotten is the fact that smoking alters the metabolism of many chemotherapy drugs, potentially adversely altering the toxicities and therapeutic benefits of the agents.46 Therefore, it is important to promote smoking cessation even after the diagnosis of lung cancer is established.47,48 Although smoking cessation is extremely difficult, patients with lung cancer tend to be highly motivated and success rates mirror that of other disease states.49 However, the individual must want to stop smoking and must be willing to work hard to achieve the goal of smoking abstinence. Nicotine replacement therapies, bupropion and varenicline (an α4β2 nicotinic acetylcholine receptor partial agonist), are approved by the U.S. Food and Drug Administration (FDA) as first-line treatments for nicotine dependence.50 Varenicline has been demonstrated to be significantly more efficacious than bupropion alone for smoking cessation.51 Furthermore, prolonged use of varenicline beyond the initial induction phase has proven useful in maintaining smoking abstinence.52 Clonidine and nortriptyline are recommended as second-line treatments.47 A systematic review of extant smoking cessation studies indicates self-help strategies alone only marginally affect quit rates, whereas individual and combined pharmacotherapies and counseling either alone or in combination can significantly increase rates of cessation.53
Biology of Lung Cancer
The specific events that trigger malignant transformation of bronchoepithelial cells are unknown in the vast majority of cases. However, it is clear that exposure to environmental carcinogens, such as those found in tobacco smoke or asbestos fibers, induce or facilitate the transformation (extrinsic component).54 The contribution of the extrinsic carcinogen on transformation is modulated by genetic variations in genes (intrinsic component) that affect aspects of carcinogen metabolism, such as the conversion of procarcinogens to carcinogens and their subsequent inactivation.55 These genetic variations occur at relatively high frequency in the population. Their contribution to an individual’s lung cancer risk is generally low, but because of their population frequency, their overall impact on lung cancer risk could be high. Epidemiological studies further suggest that a familial predisposition to lung cancer exists that is independent of tobacco smoke exposure. One study found evidence for an autosomal dominant model linked to 6q23-25,56 but other studies have proposed a complex multigene model for inherited risk.57 As mentioned above, familial clustering of lung cancer cases has been reported with an inherited T790M mutation in the EGFR gene.34,35 The identification of individuals at particularly high risk for the development of lung cancer could justify more intense screening regimens, and the identification of the responsible chromosomal loci for lung cancer susceptibility genes could allow the development of specific chemopreventive strategies.
Environmental factors, as modified by inherited modulators, likely affect specific genes by deregulating important pathways to enable the cancer phenotype. Particularly important in lung cancer are acquired abnormalities in ras, Rb, p53, Akt, LKB, and BRAF (reviewed in Fong et al58). However, the most clinically significant acquired genetic abnormalities in lung cancer are the mutation of the EGFR59–62 and the anaplastic lymphoma kinase (ALK) fusion.65–65 EGFR mutations occur in approximately 20% of lung cancer patients and are primarily in exons 19 (in-frame deletions usually of four amino acids LREA) and 20 (L858R point mutants), and result in constitutive signaling and AKT activation66 and are associated with very high response rates (60% to 90%) to the specific tyrosine kinase inhibitors gefitinib and erlotinib.67 ALK fusions have been reported in 3% to 7% of lung cancer patients and are associated with a 57% response to crizotinib.68 The EGFR and ALK abnormalities are non–overlapping. Interestingly, both EGFR mutations and ALK fusions are associated with younger age, light (<10 pack-year) and nonsmokers and adenocarcinoma histology, and the frequency of EGFR mutations is much higher in Asian than Western populations (30% to 70% vs. 10%).68–72 Response to specific EGFR or ALK inhibitors are often dramatic, and occur even in heavily pretreated patients, demonstrating that tumors with these mutations are “addicted” to the activation of this pathway. Almost all patients with these dramatic responses, however, develop progressive, resistant disease. The most common mechanism of acquired resistance to EGFR tyrosine kinase inhibitors is a second-site mutation within exon 20 that results in an amino acid substitution at position 790 in EGFR, from threonine to methionine (T790M).73,74 To date, several second-site mutations within the ALK tyrosine kinase domain have been reported as mechanisms of acquired resistance to the ALK tyrosine kinase inhibitor crizotinib.75
In spite of the incontrovertible high response rate in patients with EGFR mutations, analysis of samples from BR.21, a placebo-controlled, randomized, clinical trial of erlotinib in second-line non–small cell lung cancer (NSCLC) showed that the presence of an EGFR mutation was not associated with prolonged survival,76 but in univariate analysis, amplification of the receptor was associated with improved survival.77 Interestingly, there was a survival benefit in the entire erlotinib arm compared with placebo, as well as every subset of patients, including smokers and patients with squamous cell cancers,78 suggesting that factors other than mutation may contribute to clinical benefit, and that this was not necessarily related to objective response rates. Other assays, including serum proteomics, show potential for selecting patients likely to achieve clinical benefit.79,80
The histologic sequence of events that leads to the various forms of lung cancer is not well understood, and it is clearly different for the various histopathologic entities.81–84 Current knowledge suggests that squamous cell carcinoma arises in a relatively ordered progression that includes squamous metaplasia and carcinoma in situ (CIS). Peripheral adenocarcinomas are thought to arise from atypical adenomatous hyperplastic lesions, but this process is much more obscure, largely because this type of lesion is much less accessible by bronchoscopy. Small cell carcinoma might arise from neuroendocrine hyperplasia, but evidence in support of this hypothesis is scarce.
The pathology of premalignancy is discussed in more detail in the next section, but the molecular biology of premalignancy is of great clinical importance, not only to better understand the process of cancer development, but to provide potential therapeutic targets to intervene in this process and intermediate biomarkers to assess risk and evaluate candidate chemoprevention strategies. Premalignant lesions of the lung have been investigated for molecular alterations in NSCLC, but much less information is available for small cell lung cancer (SCLC).85 Microdissected specimens derived from normal, hyperplastic, metaplastic, dysplastic, and CIS, as well as invasive neoplastic foci of patients with lung cancer, were studied for gene mutations, promotor hypermethylation, and allele loss. Results obtained to date suggest that allele loss on chromosome 3p is the earliest event, followed by allele loss/hypermethylation on chromosome 9p and, subsequently, on chromosome 8p.84 Loss of heterozygosity at the p53 gene locus (17q13.1) is relatively rare (10%) and occurs predominantly at the dysplasia or CIS stage. Point mutations in the p53 gene86 and the EGFR gene,87 however, have been observed in morphologically normal bronchial epithelium obtained from the airways of patients with lung cancer. In contrast, K-ras gene mutations represent a late event, found only in CIS or invasive cancers.88 In addition to copy number changes and mutations, alterations in the expression of the retinoic acid receptor β have also been used as a molecular marker for premalignancy and as an intermediate biomarker for chemoprevention trials.89
The fate of morphologically or molecularly abnormal areas is currently an area of intense research. One study showed that 54% of patients with high-grade dysplastic lesions developed lung cancer within 2 years, but 80% of these arise in a different part of the lung than the CIS.90 None of the patients with low-grade dysplastic lesions progressed to cancer in this study. Of the low-grade lesions in this study, 82% spontaneously regressed, and 18% remained unchanged, and none progressed to CIS. Preliminary data also suggest that abnormal regions display reproducible molecular changes on repeat biopsies over time, and that these and additional abnormalities can be observed in tumors arising from these lesions.91
Part of the problem with the studies of preneoplasia is that we may be evaluating the microscopic appearance and molecular characteristics of the wrong population of cells. The vast majority of the respiratory epithelium (and most other tissues) is thought to be terminally differentiated and incapable of sustained replication; however, one theory holds that a small subset of the cells have unlimited, but normally tightly regulated, replicative potential. These are the so-called pulmonary stem cells,92,93 whose biology is very poorly understood. The stem cell concept may also underlie the failure of standard medical therapies to eradicate lung cancers, even when there is a clinical complete response. The theory is that therapies have been refined to induce measurable reductions in the bulk tumor mass, whereas true replicative potential exists only in a small subset of “cancer stem cells” that are not effectively targeted by current therapies. Specific isolation of these cells and development of stem cell–targeted therapies may thus not cause rapid tumor regressions, but rather result in significantly increased long-term survival.
Pathology
In recent years, significant advances in the treatment of pulmonary adenocarcinoma have affected the pathological diagnosis of lung cancer. The latest World Health Organization (WHO) classification of lung tumors from 200494,95 (Box 72-1) is a purely morphologic classification and geared towards resection specimens. Most pathological diagnoses of lung cancer, however, are rendered on small biopsies or cytologic specimens. A new multidisciplinary classification of lung adenocarcinoma has been proposed jointly by the International Association for the Study of Lung Cancer (IASLC), American Thoracic Society (ATS), and European Respiratory Society (ERS) (Box 72-2).96 Although based predominantly on histology, its purpose is to provide an integrated clinical, molecular, radiologic, and pathological approach to the classification of lung adenocarcinoma. In addition, this classification addresses the diagnosis of lung cancer in small biopsies and cytologic specimens and emphasizes the use of immunohistochemistry for the subtyping of non–small cell carcinoma.
Tissue and Cytologic Diagnosis of Lung Cancer
The diagnosis of lung cancer can be made on tissue specimens such as endobronchial and transbronchial biopsies or resected specimens, or by assessment of cytological specimens such as transbronchial or transthoracic fine-needle aspirates, bronchial brushing and washings, bronchoalveolar lavage or sputum or pleural fluid. The diagnostic yield depends on several factors including location (accessibility) of the tumor, tumor size, tumor type, or technical aspects of the diagnostic procedure, including the experience level of the bronchoscopist and pathologist.96 In general, central lesions such as squamous cell carcinomas, small cell carcinoma, or endobronchial lesions such as carcinoid tumors, are more readily diagnosed by bronchoscopic examination, whereas peripheral lesions, such as adenocarcinomas, and large cell carcinomas are more amenable to transthoracic fine-needle aspiration (FNA) or biopsy. Diagnostic accuracy for most specimens in distinguishing small cell carcinoma and non–small cell carcinomas is excellent, 97 with less accuracy for subtypes of non–small cell carcinoma96 if immunohistochemical staining is not used.
Bronchoscopic specimens include bronchial brushings, washings, bronchoalveolar lavage, and transbronchial fine needle aspiration. Of these, transbronchial FNA consistently demonstrates the highest sensitivity, surpassed only by the use of a combination of bronchoscopic specimens.96–100 Overall sensitivity for combined use of bronchoscopic methods is approximately 80%, and with tissue biopsy, the yield increases to 85% to 90%.96,97,99 In fact, transbronchial FNA is more often diagnostic than transbronchial biopsy when the lesion of interest is submucosal.96,101
Like transbronchial fine-needle aspirate specimens, transthoracic fine-needle aspirate specimens are also diagnostically useful, yielding diagnostic material in 70% to 95% of cases. Sensitivity is highest for larger lesions and peripheral tumors.102 In general, fine-needle aspirate specimens, whether transbronchial, transthoracic, or endoscopic ultrasound-guided, are superior to other specimen types.103 The superiority of fine-needle aspirate specimens is primarily a result of the higher yield of lesional tumor cells with fewer confounding factors, such as obscuring inflammation and reactive nonneoplastic cells.
Sputum cytology is inexpensive and noninvasive but has a lower yield than other specimen types because of poor preservation of the cells and more variability in acquiring a good-quality specimen. The yield for sputum cytology is highest for larger and centrally located tumors, such as squamous cell carcinoma and small cell carcinoma, although occasionally an accurate diagnosis is possible with tumors located more peripherally within the lung.96 The specificity for sputum cytology averages close to 100%, although sensitivity is generally less than 70%. The accuracy of sputum cytology improves with increased numbers of specimens analyzed and consequently, analysis of at least three sputum specimens is recommended. Sputum cytology has also been extensively studied with varying success as a screening tool for early detection of lung cancers. Sputum cytology is not currently recommended as a routine screening tool, but recent advances in molecular diagnostic techniques may result in a resurgence of its use.104–107
Squamous Cell Carcinoma
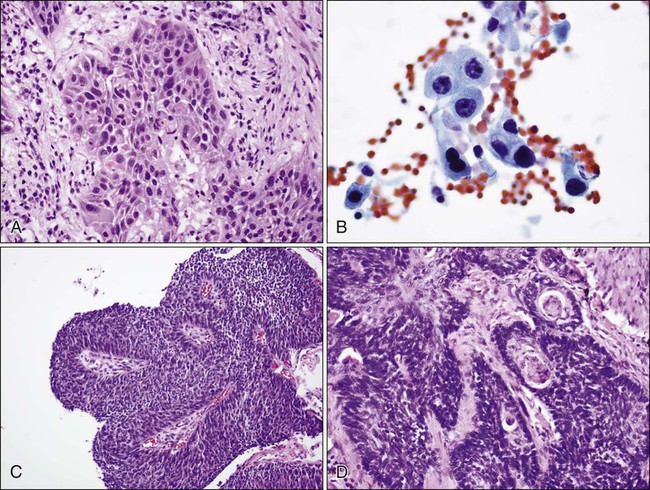
Squamous cell carcinoma (Fig. 72-3) tends to occur centrally and is highly associated with smoking. Histopathologically, squamous cell carcinoma cells show keratinization (in the form of keratin pearls or single cell keratinization) and/or intercellular bridges. These features, however, may be difficult to identify in poorly differentiated tumors. The most common pattern is one of infiltrating nests of malignant squamous cells (Fig. 72-3A), with central necrosis, often resulting in cavitation. Several important variants are described, including a papillary variant (Fig. 72-3B), which can present as an exophytic and endobronchial growth,108 and a basaloid variant (Fig. 72-3C) that can mimic other basaloid or neuroendocrine tumors histologically.109 When evaluating cytology specimens containing squamous cell carcinoma, intercellular bridges are not usually identified, but keratin can be clearly seen when present. In addition, the tumor tends to consist of sheets of cells rather than the three-dimensional groups of cells characteristic of adenocarcinomas (Fig. 72-3D).
