Cancer of the Endocrine System
David F. Schneider, Haggi Mazeh, Sam J. Lubner, Juan C. Jaume and Herbert Chen
• The incidence of thyroid cancer is increasing, and there are approximately 33,500 new cases per year in the United States.
• The incidence of differentiated thyroid cancer is 11.6 per 100,000 people per year, with a female-to-male ratio of more than 3 : 1.
• Differentiated thyroid cancer (DTC) includes papillary thyroid cancer (PTC), which accounts for 80% of all thyroid cancers, follicular thyroid cancer (FTC), which accounts for 10% to 20% of all thyroid cancers, and a rare type, Hürthle cell cancer.
• Medullary thyroid cancer (MTC) arises from the parafollicular C cells and accounts for 5% to 10% of all thyroid cancers.
• Anaplastic thyroid cancer is a rare, but rapidly fatal, form of thyroid cancer.
• Other histologic types of cancer, such as lymphoma, sarcoma, and metastatic cancers, can also be found within the thyroid.
• Known risk factors for the development of thyroid cancer include radiation exposure and iodine deficiency.
• Thyroid cancer can also run in families or exist as part of familial syndromes (Gardner, Cowden, and Werner syndromes)
• More recently, the molecular pathogenesis of thyroid cancer has been investigated. The following are the most widely studied molecular markers for DTC to date:



• Well-differentiated histology has excellent 5-year survival (>95%).
• Older age and extent of invasion are related to prognosis.
• Lymph node involvement is associated with higher recurrence, but has questionable impact on survival.
• Many staging systems exist for DTC.
• Hürthle cell adenoma—larger size (>6 cm) predicts malignancy.
• Poorly differentiated tumors—recurrence lymph node metastases are common.
• Anaplastic cancers are extremely aggressive with 5-year survival less than 5%.
• The history should include radiation exposure, family history, and compressive symptoms (dysphagia, hoarseness, pain/pressure) from enlarging tumor.
• Concerning exam findings, include a fixed mass or lymphadenopathy.
• Incidental fining on computed tomography (CT), magnetic resonance imaging (MRI), positron emission tomography (PET), and ultrasound.
• Preoperative laboratory studies include thyroid-stimulating hormone (TSH), Tg.
• Fine-needle aspiration (FNA) biopsy is a key component of the workup of thyroid nodules. The Bethesda Criteria classify FNA results and determine the risk of cancer in the nodule.
• Preoperative imaging should cervical ultrasound. CT is used when aggressive variants are suspected to assist in operative planning.
• Treatment begins with surgery. Most thyroid cancers are treated with total thyroidectomy. Compartment-oriented neck dissection is added when there is metastatic disease in the cervical lymph nodes.
• Adjuvant therapy for differentiated tumors is radioactive iodine (131I).
• Patients must be prepared for radioactive iodine ablation with a low iodine diet.
• And thyroid hormone withdrawal or recombinant human thyroid-stimulating hormone (rhTSH) if there is no evidence of metastatic disease.
• After surgery and radioactive iodine, thyroxine-suppression prevents the growth of microscopic disease.
• External beam radiation is utilized for persistent, recurrent, anaplastic, poorly differentiated tumors that are not iodine avid.
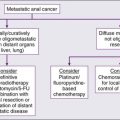
• Chemotherapy is mainly palliative for poorly differentiated or anaplastic tumors. Traditional chemotherapy has minimal response rates, but newer, targeted therapies, such as sorafenib or sunitinib, are showing promise.
• Surveillance for recurrent thyroid cancer includes measurements of TSH, Tg, and antithyroglobulin antibodies in addition to cervical ultrasound. The schedule of these tests is tailored to risk level.
• Treatment of recurrence can include external beam radiation or targeted therapies, depending on the iodine avidity of the tumor.
• Medullary thyroid cancer accounts for 5% to 10% of all thyroid cancers; 75% of cases are sporadic and 25% are familial (multiple endocrine neoplasia [MEN]-2, familial medullary thyroid carcinoma [FMTC]).
• The diagnosis is made by FNA with calcitonin washout. RET testing can identify inherited germline mutations. As in DTC, cervical ultrasound assists with operative planning. The tumor markers calcitonin and carcinoembryonic antigen (CEA) can be useful in following patients postoperatively for identifying recurrence and metastases.
• At a minimum, treatment of MTC should consist of total thyroidectomy plus central lymph node dissection. Lateral neck dissection is added when there are clinically positive nodes in the central neck and for high-risk patients.
• Traditional chemotherapy is not effective for metastatic MTC, but newer, targeted therapies for metastatic disease such as vandetanib, have shown some promise.
• The incidence of adrenocortical cancer is 1 to 2 per million people.
• Most adrenocortical cancers are sporadic, but they can also occur as part of familial syndromes such as MEN-1, Li-Fraumeni syndrome, Beckwith-Wiedemann syndrome, and Carney complex.
• Most are asymptomatic, but 40% to 60% are functional (hormone production), and this may be the presenting symptom(s).
• The diagnosis is by urinary/plasma biochemical testing and imaging: CT, fluorodeoxyglucose (FDG)-PET.
• Often, the diagnosis is not made definitively until after resection of suspicious masses, and pathology provides definitive diagnosis. FNA of adrenal masses is rarely indicated.
• Surgery is the mainstay of treatment for adrenocortical cancer and should consist of en bloc resection of the adrenal gland with adjacent organs/tissue that is involved; cardiopulmonary bypass may be necessary for caval involvement.
• Long-term surveillance, consisting of physical exams and CT scans, is necessary to monitor for disease recurrence.
• Mitotane alone or in combination with other chemotherapeutic agents improves recurrence-free survival.
• Radiotherapy may improve local control, but there are no clear recommendations.
• Hormonal control can also limit disease spread and consists of mitotane, ketoconazole, metyrapone, etomidate.
• Excision or reoperation is recommended for recurrent or metastatic disease.
• The prognosis is poor, with overall 5-year survival of less than 40%.
• The incidence of malignant pheochromocytoma is 2 to 8 per 1,000,000 adults.
• Most malignant pheochromocytomas are sporadic, but 10% are part of inherited syndromes such as MEN syndromes, neurofibromatosis type I, von Hippel-Lindau syndrome, and succinate dehydrogenase gene mutations.
• Pheochromocytomas present with the classic triad of functional tumors-headache, tachycardia, and sweating, but they are asymptomatic in more than 50%, presenting as an incidental adrenal mass.
• The diagnosis is established with urinary or plasma fractionated metanephrines and catecholamines.
• Tumors are localized with CT/MRI; metaiodobenzylguanidine (MIBG) to identify extraadrenal metastases.
• Treatment begins with surgery to resect entire gland with clear margins. Surgery can also be useful in debulking for metastatic disease.
• Metastatic or unresectable disease can be treated with 131I-MIBG or chemotherapy.
• Radiotherapy is used for palliation in bone and lymph node metastases.
• Medical therapy to prepare patients for surgery or control symptoms of catecholamine excess can include phenoxybenzamine, nicardipine, or metyrosine.
• The 5-year survival ranges from 20% to 50%.
• MEN-1 presents first with hyperparathyroidism in their 30s and 40s. Other manifestations include pituitary tumors and neuroendocrine tumors of the pancreas (such as gastrinoma) and upper gastrointestinal tract.
• MEN-1 is inherited in an autosomal-dominant fashion. Mutations in the MENIN tumor suppressor gene cause this disease, but expression is variable.
• Treatment of parathyroid hyperplasia is subtotal parathyroidectomy and bilateral cervical thymectomy.
• MEN-1 patients with pancreatic and duodenal tumors are treated with distal pancreatectomy, enucleation of pancreatic head tumors, duodenotomy, and mucosal resection of multiple duodenal tumors.
• MEN-2A is characterized by pheochromocytoma, MTC, and primary hyperparathyroidism (hyperplasia).
• MEN-2B is characterized by more aggressive MTC, pheochromocytoma, and mucosal ganglioneuromas.
• The MTC in MEN-2 syndromes arises from C-cell hyperplasia and germline RET mutations.
• Specific codon mutations in the RET gene determine the disease phenotype in MEN-2 syndromes, and help risk-stratify patients.
• Prophylactic thyroidectomy should be offered to mutation carriers; the timing of thyroidectomy is determined by the specific codon mutation.
• Pheochromocytomas are often bilateral in MEN-2 syndromes, but onset is asynchronous. Consequently, prophylactic adrenalectomy is not indicated. Those MEN-2 patients who develop bilateral disease can be treated with bilateral adrenalectomy and hormone replacement or cortical-sparing adrenalectomy.
• The incidence of carcinoid tumors is 5.25 in 100,000 people.
• Tumors are identified with specific immunohistochemical staining for NSE or chromogranin A. Chromogranin A also serves as a blood marker for the disease.
• Several classification systems exist for carcinoid tumors, including the World Health Organization (WHO) classification and European Neuroendocrine Tumor Society (ENETS) staging system.
• Carcinoids arise from the Kulchitsky cells in crypts of Lieberkühn of the gut or disseminated in the endobronchial mucosa, and are classified by location.
• The diagnosis is made definitively by tissue diagnosis, but urinary 5-hydroxyindoleacetic acid (5-HIAA), serum NSE, and chromogranin A are serum markers of the disease.
• CT or MRI can localize the carcinoid tumors, or OctreoScan can be used as these tumors have somatostatin receptors.
• The carcinoid syndrome occurs in metastatic carcinoid and presents as flushing, diarrhea, and bronchoconstriction. Right-sided valvular heart disease is also a manifestation of the disease.
• Treatment begins with resection of primary tumor with nodal metastases.
• Debulking/metastasectomy is beneficial for controlling symptoms in patients with liver disease or bulky disease.
• Radiation therapy is rarely used for primary therapy, but can be palliative.
• Antihormonal therapy consists of octapeptide analogs of somatostatin; Sandostatin LAR is a helpful, long-acting formulation octapeptide analogs of somatostatin.
• The liver is a common site for metastatic carcinoid, and there are several options for hepatic-directed therapy, including surgery and embolization (chemoembolization or radioembolization).
• Metastatic disease can also be treated with targeted agents or emerging radionuclide therapy.
Pancreatic Neuroendocrine Tumors
• Pancreatic neuroendocrine tumors (NETs) can be sporadic or inherited (MEN).
• Pancreatic NETs are diagnosed on CT/MRI imaging; ultrasound or endoscopic ultrasound (EUS) can help guide biopsy.
• The ENETS staging system is proposed to help stage pancreatic NETs.
• Insulinomas are diagnosed by fasting hypoglycemia with elevated plasma insulin levels; 10% are malignant, and surgical resection (enucleation) is curative.
• Glucagonoma is characterized by migratory necrotizing erythema, insulin-resistant diabetes, glossitis, ileus, and constipation; 50% to 80% are metastatic.
• Because of the higher rate of metastatic disease, surgical resection is curative in less than one-third of patients with glucagonoma.
• Somatostatinoma is characterized by diabetes, diarrhea, and gallbladder disorders.
• Treatment for somatostatinoma includes cytoreductive surgery and chemotherapy.
• Gastrinoma is characterized by ulcer disease in spite of adequate treatment and diarrhea.
• Gastrinoma is diagnosed by hypergastrinemia with elevated basal acid output or positive secretin test; tumors are localized with CT, MRI, or octreotide scan; these tumors are frequently metastatic.
• Targeted therapies such as everolimus and sunitinib have been FDA approved for first-line treatment of pancreatic NETs.
• The incidence of parathyroid carcinoma is 5.73 per 10 million people.
