Basics of Radiation Therapy
Elaine M. Zeman, Eric C. Schreiber and Joel E. Tepper
Introduction and Historical Perspective
• X-rays were first discovered emanating from an energized Crooke’s tube by Wilhelm Roentgen in 1895.1 In 1896, Henri Becquerel discovered that some naturally occurring elements emitted ionizing radiation.2 The radioactive elements radium and polonium were isolated and characterized by the Curies in 1898.3
• Within a year or two, ionizing radiation was in use worldwide for medical imaging and radiation therapy.
• Several types of ionizing radiation are used to treat patients; most are of the low linear energy transfer, less biologically potent varieties.
• Therapeutic x-rays (photons) and electrons are produced by linear accelerators but can also be produced by nuclear isotopes that undergo radioactive decay. These form the basis of external beam radiotherapy and brachytherapy, respectively.
• Ionizing radiation interacts with matter via several processes, the most important of which for clinical radiation therapy is Compton scattering.
• Megavoltage photons from linear accelerators have the desirable property of delivering their maximum dose at depth within the patient, thereby sparing the skin and, to some extent, other normal tissues.
The Radiobiology of Radiotherapy
• Ionization of biomolecules from the deposition of energy by photons or particles can occur directly and indirectly. The most important cellular target for radiation is DNA, with irreparable or “misrepaired” double-stranded breaks believed to be the lesions most responsible for cell killing.
• Irradiation elicits diverse cellular responses that include the sensing of DNA damage, mobilization of DNA repair proteins, repair (or attempted repair) of DNA damage, triggering of cell cycle checkpoints, and, for irreparable or mis-rejoined damage, cell death by one of several mechanisms (e.g., mitotic catastrophe, apoptosis, and senescence).
• The most commonly applied model of cell survival probability is the linear quadratic (α/β) model, with the surviving fraction of irradiated cells described by the equation 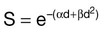 . The α/β ratio is a convenient metric for describing cellular radiosensitivity and has been adapted to describe the response of irradiated tissues as a function of time, dose, and fractionation.
. The α/β ratio is a convenient metric for describing cellular radiosensitivity and has been adapted to describe the response of irradiated tissues as a function of time, dose, and fractionation.
• DNA damage and repair were initially inferred by monitoring increases in cell survival or tissue tolerance with fractionation. These phenomena were termed sublethal and potentially lethal damage repair or recovery.
• Cells in different cell cycle phases possess different radiosensitivities; cells are most radiosensitive in the G2 and M phases of the cell cycle, and most resistant in the S phase, particularly the late S phase. Cells in the G1 phase are of intermediate radiosensitivity.
• Well-oxygenated cells are as much as three times more sensitive to radiation-induced cell killing than (severely) oxygen-deprived cells. Viable hypoxic cells that exist in many human tumors but that are mostly absent in normal tissues may be an impediment to tumor control. The elimination of such cells has been a long-standing clinical goal. Hypoxia may provide avenues for therapeutic gain through the use of hypoxia-directed therapies.
• Radiation sensitizers, particularly cytotoxic chemotherapy and, to a lesser extent, radiation protectors, aim to improve the therapeutic ratio.
• Radiation therapy is used in more than half of all patients with cancer, either as an adjuvant or neoadjuvant treatment in combination with surgery; as a definitive treatment alone or in combination with chemotherapy; as an organ-sparing therapy; or to palliate symptoms.
• Fractionation of radiation and altered fractionation schedules, such as accelerated hyperfractionated radiation therapy, make use of differences in the responses of normal and malignant tissues to irradiation to achieve higher therapeutic ratios.
• Radiation produces early effects, such as mucositis, skin erythema, or desquamation, and late effects, such as fibrosis and carcinogenesis.
Planning and Delivery of Radiation Treatment
• Patient simulation uses multiple imaging approaches to identify cancerous and healthy regions within the patient and to select appropriate beams to deliver a dose to the tumor while minimizing the dose delivered to surrounding tissues.
• Three-dimensional conformal treatment planning and delivery has permitted escalation of doses and improved sparing of normal tissues.
• Intensity-modulated radiation therapy uses varying radiation beam intensities to precisely sculpt the dose distribution around the tumor to improve the therapeutic ratio.
• Image-guided radiation therapy uses real-time and/or daily imaging to ensure that the tumor is positioned such that the radiation beams are precisely delivered to the appropriate location within the patient.
• Brachytherapy delivers extremely high-dose radiation to tumor tissue with a much lower dose to surrounding normal tissues.
• Stereotactic radiosurgery and stereotactic body radiation therapy combine a high dose per fraction with highly conformal treatment delivery to increase the therapeutic ratio while reducing treatment time.
• Proton therapy has dose distribution advantages compared with photon therapy, and it may be used to deliver high doses of radiation to tumors in close proximity to sensitive normal structures.
Overview of Radiation Physics
Interactions of Radiation and Matter
In order of roughly increasing energy, photons can interact with:
2. tightly bound inner shell electrons;
3. loosely bound outer shell electrons;
Photoelectric Effect
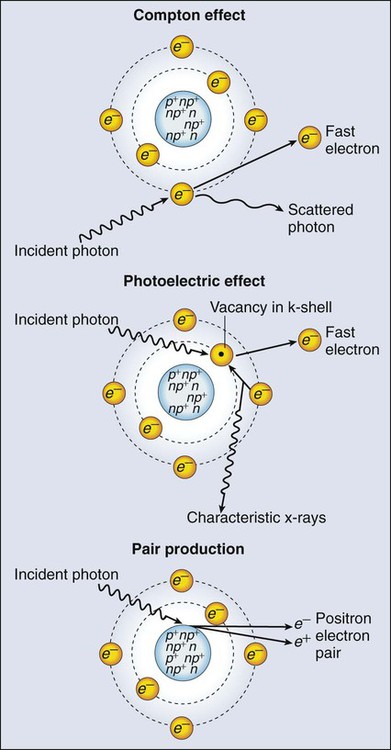
Photons having sufficient energy to ionize an atomic electron can undergo the photoelectric effect (Fig. 27-1). In this process, the photon energy is entirely absorbed. Some energy is lost to breaking the electron binding energy, and the rest is carried away as kinetic energy of the ejected electron. The probability of a photoelectric interaction scales with the cube of the atomic number (Z) and the inverse cube of the photon energy (E), making the photoelectric effect very sensitive to material type and much more prevalent for lower photon energies. The photoelectric effect is the dominant photon interaction in tissue below 30 keV.
Compton Scattering
When photon energy is significantly higher than the binding energy of an electron, the photon can scatter from the electron without being absorbed, as illustrated in Figure 27-1. The result of this interaction is a photon with reduced energy and new direction and a recoil electron with some fraction of the initial photon energy. The energy of the scattered electron varies with the scattering direction. An electron scattered in the direction of the incident photon claims most of the initial photon energy, whereas electrons scattered at greater angles have successively less energy. Compton scattering is only weakly dependent on Z and is the dominant photon interaction in tissue between 30 keV and 30 MeV.
Pair Production
Above 1.022 MeV, photons can interact in the presence of a strong nuclear field. The photon will disappear and spontaneously become an electron-positron pair (Fig. 27-1). The electron and positron will divide the initial photon energy between them to create their mass and kinetic energy. These particles will lose their energy as they interact with the surrounding materials. Upon losing all their energy, the electrons will be absorbed into an atom. The positron, on the other hand, will annihilate by interacting with a local electron, creating two 511 keV photons. (This annihilation reaction is what is detected during positron emission tomography scanning.) Pair production is the dominant atomic interaction in tissue for photons above 30 MeV and therefore has only a minor effect in radiation therapy, where energies are significantly lower.
The Generation of Therapeutic Radiation
Linear Accelerators
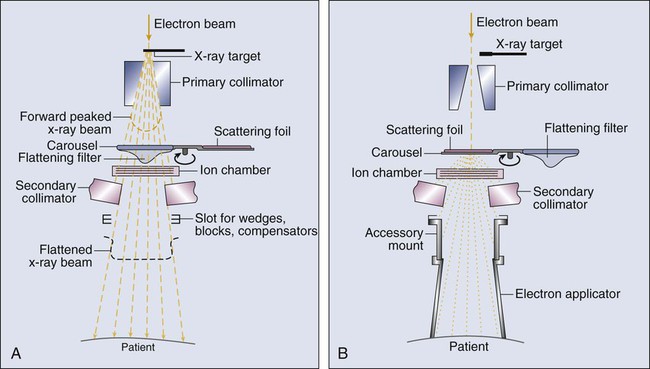
The most common modality used in radiation oncology is external beam radiation therapy. Although a small number of radiation therapy facilities generate external beams using radioactive sources such as cobalt-60, the vast majority of therapeutic electromagnetic radiation is generated in a linac. A linac is a device that accelerates charged particles (electrons) to velocities near the speed of light using oscillating electric fields to push the electrons through a series of accelerating cavities. A schematic of a linac is shown in Figure 27-2, A. Electrons are accelerated to energies typically between 4 and 18 MeV. Electric and magnetic fields focus and steer the high-energy electrons such that they strike a thin metal target that stops the electron beam, with some fraction of the electron energy converted to a spray of photons through the bremsstrahlung process. The bremsstrahlung photons, called x-rays, move approximately in the same direction as the electrons and have an energy spectrum, ranging from a few 10’s of keV up to the maximum energy of the initial electrons. The resulting photon beam then passes through a series of filters and beam-shaping elements that flatten and define the edges of the beam.