Adenocarcinoma
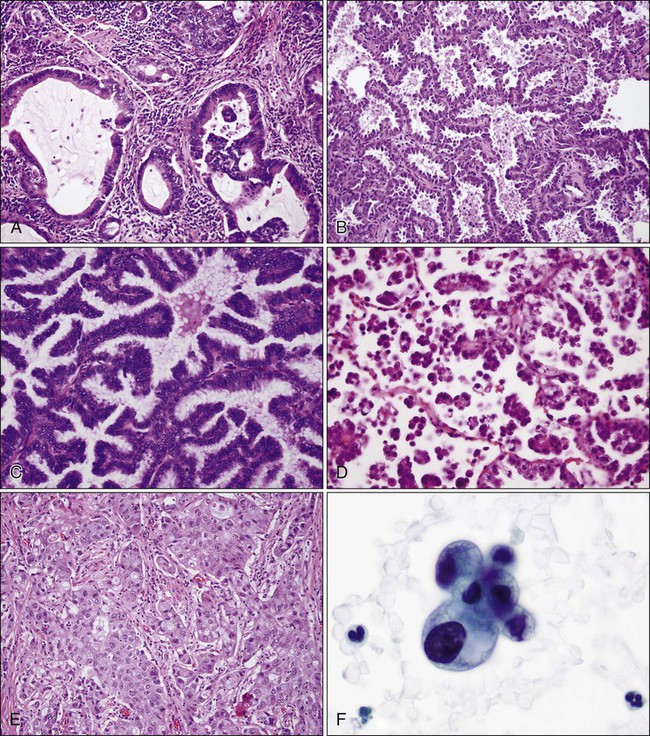
In North America and Japan, adenocarcinoma is the most common histologic type of lung cancer. As in other lung carcinomas, adenocarcinomas occur predominantly in smokers although non-smokers are more likely to develop adenocarcinoma compared with other lung cancer types. Adenocarcinomas tend to occur in the periphery of the lung, but can occur centrally, can be multifocal, or can fill an entire lobe. Radiographically, they are associated with solid opacities, ground glass opacities, or mixed patterns, generally correlating with the amount of in situ and invasive components of the tumor.110,111 On tissue sections, the diagnosis is made based on the presence of glands (acini), papillary structures, a lepidic (bronchioloalveolar) pattern, a micropapillary pattern, cellular mucin, or a solid pattern (if poorly differentiated) (Fig. 72-4A-E).95 Eighty percent of adenocarcinomas demonstrate a mixture of at least two of these patterns112 and many mixed adenocarcinomas (>20%) show a focal lepidic pattern.113,114 Of these patterns, the solid and micropapillary patterns in adenocarcinomas may predict a worse prognosis.115–121 On cytologic preparations, three-dimensional groups or glandular and papillary patterns may be identified and are diagnostic of adenocarcinoma (Fig. 72-4F). Variants of adenocarcinoma include colloid, enteric, and fetal adenocarcinomas.95,123
The 2004 WHO classification introduced the concept of atypical adenomatous hyperplasia as a precursor of some adenocarcinomas. Bronchioloalveolar carcinoma (BAC) was defined as noninvasive tumor, if the strict criteria were followed.94,95 This definition arose out of studies showing that noninvasive tumors less than 3 cm resulted in a 100% 5-year patient survival122; in addition, tumors with limited fibrosis, smaller size, and limited invasion had a better prognosis.122–125 Minimally invasive tumors, while not defined as BAC in the strictest sense, also carried an excellent prognosis.111 The IASLC/ATS/ERS classification96 proposes discontinuing the use of the term bronchioloalveolar carcinoma and instead using adenocarcinoma in situ (AIS) for nonmucinous and rarely mucinous lesions 3 cm or smaller with purely lepidic growth (tumor cells lining alveolar walls). The term minimally invasive adenocarcinoma (MIA) is to be applied to nonmucinous and rarely mucinous lesions 3 cm or smaller with invasion limited to 5 mm or less. Lepidic predominant adenocarcinoma is proposed for nonmucinous lesions if invasion is greater than 5 mm. Most tumors previously called mucinous BAC are thought to actually represent invasive tumors and are thus reclassified as invasive mucinous adenocarcinoma by the IASLC/ATS/ERS criteria. Of note, the diagnosis of AIS or MIA cannot be rendered on small biopsies or cytologic specimens, because the entire tumor has to be examined in order to exclude or measure invasion.
Atypical adenomatous hyperplasia (AAH) is a small noninvasive lesion (usually less than 5 mm) consisting of mild to moderately atypical cells lining the alveoli in the absence of an underlying inflammatory process. AAH is usually an incidental lesion, found either in lung tumor resection specimens, or on radiographic imaging.95 Because these lesions (along with BAC/AIS/MIA) preserve the underlying lung architecture and alveolar spaces, they may appear as “ground-glass opacities” radiographically.111 Although when associated with malignancy they are most often associated with adenocarcinoma of the lung, AAH has also been identified in conjunction with large cell carcinoma, squamous cell carcinoma, and metastatic tumor.95
The IASLC/ATS/ERS classification96 recommends using comprehensive histologic subtyping of adenocarcinomas to make a semiquantitative assessment of the percentages of the various histologic patterns of adenocarcinoma: acinar, papillary, micropapillary (newly introduced in this classification), lepidic, and solid. At the time of pathological diagnosis, tumors should be classified according to the predominant histologic subtype. If additional subtypes are present, they should be listed as well. Studies utilizing this classification have demonstrated prognostic significance in stage I cancers based on the predominant pattern.128,129 Adenocarcinoma in situ and minimally invasive adenocarcinoma resulted in 100% disease-free survival at 5 years. Lepidic, acinar, and papillary predominant tumors had intermediate prognosis. Solid, micropapillary and colloid predominant, and invasive mucinous and mixed mucinous/non mucinous carcinomas had the worst prognosis.129
Large Cell Carcinoma
Large cell carcinomas (LCCs) comprise less than 10% of lung carcinomas, tend to occur peripherally, and are defined as poorly differentiated carcinomas of the lung composed of large malignant cells without evidence of squamous or glandular differentiation or features of small cell carcinoma by light microscopy after examination of the entire tumor at resection (Fig. 72-5).95 LCCs usually consist of sheets of large malignant cells, often with associated necrosis. If examined on cytologic preparations, the tumor cells are arranged as single cells and in syncytial groups, again lacking squamous, glandular, papillary, or small cell carcinoma features. However, the diagnosis of LCC is not made on cytology or small biopsies because it cannot be distinguished from poorly differentiated squamous cell carcinoma or adenocarcinoma because of sampling limitations. In fact, because immunohistochemistry is now used routinely for distinguishing adenocarcinoma from squamous cell carcinoma (see immunohistochemistry below), LCC may become a less common diagnosis, reserved for tumors in which immunohistochemistry cannot reliably distinguish between adenocarcinoma and squamous cell carcinoma.130,131 By electron microscopy, some tumors histologically classified as LCC may show evidence of glandular, squamous, or neuroendocrine differentiation, while others do not.126 Variants of LCC include basaloid carcinoma, which may present as an endobronchial lesion and may resemble a high-grade neuroendocrine tumor, and lymphoepitheliomalike carcinoma, which is similar to the same-named tumor of other sites and is related to Epstein-Barr virus.
Neuroendocrine Tumors of the Lung
Neuroendocrine tumors of the lung include a spectrum of lesions from the very indolent to the most aggressive pulmonary neoplasms. The four main neuroendocrine tumors of the lung are typical carcinoid, atypical carcinoid, large cell neuroendocrine carcinoma (a subtype of LCC), and small cell carcinoma (Fig. 72-6). In general, it is helpful to think of these tumors according to their clinical characteristics. Typical and atypical carcinoid tumors are less associated with smoking history, tend to occur in younger patients, and are less aggressive, whereas small cell carcinoma and large cell neuroendocrine carcinoma occur in smokers and behave much more aggressively. Differentiating these tumors from one another histologically is usually straightforward based on morphologic features and defined criteria. In some cases, however, there can be overlap in the morphologic features. The WHO classification emphasizes mitotic count in differentiating and defining these tumors (Table 72-1). Mitotic counting is usually straightforward unless the tissue available is in the form of a small biopsy or cytologic specimen. Although the use of immunohistochemistry for Ki-67 (MIB1), a marker representing cell proliferation, is not described in the 2004 WHO classification for separating pulmonary neuroendocrine tumors, several studies in recent years have shown its utility, particularly in small biopsies and cytologic specimens in which a reliable mitotic count is difficult to obtain.133 In this context, Ki-67 staining is most useful to distinguish carcinoid (both typical and atypical) from small cell carcinoma. The Ki-67 proliferation rate for typical carcinoid is usually less than 2%, for atypical carcinoid less than 20% (and usually approximately 10%) and is greater than 25% (commonly greater than 50%) for small cell carcinoma and large cell neuroendocrine carcinoma. Despite a rather distinct histologic appearance, immunohistochemistry may also be performed to verify the neuroendocrine nature of a tumor and thus differentiate neuroendocrine tumors from other non–small cell carcinomas.127
Table 72-1
General Histologic Features of Neuroendocrine Tumors
| Diagnosis | Morphology | Mitotic Count per mm2 |
| Typical carcinoid tumor | Generally bland neuroendocrine morphology | <2 |
| Atypical carcinoid tumor | Generally bland neuroendocrine morphology ± focal necrosis | 2–10 |
| Large cell neuroendocrine carcinoma | High-grade neuroendocrine carcinoma—larger cells with nucleoli | >10 |
| Small cell carcinoma | High-grade neuroendocrine carcinoma—smaller cells and few nucleoli | >10 |
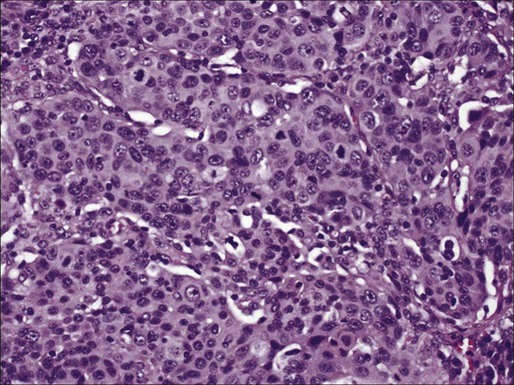
Large Cell Neuroendocrine Carcinoma
Large cell neuroendocrine carcinoma (LCNEC) is a subtype of LCC that shows neuroendocrine differentiation by light microscopy and accounts for approximately 3% of lung cancers. It is included in the differential diagnosis of neuroendocrine lung tumors (see below). LCNEC is a high-grade carcinoma showing neuroendocrine architectural features (formation of rosettes, trabeculae, organoid nests, or perilobular palisading patterns), greater than 10 mitoses/2 mm2, and positivity with neuroendocrine immunohistochemical markers.95 LCNEC can be difficult to diagnose by cytology and to differentiate from a small cell carcinoma. Features seen on cytologic specimens include evidence of neuroendocrine differentiation, such as sheets or groups of cells with peripheral palisading or rosette formation, as seen in other neuroendocrine tumors. Unlike small cell carcinoma, these tumors tend to have larger cells with prominent nucleoli.95,127–129 LCNEC is an aggressive tumor and shares several molecular abnormalities with small cell carcinoma. The prognosis for these tumors is intermediate between those of other non–small cell carcinomas and small cell carcinoma.
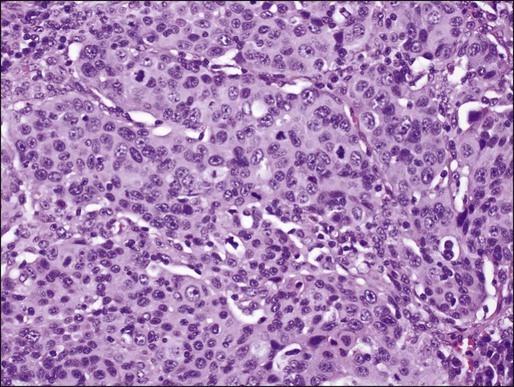
Small Cell Carcinoma
Small cell carcinoma is a poorly differentiated neuroendocrine tumor that tends to occur centrally and is highly associated with smoking. Incidence rates of small cell carcinoma are higher among men than women, but a higher percentage of lung cancers are of small cell origin among women than men.130 Small cell carcinoma consists of smaller, but clearly malignant cells with scant cytoplasm, characteristic finely granular (“salt-and-pepper”) chromatin without prominent nucleoli, and greater than 10 mitoses/2 mm2 (usually greater than 50 mitoses/2mm2) 95 (Fig. 72-7A). The tumor cells may be arranged in sheets, but often show neuroendocrine patterns such as rosettes, trabeculae, or peripheral palisading of cells along the edges of nests. Commonly, there is a spectrum of viability with necrosis and crushing of tumor cells, as well as nuclear “molding.” The cells are defined as “small,” meaning less than 21 μm in diameter (up to 2 to 3 times the size of a resting lymphocyte); however, variation in size is common in these tumors, and it is the nuclear features that allow diagnosis, not strictly cell size.131,132 The characteristic features are usually easily identified in well-preserved cytologic specimens, but may be more difficult to differentiate from other round, blue cell tumors on liquid-based cytology132 or on less-well-preserved specimens or small biopsies (Fig. 72-7B).99 As with other histologic types of lung carcinoma, small cell carcinoma may occur alone, or combined with other tumors. Combined small cell with LCNEC, LCC, adenocarcinoma, and squamous cell carcinoma have all been well documented. The differential diagnosis of small cell carcinoma includes poorly differentiated non–small cell carcinomas and neuroendocrine carcinomas, especially poorly differentiated squamous cell carcinoma and LCNEC, as well as nonepithelial tumors, such as lymphoma, small, round, blue cell tumors, and some sarcomas, for example, synovial sarcoma.