• The etiology of parathyroid cancer has recently been attributed to pericentromeric inversion resulting in overexpression of the cyclin D1 gene.
 In hyperparathyroidism-jaw tumor syndrome, there are mutations in the HRPT2 tumor suppressor gene (parafibromin).
In hyperparathyroidism-jaw tumor syndrome, there are mutations in the HRPT2 tumor suppressor gene (parafibromin).
• Clinical characteristics of parathyroid carcinoma include the constitutional symptoms of primary hyperparathyroidism, including muscle weakness, fatigue, nausea, vomiting, increased thirst, and frequent urination, in addition to bone pain and fractures.
 A neck mass occurs in 34% to 52%, but is uncommon in benign parathyroid adenomas.
A neck mass occurs in 34% to 52%, but is uncommon in benign parathyroid adenomas.
• In parathyroid carcinoma, serum calcium is quite elevated (14.6 to 15.9) with elevated serum parathyroid hormone (PTH) (commonly 10-fold higher than the upper limit of normal).
 The diagnosis is made by laboratory measurement of Ca and PTH, and then 99m-technetium sestamibi scan and neck ultrasound can localize the tumor ± washout for PTH measurement in FNA material.
The diagnosis is made by laboratory measurement of Ca and PTH, and then 99m-technetium sestamibi scan and neck ultrasound can localize the tumor ± washout for PTH measurement in FNA material.
• Pathological features of parathyroid carcinoma include local invasion and lymph node metastases.
• Treatment of parathyroid carcinoma should include en bloc resection of the parathyroid mass with ipsilateral thyroid lobe ± ipsilateral neck dissection followed by postoperative calcium and activated vitamin D supplementation.
• Medical therapy for hypercalcemia precipitated by parathyroid carcinoma should start with hydration and loop diuretics; calcimimetics (Cinacalcet) or bisphosphonates can later be added to lower the serum calcium levels.
• Adjuvant therapy includes chemotherapy, such as dacarbazine, 5-FU, cyclophosphamide; radiotherapy is of limited efficacy.
• Patients with features of hyperparathyroid-jaw-tumor syndrome or a family history should undergo genetic counseling and HRPT2 testing.
• After surgery, one-third of patients are cured, one-third have recurrence after prolonged disease-free survival, and one-third experience a short, aggressive course; the 5-year survival is 83.9%.
Thyroid Cancer
Incidence
Thyroid cancer is the most common endocrine cancer. A spectrum of biological behavior exists, ranging from indolent, well-differentiated tumors to extremely aggressive, poorly differentiated or anaplastic cancers.1,2 Thyroid cancer is the most rapidly increasing malignancy in the United States for both men and women. From 1980 to 2006, the annual U.S. thyroid cancer age-adjusted incidence rose from 4.33 to 11.03 cases per 100,000 population. This is a nearly threefold increase in incidence. The age-adjusted incidence of thyroid cancer is now 11.6 per 100,000 men and women per year.3 This increasing incidence is attributed to improved detection of smaller tumors. Despite this improved detection, the mortality rate remains unchanged at 0.5 per 100,000 population.4,5 Therefore, thyroid cancer presents a unique challenge to the treating physician to manage patient expectations, minimize potentially lifelong complications, employ the appropriate surveillance for followup, and identify patients with more aggressive, poorly differentiated forms.
Classification
The most common type of thyroid cancer is papillary thyroid cancer (PTC), representing 80% of all cases. The second most common type is follicular thyroid cancer (FTC), which represents 10% to 20% of all cases. Together, papillary and follicular cancers are termed differentiated thyroid cancer (DTC), and both arise from the thyroid follicular cells. Medullary thyroid cancer (MTC) comes from the parafollicular C cells. This neuroendocrine thyroid tumor represents 5% to 10% of all thyroid cancer cases and occurs in familial and sporadic forms. Finally, anaplastic thyroid cancer is one of the most aggressive and rapidly fatal cancers. It can develop from DTC that dedifferentiates over time or it also arises de novo.1,2,6,7 The first part of this section on thyroid cancer considers DTC, and the second part reviews MTC (Table 71-1, Fig. 71-1).
Table 71-1
Histologic Classification of Thyroid Cancers and Their Incidence
| Tumor Histology | Incidence (%) |
| Differentiated carcinomas | 81–87 |
| Papillary | |
| Follicular variant of papillary | |
| Follicular and Hürthle cell | |
| Medullary | 6–8 |
| Anaplastic | 5 |
| Lymphoma | 1–5 |
| Metastatic | <1 |
Etiology
External radiation exposure to the cervical region is one of the most well-known causes of thyroid cancer. Historically, patients received radiation treatments for enlarged tonsils or facial acne. Today, patients with cancer such as Hodgkin disease might still receive radiation treatments. In addition, children exposed to radioactive fallout from the Chernobyl (Russia) accident have demonstrated an increased incidence of thyroid cancer.8,9 Based on evidence from patients radiated for Hodgkin disease, doses of 40 Gy are potentially carcinogenic.10 Epidemiological studies report that 7% to 9% of patients who received 5 to 10 Gy external beam radiation develop thyroid cancer.11 A lag time of 10 to 20 years usually exists between exposure and diagnosis of thyroid cancer, although much shorter periods have been reported (Table 71-2).11
Table 71-2
Risk Factors for Malignancy in Nodular Thyroid
| Low Risk ↔ High Risk | |||||
| Factor | 1 | 2 | 3 | 4 | 5 |
| Age | |||||
| Elderly | • | ||||
| Child | • | ||||
| Sex | |||||
| Male | • | ||||
| Female | • | ||||
| Low-dose radiation in childhood | • | ||||
| Family history | • | ||||
| Cystic mass | • | ||||
| Solid mass | • | ||||
| Multiple masses | • | ||||
| Solitary mass | • | ||||
| Growing mass | • | ||||
| Stable mass | • | ||||
| Hot scan | • | ||||
| Cold scan | • | ||||
| Warm scan | • | ||||
| Fine-needle aspiration (−) | • | ||||
| Fine-needle aspiration (+) | • | ||||
| Associated cervical adenopathy | • | ||||
| Complete resolution in response to thyroid suppression | • | ||||
| Partial resolution in response to thyroid suppression | • | ||||
| No response to suppression | • | ||||
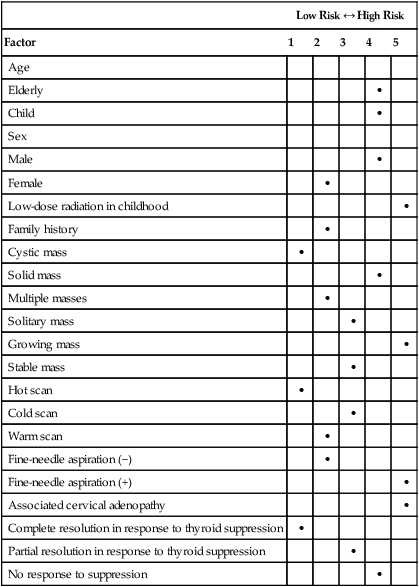
Modified from Sessions RB, Diehi WL. Thyroid cancer and related nodularity. In: Myers E, Suen J, editors. Cancer of the head and neck. 2nd ed. New York: Churchill Livingstone; 1981. p. 766.
Another environmental etiology for thyroid cancer is dietary iodine content. A higher incidence of PTC exists in regions with high dietary iodine content, such as the Pacific rim and Iceland.12 Iodine-deficient countries, in contrast, experience a higher incidence of FTC in addition to benign thyroid goiters. Many factors confound these studies that link changes in DTC rates to iodine intake. Ethnicity, selenium, goitrogen, and carcinogen intake likely play causative roles.13
Increasing investigation into molecular markers that can distinguish carcinoma from benign nodules has led to a greater understanding of the genetic alterations in thyroid cancer. For example, 70% of cancers found in Chernobyl survivors carried a RET and PTC gene (RET/PTC) rearrangement. The fusion of the tyrosine kinase encoding domain of the RET protein with a heterologous group of genes occurs in 20% to 40% cases of PTC and is called the RET/PTC rearrangement.14 RET/PTC rearrangements are frequent in small, multifocal PTCs accompanied by an inflammatory infiltrate, often seen in individuals exposed to ionizing radiation and in children.15 BRAF is a member of the RAF-MEK-ERK serine/threonine kinase-signaling cascade, and a BRAF mutation is found in 40% of PTC cases.16 The V600E BRAF point mutation or mutations in another member of this signaling pathway, RAS, are frequent in cases of poorly differentiated PTC or anaplastic thyroid cancer (ATC).17 Most BRAF substitutions keep the protein in a catalytically active form, resulting in constitutive activation of the RAF-MEK-ERK signaling cascade and constant mitogenic activity.