The dose from a photon beam is related to its intensity, defined as the number of photons per unit area. Two major effects serve to decrease the intensity of a photon beam as it passes through tissue. First, as with any photon source, the beam intensity decreases with increasing distance from the source, just as is the case for a light bulb. In addition, beam intensity decreases as photons are attenuated from the beam via various scattering and absorption effects. This process leads to a characteristic decrease in intensity versus depth that varies based on photon energy. Although the photon intensity begins decreasing immediately upon entering a material, the energy released through the photon interactions is spread over a few centimeters as the electrons scattered by the photons gradually lose their energy as they pass through the material. The resulting dose distribution is characterized by a region of rapid increase near the surface, a leveling off of dose at a depth of 1 to 3 cm, and a gradual dose falloff as depth increases. The plot of dose versus depth is called a percent depth dose curve, as shown in Figure 27-3. Because higher energy photons are more penetrating, higher energy beams will attenuate more slowly, leading to a more gradual decrease in dose with depth.
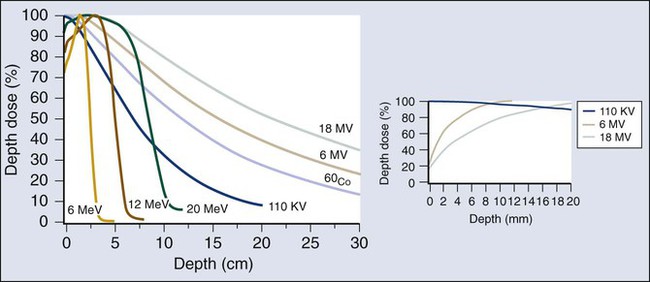
Linacs designed to produce photon beams can also be configured to produce therapeutic electron beams. Removing the photon-generating target and replacing it with a comparatively thinner electron scattering foil allows the transmission of the initial electron beam, but not without scattering the initially narrow beam into a broader distribution. Multiple filters and beam-shaping elements, as shown in Figure 27-2, B, produce an even distribution of customized shape at the surface of the patient. Electron beams lose their energy through different types of interactions than photons, leading to a different pattern of dose versus depth for electron beams. Rather than periodically removing photons from the beam through attenuation, electrons lose their energy gradually and at a relatively constant rate until the entire kinetic energy of the electron is expended and the particles simply stop. Ideally, this phenomenon would lead to a region of constant dose with increasing depth until the depth of full energy loss is reached, at which point the dose would drop abruptly to zero, with the depth of the dose drop-off being dependent on the initial electron energy. In practice, scattering and redirection of electrons within the beam lead to a mildly peaked dose plateau region and a somewhat more gradual dose falloff, as shown in Figure 27-3.
Radioactive Sources
Therapeutically useful isotopes vary in half-life and in energy of emitted particles, as shown in Table 27-1. Isotopes emitting higher energy particles can deliver significant amounts of dose farther from the radioactive source than those emitting lower energy particles. One type of radioactive source, 60Co, emits photons with an average energy of 1.25 MeV, which is sufficiently similar to photon energies found in linacs that 60Co can be used as an external source to treat targets deep in a patient. Most other therapeutically useful radioactive sources emit lower energy radiation with less penetrating power and must be placed in close proximity to the area to be treated. Sources are formed into small sealed seeds, typically 1 to 5 mm in size, and can be inserted into the treatment area on a temporary or permanent basis. The dose rate falls off very quickly with distance from a seed, both because of rapid attenuation and the rapid spread of photons as they move away from the source.
Table 27-1
Therapeutically Useful Radioisotopes
| Isotope | Half-Life Average | Energy (keV) |
| PHOTON | ||
| 226Ra | 1620 y | 830 |
| 137Cs | 30 y | 662 |
| 198Au | 2.7 d | 412 |
| 192Ir | 73.8 d | 370 |
| 125I | 60 d | 28 |
| 103Pd | 16.97 d | 21 |
| Isotope | Half-Life | Maximum Energy (keV) |
| BETA | ||
| 32P | 14.3 d | 1710 |
| 90Sr/90Y | 28.5 y/2.7 d | 550/2280 |
| 188W/188Re | 69.4 d/17 h | 350/2120 |
| 186Re | 3.8 d | 1070 |
| 62Zn/62Cu | 9.3 h/9.7 min | 660/2930 |
| 133Xe | 5.2 d | 360 |
| 131I | 8.0 d | 600 |
| 89Sr | 50.5 d | 1495 |
| 166Ho | 26.8 h | 1850 |
From Cox JD, Ang KK, editors. Radiation oncology: rationale, technique, results. 8th ed. St Louis: Mosby; 2003.
Delivery of Therapeutic Radiation
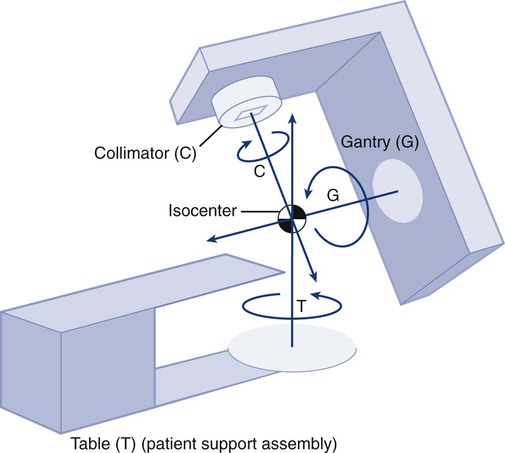
The cytotoxic properties of ionizing radiation provide an opportunity for tumor control but also require that care be exercised to limit the exposure of healthy tissue to radiation. For external beam radiation therapy, linacs are typically mounted on rotating gantries (Fig. 27-4) that allow beams to pass through the patient and the target from a variety of directions. By placing the area to be treated at or near the center of rotation, multiple beams can be made to overlap in the region of the tumor, delivering a high dose to the overlap area and a comparatively low dose to other areas. For treatment based on implanted radioactive sources, a procedure known as brachytherapy, proper dose delivery consists of designing and delivering a three-dimensional (3D) distribution of radioactive seeds within the volume to be treated, creating a high-dose region that decreases rapidly beyond the treatment volume. Both of the aforementioned treatment modalities require a 3D understanding of the patient anatomy and require:
The Radiobiology of Radiation Therapy
Mechanisms of Radiation Damage to Cells
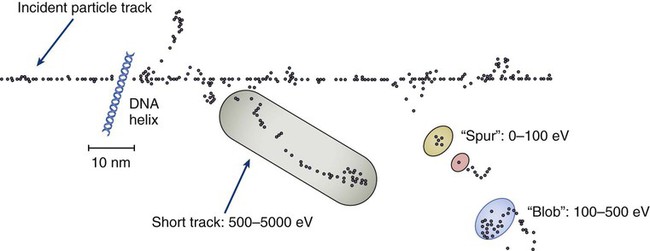
The total amount of energy imparted to a cell during the passage of ionizing radiation (expressed in units of dose, Gray, which in turn is expressed in units of energy deposited, joules per kilogram) is by itself insufficient to describe its biological consequences. For example, 1 Gy of x-rays and 1 Gy of neutrons deliver the same total energy to a cell macroscopically speaking but do not produce equivalent biological effects because it is the microscopic pattern of that energy deposition, the spacing or density of the discrete ionization events along the track of the photon or particle, that is key to determining biological effectiveness. In this example, the 1 Gy of neutrons is much more potent biologically because the average energy deposited locally along the length of each neutron’s track is higher than for x-rays. This quantity of ionization density, typically expressed in units of keV/µm, is termed the radiation’s LET. The concept of ionization density is illustrated graphically in Figure 27-5 for radiations of differing LET, using a strand of DNA drawn to scale as a representative “biomolecule.” Although most radiation therapy is performed using low LET x-rays, γ-rays, or electrons, a few institutions do use high LET neutrons or even higher LET heavy charged particles (heavy ions), such as carbon ions, for treatment. Lower total doses of these radiations are used to achieve tumor control in keeping with their greater biological potency, and especially stringent limits are placed on the amount of normal tissue incidentally irradiated out of justifiable concern for an increased frequency of complications. The use of protons for radiotherapy is also gaining in popularity; protons behave like other high LET particles in terms of their physical properties, although somewhat surprisingly, they are only marginally more potent biologically than x-rays or electrons.
