Targeting the PI3K/Akt/mTOR signaling pathway is another promising approach, especially in the lung. Tobacco carcinogens induce Akt activation and lung carcinogenesis. The Akt pathway is activated in bronchial premalignancy (both proximal airway and alveolar epithelium) in smokers and patients with lung IEN or cancer. Preclinical in vivo studies show that deguelin and myo-inositol have preventive activity in lung tumorigenesis, in part via suppressing the PI3K/Akt pathway, disrupting Hsp90 function, and inhibiting HIF-1α expression.
64–66 The kinase mammalian target of rapamycin (mTOR) is downstream of Akt, and the mTOR inhibitor CCI-779 blocked malignant progression of premalignant lesions with activated mTOR arising in the alveoli of mice that develop lung cancer because of activated K-ras.
67 The mechanism by which CCI-779 inhibited tumorigenesis was unexpected. These lesions were infiltrated with macrophages, shown immunohistochemically to have prominent activation of mTOR signaling. A similar pattern of macrophage infiltration occurred in human alveolar premalignant lesions (atypical alveolar hyperplasia). Treatment with CCI-779 induced apoptosis of macrophages, which coincided with the chemopreventive effect. In vitro, CCI-779 had no effect on LKR-13, a lung adenocarcinoma cell line derived from this mouse, whereas it did induce apoptosis of macrophages, and conditioned media from macrophages directly stimulated the proliferation of LKR-13 cells. In summary, mTOR is activated in lung premalignancy and is required for malignant progression in the lung. This kinase drives tumorigenesis in part through macrophages, a prominent component of the tumor microenvironment, and the antitumor effect of mTOR inhibition required the presence of the tumor microenvironment. These findings have two important implications: mTOR is a potentially important kinase target, and the tumor microenvironment is crucial in malignant progression and a source for novel targets in chemoprevention. An mTOR inhibitor also has reversed Akt-dependent prostatic IEN in transgenic mice.
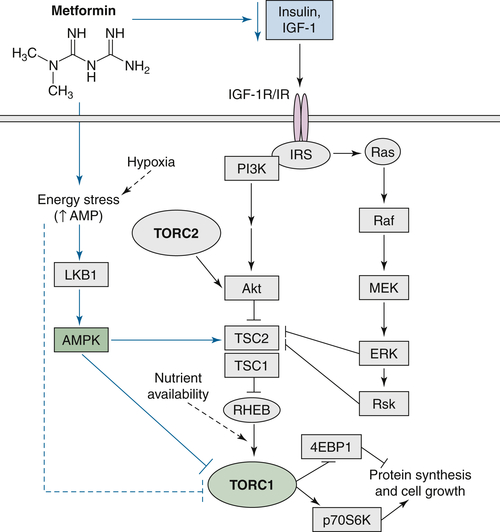
68 Metformin belongs to the biguanide class of antidiabetic drugs and activates the LKB1/AMPK axis (mediating glucose and energy homeostasis) and inhibits cancer cell viability through the inhibition of mTOR. Metformin can also downregulate mTOR and subsequent cell growth through AMPK-independent mechanisms
69 (
Figure 58-1 ). A recent study using mouse models of lung cancer to assess the protective effect of metformin suggested two possible mechanisms: decreased levels of circulating insulin and lowered energy stress leading to inhibition of mTOR.
70 Owing to the fact that studies show metformin is associated with a decreased risk of cancer incidence compared with other treatments (such as insulin) among diabetic patients,
71 metformin is rightfully garnering interest for its role in cancer prevention and therapy and supports further testing in the clinical setting.
Looking at new targets of the antineoplastic activities of metformin yields some surprising and unique mechanisms. In paraquat-treated mice, metformin reduced the levels of mitochondrial ROS in an AMPK-independent manner while also reducing DNA double-stranded breaks.
72 A recent compelling study suggests the molecular mechanism by which metformin can elicit its biologic effects in pancreatic cancer stem-like cells (CSCs) is mediated through reexpression of miRNAs and decreased expression of CSC-specific genes.
73 Indeed, these novel mechanisms may help to explain reduced cancer incidence associated with metformin therapy.
Figure 58-1 mTOR exists in the intracellular complexes TORC1 and TORC2. Growth factor signaling (through PI3K/AKT and ERK/ribosomal S6 kinase [Rsk] signaling) and energy homeostasis (through AMPK) directly lead to TSC2 phosphorylation. In vivo, metformin downregulates TORC1 via several potential mechanisms including AMPK-dependent and AMPK-independent mechanisms. IRS, insulin receptor substrate; LKB1, liver kinase B1; MEK, mitogen-activated protein kinase. (Reprinted by permission from the American Association for Cancer Research: Engleman JA, Cantley LC. Chemoprevention meets glucose control. Cancer Prev Res (Phila). 2010;3:1049-1052.)