The Ras proteins are plasma membrane guanosine triphosphatases activated by growth factor receptors. Mutations that result in their constitutive activation lead to oncogenesis. RAS mutations also occur in 20% to 50% of follicular cancers. Similar to the RET/PTC rearrangement, another interchromosomal translocation occurs in FTC. The promoter element of the gene encoding paired box 8 (PAX8) fuses with the coding sequence of the peroxisome proliferator-activated receptor γ (PPARγ) gene in 35% of FTCs.19–19 The functional consequences of the PAX8-PPARγ rearrangement remain unclear. RAS mutations are also highly prevalent in FTC, but Nikiforova et al. found that RAS and PAX8-PPARγ rearrangements are mutually exclusive, suggesting that these are two distinct molecular pathways for FTC development.17
Aside from these acquired genetic lesions, some forms of DTC are also inherited. DTC is seen in familial syndromes such as Gardner syndrome, Cowden syndrome, and Werner syndrome.20,21 Familial nonmedullary thyroid cancer (FNMTC), defined as when two or more first-degree relatives are diagnosed with DTC in the absence of another syndrome, exhibits autosomal-dominant behavior with incomplete penetrance and variable expressivity.23–23 Linkage analyses have identified several candidate genes for FNMTC, including TCO1, MNG1, fPTC/PRN, and NMTC1, but a single responsible gene has not been identified.24–27 Compared to sporadic DTC, FNMTC is more aggressive with increased recurrence, local invasion, multicentricity, and lymph node metastases. However, in the absence of a suitable genetic test, families cannot be screened, and FNMTC is difficult to distinguish from sporadic DTC.28
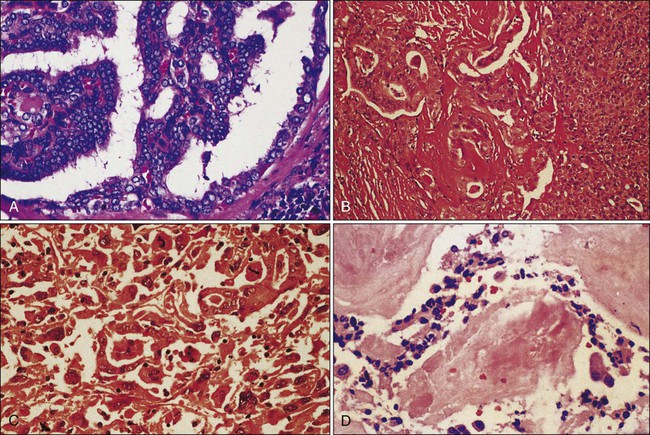
Classification and Prognosis
DTCs are divided broadly as papillary or follicular. Follicular variant PTC has features of both PTC and FTC, but is classified as a PTC subtype (see Table 71-1 and Fig. 71-1A). In general, well-differentiated PTC has an excellent prognosis with 5-year survival greater than 97%.4 Smaller tumors carry a better prognosis than larger tumors. PTC less than 1 cm in size are called papillary microcarcinomas, and have been reported in 10% to 30% of autopsy studies.31–31 In the past, these tumors were incidentally detected in thyroidectomy specimens, but they are now detected with increasing frequency by high-resolution ultrasound. They are believed to have an excellent prognosis, but some may behave more aggressively than previously appreciated, and management remains controversial.32,33
Age is another important determinant of prognosis in DTC. Older patients tend to have more poorly differentiated, aggressive variants. In these cases, death results from local invasion and extensive metastases. Therefore, the completeness of resection and extrathyroidal extension are two prognostic indicators employed in many staging systems for DTC.34,35
The role of lymph node metastases in determining DTC-specific survival remains controversial. Lymph node involvement is common in PTC, but the exact incidence of lymph node metastases depends on how it is defined. Palpable disease in the lymph nodes is present in 5% to 10% of patients with PTC, but ultrasound detects pathological lymph nodes in 30% of patients. Only 2% of patients with FTC have lymph node metastases since the route of spread is mostly hematogenous, but treatment guidelines and retrospective studies frequently consider PTC and FTC together. Routine histologic examination of lymph nodes detects DTC in 20% to 50% of patients, but when more detailed inspection is performed, up to 90% of patients with DTC will have lymph nodes with microscopic disease. Historically, lymph node involvement was felt to increase local recurrence without affecting survival, and therefore surgeons took a conservative approach to lymph node dissection for DTC. Wada et al. demonstrated that patients with pathologically positive lymph nodes had a recurrence rate of 16.3% compared to 0% in patients without pathological lymph nodes.36 Whether metastatic lymph nodes are evident preoperatively appears to be an important factor determining recurrence. For example, Ito et al. found that if metastatic lymph nodes were not seen preoperatively, then the risk of nodal recurrence was only 1.5%. Of note, in this study of 590 patients with microcarcinomas, 40% of patients had lateral neck lymph node metastases identified histologically after prophylactic neck dissection.37 Hence, lymph node metastases do affect recurrence, and clinically apparent nodes are more important than pathologically positive nodes. The impact of lymph nodes on survival is less clear. Large series and population-based studies suggest that there is a small, but significant, effect on survival.38,39
Because of their questionable effect on mortality, lymph node status is not included in all of the staging systems available for DTC. For example, the AGES system considers age, grade, extrathyroidal extension, and size.40 The AMES system uses age, distant (non-lymph node) metastases, extent of primary tumor, and size.41 Some, like the MACIS (metastases, age, complete excision, invasion, and size) system also account for the adequacy of surgical treatment.42 Alternatively, staging systems developed by the Ohio State University,43 the European Organization for Research and Treatment of Cancer (EORTC),44 the National Thyroid Cancer Treatment Cooperative Study (NTCTCS),45 and the American Joint Committee on Cancer (AJCC)46 all do consider lymph node status. The AJCC staging system is the most widely used (Table 71-3). It is also known as the TNM system because it considers tumor size (T), lymph node metastases (N), and distant metastases (M). Like many of the other thyroid cancer staging systems, it also considers age, with two different classifications for those younger and older than 45 years of age. In those younger than 45 years of age, patients with lymph node metastases are classified as stage I unless they have distant metastases (stage II) (Table 71-4).46
Table 71-3
TNM Classification of Malignant Tumors of the Thyroid Gland
| PRIMARY TUMOR (T STAGE) | |
| Tx | Tumor cannot be assessed |
| T0 | No clinical evidence of tumor |
| T1 | Tumor ≤2 cm limited to the thyroid |
| T2 | Tumor >2 cm and <4 cm limited to the thyroid |
| T3 | Tumor ≥4 cm limited to the thyroid or any tumor with minimal extrathyroid extension |
| T4a | Tumor of any size extending beyond the thyroid capsule to invade the subcutaneous soft tissues, larynx, trachea, esophagus, or recurrent laryngeal nerve |
| T4b | Tumor that invades prevertebral fascia or encases carotid artery or mediastinal vessels |
| ANAPLASTIC CARCINOMAS* | |
| T4a | Intrathyroidal—surgically resectable |
| T4b | Extrathyroidal—surgically unresectable |
| REGIONAL LYMPH NODES (N STAGE) | |
| Nx | Regional nodes cannot be assessed |
| N0 | No palpable nodes |
| N1 | Regional nodal metastases |
| N1a | Level VI nodes (pretracheal, paratracheal, prelaryngeal) |
| N1b | Metastasis to unilateral, bilateral, or contralateral cervical or superior mediastinal nodes |
| DISTANT METASTASES (M STAGE) | |
| Mx | Metastases cannot be assessed |
| M0 | No evidence of distant metastases |
| M1 | Distant metastases present |

*All anaplastic carcinomas are considered T4 tumors.
Adapted from Greene FL, Page DL, Fleming ID, et al, editors. AJCC Cancer Staging Manual. 6th ed. New York: Springer-Verlag; 2002.
Table 71-4
| Stage | TNM |
| PATIENTS < AGE 45 YEARS | |
| I | Any T, any N, M0 |
| II | Any T, any N, M1 |
| PATIENTS ≥ AGE 45 YEARS | |
| I | T1, N0, M0 |
| II | T2, N0, M0 |
| III | T3, N0, M0 |
| T1–3, N1a, M0 | |
| IVA | T4a, N0–1a, M0 |
| T1–4a, N1b, M0 | |
| IVB | T4b, any N, M0 |
| IVC | Any T, any N, M1 |
| MEDULLARY THYROID CANCER | |
| I | T1, N0, M0 |
| II | T2–3, N0, M0 |
| III | T1–3, N1a, M0 |
| IVA | T4a, N0–1a, M0 |
| T1–4a, N1b, M0 | |
| IVB | T4b, any N, M0 |
| IVC | Any T, any N, M1 |
| ANAPLASTIC CANCER | |
| IVA | T4a, any N, M0 |
| IVB | T4b, any N, M0 |
| IVC | Any T, any N, M1 |
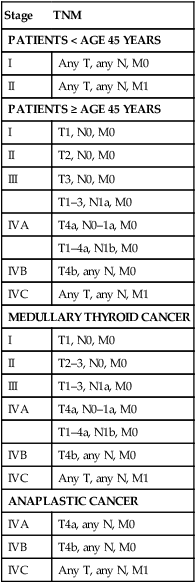
From Greene FL, Page DL, Fleming ID, et al, editors. AJCC Cancer Staging Manual. 6th ed. New York: Springer-Verlag; 2002.
It is worth noting some important ways in which FTC differs from PTC (see Table 71-1, Fig. 71-1B). Pure follicular carcinoma carries a worse prognosis than PTC. Even when the disease is confined to the thyroid, 5% to 15% of patients ultimately succumb to the disease, although survival still extends decades as in PTC.47 In addition to the prognostic factors common to DTC staging systems mentioned above, prognosis in FTC depends on the degree of capsular and vascular invasion. Minimally invasive tumors are grossly contained within the thyroid, but have microscopic foci of invasion into the capsule. Invasive tumors carry a worse prognosis and invade the capsule and vessels.50–50
Hürthle cell tumors of the thyroid are often classified with follicular cancer as they are derived from the follicular cell. Both adenomas and carcinomas of the Hürthle cell can occur, and differentiating them by cytology is difficult as it is with follicular lesions.50,51 Capsular and vascular invasion distinguish carcinoma from adenomas. Large Hürthle cell cancers (>2 cm) have a higher recurrence rate, ranging from 21% to 59%.51,52 Furthermore, Hürthle cell cancers do not always concentrate iodine. For these reasons, Hürthle cell carcinoma carries a worse prognosis compared to DTC. Adenomas have an excellent prognosis after resection and less than 2.5% demonstrate malignant behavior, but resection is recommended for larger adenomas as size is a major predictor of malignancy.52,53
As follicular or papillary cancers progress or dedifferentiate, their prognosis becomes much worse. Anaplastic cancers are at the least differentiated end of the spectrum, and represent one of the most aggressive cancers with a 5-year disease-free survival and cause-specific survival of zero percent.54 Anaplastic thyroid cancer can arise from well-differentiated tumors or it can also develop de novo (see Fig. 71-1C).54 A group of tumors falls in between well-differentiated thyroid cancers and anaplastic cancers. These cancers, called poorly differentiated thyroid cancers, are intermediate in terms of their histologic appearance and their biological behavior.1 Although the literature remains inconsistent about what constitutes poorly differentiated cancer, the best definition comes from Burman et al. that “poorly differentiated thyroid carcinoma is a concept proposed to include carcinomas of follicular thyroid epithelium that retain sufficient differentiation to produce scattered small follicular structures and some thyroglobulin, but generally lack the usual morphologic characteristics of papillary and follicular carcinoma.”55 These tumors include insular, large cell, tall cell, columnar cell, solid, and diffuse sclerosing variants.1,55 Patients with these variants tend to recur and metastasize. Furthermore, dedifferentiation of thyroid cancers leads to under expression or disordered assembly of the sodium-iodide symporter, decreasing the utility of radioactive iodine for treating micrometastatic disease or detection of metastases.56 For these reasons, poorly DTCs have a 51% disease-free survival and a 70% cause-specific survival at 5 years.1,54
Primary lymphoma of the thyroid is not as common as DTC. Older females or patients with Hashimoto thyroiditis are at highest risk for developing thyroid lymphoma.57,58 These tumors typically present as a rapidly expanding mass causing pain and compressive symptoms. Flow cytometry of cytologic specimens can sometimes make the diagnosis, but might be mistaken for Hashimoto thyroiditis. Consequently, a core biopsy is sometimes necessary when this diagnosis is suspected. Most are B-cell lymphomas treated with chemotherapy and radiation. Surgery is occasionally needed for palliation.58 Prognosis depends on the histologic subtype.
Diagnosis
Just as with a newly discovered mass anywhere else in the body, the workup of a thyroid nodule begins with a thorough history and physical exam. A strong family history of thyroid cancer or history of radiation exposure to the head and neck should raise the suspicion of thyroid cancer. Rapid growth with compressive symptoms may indicate that the thyroid nodule is thyroid lymphoma or a poorly differentiated thyroid cancer.1,2,11 On examination, malignant nodules are harder and fixed, whereas a nodule that is rubbery or soft and moves easily with deglutition is reassuring but not diagnostic of a benign nodule. Cervical lymphadenopathy also increases the likelihood that a thyroid nodule is malignant.2,59
Laboratory Studies
Because the management of patients with functional thyroid nodules differs from those with nonfunctional nodules, obtaining a thyroid-stimulating hormone (TSH) measurement early in the workup of a thyroid nodule can efficiently identify patients with a nodule and hyperthyroidism. In this subset of patients with a suppressed TSH, an 123I scan can distinguish a solitary toxic nodule from a toxic multinodular goiter and Graves disease. A solitary hyperfunctioning nodule is rarely malignant, and fine-needle aspiration (FNA) biopsy or further cancer workup is rarely necessary. The one exception is that functioning nodules in children do carry a higher risk of malignancy.60 If thyroid radionuclide scanning is undertaken, “cold” nodules should undergo FNA biopsy because 10% to 20% of “cold” nodules are malignant.61,62
Other laboratory tests can be helpful once the diagnosis of a certain type of thyroid cancer is made. For example, measuring serum thyroglobulin (Tg) in patients with DTC can assist with the long-term followup of patients treated for DTC.65–65 Although Tg can be elevated in patients with DTC, the test is not specific for diagnosing cancer, elevations in Tg can occur in benign thyroid disorders, and The American Thyroid Association guidelines do not recommend routine preoperative Tg measurement for patients with DTC. After a total thyroidectomy, however, elevations in Tg can reliably indicate recurrent or metastatic disease.64 Different threshold Tg levels can indicate recurrence depending on the concomitant TSH level. It should be emphasized, however, that there is no role for Tg measurement in the initial evaluation of thyroid nodules.
