Chapter 40 Biliary atresia
Overview
Biliary atresia (BA) remains a somewhat elusive disease, with our understanding confined as it is to its infancy but yet with the origin of BA essentially unknown. BA is potentially fatal, certainly if left to run its natural course (Hartley et al, 2009), but it can be treated effectively with expeditious surgery in a high proportion of patients. For the remainder, liver transplantation is an option, and indeed BA remains the single most common indication for liver transplant in the pediatric population.
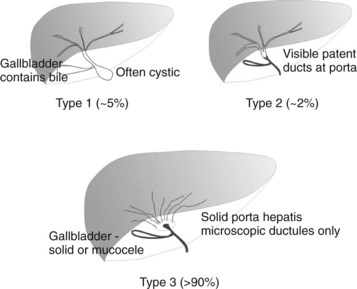
BA is an occlusive panductular cholangiopathy affecting both intrahepatic and extrahepatic bile ducts. The most common classification divides BA into three types based on the most proximal level of occlusion of the extrahepatic biliary tree (Fig. 40.1). In types 1 and 2, in which there is a degree of preservation of structure of the intrahepatic bile ducts, blunting, irregularity, and pruning occurs, and dilation may be absent, even when the duct is obstructed. In the most common type of BA, type 3, the intrahepatic bile ducts are grossly abnormal with myriad small ductules coalescing at the porta hepatis. When retrograde cholangiography is possible, this is seen as a “cloud.” In approximately 5% of cases, extrahepatic formation of cysts containing clear mucus or bile can occur in the otherwise occluded biliary tree. Such cystic biliary atresia (Caponcelli et al, 2008) is distinguishable clinically and histologically from simple obstruction in a cystic choledochal malformation, where there is preservation of an epithelial lining and retention of a normal and often dilated intrahepatic biliary tree.
Etiologic Heterogeneity
BA is not one disease, certainly not one with a single cause. In all probability it is a phenotype resulting from a number of different etiologies. Three groups can be defined clinically: 1) biliary atresia splenic malformation (BASM) syndrome (Davenport et al, 1993, 2008), 2) cystic BA (Caponcelli et al, 2008), and 3) isolated BA (Table 40.1).
Other related entities are fewer in number, but there seems to be a relationship with other gastrointestinal anomalies, such as esophageal atresia and jejunal atresia, in a small proportion (<1% of large series); occasional cases show a defined chromosomal abnormality, such as “cat-eye” syndrome and chromosome 22 aneuploidy (Allotey et al, 2008).
Developmental biliary atresia is a term we use that includes BASM and cystic BA, for which the onset is almost certainly prenatal and is evident by the time of birth (Livesey et al, 2009). The onset of occlusion in isolated BA is much more contentious, and some authorities hold that the bile duct can be normal and patent at the time of birth and can then become occluded secondarily by virally mediated damage.
Biliary Atresia Splenic Malformation Syndrome
Although the association of BA with polysplenia had been recognized for some time, clarification of what constitutes BASM (Table 40.2) is only relatively recent (Davenport et al, 1993). The constellation of other anomalies is peculiar, and the reasons for this are still obscure. The common embryologic insult may simply be timing (30 to 35 days) rather than a specific genetic defect. Key genes are important in both bile duct development (JAG1, HNF-6 [ONECUT1]; Kohsaka et al, 2002) and visceral and somatic symmetry (INVS, CFC1; Bamford et al, 2000; Shimadera et al, 2007), although correlation with the human condition is patchy. A possible genetic link has recently been reported by a French group, who found an increased frequency of mutations in the CFC1 gene (on chromosome 2) compared with controls (Davit-Spraul et al, 2008).
Table 40.2 Spectrum of Anomalies in Biliary Atresia Splenic Malformation Syndrome
| Organ System | Malformation | Notes |
|---|---|---|
| Splenic | Polysplenia, double spleen (95%); asplenia (5%) | No evidence of immune hypofunction |
| Situs determination | Inversus (50%) | |
| Venous | Preduodenal portal vein (40%), portosystemic shunt (<2%); vena cava absence (50%) | |
| Intestinal development | Malrotation (60%) | |
| Cardiac | Atrial septal defect, ventricular septal defect, tetralogy of Fallot, etc. (~40%) | ± Dextrocardia |
| Liver | Mirror image, normal, or symmetrical | Position dictated by abdominal situs |
| Biliary appearance | Atrophic or absent gallbladder | Often noninflammatory remnants with absence of common bile duct and obvious segmental appearance |
| Pancreas | Annular (<5%) | |
| Miscellaneous | Immotile cilia syndrome, sacral agenesis |
Epidemiology
A clear difference in incidence is seen across the globe. The highest is reported in Taiwan, at one in 5000 live births (Hsiao et al, 2008). In Japan, the incidence is one in 10,000 (Nio et al, 2003), with national studies from the United Kingdom and France reporting an incidence of one in 17,000 and 19,000, respectively (Chardot et al, 1999; Livesey et al, 2009; Serinet et al, 2006). Although there are no national studies in North America, smaller regional studies suggest an incidence closer to that of Europe (Yoon et al, 1997) but with some interracial variation (The et al, 2007). Significant regional differences within the United Kingdom have also been observed, again ascribed to variations in racial origin and the nature of the BA. For example, developmental BA is more common in white infants than in those of Asian origin (Livesey et al, 2009).
A distinct female predominance in seen in those with developmental BA that is not seen in the isolated BA group for unknown reasons (Davenport et al, 1993, 2006; Livesey et al, 2009). A seasonal variation in incidence has also been observed in some regionally based studies (Yoon et al, 1997; The et al, 2007), although this has not been confirmed in the larger, national studies (Chardot et al, 1999; Livesey et al, 2009).
