Cancer Immunology
• Cancer is characterized by genetic and epigenetic instability leading to multiple unique and sometimes common mutations and “ectopic” overexpression of many genes normally not expressed in the tissue of origin. These alterations provide antigens that the adaptive immune system can recognize to distinguish the cancer cell from normal cells. Thus the immune system of a patient with cancer has the potential to selectively recognize his or her cancer.
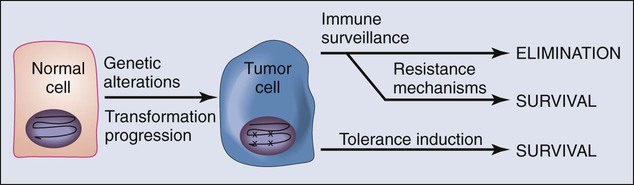
• As cancers develop, they may respond to “immune pressure” in a number of ways. They can eliminate antigens through further mutation or deletion, a process termed “editing.” Although immune editing of tumors is well characterized in mouse cancer models, it has yet to be established in human cancers. Cancers can also resist immune elimination by inducing tolerance among tumor-specific T cells, which they achieve by co-opting natural self-tolerance mechanisms.
• A major focus of recent studies in cancer immunology is the immune microenvironment. Multiple cells and molecular interactions in the tumor microenvironment inhibit antitumor immune responses. These cells include regulatory T cells and myeloid-derived suppressor cells, which accumulate in tumors. Both cell types suppress cytotoxic T-cell responses that threaten tumors with destruction.
• In addition to suppressive cells, tumor cells and myeloid cells in the tumor microenvironment express ligands for inhibitory receptors expressed by effector T cells, termed “immune checkpoints.” The best-studied checkpoint expressed in the tumor microenvironment is the PD1 pathway. Many tumors express high levels of the PD1 ligands PDL1 and PDL2, whereas tumor infiltrating lymphocytes express high levels of PD1. These inhibitory ligand/receptor interactions are prime targets for antibody blockade.
• Certain chronic immune responses can promote cancer development. In particular, T-cell responses characterized by interleukin 17 (IL-17)–producing cells, termed T helper 17 (Th17) cells, are procarcinogenic. Th17 differentiation and IL-17 production depend on STAT3 signaling. The major antitumor immune response is a Th1 response, characterized by production of interferon-γ and driven by STAT1 and STAT4 activation. Th1 and Th17 development are mutually antagonistic. These contrasting immune responses explain how certain types of immune responses can inhibit cancer, whereas other types of immune responses can promote cancer development.
• The definition of signaling pathways and molecules and cells that dampen antitumor immunity is leading directly to therapies for cancer. Anti-CTLA4 blocking antibodies have shown potent therapeutic effects in melanoma and anti-PD1 antibodies have shown potent responses in melanoma, renal cancer, and non–small cell lung cancer.
Overview
Support for this notion originally came from animal models of carcinogen-induced cancer in which it was demonstrated that a significant number of experimentally induced tumors could be rejected upon transplantation into syngeneic immunocompetent animals.1–4 Extensive studies by Prehn and Main5 on the phenomenon of tumor rejection suggested that the most potent tumor rejection antigens were unique to the individual tumor. The recent wave of cancer genomics and epigenomics illustrates the unique antigenicity of cancer cells. In addition to mutations, deletions, amplifications, and rearrangements—the ultimate neoantigens—aberrant expression of membrane molecules can provide a therapeutic window for tumor-targeted antibodies. For example, overexpression of membrane molecules including “oncogenic” growth factor receptor tyrosine kinases such as HER2/neu and epidermal growth factor receptor (EGFR) via epigenetic mechanisms has provided clinically relevant targets for one arm of the immune system—antibodies.6,7 Indeed, tumor-targeted monoclonal antibodies are an ever-growing class of cancer therapeutic agents obtaining approval from the U.S. Food and Drug Administration (FDA).
The Antigenic Profile That Distinguished Tumors From Normal Tissues
As previously described, tumors differ fundamentally from their normal cell counterparts in both antigenic composition and biological behavior. Genetic instability, a basic hallmark of cancer, is a primary generator of tumor-specific antigens. Data accrued for more than a thousand human cancers to date demonstrate unequivocally that altered genetic and epigenetic features of tumor cells indeed result in a distinct tumor antigen profile. Cancers express between 50 and 1000 missense mutations in coding regions, roughly 20% of which can potentially create neoantigenic peptides presented by at least one of the individual’s human leukocyte antigen (HLA) alleles and thus recognized by T cells.8–16 Additionally, deletions, amplifications, and chromosomal rearrangements can result in new genetic sequences resulting from juxtaposition of coding sequences not normally contiguous in untransformed cells. The vast majority of these mutations occur in intracellular proteins, and thus the “neoantigens” they encode would not be readily targeted by antibodies. However, the major histocompatibility complex (MHC) presentation system for T-cell recognition makes peptides derived from all cellular proteins available on the cell surface as peptide MHC complexes capable of being recognized by T cells.
In accordance with the original findings of Prehn and Main,5 the vast majority of tumor-specific antigens derived from mutation as a consequence of genetic instability are unique to individual tumors. The consequence of this fact is that antigen-specific immunotherapies targeted at most truly tumor-specific antigens would by necessity be patient specific. However, there are a growing number of examples of tumor-specific mutations that are shared. The three best studied examples are the Kras codon 12 G->A (found in roughly 40% of colon cancers and >75% of pancreas cancers), the BrafV600E (found in roughly 50% of melanomas), and the p53 codon 249 G->T mutation (found in ~50% of hepatocellular carcinomas).17–20 As with nonshared mutations, these common tumor-specific mutations all occur in intracellular proteins and therefore require T-cell recognition of MHC-presented peptides for immune recognition. Indeed, both the Kras codon 12 G->A and the BrafV600E mutations result in “neopeptides” capable of being recognized by HLA class I– and class II–restricted T cells.21–24
The other major difference between tumor cells and their normal counterparts derives from epigenetics.25 Global alterations in DNA methylation and chromatin structure in tumor cells results in dramatic shifts in gene expression. All tumors overexpress hundreds of genes relative to their normal counterparts, and in many cases, they turn on genes that are normally completely silent in their normal cellular counterparts. Overexpressed genes in tumor cells represent the most commonly targeted tumor antigens by both antibodies and cellular immunotherapies because, in contrast to most antigens derived from mutation, overexpressed genes are shared among many tumors of a given tissue origin or sometimes multiple tumor types. For example, mesothelin, which is targeted by T cells from vaccinated patients with pancreatic cancer,26 is highly expressed in virtually all pancreatic cancers, mesotheliomas, and most ovarian cancers.27,28 Although mesothelin is expressed at low to moderate levels in the pleural mesothelium, it is not expressed at all in normal pancreatic or ovarian ductal epithelial cells.
The most dramatic examples of tumor-selective expression of epigenetically altered genes are the so-called cancer-testis antigens.29 These genes appear to be highly restricted in their expression in the adult. Typically, they are expressed almost exclusively in germ cells of the testis and ovaries. Expression in the testis appears to be restricted to germ cells, and in fact, some of these genes appear to encode proteins associated with meiosis.32–32 Cancer-testis antigens therefore represent examples of widely shared tumor selective antigens whose expression is highly restricted to tumors. Many cancer-testis antigens have been shown to be recognized by T cells from nonvaccinated and vaccinated patients with cancer.29 From the standpoint of immunotherapeutic targeting, a major drawback of the cancer-testis antigens is that none appears to be necessary for the tumor’s growth or survival. Therefore their expression appears to be purely the consequence of epigenetic instability rather than selection and antigen-negative variants are easily selected out in the face of immunotherapeutic targeting.
A final category of tumor antigen that has received much attention encompasses tissue-specific antigens shared by tumors of similar histologic origin. Interest in this class of antigen as a tumor-selective antigen arose when melanoma-reactive T cells derived from patients with melanoma were found to recognize tyrosinase, a melanocyte-specific protein required for melanin synthesis.33,34 In fact, commonly generated melanoma-reactive T cells from patients with melanoma recognize melanocyte antigens.35 Although one cannot formally call tissue-specific antigens tumor specific, they are nonetheless potentially viable targets for therapeutic T-cell responses when the tissue is dispensable (i.e., prostate cancer or melanoma). Because melanoma is the easiest cancer from which to grow tumor-specific T cells, Anichini and colleagues36 were able to demonstrate the existence in the same patient of melanoma-specific T cells recognizing tumor-specific (presumably from mutations) and shared antigens.
From the standpoint of T-cell targeting, tumor antigens upregulated as a consequence of epigenetic alterations represent “self-antigens” and are therefore likely to induce some level of immune tolerance. However, it is now clear that the stringencies of immune tolerance against different self-antigens differ according to tissue distribution and normal expression level within normal cells. The mesothelin antigen previously described is such an example. In a recent set of clinical pancreatic cancer vaccine studies, mesothelin-specific T-cell responses were induced by vaccination with genetically modified pancreatic tumor cell vaccines, and induction of mesothelin-specific T cells correlated with ultimate disease outcome.37 Given that the immune system is capable of differential responsiveness determined by antigen levels, it is quite possible to imagine generating tumor-selective immune responses against antigens whose expression level in the tumor is significantly greater within normal cells in the tumor-bearing host. Additionally, upregulated antigens that provide physiologically relevant growth or survival advantages to the tumor are preferred targets for any form of therapy because they are not so readily selected out.
Evidence Pro and Con for Immune Surveillance of Cancer
The fundamental tenet of the immune surveillance hypothesis, which was first conceived nearly a half century ago,38,39 is that a fundamental role of the immune system is to survey the body for tumors as it does for infection with pathogens, recognizing and eliminating them based on their expression of tumor-associated antigens. In animal models, carcinogen-induced tumors can be divided into those that grow progressively (termed “progressor tumors”) and those that are rejected after an initial period of growth (termed “regressor tumors”).2,3 The phenomenon of regressor tumors was thought to represent an example of the ongoing process of immune surveillance of cancer. A corollary to the original immune surveillance hypothesis is that progressor tumors in animals (presumed to represent clinically progressing cancers in humans) fail to be eliminated because they develop active mechanisms of either immune escape or resistance (Fig. 6-1).
A fundamental prediction of the immune surveillance hypothesis is that immunodeficient individuals would display a dramatic increase in tumor incidence. After an extensive analysis of spontaneous tumor formation in immunodeficient nude mice, which have atrophic thymi and therefore significantly reduced numbers of T cells and T-cell dependent immune responses, no increased incidence of tumors was observed.40–44 The findings of these studies were taken to be a major blow to the immune surveillance hypothesis. However, a caveat to the interpretation of these results is that nude mice still produce diminished numbers of T cells via thymus-independent pathways and therefore can mediate some degree of T-cell–dependent immunity. In addition, nude mice frequently display compensatory increases in innate immunity which, as will be discussed later, may represent a potent form of antitumor immunity and could contribute to immune surveillance of cancer.