Typical Carcinoid
Typical carcinoid (TC) is a low-grade neuroendocrine neoplasm showing tumor cells arranged in organoid nests, trabeculae, or spindled patterns characteristic of neuroendocrine differentiation, but is differentiated from other neuroendocrine tumors by its bland uniform cells, lack of necrosis, and lack of mitotic activity (fewer than 2 mitoses/2mm2). TC occurs in nonsmokers, most often as an endobronchial lesion, but can occur more peripherally. Cytologic specimens show uniform bland cells with finely stippled (“salt and pepper”) chromatin. Although these tumors are known for their excellent prognosis, as many as 10% to 15% can have metastases at diagnosis.95 A tumorlet is defined as a carcinoid tumor measuring less than 5 mm. Although tumorlets are usually an incidental finding, they can be multiple, associated with carcinoid tumors or neuroendocrine hyperplasia of the bronchial epithelium.95
Atypical Carcinoid
Atypical carcinoid is an intermediate-grade tumor that has a similar neuroendocrine morphology to TC, but is more aggressive. It is defined by the presence of mitoses in the range of 2 to 10 per 2 mm2 or the presence of punctate necrosis. It has a higher risk of metastasis (up to 50% lymph node metastasis at presentation)95 than TC. Cytologically, atypical carcinoid has a similar appearance to TC, although tumor cells may show more atypia and nuclear enlargement.95
Immunohistochemistry of Lung Tumors
With the advent of different therapies for adenocarcinoma and squamous cell carcinoma, it has become crucial to distinguish the two on small biopsies and cytologic specimens when the morphologic features of each of these types of carcinoma may not be evident.141–145 Immunohistochemistry stains useful in the diagnosis of adenocarcinoma include TTF-1 (thyroid transcription factor-1, a protein that regulates transcription of genes specific for the thyroid, lung, and diencephalon) and napsin A (a member of the aspartic protease family, expressed in normal lung and kidney, adenocarcinomas from lung, and some renal carcinomas) (Fig. 72-8). Squamous cell carcinoma markers include high-molecular-weight cytokeratins, such as CK5/6, and p63 (a member of the p53 family of transcription factors). A few carcinomas may not show clear separation by immunohistochemistry and will still fall into the category of non–small cell carcinoma, not otherwise specified.
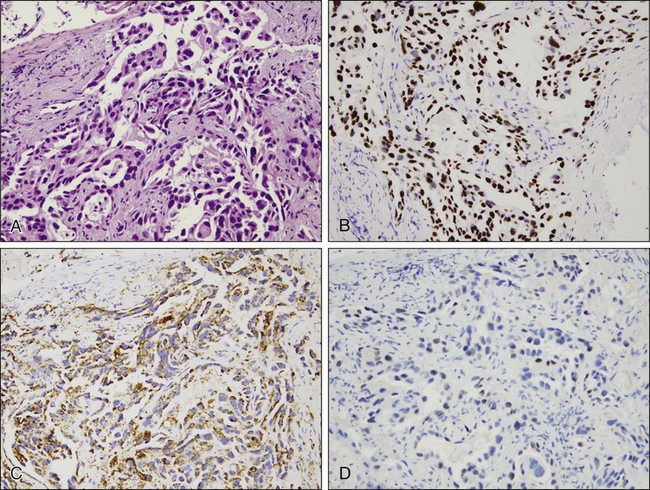
Immunohistochemistry may also be used to confirm neuroendocrine differentiation within a tumor. Neuroendocrine markers include CD56 or neural cell adhesion molecule (NCAM), synaptophysin, and chromogranin. Neuron-specific enolase (NSE) shows significant nonspecific staining of nonneuroendocrine tumors and should not be used. Most often a combination of these stains, for example, CD56, synaptophysin, and chromogranin, is used to establish a diagnosis.112 These markers support neuroendocrine differentiation, but do not distinguish between specific types of neuroendocrine tumors.
Immunohistochemistry is also helpful in distinguishing primary lung tumors from malignancies metastatic to the lung. This is especially true for distinguishing primary lung adenocarcinomas and metastatic adenocarcinomas. TTF-1, identified in tumors of thyroid and pulmonary origin, stains more than 70% of pulmonary adenocarcinomas.133 When positive, TTF-1 is a reliable indicator of a primary lung cancer provided a thyroid primary has been excluded. A negative TTF-1, however, does not exclude the possibility of a lung primary tumor. Interestingly, TTF-1 can also be positive in neuroendocrine tumors of pulmonary and extrapulmonary origin.127,134–136 Cytokeratins 7 and 20, used in combination, also assist in categorizing certain tumors. These stains are not specific for a particular site of origin but can narrow the differential diagnosis based on their pattern of expression.
Although mesothelioma can be easily identified ultrastructurally, it has historically been difficult to differentiate from adenocarcinoma through morphology and immunohistochemical staining. Several markers in the last few years have proven to be helpful including CK5/6, calretinin, and Wilms tumor gene-1 (WT-1),95 all of which are positive in mesothelioma.
Table 72-2 summarizes the common molecular alterations found in lung tumors, and Table 72-3 summarizes commonly used immunohistochemical stains.95,109,112,127,133,134,137–142
Table 72-2
Examples of Common Molecular Alterations in Lung Tumors
| Diagnosis | Common Molecular Alterations |
| Squamous preneoplasia | LOH—3p, 9p21,8p21-23, aneuploidy, methylation |
| Atypical adenomatous hyperplasia | LOH—3p,9p aneuploidy K-ras codon 12 mutation |
| Adenocarcinoma | p53 mutation p16 mutation/inactivation K-Ras (42%); smokers more common EGFR overexpression (40%) EGFR mutation Her2/neu, COX-2 overexpression, ALK rearrangement, ROS1 fusion, etc. |
| Squamous cell carcinoma | p53 mutation p16 inactivation Allelic loss 3p EGFR overexpression (80%) |
| Large cell carcinoma | K-Ras, p53, loss p16 |
| Large cell neuroendocrine carcinoma | p53 bcl-2 overexpression Rb mutation 3p21, FHIT, 3p22-24,5q21,9p21 |
| Small cell carcinoma | Rb mutation (80+%) p53 mutation 50-80% BCL-2 expression 3p21, FHIT, 3p22-24,5q21,9p21 |
EFGR, Epidermal growth factor receptor; LOH, Loss of heterozygosity.
Table 72-3
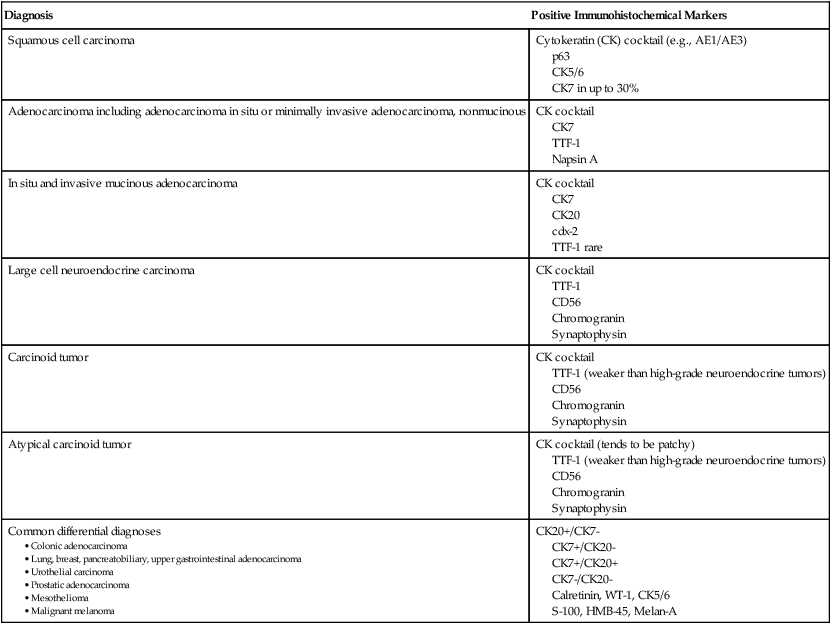
Common Immunohistochemical Markers Used in the Diagnosis of Lung Tumors
| Diagnosis | Positive Immunohistochemical Markers |
| Squamous cell carcinoma | Cytokeratin (CK) cocktail (e.g., AE1/AE3) p63 CK5/6 CK7 in up to 30% |
| Adenocarcinoma including adenocarcinoma in situ or minimally invasive adenocarcinoma, nonmucinous | CK cocktail CK7 TTF-1 Napsin A |
| In situ and invasive mucinous adenocarcinoma | CK cocktail CK7 CK20 cdx-2 TTF-1 rare |
| Large cell neuroendocrine carcinoma | CK cocktail TTF-1 CD56 Chromogranin Synaptophysin |
| Carcinoid tumor | CK cocktail TTF-1 (weaker than high-grade neuroendocrine tumors) CD56 Chromogranin Synaptophysin |
| Atypical carcinoid tumor | CK cocktail (tends to be patchy) TTF-1 (weaker than high-grade neuroendocrine tumors) CD56 Chromogranin Synaptophysin |
| Common differential diagnoses |
CK7+/CK20-
CK7+/CK20+
CK7-/CK20-
Calretinin, WT-1, CK5/6
S-100, HMB-45, Melan-A
Molecular Alterations
Although numerous molecular alterations have been described in lung cancer throughout the years, more recently, the role of so-called “driver” mutations in NSCLC, particularly adenocarcinoma, has received much attention, in view of their potential use as therapeutic targets.154–154 “Driver” mutations lead to activation of intracellular signaling proteins that induce and sustain tumorigenesis. KRAS mutations, which occur in approximately 25% of lung adenocarcinomas, have been described for many years.155 The first mutation that had therapeutic implications was identified in the EGFR gene of patients with adenocarcinomas that showed response to treatment with EGFR tyrosine kinase inhibitors (TKIs).156,157 EGFR mutations occur in approximately 10% of lung cancers in the United States and in 35% of lung cancers in Asia.59,61,62 Patients with EGFR mutant tumors have a better prognosis in general compared to those with EGFR wild-type tumors.158 EGFR mutations are more commonly found in tumors from female, never smokers, with adenocarcinoma histology. Although most of the EGFR mutations confer sensitivity to treatment with EGFR TKIs (including the two most common: exon 19 deletions and L858R substitution in exon 21), a few mutations, such as insertions in exon 20 and the T790M mutation, also in exon 20, are associated with resistance to treatment with EGFR TKIs.159 The T790M mutation is associated with approximately 1% of primary resistance, but more than 50% of acquired resistance to EGFR TKI treatment.154
More recently, a mutation was described in the ALK gene that causes inversion of ALK with resulting fusions with other genes, particularly EML-4, resulting in oncogenic constitutive activation of ALK.160 This mutation occurs in 3% to 7% of adenocarcinomas and tends to occur in younger male patients who are light smokers or who have never smoked. An ALK TKI (crizotinib) has been approved by the FDA for treatment of patients with tumors that harbor this rearrangement.161 The recommended method to detect ALK mutations is fluorescent in situ hybridization (FISH) using a dual-color break-apart probe.
Many authors have attempted to correlate the histologic subtype of adenocarcinoma with the different known mutations.162–165 The only strong correlation has been between invasive mucinous adenocarcinoma (mucinous bronchioloalveolar carcinoma in the WHO classification) and KRAS mutation (80% to 100%).164,165
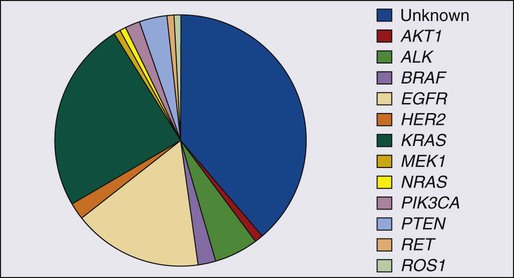
In addition to EGFR, KRAS, and ALK mutations, other “driver” mutations have been discovered, such as BRAF, PIK3CA, NRAS, AKT1, MET, NRAS, MEK1 (MAP2K1), and ROS1 (Fig. 72-9).154,166 Each of these mutations occurs in less than 3% of lung adenocarcinomas. The great majority of the “driver” mutations are mutually exclusive and there are ongoing clinical studies for their specific inhibitors. Most of these mutations are present in adenocarcinoma; however, mutations that may be linked to future targeted therapies in squamous cell carcinomas are emerging. These include FGFR1, DDR2, and PIK3CA.167–169
The most commonly used method to detect EGFR mutations is direct gene sequencing. Some institutions use SNaPshot technology, a multiplexing technique that allows identification of more than 50 mutations using the same amount of tumor used for direct sequencing of specific genes.170 For all molecular methods, tissue evaluation for tumor adequacy and tumor selection should be done by a pathologist to ensure successful testing.
Early Detection and Screening
Early detection is a process that involves screening tests, surveillance, and diagnosis, and also implies early treatment,143 whereas screening is defined as the systematic testing of asymptomatic individuals for preclinical disease.144 The purpose of screening is to prevent or delay the development of advanced disease in patients with preclinical disease through early detection and treatment. Screening presumes that a test or series of tests will identify asymptomatic persons at risk for a specific disease and that a positive result leads to further testing to establish definitively the presence or absence of disease.144 The monitoring of these subjects (surveillance) intends to detect the disease early and to treat it early. Under ideal circumstances, early intervention should change the course of the disease once the diagnosis is established, resulting in a decrease in disease-related mortality (the number of disease-specific deaths relative to the total number of persons evaluated). In addition, screening should apply to large populations that would benefit from early detection (therefore with a chance of survival for longer than 5 years). Finally, screening should cause no harm and be cost-effective.145 (See Chapter 23.)