Pathophysiology
The nature of the mononuclear infiltrate has assumed a degree of importance, and many authors suggest this is the specific primary destructive element of BA (Davenport et al, 2001; Mack et al, 2007). Although not necessarily uniformly applied across the spectrum of BA, the infiltrate is largely composed of CD4+ T lymphocytes, specifically Th1 (Mack et al, 2004) and CD56+ natural killer (NK) cells (Davenport et al, 2001; Shivakumar et al, 2009), which exhibit markers for proliferation (CD71+) and activation (particularly LFA-1+ but also CD25+). Infiltrating CD8+ cells appear to lack markers of activation—such as perforin, granzyme B, and Fas ligand (Ahmed et al, 2001)—in contrast to other diseases such as primary biliary cirrhosis (see Chapter 9).
Expression of cell adhesion molecules, proteins involved in cell-cell binding, is an important precursor to an immune-active cell infiltrate. Both interstitial cell adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1 are important in binding circulating leukocytes by interaction with cell-surface integrins, and they are expressed on sinusoidal and biliary epithelium of infants with BA (Dillon et al, 1997; Davenport et al, 2001).
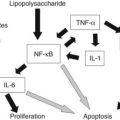
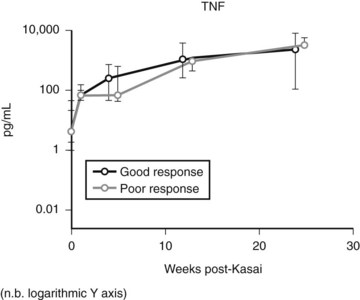
Expression of soluble(s) ICAM-1 and sVCAM-1 in the circulation is also increased (Minnick et al, 1998; Davenport et al, 2005), as is seen in other immunologically mediated liver diseases such as primary biliary cirrhosis and sclerosing cholangitis; levels of sVCAM-1 at the time of presentation have been shown to be prognostic (Davenport et al, 2005). Increasing levels of various other cytokines (interleukin [IL]-2, tumor necrosis factor [TNF]-α, IL-18) can also be shown postoperatively (Fig. 40.2), but most discriminate poorly between patients who would clear their jaundice and those who would not; conversely, some cytokines (IL-2, interferon [IFN]-γ, IL-4, IL-10, TNF-α, and sICAM-1) were better predictors of subsequent need for early transplantation (Narayanaswamy et al, 2007).
Resident (Kupffer cells) or systemic/recruited macrophages and monocytes seem to play a dual role in BA, as both the presenters of antigenic material in the first place and as the initiating driver for fibrosis in the development of chronic liver disease. Tracy and colleagues (1996) first showed increases in resident macrophages (CD68+) with marked expression of the lipopolysaccharide receptor CD14+. Increased levels of both CD68+ cells and circulating markers of CD68+, TNF-α, and IL-18, have been shown to predict outcome post-Kasai (Kobayashi et al, 1997; Narayanaswamy et al, 2007).
Viral Hypothesis of Biliary Atresia
One suggested cause of BA is infection with hepatotropic viruses, such as rotavirus, reovirus, and cytomegalovirus, either as an indirect trigger of an abnormal immune-mediated reaction or as an actual pathologic agent. The evidence is largely based on animal models in which intraperitoneal innoculation on the first day of life is able to mimic postnatal biliary inflammation (Riepenhoff-Talty et al, 1993; Petersen et al, 1998). Subsequent translation into humans has been more problematic. Serologic evidence of viremia is contradictory (Morecki et al, 1982; Tarr et al, 1996), and isolation of both viral RNA and DNA have (Riepenhoff-Talty et al, 1996; Tyler et al, 1998; Fischler et al, 1999) and have not (Brown et al, 1988; Steele et al, 1995; Bobo et al, 1997; Jevon & Dimmick, 1999) been identified by PCR techniques.
The most recent study is one from Hannover, Germany, in which liver biopsies from 74 infants obtained at the time of portoenterostomy were tested for a panel of DNA and RNA hepatotropic viruses (Rauschenfels et al, 2009). One or more viruses was detected in about one third of infants, and the rate of detection increased with infant age, which suggested that observed viral infection was more likely to be a secondary finding rather than a specific cause.
One problem with the viral hypothesis is the lack of an explanation as to why such ubiquitous organisms only damage the biliary tree in such a small fraction of infants and children. It may be that such infants are in some way susceptible to cholangiocyte damage and require a (viral) trigger to set off an immune-mediated destructive process (Mack, 2007).
RNA gene expression studies in rotavirus-induced murine models have shown upregulation of many proinflammatory genes, among them the IFN-γ inducers IRF7 and IRF9 (Carvalho et al, 2005). Similar gene-expression profiling in human BA samples has also shown overexpression of immune regulatory genes and differences between isolated and syndromic forms (Bezerra et al, 2002; Zhang et al, 2004).
A number of studies have shown different but specific elements of genetic variation between patients with isolated BA and unaffected control subjects. Thus, susceptibility may be imparted by polymorphisms in a number of relevant genes. Examples of this include vascular endothelial growth factor (VEGFA; Lee et al, 2010), macrophage migration inhibitory factor (MIF, Arikan et al, 2006), and intercellular adhesion molecules (ICAMs; Arikan et al, 2008).
Most recently, maternal microchimerism, a novel mechanism of immune damage, has been suggested based on the observation that male infants with BA have a threefold increase in cells of maternal origin in their livers (Hayashida et al, 2007). These were later shown to be maternal-origin chimeric CD8+ T cells and CD45 NK cells capable of initiating immune cholangiolar damage (Muraji et al, 2008).
Clinical Features
The cardinal features of BA are conjugated jaundice persisting beyond 14 days of age, pale and unpigmented stools, and dark urine in an otherwise healthy, term neonate. Liver fibrosis and cirrhosis are later developments, even in infants with intrauterine BA evident at birth (Makin et al, 2009); therefore ascites and marked hepatosplenomegaly are rarely seen before 3 months. Fat malabsorption occurs by stealth, and these infants will fall behind in their growth (Livesey et al, 2009). Fat malabsorption can also cause fat-soluble vitamin K deficiency, and some infants (<3%) will be seen with a possibly catastrophic bleeding tendency and a raised international normalized ratio (INR).