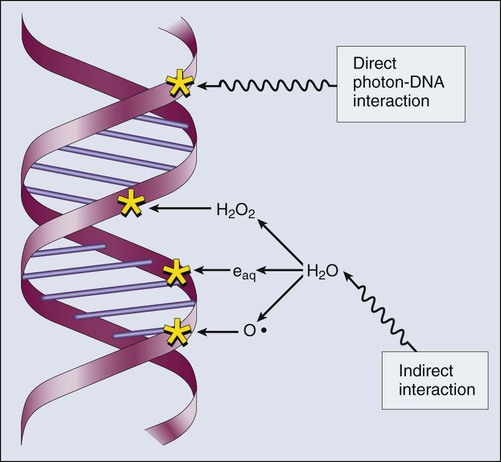
Whether the ionization of a particular molecule results in a measurable biological effect depends on a number of factors, including how important the molecule is to the continued survival and function of the cell, how many copies of the molecule are normally present in the cell, and to what extent and how the cell responds to the loss of working copies. DNA is arguably the most important cellular macromolecule and one that is present only as a single, double-stranded copy, and thus an energy deposition event occurring directly in DNA certainly could affect a cell’s continued survival and functioning. Accordingly, much attention has been focused historically on understanding radiation-induced DNA damage and its repair and the consequences when that damage is either irreparable or “misrepaired.” That said, many other molecules in the cell may be less crucial to survival yet are much more abundant than DNA and therefore have a much higher probability of being ionized. By far, the most abundant molecule in the cell is water. Free radicals formed by the radiolysis of water (the hydroxyl radical, •OH, in particular) are capable of adding to the DNA damage resulting from direct energy absorption by migrating to the DNA and damaging it indirectly, as illustrated in Figure 27-6. This mechanism is referred to as “indirect radiation action” to distinguish it from “direct radiation action”4 previously described. Approximately 30% of the total DNA damage produced by a given dose of x-rays is from the direct effect, and 70% is from the indirect effect.4
Complex macromolecules like DNA that have been ionized and converted to free radicals undergo a series of chemical transmutations in an attempt to rid themselves of unpaired electrons, many of which involve the further breakage of chemical bonds. These broken bonds can result in the modification or loss of a DNA base or an entire nucleotide, a cross-linking of the two DNA strands, or a scission of the sugar phosphate backbone involving either one or both strands. Luckily, DNA is unique in that it is the only cellular macromolecule with its own repair system, and most of this damage can be repaired efficiently and typically with very high fidelity. Under certain circumstances, however, the cell’s attempts to repair these lesions may result in large segments of DNA being lost, rearranged, exchanged, or rejoined in inappropriate ways, so-called “misrepair.” In other cases, repair is impossible because of the complex nature of the damage itself, particularly when both DNA strands are involved (e.g., a DNA double-stranded break), or its location in the genome in time and space. It is this residual DNA damage that manifests itself as chromosome aberrations the next time the cell attempts to go through mitosis and that usually leads to cell death. In fact, the radiation dose response for the production of asymmetrical, exchange-type chromosome aberrations mirrors the shape of the corresponding cell survival curve.5
We now know that several different DNA repair pathways exist in mammalian cells, including single-step reactions that directly reverse certain simple types of damage, single and multistep base excision and resynthesis processes, and multistep pathways to “clean up,” resynthesize, and ligate single or double-stranded breaks in the DNA backbone.6 Which one (or more) of these repair pathways is activated depends on a number of factors, including the type of lesion produced, the physical location of the lesion in the genome (e.g., in the coding region of a gene versus in apparently noncoding DNA), the functional/temporal location of the lesion (i.e., in an actively replicating or transcribing gene versus an inactive one), and, critically, the overall repair competence of the cell in which the lesion was created.7
For a more in-depth discussion of the molecular biology, biochemistry, and regulation of DNA repair, please refer to Chapter 10.
Many bacterial, yeast, rodent, and human genes involved in DNA repair processes have been identified and cloned,8 with many of the encoded proteins functioning as components of large repair complexes. Some of these proteins are interchangeable and participate in different DNA repair and replication pathways, whereas others are unique to specific types of repair. In fact, some are not directly involved with repair per se but rather link DNA repair to other cellular functions, including damage sensing, cell cycle checkpoint control, chromatin remodeling, and apoptosis.9 Inactivation or loss of any of these myriad proteins can lead to dysfunction of the DNA damage response, which in turn can precipitate diverse clinical syndromes of varying severity including immunodeficiency, neurodegenerative disorders, infertility, premature aging, hypersensitivity to DNA damaging agents, and cancer proneness.10
It is of particular interest that most cancer cells harbor one or more defects in the DNA damage response. Further, because different DNA repair pathways can share common components and functions under normal circumstances, and in some cases compensate for defects in other pathways, it may be that the clinical strategy of DNA repair inhibition will have a greater effect on the tumor than on normal tissues.10 The approach of combining DNA repair inhibitors with radiation to produce selective tumor radiosensitization is discussed further later in this chapter.
The Molecular Biology of Cellular Radiation Responses
The first activated oncogene identified to confer radiation resistance was NRAS (see references 11 and 12). Ras proteins, products of RAS family member genes, are small guanosine triphosphatases activated by upstream receptor tyrosine kinases. They transduce signals to a complex downstream cascade of protein kinases that have the net effect of promoting cell survival (for example, by the suppression of apoptotic cell death pathways) and unlimited proliferation (secondary to, for example, the downregulation of antiproliferative proteins), two of cancer’s hallmark phenotypes. When the ras protein is mutated, however, as is the case in approximately 30% of all human cancers, these pathways become overactive and no longer responsive to the normal regulatory mechanisms that antagonize the ras pathway and reign in excessive proliferation. This situation is exacerbated in many tumors by the concurrent loss of function of tumor suppressor proteins.
Ras became, accordingly, a desirable target for molecular cancer drug development.13 The general approach to targeting ras focused on its need for posttranslational modification through prenylation to become activated.14 Despite encouraging preclinical results with a class of prenylation-inhibiting agents known as farnesyl transferase inhibitors—drugs that, when used alone or combined with radiation, produced sensitization of tumor cells with constitutively active ras pathways—these drugs did not meet expectations in clinical trials because of alternative prenylation pathways.13
Subsequent drug development focused on inhibiting proteins upstream of ras, in particular, members of the human epidermal growth factor receptor (EGFR) family of receptor tyrosine kinases that transduce growth signals through ras and other signaling proteins.15,16 These transmembrane glycoproteins are activated through binding of ligands belonging to the EGF family of peptide growth factors.
Cetuximab, a human-murine chimeric immunoglobulin G1 monoclonal antibody raised against the EGFR, is among the more successful molecularly targeted drugs, yielding improved outcomes in squamous cell head and neck cancers when combined with radiation therapy.17 Details of its mechanism of action and clinical use are discussed later in this chapter. Other classes of drugs that target EGFR include the small molecule tyrosine kinase inhibitors gefitinib and erlotinib, which inhibit EGFR phosphorylation and its downstream cascade by blocking the intracellular catalytic domain of the receptor.18 These drugs have shown some efficacy in selected patients with treatment-refractory, advanced non–small cell lung cancer (NSCLC).19
A second molecular target of interest is vascular endothelial growth factor (VEGF) and/or its cell surface receptor. VEGF is the most potent of the proangiogenic endothelial cell proliferation stimulators and chemoattractants and plays a pivotal role in promoting tumor survival by stimulating the growth of new blood vessels derived from the host vasculature. This phenotype of sustained angiogenesis is a hallmark property of cancer. Several isoforms of VEGF bind to the corresponding receptors VEGFR1, VEGFR2, and VEGFR3 (encoded, respectively, by the genes FLT1, KDR, and FLT4).20
Bevacizumab, a recombinant humanized monoclonal antibody, is the first molecularly targeted antiangiogenic drug to gain approval for clinical use by the U.S. Food and Drug Administration. This antibody binds to VEGF, with the net effect of eliminating signaling to vascular endothelial cells to initiate angiogenesis.21,22 The clinical use of bevacizumab in combination with radiation therapy is discussed later in this chapter.
Cell Survival and Tissue Dose-Response Curves
Death as a permanent, irreversible cessation of vital functions is not the same as what constitutes “death” to the radiation biologist or oncologist. For proliferating cells, including those maintained in vitro and the stem cells of both normal tissues and tumors in vivo, cell death in the radiobiological sense refers to a loss of reproductive integrity or clonogenicity, that is, an inability to sustain proliferation indefinitely. It is important to note that the term “clonogenic death,” as first described more than 50 years ago, is operationally defined and today serves as a catch-all term encompassing various mechanistic ways that cells die, all of which culminate in a cell losing its ability to divide indefinitely. These modes of cell death include mitotic catastrophe (the most common form of cell death after radiation exposure), apoptosis, autophagy, necrosis, senescence, and, strictly speaking, differentiation as well, to the extent that differentiated cells lose their ability to divide.23 Another noteworthy feature of clonogenic death is that it does not necessarily preclude the possibility that a cell may remain physically intact, metabolically active, continue its tissue-specific functions, and even divide a limited number of times after irradiation.
The first report of a quantitative measure of intrinsic radiosensitivity for a human cell line (HeLa, derived from a cervical carcinoma) was published by Puck and Marcus in 1956.24 For different doses of x-rays, the reproductive integrity of HeLa cells was measured by their ability to form macroscopic colonies of at least 50 cells (corresponding to approximately six successful postirradiation cell divisions) on Petri dishes. The HeLa cell survival curve, in which the log of the surviving fraction of cells was plotted as a function of the radiation dose, was characterized by a roughly exponential dose response at intermediate to high doses and a bending, “shoulder” region at low doses where cell killing was less effective. This phenomenon is illustrated graphically in the upper panel of Figure 27-7. Radiation survival curves for hundreds of cell types derived from mammalian tumors and normal tissues have been generated in the years since the pioneering work of Puck and Marcus,24 and most are qualitatively similar to the original HeLa survival curve (see lower panel of Fig. 27-7).