Until recently, lung cancer screening efforts using periodic chest x-rays coupled with regular assessment of sputum cytology failed to demonstrate a decrease in lung cancer-related mortality.146–149 Details of these trials have been extensively reviewed elsewhere.150 Ironically, more lung cancers were diagnosed in the screening arms of these trials. The lung cancers identified in the screened population were often found at early stages, allowing more patients in the screened arms to go to definitive surgery. Nonetheless, there was not an improvement in lung cancer-related mortality, which is considered a requirement to validate potential screening methods.150 Several hypotheses have been advanced to explain the findings including flawed trial design, lead-time bias, length-time bias and over diagnosis bias.151,152 Briefly, these biases can be defined as follows: lead-time bias implies that earlier detection could result in longer survival from the time of diagnosis even if death is not delayed. Length time bias occurs when screening examination detects slow-growing cancers. In other words, the slower the growth of the neoplasm, the longer it is present without symptoms and the greater the likelihood of detection. Over diagnosis bias refers to the phenomenon of detecting a lung cancer that would otherwise have remained subclinical before death from other causes. Overdiagnosis is of some concern in lung cancer screening because newer screening modalities can identify small nodules of unknown clinical significance. Mayo Clinic investigators found a persistence of excess lung cancer cases in the intervention arm of their original screening after an additional 16 years of follow up,152 providing support for some overdiagnosis in lung cancer screening. In recent years, the field has advanced dramatically. The progress is summarized below and so are the many challenges it brings along.
The High-Risk Population, a Susceptible Subgroup
As noted, a challenging problem in screening for lung cancer is the definition of a “high-risk” population—the population that could best benefit from lung cancer screening.153 Individuals “at risk” include current and former smokers, those with specific various occupational exposures (e.g., asbestos; see epidemiology), the presence of airflow obstruction, a family history of lung cancer, older individuals, and those with a prior history of a cancer of the aerodigestive tract. Individuals who smoke expose their entire aerodigestive tract to multiple carcinogens and, therefore, it is not surprising that this population experiences a high rate of second primary tumors, estimated to be between 1% and 4% per patient per year.156–156 However, mere recognition of these features is not sufficient to identify “high-risk” individuals. For example, although smoking is an obvious risk factor for lung cancer—the cumulative risk of dying from lung cancer for a life-long smoker is estimated to be approximately 16% in men and approximately 10% in women157—the risk of developing lung cancer varies greatly among individual current and former smokers.158 For example, in the Carotene and Retinol Efficacy Trial, a large, randomized trial of lung cancer prevention, the 10-year cancer risk among current and former smokers ranged from less than 1% to 15%.158 An example of an individual with a relatively low risk of developing lung cancer would be a 51-year-old woman who smoked one pack per day for 28 years and quit 9 years earlier. By contrast, a 68-year-old man who had smoked two packs per day for 50 years and continued to smoke might have a 15% risk of lung cancer.158 Both of these individuals would be potentially eligible for screening and yet the “payoff” may be quite different. This fact suggests that an accurate risk prediction model may facilitate greatly the admission of subjects to screening (or chemoprevention) trials.158,159 Many attempts to improve these prediction models have been published recently, trying to bring demographic and clinical,158,160–162 and now molecular biomarkers, such as DNA SEX6L polymorphism163 or DNA sputum cytometry,161 or imaging biomarkers, into the equation, such as the presence of a nodule or emphysema at baseline.164
Relative to smokers with normal lung function, smokers with COPD have a four- to sixfold elevated risk of lung cancer, making COPD the greatest clinical risk factor in ever smokers.165–168 Low-dose chest computerized tomography (LDCT) screens provide an in vivo assessment of both biologically relevant features of indeterminate nodules and extent of injury reflected by emphysema. The integration of biological and imaging-based biomarkers to individualize lung cancer risk not only identify individuals most likely to benefit from screening, and perhaps more intensive screening, but also could identify individual smokers who are at great risk of developing cancer and further convince them to stop smoking, which is the first and most effective step in the prevention of lung cancer and all other smoking-related diseases.
Cytologic atypia in sputum samples reflect the abnormalities that develop in the bronchial epithelium. Moreover, the presence of cytologic atypia has been shown to predict lung cancer risk.169 The presence of moderate dysplasia or worse cytologic atypia was associated with an increased risk of developing lung cancer in a cohort of heavy smokers with airflow obstruction (adjusted hazard ratio of 2.8).170 This translates into a cumulative lung cancer incidence of 10% at 3 years and 20% at 6 years. Given the limitations of cytologic evaluation of sputum samples, however, the study of molecular abnormalities in the sputum (e.g., methylation patterns of specific genes involved in lung cancer progression and cytogenetic alterations) may strengthen the assessment of risk for lung cancer in this population.
Molecular epidemiology may be used to identify patients at risk for developing lung cancer through the identification of specific genes, single-nucleotide polymorphisms, or other genetic traits associated with increased susceptibility for lung cancer.171 Although many genotypes show increased risk (relatively low odds ratios) with lung cancer (e.g., CYP2A6, thymidylate synthase, GSTM1, XPA), their large number and low penetrance make targeted interventions extremely challenging. Assessing genetic susceptibility for lung cancer may allow the identification of susceptible subgroups most likely to be ideal candidates for early detection. With the exception of the rare EGFR T790M polymorphism, autosomal-dominant genes have not been found in association with family history of lung cancer. There is epidemiological evidence demonstrating a 2.5 times increased risk in any patient with a family history of lung cancer (two affected first-degree relatives) after controlling for smoking.172 A region on chromosome 6q23-25 has been identified as a locus of susceptibility with a maximum heterogeneity logarithm of odds (LOD) score of 2.79, 3.47, and 4.26 for families with three, four, five, or more affected individuals.56 Further complicating the picture is the role of the environment in modifying these specific genes and the interaction of genes between each other and evidence to support a genetic predisposition to smoking addiction.173,174 If markers of these genetic predispositions can be firmly established, intensive intervention to alter risk factors in these selected populations may alter their clinical outcome.
The Imaging Approach
The last decade has witnessed a dramatic improvement in technology allowing for faster, higher-resolution imaging of the chest with reduced radiation exposure. The computed tomography (CT) scanner first became available in the 1970s but was impractical for screening because of its slow speed and high radiation dose. In the mid-1990s low-dose scanners capable of imaging the chest in less than 15 seconds using radiation doses equivalent to 10 radiographs became available, opening the door to their potential use for screening.175 Moreover, new bronchoscopic methods also demonstrate promise for the detection of preinvasive lesions.176 Other imaging modalities, such as positron emission tomography (PET) scanning, are able to provide metabolic information on lesions and can be combined with CT scan to give detailed resolution.
Low-Dose Spiral Computed Tomography Scan
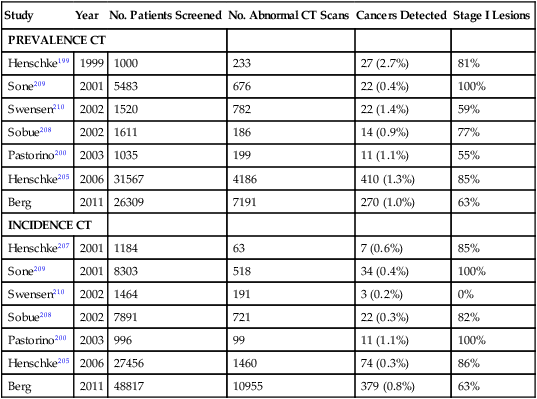
In recent years there has been a substantial increase in the use of spiral CT scans to screen for lung cancers in former and current smokers.143,150 Spiral CT screening allows for a rapid and comprehensive evaluation of the lungs and is attractive because of its potential increased sensitivity, low radiation exposure, and potential cost-effectiveness.143 Multiple pilot studies that used spiral CT scans to screen for lung cancers have shown that this technology can identify a higher percentage of early stage lung cancer compared with conventional imaging studies.177–180 These results are summarized in Table 72-4 and discussed in detail elsewhere.183–183 Lung cancer prevalence rates range between 0.4% and 2.7%, depending on the population screened. In general, these prevalence rates are significantly higher than that reported using conventional imaging studies. The mean diameter of screened detected cancers ranges between 14 and 21 mm. Incidence rates based on detection of new malignancies at annual repeat screening range from 0.07% to 1.1%.143,184 Notably, up to 85% of CT-screened detected lung cancers are clinical stage I lesions. By contrast, only 15% of lung cancers diagnosed through routine clinical care are found to be stage I.143 Because stage I lung cancer is the most curable form of this disease, a high frequency of detection of stage I tumors is considered a necessary (though not sufficient) indication of a favorable screening outcome.143
Table 72-4
Results of Prevalence and Incidence Screens in CT Screening Trials
| Study | Year | No. Patients Screened | No. Abnormal CT Scans | Cancers Detected | Stage I Lesions |
| PREVALENCE CT | |||||
| Henschke199 | 1999 | 1000 | 233 | 27 (2.7%) | 81% |
| Sone209 | 2001 | 5483 | 676 | 22 (0.4%) | 100% |
| Swensen210 | 2002 | 1520 | 782 | 22 (1.4%) | 59% |
| Sobue208 | 2002 | 1611 | 186 | 14 (0.9%) | 77% |
| Pastorino200 | 2003 | 1035 | 199 | 11 (1.1%) | 55% |
| Henschke205 | 2006 | 31567 | 4186 | 410 (1.3%) | 85% |
| Berg | 2011 | 26309 | 7191 | 270 (1.0%) | 63% |
| INCIDENCE CT | |||||
| Henschke207 | 2001 | 1184 | 63 | 7 (0.6%) | 85% |
| Sone209 | 2001 | 8303 | 518 | 34 (0.4%) | 100% |
| Swensen210 | 2002 | 1464 | 191 | 3 (0.2%) | 0% |
| Sobue208 | 2002 | 7891 | 721 | 22 (0.3%) | 82% |
| Pastorino200 | 2003 | 996 | 99 | 11 (1.1%) | 100% |
| Henschke205 | 2006 | 27456 | 1460 | 74 (0.3%) | 86% |
| Berg | 2011 | 48817 | 10955 | 379 (0.8%) | 63% |
The Early Lung Cancer Action Program (ELCAP) was a large lung cancer screening trial started in the 1990s utilizing chest CT imaging.178 It showed an improved detection rate and survival of early stage lung cancers. International Early Lung Cancer Action Program (IELCAP) investigators reported a 92% 10-year survival rate among screen-detected clinical stage I lung cancer patients who underwent surgical resection within 1 month after diagnosis.185 In a separate report, IELCAP investigators reported that the cancers identified by CT screening met standard criteria for full-fledged aggressive lung cancer.113 Although these data are intriguing and encouraging, long-term followup of these patients will be very important to exclude lead-time bias (simply diagnosing the cancer earlier, but not altering its outcome) as a confounder for an improved survival and a reduced disease-related mortality. These results prompted the design of a large randomized National Lung Screening Trial (NLST). The NLST was a prospective comparison of spiral CT and standard chest x-ray in 50,000 current or ex-smokers, between the ages of 55 and 74 years, designed to determine the best strategy to reduce lung cancer-related mortality. A similar study is ongoing in the Netherlands and Belgium (the NELSON trial) comparing CT scanning with standard of care among 20,000 subjects with history of heavy smoking.186,187 Exciting results of the recently completed NLST showed a 20% reduction in lung cancer-specific mortality using low-dose CT screening for patients at high risk for lung cancer after a median follow up of 6.5 years, compared with chest x-ray.180 This is the first large, randomized, screening study of lung cancer by low-dose chest CT to show an improvement in overall survival, thus giving new hope that broader application of this technology will result in a significant reduction in the death rate for this cancer. The results of this trial were further strengthened by the results of the Prostate, Lung, Colorectal, and Ovarian (PLCO) cancer study, which reported that annual screening with chest radiograph did not reduce lung cancer mortality compared with usual care.188 However, all screening effort comes at some cost. In the NLST, only 4% of the nodules detected ended up being malignant (a 96% false-positive rate). Twenty-four percent of patients who had a high enough clinical suspicion of cancer to undergo a diagnostic surgical procedure (mediastinoscopy, thoracoscopy, or thoracotomy) had benign disease.180 As LDCT screening for lung cancer evolves, better predictive models that incorporate biomarkers are needed to determine which patients have lung cancer. New imaging and molecular biomarkers are considered to be front runners in the field of early detection.159 In summary, LDCT screening, though yet to be broadly adopted, has been shown to reduce lung cancer mortality in a large, U.S.-based clinical trial, and is now recommended by several major medical organizations in specific high-risk groups within the United States.
Among the unanswered questions with LDCT screening are the duration of screening, the interval between tests, the definition of the high-risk individuals, the management of indeterminate pulmonary nodules, the cost-effectiveness analysis,189 the benefit of computerized characterization of lung nodules and lung parenchyma over routine visual analysis, and the assessment of the radiation risk associated with LDCT screening. Although the amount of radiation exposure from a low dose spiral CT scan is relatively modest (roughly equal to 10 chest x-rays or one-tenth that experienced with a regular chest CT scan) it is not an insignificant issue,185,186 and if applied to very low-risk populations, some calculations actually show a theoretically increased risk of lung cancer.