A proportion of infants (approximately 5%) will be seen initially with an abnormal antenatal ultrasound (US) scan (Caponcelli et al, 2008). These will be shown to have cystic BA, with the “cyst” evident from at least 20 weeks’ gestation. This has no untoward effects during pregnancy but clearly needs thorough investigation postnatally. The differential diagnosis for these infants, if the cyst is actually recognized as of hepatobiliary origin, is that of a cystic choledochal malformation, better known as a choledochal cyst (Hinds et al, 2004).
Diagnostic Workup
US examination is a key part of the workup in order to exclude other possible surgical diagnoses (choledochal malformation, inspissated bile syndrome), and evidence of at least intrahepatic bile duct dilation should be visible (Davenport et al, 2003). Although suggestive and supportive, US is uncommonly diagnostic of BA and typically shows a shrunken, atrophic gallbladder with no evidence of filling between feeds. Some US signs do appear more reliable, such as the “triangular cord sign” (Park et al, 1997). This suggests a sonographic appearance of the proximal solid biliary remnant in front of the bifurcation of the portal vein, and advocates quote accuracy rates of greater than 80% (Humphrey & Stringer, 2007).
Direct cholangiography is becoming more prevalent in larger centers with direct access to endoscopic retrograde cholangiopancreatography (ERCP) (Shanmugam et al, 2009; Petersen et al, 2009). It requires a sufficiently small endoscope and a level of familiarity with the procedure, as failure to cannulate the bile duct are features of both BA and an inexperienced operator. Laparoscopy with or without liver biopsy offers an alternative to ERCP, and it may be the prelude to the definitive procedure. Direct puncture of the gallbladder is relatively straightforward to show presence or absence of bile and as an access point for a cholangiogram. This can be coupled with liver biopsy.
Surgery: Kasai Portoenterostomy
Preoperative preparation for a Kasai portoenterostomy includes normalizing the coagulopathy and possibly providing a bowel preparation regimen.* An intravenous perioperative antibiotic regimen active against aerobic and anaerobic bowel flora is standard.
Mobilization
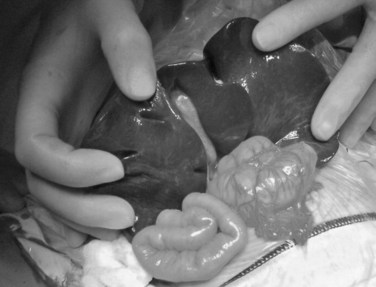
The liver should be fully mobilized by dividing the falciform, coronary, and triangular ligaments such that the organ can then be everted outside of the abdominal cavity (Fig. 40.3). This allows full exposure of the porta hepatis and facilitates dissection; it also reduces venous return, and an increase in intravenous volume support is always needed, together with close communication between surgeon and anesthesiologist.
Portal Dissection
Initially the gallbladder is mobilized and the distal CBD divided (Fig. 40.4A), with the dissection consisting of elevation of the proximal pyramidal remnant from the right hepatic artery and bifurcation of portal vein (Fig. 40.4B). Small veins to the porta hepatis need division and facilitate downward traction of the portal vein and exposure of the posterior part of the portal plate and caudate lobe. On the left side, the recessus of Rex, where the umbilical vein joins the left portal vein, should be exposed; an isthmus of liver parenchyma from segment III to IV may need division by coagulation diathermy to achieve this. On the right side, remnant biliary tissue can be identified almost circumferentially around the right vascular pedicle. The division into right anterior and posterior pedicles should be visualized, and the posterior pedicle’s biliary elements should also be incorporated into the subsequent Roux loop. Excision of remnants flush with the liver capsule is accomplished by developing a plane between solid white biliary remnant and the underlying liver (Fig. 40.4C); excising liver parenchyma itself does not seem to improve bile drainage. All the denuded area of the porta hepatis needs to be incorporated into the Roux loop as the portoenterostomy. Some authors (Gittes & Koybayashi, 2007) have suggested a role for frozen section of the portal plate to determine that the observed ductules are large enough. My view is that there should be nothing left to resect from the initial dissection. Burrowing into liver parenchyma itself seems pointless, possibly because subsequent scarring obliterates any exposed ductules.
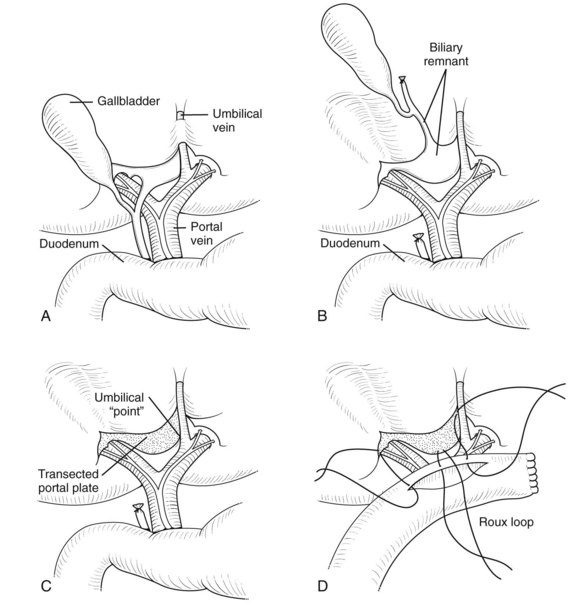
Roux Loop and Portoenterostomy
A standard retrocolic Roux loop measuring 40 to 45 cm should be constructed (Fig. 40.4D). The jejunojejunostomy should lie about 10 cm from the ligament of Treitz and can be stapled or sutured. The proximal anastomosis must be wide (approximately 2 cm), and as such, an end-to-side tension-free arrangement is preferred. Fine, precise sutures (e.g., 6-0 PDS) at the edge of the portal plate are satisfactory. Remnant ductules are concentrated in the right and left recesses, and it is important that all parts of the transected plate are incorporated.