Fine-Needle Aspiration Biopsy
FNA biopsy remains the gold standard for evaluating thyroid nodules. Most clinical practice guidelines recommend FNA biopsy for nodules greater than 1 cm in largest dimension.61,64,66 When the FNA result is clearly benign or malignant, then the decision for further treatment, including thyroidectomy, becomes evident. The false-negative rate for FNA biopsy is 1% to 3% (Table 71-5). The false-negative rate increases to 10% to 15% when the nodule is large (>4 cm).68–68 Other clinical scenarios where the clinician should not always trust a benign FNA result include patients with a family history of thyroid cancer, patients with a history of radiation exposure, and cystic nodules.69 Ultrasound guidance can improve the accuracy of FNA biopsy because ultrasound can confirm that the nodule is actually being sampled and target the most suspicious portions of the nodule (i.e., the wall of a cyst). This is especially true for nonpalpable or posteriorly located nodules.70,71
Table 71-5
The Bethesda System for Thyroid Cytopathology
| Category | Risk of Malignancy (%) | Recommended Management |
| Nondiagnostic or unsatisfactory | 1–4 | Repeat FNA with ultrasound guidance |
| Benign | 0–3 | Clinical followup |
| Atypia of undetermined significance (AUS) or follicular lesion of undetermined significance (FLUS) | 5–15 | Repeat FNA* |
| Follicular neoplasm or suspicious for follicular neoplasm | 15–30 | Lobectomy |
| Suspicious for malignancy | 60–75 | Lobectomy ± frozen section or total thyroidectomy |
| Malignant | 97–99 | Total thyroidectomy |
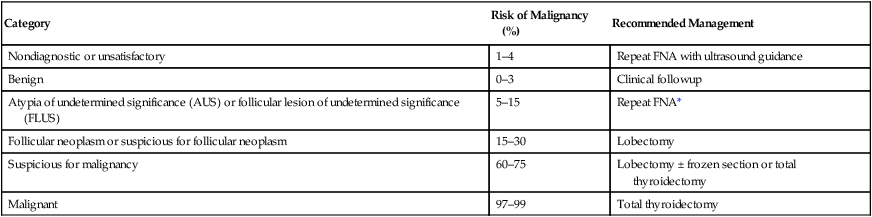
*Lobectomy also can be considered depending on clinical or sonographic characteristics.
FNA results are classified according to the Bethesda criteria that indicate the risk of malignancy (see Table 71-5). One of the limitations of cytology in evaluating thyroid nodules is that it cannot distinguish between adenoma and carcinoma for follicular lesions.61,67,68 Therefore, lobectomy with permanent histology may be the best way to make a definitive diagnosis in follicular or indeterminate lesions. Many centers have turned to molecular analysis of FNA specimens to help distinguish follicular lesions. Cytology specimens are analyzed for a panel of mutations, including BRAF, RAS, RET/PTC, PAX8-PPARγ rearrangements. Although this is an exciting area of research, the clinical utility of the various gene panels has varied and more prospective data is needed.16–18,54,72
Imaging
Not only can ultrasound improve the accuracy of FNA biopsy, but it is also an important tool in evaluating thyroid nodules as it is used to measure size and features of the nodule and it can also identify additional nonpalpable nodules. Ultrasound alone can increase the clinician’s suspicion for malignancy if the nodule has fine microcalcifications, irregular borders, or chaotic vascular patterns. In addition, ultrasound evaluates the lymph nodes in both the central and lateral neck compartments, which may prompt additional FNA biopsy of suspicious lymph nodes or alter the surgical plan. Although ultrasound is highly operator-dependent, it is noninvasive and does not involve any radiation or contrast risk to the patient. High-resolution ultrasound can also demonstrate extracapsular invasion and subtle lymph node involvement.70,73–78 Consequently, ultrasound is the preferred method to evaluate the thyroid and cervical lymph nodes. Although routine screening is not recommended for all patients, those with a strong family history or radiation exposure can undergo ultrasound screening for thyroid nodules. Positron emission tomography (PET) and/or computed tomography (CT) scan are helpful for identifying lung or bone tumors in patients at risk for metastases.75,79
Highly aggressive cancers that may invade local structures, extend into the chest, or demonstrate poorly differentiated cytology, require careful preoperative planning. In this case, CT becomes a helpful preoperative imaging study to help plan en bloc resection of other organs aside from the thyroid, understand the extent of vascular involvement, determine if a thoracic incision is necessary, and plan for reconstruction.1,75,79,80
Treatment
Surgery
The extent of surgery for DTC remains controversial. This is especially true for small, encapsulated, well-differentiated tumors, and tumors less than 1 cm in size (microcarcinomas). These are discussed further below, but for most DTC ≥1 cm diagnosed preoperatively, most clinicians recommend a total thyroidectomy.64 The rationale for total thyroidectomy is based on tumor biology and current treatment modalities. DTC, especially PTC, tends to be multicentric, with up to 80% of patients having multiple tumor foci and bilateral disease in 60% when a thorough pathological examination of the contralateral lobe is performed.7,32,75 A total thyroidectomy as the initial procedure obviates the need for reoperative surgery to remove the contralateral lobe should a recurrence become detected. Second, experienced thyroid surgeons can safely perform a total thyroidectomy with permanent complications, such as recurrent laryngeal nerve injury and hypoparathyroidism, occurring at a rate of less than 2%.80,81 Radioactive iodine therapy for ablating microscopic disease becomes most effective when the thyroid remnant is small or absent. Tg measurement and radioiodine whole-body scanning are highly sensitive modalities for detecting recurrent or metastatic disease, but these two methods are most effective when no thyroid tissue remains in the neck.2,75
Most low-risk cancers carry an excellent prognosis regardless of the extent of thyroidectomy, and there are no randomized prospective trials comparing total thyroidectomy to thyroid lobectomy in this group of patients. In addition, radioiodine may have limited utility in low-risk patients.7,32 For these reasons, some researchers favor thyroid lobectomy in low-risk patients. For example, Shaha et al. have reported 20-year followup on 465 patients with low-risk DTC. Although the lobectomy group had more local recurrence compared to the total thyroidectomy group (4% vs. 1%), there was no statistical significance.82 Similarly, other groups have also failed to demonstrate any significant effect on survival.85–85 In contrast, large retrospective series have demonstrated improvement in recurrence for total thyroidectomy compared to lesser operations.86–89 In a frequently cited study, Mazzaferri et al. reported on 1355 patients with a mean followup of 15.7 years. Patients treated with total thyroidectomy experienced significant improvements in recurrence rate (26% vs. 40%, P < 0.02) and mortality rate (6% vs. 9%, P = 0.02) compared to lesser resections.86 Although some researchers have questioned the accuracy of risk-stratification and accounting for complications in these retrospective studies, current guidelines still recommend a total or near-total thyroidectomy for small (<4 cm), unifocal, well-differentiated tumors with no lymph node metastases, or extrathyroidal extension.64
Another hotly debated topic related to the extent of initial surgery for DTC is the role of prophylactic central neck dissection. Although the 2006 American Thyroid Association guidelines stated that routine prophylactic central neck dissection should be considered for patients with DTC,90 the most recent guidelines have been revised to recommend that “prophylactic central neck dissection may be performed, especially in patients with advanced primary tumors” and “total thyroidectomy without prophylactic central neck dissection may be appropriate for small (T1 or T2), noninvasive, clinically node-negative patients.”64
The central neck lymph nodes are also classified as level VI lymph nodes and include the paratracheal, perithyroidal, and precricoid lymph nodes. These nodes are found along and behind the recurrent laryngeal node, and frequently surround the lower parathyroid gland. Although the level VI lymph nodes contain macroscopic disease in 10% of cases, when they are removed prophylactically, 32% to 69% of patients will have microscopic metastases.93–93
Proponents of prophylactic central neck dissection argue that the initial operation is the safest time to remove central neck lymph nodes to prevent local recurrences and the complications associated with reoperative surgery in the central neck. Wada et al. found the recurrence rate in patients treated with therapeutic lymph node dissection to be 21%, whereas patients who underwent prophylactic neck dissection experienced a recurrence rate of only 0.43%. Importantly, those patients without clinically overt nodal disease who did not undergo prophylactic central neck dissection also experienced a very low recurrence rate of 0.65%. Hence, the absolute differences in recurrence are miniscule.36 Several other studies also support the concept that microscopically positive lymph nodes rarely progress to recurrence, especially after postoperative radioactive iodine ablation.96–96 Clinically evident lymph node metastases place patients at higher risk for recurrence, and these patients clearly benefit from therapeutic lymph node dissection. Prophylactic central neck dissection reduces an already low recurrence rate, potentially eliminates or reduces the need for radioactive iodine, but is also associated with risks such as hypoparathyroidism. The risk-to-benefit ratio may favor prophylactic central neck dissection in a subset of patients, but the putative risk factors that define such a subset remains unknown.75,97–99
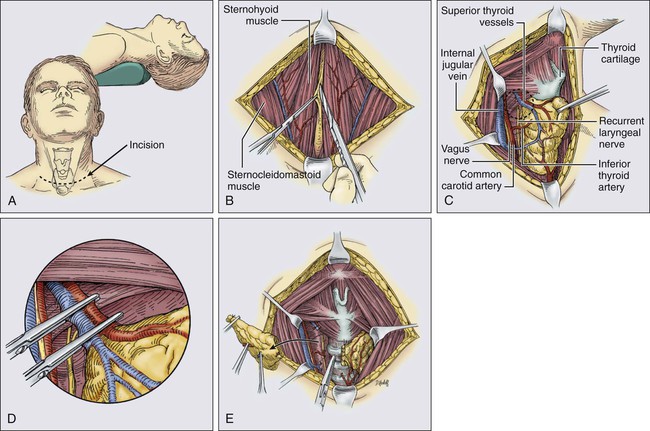
Thyroidectomy begins with proper positioning in the semirecumbent position with the neck extended (Fig. 71-2). A transverse, curvilinear incision is made in a suitable skin crease at or beneath the level of the cricoid cartilage. Traditionally, a Kocher collar incision was utilized, but this requires a very large dissection superiorly to reach the upper pole of the thyroid, placing the patient at risk for postoperative seroma. Intraoperative ultrasound can help assess the upper extent of the gland and place the incision appropriately. Superior and inferior subplatysmal flaps are raised to create a working space around the thyroid. The strap muscles are divided in the median raphe and are retracted laterally. It is rarely necessary to transect the strap muscles, but this can be accomplished if the tumor is large or adherent to the overlying muscles. The perithyroidal soft tissue is swept off the gland bluntly to identify the boundaries of the thyroid. Because the thyroid is most fixed at the upper pole, these vessels are divided first. Much of this can be accomplished using energy devices such as the Harmonic Scalpel or LigaSure, but larger vessels may require clips and/or ties. The upper pole vessels should be ligated close to the thyroid capsule to avoid injuring the external branch of the superior laryngeal nerve. In addition, the surgeon should remain vigilant for the upper parathyroid gland, as it is frequently located near the upper pole vessels. Next, the thyroid gland is reflected medially. To accomplish this, the middle thyroid vein is ligated. In addition, dividing the thyroid isthmus can also facilitate medial rotation, assuming that the tumor is not located within the isthmus. Before dividing any structures along the medial border of the gland, the recurrent laryngeal nerve must be identified and its course dissected. The nerve is found medial to the upper parathyroid gland and lateral to the lower parathyroid. The parathyroid glands must also be identified and dissected free from the thyroid on an intact vascular pedicle. Once the recurrent laryngeal nerve is identified, the branches of the inferior thyroid artery can be divided along the thyroid capsule. The inferior pole also is mobilized by a combination of blunt dissection and ligation of the inferior thyroid artery. The thyroid is then dissected off the anterior surface of the trachea using electrocautery or other energy devices to divide the small vessels contained within the ligament of Berry. Performing the identical procedure on the contralateral lobe completes a total thyroidectomy.