A major challenge confronting advocates of CT screening is the high false-positive rate.190 On initial screening of at-risk populations, false-positive rates range between 10% and 20% of the screened population but can be as high as 50%.191–194 Positive predictive values range from 2.8% to 11.6%.191–194 False positives can have a substantial impact on patients through the expense and risk of unneeded further invasive and morbidity-associated evaluation,195 as well as creating uncertainty and emotional stress. False-positive rates and positive predictive values are somewhat improved in annual follow up CT scans, but there is still significant room for improvement. Based on extant data, it appears that nodules smaller than 5 mm are unlikely to be cancerous and those 5 to 10 mm in diameter (25% to 40% of noncalcified nodules detected) are of uncertain significance.196 The management of patients with these nodules usually consists of repeated CT scans over time to see if the nodules grow, attempted fine-needle aspirates, or surgical resection.
Finally, CT scanning has also shown promise in the detection of some preinvasive lesions. High-resolution CT has proven to be sensitive enough to detect ground-glass opacities, some of which may represent inflammatory lesions, adenocarcinoma in situ, or AAH, a presumed precursor lesion to adenocarcinoma.197–200 Because adenocarcinoma is now the most common histologic type of lung cancer in the United States, there is some hope that CT may be able to improve survival for these patients by detecting a precursor lesion before its transformation to invasive carcinoma. Chest CT scanning, however, is not yet sensitive enough to detect preinvasive epithelial lesions of the bronchial tree with squamous differentiation, likely precursors of squamous carcinoma of the lung.
Positron Emission Tomography Scan
Many CT screening protocols now use PET-CT scan as a method of reducing this high false-positive rate. The data in this application remains controversial. PET scanning attempts to identify malignancy based on glucose metabolism by measuring the uptake of fluorodeoxyglucose (FDG) 18F. Lung cancers will preferentially take up 18FDG and appear as a “hot spot”. To date, PET has been mostly used for staging and detection of metastases in lung cancer201,202 and in the diagnosis of nodules larger than 15 mm in diameter.179,203 In combination with another imaging modality, PET may provide information about the metabolic state of lung nodules. Combined 18FDG-PET-CT scan has been shown to improve the accuracy of staging in lung cancer compared with visual correlation of PET and CT or either study alone.204 The added information gained from PET and combined PET-CT may help reduce the high false-positive rates seen in trials using CT alone. A trial of low-dose CT, used in combination with PET scan of 1035 patients, resulted in only six false positives, defined as a surgical biopsy of a benign nodule, out of 27 surgical biopsies of suspicious nodules.179 There are important limitations to this study, however. First, lung nodules less than or equal to 5 mm in size were followed, with repeat CT in 1 year, but had no other intervention. Second, PET scanning was only performed on larger nodules (≥7 mm). Typically, PET scan has not performed well in the identification of small nodules (<15 mm), so its usefulness in intermediate lesions remains in question, in particular in areas with high prevalence of pulmonary fungal infections.205 There is no evidence of a survival benefit from using PET scan as part of a screening protocol.
In fact, PET has limited usefulness in detection of lesions smaller than 1 cm and has shown to be of limited value in adenocarcinoma, particularly very well differentiated subtypes.206,207 Each method of evaluation is costly, and some have morbidity.195 The impact of waiting to assess nodule growth on patient outcome is also not clear, but can only decrease curability. Even for patients with disease that is highly suspicious for lung cancer on clinical grounds, there is a 10% to 20% incidence of “futile thoracotomies” in which the suspicious lesion is found to be benign208 and the patient unnecessarily incurred the morbidity and potential mortality of a thoracotomy.211–211 Eliminating unnecessary surgeries should be one of our priorities, although this may not be feasible without the development of other strategies of early detection.
Other Imaging Techniques
Fluorescence endoscopy uses differences in the autofluorescence characteristics of normal and neoplastic epithelium to localize lesions. Fluorescence bronchoscopy has been shown to be more sensitive than white light bronchoscopy in the detection of preneoplastic lesions in many studies, including a randomized trial.176,212–214 A randomized trial demonstrated that the use of the LIFE (lung imaging fluorescent endoscopy) procedure resulted in a 46.9% absolute increase in the sensitivity of detecting moderate dysplasia or worse in high-risk patients when compared with white light bronchoscopy, although the specificity was worse.176 Fluorescence bronchoscopy has not been proven as a validated method of early detection of lung cancer, but it is a very important research tool.
New imaging techniques are currently being developed to help improve not only the resolution of current imaging techniques, but also to image lesions based on biological activity. Both magnetic resonance and gamma camera techniques are being tested as molecular imaging tools.217–217 Near-infrared Raman spectroscopy,218 optical coherence tomography,219 and confocal microscopy use optical differences within tissue to allow imaging of individual cell nuclei. This technology is being adapted for use during endoscopic exams and may be able to provide real-time histologic evaluation of bronchial mucosa.
Biofluids-Based Biomarkers for Lung Cancer
The underlying premise of biofluids-based biomarker research is that molecular alterations of tumor cells lead to the synthesis and shedding of distinct molecular species that can be detected in biofluids. Biofluids-based detection strategies are an attractive approach for screening, namely because of their ease of acquisition. Biofluids, including peripheral blood and its components (circulating cells, plasma, and serum), exhaled breath condensate (EBC), urine, and sputum, offer noninvasive access to large quantities of samples available for analysis. These alterations can lead to the generation of disease specific molecular species such as altered or methylated DNA, overexpressed messenger RNA (mRNA), microRNA, or proteins that can potentially be released into the extracellular microenvironment at a rate that is dependent on multiple variables but mainly tumor growth and shedding rates.220 Therefore, molecular analyses of early stage lung cancer–related biofluids represent an attractive choice for the discovery and validation of diagnostic biomarkers. The success of this approach will also depend on our ability to not only improve the specificity of candidate biomarkers but also on the sensitivity of detection assays. Although various serum biomarkers have been investigated in lung cancer, none has proved useful in general clinical practice, mainly because of the lack of sufficient sensitivity and specificity as recently reviewed.159 For example, cytokeratin fragment antigen 21.1 (Cyfra-21.1), a marker of cytokeratin,223–223 carcinoma embryonic antigen (CEA),222 and tissue polypeptide antigen (TPA)221 were found to have relatively poor sensitivity (31% to 64%) when specificity limits of 95% were set. In addition, most markers reach better sensitivity in advanced disease stages as compared with stage I lung cancer. Thus, their use for early diagnosis or screening has not had an impact on patient care in the clinic. Blood-derived biomarkers used in combination with other clinical, imaging or molecular tools might play an important role in early detection,224 in monitoring response to therapy, in risk assessment of recurrence and in prognosis, especially in intermediate-risk nodules in low-risk individuals and in nodules that are PET negative.
Clinical Presentation and Staging of Lung Cancer
Presenting Signs and Symptoms
Symptoms, signs, and laboratory test abnormalities relating to lung cancer can be classified as those caused directly by the primary lesion, those related to intrathoracic spread or to distant metastasis, and those related to paraneoplastic syndromes.227–227 The prototypical lung cancer patient is a current or former smoker of either gender, usually in the seventh decade of life, who presents with symptoms attributable to bulky intrathoracic disease (i.e., cough, dyspnea, chest pain, hoarseness, and/or hemoptysis) or distant metastases (e.g., bone pain, central nervous system [CNS] symptoms).226,228 Constitutional symptoms may include weakness, anorexia, weight loss, and, rarely, fever.226,228 Apart from the brevity of symptom duration, these parameters fail to clearly distinguish SCLC from NSCLC or even from neoplasms metastatic to the lungs.229 Lung cancer arising in a lifelong never-smoker is more common in women and certain ethnic groups and tends to be an adenocarcinoma.230–233 Such patients also tend to be slightly younger than their smoking counterparts at the time of diagnosis.230 However, the clinical presentation of lung cancer in never-smokers tends to mirror that of current and former smokers even though lung cancers arising in never-smokers appear to be biologically distinct, as evidenced by the differing molecular abnormalities found in tumors derived from smokers and nonsmokers.232 The prognosis of lung cancer in never-smokers is generally improved compared with cancers in current or former smokers regardless of stage.41,230,234 This improved prognosis may be because of the better overall condition of these patients, the higher frequency of actionable genetic abnormalities in tumors from these patients (discussed below), or other as yet unknown reasons.
Cough, dyspnea, and chest discomfort are the most common presenting symptoms in lung cancer (Table 72-5).225 A history of chronic cough with or without hemoptysis in a current or former smoker with COPD aged 40 years or older should prompt a thorough investigation for lung cancer, even in the face of a normal chest x-ray.225 A persistent “pneumonia” without constitutional symptoms and unresponsive to repeated courses of antibiotics also should prompt an evaluation for an underlying cause (e.g., an occult endobronchial lesion), especially if the patient is a current or former smoker. Less frequently, patients present with hemoptysis, which rarely is massive and is usually described as streaks of fresh or old blood in sputum. The spread of disease within the chest may result in hoarseness secondary to recurrent laryngeal nerve paralysis and less frequently phrenic nerve paralysis. The latter is associated with an elevated hemidiaphragm on standard chest x-ray. Chest wall involvement is commonly accompanied by pain that in turn may serve as a more accurate indicator of chest wall invasion than radiographic studies. Chest wall pain is usually related to either direct invasion of the pleura or chest wall by the primary tumor, or caused by a rib metastasis. Tenderness may be elicited at the site of rib involvement and, rarely, a soft-tissue mass can be palpated.225 The chest pain may have a pleuritic component if there is pleural involvement. The disappearance of pleuritic chest pain may signify the development of a pleural effusion that in turn may cause shortness of breath or worsen existing dyspnea. Venous distention of the neck and chest wall, cyanosis, facial plethora, and upper extremity edema may indicate obstruction of the superior vena cava, which, today, is most commonly seen with SCLC.225,235 Although the heart and other mediastinal structures are often involved with tumor at postmortem examination, only rarely does this involvement serve as the source of a presenting symptom.
Table 72-5
Presenting Sign and Symptoms of Lung Cancer
| Symptom and Signs | Range of Frequency |
| Cough | 8%–75% |
| Weight loss | 0%–68% |
| Dyspnea | 3%–60% |
| Chest pain | 20%–49% |
| Hemoptysis | 6%–35% |
| Bone pain | 6%–25% |
| Clubbing | 0%–20% |
| Fever | 0%–20% |
| Weakness | 0%–10% |
| Superior vena cava obstruction | 0%–4% |
| Dysphagia | 0%–2% |
| Wheezing and stridor | 0%–2% |
Data from Beckles MA, Spiro SG, Colice GL, Rudd RM. Initial evaluation of the patient with lung cancer: symptoms, signs, laboratory tests, and paraneoplastic syndromes. Chest 2003;123:97-104.
Approximately one-third of patients present with symptoms as a result of distant metastases.225 The most common sites of distant metastasis from lung cancer are the bones; liver, adrenal glands, and intra-abdominal lymph nodes; brain and spinal cord; and lymph nodes and skin. Lung cancer can metastasize to virtually any bone with pain being the primary presenting symptom in up to 25% of patients at diagnosis.225 Similarly, liver metastases are common at initial presentation in both SCLC and NSCLC. However, liver function test results are seldom abnormal until the metastases are numerous and large. Hepatic metastases most commonly produce symptoms of weakness and weight loss. Adrenal lesions (only rarely associated with adrenal insufficiency) and paraaortic lymph node metastases are most commonly seen with SCLC. Intracranial metastases at presentation are most commonly in SCLC and adenocarcinomas of the lung. Presenting symptoms may include headache, nausea and vomiting, focal neurologic symptoms or signs, seizures, confusion, and personality changes.
The stigmata of COPD in smokers may be the only findings on physical examination, or one may detect lymphadenopathy, hepatomegaly, bone tenderness, or abnormal neurologic findings. Digital clubbing and hypertrophic osteoarthropathy (HPO) may be associated with any histologic subtype of lung cancer but are most frequently associated with squamous cell and adenocarcinoma, and are least likely to occur in a patient with small cell carcinoma.225,236 Digital clubbing is more common than HPO and the latter is characterized by painful symmetrical arthropathy and periosteal new bone formation of the distal limbs. Its mechanism of development is unknown. In a published series of 111 consecutive lung cancer patients, clubbing was noted in 29%, with an incidence of 35% in NSCLC and only 4% in SCLC.236 Up to 20% of patients present with palpable lymphadenopathy in the supraclavicular fossa during the course of the disease. Subcutaneous metastases, although rare, portend a poor prognosis but serve as a readily accessible source of diagnostic material.
Paraneoplastic Disorders
Although many of the symptoms of NSCLC and SCLC are attributable to mass effect and direct impingement upon vital organs, less commonly patients with lung cancer present with symptoms related to hypercalcemia,237 hyponatremia,229,238 Cushing syndrome,239 Lambert-Eaton syndrome, and other neurologic disorders.242–242 These so-called paraneoplastic phenomena can be seen in any histologic type of lung cancer, but are most frequently associated with SCLC. In general, a majority of these paraneoplastic phenomena fall into endocrine or neurologic categories.245–245 (See Chapter 39.)
Hypercalcemia
Hypercalcemia of malignancy (HCM) is the most common life-threatening metabolic complication of malignancy, affecting approximately 10% to 20% of patients with advanced cancer. Hypercalcemia may be associated with or caused by production of a parathyroid hormone-related peptide.237 The incidence of HCM varies widely by cancer type, but occurs most frequently in patients with multiple myeloma and carcinomas of the lung, breast, kidney, and head and neck. With respect to lung cancer, squamous cell carcinoma is the most common histologic subtype. Clinical symptoms of HCM include nausea, vomiting, abdominal pain, constipation, polyuria and thirst, and altered mental status. HCM may lead to renal failure. The early symptoms of nausea, vomiting and constipation may be easily confused with the initiation of narcotics for pain control. (See Chapter 37.)
