html xmlns=”http://www.w3.org/1999/xhtml”>
52
Cancer Immunotherapy with Vaccines and Checkpoint Blockade
Therapeutic vaccination for cancer continues to be a major approach to the overall immunotherapy of cancer. Historically, interest in cancer immunology stemmed from the perceived potential activity of the immune system as a weapon against cancer cells. In fact, the term magic bullet, commonly used to describe many visions of cancer therapy, was coined by Paul Ehrlich in the late 1800s in reference to antibodies targeting both microbes and tumors. Central to the concept of successful cancer immunotherapy are the dual tenets that tumor cells express an antigenic profile distinct from their normal cellular counterparts and that the immune system is capable of recognizing these antigenic differences. Support for this notion originally came from animal models of carcinogen-induced cancer in which it was demonstrated that a significant number of experimentally induced tumors could be rejected on transplantation into syngeneic immunocompetent animals. 1–4 Extensive studies by Prehn on the phenomenon of tumor rejection suggested that the most potent tumor rejection antigens were unique to the individual tumor. 5
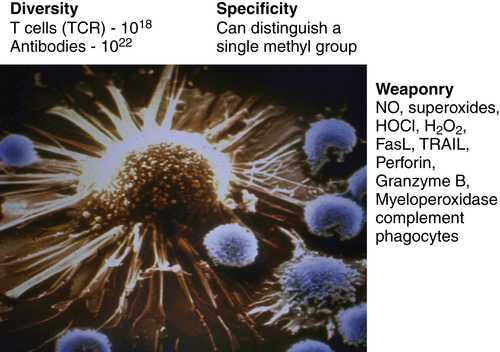
Figure 52-1 The immune system as the perfect anti-cancer weapon Shown in the figure is a single cytotoxic T lymphocyte specific for a tumor antigen expressed by the tumor cell it is about to kill. Although other lymphocytes contact the tumor cell, if they do not express a T-cell receptor specific for a peptide derived from a protein in the tumor cell and presented on MHC class I, they will ignore the tumor cell. Fundamentally, the immune system is endowed with all of the assets desired to specifically eliminate cancer cells while having a minimal effect on normal cells. The tremendous diversity of T-cell receptors and antibodies affords the adaptive immune system both specificity and adaptability. The MHC transport system that carries peptides from degraded proteins to the cell surface allows T cells to recognize protein antigens expressed anywhere in the cell. In addition, the immune system can produce more than 20 cytocidal molecules with diverse mechanisms of killing once activated.
Since the original reports of Jenner over two centuries ago, prophylactic vaccination against infectious diseases has been one of the most influential medical interventions. Cancer vaccination, an immunotherapy approach applied to patients with established cancer, has tremendous potential based on the ability of both T cells and antibodies to specifically recognize cancer antigens and kill cancer cells expressing these antigens. However, at the time of this writing, only one human cancer vaccine has received U.S. Food and Drug Administration (FDA) approval, despite multiple Phase III clinical trials over the past two decades. Despite the clinical failures of cancer vaccines to date, continuing molecular definition of tumor-specific and tumor-selective antigens, new vaccine platforms that selectively target and activate dendritic cells, and preclinical results with combinations of vaccination together with other immune modulators have generated renewed optimism that cancer vaccination will ultimately take its place among the pantheon of cancer therapies.
As cancer genetics and genomics have exploded over the past decade, it is now quite clear that altered genetic and epigenetic features of tumor cells indeed result in a distinct tumor antigen profile. Overexpression of “oncogenic” growth factor receptor tyrosine kinases such as HER2/Neu and epidermal growth factor receptor (EGFR) via epigenetic mechanisms has provided clinically relevant targets for one arm of the immune system—antibodies. 6,7 Indeed, monoclonal antibodies are the fastest growing single class of cancer therapeutics based on successful new FDA approvals. In striking contrast, cellular immunotherapy of cancer has been quite disappointing in establishing therapeutic success in clinical trials to date. Emerging insights about the nature of the interaction between the cancer and the immune system have led us to understand why cell-based cancer immunotherapy approaches such as therapeutic vaccines have been less potent against established cancer than originally imagined. In general, we have learned that tumors employ mechanisms of tolerance induction to turn off T cells specific for tumor-associated antigens. Oncogenic pathways in tumors result in the elaboration of factors that organize the tumor microenvironment in ways that are quite hostile to antitumor immune responses.