Postoperative Complications and Outcome
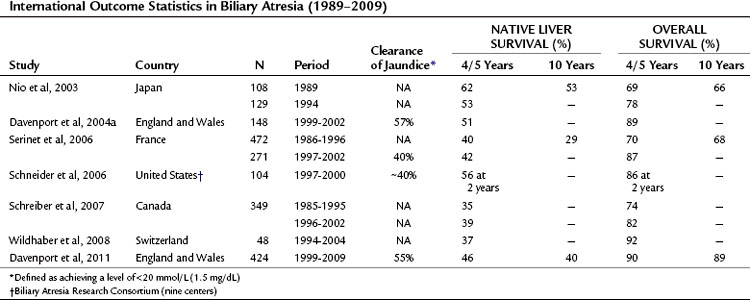
The aims of portoenterostomy are to restore bile flow and reduce bilirubin levels to normal. This is achievable in isolated BA in about 50% to 60% of infants (Davenport et al, 2004a, 2008); and should halt the progression of liver fibrosis. Table 40.3 illustrates current outcome statistics derived from national studies. Our own studies have shown that in a centralized system with experienced surgeons, the clearance of jaundice can be achieved after Kasai portoenterostomy in about 50% to 55% of patients, with translation into a 5- and 10-year native liver survival of approximately 50% and 45%, respectively. Overall survival depends heavily on the access to efficient liver transplant programs and can be approximately 90% at both 5 and 10 years (Davenport et al, 1997, 2004a, 2009). If success is achieved after either portoenterostomy or transplantation, children can grow and develop normally to adulthood with minimal medical support.
The most common complication is that the portoenterostomy does not work, and therefore it will have no effect on restoration of bile flow, and jaundice will worsen and lead to end-stage liver disease. For these infants, transplantation will be required for survival, typically within the first 2 years of life (Schneider et al, 2006; see Chapters 71 and 97A). Such infants have increasingly evident portal hypertension with splenomegaly, ascites, and variceal formation. They fail to thrive and have reduced synthetic liver function with falling levels of serum albumin and increasing evidence of a vitamin K–independent coagulopathy (DeRusso et al, 2007). This is a crucial period within which most deaths occur as a result of biliary atresia, and unfortunately most are already on a transplant waiting list.
Cholangitis
Cholangitis is presumed to occur by bacterial ascent from the Roux loop via patent biliary ductules (see Chapter 43). It is therefore uncommon in those who have no effect from Kasai surgery. Nonetheless, it may occur in 30% to 50% and is most frequent within a year of surgery (Ecoffey et al, 1987). Clinically, cholangitis is characterized by worsening jaundice, fever, and acholic stools. The diagnosis may be confirmed by blood culture or by percutaneous liver biopsy, but it is important to treat suspected cases early with broad-spectrum antibiotics effective against gram-negative organisms (e.g., ceftazidime, amoxycillin, piperacillin). Sometimes recurrent cholangitis may be problematic, which can be associated with intrahepatic cyst formation (Bu et al, 2002), presumably acting as a bacterial nidus. Most patients respond to prolonged antibiotic prophylaxis, either with intravenous broad-spectrum antibiotics via an indwelling vascular access device or with oral nonabsorbable antibiotics (e.g., neomycin). If cholangitis still recurs despite these measures, liver transplantation should be considered.
Sometimes cholangitis occurs some years after the portoenterostomy in children with otherwise good liver function. It is important in these cases to exclude a mechanical Roux loop obstruction, typically at the level of the mesocolon, because corrective surgery for obstruction is relatively simple (Houben et al, 2006). Percutaneous transhepatic cholangiography and radionuclide hepatic imaging may be useful for diagnosis.
Portal Hypertension
Elevated portal venous pressure can be shown in virtually all infants at the time of the Kasai operation (Duché et al, 2006). However, whether it persists or regresses depends on the degree of established fibrosis and, most importantly, the response to surgery (Kang et al, 1993). About 60% of children who survive to 2 years will have endoscopic evidence of esophageal varices, although only approximately half of these will ever bleed. The severity and degree of the varices themselves are not necessarily related to the original degree of liver fibrosis or to the number of episodes of cholangitis (Kang et al, 1993). Endoscopic surveillance of the upper gastrointestinal tract in children with treated biliary atresia has been suggested from about the age of 2 years because of this unpredictability of variceal formation (see Chapter 71).
The age at first hemorrhage is typically approximately 2 to 3 years, and it manifests as sudden hematemesis and melena from rupture of an esophageal varix. Ectopic intestinal variceal bleeding may also occur, usually in an older child with relatively normal liver function. The initial treatment of bleeding varices is supportive, with restoration of blood volume and correction of platelet deficiency and any coagulopathy. Drugs such as octreotide, or more recently terlipressin, may be used be used to reduce portal venous pressure. Banding has become the most efficacious technique to control varices, although it may be limited by patient size (Hall et al, 1988). For infants and small children, endoscopic sclerotherapy is still effective (Stringer et al, 1989), using sclerosants such as ethanolamine or sodium tetradecyl sulfate. Even with various pharmacologic and endoscopic alternatives, sometimes the only means of variceal control is a Sengstaken or equivalent pattern tube. Transjugular intrahepatic portosystemic shunt (TIPS) as a method of acutely reducing portal hypertension is also possible in children with BA, but it should be regarded only as a “bridge” to transplantation (Johnson et al, 1996; Cao et al, 1997), and it is not widely available.
Hepatopulmonary Syndrome and Portopulmonary Hypertension
Hepatopulmonary syndrome (HPS) is characterized by cyanosis, hypoxia, and clubbing and is due to shunting across the pulmonary vascular bed, likely caused by failure to deactivate vasoactive substances, possibly endothelin-1, by the damaged liver (Iqbal et al, 2008). Patients with HPS have higher levels of exhaled nitric oxide, presumably the final link in this chain. It is more commonly seen in those with BASM, suggesting a congenital element (Davenport et al, 2004a), and it can occur even in those who are anicteric. The diagnosis is made by arterial blood gas estimation showing significant hypoxia with no improvements seen on increasing ambient oxygen. Radionuclide lung scans with macroaggregated tagged albumin can be used to quantify the degree of shunting and can range from 4% to 50% (Al-Hussaini et al, 2010).