Hyponatremia and the Syndrome of Inappropriate Antidiuretic Hormone
The inappropriate secretion of antidiuretic hormone, or arginine vasopressin, with its resultant euvolemic, refractory, hyposmolar hyponatremia, is observed in up to 15% of SCLC patients.238,246 However, up to one third of patients with hyponatremia have no evidence of ectopic arginine vasopressin production.247 In such cases hyponatremia may be caused by ectopic production of atrial natriuretic peptide (ANP). SCLC is the most common malignant cause of acute or chronic syndrome of inappropriate antidiuretic hormone (SIADH).248 The presence of SIADH does not correlate with clinical stage, distribution of metastatic sites, or gender, nor does SIADH influence response to chemotherapy or overall survival as an independent variable.238 As with most paraneoplastic syndromes, the best therapy for SIADH is effective treatment of the underlying SCLC. SIADH typically resolves within 1 to 4 weeks of initiating chemotherapy in the vast majority of cases.238 While waiting for the effects of chemotherapy, serum sodium can usually be managed and maintained above 128 mEq/L via strict fluid restriction alone.249 Demeclocycline, which blocks the action of vasopressin at the level of the renal tubule, can be a useful adjunctive measure when fluid restriction alone is insufficient to restore sodium level.250,251 The starting dose is 150 mg four times a day, but may need to be increased to 1200 mg. Tolvaptan, an oral vasopressin V2-receptor nonpeptide antagonist, also is effective in increasing serum sodium concentrations in patients with euvolemic and hypervolemia hyponatremia.252 Of note, patients with ectopic ANP secretion do not respond to fluid restriction.247 In fact, fluid restriction may actually worsen hyponatremia if sodium intake is not concomitantly increased. Accordingly, if hyponatremia fails to improve or worsens after 3 to 4 days of adequate fluid restriction, plasma levels of AVP and ANP should be measured to determine whether inappropriate secretion of antidiuretic hormone (ADH) or ANP is the causative syndrome.247
Ectopic Adrenocorticotropic Hormone Production
Cushing syndrome may be caused by ectopic secretion of adrenocorticotropic hormone (ACTH) from a nonpituitary tumor resulting in bilateral adrenocortical hyperplasia and hypercortisolemia.253 Neuroendocrine lung tumors, including SCLC and pulmonary carcinoids, account for approximately half of the cases of ectopic ACTH-producing tumors.239,254 Although hypercortisolemia has been documented in up to 50% of SCLC cases, only 2% to 5% of SCLC patients have the characteristic clinical features of Cushing syndrome.239 Unlike Cushing disease, the onset of symptoms in ectopic ACTH is often abrupt as a result of the characteristic rapid growth of SCLC. Consequently, the classical features of Cushing disease—a buffalo hump, striate, and moon faces—are frequently absent. By contrast, hypokalemic alkalosis, hypertension, hyperglycemia, and, rarely, edema and muscle wasting are common.239 The effect of Cushing syndrome on survival is unclear, although some investigators hold that its onset heralds a more aggressive tumor behavior.239 Treatment with standard medications, such as metyrapone and ketoconazole, is largely ineffective because of extremely high cortisol levels.255 Some patients require bilateral adrenalectomy to control symptoms.253 The most effective strategy for management of the Cushing syndrome is effective treatment of the underlying SCLC.239
Neurologic Paraneoplastic Syndromes
The paraneoplastic neurologic disorders are a diverse group of diseases characterized by the presence of neurologic dysfunction in the setting of a remote cancer.244,245 They are often the result of production of antibodies that react with both the small cell cancer cells and with normal host tissue. Well-described syndromes include the Lambert-Eaton myasthenic syndrome, paraneoplastic encephalomyelitis and sensorimotor neuropathy and paraneoplastic cerebellar degeneration.242,256 Less-frequent abnormalities include subacute sensory neuropathy, autonomic disturbances, myelopathies, progressive encephalopathy, and visual paraneoplastic syndromes.241,244,257
Lambert-Eaton myasthenic syndrome (LEMS) is caused by autoantibodies directed against presynaptic voltage-gated P/Q calcium channels.242,256 The P/Q calcium channel autoantibodies decrease calcium entry into the presynaptic terminal, which prevents binding of vesicles to the presynaptic membrane and acetylcholine release. Patients with this disorder present with proximal muscle weakness, usually in the lower extremities, occasional autonomic dysfunction, and rarely with cranial nerve symptoms or involvement of the bulbar or respiratory muscles. Depressed deep tendon reflexes are frequently present. As contrasted to patients with myasthenia gravis, strength improves with serial effort. The diagnosis is confirmed by electrophysiologic testing, which demonstrates small compound muscle action potentials and facilitation with exercise or 20-Hz repetitive stimulation. A serum test for voltage-gated calcium channel antibodies, estimated to occur in 5% of patients with SCLC, is commercially available. Plasma exchange and intravenous immunoglobulin can provide short-term benefit, and 3,4-diaminopyridine, which enhances the release of acetylcholine from presynaptic terminals,258 prednisone, and azathioprine can provide limited long-term benefit.259 Some patients who respond to chemotherapy will have resolution of the neurologic abnormalities, and this is the initial treatment of choice.
Paraneoplastic encephalomyelitis and sensory neuropathies, cerebellar degeneration, limbic encephalitis, and brainstem encephalitis occur in SCLC in association with a variety of antineuronal antibodies such as anti-Hu, anti-CRMP5, and ANNA-3.241,244,260 These antibodies have been found in up to 25% of SCLC patients, although not always in association with a clinically obvious neurologic disorder.244 These disorders may predate the diagnosis of SCLC.244 Most paraneoplastic neuropathies are sensorimotor and axonal, symmetric in distribution and frequently disabling.244 Limbic encephalitis is characterized by degeneration of neurons in the medial temporal lobe with clinical features that include behavioral changes, hallucinations, short-term memory loss, anosmia, ageusia, and dementia.261 Symptoms of brainstem encephalitis include vertigo, nystagmus, oscillopsia, ataxia, diplopia, dysarthria, and dysphagia reflecting the predominant involvement of the floor of the fourth ventricle and inferior olives.262 Some patients develop respiratory insufficiency requiring assisted ventilation. Cerebrospinal fluid (CSF) studies may show pleocytosis and elevated protein levels. Magnetic resonance imaging (MRI) brain scans are typically normal. As with voltage-gated calcium channel antibodies, the presence of anti-Hu antibodies does not correlate with neurologic symptoms nor with an improved prognosis.260,263
Paraneoplastic cerebellar degeneration, manifesting with ataxia, dysarthria, and nystagmus, may be associated with anti-Hu, anti-Yo or P/Q calcium channel autoantibodies.243,264,265 Patients often present with loss of coordination that usually starts on one side and rapidly progresses over days to weeks to involve both sides equally.266 Additional presenting symptoms include limb and truncal ataxia, lack of coordination, dysarthria, and nystagmus. More rarely, patients experience opsoclonus, myoclonus, memory disturbances, pyramidal signs, sensory disturbances, or hyporeflexia. After progressing for a few weeks, the symptoms stabilize, leaving the patient in a severely disabled state. On examination, patients may be unable to stand without assistance as a consequence of severe truncal and neck ataxia with markedly ataxic gait. Ocular findings may include horizontal or vertical nystagmus, dysconjugate gaze, ocular dysmetria, and opsoclonus.265 Speech also can be affected severely, presenting initially as mild dysarthria and progressing to incomprehensible words in severe cases. Mild deterioration of mental status also may be seen, but marked changes in mental status are not compatible with this diagnosis.265 Treatment may include steroids, plasmapheresis, and chemotherapy.243,264 However, treatment of the tumor and/or immunomodulation does not alter the course of paraneoplastic cerebellar degeneration, but may improve LEMS symptoms. Death is frequently from neurologic complications. Paradoxically, the tumor frequently remains localized to the chest or not detected.265
Diagnostic Work Up and Staging
Assessment of Intrathoracic Disease
Accurate clinical staging of lung cancer is extremely important because treatment options and prognosis are dictated by stage at presentation.267 The most significant dividing line is between those patients who are candidates for curative-intent (surgery or definitive chemoradiation) and those who are not suitable for these but who will benefit from chemotherapy, radiation therapy, or both. Staging with regard to a patient’s potential for surgical resection is most applicable to NSCLC. The basis for staging NSCLC is the TNM (tumor–nodes–metastases) system as described in the American Joint Committee on Cancer (AJCC) seventh edition32 (Table 72-6),33,34,653–657 whereas for SCLC a more simplified staging classification is often used (described later). From a practical standpoint, metastases to the hilar or mediastinal lymph nodes are reflected in the “N” designator.267 A new international lymph node map was developed as part of this international staging process.655 Determination of metastases to mediastinal lymph nodes constitutes a critical point in staging and treatment recommendations.267 Patients with mediastinal node metastases may also be selected for multidisciplinary therapy including induction therapy (either chemotherapy, or chemoradiotherapy) followed by resection.
Table 72-6
New TNM Classification of Lung Cancer
| PRIMARY TUMOR | |
| Tx | Tumor that cannot be assessed or is not detected radiologically or bronchoscopically but is proven histopathologically (malignant cells in bronchopulmonary secretions) |
| T0 | No evidence of primary tumor |
| Tis | Carcinoma in situ |
| T1a | ≤2 cm |
| T1b | >2 cm and ≤3 cm |
| No local invasion Airway location: in lobar bronchus or more distal airways |
|
| T2a | >3 cm and ≤5 cm |
| T2b | >5 cm and ≤7 cm |
| Involvement of visceral pleura Airway location: involvement of the main bronchus (distance to the carina is ≥2 cm) or presence of atelectasis or obstructive pneumonitis that extends to hilar region but does not involve the entire lung |
|
| T3 | Tumor >7 cm with any of the following: |
Data from Edge SB, Byrd DR, Compton CC, et al. AJCC Cancer Staging Manual. 7th ed. New York: Springer; 2009.
Several noninvasive imaging techniques are available to aid in identifying the extent of disease both within and outside of the chest. Some lung cancers are detected by plain chest x-ray (Fig. 72-10). However, the chest x-ray is not sufficiently sensitive to accurately assess the mediastinum. Accordingly, a chest CT scan should be performed in all patients with known or suspected NSCLC. The chest CT scan provides anatomic detail that better identifies the location of the tumor, its proximity to local structures, and whether or not lymph nodes in the mediastinum are enlarged (Fig. 72-10). Unfortunately, the accuracy of chest CT scanning in differentiating benign from malignant lymph nodes in the mediastinum based on node size is low. The most commonly used size criterion is a short axis diameter of ≥1 cm on a transverse CT scan as an indicator of a “suspicious” lymph node. Whole-body PET scanning provides functional information on metabolic activity, is more sensitive and specific than chest CT scanning for staging lung cancer in the mediastinum (Fig. 72-11), and can detect occult metastases. Positive findings of PET scans can occur from nonmalignant etiologies (e.g., infections), so that tissue sampling to confirm the suspected malignancy is strongly recommended.34,267 MRI can be useful in selected circumstances, such as superior sulcus tumors to rule out brachial plexus involvement, but in general does not play a major role in NSCLC staging. Of course abnormalities detected by any of the aforementioned imaging studies are not necessarily cancer. Unless overwhelming evidence of metastatic disease is present on imaging studies, curative-intent therapy should not be withheld based on imaging studies alone, but putative metastatic disease should be documented by tissue confirmation of malignancy.
Assessment of Extrathoracic Disease
The best initial predictor of metastatic disease remains a careful history and physical examination of the patient. If signs, symptoms, or findings from the physical examination suggest the presence of malignancy, then sequential imaging, starting with the most appropriate study based on the clues obtained by the clinical evaluation should be performed. If the findings from the clinical evaluation are negative, then additional imaging studies such as a CT scan of the head, a bone scan, or an abdominal CT scan are often unnecessary, and the search for metastatic disease is complete. Clinical findings suggestive of metastatic disease are listed in Table 72-7. Patients with abnormal clinical evaluations should undergo imaging for extrathoracic metastases. Site-specific symptoms warrant directed evaluation of that site with the most appropriate study (e.g., head CT scan, bone scan, and abdominal CT scan). More controversial is how one should assess patients with known clinical stage III disease. Additional imaging may be required given the high frequency of brain metastases, for example.267

GGT, γ-Glutamyltransferase; SGOT, serum glutamic-oxaloacetic transaminase.
Data from Silvestri GA, Tanoue LT, Margolis ML, Barker J, Detterbeck F. The noninvasive staging of non–small cell lung cancer: the guidelines. Chest 2003;123(1 Suppl):147S–156S.
Solitary Pulmonary Nodule
The approach to a patient with a solitary pulmonary nodule is based on an estimate of the probability of cancer, determined according to the size of the nodule, the presence or absence of a history of smoking, the patient’s age, and characteristics of the nodule’s margins on CT imaging.35,658 Mayo Clinic investigators found reported clinical characteristics (age, cigarette-smoking status, and prior cancer diagnosis 5 or more years ago) and three radiologic characteristics (diameter, spiculation, and upper lobe location) were independent predictors of malignancy.64,659 Figure 72-12 is an efficient algorithm for assessing these lesions.