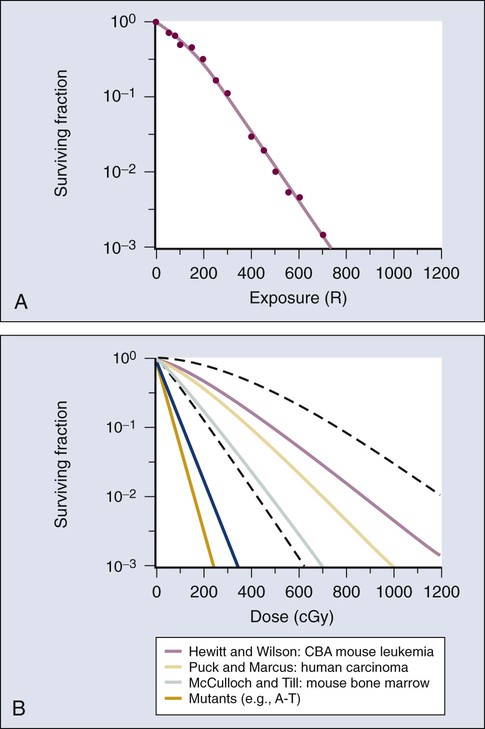
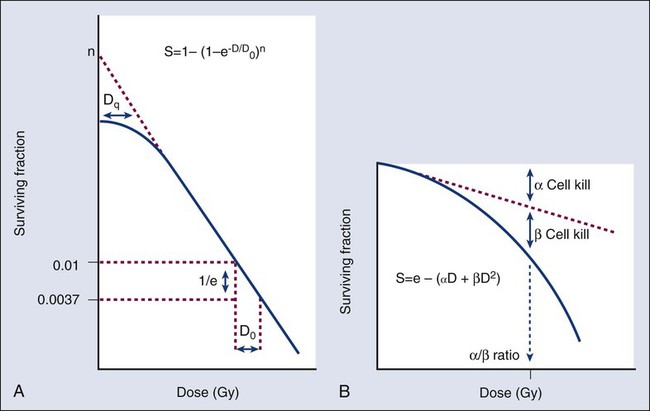
Mathematical models were developed to fit the cell survival data, with survival curve theory originating in a consideration of the physics of energy deposition in matter by ionizing radiation. An assumption inherent to target theory was that a biological response (cell killing in this case) resulted from critical “targets” receiving random “hits”25 in a probabilistic manner. Further, for cell survival curves with shoulders, each target was envisioned as requiring more than one hit to elicit the response, that is, that “sublethal” damage had to accumulate first before the cell would be killed. One mathematical expression derived from target theory that provided a fairly good fit to survival data was:
< ?xml:namespace prefix = "mml" />
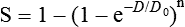
Over time, it became apparent that some features of this model were inadequate,26 not the least of which was that its basis was the probabilistic nature of energy deposition in matter by ionizing radiation and not anything biologically based. For example, target theory was not concerned per se with which biomolecules in the cell were the purported “targets” of radiation damage, what the nature of the damage was in a molecular sense, or how the cell responded to it. (Of course, it was not lost on radiation biologists of the day that at least one cellular target was likely to be the DNA contained in chromosomes.24)
A different and more biology-based interpretation of the dose response for radiation-induced cell killing was proposed by both Kellerer and Rossi27 and Chadwick and Leenhouts.28 The linear-quadratic (LQ) or “alpha-beta” equation,
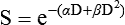
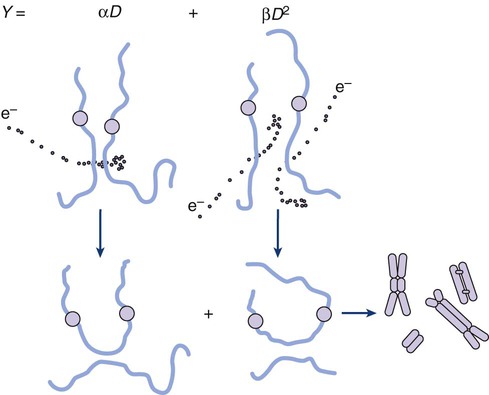
was shown to fit cell survival data quite well, particularly in the low-dose region of the curve where the target theory model often failed.26 In this expression, S is again the fractional cell survival following a dose (D), α is the rate of cell kill by a single-hit process, and β is the rate of cell kill by a two-hit mechanism. Implicit in the LQ model was that (borrowing the language of target theory for comparative purposes only) DNA (or a chromosome) was the target, and the hits corresponded to irreparable or misrepaired lesions produced by either one or two radiation tracks traversing the cell nucleus (Fig. 27-8).
A comparison of the features and parameters of the target theory and LQ survival curve models is shown in Figure 27-9.
To bridge the gap between the radiation responses of single cells grown in culture and tissues or tumors in a laboratory animal or human patient, several ingenious methods were developed to measure, or at least estimate, the radiation sensitivity of intact normal tissues and tumors in vivo.31–31 Some of these assays used the reproductive integrity of cells as an end point, similar in principle to the in vitro survival curve assay, but in which the animal essentially served as its own Petri dish. One classic example of an in vivo clonogenic assay is the spleen colony assay of Till and McCulloch,30 which originally was developed as a model system for the study of bone marrow transplantation. These authors determined that lethally irradiated mice could be “rescued” by a bone marrow transplant and that the transplanted, viable bone marrow stem cells were noted to form discrete nodules or colonies in the spleens of irradiated animals. By extension, then, a mouse’s spleen could be used as a pseudo Petri dish, with the number of clonogenic bone marrow colonies countable as a function of the radiation dose that the donated bone marrow received prior to transplantation. Assays such as this showed that the radiosensitivity of individual cells (tumor cells or normal cells) was largely unchanged whether the cells were irradiated in relative isolation in Petri dishes or as parts of a more complex tissue containing many different, interacting cell types in 3D contact.
Two examples of nonclonogenic assays, one for normal tissues and one for tumors, are worth mentioning, especially because aspects of both are used routinely in the clinic, if not in the exact same way as the laboratory assay. One of the first nonclonogenic methods developed to assess normal tissue radioresponse was the skin reaction assay.32 (Pigs were used in the original studies because their skin is similar to that of humans in several key respects, although rodents have also been used.) An ordinate scoring system was used to quantify the severity of the skin reaction; for example, a skin score of “1” might correspond to mild erythema, whereas a score of “4” might correspond to confluent moist desquamation over more than half of the irradiated area. Then, when comparing different time, dose, and fractionation schedules, any combination that resulted in the same skin reaction score was assumed to correspond to an equivalent amount of cell killing. In this way, and by collecting information for different severities of skin reactions, a dose-response curve could be generated. A common nonclonogenic assay of tumor response to radiotherapy is the regrowth delay assay.33 In this assay, the tumor’s dimensions (or volume) are measured periodically as a function of time after irradiation, with the degree of tumor shrinkage assumed to be a reflection of the fraction of clonogenic tumor cells killed. Dose-response curves are generated by plotting the amount of growth delay (in days) as a function of radiation dose.
Modifiers of Radiation Sensitivity
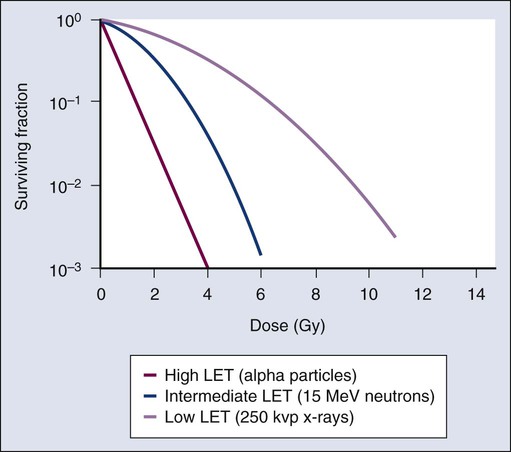
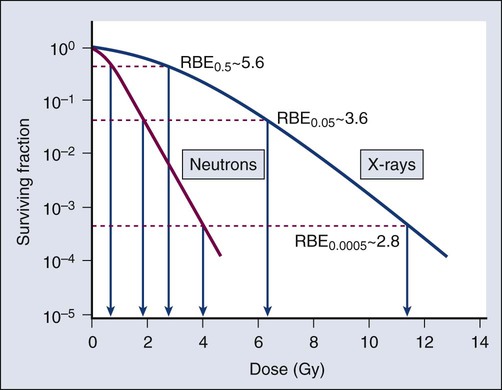
As discussed previously, factors both intrinsic and extrinsic to the cell can alter its radiosensitivity. In addition to the intrinsic, genetic determinants of radiation sensitivity, other physical and chemical modifiers can also play important roles. The type of radiation used for treatment can be considered a physical modifier of radiosensitivity to the extent that high LET types of radiation (e.g., neutrons and heavy ions) are more biologically effective for a given unit of dose than low LET types (e.g., x-rays and electrons). Representative dose-response curves for radiations with different LETs are shown in Figure 27-10. In light of these differences in biological potency, the term relative biological effectiveness (RBE) has been coined to compare and contrast two radiation beams of different LET. RBE is defined as the ratio of doses of a known type of low LET radiation (historically, 250 kVp x-rays were the standard, but others can also be used) to that of a higher LET radiation to yield the same biological end point. RBE is highly variable and depends on several irradiation parameters, including the type of radiation, total dose, dose rate, dose fractionation pattern, and the biological effect being assessed. An example of how RBE values are obtained from cell survival curves is shown in Figure 27-11.
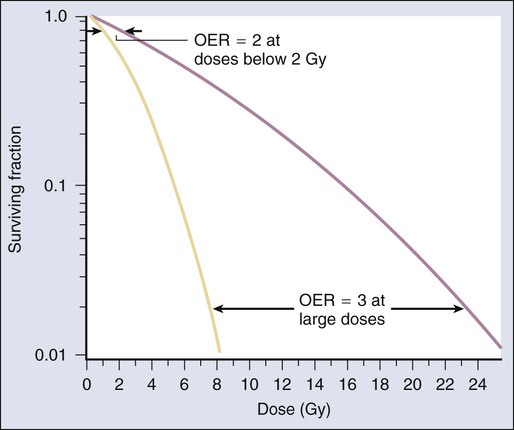
Chemical modifiers of radiation response are also important, and perhaps the “chemical” of greatest significance in this regard is molecular oxygen, a potent radiosensitizer. Mechanistically speaking, the reason that well-oxygenated cells are more sensitive to radiation than cells relatively lacking in oxygen is that oxygen readily participates in the free radical reactions that occur in the micro- to milliseconds after irradiation and has the net effect of enhancing the radiation damage to cellular macromolecules. Poorly oxygenated cells experience less enhancement of radiation injury and are therefore more radiation resistant. This phenomenon is termed the oxygen effect. The relative resistance of poorly oxygenated cells compared with well-oxygenated ones can be expressed in terms of an oxygen enhancement ratio (OER). The OER is the ratio of doses to produce the same biological effect under low versus normal conditions of oxygenation. The OER typically ranges between 2.5 and 3.0 for large single doses of x-rays or γ-rays and 1.5 to 2.0 when multiple small dose fractions are used.34 Further, the oxygen effect is reduced or eliminated as the LET of the type of radiation increases; OERs of 1.5 to 2.0 are obtained for radiations of intermediate LET, and an OER of 1.0 (i.e., no oxygen effect) is obtained for high LET radiations. Representative radiation survival curves generated in the presence or relative absence of oxygen are shown in Figure 27-12.