Epidemiological studies of patients with heritable immunodeficiencies revealed a significantly increased risk of certain cancers that are distinct from the epithelial cancers commonly observed in normal immunocompetent adults.47–47 Many of these cancers are also observed in patients receiving a transplant who are undergoing chronic pharmacologic immune suppression, as well as in patients with human immunodeficiency virus/acquired immunodeficiency syndrome whose immune system is depressed. The most common cancers in these individuals include lymphoblastic lymphomas and Kaposi sarcoma; however, certain epithelial cancers, such as stomach cancer, are also observed at increased frequency. A common theme for most cancers observed in immunodeficient individuals is a microbial stimulus. Most lymphoplastic lymphomas are Epstein-Barr virus–associated lymphomas,48 and Kaposi sarcoma is a result of infection with Kaposi sarcoma herpesvirus.49 Other virus-associated cancers such as cervical cancer (from human papillomavirus)50,51 are also observed at increased frequency. It is now appreciated that stomach cancer is associated with ulcer disease related to infection with the bacterium Helicobacter pylori.52 From these studies, the notion emerged that immune surveillance indeed protects persons against certain pathogen-associated cancers by either preventing infection or altering chronic infection by viruses and other microbes that can eventually induce cancer. The findings of these studies were taken to represent evidence that the common nonpathogen-associated cancers most commonly seen in adults in developed countries (e.g., prostate cancer, colon cancer, and lung cancer) are not subject to immune surveillance.
Two caveats to this interpretation should be noted, however. First, detailed epidemiological analyses of immunodeficient individuals were performed at a time when these patients rarely lived beyond their 20s and 30s, when cancer incidence normally increases most significantly. It is therefore possible that a more subtle cumulative increased incidence of common nonpathogen-associated cancers would have been observed had these persons lived further into adulthood. Indeed, more recent analyses definitively demonstrate an increase incidence of some nonpathogen-associated cancers in immunodeficient individuals, particularly melanoma.53 In addition to epidemiological data, dramatic anecdotal examples are difficult to ignore. It has been reported that in patients who received kidneys from a cadaver donor that had been in complete remission from a melanoma prior to organ donation, metastatic melanoma of donor origin developed rapidly after the transplant.56–56 These results indicate that at least for some nonpathogen-associated tumors, the immune system can play a significant role in maintaining the micrometastatic disease in a dormant state. Whether this principle applies to nonpathogen-associated human tumors besides melanoma remains to be demonstrated.
Several recent studies reevaluating tumor immune surveillance in genetically manipulated mice have revealed clear-cut evidence that various components of the immune system can at least modify, if not eliminate, both carcinogen-induced and spontaneously arising cancers. In a series of studies, Schreiber and colleagues59–59 reexamined cancer incidence in mice rendered immunodeficient via genetic knockout of either the RAG2 gene (deficient in both B and T cells), the γ-interferon receptor gene (IFNGR1), the STAT1 gene, or the type 1 interferon receptor gene (IFNAR1). When these knockout mice were either treated with carcinogens or crossed onto a cancer-prone p53 knockout background, the incidence of cancers was modestly but significantly increased relative to nonimmunodeficient counterparts when observed over an extended period (longer than 1 year). Transplantation studies demonstrated that direct γ-interferon (γ-IFN) insensitivity by the developing tumors played a significant role in the defect in immune surveillance. Interestingly, in contrast to IFNGR1 knockout mice, the mechanism for increased tumor incidence in tumors in IFNAR1 knockout mice did not involve sensitivity by the tumor to type-1 IFNs but rather reflected the role of the type-1 IFNs in induction of innate and adaptive immunity. Even animals not crossed onto a cancer-prone genetic background or treated with carcinogens developed an increased incidence of invasive adenocarcinomas when observed over their entire life span. Furthermore, a broader spectrum of tumors developed in γ-IFN × RAG2 double-knockout mice than in RAG2 knockout mice. All of the tumors that arise in these genetically manipulated immunodeficient animals behave as regressor tumors when transplanted into immunocompetent animals. These findings indeed suggest that tumors that arise in immunodeficient animals would have been eliminated had they arisen in immunocompetent animals. The relatively subtle effects on tumorigenesis, requiring observation over the life span of the animal, suggests that the original concept of immune surveillance of tumors arising on a daily basis is in fact not correct. Instead, it is clear that the presence of a competent immune system “sculpts” the tumor through a process that has been termed “immunoediting.”
The immunoediting hypothesis has been somewhat controversial, with differing outcomes in different animal models. One of the caveats in the interpretation of these studies comes from the work of Enzler and Dranoff,60 who studied mechanisms of increased tumorigenesis in granulocyte-macrophage colony-stimulating factor (GM-CSF) × γ-IFN double-knockout mice. Although they observed an increase in gastrointestinal and pulmonary tumors, they noted that such animals harbored infection with a particular bacterium not normally observed in immunocompetent animals. Maintenance of these double-knockout mice on antibiotics essentially eliminated the increased rate of tumor formation. Thus it is possible that some of the increased tumor rates in genetically immunodeficient animals could be related to unappreciated chronic infections that develop in these animals, which are not housed under germ-free conditions. Nonetheless, although the classic concepts of immune surveillance of cancer remain unsupported by experimental evidence, studies on tumorigenesis in genetically manipulated immunodeficient mice suggest that developing tumors must actively adapt themselves to their immune microenvironment to exist within the context of a competent immune system.
One of the approaches to test the immune surveillance and immunoediting of endogenously arising tumors has been to combine genetically engineered autochthonous tumor models with T-cell receptor transgenic models expressing defined marked T cells specific for a tumor antigen (either the transgenic oncogenic driver protein in the tumors or an antigen co-expressed with the oncogenic driver). In these models, tumor growth can be monitored in immunodeficient versus immunocompetent mice, as well as expression of the cognate tumor antigen recognized by the transgenic T-cell receptor. In such a tumor model driven by Kras and p53 loss, tumors emerging in immunocompetent mice either lost antigen or MHC presentation capacity, unlike tumors emerging in immunodeficient mice.61. In contrast, in a mouse model of spontaneous random oncogene activation, antigen-specific tolerance was generated in immunocompetent mice without evidence for antigen loss.62 In some models, an intermediate result has been observed. For example, under some circumstances, endogenous immune responses can establish an equilibrium state with the tumor in which the tumor is prevented from outgrowth in immunocompetent mice but is not completely eliminated.63 Ultimately, given that experimental outcomes are different in different models (i.e., tolerance vs. surveillance vs. editing vs. equilibrium), it will be important to ascertain which mechanisms are operative in particular human cancers.
Innate Mechanisms of Tumor Immune Surveillance
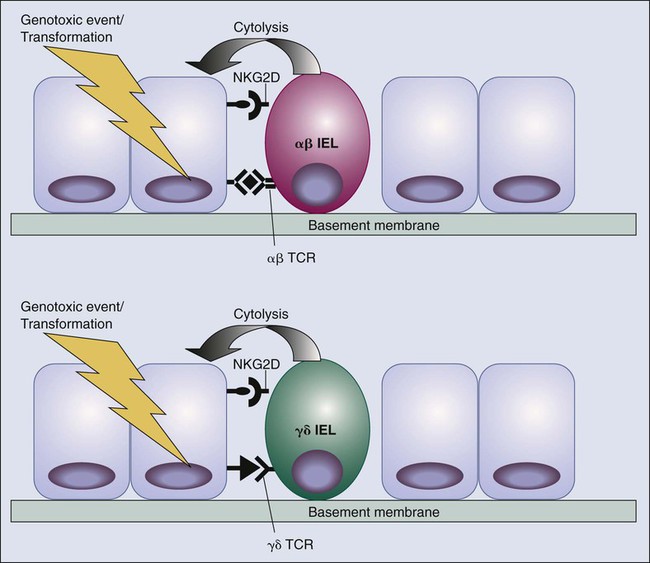
Although much emphasis has been placed on the role of adaptive immunity, particularly of conventional T cells, in immune surveillance of cancer, a confluence of more recent findings points to innate immunity and epithelial immunity in the immunologic sensing of carcinogenic events in the skin, gut, and possibly other sites. Much of the evidence focuses on the NKG2D receptor. NKG2D was originally defined as an activating natural killer (NK) receptor.66–66 Most NK receptors appear to be inhibitory when engaged; this inhibition is often associated with immunoreceptor tyrosine kinase–based inhibitory motif (ITIM) domains in the cytoplasmic tails. ITIMs provide docking sites for phosphatases that oppose the activity of tyrosine kinases involved in lymphocyte activation. NK activation status is a balance between engagement of activating and inhibitory receptors. NKG2D, the best-studied activating receptor on NK cells, is somewhat unusual in that it does not contain an immunoreceptor tyrosine kinase–based activating motif (ITAM) and is associated with an adaptor molecule, DAP 10, that contains neither conventional ITIMs nor ITAMs.67 Instead, DAP 10 contains a KYXXM motif that appears to bind to phosphatidylinositol (PI)-3 kinase upon phosphorylation of the tyrosine in this motif. NKG2D is expressed on all NK cells, as well as on some αβ and γδ T cells. Beyond NK cells, NKG2D is expressed at high levels on a number of subsets of intraepithelial lymphocytes (IELs). IELs represent a distinct population of lymphocytes residing in epithelial tissues that display features of both adaptive and innate immune responses.68–72 They are thought to represent a major first line of defense against pathogens attempting to invade across epithelial linings exposed to the environment (i.e., skin, gut, and respiratory tract). Fifty percent of the IELs of the gut express the γδ TCR (normally expressed by <3% of circulating T cells), whereas the other 50% express the common αβ TCR. γδ TCR-expressing IELs in different compartments express a very restricted repertoire and are thought to recognize certain types of microbial antigens or potentially self-antigens associated with stress or inflammatory responses to microbial infection. Even the αβ TCR-expressing IELs have an extremely restricted TCR repertoire similar to invariant NK T cells. A significant subset of gut IELs express TCR that utilize a particular Vα and Vβ and are thought to recognize a limited subset of microbial or self nonpeptide antigens presented by nonclassical class I MHC molecules. Thus NKG2D expression marks diverse subsets of lymphocytes that, although expressing different families of recognition receptors, act as components of innate immunity in that they recognize a stereotypical set of antigens associated with infection or stress (see later discussion).