Portopulmonary hypertension is much less common and may share the same underlying vascular mechanism as HPS. Although it is often silent and perhaps only detected because of cardiomegaly on a chest radiograph, it can be a cause of sudden death and is associated with systolic pulmonary arterial pressures greater than 40 mm Hg. Although no real therapy is available for the pulmonary vascular anomaly, it is a definite indication for liver transplantation and is usually reversible, albeit with an increased risk of complications (Iqbal et al, 2008).
Liver Malignancy
The cirrhotic liver of treated BA is potentially premalignant, and there are several reports (Kohno et al, 1995; Iida et al, 2009; Hol et al, 2008) and a single series of three cases (Tatekawa et al, 2001). This of course may be due simply to the relatively small numbers of long-term survivors, but perhaps it implies only a modest risk. Malignant change may include hepatocellular carcinoma (Hol et al, 2008; Iida et al, 2009; see Chapter 80) or cholangiocarcinoma (Kulkarni & Beatty, 1977; see Chapter 50A, Chapter 50B, Chapter 50C, Chapter 50D ), but more benign changes, such as focal nodular hyperplasia (Okugawa et al, 2008), may also occur. Although the youngest reported case was diagnosed in a patient 10 months old, but most are seen during later childhood or adolescence as a nodule or mass on routine US screening of the liver. Because of the risk for malignant change, serum α-fetoprotein levels should probably be part of the routine follow-up for such children, although clearly in some, these levels will still be normal.
Areas of Debate and Controversy in Biliary Atresia
Laparoscopy
The open technique described above requires precision, maximal exposure of the portal plate, and delicate suturing. This can be approximated by laparoscopic or robotic means, and small series have been reported (Esteves et al, 2002; Lee et al, 2004; Dutta et al, 2007; Ayuso et al, 2008). However, now that the first wave of enthusiasm has perhaps abated, it appears that the results are not comparable to the open technique (Wong et al, 2008). The only outcomes that matter are clearance of jaundice and preservation of the native liver; other outcomes, such as early feeding and better cosmesis, are irrelevant. In many centers, there is currently a self-imposed moratorium, until the reasons for failure become clearer or techniques improve.
Adjuvant Steroids
The inflammatory nature of BA has been emphasized above, and postoperative steroids seem a logical approach to this. In addition, most steroids have a chloretic action, although actual evidence of effect is scant. A number of small, uncontrolled studies from the United States and Japan suggesting benefit have been published (Meyers et al, 2004; Kobayashi et al, 2005). Our large, randomized, double-blind, placebo-controlled trial starting with 2 mg/kg/day showed a significant early effect at 1 month after operation with a lower bilirubin (Davenport et al, 2007), but this did not translate to a diminished need for transplantation or to a significant difference in the proportion with complete clearance of jaundice. Further studies from Germany using a higher starting dose (10 mg/kg/day) showed no difference, although their overall clearance rates were poor (27% were jaundice free at 2 years; Petersen et al, 2008).
Effect of Age at Surgery
It is axiomatic that once cirrhosis is established, the outcome will be worse. And because cirrhosis takes time to develop, it is logical to think that the earlier the surgery, the better the outcome. However, given that we do not know when biliary obliteration starts in isolated BA, trying to identify a relationship with postnatal age may be difficult. Large, multicenter series with multiple surgeons have been unable to show a statistically significant relationship (Davenport et al, 2004a).
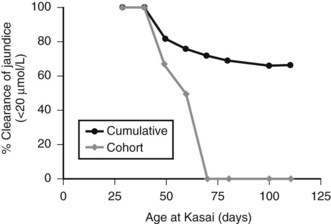
Figure 40.5 illustrates this for isolated BA derived from a single center with only two surgeons, and it uses clearance of jaundice (<20 Umol/L) as a measure of outcome. This shows that there is no discernable difference up to 100 days, and certainly no evidence of any “cutoff.” By contrast, Figure 40.6 shows the same analysis for one of the developmental forms, cystic BA, and here a relationship is obvious, with an impaired outcome the older the infant is at surgery (Davenport et al, 2008).
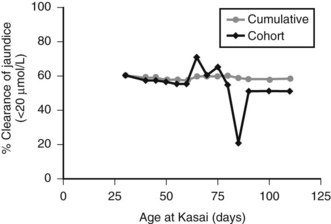
FIGURE 40.5 Relationship of age at surgery to clearance of jaundice in infants with isolated biliary atresia (n = 177).
(Modified from Davenport et al, 2008: Surgical outcome in biliary atresia: etiology affects the influence of age at surgery. Ann Surg 247:694-698.)
Is there any benefit of surgery in those infants who come to surgery after 100 days? By now, this should be relatively infrequent in developed countries, but it still occurs. Such infants are much more likely to have established cirrhosis, and the prognosis is, without doubt, worse. Nevertheless, a Kasai portoenterostomy as the first step may be reasonable and has been associated with long-term jaundice-free survival in our series (Davenport et al, 2004b). The alternative is a primary liver transplant.
Kasai Variations
Although most surgeons operating today follow the original principles set out by Kasai and Suzuki (1959), most likely use a much wider portal dissection to try and incorporate as much as possible of the transected portal plate. There have been published variations in the construction of the Roux loop. Typically these were developed to reduce the postoperative incidence of cholangitis, and it included bringing the Roux loop out as a stoma and creating an antireflux valve (Tanaka et al, 1991; Nakajo et al, 1990). Hindsight suggests these have no real value and serve to impair the later transplant procedure, should it become necessary (Ogasawara et al, 2003). Use of the appendix as a conduit to the duodenum has also been described (Crombleholme et al, 1989), but again, later reports suggest no value (Lee et al, 2007; Tsao et al, 2003).