Not only is the cancer capable of inducing potent tolerance among tumor-specific T cells, we now know that there are distinct forms of inflammatory and immune responses that are procarcinogenic. Thus, two frontiers in cancer immunology are the elucidation of how the tumor organizes its immune microenvironment and the nature of immune responses that are anticarcinogenic versus procarcinogenic. As the receptors, ligands, and signaling pathways that mediate immune tolerance and immune-induced procarcinogenic events are elucidated, these factors and pathways can be selectively inhibited by both antibodies and drugs in a way to shift the balance to antitumor immune responses. This chapter outlines the major features of tumor–immune system interactions and set the stage for molecularly based approaches to manipulate immune responses for successful cancer therapy. The clinical results over the past few years, particularly with checkpoint blockade, validate clinically the tremendous potential of the immune system to destroy cancer cells (Figure 52-1 ).
Indeed, adoptive T-cell transfer trials using ex vivo expanded tumor-specific T cells have demonstrated clear proof of the principle that activated tumor-specific T cells can induce tumor regressions, even in patients with bulky metastatic cancer. Because adoptive T-cell transfer is expensive, labor intensive, and extremely difficult to standardize, it is an immunotherapy approach that is difficult to broadly export. Most cancer immunotherapy efforts, including those that involve vaccination, seek to activate and expand tumor-specific T cells in vivo via various manipulations involving standardized reagents. The major barriers to be overcome are induction of tolerance among tumor-specific T cells and a tumor microenvironment that has developed to resist infiltration and attack by activated tumor-specific T cells. Although these two barriers represent significant hurdles to successful cancer immunotherapy, the elucidation of specific molecular mechanisms for tolerance induction as well as immune inhibition within the tumor microenvironment have led to the generation of specific combinatorial approaches to cancer therapy.
Cancer Antigens—the Difference between Tumor and Self
Tumors reflect the biologic and antigenic characteristics of their tissue of origin but also differ fundamentally from their normal-cell counterparts in both antigenic composition and biologic behavior. Both these elements of cancer provide potential tumor-selective or tumor-specific antigens as potential targets for cancer vaccination specifically and antitumor immune responses in general. Genetic instability, a basic hallmark of cancer, is a primary generator of tumor-specific antigens. The most common genetic alteration in cancer is mutation arising from defects in DNA damage repair systems of the tumor cell. 8–15 Recent estimates from genome-wide sequencing efforts suggest that every tumor contains a few hundred mutations in coding regions. 16 In addition, deletions, amplifications, and chromosomal rearrangements can result in new genetic sequences resulting from the juxtaposition of coding sequences that are not normally contiguous in untransformed cells. The vast majority of these mutations occur in intracellular proteins, and thus the “neoantigens” they encode would not be readily targeted by antibodies. However, the major histocompatibility complex (MHC) presentation system for T-cell recognition makes peptides derived from all cellular proteins available on the cell surface as peptide MHC complexes capable of being recognized by T cells. Based on analysis of sequence motifs, it is estimated that roughly one third of the mutations identified from genome sequencing of 22 breast and colon cancers 16 were capable of binding to common human leukocyte antigen (HLA) alleles based on analysis of sequence motifs.
In accordance with the original findings of Prehn, 5 the vast majority of tumor-specific antigens derived from mutation as a consequence of genetic instability are unique to individual tumors. The consequence of this fact is that antigen-specific immunotherapies targeted at most truly tumor-specific antigens would by necessity be patient specific. However, there are a growing number of examples of tumor-specific mutations that are shared. The three best studied examples are the Kras codon 12 G→A (found in roughly 40% of colon cancers and more than 75% of pancreas cancers), the BrafV599E (found in roughly 70% of melanomas), and the P53 codon 249 G→T mutation (found in about 50% of hepatocellular carcinomas). 17–20 As with nonshared mutations, these common tumor-specific mutations all occur in intracellular proteins and therefore require T-cell recognition of MHC-presented peptides for immune recognition. Indeed, both the Kras codon 12 G→A and the BrafV599E mutations result in “neopeptides” capable of being recognized by HLA class 1– and class II–restricted T cells. 21–24
The other major difference between tumor cells and their normal counterparts derives from epigenetics. 25 Global alterations in DNA methylation as well as chromatin structure in tumor cells result in dramatic shifts in gene expression. All tumors overexpress hundreds of genes relative to their normal counterparts and, in many cases, turn on genes that are normally completely silent in their normal cellular counterparts. Overexpressed genes in tumor cells represent the most commonly targeted tumor antigens by both antibodies and cellular immunotherapies. This is because, in contrast to most antigens derived from mutation, overexpressed genes are shared among many tumors of a given tissue origin or sometimes multiple tumor types. For example, mesothelin, which is targeted by T cells from vaccinated pancreatic cancer patients, 26 is highly expressed in virtually all pancreatic cancers, mesotheliomas, and most ovarian cancers. 27,28 Although mesothelin is expressed at low to moderate levels in the pleural mesothelium, it is not expressed at all in normal pancreatic or ovarian ductal epithelial cells.