The first evidence that the NKG2D receptor might play a role in tumor immune surveillance came from the finding that normal colonic epithelium and a significant proportion of tumors could express the two defined human ligands for NKG2D: MICA and MICB. MICA and MICB, which represent nonclassical MHC class I–type molecules whose structure demonstrates no antigen-binding grove characteristic of most MHC molecules, are stress-induced proteins whose genes contain stress response elements in their promoters.73,74 Gasser and colleagues75 have demonstrated that upregulation of MICA/B is induced through the ATM/ATR/Chk1 pathway of DNA damage recognition. An analysis in human cancer suggested a correlation between expression of MICA/B and infiltration of certain subsets of γδ T cells that express NKG2D. Initially it was proposed that MICA and MICB were direct ligands for specific γδ receptors themselves as well as NKG2D,76,77 but this idea is controversial. MICA and MICB do not have any murine orthologs, but murine NKG2D does bind to products of the retinoic acid–inducible gene family, RAE1αε, as well as the product of the H60 gene. ULBP3 is an additional NKG2D ligand to be described78,79 and appears to represent the human homologue to the RAE1 molecules in mice. These NKG2D ligands appear to be involved in immune recognition and possibly tumor surveillance of mice.82–82 Recognition and killing of murine skin keratinocytes or intestinal epithelial cells by γδ IEL require expression of NKG2D ligands and are blocked by anti-NKG2D antibodies. Transfection of murine tumors with genes encoding NKG2D ligands renders them susceptible to NKG2D-dependent killing by NK cells. Emerging data on NKG2D function on IELs together with the potentially stress-induced nature of its ligands suggests that the IEL system of immune surveillance may indeed be relevant to carcinogenesis, as well as infectious challenges.62 The major initiating event of carcinogenesis in the skin—ultraviolet light—is a potent source of DNA damage, which, as previously mentioned, has been shown to induce NKG2D ligands via the ataxia telangiectasia (ATM) pathway. Thus, in addition to endogenous killers of genome damaged cells, such as p53, IELs and NK cells may represent an extrinsic sensor of DNA damage and genotoxic stress via recognition of cells that have upregulated NKG2D ligands (Fig. 6-2).
As with the case of classic immune surveillance mediated by classical T cells, the emergence of a clinically evident cancer implies that the tumor has developed a mechanism to circumvent or evade any innate immune surveillance systems. In the case of the NKG2D system, Groh and colleagues83 have provided suggestive evidence that tumors can shed MICA/B in a soluble form as a means of evading NKG2D-dependent recognition. They demonstrated that certain tumors are associated with high levels of shed MICA/B and that soluble MICA/B binds to and downmodulates NKG2D on NK cells, thereby acting as an antagonist to NKG2D activation via cell surface–bound MICA/B. Although this mechanism remains to be proven as a true evasion system for NKG2D-dependent tumor recognition, it points out the diversity of mechanisms that tumors use to evade immune recognition. It also points out straightforward approaches to block these evasion systems. If indeed soluble MICA/B represents a mechanism for tumor immune evasion of innate immune recognition, antibodies that would bind to and clear soluble MICA/B but not block the interaction between cell membrane MICA/B and NKG2D on NK cells could potentially restore the capacity of NK cells to recognize MICA/B-expressing tumors. Further evidence for the importance of soluble MICA/B as a mechanism for escape from innate tumor surveillance comes from the finding that antitumor clinical responses to vaccination of patients with melanoma can be associated with the generation of humoral immune responses that “neutralize” soluble MICA/B.84
Immune Tolerance and Immune Evasion—the Immune Hallmarks of Cancer
The first direct evidence for induction of T-cell tolerance by tumors was provided by Bogen and colleagues,85,86 who examined the response of TCR transgenic T cells specific for the idiotypic immunoglobulin expressed by a murine myeloma tumor. They first demonstrated induction of central tolerance to the myeloma protein followed by peripheral tolerance. With use of influenza hemagglutinin (HA) as a model tumor antigen, it was demonstrated that adoptively transferred HA-specific TCR transgenic T cells were rapidly rendered anergic by HA-expressing lymphomas and HA-expressing renal carcinomas.87,88 Tolerance induction has been demonstrated in both the CD4 and CD8 compartment. In general, initial activation of tumor-specific T cells is commonly observed; however, the activated state of T cells is typically not sustained, with failure of tumor elimination as a frequent consequence. Tolerance induction among tumor antigen–specific T cells is an active process involving direct antigen recognition, although in some murine systems, tolerance to tumors appears to be associated with failure of antigen recognition by T cells; that is, the immune system “ignores” the tumor.89,90 Beyond studies on transplantable tumors, more recent analyses of immune responses to tumor antigens in tumor transgenic mice that have developed spontaneous cancer have further emphasized the capacity of spontaneously arising tumors to induce tolerance among antigen-specific T lymphocytes. In a model of prostate tumorigenesis, Drake and colleagues91 evaluated CD4 responses to HA and double transgenic animals expressing HA and SV40 T antigen under control of the prostate-specific probasin promoter. Development and progression of prostate tumors did not result in enhanced activation of adoptively transferred HA-specific T cells. Tolerance to HA as a normal prostate antigen occurred largely through ignorance because no evidence for antigen recognition by HA-specific T cells was found. However, increased recognition was observed upon either androgen ablation (which causes massive apoptosis within the prostate) or development of prostate cancer. Nonetheless, enhanced antigen recognition was not accompanied by activation of effector functions such as γ-IFN production. Analysis of the consequences of transformation in additional tumor transgenic mouse systems has also been performed. Willimsky and Blankenstein62 evaluated T-cell responses and rejection in a model of sporadic induction of tumors associated with expression of a tumor-specific antigen only at the time of transformation. They found that preimmunization of mice against the tumor-associated antigen prevented the development of tumors. However, nonimmunized mice developed spontaneous tumors without any significant evidence of natural immune surveillance in the absence of preimmunization. They further demonstrated that an initial antigen-dependent activation of tumor-specific T cells could be observed at the time of spontaneous tumor induction but that this recognition ultimately resulted in an anergic form of T-cell tolerance similar to that observed by Drake and colleagues91 in the prostate system.
The capacity of spontaneously arising tumors to tolerize T cells has not been uniformly observed. A contrasting result by Nguyen and colleagues92 was observed when lymphocytic choriomeningitis virus (LCMV) GP33-specific TCR transgenic CD8 T cells were adoptively transferred into double transgenic mice expressing both SV40 T antigen and LCMV GP33 under control of the rat insulin promoter. Pancreatic islet cell tumors that express GP33 develop in these animals. Nguyen and colleagues92 found that as tumors progressed in the mice, enhanced T-cell activation occurred. It was demonstrated through bone marrow chimera experiments that CD8 T-cell activation occurs exclusively via cross presentation in the draining lymph nodes. Despite the activation of tumor-specific T cells, the tumors grew progressively, indicating that the degree of immune activation induced by tumor growth was insufficient to ultimately eliminate the tumors. These results suggest that developing tumors can induce immune responses but may titrate their level of immune activation to one that ultimately does not “keep up” with tumor progression. Such a circumstance is one that is highly susceptible to the immune editing concept put forward by Schreiber and colleagues57 in which the tumor edits itself genetically to maintain a sufficient level of resistance to induced immune responses. In the case of the LCMV GP33 T-antigen transgenic mice, because neither anergic nor deletional tolerance was observed, animals treated with the dendritic cell stimulatory anti-CD40 antibody demonstrated significant slowing of tumor growth. Thus it may be possible under some circumstances to shift the balance between tumor immune evasion and tumor immune recognition by agents that affect the overall activation state of either antigen-presenting cells or T cells (see later discussion).
It has been more difficult to obtain definitive evidence that human cancers tolerize tumor-specific T cells because humans cannot be manipulated the way mice are. However, the T cells that are grown out of patients with cancer tend to be either of low affinity for their cognate antigen or recognize antigens that bind poorly to their presenting HLA (human MHC) molecule, resulting in inefficient recognition by T cells. Recently, the first crystal structure of the TCR-peptide-MHC trimolecular complex has been solved for an MHC class II–restricted human tumor antigen.93 Interestingly, the orientation of the TCR, which is of low affinity for the peptide-MHC complex, is distinct from trimolecular complexes for viral (foreign) antigens and is partially similar to trimolecular complexes for a self-antigen. Thus there may be fundamental structural features of tumor antigen recognition that lie between those of foreign antigen and self-antigen recognition.
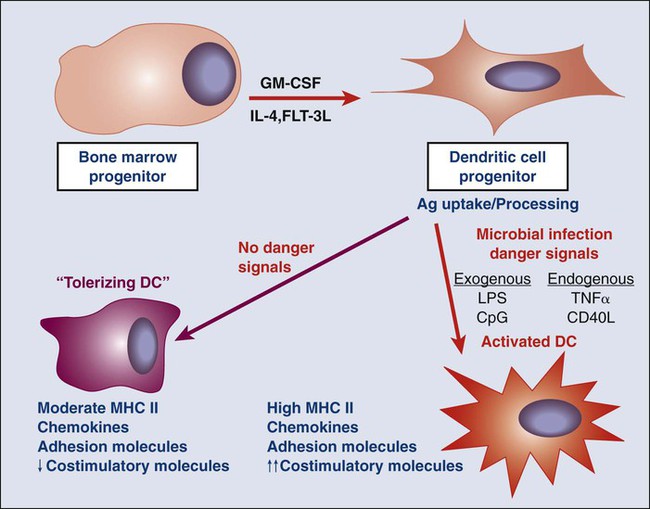
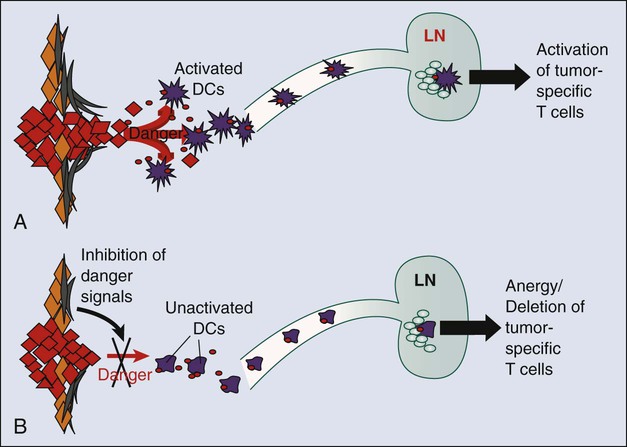
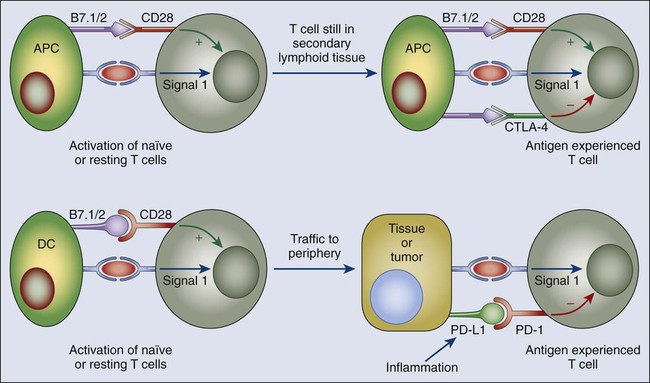
As will be discussed later, one of the features of the tumor microenvironment that is likely central to the capability of tumors to tolerize tumor-specific T cells is the immature or inactive state of tumor-infiltrating dendritic cells (DCs). DCs are the major antigen-presenting cells (APCs) that present peptides to T cells to initiate adaptive immune responses. In the context of infection, microbial ligands or endogenous “danger signals” associated with tissue destruction activated DCs to a state whereby they present antigens to T cells together with co-stimulatory signals that induce T-cell activation and development of effecter function. However, in the absence of microbial products or danger signals, DCs remain in an immature state in which they can still present antigens to T cells but without co-stimulatory signals. These immature DCs function as “toleragenic” DCs, inducing a state of antigen-specific T-cell unresponsiveness (termed “anergy”; Fig. 6-3). It is thought that steady-state presentation of self-antigens by immature DCs is an important mechanism of peripheral self-tolerance. Thus if a tumor is able to produce factors that inhibit local DCs from becoming activated in response to the endogenous danger signals associated with tissue invasion, it could shift tumor-specific T cells from a state of activation (Fig. 6-4, A) to one of tumor-specific tolerance (Fig. 6-4, B).