Liver Transplantation
The first ever attempt at human transplantation occurred in Denver, Colo., by Thomas Starzl on March 1, 1963 (Starzl et al, 1963; see Chapter 97A, Chapter 97B, Chapter 97C, Chapter 97D, Chapter 97E ). The patient was a ventilator-bound child born with BA who, because of complications related to severe bleeding, did not survive the operation; nonetheless, a milestone had been passed. Still, very few similar operations were carried out over the next 20 years, until the advent and increasing availability of cyclosporine during the 1980s made effective immunosuppression possible and transplantation a realistic option.
It is still a surgically challenging procedure with reported early survival rates (1 to 3 years) from 85% to 94% and later (5 to 10 years) survival from 78% to 87% (Diem et al, 2003; Barshes et al, 2005; Fouquet et al, 2005; Cowles et al, 2008). There is, of course, the expectation of life-long immunosuppression; but in return, transplant offers the prospect of an excellent quality of life with normal liver function, and beyond the immediate postoperative period, expectation of long-term survival.
Ahmed AF, et al. CD8+ T cells infiltrating into bile ducts in biliary atresia do not appear to function as cytotoxic T cells: a clinicopathological analysis. J Pathol. 2001;193:383-389.
Al-Hussaini A, et al. Long-term outcome and management of hepatopulmonary syndrome in children. Pediatr Transplant. 2010;14:276-282.
Arikan C, et al. Positive association of macrophage migration inhibitory factor gene-173G/C polymorphism with biliary atresia. J Pediatr Gastroenterol Nutr. 2006;42:77-82.
Arikan C, et al. Polymorphisms of the ICAM-1 gene are associated with biliary atresia. Dig Dis Sci. 2008;53:2000-2004.
Allotey J, et al. Congenital bile duct anomalies (biliary atresia) and chromosome 22 aneuploidy. J Pediatr Surg. 2008;43:1736-1740.
Ayuso L, et al. Laparoscopic Kasai portoenterostomy: present and future of biliary atresia treatment [in Spanish]. Cir Pediatr. 2008;21:23-26.
Bamford RN, et al. Loss-of-function mutations in the EGF-CFC gene CFC-1 are associated with human left–right laterality defects. Nat Genet. 2000;26:365-369.
Barshes NR, et al. Orthotopic liver transplantation for biliary atresia: the U.S. experience. Liver Transp. 2005;11:1193-1200.
Bezerra JA, et al. Genetic induction of proinflammatory immunity in children with biliary atresia. Lancet. 2002;360:1653-1659.
Bobo L, et al. Lack of evidence for rotavirus by polymerase chain reaction/enzyme immunoassay of hepatobiliary samples from children with biliary atresia. Pediatr Res. 1997;41:229-234.
Brown WR, et al. Lack of correlation between infection with Reovirus type 3 and extrahepatic biliary atresia. J Pediatr. 1988;113:670-676.
Bu LN, et al. Multiple intrahepatic biliary cysts in children with biliary atresia. J Pediatr Surg. 2002;37:1183-1187.
Cao S, et al. Emergency transjugular portosystemic shunt in an infant: a case report. J Pediatr Surg. 1997;32:125-127.
Caponcelli E, Knisely AS, Davenport M. Cystic biliary atresia: an etiologic and prognostic subgroup. J Pediatr Surg. 2008;43:1619-1624.
Carvalho E, et al. Analysis of the biliary transcriptome in experimental biliary atresia. Gastroenterology. 2005;129:713-717.
Chardot C, et al. Epidemiology of biliary atresia in France: a national study 1986-96. J Hepatol. 1999;31:1006-1013.
Cowles RA, et al. Timing of liver transplantation in biliary atresia: results in 71 children managed by a multidisciplinary team. J Pediatr Surg. 2008;43:1605-1609.
Crombleholme TM, et al. Biliary appendico-duodenostomy: a nonrefluxing conduit for biliary reconstruction. J Pediatr Surg. 1989;24:665-667.
Davenport M, et al. The biliary atresia splenic malformation syndrome. Surgery. 1993;113:662-668.
Davenport M, et al. Biliary atresia: the King’s College Hospital experience (1974-1995). J Pediatr Surg. 1997;32:479-485.
Davenport M, et al. Immunohistochemistry of the liver and biliary tree in extrahepatic biliary atresia. J Pediatr Surg. 2001;36:1017-1025.
Davenport M, et al. The spectrum of surgical jaundice in infancy. J Pediatr Surg. 2003;38:1471-1479.
Davenport M, et al. Seamless management of biliary atresia: England & Wales 1999-2002. Lancet. 2004;363:1354-1357.
Davenport M, et al. The outcome of the older (> 100 days) infant with biliary atresia. J Pediatr Surg. 2004;39:575-581.
Davenport M, et al. Soluble adhesion molecule profiling in preoperative infants with biliary atresia. J Pediatr Surg. 2005;40:1464-1649.
Davenport M, et al. The biliary atresia splenic malformation syndrome: a 28-year single-center retrospective study. J Pediatr. 2006;149:393-400.
Davenport M, et al. Randomized, double-blind, placebo-controlled trial of corticosteroids after Kasai portoenterostomy for biliary atresia. Hepatology. 2007;46:1821-1827.
Davenport M, et al. Surgical outcome in biliary atresia: etiology affects the influence of age at surgery. Ann Surg. 2008;247:694-698.
Davenport M, et al: Biliary atresia: results of centralization and a new benchmark. J Pediatr Surg. In press.
Davit-Spraul A, et al. CFC1 gene involvement in biliary atresia with polysplenia syndrome. J Pediatr Gastroenterol Nutr. 2008;46:111-112.
DeRusso PA, et al. Growth failure and outcomes in infants with biliary atresia: a report from the Biliary Atresia Research Consortium. Hepatology. 2007;465:1632-1638.
Diem HV, et al. Pediatric liver transplantation for biliary atresia: results of primary grafts in 328 recipients. Transplantation. 2003;75:1692-1697.