The most dramatic examples of tumor-selective expression of epigenetically altered genes are the so-called cancer-testis antigens. 29 These genes appear to be highly restricted in their expression in the adult. Many are expressed selectively in the testes of males and are not expressed at all in females. Expression in the testis appears to be restricted to germ cells, and in fact some of these genes appear to encode proteins associated with meiosis. 30–32 Cancer-testis antigens therefore represent examples of widely shared tumor-selective antigens whose expression is highly restricted to tumors. Many cancer-testis antigens have been shown to be recognized by T cells from nonvaccinated and vaccinated cancer patients. 29 From the standpoint of immunotherapeutic targeting, a major drawback of the cancer-testis antigens is that none appears to be necessary for the tumors’ growth or survival. Therefore, their expression appears to be purely the consequence of epigenetic instability rather than selection, and antigen-negative variants are easily selected out in the face of immunotherapeutic targeting.
A final category of tumor antigen that has received much attention encompasses tissue-specific antigens shared by tumors of similar histologic origin. Interest in this class of antigen as a tumor-selective antigen arose when melanoma-reactive T cells derived from melanoma patients were found to recognize tyrosinase, a melanocyte-specific protein required for melanin synthesis. 33,34 In fact, the most commonly generated melanoma-reactive T cells from melanoma patients recognize melanocyte antigens. 35,36 Although one cannot formally call tissue-specific antigens tumor-specific, they are nonetheless potentially viable targets for therapeutic T-cell responses when the tissue is dispensable (i.e., prostate cancer or melanoma).
From the standpoint of T-cell targeting, tumor antigens upregulated as a consequence of epigenetic alterations represent “self-antigens” and are therefore likely to induce some level of immune tolerance. However, it is now clear that the stringencies of immune tolerance against different self-antigens differ according to tissue distribution and normal expression level within normal cells. The mesothelin antigen described earlier is an example. In a recent set of clinical pancreatic cancer vaccine studies, mesothelin-specific T-cell responses were induced by vaccination with genetically modified pancreatic tumor cell vaccines, and induction of mesothelin-specific T cells correlated with ultimate disease outcome. 37 Given that the immune system is capable of differential responsiveness determined by antigen levels, it is quite possible to imagine generating tumor-selective immune responses against antigens whose expression level in the tumor is significantly greater within normal cells in the tumor-bearing host. In addition, upregulated antigens that provide physiologically relevant growth or survival advantages to the tumor are preferred targets for any form of therapy because they are not so readily selected out.
Beyond the antigenic differences between tumor cells and normal cells, there are important immunologic consequences to the distinct biological behavior of tumor cells relative to their normal counterparts. Whereas uncontrolled growth is certainly a common biological feature of all tumors, the major pathophysiologic characteristics of malignant cancer responsible for morbidity and mortality are their ability to invade through natural tissue barriers and ultimately to metastasize. Both of these characteristics, never observed in nontransformed cells, are associated with dramatic disruption and remodeling of tissue architecture. Indeed, the tumor microenvironment is quite distinct from the microenvironment of normal tissue counterparts. One of the important consequences of tissue disruption, even when caused by noninfectious mechanisms, is the elaboration of pro-inflammatory signals. These signals, generally in the form of cytokines and chemokines, are potentially capable of naturally initiating innate and adaptive immune responses. Indeed, the level of leukocyte infiltration into the microenvironment of tumors tends to be significantly greater than the leukocyte component of their normal-tissue counterparts. Cancers are therefore constantly confronted with inflammatory responses as they invade tissues and metastasize. In some circumstances these inflammatory and immune responses can potentially eliminate a tumor—so-called immune surveillance. However, as discussed later, oncogenic pathways in the tumor appear to organize the immunologic component of the microenvironment in a fashion that not only protects the tumor from antitumor immune responses but can qualitatively shift immune responses to those that actually support and promote tumor growth. It is these elements of the cancer–immune system interaction that will be the central targets of future immunotherapeutic strategies.