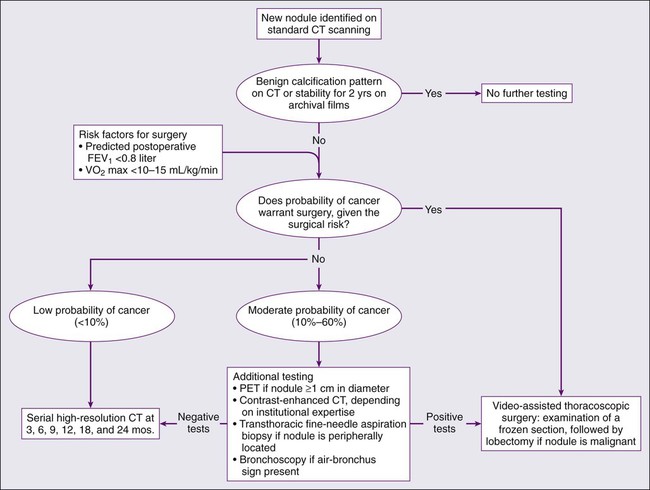
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]72
Cancer of the Lung
Non–Small Cell Lung Cancer and Small Cell Lung Cancer
Leora Horn, Rosana Eisenberg, David Gius, Katherine N. Kimmelshue, Pierre P. Massion, Joe Bill Putnam, Clifford G. Robinson and David P. Carbone
• Constitutes 80% to 85% of new cases of lung cancer in North America.
• Most frequent histologies: adenocarcinoma, squamous cell carcinoma, and large cell carcinoma.
• Leading cause of cancer-related death in the United States for both men and women; represents one of the most preventable forms of cancer death.
• Tobacco use causes 80% of lung cancer deaths.
• Low-dose computed tomography (CT) scan in high-risk individuals is associated with high sensitivity but low specificity for detection of early stage disease.
• Screening remains controversial.
• History and physical examination, routine hematologic and biochemical testing.
CT scan (with contrast) evaluating lungs, mediastinum, liver, and adrenals
Positron emission tomography (PET) scan
If patient has locally advanced or metastatic disease, add magnetic resonance imaging (MRI) of brain
If PET is not available, add bone scan
• Bronchoscopy and/or CT-guided and/or ultrasound-guided biopsy.
• If disease is clinically more advanced, use the least invasive biopsy procedure and sample to confirm advanced disease.
• Mediastinal node evaluation:
If disease is in early stage (I or II) and PET of mediastinum is negative, proceed directly to surgery. If PET is positive, proceed to biopsy via endobronchial ultrasound (EBUS), or endoscopic ultrasound (EUS), or mediastinoscopy, depending on node location.
Stereotactic body radiotherapy in selected cases.
Postoperative chemotherapy prolongs survival in patients with a good performance status.
Stereotactic body radiotherapy in selected cases.
Postoperative chemotherapy useful in resected IIIA in patients with a good performance status.
Concurrent chemotherapy combined with radiation therapy in patients with a good performance status.
Concurrent chemotherapy combined with radiation is superior to radiation alone; increased toxicity limits this approach in frail patients.
Clinical trials of new approaches should be a high priority.
Primary chemotherapy with platinum-based doublet improves the quality and quantity of life.
All tumors should be sent for molecular testing.
No advantage for triplet therapy.
Non–platinum-based therapy is appropriate in selected patients.
Single-agent therapy reserved for select populations.
There is a clear need for more clinical trials at this stage.
• Second- or third-line therapy
Further treatment with any of several agents is appropriate with progression after chemotherapy if patient sustains a good performance status; docetaxel, pemetrexed, erlotinib, and crizotinib are all U.S. Food and Drug Administration (FDA)-approved.
• Accounts for approximately 15% of new cases of lung cancer each year in United States; incidence is decreasing.
• Subtypes include small cell carcinoma and combined small cell carcinoma (most often combined with squamous cell carcinoma, adenocarcinoma or large cell carcinoma).
• History and physical examination, routine histologic and biochemical testing.
CT scan (with contrast) evaluating lungs, mediastinum, liver, and adrenals.
PET useful with clinically early stage disease.
Brain evaluation (MRI preferred).
If there are signs or symptoms of bone involvement, a bone scan or PET scan is recommended.
Mediastinal node evaluation is not required unless as a convenient site for biopsy.
Concurrent etoposide plus cisplatin and radiation to the intrathoracic disease.
Prophylactic cranial irradiation in complete (or near complete) responders.
Etoposide (or irinotecan) plus cisplatin or carboplatin.
No evidence a third agent or dose intensification improves outcome.
Research studies are a clear priority given the lack of progress in this arena.
Prophylactic cranial irradiation in complete (or near complete) responders.
Consider consolidative radiotherapy (RT) to primary in complete responders.
Second-line therapy useful mainly in patents who have experienced a longer treatment-free interval and a good performance status.
Topotecan FDA-approved; however, any of several chemotherapy agents offer short-term benefit.
Research protocols are a preferred choice for patients with disease recurrence.
Introduction
Although reports of pulmonary malignancies date to antiquity, lung cancer is largely a disease of modern man. Prior to 1900, lung cancers were viewed as “matters of medical curiosity not known to be in any degree influenced by medicine and too rare to be of much practical importance.”1 By the mid-20th century, following the introduction of cheap, mass-produced cigarettes, however, lung cancer had become epidemic and firmly established as the leading cause of cancer-related death in North America and Europe.2 It should not be forgotten that lung cancer is potentially one of the most preventable of all of the major malignancies afflicting man. Its primary cause is tobacco smoke.3 King James I was among the first to chronicle the adverse health effects of tobacco smoke,4 but it was Raymond Pearl’s landmark 1938 report that conclusively established the devastating impact smoking has on longevity.5 It would be another decade before tobacco smoking was firmly established as a causative agent of lung cancer6,7 and nearly 30 more years before the emergence of the U.S. Surgeon General’s initial report of the ill effects of tobacco smoking. Regrettably it would be yet another 3 decades before the tobacco industry publicly acknowledged this obvious truth, but only after a long, drawn out battle of misinformation and deception,8 ironically helped along by the unwitting (perhaps) complicity of physicians.9 With the belated recognition of the etiologic role of tobacco smoke the incidence of lung cancer has started to decline in North America and parts of Europe. For the most part the decline is seen most clearly in men. Only recently has this decline become apparent in women in the United States, following a similar decline among men 15 to 20 years prior.10 In short, the story of lung cancer is replete with controversy,11 politics,12 pessimism,13 and, more recently, guarded optimism.14 This chapter focuses on our perceptions of modern-day management of lung cancer with an emphasis on advances made over the past 5 years.
Epidemiology
Lung cancer was an uncommon disease in the early part of the 20th century but then began an epidemic rise to far surpass all other cancers in numbers and rates of death.15 Lung cancer is the leading cause of cancer-related mortality with an estimated 244,180 new cases and 164,770 deaths anticipated in 2012.16 Lung cancer is relatively rare in persons younger than age 40 years, but rates rise steadily until age 80 years and then taper off; the projected lifetime probability of developing lung cancer is estimated to be approximately 8% among males and approximately 6% among females.17 The incidence of lung cancer varies by racial and ethnic group, with the highest age-adjusted incidence rates among African American men (Fig. 72-1). The excess in age-adjusted rates among African Americans occurs only among men, but examinations of recent age-specific rates show that before age 50 years, mortality from lung cancer is more than 25% higher among African American women compared with white women.17 Incidence and mortality rates among Hispanic, Native, and Asian Americans are only 40% to 50% those of whites.
The trends in overall lung cancer rates mask differences in temporal patterns according to lung cancer cell type. Figure 72-2 shows changes in age-adjusted incidence rates for squamous cell carcinoma and for adenocarcinoma during 1973 to 2003 based on data from nine continuing cancer registries in the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) program of cancer registration. Among men, squamous cell cancers predominated in the first two thirds of this period. The decline in incidence of lung cancer among men was first apparent for squamous cell tumors. Decreases began in the early 1980s so that by the mid-1990s rates of squamous cell carcinoma among men had dropped below those for adenocarcinoma, which did not peak until over a decade later. Among women, adenocarcinomas have been more common than squamous cell carcinomas during the past 3 decades. The adenocarcinoma excess among women has become more pronounced over time, as rates of squamous cell cancers increased steadily through the 1980s before beginning to decline, whereas adenocarcinoma did not plateau until about a decade later. Although not shown, rates of small cell carcinoma, the third most frequent cell type, tended to parallel those for squamous cell cancer among both sexes. Trends for other cell types of lung cancer, including large cell carcinomas, tend to be intermediate between those of squamous cell carcinomas and adenocarcinomas. As discussed later in the chapter, although 5-year relative survival rates for lung cancer have improved over time, the survival rates are low, currently approximately 15% overall.17 Some variation exists by sex, race, cell type, and molecular status, with slightly higher survival among whites than blacks and females than males, but for no group does the overall 5-year relative survival exceed 20%.
Risk Factors
The cause of the large majority (80%) of lung cancers is cigarette smoking.18,19 There is a massive compilation of scientific evidence from epidemiological studies conducted around the world since the 1950s demonstrating the link between smoking and lung cancer. Epidemiological research has also revealed that several other factors have been implicated as causes of lung cancer, although none to the extent of tobacco.20,21 Cigarette smokers have been shown to have large increases in the risk of lung cancer. A recent deep-sequencing study suggested that one genetic mutation is induced for every 15 cigarettes smoked. Numerous investigations typically show 10 fold or greater increases in risk of this cancer among smokers compared with those who have never smoked.18,19 One of the largest studies is the American Cancer Society’s prospective cohort study of more than 1 million Americans in which a more than 20 fold increase of lung cancer has been observed among men who were current smokers at the start of the followup in the early 1980s.18,19 In contrast, even the most prolonged and intense exposures to asbestos, perhaps the most prominent occupational cause of lung cancer, are associated with no more than about fivefold increase in risk of lung cancer.22
Risks of lung cancer are lower among persons who quit smoking than among those who continue smoking.18,19 The reductions in risk indicate that quitting smoking is beneficial (and conversely that continuing to smoke is harmful). Risk among former smokers on average is less than one-half that of those who continue to smoke. In the American Cancer Society cohort study cited above, former smokers had a ninefold increase in lung cancer compared with men who had never smoked versus the 20 fold excess in those who continued to smoke.18,19 Such relative reductions have been consistently seen, with the size of the reduction in risk increasing the longer the person has quit smoking, although generally even long-term former smokers have higher risks of lung cancer than those who never smoked.18,19
Cigarette smoking has been shown to increase risk of all the major lung cancer cell types.18–21 The magnitude of the increase varies by histologic type, however, with highest risks for squamous cell, small cell, and large cell carcinomas of the lung. Some early studies tended to show only small increases in risk of adenocarcinoma among smokers, but more recent studies indicate that the excess of lung adenocarcinoma is substantial.18,19,21
Cigarette smoke has also been implicated in increasing risk of lung cancer among nonsmokers. A 2006 update of the Surgeon General’s report declared that there is sufficient evidence to list passive smoking as an established cause of lung cancer and called for further control of environmental tobacco smoke (ETS) exposures.23 The risk from ETS is far less than from active smoking, with approximately a 20% to 30% increase in lung cancer observed among nonsmokers married for many years to smokers, in comparison to the 2000% increase among continuing active smokers. Nevertheless, cancer-control activities based on the knowledge that ETS exposure may convey an increased risk of lung cancer have helped reduce exposures in public places and have also provided additional incentive for smokers to quit the habit.
Although cigarette smoking is the dominant cause of lung cancer, several other risk factors for this cancer have been identified.20,21 These include occupational exposures to asbestos and other workplace agents, some of which have been evaluated for nearly as long as cigarette smoking. Among the occupational agents considered as known lung carcinogens are arsenic, bischloromethyl ether, hexavalent chromium, mustard gas, nickel (as found in certain nickel refining processes), and polycyclic aromatic hydrocarbons.20,21,24 Several other occupational exposures have been associated with increased rates of lung cancer, but the causal nature of the association is not clear. Epidemiological studies have attempted to assess the potentially synergistic interrelationship between certain workplace exposures and smoking. Risk of lung cancer among men exposed to asbestos who also smoked was originally thought to be exceptionally high (with early reports of 50 fold or greater excesses compared with unexposed nonsmokers), but recent modeling of larger data pools suggests that asbestos and tobacco combine to enhance lung cancer risk in a less than multiplicative manner.25 Occupational observations have also provided clues to the mechanisms of lung cancer induction. Risk of lung cancer among asbestos-exposed workers, for example, is increased primarily among those with underlying asbestosis, raising the possibility that the scarring and inflammation produced by this fibrotic nonmalignant lung disease may, in many cases (though likely not in all), be the trigger for asbestos-induced lung cancer.26
Increased risks of lung cancer have also been associated with other factors. Diet and nutrition are thought to be involved, as numerous investigations have shown somewhat higher risks of this cancer among those with low fruit and vegetable intake during adulthood.20,21 The early observational studies led to hypotheses that specific nutrients, in particular retinoids and carotenoids, might have chemopreventive effects for lung cancer. Randomized clinical trials were launched, but failed to validate this hypothesis when reports from interventions involving supplementation with beta-carotene in trials both in Finland and the United States found increased rather than decreased incidence of lung cancer among the supplemented.27,28 The current consensus regarding diet and lung cancer remains muddled, with a minor role for nutritional factors likely, but difficult to assess epidemiologically. Ionizing radiation has been established as a lung carcinogen, most convincingly demonstrated from studies showing modestly increased rates of this cancer among persons exposed to the atomic bombs of Hiroshima and Nagasaki and large excesses among workers exposed to α irradiation from radon in underground uranium mining.20,21 Extrapolations from the high exposures in mines to low-level radon exposures in homes, as well as direct observations from case-control studies assessing measured levels in homes, suggest that prolonged radon exposures above the recommended remedial levels might impart a risk of lung cancer equal to or greater than that of ETS.29 Prior lung diseases, such as asbestosis (mentioned above), chronic bronchitis, emphysema, and tuberculosis have also been linked to increased risks of lung cancer. Although smoking itself is a cause of the chronic obstructive pulmonary diseases (COPDs), the link between chronic bronchitis and emphysema and lung cancer persists after adjustment for smoking, with as much as double the smoking-adjusted cancer risk among those with COPD.20,21,30
Familial clustering of lung cancer has been observed, raising the possibility of inherited traits that may increase risk among some individuals, with risk about doubled in families with prior lung cancer.20,21 Individuals with inherited mutations in retinoblastoma and p53 (Li-Fraumeni syndrome) genes may develop lung cancer. Genome-wide association studies have recently identified four genetic loci for lung cancer risk, including two on 5p15 (TERT-CLPM1L), 15q25.1 (CHRNA5-CHRNA-3 nicotinic acetylcholine receptor subunits) and 6p21 (BAT3-MSH5).33–33 A rare germline mutation involving the epidermal growth factor receptor (EGFR) within exon 20 that results in an amino acid substitution at position 790 from threonine to methionine (T790M) has been linked to lung cancer susceptibility in never smokers.34,35 Smoking also clusters within families, so some of the familial aggregation of lung cancer may be smoking related. Nevertheless, there appear to be multiple genetic factors that help determine the way in which individuals metabolize, detoxify, repair, or otherwise respond to lung carcinogens, including the carcinogens in cigarette smoke, as discussed in more detail later in this chapter.