Dillon PW, et al. Differential expression of the major histocompatibility antigens and ICAM-1 on bile duct epithelial cells in biliary atresia. Tohoku J Exp Med. 1997;181:33-40.
Duché M, et al. Prognostic value of portal pressure at the time of Kasai operation in patients with biliary atresia. J Pediatr Gastroenterol Nutr. 2006;43:640-645.
Dutta S, Woo R, Albanese CT. Minimal access portoenterostomy: advantages and disadvantages of standard laparoscopic and robotic techniques. J Laparoendosc Adv Surg Tech A. 2007;17:258-264.
Ecoffey C, Rothman E, Bernard O. Bacterial cholangitis after surgery for biliary atresia. J Pediatr. 1987;111:824-829.
Esteves E, et al. Laparoscopic Kasai portoenterostomy for biliary atresia. Pediatr Surg Int. 2002;18:737-740.
Fischler B, et al. Cytomegalovirus DNA detection on Guthrie cards in patients with neonatal cholestasis. Arch Dis Child. 1999;80:F130-F134.
Fouquet V, et al. Long-term outcome of pediatric liver transplantation for biliary atresia: a 10-year follow-up in a single center. Liver Transpl. 2005;11:152-160.
Gittes GK, Koybayashi H. Biliary atresia. Blumgart, LH, editor. Blumgart’s Surgery of the Liver, Biliary Tract, and Pancreas, 4th ed, Philadelphia: Elsevier, 2007.
Hall RJ, Lilly JR, Stiegmann GV. Endoscopic esophageal varix ligation: technique and preliminary results in children. J Pediatr Surg. 1988;23:1222-1223.
Hartley JL, Davenport M, Kelly DA. Biliary atresia. Lancet. 2009;374:1704-1713.
Hayashida M, et al. The evidence of maternal microchimerism in biliary atresia using fluorescent in situ hybridization. J Pediatr Surg. 2007;42:2097-2101.
Hinds R, et al. Antenatal presentation of biliary atresia. J Pediatr. 2004;144:43-46.
Hol L, et al. Hepatocellular carcinoma complicating biliary atresia after Kasai portoenterostomy. Eur J Gastroenterol Hepatol. 2008;20:227-231.
Houben C, Phelan S, Davenport M. Late-presenting cholangitis and Roux loop obstruction after Kasai portoenterostomy for biliary atresia. J Pediatr Surg. 2006;41:1159-1164.
Hsiao CH, et al. Universal screening for biliary atresia using an infant stool color card in Taiwan. Hepatology. 2008;47:1233-1240.
Humphrey TM, Stringer MD. Biliary atresia: US diagnosis. Radiology. 2007;244:845-851.
Iida T, et al. Hepatocellular carcinoma in a 10-month-old biliary atresia child. Pediatr Transplant. 2009;13:1048-1049.
Iqbal CW, et al. Liver transplantation for pulmonary vascular complications of pediatric end-stage liver disease. J Pediatr Surg. 2008;43:1813-1820.
Jevon GP, Dimmick JE. Biliary atresia and cytomegalovirus infection: a DNA study. Pediatr Dev Pathol. 1999;2:11-14.
Johnson SP, et al. Transjugular portosystemic shunts in pediatric patients awaiting liver transplantation. Transplantation. 1996;62:1178-1181.
Kang N, et al. Hepatic histology and the development of oesophageal varices in biliary atresia. J Pediatr Surg. 1993;28:63-66.
Kasai M, Suzuki M. [A new operation for “non-correctable” biliary atresia: hepatic portoenterostomy]. Shujitsu (in Japanese). 1959;13:733-739.
Kobayashi H, et al. Hepatic overexpression of MHC class II antigens and macrophage-associated antigens (CD68) in patients with biliary atresia of poor prognosis. J Pediatr Surg. 32, 1997. 596–593
Kobayashi H, et al. Optimum prednisolone usage in patients with biliary atresia post-portoenterostomy. J Pediatr Surg. 2005;40:327-330.
Kohno M, et al. Hepatocellular carcinoma complicating biliary cirrhosis caused by biliary atresia: report of a case. J Pediatr Surg. 1995;30:1713-1716.
Kohsaka T, et al. The significance of human jagged 1 mutations detected in severe cases of extrahepatic biliary atresia. Hepatology. 2002;36:904-912.
Kulkarni PB, Beatty EJr. Cholangiocarcinoma associated with biliary cirrhosis due to congenital biliary atresia. Am J Dis Child. 1977;131:442-444.
Lee H, et al. Initial experience with complex laparoscopic biliary surgery in children: biliary atresia and choledochal cyst. J Pediatr Surg. 2004;39:804-807.
Lee HC, et al. Genetic variation in the vascular endothelial growth factor gene is associated with biliary atresia. J Clin Gastroenterol. 2010;44:135-139.
Lee KD, et al. Long-term outcome of hepatic portoduodenostomy with interposition of the ileocecoappendix for biliary atresia. Pediatr Surg Int. 2007;23:935-938.
Livesey E, et al. Epidemiology of biliary atresia in England and Wales (1999-2006). Arch Dis Child Fetal Neonatal Ed. 2009;94:F451-F455.
Mack CL. The pathogenesis of biliary atresia: evidence for a virus-induced autoimmune disease. Semin Liver Dis. 2007;27:233-242.
Mack CL, et al. Biliary atresia is associated with CD4+ Th1 cell–mediated portal tract inflammation. Pediatr Res. 2004;56:79-87.
Mack CL, et al. Oligoclonal expansions of CD4+ and CD8+ T-cells in the target organ of patients with biliary atresia. Gastroenterology. 2007;133:278-287.
Makin E, et al. Congenital biliary atresia: liver injury begins at birth. J Pediatr Surg. 2009;44:630-633.
Meyers RL, et al. High-dose steroids, ursodeoxycholic acid and chronic intravenous antibiotics improve bile flow after Kasai procedure in infants with biliary atresia. J Pediatr Surg. 2004;38:406-411.