Although “skip” metastases directly to the lateral compartment can occur in PTC, the central neck nodes (level VI) are usually the first nodes to receive drainage from the thyroid (Fig. 71-3). The boundaries of the central neck are the carotid sheathes laterally, the hyoid bone superiorly, and the innominate artery inferiorly.100 Lymphadenectomy in this area requires skeletonizing the recurrent laryngeal nerve along its entire cervical course, and removing all the fibrofatty tissue along the trachea. Frequently, the lower parathyroid is invested in this tissue and becomes devascularized with this dissection.97
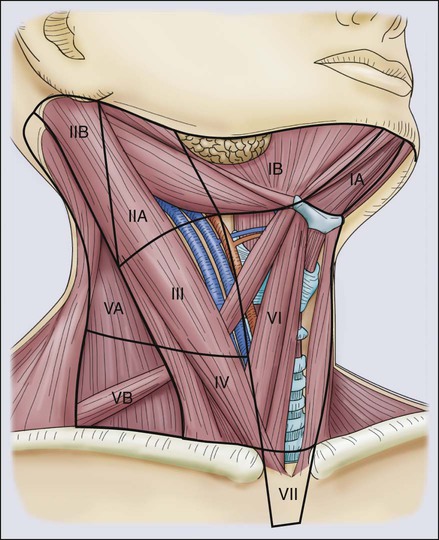
A lateral neck dissection usually involves dissection of levels II, III, and IV (see Fig. 71-3). This dissection puts the spinal accessory, phrenic, vagus, cervical sensory, sympathetic trunk, hypoglossal, greater auricular, and the marginal mandibular branch of the facial nerves at risk. The extent of node dissection should be guided by preoperative and intraoperative ultrasound findings. Usually, the great vessels can be preserved, but more aggressive tumors can invade the internal jugular vein, and it should be sacrificed in this scenario. In addition, to nerve injury, chyle leak is another complication from performing lateral neck dissection.75,80
Radioactive Iodine
Remnant ablation with radioactive iodine is the standard adjuvant treatment for selected patients with DTC. It can only be administered after a total or near-total thyroidectomy, otherwise the radioactive isotope will be absorbed by the remnant thyroid and not destroy any micrometastatic disease as intended. Radioactive iodine is administered 1 to 3 months postoperatively as 131I as sodium iodide in an oral form whose half-life is 7 to 8 days (Figs. 71-4 and 71-5). Consensus guidelines recommend a dose of 30 to 100 mCi for patients with low-risk tumors and higher doses (100 to 200 mCi) for patients with residual disease, suspected microscopic disease, or more aggressive histologic subtypes (i.e., tall cell, columnar cell, or insular variants).64,101–103 To stimulate intracellular uptake of the isotope, the TSH concentration should be at least as high as 30 mU/L. There are two methods for achieving such an elevation in TSH. The traditional method requires the patient to withdraw from thyroid hormone replacement over 4 to 6 weeks.2,104 A newer method is to administer recombinant human TSH (rhTSH). rhTSH is administered in the form of intramuscular injections on two consecutive days followed by radioactive iodine on the third day. The advantage of this method is that the patient does not experience an extended period of hypothyroidism as with hormone withdrawal. However, long-term data on the effectiveness of rhTSH compared to traditional withdrawal are lacking, although it appears effective for low-risk patients. The U.S. Food and Drug Administration (FDA) approved rhTSH for thyroid remnant ablation in patients who do not have evidence of metastatic disease.105,106 In addition to making the TSH rise, clinicians should also prepare patients by instructing them to follow a low-iodine diet for 1 to 2 weeks prior to radioactive iodine treatment. This diet requires patients to avoid foods that contain iodized salt, dairy products, eggs, seafood, soybeans or soy-containing products, and foods colored with red dye #3.102,103
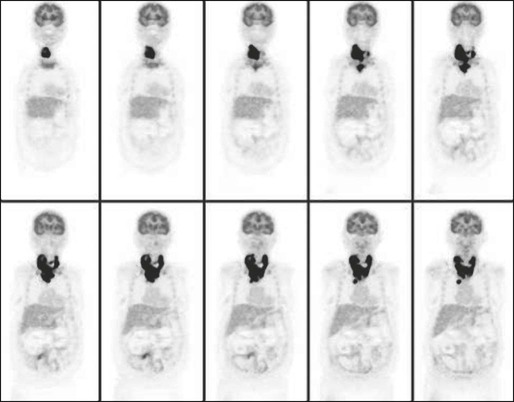
Although some studies show no benefit to radioactive iodine therapy,107,108 other studies demonstrate a reduction in locoregional recurrences and distant metastases.86,87 As with the controversy over the extent of thyroidectomy, the benefit of radioactive iodine for low-risk patients remains unclear.104 The most recent American Thyroid Association (ATA) guidelines recommend remnant ablation for all but the lowest-risk patients (unifocal, well-differentiated tumor, <1 cm in size, and confined to the thyroid gland without lymph node metastases).64 The National Comprehensive Cancer Network (NCCN) guidelines require a more thorough evaluation for the extent of remaining disease after thyroidectomy with a radioiodine scan 1 to 12 weeks postoperatively. Radioactive iodine ablation is not recommended if the stimulated Tg is less than 1 ng/mL and the radioiodine scan is negative.101
Recently, some studies have shown an increase in the risk of developing secondary malignancies after radioactive iodine therapy. This has been examined using the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) database. Brown et al. found that patients treated for DTC had significantly higher rates of nonthyroid second primary malignancies than expected in the general population. Although the excess risk was relatively small, it was greater in the subset of patients who were treated with radioactive iodine.109 Iyer et al. specifically examined low-risk patients (T1N0) treated with radioactive iodine and found that their excess absolute risk was 4.6 excess cases per 10,000 person years at risk.110 As discussed above, radioactive iodine clearly benefits patients with larger tumors and metastatic disease, but the increased risk of secondary malignancies in low-risk patients where the long-term benefit of radioactive iodine is questionable means that careful patient selection for radioactive iodine treatment is necessary.
Hematologic malignancies are the most common secondary malignancies after radioactive iodine, but there is also an association with kidney, breast, bladder, skin, and salivary gland cancers.113–113 The more commonly noted side effects after radioiodine treatment include dry mouth, mouth pain, salivary gland swelling (sialadenitis), altered smell and taste, conjunctivitis, and fatigue. Women should not be pregnant at the time of treatment nor should they become pregnant for at least 6 months following treatment. Similarly, men should avoid conception for at least 6 months following treatment.103,112,114,115
Thyroxine Suppression
Because all cells of follicular origin depend on TSH for growth, TSH suppression through the administration of supraphysiologic doses of levothyroxine (T4) remains an important strategy for maintaining disease-free survival and overall survival.116,117 For high-risk patients with incomplete resection, tumor invasion into adjacent structures, or distant metastases, their physician should initially titrate levothyroxine dosing to a TSH less than 0.1 mU/L. Lower-risk patients should be dosed to a TSH at or slightly below the lower limit of normal (0.1 to 0.5 mU/L).64,101 Once patients remain disease-free for at least 2 years, their TSH suppression can be liberalized to within the reference range. Patients with persistent disease should be kept at a TSH less than 0.1 mU/L indefinitely. TSH suppression carries risks of arrhythmias, anxiety, and osteoporosis. The risks and benefits should be carefully considered, particularly in older patients. Because of the risk of bone loss, the NCCN guidelines recommend daily calcium and vitamin D supplementation for patients on TSH suppression.101
External Beam Radiation
Although 131I is the preferred adjuvant therapy for thyroid carcinoma, external beam radiation sometimes plays a role in treating this disease. Persistent, recurrent, anaplastic, or poorly differentiated tumors may fail to take up 131I. Treatment of anaplastic thyroid tumors almost always includes external beam radiation as these tumors often cannot be completely resected and do not concentrate iodine. Although no improvement in overall survival has ever been documented, external beam radiation is often given after resection of poorly differentiated tumors to reduce the risk of local relapse.118 The group at Memorial Sloan Kettering Cancer Center has found that up to 85% of poorly differentiated tumors display some iodine avidity, and therefore treatment with radioactive iodine may remain worthwhile. Patients with incompletely resected tumors, unresectable disease, and locoregional recurrence in a previously operated field may benefit from external beam radiation.1,118,119
Chemotherapy
Because radioactive iodine often can be effective treatment for well-differentiated tumors that have metastasized, cytotoxic chemotherapy has not been extensively evaluated for metastatic thyroid cancers. For large burden of disease, anaplastic cancers, or poorly differentiated tumors that are not iodine avid, chemotherapy becomes an important treatment component after surgery or if the tumor is not resectable. In these situations, chemotherapy confers minimal effects as these tumors hold a very poor prognosis. Historically, doxorubicin was the most effective single agent. Combination therapy with doxorubicin and cisplatin resulted in modest objective response rates.120,121 Newer, targeted therapies have shown some promise. Small-molecule tyrosine kinase inhibitors (such as sorafenib or sunitinib) and antibodies (anti-vascular endothelial growth factor [VEGF]) should be considered in the context of ongoing clinical trials.101,122–124
Recurrence
For most DTC, the long-term survival exceeds 95%. Even though mortality remains quite low, recurrence rates exceed mortality rates. For low-risk tumors, the recurrence rates range from 2% to 10%.125 Higher-risk tumors (larger size, extrathyroidal extension, cervical lymph node metastases) carry recurrence rates of 21% to 68%.37,125 For these reasons, a tailored approach to followup and treatment of recurrent disease best serves patients with DTC.
Surveillance
The original tumor characteristics and operative findings dictate the followup schedule for DTC. Surveillance consists of measuring serum Tg, TSH, and anti-Tg antibodies in addition to imaging. Cervical ultrasound is a highly sensitive test for detecting metastatic lymphadenopathy. Elevations in Tg in the absence of cervical disease seen on ultrasound suggest distant metastases.35,64,101 Whole-body radioiodine scanning is sensitive for detecting iodine-avid bone or pulmonary metastases.63,66 Poorly differentiated, aggressive tumors will not concentrate iodine, but can be detected by CT scan or FDG-PET(see Fig. 71-5).56
The frequency of surveillance depends on the original tumor characteristics, and the AJCC stage.46 For lower-risk tumors, physical exam, cervical ultrasound, and measurement of TSH, Tg, and anti-Tg antibodies should be performed every 12 months. These studies can be scheduled 3 to 6 months after the initial radioactive iodine treatment in patients with high-risk tumors.64,101,124
Treatment of Recurrent Disease
Recurrence in the neck or cervical lymph nodes is best treated surgically. Radioactive iodine cannot ablate bulky nodal disease. Following surgical resection, radioiodine can effectively ablate micrometastatic disease throughout the body. External beam radiation should be considered for recurrent disease that is unresectable or not iodine avid (Fig. 71-6).