Smoking Cessation
Given the undeniable link between cigarette smoking and lung cancer,3 it is incumbent upon physicians to promote tobacco abstinence and help their patients who smoke to stop smoking.36 Smoking cessation, even well into middle age, can minimize an individual’s subsequent risk of lung cancer, and stopping before middle age avoids more than 90% of the risk attributable to tobacco.37,38 By contrast, there is little health benefit realized by simply “cutting back.”39 Among victims of lung cancer smoking cessation is associated with improved survival,40–43 fewer side effects from therapy,44 and an overall improvement in quality of life.45 Often forgotten is the fact that smoking alters the metabolism of many chemotherapy drugs, potentially adversely altering the toxicities and therapeutic benefits of the agents.46 Therefore, it is important to promote smoking cessation even after the diagnosis of lung cancer is established.47,48 Although smoking cessation is extremely difficult, patients with lung cancer tend to be highly motivated and success rates mirror that of other disease states.49 However, the individual must want to stop smoking and must be willing to work hard to achieve the goal of smoking abstinence. Nicotine replacement therapies, bupropion and varenicline (an α4β2 nicotinic acetylcholine receptor partial agonist), are approved by the U.S. Food and Drug Administration (FDA) as first-line treatments for nicotine dependence.50 Varenicline has been demonstrated to be significantly more efficacious than bupropion alone for smoking cessation.51 Furthermore, prolonged use of varenicline beyond the initial induction phase has proven useful in maintaining smoking abstinence.52 Clonidine and nortriptyline are recommended as second-line treatments.47 A systematic review of extant smoking cessation studies indicates self-help strategies alone only marginally affect quit rates, whereas individual and combined pharmacotherapies and counseling either alone or in combination can significantly increase rates of cessation.53
Biology of Lung Cancer
The specific events that trigger malignant transformation of bronchoepithelial cells are unknown in the vast majority of cases. However, it is clear that exposure to environmental carcinogens, such as those found in tobacco smoke or asbestos fibers, induce or facilitate the transformation (extrinsic component).54 The contribution of the extrinsic carcinogen on transformation is modulated by genetic variations in genes (intrinsic component) that affect aspects of carcinogen metabolism, such as the conversion of procarcinogens to carcinogens and their subsequent inactivation.55 These genetic variations occur at relatively high frequency in the population. Their contribution to an individual’s lung cancer risk is generally low, but because of their population frequency, their overall impact on lung cancer risk could be high. Epidemiological studies further suggest that a familial predisposition to lung cancer exists that is independent of tobacco smoke exposure. One study found evidence for an autosomal dominant model linked to 6q23-25,56 but other studies have proposed a complex multigene model for inherited risk.57 As mentioned above, familial clustering of lung cancer cases has been reported with an inherited T790M mutation in the EGFR gene.34,35 The identification of individuals at particularly high risk for the development of lung cancer could justify more intense screening regimens, and the identification of the responsible chromosomal loci for lung cancer susceptibility genes could allow the development of specific chemopreventive strategies.
Environmental factors, as modified by inherited modulators, likely affect specific genes by deregulating important pathways to enable the cancer phenotype. Particularly important in lung cancer are acquired abnormalities in ras, Rb, p53, Akt, LKB, and BRAF (reviewed in Fong et al58). However, the most clinically significant acquired genetic abnormalities in lung cancer are the mutation of the EGFR59–62 and the anaplastic lymphoma kinase (ALK) fusion.65–65 EGFR mutations occur in approximately 20% of lung cancer patients and are primarily in exons 19 (in-frame deletions usually of four amino acids LREA) and 20 (L858R point mutants), and result in constitutive signaling and AKT activation66 and are associated with very high response rates (60% to 90%) to the specific tyrosine kinase inhibitors gefitinib and erlotinib.67 ALK fusions have been reported in 3% to 7% of lung cancer patients and are associated with a 57% response to crizotinib.68 The EGFR and ALK abnormalities are non–overlapping. Interestingly, both EGFR mutations and ALK fusions are associated with younger age, light (<10 pack-year) and nonsmokers and adenocarcinoma histology, and the frequency of EGFR mutations is much higher in Asian than Western populations (30% to 70% vs. 10%).68–72 Response to specific EGFR or ALK inhibitors are often dramatic, and occur even in heavily pretreated patients, demonstrating that tumors with these mutations are “addicted” to the activation of this pathway. Almost all patients with these dramatic responses, however, develop progressive, resistant disease. The most common mechanism of acquired resistance to EGFR tyrosine kinase inhibitors is a second-site mutation within exon 20 that results in an amino acid substitution at position 790 in EGFR, from threonine to methionine (T790M).73,74 To date, several second-site mutations within the ALK tyrosine kinase domain have been reported as mechanisms of acquired resistance to the ALK tyrosine kinase inhibitor crizotinib.75
In spite of the incontrovertible high response rate in patients with EGFR mutations, analysis of samples from BR.21, a placebo-controlled, randomized, clinical trial of erlotinib in second-line non–small cell lung cancer (NSCLC) showed that the presence of an EGFR mutation was not associated with prolonged survival,76 but in univariate analysis, amplification of the receptor was associated with improved survival.77 Interestingly, there was a survival benefit in the entire erlotinib arm compared with placebo, as well as every subset of patients, including smokers and patients with squamous cell cancers,78 suggesting that factors other than mutation may contribute to clinical benefit, and that this was not necessarily related to objective response rates. Other assays, including serum proteomics, show potential for selecting patients likely to achieve clinical benefit.79,80
The histologic sequence of events that leads to the various forms of lung cancer is not well understood, and it is clearly different for the various histopathologic entities.81–84 Current knowledge suggests that squamous cell carcinoma arises in a relatively ordered progression that includes squamous metaplasia and carcinoma in situ (CIS). Peripheral adenocarcinomas are thought to arise from atypical adenomatous hyperplastic lesions, but this process is much more obscure, largely because this type of lesion is much less accessible by bronchoscopy. Small cell carcinoma might arise from neuroendocrine hyperplasia, but evidence in support of this hypothesis is scarce.
The pathology of premalignancy is discussed in more detail in the next section, but the molecular biology of premalignancy is of great clinical importance, not only to better understand the process of cancer development, but to provide potential therapeutic targets to intervene in this process and intermediate biomarkers to assess risk and evaluate candidate chemoprevention strategies. Premalignant lesions of the lung have been investigated for molecular alterations in NSCLC, but much less information is available for small cell lung cancer (SCLC).85 Microdissected specimens derived from normal, hyperplastic, metaplastic, dysplastic, and CIS, as well as invasive neoplastic foci of patients with lung cancer, were studied for gene mutations, promotor hypermethylation, and allele loss. Results obtained to date suggest that allele loss on chromosome 3p is the earliest event, followed by allele loss/hypermethylation on chromosome 9p and, subsequently, on chromosome 8p.84 Loss of heterozygosity at the p53 gene locus (17q13.1) is relatively rare (10%) and occurs predominantly at the dysplasia or CIS stage. Point mutations in the p53 gene86 and the EGFR gene,87 however, have been observed in morphologically normal bronchial epithelium obtained from the airways of patients with lung cancer. In contrast, K-ras gene mutations represent a late event, found only in CIS or invasive cancers.88 In addition to copy number changes and mutations, alterations in the expression of the retinoic acid receptor β have also been used as a molecular marker for premalignancy and as an intermediate biomarker for chemoprevention trials.89
The fate of morphologically or molecularly abnormal areas is currently an area of intense research. One study showed that 54% of patients with high-grade dysplastic lesions developed lung cancer within 2 years, but 80% of these arise in a different part of the lung than the CIS.90 None of the patients with low-grade dysplastic lesions progressed to cancer in this study. Of the low-grade lesions in this study, 82% spontaneously regressed, and 18% remained unchanged, and none progressed to CIS. Preliminary data also suggest that abnormal regions display reproducible molecular changes on repeat biopsies over time, and that these and additional abnormalities can be observed in tumors arising from these lesions.91
Part of the problem with the studies of preneoplasia is that we may be evaluating the microscopic appearance and molecular characteristics of the wrong population of cells. The vast majority of the respiratory epithelium (and most other tissues) is thought to be terminally differentiated and incapable of sustained replication; however, one theory holds that a small subset of the cells have unlimited, but normally tightly regulated, replicative potential. These are the so-called pulmonary stem cells,92,93 whose biology is very poorly understood. The stem cell concept may also underlie the failure of standard medical therapies to eradicate lung cancers, even when there is a clinical complete response. The theory is that therapies have been refined to induce measurable reductions in the bulk tumor mass, whereas true replicative potential exists only in a small subset of “cancer stem cells” that are not effectively targeted by current therapies. Specific isolation of these cells and development of stem cell–targeted therapies may thus not cause rapid tumor regressions, but rather result in significantly increased long-term survival.
Pathology
In recent years, significant advances in the treatment of pulmonary adenocarcinoma have affected the pathological diagnosis of lung cancer. The latest World Health Organization (WHO) classification of lung tumors from 200494,95 (Box 72-1) is a purely morphologic classification and geared towards resection specimens. Most pathological diagnoses of lung cancer, however, are rendered on small biopsies or cytologic specimens. A new multidisciplinary classification of lung adenocarcinoma has been proposed jointly by the International Association for the Study of Lung Cancer (IASLC), American Thoracic Society (ATS), and European Respiratory Society (ERS) (Box 72-2).96 Although based predominantly on histology, its purpose is to provide an integrated clinical, molecular, radiologic, and pathological approach to the classification of lung adenocarcinoma. In addition, this classification addresses the diagnosis of lung cancer in small biopsies and cytologic specimens and emphasizes the use of immunohistochemistry for the subtyping of non–small cell carcinoma.
Tissue and Cytologic Diagnosis of Lung Cancer
The diagnosis of lung cancer can be made on tissue specimens such as endobronchial and transbronchial biopsies or resected specimens, or by assessment of cytological specimens such as transbronchial or transthoracic fine-needle aspirates, bronchial brushing and washings, bronchoalveolar lavage or sputum or pleural fluid. The diagnostic yield depends on several factors including location (accessibility) of the tumor, tumor size, tumor type, or technical aspects of the diagnostic procedure, including the experience level of the bronchoscopist and pathologist.96 In general, central lesions such as squamous cell carcinomas, small cell carcinoma, or endobronchial lesions such as carcinoid tumors, are more readily diagnosed by bronchoscopic examination, whereas peripheral lesions, such as adenocarcinomas, and large cell carcinomas are more amenable to transthoracic fine-needle aspiration (FNA) or biopsy. Diagnostic accuracy for most specimens in distinguishing small cell carcinoma and non–small cell carcinomas is excellent, 97 with less accuracy for subtypes of non–small cell carcinoma96 if immunohistochemical staining is not used.
Bronchoscopic specimens include bronchial brushings, washings, bronchoalveolar lavage, and transbronchial fine needle aspiration. Of these, transbronchial FNA consistently demonstrates the highest sensitivity, surpassed only by the use of a combination of bronchoscopic specimens.96–100 Overall sensitivity for combined use of bronchoscopic methods is approximately 80%, and with tissue biopsy, the yield increases to 85% to 90%.96,97,99 In fact, transbronchial FNA is more often diagnostic than transbronchial biopsy when the lesion of interest is submucosal.96,101
Like transbronchial fine-needle aspirate specimens, transthoracic fine-needle aspirate specimens are also diagnostically useful, yielding diagnostic material in 70% to 95% of cases. Sensitivity is highest for larger lesions and peripheral tumors.102 In general, fine-needle aspirate specimens, whether transbronchial, transthoracic, or endoscopic ultrasound-guided, are superior to other specimen types.103 The superiority of fine-needle aspirate specimens is primarily a result of the higher yield of lesional tumor cells with fewer confounding factors, such as obscuring inflammation and reactive nonneoplastic cells.
Sputum cytology is inexpensive and noninvasive but has a lower yield than other specimen types because of poor preservation of the cells and more variability in acquiring a good-quality specimen. The yield for sputum cytology is highest for larger and centrally located tumors, such as squamous cell carcinoma and small cell carcinoma, although occasionally an accurate diagnosis is possible with tumors located more peripherally within the lung.96 The specificity for sputum cytology averages close to 100%, although sensitivity is generally less than 70%. The accuracy of sputum cytology improves with increased numbers of specimens analyzed and consequently, analysis of at least three sputum specimens is recommended. Sputum cytology has also been extensively studied with varying success as a screening tool for early detection of lung cancers. Sputum cytology is not currently recommended as a routine screening tool, but recent advances in molecular diagnostic techniques may result in a resurgence of its use.104–107