Minnick KE, Kreisberg R, Dillon PW. Soluble ICAM-1 (sICAM-1) in biliary atresia and its relationship to disease activity. J Surg Res. 1998;76:53-56.
Morecki R, et al. Biliary atresia and reovirus type 3 infection. N Engl J Med. 1982;307:481-484.
Muraji T, et al. Maternal microchimerism in underlying pathogenesis of biliary atresia: quantification and phenotypes of maternal cells in the liver. Pediatrics. 2008;121:517-521.
Nakajo T, et al. Intussusception-type antireflux valve in the Roux-en-Y loop to prevent ascending cholangitis after hepatic portojejunostomy. J Pediatr Surg. 1990;25:311-314.
Narayanaswamy B, et al. Serial circulating markers of inflammation in biliary atresia—evolution of the post-operative inflammatory process. Hepatology. 2007;46:180-187.
Nio M, et al. Five- and 10-year survival rates after surgery for biliary atresia: a report from the Japanese Biliary Atresia Registry. J Pediatr Surg. 2003;38:997-1000.
Ogasawara Y, et al. The intussusception antireflux valve is ineffective for preventing cholangitis in biliary atresia: a prospective study. J Pediatr Surg. 2003;38:1826-1829.
Okugawa Y, et al. Focal nodular hyperplasia in biliary atresia patient after Kasai hepatic portoenterostomy. Pediatr Surg Int. 2008;24:609-612.
Park WH, et al. A new diagnostic approach to biliary atresia with emphasis on the ultrasonographic triangular cord sign: comparison of ultrasonography, hepatobiliary scintigraphy, and liver needle biopsy in the evaluation of infantile cholestasis. J Pediatr Surg. 1997;32:1555-1559.
Petersen C, Grasshoff S, Luciano L. Diverse morphology of biliary atresia in an animal model. J Hepatol. 1998;28:603-607.
Petersen C, et al. Postoperative high-dose steroids do not improve mid-term survival with native liver in biliary atresia. Am J Gastroenterol. 2008;103:712-719.
Petersen C, et al. Endoscopic retrograde cholangiopancreaticography prior to explorative laparotomy avoids unnecessary surgery in patients suspected for biliary atresia. J Hepatol. 2009;51:1055-1060.
Rauschenfels S, et al. Incidence of hepatotropic viruses in biliary atresia. Eur J Pediatr. 2009;168:469-476.
Riepenhoff-Talty M, et al. Group A rotaviruses produce extrahepatic biliary obstruction in orally inoculated newborn mice. Pediatr Res. 1993;33:394-399.
Riepenhoff-Talty M, et al. Detection of group C rotavirus in infants with extrahepatic biliary atresia. J Infectious Dis. 1996;174:8-15.
Schneider BL, et al. Biliary Atresia Research Consortium: a multicenter study of the outcome of biliary atresia in the United States, 1997 to 2000. J Pediatr. 2006;148:467-474.
Schreiber RA, et al. Biliary atresia: the Canadian experience. J Pediatr. 2007;151:659-665.
Serinet MO, et al. Management of patients with biliary atresia in France: results of a decentralized policy 1986-2002. Hepatology. 2006;44:75-84.
Shanmugam NP, et al. Selective use of endoscopic retrograde cholangiopancreatography in the diagnosis of biliary atresia in infants younger than 100 days. J Pediatr Gastroenterol Nutr. 2009;49:435-441.
Shimadera S, et al. The INV mouse as an experimental model of biliary atresia. J Pediatr Surg. 2007;42:1555-1560.
Shivakumar P, et al. Neonatal NK cells target the mouse duct epithelium via Nkg2d and drive tissue-specific injury in experimental biliary atresia. J Clin Invest. 2009;119:2281-2290.
Starzl TM, et al. Homotransplantation of the liver in humans. Surg Gynecol Obstet. 1963;117:659-676.
Steele MI, et al. Reovirus type 3 not detected by reverse transcriptase-mediated polymerase chain reaction analysis of preserved tissue from infants with cholestatic liver disease. Hepatology. 1995;21:696-702.
Stringer MD, Howard ER, Mowat AP. Endoscopic sclerotherapy in the management of esophageal varices in 61 children with biliary atresia. J Pediatr Surg. 1989;24:438-442.
Tanaka K, et al. A valved hepatic portoduodenal intestinal conduit for biliary atresia. Ann Surg. 1991;213:230-235.
Tarr PI, Haas JE, Christie DL. Biliary atresia, cytomegalovirus and age at referral. Pediatrics. 1996;97:828-831.
Tatekawa Y, et al. Liver transplantation for biliary atresia associated with malignant hepatic tumors. J Pediatr Surg. 2001;36:436-439.
The NS, et al. Risk factors for isolated biliary atresia, National Birth Defects Prevention Study, 1997-2002. Am J Med Genet A. 2007;143A:2274-2284.
Tracy TF, et al. The inflammatory response in pediatric biliary disease: macrophage phenotype and distribution. J Pediatr Surg. 1996;31:121-125.
Tsao K, et al. Comparison of drainage techniques for biliary atresia. J Pediatr Surg. 2003;38:1005-1007.
Tyler KL, et al. Detection of reovirus RNA in hepatobiliary tissues from patients with extrahepatic biliary atresia and choledochal cysts. Hepatology. 1998;27:1475-1482.
Wildhaber BE, et al. Biliary atresia: Swiss national study, 1994-2004. J Pediatr Gastroenterol Nutr. 2008;46:299-307.
Wong KK, et al. Should open Kasai portoenterostomy be performed for biliary atresia in the era of laparoscopy? Pediatr Surg Int. 2008;24:931-933.
Yoon PW, et al. Epidemiology of biliary atresia: a population-based study. Pediatrics. 1997;101:729-730.
Zhang DY, et al. Coordinate expression of regulatory genes differentiates embryonic and perinatal forms of biliary atresia. Hepatology. 2004;39:954-962.