Immunologic Characteristics of the Tumor Microenvironment
Ultimate understanding of the relationship between the tumor and the host immune system requires elucidation of local cross talk at the level of the tumor microenvironment. As mentioned at the outset, the hematopoietic/immune system is a major component of the tumor microenvironment. The systemic tolerance to tumor antigens begins with events that occur in this microenvironment. Beyond mechanisms that skew tumor-specific T cells toward immune tolerance, the tumor microenvironment is replete with mechanisms that dampen antitumor immune responses locally (Fig. 6-5), which represents an important barrier to successful immunotherapy even when activated effector responses can be generated with vaccines. As the specific cells and molecules within the tumor microenvironment that mediate this hostile immune environment are elucidated, inhibitors are being developed and tested to use as adjuncts to vaccination that will allow activated immune cells to function more effectively within the tumor microenvironment.
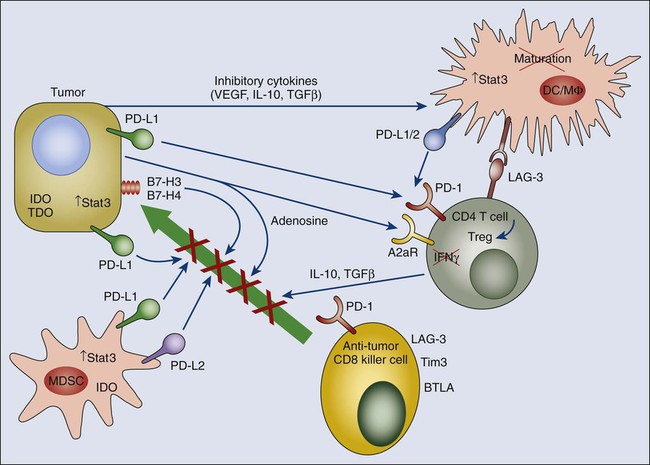
Regulatory T Cells and Cancer
During the past 10 years, regulatory T (Treg) cells have emerged as a central player in maintenance of the tolerant state, as well as general downregulation of immune responses to pathogens.94,95 Not surprisingly, they appear to play a role in tolerance to tumor antigens, as well as the resistance of tumors to immune-mediated elimination.96,97 In contrast to the ephemeral CD8 suppressor cells of the 1970s that failed to withstand experimental scrutiny, the more recently defined CD4+ Treg cells are characterized by expression of a central master regulatory transcription factor—Foxp3—whose role in the gene expression programs of regulatory T cells is being actively studied.98 Although CD4+ Treg cells selectively (but not specifically) express a number cell membrane molecules, including CD25, neuropilin, glucocorticoid-induced tumor necrosis factor receptor, and lymphocyte activation gene (LAG3),95,99–101 their overall genetic program and inhibitory capacity is absolutely dependent on sustained expression of Foxp3.102,103 Mechanisms of immune suppression by Treg cells vary and include production of inhibitory cytokines such as IL-10 and transforming growth factor (TGF)–β. In addition, a recently described IL-12 family “hybrid” cytokine, IL-35, consisting of the alpha subunit of IL-12 and the beta subunit of IL-27, has been discovered to be made by Treg and to mediate suppression of certain autoimmune responses.104 However, its role in Treg-mediated cancer immunity has not yet been documented. In keeping with the emerging appreciation that tumors are by nature highly tolerogenic, numerous murine studies have demonstrated that Treg cells expand in animals with cancer and significantly limit the potency of antitumor immune responses, either natural or vaccine induced. For example, in a study by Sutmuller et al.,105 a combination of GM-CSF–transduced tumor vaccine plus anti-CTLA4 antibodies was much more effective at eliminating established tumors when animals were treated with anti–IL-2 receptor alpha antibodies to eliminate CD4+ regulatory T cells. It is now appreciated that treatment with low-dose cytoxan is a relatively simple and reasonably effective way to temporarily eliminate cycling Treg cells.106–109 Treg depletion appears to be a major mechanism by which pretreatment with low-dose cytoxan prior to vaccination can significantly enhance the capacity of vaccines to break tolerance. As new cell membrane molecules that define Treg cells are identified, the capacity to block Treg cell activity with antibodies to these molecules presents new opportunities for immunotherapeutic strategies to break tolerance to tumor antigens.
Numerous studies have shown that Foxp3+ CD4 T cells represent a much higher proportion of tumor-infiltrating lymphocytes in human tumors relative to what is found in peripheral blood (about 5%). In some cases, these cells have been shown to suppress in vitro T-cell responses when mixed into the culture, but this finding does not prove their in vivo role in suppressing antitumor immunity. The best evidence for the in vivo role of Treg cells in suppressing antitumor immunity was an extensive study correlating Treg cell number in resected ovarian cancers with ultimate clinical outcome. Patients with greater numbers of Treg cells (defined by Foxp3 expression and high CD25 expression) had a worse outcome.96 Analysis of correlations between Treg infiltration and clinical outcome was also performed in a cohort of patients with colorectal cancer. In that cohort, Treg gene signatures did not correlate with clinical outcome. However, actual Treg cell numbers were not evaluated in that study. A number of clinical trials have been performed using a toxin-conjugated IL-2 reagent that would bind CD25 and selectively kill Treg cells. Although the results of these trials have been variable, a recent report of objective responses in patients with melanoma110 suggests that this approach has some clinical promise.
Immature Myeloid Cells/Myeloid-Derived Suppressor Cells in Cancer
Immature myeloid cells (iMCs),111,112 often termed “myeloid-derived suppressor cells” (MDSCs),113–116 represent a cadre of myeloid cell types, somewhat overlapping with tumor-associated macrophages, that share the common feature of inhibiting both the priming and effector function of tumor-reactive T cells. It is still not clear whether these myeloid cell types represent distinct lineages or different states of the same general immune inhibitory cell subset. In mice, iMCs and MDSCs are characterized by co-expression of CD11b (considered a macrophage marker) and Gr1 (considered a granulocyte marker) while expressing low or no MHC class II or the CD86 co-stimulatory molecule. In humans they are defined as CD33+ but lacking markers of mature macrophages, DCs, or granulocytes and are DR−. A number of molecular species produced by tumors tend to drive iMC/MDSC accumulation. These species include IL-6, CSF1, IL-10, and gangliosides. IL-6 and IL-10 are potent inducers of STAT3 signaling, which has been shown to be important in iMC/MDSC persistence and activity.
Immune Inhibitory Molecules Expressed in the Tumor Microenvironment
Myeloid cells of multiple type in the tumor microenvironment express a number of enzymes whose metabolic activity ultimately results in inhibition of T-cell responses within the tumor microenvironment. Metabolic products include the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS). Nitrous oxide production by iMCs/MDSCs as a result of arginase and inducible nitric oxide synthase activity has been well documented, and inhibition of this pathway with a number of drugs can mitigate the inhibitory effects of iMCs/MDSCs. ROS, including H2O2, have been reported to block T-cell function associated with the downmodulation of the ζ chain of the TCR signaling complex,117 a phenomenon well recognized in T cells from patients with cancer and associated with generalized T-cell unresponsiveness.
Another mediator of T-cell unresponsiveness associated with cancer is the production of indolamine-2,3 dioxygenase (IDO).118 IDO appears to be produced by DCs either within tumors or in tumor-draining lymph nodes. Interestingly, IDO in DCs has been reported to be induced via backward signaling by B7-1/2 upon ligation with CTLA4.119,120 The major IDO-producing DC subset is either a plasmacytoid DC or a plasmacytoid DC–related cell that is B220+121; however, IDO has been subsequently shown to be expressed by multiple cell types in the immune microenvironment, including tumor cells themselves.122 IDO appears to inhibit T-cell responses through catabolism of tryptophan. Activated T cells are highly dependent on tryptophan and are therefore sensitive to tryptophan depletion. Thus Munn and colleagues118 have proposed a bystander mechanism whereby DCs in the local environment deplete tryptophan via IDO upregulation, thereby inducing metabolic apoptosis in locally activated T cells. IDO has two isoforms, IDO1 and IDO2, which are encoded by distinct genes. The role of IDO2 in human cancer is still unclear; a major IDO2 polymorphism in humans encodes an inactive enzyme. A second tryptophan-metabolizing enzyme is tryptophan dioxygenase, which is upregulated commonly in human cancers and may inhibit antitumor responses within the microenvironment similarly to IDO.123 Finally, there has been greater appreciation that a major product of IDO and tryptophan dioxygenase metabolism of tryptophan—kynurenine—has potent effects on T-cell differentiation. Under some circumstances, kynurenine can promote Treg development,124 and under other circumstances, it can promote development of a class of a subset of T cells termed T-helper 17 (Th17),125 known for its production of IL-17 and for its procarcinogenic properties (see later discussion). Ultimately, the relative role of tryptophan depletion versus kynurenine production in modulating the immune microenvironment remains to be determined.
TGF-β—A Major Inhibitory Cytokine in the Tumor Microenvironment
A major inhibitory cytokine produced by many cell types that has been implicated in blunting antitumor immune responses is TGF-β, which is produced by a variety of cell types, including tumor cells, and which has pleiotropic physiological effects. For most normal epithelial cells, TGF-β is a potent inhibitor of cell proliferation, causing cell cycle arrest in the G1 stage.126 In many cancer cells, however, mutations in the TGF-β pathway confer resistance to cell cycle inhibition, allowing uncontrolled proliferation. Additionally, in cancer cells, the production of TGF-β is increased and may contribute to invasion by promoting the activity of matrix metalloproteinases. In vivo, TGF-β directly stimulates angiogenesis; this stimulation can be blocked by anti–TGF-β antibodies.127 A bimodal role of TGF-β in cancer has been verified in a transgenic animal model using keratinocyte-targeted overexpression.128 Initially, these animals are resistant to the development of early-stage or benign skin tumors. However, once tumors form, they progress rapidly to a more aggressive spindle-cell phenotype. Although this clear bimodal pattern of activity is more difficult to identify in a clinical setting, it should be noted that elevated serum TGF-β levels are associated with poor prognosis in a number of malignancies, including prostate cancer,129 lung cancer,130 gastric cancer,131 and bladder cancer.132
From an immunologic perspective, TGF-β possesses broadly immunosuppressive properties, and widespread inflammatory pathology and corresponding accelerated mortality develop in TGF-β knockout mice.133 Interestingly, a majority of these effects seem to be T-cell–mediated, because targeted disruption of T-cell TGF-β signaling also results in a similar autoimmune phenotype.134 Recent experiments by Chen et al.135 rather convincingly demonstrated a role for TGF-β in Treg-mediated suppression of CD8 T-cell antitumor responses. In these experiments, adoptive transfer of CD4+ CD25+ regulatory T cells inhibited an antitumor CD8 T-cell effector response, and this inhibition was ameliorated when the CD8 T cells came from animals with a dominant negative TGF-β1 receptor.