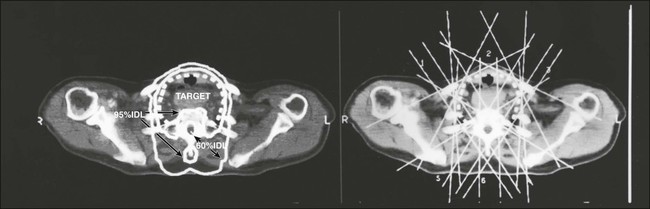
Medical treatment for recurrent or metastatic disease consists of maintaining TSH suppression. Depending on the location of recurrence, health of the patient, tumor risk stratification, and patient preference, patients with metastatic disease can be referred for experimental protocols using targeted therapies, undergo traditional cytotoxic chemotherapy, or pursue watchful waiting and supportive care.124 A multidisciplinary team experienced with metastatic thyroid cancers best handles these decisions.
Medullary Thyroid Cancer
MTC accounts for 5% to 10% of all thyroid cancers. Unlike DTC, MTC arises from the parafollicular C cells instead of the follicular epithelium (see Fig. 71-1D). Hence, MTC is a neuroendocrine tumor, and shares some properties common among neuroendocrine cancers, including secretion of peptide hormones such as calcitonin, serotonin, or vasoactive intestinal peptide. Most cases of MTC are sporadic, but 25% are a result of germline genetic mutations. Hereditary cases occur either in isolation (FMTC) or as part of the multiple endocrine neoplasia (MEN) syndrome type 2 (2A or 2B).128–128
Diagnosis
Although any thyroid nodule could potentially harbor MTC, historical features that may alert the physician to the potential for MTC include a family history of MTC, pheochromocytoma, hyperparathyroidism, or other manifestations of MEN-2 syndromes.6 As in evaluating all thyroid nodules, neck ultrasound and FNA play a major role in diagnosing MTC. Hereditary cases are often detected through genetic screening to identify germline mutations in the RET gene. Almost all sporadic cases present with a palpable neck mass and this could be either the thyroid mass or a metastatic lymph node. Lymph nodes metastases occur in 35% to 50% of patients at initial diagnosis.129 Therefore, ultrasound evaluation of the central and lateral neck compartments for suspicious lymph nodes becomes a crucial component to the initial diagnosis.130
Because the parafollicular C cells are concentrated in the upper, posterior portion of each thyroid lobe, many MTCs arise in a posterior location, causing symptoms such as hoarseness or dysphagia as a result of compression of local structures. If there is any concern for vocal cord function, then direct laryngoscopy should be performed preoperatively.129 Markedly elevated calcitonin levels can cause symptoms such as flushing, diarrhea, and weight loss.131
FNA characteristics of MTC include the presence of stromal amyloid without thyroid follicles. Because spindle-shaped cells may be seen, MTC can be mistaken for parathyroid carcinoma or anaplastic thyroid cancer unless the specimen is stained for calcitonin, chromogranin A, or carcinoembryonic antigen (CEA)—substances produced by MTC that confirm the diagnosis. A more sensitive technique than immunohistochemistry on cytology specimens is to measure the calcitonin level in the washout fluid from an FNA.132 Additionally, the presence of calcitonin messenger RNA (mRNA) has been performed when the cytologic or histologic diagnosis remains unclear.133
Several serum markers can confirm the diagnosis of MTC, and are useful in following patients for recurrence and metastases. Calcitonin is commonly elevated in patients with MTC. Although a small percentage of normal patients will have some elevation in calcitonin, patients with a diagnosis of MTC typically exhibit levels above 100 pg/mL. In borderline cases, the diagnosis can be clarified by stimulating the calcitonin with either intravenous calcium gluconate or pentagastrin. Before the advent of genetic testing, these stimulated measurements were used to screen patients at high risk for MTC.134 The degree of calcitonin elevation correlates with tumor burden, with nodal metastases found at basal calcitonin levels of 10 to 40 pg/mL, and distant metastases found with calcitonin levels greater than 150 pg/mL. Patients with calcitonin levels greater than 3000 pg/mL are likely to have widely metastatic disease, and are unlikely to experience a cure.135
Preoperative measurement of serum CEA can also help risk-stratify patients. Overall, CEA elevations occur in more than 50% of patients with MTC, but a preoperative serum CEA greater than 30 ng/mL highly predicts the inability to cure the patient with surgery.136 CEA levels above 100 ng/mL may signify extensive lymph node and distant metastases. Following CEA levels postoperatively can also monitor disease progression. An increasing CEA level in the presence of a stable calcitonin is associated with a worse prognosis as it may indicate tumor dedifferentiation and distant metastases. Other markers, such as chromogranin A and serotonin, may be elevated in patients with MTC, as with many other neuroendocrine tumors, but calcitonin and CEA are the most useful for following MTC patients long-term.129,137,138
Genetic testing plays an important part of the initial management because it identifies familial disease and risk stratifies patients. Germline mutations in the RET gene characterize familial disease.139 A small percentage of apparently “sporadic” disease will also carry germline RET mutations, but truly sporadic cases frequently harbor somatic RET mutations. Commercial testing is performed through polymerase chain reaction (PCR) amplification of the patient’s germline DNA obtained from the patient’s white blood cells. A spectrum of tumor aggressiveness exists among the various RET mutations, and the timing of prophylactic thyroidectomy is based on the specific mutation. Once a patient tests positive for a germline RET mutation, the patient should be carefully counseled regarding the risk to other family members and the patient’s children. At-risk family members should be identified and also tested so that prophylactic thyroidectomy can be offered at the appropriate time. Although some overlap exists for genetic mutations associated with MEN-2A and familial MTC, distinct mutations are usually associated with MEN-2B.140,141
Treatment
Complete surgical excision is the treatment of choice for MTC. The minimum extent of surgery for patients with clinically apparent disease is a total thyroidectomy with bilateral central neck dissection. Eighty-one percent of patients with palpable disease have central neck lymph node metastases, and the addition of central neck dissection improves cure rates over total thyroidectomy alone in patients with clinically evident disease at presentation.142,143 The initial approach to lateral neck lymph nodes continues to evolve. Historically, the initial surgical treatment included an ipsilateral lateral compartment neck dissection because up to 80% of patients will have ipsilateral nodal metastases.144 However, current guidelines recommend performing an ipsilateral lateral neck dissection if ultrasound or physical exam detects lymphadenopathy in the lateral neck, central compartment lymph nodes are involved, or when the primary tumor is greater than 1 cm.130 Contralateral lateral neck dissection is added when patients have bilateral tumors or there is extensive lymph node disease on the ipsilateral side. Because some patients often require extensive neck dissection, these procedures are often staged.129,130 Unlike DTC where micrometastatic disease can be effectively treated with radioactive iodine ablation, the only effective treatment for MTC is complete surgical resection. Therefore, all evident disease must be resected for the best long-term cure.
Prophylactic thyroidectomy is recommended for at risk family members in hereditary MTC. Current recommendations for the timing of prophylactic thyroidectomy balance the need to remove the at-risk organ prior to it developing clinically apparent disease with the risks of surgery. In hereditary MTC, an age-related progression exists from C-cell hyperplasia to carcinoma, and, ultimately, nodal metastases. The optimal timing of prophylactic thyroidectomy depends on the risk level of the RET mutation. In general, current guidelines recommend operating on children with MEN-2A and familial MTC by age 5 years while those with MEN-2B should be operated on before 6 months of age.145 Prophylactic surgery should consist of at least a total thyroidectomy. The role of prophylactic lymph node dissection in familial disease remains controversial. Lymph node metastases are present in 6% of screened patients,146 and therefore, some argue that prophylactic central lymph node dissection should be performed. Opponents to this approach state that with a normal preoperative ultrasound, normal calcitonin (basal and/or stimulated), and a normal CEA, the risk of occult nodal disease is very low and do not outweigh the risks of a central neck dissection such as permanent hypoparathyroidism.6,142,146 Because any complications resulting from prophylactic surgery become lifelong problems for the patient, experienced surgeons should perform prophylactic surgery. Prior to proceeding with surgery, the surgeon should screen patients with hereditary disease for associated conditions such as pheochromocytoma (MEN-2A and B) and hyperparathyroidism (MEN-2A).141,146
A spectrum of disease severity exists for both hereditary and sporadic MTC, and, therefore, the natural history of MTC varies widely. Distant metastases in the lung, liver, or bone can arise and lead to death quite quickly. On the other hand, many patients live with a large tumor burden and very high calcitonin levels with few symptoms. Others suffer from intractable diarrhea. In this case, cytoreductive surgery or somatostatin analogs like octreotide can palliate severe symptoms.147
Conventional chemotherapy regimens with doxorubicin, dacarbazine, capecitabine, and 5-fluorouracil have demonstrated limited efficacy in patients with MTC. Newer, targeted therapies block the RET receptor tyrosine kinase or its multiple downstream pathways, such as the extracellular signal-related kinase (ERK), phosphatidylinositol 3-kinase (PI3K)/Akt, p38 mitogen-activated protein kinase (MAPK), and c-Jun N-terminal kinase pathways.72,129,148 Some of these tyrosine kinase inhibitors inhibit multiple signaling pathways simultaneously.72,149 These targeted therapies are currently being evaluated in multicenter trials.149,150
Recently, the FDA approved one of these targeted therapies, vandetanib, for treatment of metastatic MTC. Vandetanib is a small-molecule inhibitor of the VEGF receptor, epidermal growth factor (EGF) receptor, and the RET tyrosine kinase.151 In a randomized controlled clinical trial, patients treated with vandetanib experienced a median progression-free survival of 22.6 months compared to 16.4 months in patients treated with placebo.149
Adrenocortical Cancer
Incidence
Adrenocortical cancer (ACC) is a rare endocrine malignancy with an incidence of 1 to 2 per million people and causing less than 0.2% of all cancers deaths.152,153 Following anaplastic thyroid cancer, ACC is the second most aggressive endocrine cancer. Although ACC can develop at any age, it occurs in a bimodal age distribution: less than 5 years of age and the fourth and fifth decades of life.154 Women are affected more than men with a ratio of 1.5 : 1 and are more likely to have a functional tumor.155 The incidence of ACC also varies between countries and the highest incidence is in southern Brazil with 10 to 15 times higher incidence in children.156
Pathogenesis
Most ACCs arise sporadically, and the pathogenesis is not completely understood. It is not known if ACC develops from hyperplastic or adenomatous adrenal nodules. However, several genetic alterations have been identified with the most common mutation involving over expression of the insulin-growth factor (IGF) gene. Other growth factor and growth factor receptor mutations have also been identified in sporadic ACC.153,157 Familial tumor syndromes that involve ACC include multiple endocrine neoplasia 1 (MEN-1, 11q13), Li-Fraumeni syndrome (17p13), Beckwith-Wiedemann syndrome (11p15.5 and 15q11–13), and Carney complex (17q23–24 or 2p16).153,158,159
Clinical Presentation
Most ACC patients are asymptomatic until the tumor reaches a size that causes compression of nearby structures, local invasion, or distant metastasis. Although symptoms are vague, they can include fever, early satiety, weight loss, pain, anemia, nausea, and fatigue. The presence of these symptoms in the setting of ACC is ominous. The mean duration from the onset of symptoms to the diagnosis of ACC varies from 6 to 16 months and appears to be independent of whether the tumor is functional.154–154