Hypoxic cell radiosensitizers, exogenous chemicals that mimic the radiation damage–enhancing effects of oxygen, have been used clinically in an attempt to combat the relative radiation resistance of tumors that contain a clonogenic fraction of poorly oxygenated, hypoxic cells. A second approach to the problem posed by tumor hypoxia is to combine radiotherapy with a drug that, rather than sensitizing hypoxic cells, kills them outright. These drugs are called “bioreductive” because their toxic effects only occur secondary to the reductive metabolism common in cells relatively lacking in oxygen.35,36 These drugs are discussed further later in this chapter.
One of the first studies demonstrating an association between directly measured oxygen tension in tumors and clinical outcome was published in 1988 by Gatenby et al.37 An oxygen-sensing electrode was inserted into patients’ tumors (advanced squamous cell carcinomas of the head and neck), and multiple readings of partial pressure of O2 were taken at different depths along the probe’s track; the arithmetic mean pO2 value for each tumor positively correlated with local control rate, as did the tumor volume–weighted pO2 value. A high tumor oxygen tension was associated with a high complete response rate and vice versa. Several years later, comparable oxygen electrode results for uterine cervix cancers were obtained by Höckel et al.38 They made the important discovery that tumor hypoxia was a negative prognostic indicator in general, regardless of whether the patient had received radiation therapy, suggesting that radiation resistance was only one aspect of the hypoxia problem. A large meta-analysis of the relationship between tumor oxygenation status measured with oxygen electrodes and clinical outcome was published more recently for advanced head and neck tumors,39 with comparable findings of improved overall survival in patients whose tumors were less hypoxic.
Another method of directly identifying and quantifying hypoxic cells in tumors is through the use of “hypoxia markers.” Both exogenous and endogenous markers for tumor hypoxia are available and can be studied in relation to each other, the tumor vasculature, or other “markable” features of the tumor microenvironment (e.g., the proliferation status of tumor cells). Exogenous markers consist of injectable drugs or chemicals that are bioreducible only under hypoxic conditions, causing them to bind to cellular proteins. These bound metabolites can be marked radioactively or with antibodies and can be visualized using several different techniques, such as autoradiography40 or fluorescence immunohistochemistry.41 Two exogenous markers studied extensively in both preclinical cell and animal systems, as well as in human patients, are pimonidazole hydrochloride42,43 and EF-5.44 Both are of the chemical class known as nitroimidazoles that were originally studied as possible radiosensitizers of hypoxic cells (e.g., see references 45 and 46, as well as the discussion later in this chapter) but were serendipitously found to also have the property of bioreduction and binding to cellular macromolecules under hypoxic conditions. This facilitated their use as hypoxia markers. Endogenous markers, on the other hand, consist of genes and proteins that are upregulated as part of the cellular stress/adaptive response to hypoxic conditions and whose expression levels can, with caveats, be used as surrogates for tissue oxygenation status.47,48 Commonly studied endogenous hypoxia markers include the hypoxia-inducible factor 1–alpha (HIF-1α, a transcription factor),48 the enzyme carbonic anhydrase IX (CA-9 or CAIX),49 glucose transporter–1 (GLUT1),52–52 and osteopontin.53
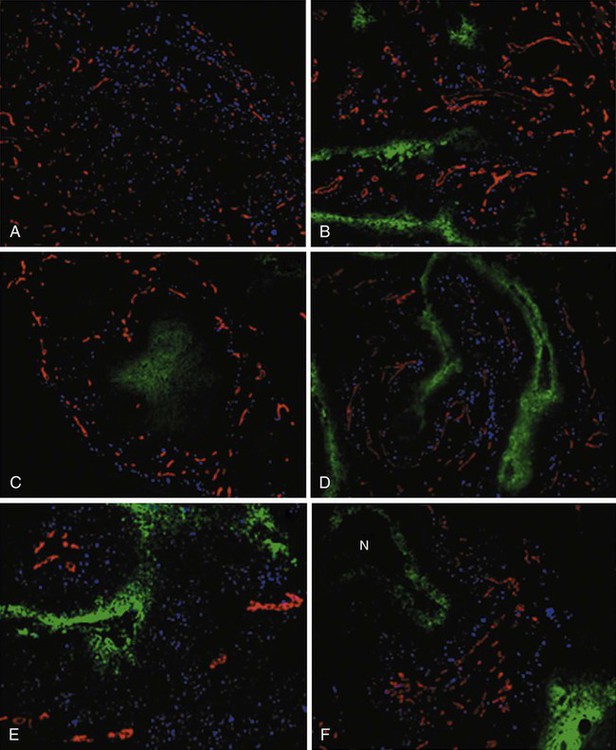
Figure 27-13 illustrates the potential of deriving “geographic” information about the extent and location of hypoxia in six different human tumors, using the exogenous marker pimonidazole hydrochloride. Simultaneously marking other tumor features, the location of blood vessels and proliferating cells in these examples, provides additional information about the tumor microenvironment.
Clinical Radiation Oncology
Therapeutic Ratio
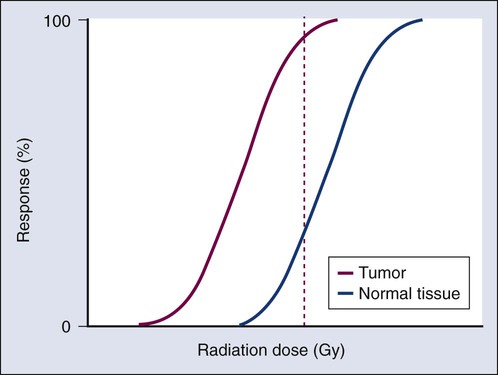
The concept of therapeutic ratio reflects the ability to estimate the likelihood of complication and tumor control in a given situation. At a given dose, these probabilities can be estimated (although usually not very accurately in clinical practice). Advances in radiation oncology can be made by moving the normal tissue complication curve to the right (as illustrated in Figure 27-14) so as to produce fewer complications at the same radiation dose, or by moving the tumor control curve to the left, so that less physical dose is needed to control the tumor (while also producing fewer complications).
The Biology of Fractionation
In the earliest days of radiotherapy, around the turn of the twentieth century, both x-rays (generated by applying an electric current across an x-ray tube1) and radium (a naturally occurring radioactive element2,3) were used for cancer treatment. Because of the greater availability and radiation output of x-ray tubes, delivering one or a few large single doses in short treatment times was convenient and efficient. This “massive dose technique”54 was a common way of administering radiation therapy from about 1900 through the 1920s. Unfortunately, normal tissue complications were severe, and to make matters worse, the rates of local tumor control were poor.
Meanwhile, radium therapy was used more extensively in France. Because of the low activity sources available, radium applications involved longer overall treatment times to reach comparable total doses as used in x-ray therapy. Although extended treatment times were less convenient, clinical results were frequently superior. Perceiving that the lengthening of the overall time was the critical factor and armed with animal experiments convincingly demonstrating the superiority of longer treatment times,55 French physicians began to experiment with the use of multiple, smaller x-ray doses delivered over extended periods in human patients.56 Clinical outcomes were improved to such an extent that fractionated radiation therapy using many small dose increments spread over several weeks’ time became the standard of care and has largely remained so to the present day. Therapy using radioactive sources (such as the aforementioned radium therapy pioneered by the French) also has continued to the present day, evolving into today’s practice of brachytherapy using both high and low activity radioactive sources capable of delivering a range of dose rates.
What was lacking in these early days of radiotherapy, however, and arguably for decades thereafter, was a biological basis for dose-rate/dose-fractionation effects. Radiobiologists studied dose-rate effects extensively in the laboratory in an attempt to better elucidate the biological factors involved in radiotherapy response; however, the clinical community was often unaware of these laboratory studies. This lack of cross talk between biologists and clinicians began to change with the publication in 1975 of a seminal book chapter entitled The Four R’s of Radiotherapy.57 The chapter was an attempt to explain the biological basis of fractionation by describing in simple terms the key radiobiological phenomena thought to affect the outcome of fractionated radiotherapy: Repair, Repopulation, Reoxygenation, and Redistribution.
Repair
The tenets of target theory suggested that the shoulder region of the radiation survival curve indicated that “hits” had to accumulate prior to cell killing. Elkind and Sutton58 sought to better characterize how this damage accumulated and how the cell processed it. Even in the absence of detailed information about DNA damage and repair at that time—the structure of DNA had only been elucidated a few years earlier—a few facts could be gleaned. First, the hits that were part of the damage accumulation process, yet did not in and of themselves produce cell killing, were, by definition, sublethal. Further, this sublethal damage (SLD) only became lethal if and when it interacted with additional SLD. Elkind and Sutton conducted experiments that deliberately interfered with the damage accumulation process by delivering part of the intended radiation dose, inserting a radiation-free interval, and then delivering the remainder of the dose in what was called a “split-dose” experiment.