Co-Inhibitory Ligands and Receptors That Downmodulate Tumor Immunity
Without question, the major molecules to be successfully targeted in clinical cancer immunotherapy are the growing class of ligand-receptor pairs, commonly referred to as immune checkpoints. In considering the mechanism(s) of action of inhibitors of various checkpoints, it is critical to appreciate the diversity of immune functions that they regulate. For example, the two immune checkpoint receptors that have been most actively studied in the context of clinical cancer immunotherapy, CTLA4 (CD152) and PD1 (CD279), regulate immune responses at very different levels and by very different mechanisms (Fig. 6-6). The clinical activity of blocking antibodies for each of these receptors implies that antitumor immunity can be enhanced at multiple levels and that combinatorial strategies can be intelligently designed, guided by mechanistic considerations and preclinical models. This section will focus particular attention on the CTLA4 and PD1 pathways, because they were the two checkpoints whose inhibition has revolutionized clinical cancer immunotherapy. However, it is important to emphasize that multiple additional checkpoints represent promising targets for therapeutic blockade based on preclinical experiments with inhibitors. These checkpoints are under active development.
The CTLA4 Checkpoint—A Global Regulator of T-Cell Activation
CTLA4, the first immune checkpoint receptor to be clinically targeted, is expressed exclusively on T cells, where it primarily regulates the amplitude of the early stages of T-cell activation. CTLA4 knockout mice die within 3 weeks from immune destruction of multiple organs, which attests to the critical role of CTLA4 as an inhibitory regulator of T-cell–dependent immune responses. Primarily, CTLA4 counteracts the activity of the T-cell co-stimulatory receptor CD28.138–138 CD28 does not affect T-cell activation unless the TCR is first engaged by cognate antigen. Once antigen recognition occurs, CD28 signaling strongly amplifies the TCR signal to activate T cells. CD28 and CTLA4 share identical ligands: CD80 (B7.1) and CD86 (B7.2).139–143 Because CTLA4 has a much higher overall affinity for both ligands, its expression on the surface of T cells dampens the activation of T cells by both outcompeting CD28 in binding CD80 and CD86, as well as actively delivering inhibitory signals to the T cell.144–149 The specific signaling pathways by which CTLA4 blocks T-cell activation are still under investigation, although a number of studies suggest that activation of the phosphatases SHP2 and protein phosphatase 2 (PP2A) are important in counteracting kinase signals induced by TCR and CD28.137 However, CTLA4 also confers “signaling independent” T-cell inhibition through sequestration of CD80 and CD86 from CD28 engagement, as well as active removal from the APC surface.150 The central role of CTLA4 in maintaining T-cell activation in check is dramatically demonstrated by the systemic immune hyperactivation phenotype of CTLA4 knockout mice.151,152
Even though CTLA4 is expressed by activated CD8 killer T cells, the major physiological role of CTLA4 appears to be through distinct effects on the two major subsets of CD4 T cells—downmodulation of helper T-cell activity and enhancement of regulatory T-cell suppressive activity.136,153,154 CTLA4 blockade results in a broad enhancement of immune responses dependent on helper T cells and, conversely, CTLA4 engagement on Treg cells enhances their suppressive function. CTLA4 is a target gene of the transcription factor Foxp3,98,155 the expression of which determines the Treg lineage,156,157 and Treg cells therefore express CTLA4 constitutively. Although the mechanism by which CTLA4 enhances the inhibitory function of Treg cells is not known, Treg-specific CTLA4 knockout or blockade significantly inhibits their ability to regulate both autoimmunity and antitumor immunity.153,154 Thus in considering the mechanism of action for CTLA4 blockade, both enhancement of effector CD4 T-cell activity and inhibition of Treg-dependent immune suppression are likely important factors.
Biology of the PD1 Checkpoint—A Pathway That Functions Within the Tumor Microenvironment
In contrast to CTLA4, the major role of PD1 is to limit the activity of T cells in the peripheral tissues at the time of an inflammatory response to infection and to limit autoimmunity (Fig. 6-6).158–164 This role translates to a major immune resistance mechanism within the tumor microenvironment.167–167 PD1 expression is induced when T cells become activated.159 When engaged by one of its ligands, PD1 inhibits kinases involved in T-cell activation via the phosphatase SHP2,158 although additional signaling pathways are also likely induced, and because PD1 engagement inhibits the TCR stop signal, this pathway could modify the duration of T-cell/APC or T-cell/target cell contact.168 Similar to CTLA4, PD1 is highly expressed on Treg cells, where it may enhance their proliferation in the presence of ligand.169 Because many tumors are highly infiltrated with Treg cells that likely further suppress effector responses, PD1 pathway blockade may also enhance antitumor responses by diminishing the number and/or suppressive activity of intratumoral Treg cells.
The two ligands for PD1 are B7-H1/PD-L1 (CD274) and B7-DC/PD-L2 (CD273).158,170–172 These B7 family members share 37% sequence homology and arise via gene duplication, positioning them within 100 kB of each other in the genome.172 Recently, an unexpected molecular interaction between PD-L1 and CD80 was discovered,173 whereby CD80 expressed on T cells (and possibly APCs) can potentially behave as a receptor rather than a ligand, delivering inhibitory signals when engaged by B7-H1174,175; the relevance of this interaction in tumor immune resistance has not yet been determined. Finally, genetic evidence from PD1-deficient T cells suggests that both B7-H1/PD-L1 and B7-DC/PD-L2 may bind to a co-stimulatory receptor expressed on T cells174; these complex binding interactions are reminiscent of the CD80/CD86 ligand pair, which binds the co-stimulatory CD28 expressed on resting T cells and the inhibitory CTLA4 expressed on activated T cells, although, as previously stated, PD1 predominantly regulates effector T-cell activity within tissue and tumors, whereas CTLA4 predominantly regulates T-cell activation (Fig. 6-6). Understanding the role of these various interactions in given cancer settings is highly relevant for selection of both antibodies and recombinant ligands for use in the clinic.
PD1 is more broadly expressed than is CTLA4; it is induced on other activated non–T lymphocyte subsets, including B cells and NK cells,176,177 limiting their lytic activity. Thus although PD1 blockade is typically viewed as enhancing the activity of effector T cells in tissues and in the tumor microenvironment, it likely also enhances NK activity in tumors and tissues and may also enhance antibody production either indirectly or through direct effects on PD1+ B cells.178
In addition, chronic antigen exposure, such as occurs with chronic viral infection and cancer, can lead to high levels of persistent PD1 expression, which induces a state of exhaustion or anergy among cognate antigen-specific T cells. This state, which has been demonstrated in multiple murine and human chronic viral infections, appears to be partially reversible by PD1 pathway blockade.179 Finally, although the PD1 pathway plays its major role in limiting immune effector responses in tissues (and tumors), it can also shift the balance from T-cell activation to tolerance at early stages in T-cell responses to antigen within secondary lymphoid tissues (i.e., at a similar point as CTLA4). Taken together, these findings imply a complex set of mechanisms of action for PD1 pathway blockade.
PD1 is expressed on a large proportion of tumor-infiltrating lymphocytes (TILs) from many different tumor types.180,181 Some of the enhanced PD1 expression among CD4 TILs reflects a generally high level of PD1 on Treg cells, which, as previously noted, can represent a large fraction of intratumoral CD4 T cells. Increased PD1 expression on CD8 TILs may either reflect an anergic/exhausted state, as has been suggested by decreased cytokine production by PD1+ versus PD1(−) TILs from melanomas.180
Just as PD1 is highly expressed on TILs from many cancers, the PD1 ligands are commonly upregulated on many different human tumors.166,182 On solid tumors, the major PD1 ligand to be expressed is B7-H1/PD-L1. Forced expression of B7-H1/PD-L1 on murine tumors inhibits local antitumor T-cell responses.166,183 Indeed, this combination of findings provides the basis for PD1 pathway blockade to enhance antitumor effector function in the tumor microenvironment. As immunohistochemistry techniques and flow cytometry analysis of surface expression have been implemented, it has become clear that the selective upregulation of PD1 ligands in various human tumor types is heterogeneous at a number of levels.167 Expression patterns of PD1 ligands may very well be critical in choosing suitability for therapeutic blockade of this pathway because its primary role in cancer is thought to be immune inhibition within the tumor microenvironment and PD1 only inhibits lymphocyte function when it is engaged by cognate ligand.
Initially, the majority of melanoma, ovarian, and lung cancer samples were reported to have high expression of B7-H1/PD-L1,166,183,184 and subsequently, many other human cancers were reported to upregulate B7-H1/PD-L1. In addition to tumor cells, B7-H1/PD-L1 is commonly expressed on myeloid cells in the tumor microenvironment.187–187 An initial report in renal cancer demonstrated that expression of B7-H1/PD-L1 on either tumor cells or infiltrating leukocytes in primary tumors predicted a worse prognosis—that is, decreased overall survival relative to B7-H1/PD-L1(-) tumors.188 Since that report, analyses of various tumors have suggested that B7-H1/PD-L1 status can either correlate with poor prognosis or better prognosis or show no correlation with prognosis.167,189–193 Variability in immunohistochemistry technique, cancer type, stage of cancer analyzed (most analyses are of primary, not metastatic lesions), and treatment history in the analyzed cohort all likely contribute to the wide range of reported outcomes.