In 40% to 60% of the patients, ACC results in increased hormone production (functional) that may cause symptoms and assist in correctly diagnosing these aggressive tumors. Because most ACCs are inefficient in mature steroidogenesis, they can secrete a variety of steroid precursors that result in clinical or subclinical syndromes. As many as 75% of the ACCs are associated with subclinical Cushing syndrome that can be diagnosed on biochemical testing.157,160 Clinical Cushing syndrome with the classic symptoms may be present in up to 50% of the patients with functional ACC. Virilization caused by excessive androgen steroids or androgenic precursors is identified in approximately 20% of the patients, and the combination of both clinical Cushing syndrome with virilization is present in 10% of the functional ACCs. Cushing syndrome with virilization almost always indicates ACC. Other clinical manifestations include feminization (5% to 8%), hypoglycemia (<5%), and hypokalemic alkalosis (<5%).153,161
Metastases are present in 20% to 50% of the patients with ACC at the time of diagnosis with lungs (40% to 50%), liver (40%), and lymph nodes (20% to 30%) being the most common sites. Other less common sites include the bones, spleen, pancreas, and diaphragm.153,161,162
Diagnosis
The correct diagnosis of ACC is important for appropriate management; however, preoperative diagnosis of ACC is complex. At present, the diagnosis relies on clinical assessment, urinary and plasma biochemical tests, and imaging studies. Clinical suspicion should be raised in any patient with adrenal Cushing syndrome with an adrenal mass, increased urinary 17-ketosteroids, age less than 20 years, or in patients with an adrenal mass, increased urinary 17-ketosteroids, virilization/feminization, weight loss, anemia, or fever.153,161
Hormonal workup includes plasma adrenocorticotropic hormone, cortisol, and urinary 17-ketosteroids, with or without a dexamethasone suppression test. Adrenal incidentalomas should also be evaluated by plasma or urinary metanephrines to rule out pheochromocytoma, and by rennin and aldosterone levels in patients with hypertension to rule out aldosteronoma.163
The role of imaging in the evaluation of adrenal mass is valuable for assessing the tumor size and other specific imaging characteristics. It has been demonstrated that 92% of the ACCs are larger than 6 cm in size,164 and that the risk of malignancy increases with adrenal tumor size as follows: 2% for tumors smaller than 4 cm, 6% for tumors that are 4 to 6 cm, and 25% for tumors larger than 6 cm.160,165 Furthermore, adrenal masses that increase in size within a 6 months period are also suggestive of malignancy.153 CT is the most useful imaging study to evaluate adrenal tumor size, adjacent organ involvement, and resectability. Irregular borders, heterogeneity, calcifications, Hounsfield units greater than 25, and delayed washout of less than 50% all suggest malignancy. When performed correctly, a washout of less than 50% carries a sensitivity and specificity of 100%.153,161,163,166 With gadolinium-enhanced magnetic resonance imaging (MRI), ACC has increased intensity on T2-weighted images. Other suspicious features on MRI include peripheral enhancement, central necrosis, and internal hemorrhage. The overall MRI sensitivity and specificity are 81% to 89% and 92% to 99%, respectively.167
Two other imaging modalities show encouraging results. The reported sensitivity of FDG-PET is 100% for identifying ACC and 11C-metomidate-PET (MTO-PET) is an emerging adrenal imaging technique with similar accuracy.168,169
FNA is a tool that should be discouraged for the diagnosis of ACC because of the high false-negative rate.153,163,170 The definitive diagnosis of ACC is by pathology. Grossly, these are large tumors that may invade the adjacent organs and large blood vessels, such as the inferior vena cava. The tumors are yellow to tan, with areas of necrosis and hemorrhage. In the absence of metastatic disease, local, capsular, or vascular invasion, the diagnosis of malignancy may be difficult. The Weiss criteria incorporate nine histologic features that help distinguish between malignant and benign tumors. These features include high nuclear grade (III or IV), mitotic rate greater than 5/50 high-power fields, atypical mitoses, diffuse architecture, microscopic necrosis, 25% or fewer clear cells, capsular invasion, sinusoidal invasion, and venous invasion. A score of less than or equal to 2 is classified as benign, whereas a score greater than 3 is considered malignant. Evaluation of molecular markers such as microRNAs, IGF-2 overexpression, increased Ki-67, and specific genetic variations may prove to be a more accurate diagnostic tool.154,157,160,170
Treatment
Primary Disease
Surgery
Surgical resection of the primary tumor is the only means to achieve cure and every attempt must be made to identify the patients that are candidates for curative resection.153,160,162 Patients with known or suspected ACC should be referred to highly experienced surgeons. The surgeon can evaluate the size and local invasiveness of the tumor and decide on the approach and extent of surgery. Tumors that are localized to the adrenal gland should be resected with adequate margins. This can be done via open (anterior or posterior) or laparoscopic approach. Although the role of laparoscopy for the resection of ACC is controversial, surgeons are expanding the indications for laparoscopy for localized tumors with comparable outcomes.171 At present, there are no randomized control trials that evaluate the role of laparoscopic surgery for ACC.
Prior to incision, patients should be given a stress dose of steroids as the contralateral gland may be suppressed.153 At surgery, every attempt must be made to achieve clear surgical margins, and, in cases of locally advanced disease, en bloc resection of involved organs should be performed. Regional lymph node dissection should be performed for involved lymph nodes, and such involvement should be assessed in every patient with known ACC.
Advanced ACCs may involve the kidney, pancreas, spleen, liver, diaphragm, renal vein, and inferior vena cava. Tumors may even extend through the inferior vena cava to the right atrium and the superior vena cava. For such advanced tumors, multidisciplinary teams are of upmost importance, as cardiopulmonary bypass or hypothermic circulatory arrest is often required.152,160,161 Table 71-6 outlines the staging of ACC.160
Table 71-6
| TNM | ||
| T | T1 | Size ≤5 cm, no local invasion |
| T2 | Size >5 cm, no local invasion | |
| T3 | Presence of local invasion without involvement of adjacent organs | |
| T4 | Invasion of adjacent organs | |
| N | N0 | No positive lymph nodes |
| N1 | Positive lymph nodes | |
| M | M0 | No distant metastases |
| M1 | Distant metastases | |
| STAGING | ||
| I | T1, N0, M0 | |
| II | T2, N0, M0 | |
| III | T3, N0, M0 T1-2, N1, M0 |
|
| IV | T4, N0, M0 T3, N1, M0 T1-4, N0-1, M1 |
|

Postoperative surveillance should be continued for many years as recurrences have been documented as long as 12 years after resection. For patients with elevated dehydroepiandrosterone sulfate (DHEAS) at presentation the postoperative levels may be used for surveillance. Patients with nonfunctional tumors should be evaluated at regular intervals by physical examination and CT scan.152,153,161
Mitotane
Mitotane (o,p′-DDD or 1,1-dichloro-2(o-chlorophenyl)-2-(p-chlorophenyl)ethane) is a pesticide isomer that is directly toxic to adrenocortical cells. Its exact mechanism is yet to be elucidated; however, it leads to mitochondrial disruption and adrenocortical cell necrosis.153,160 Although the first use in humans was described in 1959, there are no randomized controlled trials that show benefit with mitotane treatment over control. Retrospective studies found that mitotane improves recurrence-free survival in the adjuvant setting. Recurrence rate in patients treated with mitotane was 49% to 55% as compared to 70% to 90% in the control groups.172,173 The effect of mitotane alone on disease-free survival is yet to be established. There is an ongoing prospective randomized study in Europe that will evaluate the efficacy of adjuvant mitotane therapy in patients with low to moderate recurrence risk.
Most patients are started at a dose of 10 g/day and then maintained on 1 to 5 g/day, as the therapeutic window of mitotane is narrow, and toxicity may be evident when doses reach 8 to 10 g/day. Side effects of mitotane are predominantly gastrointestinal, neurologic, and cutaneous. Gastrointestinal side effects occur in approximately 80% of patients and include anorexia, nausea and vomiting, and diarrhea. Neuromuscular toxicity occurs in 4% to 60% of the patients, and the common symptoms are lethargy and somnolence. Vertigo is observed in 15% of the patients. Leucopenia and liver toxicity are less common.174,175 These side effects are often severe enough to require discontinuation of treatment. Tumor regression occurs in approximately 25% of the cases and it is rarely apparent before 6 weeks of treatment.176
Chemotherapy
The role of chemotherapy for the management of ACC is under investigation. As many as 20% of the patients present with unresectable tumors, and the prognosis of such patients is dismal. Mitotane alone or with the combination of chemotherapy remains the standard treatment for unresectable or metastatic disease. A recent randomized control study compared 149 patients treated with mitotane and streptozocin (M-SZ) to 148 patients treated with mitotane, etoposide, doxorubicin, and cisplatin (M-EDP). Patients in the M-EDP group had a significantly higher response rate than those in the M-SZ group (23% vs. 9%) and longer median progression-free survival (5 vs. 2 months). There was no significant difference in the overall survival (15 vs. 12 months).177
Radiotherapy
Radiotherapy to the tumor bed has also been used as adjuvant treatment after ACC resection. A small retrospective study (n = 14 in each arm) identified a local recurrence reduction from 79% to 19% with adjuvant radiotherapy; however, this did not translate to improved survival.178 At present there are no clear recommendations regarding patient selection, indication, or doses.179
Hormonal Control
Patients with heavy tumor burden often suffer from symptoms associated with excessive hormonal secretion. Several different drugs may alleviate these symptoms. As mentioned above, mitotane is toxic to adrenocortical cells. Ketoconazole inhibits steroid synthesis via inhibition of C17-20 desmolase. Ketoconazole toxicity includes many side effects, and because it inhibits cytochrome P450, it may interfere with doxorubicin and etoposide. Metyrapone reduces cortisol and aldosterone production via inhibition of cortical 11β-hydroxylation. Toxicity is more tolerable than ketoconazole, and metyrapone is also a cytochrome P450 inhibitor. Etomidate also inhibits 11β-hydroxylase and cholesterol side-chain cleavage, and thus reduces cortisol and aldosterone synthesis.153
Recurrent or Metastatic Disease
As many as 20% to 50% of the patients present with metastatic disease, and 50% to 90% of these patients will recur.152,160,161 Patients who have resectable recurrent or metastatic disease should be referred to highly experienced centers for excision because limited disease burden can be resected with acceptable morbidity. Patients undergoing complete resection for recurrent or metastatic disease had a significantly improved median survival of 74 months, compared to 16 months for those with incomplete resection.180 Patients with unresectable recurrent or metastatic disease should be treated with mitotane alone or combination chemotherapy even though the effect on overall survival is not proven.153
Prognosis
ACC carries a poor prognosis with a median survival of 38 months and an overall 5-year survival rate of less than 40%.152,153,161 Prognosis mainly depends on the tumor stage at presentation and varies dramatically between studies. Table 71-7 summarizes the median survival and 5-year survival for the different stages.152,153,160 Multiple variables have been shown to carry poorer outcome and include tumor size over 12 cm, age older than 55 years, nodal or distant metastases at time of initial presentation, adjacent organ invasion, poorly differentiated histology, high mitotic rate, atypical mitoses, and tumor necrosis.154,181
Table 71-7
Median and 5-Year Survival ACC Stages
| Median Survival (months) | 5-Year Survival | |
| Stage I | 61–110 | 62–92% |
| Stage II | 23–98 | 58–87% |
| Stage III | 7–17 | 18–40% |
| Stage IV | <12 | 0–14% |
Malignant Pheochromocytoma
Incidence