The overall surviving fraction of cells after a moderate to high radiation dose was higher if that dose was split into two fractions with a time interval in between than if it was delivered as a single dose. This phenomenon is illustrated graphically in the right panel of Figure 27-15. This finding suggested that the cells that survived the initial dose fraction had “repaired” some of the damage during the radiation-free interval. Therefore this damage was no longer available to interact with the damage inflicted by the second dose fraction, and thus a higher cell-surviving fraction resulted. Had the total dose been delivered as a single fraction, more damage in total would have accumulated, some SLD would have been converted to lethal damage, and the cell-surviving fraction would have been lower. The results of split-dose experiments turned out to be crucial to the understanding of why and how fractionated radiation therapy works the way it does, that is, that fractionation/protraction of a total radiation dose reduces its toxicity, in large part because of SLD repair. This phenomenon is termed the dose-fractionation or dose-rate effect, and it occurs after low LET radiation exposure in an undiminished manner regardless of the number of dose fractions.
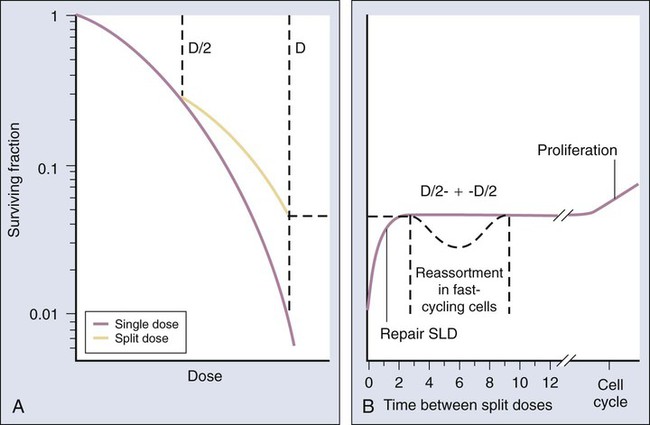
When considering complete cell survival curves, SLD repair manifests itself as a return of the low-dose, shoulder region of the radiation survival curve. After an initial radiation dose and an adequate time interval for repair to occur, the response of surviving cells to graded additional doses is nearly identical to that obtained from cells without previous radiation exposure (illustrated in the left panel of Fig. 27-15).
Bedford and Hall59,60 generated in vitro survival curves for HeLa cells irradiated at various dose rates and found that the killing effectiveness per unit dose decreased as the dose rate decreased, to a point. A limit to this dose rate effect was reached; that is, further lowering of the dose rate did not produce a further decrease in toxicity. This finding is consistent with the idea that survival curves have negative initial slopes, as described by the “αD” component of the LQ survival curve equation. A further implication is that small differences in initial slopes of survival or dose-response curves for different tissues could be magnified into large differences when many small dose fractions or continuous low dose rates were used.
Repopulation
The sparing effects of fractionated external beam and brachytherapy are largely attributed to the repair of SLD; however, other factors can be involved as well. Repopulation is defined as a compensatory increase in cell proliferation in tissues in response to an injury that results in a large amount of cell killing. Stem cells (known or suspected) of normal tissues and tumors can begin to proliferate during and after a course of radiation therapy,61 although the mechanism(s) the tissue uses to affect this process and the time it takes to commence varies with the tissue. Some tissues, however, do not seem to be able to mount a repopulation response, or at least, not a prompt or robust one, presumably because they only possess very small numbers of stem cells, if any.
For tumors, especially carcinomas, the prevailing view (although it is not without its detractors) is that compensatory repopulation does not begin until approximately a month into the course of treatment.62 One clinical implication of this view is that overall treatment times should be kept as short as practically possible, because treatment time much longer than a month would allow for more tumor cell repopulation and therefore less treatment effectiveness. Some clinical data (mostly culled from fractionation studies involving treatment of head and neck cancers) suggest that up to a third of a typical 1.8 to 2.0 Gy daily dose fraction is wasted as a result of repopulation62 and that for each day of treatment beyond the time that repopulation begins, as much as 1% of local tumor control is lost.
Reoxygenation
Tumor blood vessels tend to be abnormal both structurally, functionally, and physiologically as a consequence of the dysregulated angiogenesis characteristic of tumors63,64 (see also Chapter 8). One consequence is that tumors can develop microregions relatively lacking in nutrients and oxygen. And although prolonged oxygen deprivation, that is, anoxia, will eventually kill even the hardiest of tumor cells, hypoxia—a state of very low tissue oxygenation on the order of 0.5% oxygen or less (corresponding to a partial pressure of oxygen of about 10 mm Hg)65—is potentially survivable, particularly when it is intermittent or cyclic in nature. In contrast, the structure and function of the vasculature of normal tissues is such that it specifically avoids (in nearly all cases) the development of such gradients of oxygen and nutrients. Thus hypoxia has a built-in specificity for tumors, which, ironically, is an attractive feature clinically, where differences between normal tissues and tumors might be exploitable.
Unfortunately, as previously discussed, maintenance under hypoxic conditions renders cells more radiation resistant by upward of a factor of three (i.e., three times the total dose would be required to achieve the same biological end point). Thus a tumor containing even the smallest fraction of clonogenic, hypoxic cells could easily make or break the success of radiation therapy that is necessarily limited by the response of the aerobic, and therefore more radiosensitive, normal tissues.66 Many types of experimental solid tumors in laboratory rodents and spontaneous tumors in veterinary patients and humans studied to date show an average pretreatment hypoxic fraction of approximately 15% to 20%, although the range of values is quite broad,45,67 as might be expected given intra- and intertumor heterogeneity.
Of course, tumor hypoxia would not constitute a barrier to clinical success if, during the course of fractionated radiotherapy, the fraction of hypoxic cells decreased, ideally to zero. This process is called reoxygenation, and it has been studied extensively in experimental rodent tumors (e.g., see reference 68), and to a limited extent in human tumors as well (e.g., see reference 64).
The reoxygenation process can be either slow (days to weeks) or fast (hours), and it can be complete or incomplete, depending on the type and extent of hypoxia present in the tumor. Slower reoxygenation might be expected to occur in cases in which the type of hypoxia is chronic, such as might occur in cells that are multiple cell diameters away from vasculature and near or beyond the diffusion distance of oxygen (approximately 70 µm34,44). Physiologically speaking, the main ways that reoxygenation could occur under these circumstances is for aerobic cells closer to vasculature to decrease their oxygen consumption, which would occur if such cells are killed in large numbers by the radiation, or for the tumor mass as a whole to shrink in size, which would have the net effect of bringing the chronically hypoxic cells closer in to the existing vasculature. Some, although not all, human tumors shrink at least somewhat during the course of radiotherapy. Conversely, in cases in which hypoxia is intermittent or cyclic, reoxygenation could occur on the order of minutes to hours, as has been noted for many experimental rodent tumors.69,70
Redistribution
It was the pioneering work of Terasima and Tolmach71 during the 1960s that led first to the development of a relatively simple method of obtaining populations of cells synchronized with respect to cell cycle phase, followed by the generation of radiation survival curves for each phase.
After a single dose of approximately 7 Gy of x-rays, Chinese hamster V79 cells in culture were noted to be most radioresistant in (late) S phase. Cells in G1 phase were of intermediate radiosensitivity (i.e., more resistant at the beginning of the phase but somewhat more sensitive toward the end of the phase), and G2 cells were increasingly sensitive as they moved toward the most radiosensitive M phase. This phenomenon was termed the cell cycle “age response” for ionizing radiation71 and is a general feature of mammalian cell radiation response.72 (The original work by Terasima and Tolmach71,73 also showed this feature to be true for human HeLa cells.) Differences in radiosensitivity for cells in each phase were largely attributable to changes in the shoulder regions of their respective survival curves, with late S-phase cells characterized by broad survival curve shoulders, and mitotic cells showing strictly exponential cell killing with no survival curve shoulders (Fig. 27-16). It should also be noted that when high LET radiations such as neutrons are used in place of x-rays, the age response variation through the cell cycle is significantly reduced or eliminated.
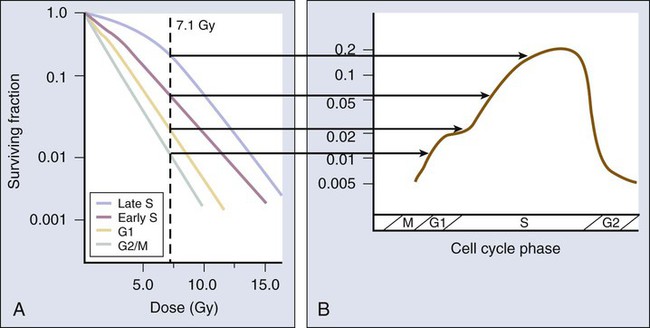
Because of the age response through the cell cycle for low LET radiations, an initially asynchronous population of cells surviving a dose of radiation becomes enriched with more resistant cells (S-phase, and to a lesser extent, G1-phase cells). This partial synchrony decays rapidly, however, in part because of radiation-induced perturbations in cell cycle progression and also because of natural variations in cell cycle phase durations. Such cells are said to have “redistributed,”74 with the net effect of sensitizing the population as a whole to subsequent dose fractions compared with what would have been expected had the cells remained in their resistant phases. The redistribution process can be demonstrated in rapidly growing cells in vitro but tends to be dampened in vivo because of the longer (and dispersed) cell cycle times and lower fractions of actively proliferating cells in tumors. Attempts to take advantage of redistribution clinically, however, such as by carefully timing each dose fraction to correspond to times when tumor cells would be expected to be most radiosensitive, have been unsuccessful.75 In fact, it has been difficult to demonstrate that redistribution has occurred in tumors (or normal tissues, for that matter) at all, other than by indirect inference, that is, that increasing resistance to each subsequent dose fraction during radiotherapy does not occur.