Although most of the analyses of PD1 ligand expression has focused on B7-H1/PD-L1, B7-DC/PD-L2 has also been reported to be upregulated on a number of tumors. B7-DC/PD-L2 is highly upregulated on certain B-cell lymphomas such as primary mediastinal, follicular cell B-cell lymphoma and Hodgkin disease.191 Upregulation in these lymphomas is commonly associated with gene amplification or rearrangement to the CIITA locus, which is highly transcriptionally active in B-cell lymphomas.194
Given the heterogeneity of expression and potential relevance as a biomarker for blockade of the PD1 pathway, it is important to understand the signals that induce expression of PD1 ligands on tumor cells and also hematopoietic cells within the tumor microenvironment. Two general mechanisms for regulation of B7-H1/PD-L1 have emerged: innate and adaptive (Fig. 6-7). For some tumors such as glioblastoma, it has been demonstrated that PD-L1/B7-H1 is driven by constitutive oncogenic signaling pathways in the tumor cell. Expression on glioblastomas is enhanced upon deletion or silencing of phosphatase and tensin homologue, implicating the PI3K-AKT pathway.195 Similarly, constitutive anaplastic lymphoma kinase signaling, observed in certain lymphomas and occasionally in lung cancer, has been reported to drive PD-L1/B7-H1 expression via STAT3 signaling.196
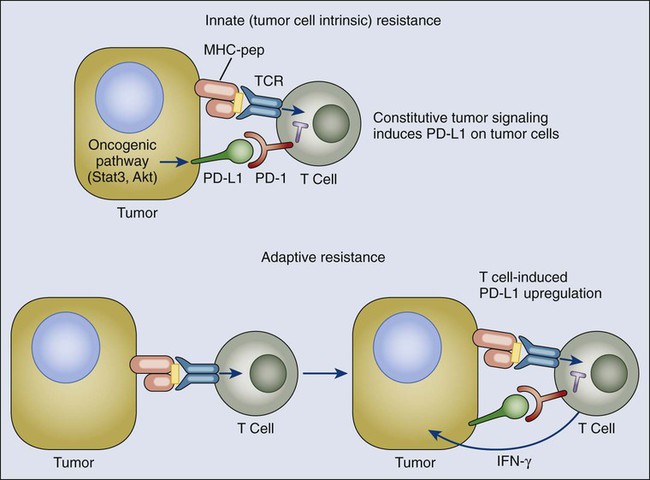
The alternative mechanism for PD-L1/B7-H1 upregulation on tumors that has emerged from both clinical and preclinical studies reflects their adaptation to endogenous tumor-specific immune responses—a process termed “adaptive resistance.”167 In adaptive resistance, the tumor utilizes the natural physiology of the PD1 ligand induction for tissue protection in the face of an immune response to infection in order to protect itself from an antitumor response. Expression of PD-L1/B7-H1 as an adaptive response to endogenous antitumor immunity can occur because it is induced on most cancers in response to IFNs—predominantly γ-IFN—similarly to epithelial and stromal cells in normal tissues.199–199 This mechanism represents an alternative to the conventional drug resistance mechanisms that involve mutation of drug targets. It also contrasts with mechanisms of viral immune escape that involve mutation of immunodominant epitopes. The mechanism of adaptive resistance intrinsically implies that immune surveillance does exist even in some advanced cancers but that the tumor ultimately resists immune elimination by upregulating ligands for inhibitory receptors on tumor-specific lymphocytes that turn off antitumor responses within the tumor microenvironment.
A number of preclinical and clinical studies support the adaptive resistance hypothesis. Gajewski and colleagues200 have demonstrated that melanomas can be roughly divided into “inflammatory” and “noninflammatory” categories defined by expression of multiple inflammatory genes, including those involved in the interferon pathway. A recent study in persons with melanoma demonstrated a very high correlation between cell-surface PD-L1/B7-H1 expression on tumor cells and both lymphocytic infiltration and intratumoral γ-IFN expression. This correlation was not only seen among tumors but within individual B7-H1/PD-L1+ tumors at the regional level, in which regions of lymphocyte infiltration were exactly the regions where B7-H1/PD-L1 was expressed on both tumor cells and infiltrating leukocytes.167
Additional Checkpoints Participate in Tumor Immune Resistance and Tolerance
Successful clinical outcomes of CTLA4 and PD1 pathway targeting have garnered great interest in a number of additional checkpoints (Fig. 6-8). Basic immunologic studies have demonstrated that a number of checkpoint receptors are expressed coordinately under circumstances of tolerance to self-antigens and chronic infections, as well as in inflammatory settings. In addition to defined lymphocyte inhibitory receptors, a number of B7-family inhibitory ligands—in particular B7-H3 (CD276) and B7-H4—do not yet have defined receptors, but murine knockout experiments support an inhibitory role for both these molecules.201 In addition, they are upregulated on tumor cells or tumor-infiltrating cells.202 B7-H3 appears to be upregulated on endothelial cells of the tumor vasculature, and B7-H4 has been reported to be expressed on tumor-associated macrophages.201 Preclinical tumor models have been used to demonstrate that blockade of many of these individual immune checkpoint ligands or receptors can enhance antitumor immunity and dual blockade of coordinately expressed receptors can produce additive or synergistic antitumor activity. Inhibitors for a number of these immune checkpoint targets are either entering the clinic or are under active development. Those described later are targets with currently available blocking antibodies or small molecule inhibitors but do not represent a comprehensive list.
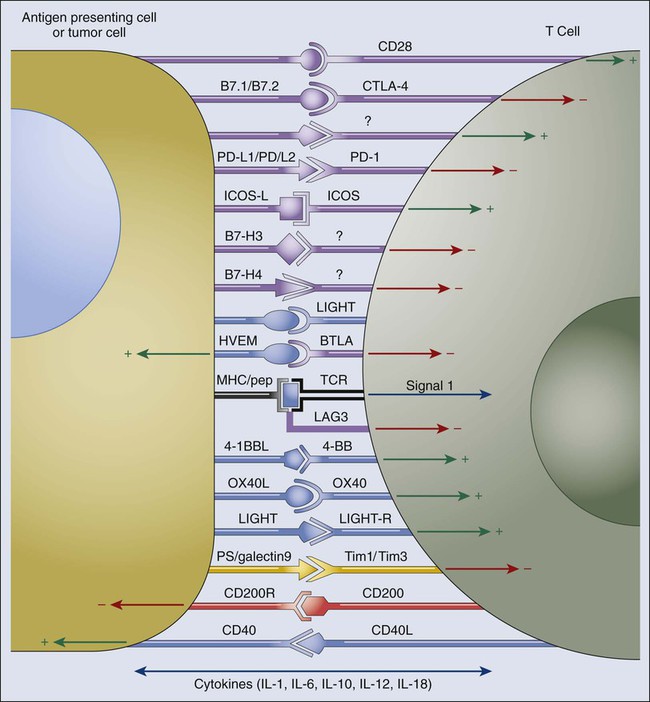
LAG3 (CD223), 2B4 (CD244), B and T lymphocyte attenuator (BTLA and CD272), Tim3, A2aR, and the family of killer inhibitory receptors have each been associated with inhibition of lymphocyte activity and in some cases induction of lymphocyte anergy. Antibody targeting of these receptors, either alone or in combination with a second immune checkpoint blocker, has been shown to enhance antitumor immunity in animal models of cancer. Because many tumors express multiple inhibitory ligands and TIL express multiple inhibitory receptors, many opportunities exist to enhance antitumor immunity via dual or triple blockade of immune checkpoints. Although human blocking antibodies specific for a number of these “second-generation” inhibitory receptors are under development, none has entered the clinic at this time. Most of these receptors are induced upon T-cell activation, in keeping with the biological theme that they play roles in feedback inhibition of T-cell responses when their cognate ligands are present. In addition to providing inhibitory signals to activated effector T cells, some of these receptors such as LAG3 are highly expressed on Treg cells, where they are important to amplify their inhibitory activity.100 This finding implies that as with CTLA4 and PD1, these receptors play a dual role in ultimately inhibiting effector immune responses and blocking antibodies and therefore have multiple potential mechanisms of action.
LAG3 was cloned more than 20 years ago as a CD4 homologue,203 but its function in the immune checkpoint was only defined in 2005 when it was shown to play a role in enhancing Treg function.100,204 LAG3 also inhibits CD8 effector function independently of its role on Treg cells.205 The only known ligand for LAG3 is MHCII, which is upregulated on some epithelial cancers (generally in response to IFN-γ) but is also expressed on tumor-infiltrating macrophages and dendritic cells. The role of the LAG3/MHCII interaction in LAG3-mediated inhibition of T-cell responses is unclear because anti-LAG3 antibodies that do not block the LAG3/MHCII interaction nonetheless enhance T-cell proliferation and effector function in vitro and in vivo. The MHC class II interaction of LAG3 may be most important for its role in enhancing Treg function. LAG3 is one of a number of immune checkpoint receptors coordinately upregulated on both Treg cells and anergic T cells, and simultaneous blockade can result in enhanced reversal of this anergic state relative to blockade of either receptor. In particular, PD1 and LAG3 are commonly co-expressed on anergic or exhausted T cells.206,207 Dual blockade of LAG3 and PD1 provide synergy in reversing anergy among tumor-specific CD8 T cells, as well as virus-specific CD8 T cells in the setting of chronic infection. Dramatic evidence of the effects of coordinate T-cell inhibition by PD1 and LAG3 comes from PD1/LAG3 double knockout mice, which completely reject even poorly immunogenic tumors in a T-cell–dependent fashion but also develop autoimmune syndromes that are ultimately fatal much more quickly than PD1 or LAG3 single knockouts (although not as quickly as CTLA4 knockouts).208 These findings emphasize the balance between antitumor effects and autoimmune side effects that must be taken into consideration in all of the immune checkpoint blockade strategies.
Tim3, the ligand of which is galectin-9 (a galectin reported to be upregulated in a number of cancer types such as breast cancer), inhibits Th1 responses,209 and anti-Tim3 antibodies enhance antitumor immunity.210 Tim3 has also been reported to be co-expressed with PD1 on tumor-specific CD8 T cells, and dual blockade of both molecules significantly enhances the in vitro proliferation and cytokine production of human T cells when stimulated by the cancer-testes antigen NY-ESO-1. In animal models, coordinated blockade of PD1 and Tim3 was reported to enhance antitumor responses and tumor rejection under circumstances where only modest effects from blockade of each individual molecule were observed.213–213
B- and T-lymphocyte attenuator (BTLA) was first identified as an inhibitory receptor on T cells based on enhanced T-cell responses observed in the BTLA knockout mice.214 Subsequently, herpes virus entry mediator (HVEM), which is expressed on certain tumor cell types (i.e., melanoma), as well as tumor-associated endothelial cells, was demonstrated to be the BTLA ligand,215 which is a rare case in which a TNF family member interacts with an immunoglobulin supergene family member. BTLA expression on activated virus-specific CD8 T cells is relatively low, but it has been demonstrated to be much more highly expressed on TILs from patients with melanoma. BTLAhi T cells are inhibited in the presence of its ligand, HVEM. Thus BTLA may also be a relevant inhibitory receptor for T cells in the tumor microenvironment.216 The system of HVEM interacting molecules is complex; two additional interacting molecules, CD160 (an immunoglobulin superfamily member) and LIGHT (a TNF family member) appear to mediate inhibitory and co-stimulatory activity, respectively. It also appears that signaling can be bidirectional depending on the specific combination of interactions. The complexity of this system makes therapeutic inhibition strategies less straightforward than other inhibitory receptors or ligands, although dual blockade of BTLA and PD1 clearly enhances antitumor immunity.217
The A2a receptor for adenosine inhibits T-cell responses, in part by driving CD4 T cells to express Foxp3 and develop into Treg cells.218 Knockout of this receptor results in enhanced and sometimes pathological inflammatory responses to infection. This receptor is particularly relevant in tumor immunity because the rate of cell death in tumors from cell turnover is high and dying cells release adenosine. In addition, Treg cells express high levels of the exoenzymes CD39, which converts extracellular adenosine triphosphate to adenosine monophosphate, and CD73, which converts adenosine monophosphate to adenosine.219 Given that A2a receptor engagement by adenosine drives T cells to become Treg cells, this can produce a self-amplifying loop within the tumor. Indeed, tumors grow more slowly in A2aR knockout mice, and tumor vaccines are much more effective against established tumors in these mice.220 A2aR can be inhibited either by antibodies that block adenosine binding or by adenosine analogues, some of which are fairly specific for A2aR. Although these drugs have been used in clinical trials for Parkinson disease, they have not yet been tested clinically in patients with cancer.