Pheochromocytomas (PCCs) are rare adrenal tumors that secrete catecholamine and are derived from the chromaffin cells of the adrenal medulla. Paragangliomas are similar tumors that are located at extraadrenal sites along the sympathetic chain (organ of Zuckerkandl, pelvis, abdomen, mediastinum, and neck).182,183 The estimated prevalence of PCC is 1 : 2500 to 1 : 6500 in Western countries with an incidence of 2 to 8 per 1 million adults. The gender distribution of PCC is nearly equal and the peak age of occurrence is the third to fifth decades of life; however, familial PCC occurs at an earlier age.184–184
The rate of malignancy differs between PCC and extraadrenal paragangliomas. Although only 3% to 13% of the PCCs are malignant, the estimated rate for paragangliomas is 15% to 35%.183,185 Some studies suggest that the rate of malignancy increases with older patient age and larger tumors. Specific mutations in the succinyl dehydrogenase gene family (SDHB) have a higher rate of extraadrenal tumors, and the malignancy rate approaches 50%.183,186
Pathogenesis
Most PCCs are sporadic tumors; however, up to 24% develop from hereditary germline mutations. Ten percent of the PCCs are associated with multiple endocrine neoplasia (MEN-2A or -2B) with a mutation in the RET gene (10q11.2), and these are more frequently bilateral with a low (3%) malignancy rate. A mutation in the von Hippel-Lindau gene (3p25-26) is associated with 5% malignancy rate. PCC may be a part of neurofibromatosis (NF) type 1 with a mutation in NF1 gene (17q11.2). The malignancy rate in these patients is 11%. The above-mentioned SDH gene family differs in their malignancy rate. Mutation in the SDHB gene (1q36.13) is associated with a 50% to 66% malignancy rate, and mutation in SDHD gene (11q23) is associated with less than 3% malignancy.183,187,188
Clinical Presentation
More than 50% of malignant PCCs may be nonfunctional, and their diagnosis is made only after resection and pathology evaluation. Functioning malignant PCCs present with the same secretory pattern of benign tumors. The classic triad of symptoms in patients with PCC includes episodes of headache, tachycardia, and sweating. Most of the patients with PCC do not have all three classic symptoms. Paroxysmal hypertension may be observed in nearly 50% of the patients.189,190 Other signs and symptoms include papilledema, weight loss, polyuria, polydipsia, orthostatic hypotension, hyperglycemia, leukocytosis, increased erythrocyte sedimentation rate, constipation, and psychiatric disorders.193–193 Cardiomyopathy and pulmonary edema may be caused by the catecholamine excess.194 Symptoms and signs may be exacerbated or triggered by stress or consumption of tyramine.191,193 As many as 50% of the patients with MEN-2 are asymptomatic at presentation, and a similar finding has been observed with PCC associated with von Hippel-Lindau disease, as 35% of these patients lack symptoms. This low rate of symptomatic patients may be a result of early detection by screening of at-risk patients.195,196
PCC should be suspected in patients who have one or more of the following:
• Self-limited palpitations, tremor, or pallor
• Hypertension resistant to medical treatment or onset before age 20 years, with or without atypical diabetes mellitus
• Hypertensive response to stress (surgery, anesthesia, other procedure)
• Presence of other syndrome-associated disease in patient of family member:
• MEN-2—MTC; primary hyperparathyroidism; mucosal neuromas; marfanoid habitus
• von Hippel-Lindau—angiomatosis; hemangioblastomas; pancreatic cysts; café-au-lait spots; renal cell carcinoma
• NF-1—neurofibromas; café-au-lait macules; freckles; optic glioma; Lisch nodules; osseous lesions
• SDH—renal cell carcinoma; gastrointestinal stromal tumors; testicular seminoma
• Carney syndrome—pulmonary chondromas and gastric stromal tumor
Diagnosis
The diagnosis of PCC is confirmed with the measurement of urinary or plasma fractionated metanephrines and catecholamines that are highly accurate for functional PCCs. The sensitivity and specificity of each test vary between institutions. It has been suggested that plasma-fractionated metanephrines is the superior test.182,193,197 Other biochemical diagnostic tests include clonidine suppression test, plasma catecholamines, chromogranin A, neuropeptide Y, vanillylmandelic acid, and provocative testing; however, because of variable accuracy, these tests have not gained wide acceptance.182,193,197
Imaging studies to localize the tumor should follow biochemical diagnostic confirmation. Approximately 90% of the tumors are PCCs within the adrenal and 95% of the tumors are within the abdomen. Both CT and MRI are highly sensitive (97% to 100%), but the specificity is only 60% to 75%. For malignant PCC, these imaging modalities may identify local invasion or, occasionally, distant metastases.130,184,193,197
123I-metaiodobenzylguanidine (123I-MIBG) is a compound resembling norepinephrine that is taken up by adrenergic tissue. It is highly specific (>96%) and may be used when CT or MRI are negative or to identify extraadrenal metastases when malignancy is suspected.198 OctreoScan and FDG-PET constitute the third-line imaging modalities and are of benefit when malignancy is suspected. PET with 6-[18F]-fluorodopamine ([18F]-DOPA) can detect extraadrenal PCCs and neck paragangliomas; however, this new imaging modality is not yet available at all centers.130,197
Although the risk of malignancy increases with size for all PCCs, size alone is not a reliable predictor of malignancy in local disease. Because pleomorphism, nuclear atypia, abundant mitotic figures, and even capsular invasion may be observed in benign PCCs, the diagnosis of malignancy may be difficult on pathology. Several other methods have been evaluated to differentiate between benign and malignant PCC. Chromogranin A (CgA) has been shown to be significantly increased in malignant tumors. Several genetic markers such as SDHB, MIB-1, nm23-H1, tissue inhibitor of metalloproteinase-4 (TIMP-4), BRMS-1, TXNIP, CRSP-3, and E-cadherin (E-cad) are also dysregulated in malignant PCC. Tissue markers such as cyclooxygenase-2, secretogranin II-derived peptide, heat shock protein 90, hTERT, and Ki-67 have all been evaluated with encouraging results.182,183,199 Lastly, Thompson created a PCC of the adrenal gland scaled score (PASS) based on histology. The following items and their values (in parentheses) were given to each tumor that together, if present, would determine the PASS score: large nests or diffuse >10% growth (2), central or confluent tumor necrosis (2), high cellularity (2), cellular monotony (2), tumor cell spindling (2), greater than 3 of 10 high-power field (HPF) mitotic figures (2), atypical mitotic figures (2), extension into adipose tissue (2), vascular invasion (1), capsular invasion (1), profound nuclear pleomorphism (1), and nuclear hyperchromasia (1). The PASS score was later validated on another cohort and it was identified that all cases with a PASS less than 4 were benign whereas all malignant cases had a PASS greater than 6.200,201 At present, only the presence of distant metastases or invasion into adjacent organs confirms the diagnosis of malignancy.
Treatment
The management approach to malignant PCCs generally follows the algorithm for adrenocortical cancer.
Surgery
Surgery should be offered to all patients with PCC unless the tumor is deemed unresectable. At surgery, every attempt must be made to resect the entire adrenal gland with clear margins. Surgical debulking is considered the mainstay of therapy even for metastatic PCCs with the rational of reducing the circulating catecholamines levels and to increase the uptake of future 131I-MIBG treatment.130,182,185,192
Adrenalectomy should be performed for patients with PCC, and extraadrenal resection should be offered to patients with paragangliomas. The transabdominal laparoscopic approach is the most commonly used, and posterior retroperitoneal surgery may be performed for smaller bilateral tumors. Highly experienced surgeons may perform laparoscopy, and it can be safely completed even for tumors larger than 6 cm.202 A low threshold for open surgery should be practiced for locally advanced tumors and paragangliomas in complex locations.189,203 Recently, minimally invasive robotic adrenalectomy has been used with comparable outcomes to laparoscopy. Surgical survival rates are 98% to 100%; however, the morbidity associated with surgery is more common and adverse perioperative complications occur in 23% to 37% of the patients.185,202,204,205
Resectable recurrent disease or isolated metastases are optimally treated with surgical resection or debulking of tumor burden especially if the patient is symptomatic.130,182
131I-MIBG
MIBG is structurally similar to noradrenaline and is thus concentrated into chromaffin cells. This may be utilized for imaging and the addition of radioactive iodine–labeled MIBG can be an effective therapeutic option. This mode of therapy is completely useless in malignant PCCs that do not take up MIBG. Importantly, external beam radiation abolishes the ability of malignant PCC to uptake MIBG and therefore patients who have been treated with external beam radiation cannot be treated with MIBG. Tumor response following 131I-MIBG treatment is 30% to 67% and biochemical response is 45% to 67%. Symptomatic relief may be achieved in 76% to 89% of the patients. The effect of 131I-MIBG on survival is not clearly established because it is clearly not curative. Its high side-effect profile, mainly myeloablative, makes it less than ideal, but it still remains the first line of adjuvant therapy.182,183,185
Chemotherapy
In 1988,18 patients were treated at the National Institutes of Health (NIH) with combined cyclophosphamide, vincristine, and dacarbazine (CVD therapy). In a 22-year followup, the tumor response rate was 55% and the biochemical response was 72%. Patients who are candidates for CVD therapy should be started on α blockade, as the treatment causes catecholamine release. Because CVD therapy has no proven survival benefit, it is reserved to patients who did not respond to 131I-MIBG or symptomatic patients with unresectable disease.182,183,206
Radiotherapy
Malignant PCC is considered a radioresistant tumor; however, the main benefit of radiation therapy is to control pain and symptoms associated with bone and lymph node metastases. CyberKnife radiation and radiofrequency ablation are the most commonly used methods for bone metastases, but more studies are needed to determine its efficacy in malignant PCC.182,183
Radiolabeled somatostatin analog [DOTA-Tyr(3)]-octreotide (DOTATOC) has shown some response with no major adverse events. It may be used in patients with somatostatin receptor–positive PCC.207
Hormonal Control
Unresectable, recurrent, and metastatic patients may suffer severely from the excessive catecholamine secretion. These patients have several treatment options. Phenoxybenzamine is an irreversible, long-acting, nonselective, α-adrenergic receptor blocker that is mostly used for preoperative preparation of these patients, but which can also be used for symptom control. Nicardipine is a long-acting calcium channel blocker that blocks the transport of norepinephrine-mediated calcium into vascular smooth muscle. Metyrosine inhibits the synthesis of catecholamines by blocking the enzyme tyrosine hydroxylase. The use of one of these drugs, or a combination, may contribute to symptom control in patients with functional, unresectable, malignant PCC.130,185,208
Future Drugs
The discovery of molecular pathways and genetic mutations that characterize malignant PCC has resulted in several phase I and phase II clinical trials. Multikinase inhibitors, tyrosine kinase receptor inhibitors, mammalian target of rapamycin (mTOR) inhibitors, HIF inhibitors, antiangiogenic agents, and HER-2/neu inhibitors are among the agents that are currently under investigation. Clearly, novel treatments are needed for the treatment of malignant PCC and paraganglioma.183
Prognosis
As metastases or local recurrences after resection of “benign” disease may present even 20 years after resection, long-term followup is indicated in all patients with PCC. Plasma metanephrines should be obtained at regular intervals for life. In high-risk patients, these intervals should initially be shorter.182
It is difficult to predict the prognosis of patients with malignant PCC. Five-year survival rates are only 20% to 50%. Survival rates decrease with increasing tumor size; however, there is no difference in overall survival between malignant PCC and paraganglionas.182,184,200,209