Traditional Radiosensitizers
Sensitizers of Proliferating Cells
Halogenated analogs of the DNA precursor thymidine, such as bromodeoxyuridine (BrdUdR) and iododeoxyuridine (IdUdR), can be incorporated into the DNA of actively proliferating S-phase cells in place of thymidine because of close structural similarities between the compounds. Cells that contain DNA substituted with these compounds are more radiosensitive than their unsubstituted counterparts, with the amount of sensitization directly proportional to the fraction of thymidine replaced.76 In general, the radiosensitization takes the form of a decrease in or elimination of the shoulder region of the radiation survival curve. The molecular basis of the ability of BrdUdR and IdUdR to radiosensitize the cells that incorporate the drugs into their DNA remains somewhat unclear. However, it is likely that both the formation of more complex radiation-induced lesions caused by the presence of the halogen atoms and an interference in DNA repair are involved.77
The clinical use of halogenated pyrimidines began in the late 1960s, with a major clinical trial in persons with head and neck cancer.78 Although BrdUdR did improve the tumor response to radiotherapy, it also produced severe oral mucositis as, in retrospect, might have been expected given that the mucosa also contains rapidly proliferating cells that would have been radiosensitized. As a result, the therapeutic ratio did not improve. Subsequently, these drugs were evaluated in more appropriate disease sites such as brain tumors and sarcomas, which consist of rapidly growing tumor cells against a background of slowly proliferating or nonproliferating cells of the surrounding normal tissues.79,80 Unfortunately, these drugs did not improve outcomes sufficiently to warrant their adoption for routine clinical use.
Sensitizers of Hypoxic Cells
The increased radiosensitivity of cells in the presence of oxygen is due to oxygen’s affinity for the electrons produced by the ionization of biomolecules. Molecules other than oxygen also have this chemical property, known as “electron affinity,”81 some of which are not consumed by the cell for other purposes, as is oxygen. Assuming that such an oxygen-mimetic compound is not otherwise used by the cell, it should diffuse further from capillaries and reach hypoxic regions of a tumor, sensitizing the hypoxic cells to radiation.
One class of compounds that represented a reasonable trade-off between efficiency as a radiosensitizer (i.e., compared with oxygen) and diffusion effectiveness was the nitroimidazoles, which include the drugs misonidazole, etanidazole, and nimorazole, all of which were the subjects of intensive preclinical and clinical studies over several decades. Unfortunately, the nitroimidazoles were found to cause adverse effects in normal tissues (in particular, peripheral neuropathy), which curtailed their clinical utility.82,83 In fact, the only one of this class of compounds currently approved for clinical use is nimorazole, which is used in Europe for subgroups of patients with advanced head and neck cancer.84
Another class of hypoxic cell radiosensitizers does not sensitize cells per se but rather kills them outright. This action produces the net effect of (apparent) sensitization of the tumor because of the elimination of an otherwise radioresistant subpopulation of cells. Agents selectively toxic to hypoxic cells are termed “bioreductive drugs,” with the organic nitroxide tirapazamine (formerly SR 4233)34,85 being the lead compound.
In preclinical models, tirapazamine has been shown to outperform the nitroimidazole radiosensitizers using clinically relevant fractionated radiotherapy86 and with some chemotherapy drugs either alone or in chemoradiotherapy regimens.87 In humans, more than 20 clinical trials are ongoing or complete, including randomized phase 2 and 3 trials with tirapazamine combined with radiotherapy and/or chemotherapy (particularly, cisplatinum) for advanced head and neck, cervix, and lung cancers. Although tirapazamine has improved outcomes for some standard regimens, results overall have been mixed (e.g., see references 88 and 89). Thus although the drug has in no way become a staple of clinical practice, its activity against some tumors warrants further investigation, as does the search for more effective agents from the same or similar chemical classes.
Traditional Radioprotectors
In theory, if normal tissues could be made to tolerate higher total doses of radiation through the use of radioprotectors, then the relative radioresistance of hypoxic tumor cells would no longer limit treatment. One such agent, amifostine, is a compound containing thiol moiety that was developed by the U.S. Army for use as a radiation protector for soldiers on a nuclear battlefield. It subsequently was repurposed for clinical use. Chemically, amifostine is an analog of naturally occurring radioprotective sulfhydryl compounds found in cells, such as cysteine, cysteamine, and glutathione.90 Like those antioxidant compounds, amifostine’s mechanism of action involves either the scavenging of free radicals produced by ionizing radiation, radicals that otherwise could go on to damage other cellular macromolecules, or reacting with oxygen, thereby preventing “fixation” of damage that has already been registered. Amifostine can also detoxify other reactive species, and because of this mechanism, the drug can also be used as a chemoprotective agent.91
Amifostine is approved for use in patients receiving radiotherapy for head and neck cancer to reduce xerostomia as a result of exposure of the parotid glands.92 It is also indicated for the reduction of renal toxicity associated with cisplatin chemotherapy in patients with advanced ovarian and NSCLC.93
Chemotherapy Drugs as Radiosensitizers
It has been known for decades that certain pharmaceutical agents can increase the tumor cell kill produced by radiation therapy.94 One of the earlier approaches used in this manner, and one that is still very commonly used, is the combination of a fluoropyrimidine (typically 5-fluorouracil or, more recently, capecitabine) with radiation therapy for a variety of tumors, but especially those of the GI tract.95 The underlying mechanism(s) of action of these fluorinated pyrimidines as radiation sensitizers is still not completely clear, but it most likely involves some combination of the drug’s selective toxicity to otherwise radioresistant S-phase cells, interference with DNA synthesis and repair, and ability to dysregulate the cell cycle.94 At present a fluoropyrimidine is used routinely in the therapy of most tumors of the GI tract, including esophageal, gastric, pancreatic, rectal, and anal cancers. In the past, the combination had been used in head and neck and uterine cervix squamous cell carcinomas, although that practice is much less common at present. The details of these combinations are described more fully in the individual disease site chapters in this book. However, it should be noted that local control has been consistently improved by this approach and has had a beneficial effect on survival in some of these diseases.
Another drug commonly used with concurrent therapy is cisplatinum, an inorganic compound consisting of a coplanar complex capable of producing both inter- and intrastrand DNA and protein cross-links.94 The cross-linking produced by cisplatinum most likely interferes with cellular repair of radiation damage, particularly DNA double-stranded breaks, resulting in radiation sensitization,96 although as in the case of the fluoropyrimidines, additional mechanisms of action have also been proposed.94 This combination is routinely used for head and neck and uterine cervix cancers (e.g., as described in references 97 and 98), with a major impact on long-term outcome. Mitomycin C is routinely used in the therapy of anal cancer,99 temozolomide is in standard use in the therapy of high-grade gliomas,100 and various other combination therapies have also been explored.
Molecularly Targeted Drugs and Biologics
Investigators have a great interest in the use of biological therapies in combination with radiation therapy. Although preclinical data have been generated for quite a few of these agents, with even newer compounds constantly being developed, the only two that have gone through substantial clinical testing and entered the treatment mainstream are anti-EGFR inhibitors used to treat squamous cell carcinomas of the head and neck17 and anti-VEGF therapies in the treatment of GI tract tumors.101
The use of cetuximab, a monoclonal antibody raised against the EGFR, has been shown to improve outcomes in head and neck cancers when combined with radiation therapy17 and is being explored in a variety of other clinical situations. Cetuximab competitively binds to the extracellular domain of EGFR, preventing activation of the receptor by endogenous ligands, which abrogates its near-constant signaling to cancer cells to proliferate. The antibody-receptor complex is then internalized and degraded, resulting in a downregulation of EGFR expression relative to the overexpression often displayed by diseases such as squamous cell cancers of the head and neck.
Bevacizumab exploits a different cancer hallmark, namely the ability of tumors to continuously recruit new blood vessels from surrounding normal tissues. This recruitment is accomplished through the production and release into their microenvironment by tumor cells of large quantities of (among other proangiogenic factors) VEGF, the principal stimulant of endothelial cells of the host vasculature to reproduce and migrate into the tumor mass, forming new—if immature and aberrant—blood vessels. The observation that tumors cannot grow beyond a size of approximately 2 mm3 without the support of neovascularization102 has led to accelerated development of angiogenesis-inhibiting agents, many of which target either VEGF or its endothelial cell surface receptor.103,104
Bevacizumab is a humanized monoclonal antibody raised against VEGF.105 The binding of the antibody to VEGF prevents it from binding to its receptor on the surface of vascular endothelial cells, which has the net effect of inhibiting the downstream signaling events that cause these cells to begin proliferating and forming vessel “sprouts” that migrate toward the tumor mass. At first blush, therapy that blocks new blood vessel formation might be expected to create more tumor hypoxia, and therefore more radiation resistance. However, findings of both preclinical and clinical studies with bevacizumab and related inhibitors of angiogenesis show that they most often promote radiation (and chemotherapy) sensitivity, not resistance. One hypothesis to explain this observation is that antiangiogenic therapy actually “normalizes” tumor vasculature106 by selectively pruning away small, immature, abnormal tumor blood vessels, leaving the larger, most “normal-like” vessels behind. This process would lead to improved tumor oxygenation and better access of chemotherapy agents.
Bevacizumab had been shown to prolong both overall survival and progression-free survival in patients with advanced colorectal cancer when added to a standard chemotherapy regime consisting of irinotecan, 5-fluorouracil, and leucovorin.107,108 In persons with rectal cancer,109 bevacizumab has been evaluated in combination with radiation therapy, and although initial reports were encouraging, with excellent early responses, it remains unclear whether the combination will improve clinical efficacy. Also, despite prolonged progression-free survival, no clear overall survival benefit of bevacizumab was seen in large clinical trials for NSCLC,110 renal cell carcinoma,111 and metastatic breast cancer.112 In response to the disappointing results in persons with metastatic breast cancer, the U.S. Food and Drug Administration has recently withdrawn its approval of bevacizumab as a first-line therapy, concluding that the drug’s benefit did not outweigh its adverse effects, which can include hypertension, proteinuria, bleeding episodes, and thrombotic events.113 Continued interest exists in trying to explore the vascular normalization hypothesis in a variety of tumor types.