Killer inhibitory receptors are a broad category of inhibitory receptors that can be divided into two classes based on structure: killer immunoglobulin receptors and C-type lectin receptors, which are type II membrane receptors.223–223 These receptors were originally described as critical regulators of the killing activity of NK cells, although many are expressed on T cells and APCs.224 The importance of their inhibitory role on T cells and APCs (i.e., DCs) is less well studied, but the resulting activation of NK cells can provide potent antitumor activity. Many of the killer inhibitory receptors are specific for subsets of HLA molecules and possess allele specificity. However, other receptors recognize broadly expressed molecules; for example, the C-type lectin receptor KLRG1 recognizes e-cadherin. The potential value of NK cells in antitumor responses when their inhibitory receptors are not appropriately engaged is best exemplified by the significantly enhanced graft-versus-tumor effects in allogeneic bone marrow transplants elicited by mismatches between donor NK inhibitory receptors and recipient HLA alleles. The big question in therapeutic blockade of NK inhibitory receptors is which among the more than 20 receptors should be targeted.
Inhibition of Antitumor Responses Versus Induction of Tumor Antigen–Specific Tolerance
One of the unresolved issues in the study of tumor immune evasion relates to the mechanisms by which tumors induce antigen-specific T-cell tolerance. Although many of these mechanisms, including Treg and iMC/MDSC–dependent mechanisms, IDO, ROS, RNS, and TGF-β, immune checkpoints clearly inhibit priming of T-cell responses and/or tumor killing by activated effector T cells, it remains to be definitively determined which processes actively induce antigen-specific T-cell tolerance that has been documented in transgenic models. Self-tolerance induction for peripheral tissue antigens is now thought to involve specific presentation of tissue-specific antigens to mature T cells in the absence of appropriate co-stimulatory signals. Similar mechanisms are likely operative in the case of tumor-induced tolerance. Originally, the relevant co-stimulatory signals were envisioned to be provided by B7 family co-stimulatory molecules expressed by DCs.138 It is now becoming clear that additional proinflammatory cytokines such as interferons, IL-12, and TNF are critical in the distinction between effector T-cell induction and tolerance induction.
An emerging concept is that immature or not fully matured dendritic cells are critical in presenting self-antigens to induce T-cell tolerance in the absence of Toll-like receptor (TLR)-mediated danger signals associated with infection.225,226 Unquestionably, dendritic cells found within the tumor microenvironment have a relatively immature, unactivated phenotype characterized by low levels of proinflammatory cytokine production and CD86 and surface MHC class II expression. As previously described, a major inhibitory signaling pathway induced in tumor-infiltrating DCs is the STAT3 pathway, which, when activated, strongly antagonizes TLR and CD40-mediated DC activation. As mentioned, tumor-derived factors such as IL-10, IL-6, and vascular endothelial growth factor (in part induced by STAT3 signaling in the tumor cell) can induce STAT3 activation in DCs. As described in the previous section, constitutive BRAF signaling in melanoma cells has additionally been shown to induce release of factors that inhibit DC activation.227 These immature “activation-inhibited” DCs clearly represent a prime candidate for the induction of tumor-specific T-cell tolerance.
Whether iMCs/MDSCs represent a distinct intertumoral cell subset capable of presenting antigens to T cells in a toleragenic fashion remains an open question.228 A recent report suggested that iMCs loaded with antigen and adoptively transferred into mice can induce antigen-specific T-cell tolerance. Finally, it has been suggested that IDO-expressing DCs can induce antigen-specific T-cell tolerance because IDO-mediated tryptophan selectively kills or inhibits proliferation of activated T cells.229 According to this model, IDO-expressing DCs would present antigen to T cells, inducing activation followed by activation-associated cell death mediated by depletion of local tryptophan stores by the IDO in the presenting dendritic cells. As described later, regulatory T cells play an additional important role in induction of or maintenance of tumor antigen–specific T-cell tolerance. Whether Treg cells mediate T-cell tolerance independently from immature or toleragenic APCs or whether the two mechanisms are completely interrelated (i.e., toleragenic DCs inducing a Treg phenotype among antigen-specific T cells and antigen-specific Treg cells acting upon DCs to enhance their toleragenic capacity) remains to be definitely determined.
The Nature of Immune Responses That Promote Cancer Formation
The best-studied signaling molecule/transcription factors involved in promoting inflammation or immune-induced carcinogenesis are nuclear factor (NF)-κB and STAT3, which was originally discovered as the major signal transducer for the IL-6 receptor. The first direct evidence for a mechanistic link between IL-6/STAT3 signaling and human cancer was shown in multiple myeloma, where IL-6 drives JAK and STAT3 activation, which in turn promotes tumor cell survival through upregulation of antiapoptotic genes.230 Activation of STAT3 in patients with multiple myeloma is also observed in bone marrow stromal cells, which is a major source of IL-6 that induces persistent activation of STAT3 in the tumor cells, thereby establishing a feed-forward loop.231 Chronic inflammatory conditions that promote tumor formation can also be attributed to genetic alterations directly affecting STAT pathways.232–235 The importance of gain-of-function mutations in the IL6ST gene, which encodes the gp130 subunit of the IL-6 receptor, has been demonstrated in human inflammatory hepatocellular adenomas.235 Constitutively activated mutant gp130 causes persistent activation of JAK and STAT3 in the absence of cytokine ligands. A critical role of STAT3 in inflammation-induced adenocarcinomas was also demonstrated in a transgenic mouse model harboring a constitutively activated gp130 mutant gene in epithelial cells.236 These studies suggest that persistent activation of the IL-6/gp130/JAK/STAT3 signaling pathway is an important contributor to inflammation-induced cancers.
In addition to STAT3, NF-κB signaling has been shown to be critical in inflammation/immune procarcinogenesis because of its central role in mediating inflammatory signals. By conditionally ablating the inhibitor of NFκB (IκB) kinase β (IKKβ), the upstream activator of NF-κB, mouse studies have demonstrated the important role of NF-κB signaling and NF-κB–induced IL-6 in mediating colorectal cancer resulting from inflammatory bowel disease and ulcerative colitis induced by carcinogens.237 An elevated level of IL-6 is also observed in human colorectal cancer, suggesting the importance of IL-6 in mediating inflammatory conditions associated with this cancer.238
More recent studies in carcinogen-induced, colitis-associated colorectal cancer in mouse models lacking both STAT3 alleles provided direct evidence that STAT3 signaling is required for inflammation-induced cancer.239,240 These studies further showed that IL-6, which is primarily produced by bone marrow–derived myeloid cells, and gp130 overactivation led to STAT3 activation in both inflammatory cells and in epithelial cells from which the tumors arose. Activation of STAT3 induced upregulation of key genes involved in cell proliferation and survival and increased nuclear localization of β-catenin, which is known to have a role in colorectal carcinogenesis.239
Inflammatory conditions caused by environmental and other etiologic factors associated with increased cancer risk, including infection, can also activate the IL-6/JAK/STAT3 pathway. A number of infectious agents known to cause inflammation-induced cancer also involve STAT3 activation and likely depend on STAT3 for their oncogenic potential. For instance, infection with H. pylori, which is associated with gastric cancer, activates STAT3 through its cytotoxin-associated gene A in host cells.241 Many tumor viruses are also known to activate STAT3 by various distinct mechanisms, including hepatitis B virus,242 human papillomavirus,243 human T-cell leukemia virus–1,244 and Epstein-Barr virus.245 A human enterotoxigenic strain of Bacteroides fragilis (termed “ETBF”) has been shown to induce colitis in both mice and humans; ETBF colonization of mice results in STAT3 activation in the colon and dramatically increases colon tumorigenesis in Min mice.246 In some cases, STAT3 can be first activated, leading to subsequent activation of IL-6/JAK, which further induces STAT3 activity. For example, both lipopolysaccharide (which mimics bacterial infection) and live bacteria are able to activate STAT3, resulting in production of IL-1β and IL-6,247 both of which are major mediators of inflammation-induced cancer. Lipopolysaccharide is known to act through TLR4, and it has been shown that engagement of TLR4 and TLR9 can directly activate STAT3.248 These and other studies provide a potential mechanistic explanation for why TLRs, which are the sensors of infection, can promote tumor growth.
One of the most debated topics in cancer biology is whether the immune system suppresses or promotes cancer. Many lines of experimental data suggest a critical role of the immune system in controlling tumor incidence and growth, and in certain human cancers or subsets of cancers, the presence of immune cells predicts better prognosis.57,63,249–253 Much evidence suggests that innate immune cells, such as macrophages and NK cells, can destroy tumor cells when appropriately activated.254 T cells from the adaptive arm of the immune system can also attack tumors when stimulated by activated dendritic cells (DCs) in the context of a Th1 immunologic milieu. However, M2 macrophages associated with tumors can promote cancer growth, in part through their ability to secrete angiogenic, metastatic, and growth factors255,256 (described later). The related iMC/MDSC population, which are increased in cancer, not only suppress antitumor immunity but also directly contribute to tumor growth.257
The distinction between immune responses that inhibit versus promote cancer is to a large extent regulated by members of the STAT family of proteins. In particular, STAT1 activation leads to IL-12 production, and STAT4 activation induced by IL-12R engagement in turn promotes Th1 responses that produce IFN-γ, whose receptor itself signals through STAT1 to enhance antitumor responses via activation of NK cells, M1 macrophages, and CD8+ T-cell–mediated cytolytic activity.258,259 Ablating the STAT3 gene in immune cells was shown to boost expression of Th1 mediators,260,261 leading to STAT1 protein upregulation and enhanced antitumor immune responses.227,262–264 Both STAT3 and STAT6 have been implicated as promoters of immunosuppressive MDSCs.257,265 Recent evidence points to a critical role of STAT3 in mediating the cancer-promoting properties of tumor-associated MDSCs, as well as macrophages.255,265–267 Specifically, STAT3 activity in tumor-associated myeloid cells is crucial for upregulating proangiogenic factors and promoting tumor angiogenesis.267 STAT3 and STAT5 signaling also have a major role within T cells in the expansion of T-regulatory cells,262,268,269 which promote tumor progression by inhibiting antitumor immune responses mediated by Th1-type CD4 T cells and CD8 cytotoxic T cells.270
Tumor Promotion Via the IL-23/Th17 Axis
New insights into STAT-dependent regulation of the balance between tumor-promoting versus antitumor immune responses come from the recent discovery of a distinct T-helper cell subset termed Th17; Th17 cells have been implicated in protective immunity to intestinal and pulmonary bacterial infections, as well as pathological immunity in a number of autoimmune diseases.271 Th17 cells are defined by secretion of IL-17A but not IFN-γ (the signature cytokine of Th1 cells) or IL-4 (the signature cytokine of Th2 cells). Although Th1 development is initiated by type 1 interferons and further promoted by IL-12, Th17 development requires a combination of IL-6 and TGF-β and is further promoted by the IL-12 family member, IL-23.254,272 IL-23 and IL-12 share one subunit (p40), and each possess a unique subunit (p19 for IL-23 and p35 for IL-12). STAT3 activation is central to Th17 development273 and function both as the major IL-6R–dependent transcription factor within the T cell and also as a transcriptional activator of the IL-23 p19 gene in macrophages and DCs.268 Recent evidence supports a cancer-promoting role for both IL-23 and Th17 responses. In an initiator-promoter (dimethylbenzyl[α]anthracene) treatment followed by skin painting with tissue plasminogen activator cutaneous carcinogenesis model, IL-23 knockout resulted in decreased skin tumor formation, whereas IL-12 knockout resulted in increased tumor formation.274 In vivo IL-23R antibody blockade also decreased growth rates of some transplanted tumors. Inducible myeloid-specific STAT3 gene ablation abrogated IL-23 production by tumor-infiltrating macrophages while inducing release of IL-12 by tumor-infiltrating dendritic cells.268 In addition to enhancing the activity of tumor-infiltrating regulatory T cells, IL-23 also enhances intratumoral Th17 activity, which can further promote tumor growth. For example, some transplanted tumors grow more slowly in IL-17 null mice and display decreased intratumoral STAT3 activity and increased intratumoral IFN-γ.275
In a number of experimental carcinogenesis models, knockout or in vivo antibody blockade of IL-23 and/or IL-17 diminishes carcinogenesis, implicating this STAT3-dependent immune axis in cancer promotion. In the ETBF bacterial procarcinogenesis model previously described, STAT3 activation is required for IL-17 production by lamina propria CD4 T cells, and IL-17 blockade abrogates most of the colon tumorigenesis induced by ETBF colonization.246 The naturally occurring small intestinal tumor formation seen in non-ETBF colonized Min mice is also dramatically diminished when Min mice are crossed onto an IL-17 knockout background.276 In the classic DMBA-TPA skin carcinogenesis model, tumor formation is likewise dramatically reduced in IL-17 knockout mice just as in the IL-23p19 knockout mice.277 Although requirements for IL-17 and IL-23 are coordinately observed in many carcinogenesis models, the methylcolanthrene sarcomagenesis model (tumor formation after intramuscular injection with methylcholanthrene) appears to depend on IL-23 but not IL-17.278 Thus IL-23–driven carcinogenesis is not always strictly due to its role in maintaining Th17 responses.
In all of these models, STAT3 activation appears to be a common denominator. In addition to promotion of IL-23/Th17 responses, part of the procarcinogenic activity of STAT3 relates to its antagonism of STAT1,262,279,280 the major signaling pathway activated by type 1 interferons, as well as the Th1 cytokine, IFN-γ.252,281 Both type 1 interferons and IFN-γ mediate antitumor immunity via STAT1-dependent innate effectors (such as NK cells), Th1 pathways, and cytotoxic T lymphocyte activation267 (Fig. 6-9). The multiple mechanisms by which STAT3 induces cancer inflammation and inhibits antitumor immunity positions STAT3 as a promising molecular target that could be targeted to “reeducate” the immune system for cancer therapy.
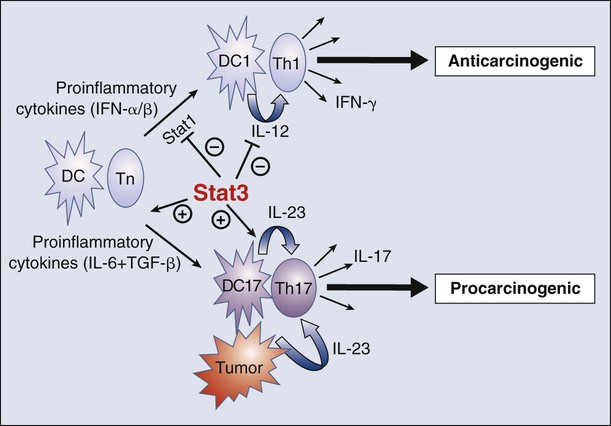
Although the procarcinogenic role of Th17 has been established in a number of animal models, the role of Th17 responses in growth and metastasis of established cancers is much less clear. The growth of B16 melanoma after subcutaneous injection is slower in IL-17 knockout mice and faster in IFN-γ knockout mice.275 In this model, IL-17 drives STAT3 activity in the tumor through an indirect mechanism of IL-16 upregulation. However, in an adoptive T-cell transfer model for treatment of pulmonary B16 metastases, therapeutic activity was diminished in the absence of IL-17.282 In other transplantable tumor models, tumors grew more quickly in IL-17 knockout mice.283 Taken together, the variable findings in different transplantable tumor models suggest that the role of IL-17 (and associated Th17 responses) in growth of established tumors is context dependent.
The role of Th17 responses in human cancer is an area of intense interest. Of note, certain chronic inflammatory conditions characterized by high IL-23 and IL-17, such as Crohn colitis, are associated with a high incidence of carcinogenesis. Patients with Crohn disease of the colon have a five- to tenfold higher incidence of colon cancer development compared with the general population, and cancer of the small intestine (which is very rare among the general population) can develop in patients with Crohn disease of the small intestine.284 Regarding the role in established cancers, Tosolini and colleagues285 analyzed gene expression among human primary colon cancers for gene signatures of Th1 versus Th2 versus Th17 versus Treg and found that the only signature with statistically significant prognostic significance for overall survival (generally a function of development of distant metastases) was a Th17 signature, which was an independent predictor of poor survival. In that study, a Th1 signature was associated with better survival, but the correlation did not reach statistical significance.
As the roles of Th1 responses in antitumor immunity and Th17 responses in carcinogenesis are becoming established, Th2 responses, which normally mediate parasitic immunity and also allergic disease, have been shown to play a potential role in promotion of metastasis. DeNardo and colleagues286 have shown that the metastasis-promoting CD4 T-cell response in an MMTV-PyMT transgenic model of breast cancer is mediated by Th2 responses that produce IL-4 and IL-13. In vivo blockade with anti-IL-4 antibodies significantly diminishes the number of metastasis. As with T cells, there is great plasticity in macrophage activation programs leading to distinct effector functions. In part instructed by helper T cells, macrophage activation programs have been divided into M1 type (Th1/IFN-γ instructed) and M2 type pathway (Th2/IL-4 and IL-13 instructed). M1 macrophages are characterized by expression of genes encoding the nitrous oxide–producing inducible nitric oxide synthase–2 enzyme and the cytokine IL-12, which further amplifies Th1 responses. As previously mentioned, M1 macrophage activation is thought to be one of the mediators of Th1-orchestrated antitumor immunity. In the MMTV-PyMT model, lung metastases were dependent on Th2 responses and M2 macrophages, whose activity is IL-4 and IL-13 dependent. M2 macrophages promoted lung metastases via TGF-β, which, as previously mentioned, suppress antitumor immune responses and EGFR ligands, which promote tumor growth and possibly invasiveness.286
Implications for Cancer Immunotherapy
Advances in understanding of DC biology led to the first successful randomized phase 3 cancer vaccine trial that used a putative DC vaccine, sipuleucel-T, to treat patients with advanced hormone-resistant prostate cancer.287 This vaccine is based on the concept that optimal T-cell activation requires antigen processing and presentation by a specialized cell—the DC—with the capacity to concomitantly deliver strong co-stimulatory signals in the form of membrane ligands and secreted cytokines. Sipuleucel-T is a patient-specific vaccine produced by transiently incubating the patient’s own peripheral blood mononuclear cells with a fusion protein consisting of prostatic acid phosphatase, a prostate/prostate cancer–specific antigen linked to the DC growth, and differentiation factor, GM-CSF. A 4-month overall survival benefit relative to the control arm (uncultured peripheral blood mononuclear cells without prostatic acid phosphatase–GM-CSF fusion protein) in the absence of objective tumor regressions or effect on time to progression emphasizes a developing clinical paradigm in immunotherapy: that immunotherapy can potentially provide overall survival benefits that are not reflected in progression-free survival or objective response rate. The survival benefit of sipuleucel-T ultimately led to FDA approval in 2010.
The most exciting clinical advances in cancer immunotherapy to emerge directly from basic studies in cancer immunology have been blockade of the CTLA4 and PD1 immune checkpoints. Encouraged by murine studies demonstrating antitumor effects of CTLA4 blockade, human anti-CTLA4 antibodies were developed and one antibody, ipilimumab, was shown in a cohort of patients with advanced melanoma to produce a roughly 10% objective response rate according to standard Response Evaluation Criteria In Solid Tumors clinical criteria.288 Although systemic IL-2 also induces tumor regressions in persons with melanoma, a number of the responders to ipilimumab (now called Yervoy) were patients whose tumor had not responded to IL-2 treatment. As might have been predicted from the phenotype of the murine knockouts, anti-CTLA4 produced significant immune-related toxicity in 25% to 30% of patients, commonly involving the skin, liver, or colon. Nonetheless, ongoing clinical development of ipilimumab resulted in two successful phase 3 trials in patients with advanced melanoma and FDA approval in 2011.289,290
A number of interesting clinical insights came from the clinical experience with ipilimumab. First, the kinetics of clinical responses to anti-CTLA4 (i.e., tumor shrinkage) tend to be much slower than chemotherapy or tyrosine kinase inhibitors and, in some patients, tumor regression is proceeded by apparent progression as assessed by computed tomography or magnetic resonance imaging scans. This finding suggests a mechanism of action in which ultimate tumor regression is preceded by immune activation and tumor infiltration of activated lymphocytes. Additionally, although formal regressions are induced in a relatively small proportion of patients and complete responses as assessed by computed tomography or magnetic resonance imaging are rare (<5%), roughly 20% of patients treated with only four doses of ipilimumab remained alive more than 4 years after therapy (as opposed to ~5% in the control group). These clinical findings led to the development of “immune response criteria” to help clinicians evaluate the efficacy of this new class of immunotherapy in their patients.291
In the case of PD1, the basic discoveries that this pathway downmodulated immune responses in the tissue, together with the upregulation of PD1 ligands in human tumors, motivated clinical testing in the mid 2000s. Despite the fact that most of the patients on the initial phase 1 trial were end stage, a number of dramatic clinical responses were observed in multiple cancer types with clinically significant toxicity in only one patient.2 This finding led immediately to larger clinical trials of both anti-PD1 and anti-PD-L1, culminating in the demonstration of major clinical efficacy, not only in advanced chemotherapy-refractory melanoma, but also renal cancer and lung cancer.292,293 The durability of the responses, even after cessation of treatment, far exceeds that of chemotherapy or tyrosine kinase inhibitors. These findings reinforce the notion that a “reeducated” immune system not only has memory but also can adapt to maneuvers by the tumor to develop resistance. Just as predicted from the mouse knockout studies, toxicity from anti-PD1 or anti-PD-L1 therapy is lower than from anti-CTLA4 therapy and also is of a different distribution than anti-CTLA4 therapy (colitis is most common with anti-CTLA4 therapy and pneumonitis is more common for anti-PD1 therapy). Consistent with the important role of the PD1 pathway in tissue protection under physiological conditions and tumor immune resistance in cancer, PD-L1 expression on tumor cells correlates highly with clinical response to anti-PD1, suggesting that this could be a “biomarker” potentially applicable for patient selection.5 At least six different companies are currently developing antibodies or recombinant molecules that target the PD1 pathway.