Because many newer biological agents have been shown to have radiation-sensitizing properties in vitro and in animal systems, investigators have a great interest in translating this information to the clinic. For example, one class of drugs currently in clinical trials is the poly(adenosine diphosphate–ribose) polymerase or “PARP” inhibitors. The PARP family of proteins participate in a number of cellular functions, including detecting the presence of DNA strand breaks and activating signaling pathways that recruit and mobilize DNA repair proteins to the site of the damage.114 PARP1 is by far the most abundant member of this family and has been the most frequently targeted for inhibition. PARP inhibitors have been evaluated in preclinical models as potential radiation sensitizers, given that unrepaired or mis-rejoined DNA double-strand breaks are considered the lethal lesions with respect to the cytotoxic effects of radiation and that PARP inhibition might be expected to enhance this damage and increase toxicity.
DNA repair inhibition will only be clinically useful insofar as it is selective for tumors, because inhibiting repair in irradiated normal tissues could have adverse results. Fortuitously, PARP inhibition was found to be selective for tumors, in particular the tumors whose cells were already defective in at least one DNA repair pathway, as is the case for cells that carry mutations in the BRCA1 or BRCA2 genes, for example.115,116 Because cells from normal tissues have intact alternate or “salvage” pathways for DNA repair, the PARP inhibitors should be less toxic. Phases 1 and 2 clinical trials of PARP inhibitors are currently under way for brain tumors.
Normal Tissue Toxicity
Tolerance Doses
Second, although it is generally true that tissues known to be composed of radiosensitive cells have lower tolerance doses (e.g., the tolerance dose for a 5% risk of radiation-induced bone marrow aplasia within 5 years of treatment, the “TD5/5,” is ≈2.5 Gy) compared with the tolerance doses for tissues containing more radioresistant cells (e.g., the TD5/5 for rectal stenosis or fistula formation is ≈60 Gy),45 this situation is not universally the case because radiosensitivity is not the sole determinant of radiation response. Early and late effects in irradiated normal tissues do result from the killing of critical target cells either directly or indirectly; however, the “process” that culminates in the complication is both complex and dynamic, involving radiation-inducible gene expression (including production of cytokines and growth factors), cellular signaling cascades, different modes of cell death, and compensatory proliferative responses.
Tolerance dose data for representative early- and late-responding normal tissues are shown in Table 27-2. Data are included for select normal tissues from three seminal publications (each about 20 years apart) that compiled such information based on exhaustive reviews of the literature and updated to reflect new knowledge about the etiology of normal tissue complications and changes in treatment technique over the years.
Table 27-2
Radiation Tolerance Dose and Complication Frequency Data for Select Normal Tissues
| Tissue | Early/Late | End Point | Irradiation and Volume Details | Tolerance Dose or Complication Frequency | Author |
| Lung | Early | Pneumonitis | Conventional fractionation; >½ of organ irradiated | TD5/5 ≈20 Gy | Rubin and Casarett (1968) |
| Conventional fractionation; ⅓ or whole organ irradiated | TD5/5 (⅓) ≈45 Gy TD5/5 (whole) ≈17.5 Gy |
Emami (1991) | |||
| Conventional fractionation; 3D-CRT; whole organ irradiated | 7 Gy ≈5% complication frequency 20 Gy ≈20% complication frequency 27 Gy ≈40% complication frequency |
QUANTEC (2010) | |||
| Rectum | Late | Ulcer/perforation/fistula/stricture | Conventional fractionation | TD5/5 ≈65 Gy | Rubin and Casarett (1968) |
| Conventional fractionation; 100 cm3 field | TD5/5 ≈60 Gy | Emami (1991) | |||
| Conventional fractionation; 3D-CRT | V50 <50%: <15% complication frequency V65 <25%: <15% complication frequency V75 <15%: <15% complication frequency |
QUANTEC (2010) | |||
| Spinal Cord | Late | Myelopathy | Conventional fractionation; 5- to 10-cm field | TD5/5 ≈50 Gy | Rubin and Casarett (1968) |
| Conventional fractionation; 5- or 20-cm field | TD5/5 (5 cm) ≈50 Gy TD5/5 (20 cm) ≈47 Gy |
Emami (1991) | |||
| Conventional fractionation; 3D-CRT; partial organ irradiation, including full cord cross section | Dmax = 50 Gy: <1% complication frequency Dmax = 60 Gy: 6% complication frequency Dmax = 69 Gy: 50% complication frequency |
QUANTEC (2010) |
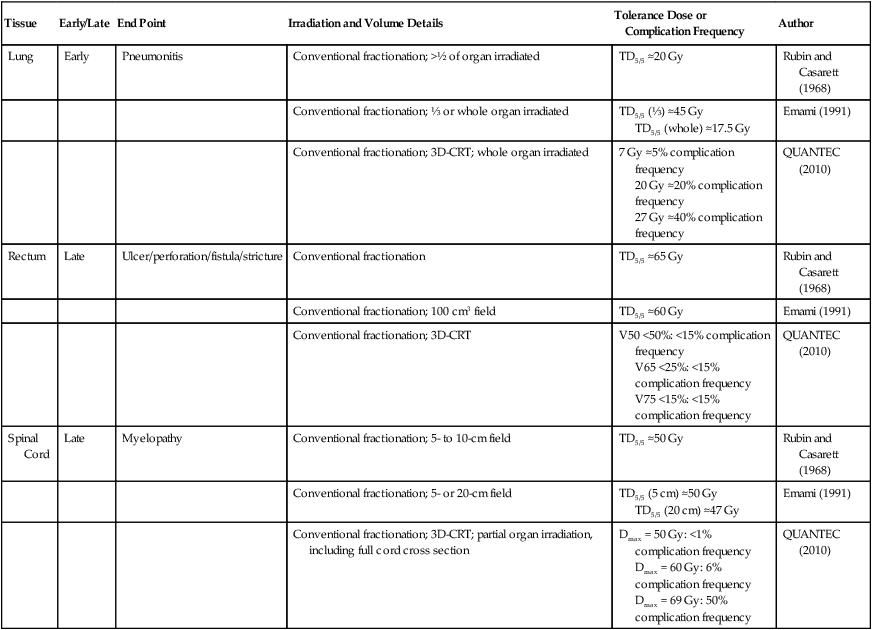
Data from Rubin P, Casarett GW. Clinical radiation pathology, vol. 1. Philadelphia: Saunders; 1968; Emami B, Lyman J, Broun A, et al. Tolerance of normal tissue to therapeutic irradiation. Int J Radiat Oncol Biol Phys 1991;21:109–22; and Marks LB, Yorke ED, Jackson A, et al. Use of normal tissue complication probability models in the clinic. Int J Radiat Oncol Biol Phys 2010;76(3 Suppl.):S10–9.
Radiation Carcinogenesis
With increasing numbers of long-term cancer survivors, one late normal tissue complication of radiotherapy that is of particular concern is the production of second malignancies. Leukemia accounts for about 20% of these second cancers, with the remainder presenting as solid tumors that develop in and around the previously irradiated site.117,118 Large epidemiological studies have assessed the breast and lung cancer risk in Hodgkin lymphoma survivors119 and leukemia and sarcomas in cervical cancer survivors.120 Certain subpopulations of previously irradiated patients are at an even higher risk than the majority, including children and young adults (who would be expected to live long enough for such second cancers to develop, and, in the case of children, who are more radiosensitive than adults to begin with), immunocompromised individuals, and persons with a known genetic predisposition to cancer. Large studies of previously irradiated patients demonstrate that the risk of late cancer developing in an incidentally irradiated organ will very roughly double the baseline risk.121 Fortunately, the baseline risk in an individual organ is quite low, and thus the absolute impact of a radiation-induced cancer is also low, assuming there is clinical benefit in the original therapy. However, the impact of a second cancer occurring many years after a prior cancer developed can be severe.
Volume Effects
By convention, normal organs have been divided into what are called “serial” and “parallel” organs, by analogy with electrical circuits.122 A serial organ is one in which a severe injury at any anatomic point in the normal structure will produce a severe functional loss. The classic example of a serial organ is the spinal cord. If there is major damage to the cord at any level, from a small dosimetric hot spot, for example, all function can be lost below that level. Examples of parallel organs are the kidneys and the liver. Radiation injury to a portion of one of these organs will generally just decrease its function by an amount approximately equal to the percent of the organ that is destroyed but often will not produce a major functional deficit unless the irradiated volume is large.
Fractionation Sensitivity
In multifraction experiments assessing skin reactions in mice, Douglas and Fowler123 analyzed their data using what was then a novel method that allowed the derivation of values for the ratio α/β, parameters of the LQ formula. This new approach to isoeffect analysis focused on the shape of dose-response curves, particularly in the low-dose region, and by extension, that the critical parameter in radiotherapy planning was the size of the dose per fraction. More widespread use of this technique in subsequent years revealed that in most cases, a systematic difference was found between early- and late-responding normal tissues and tumors in their responses to different fractionation patterns. The α/β ratios tended to be low for late-responding normal tissue end points (on the order of 1 to 6 Gy, with an average of about 3 Gy), and high for early-responding normal tissue and tumor end points (usually 7 to 20 Gy, with an average of about 10 Gy). Table 27-3 shows α/β ratios determined for a variety of human normal tissues and tumors; worth noting are the few tumor types (melanoma and prostate cancer for example) that are exceptions to the general trend and are characterized by low α/β ratios.
Table 27-3
Representative α/β Ratios for Human Normal Tissues and Tumors
| Tissue Type (and End Point) | α/β Ratio (±95% Confidence Interval) |
| EARLY-RESPONDING NORMAL TISSUES | |
| Skin: Erythema | 10.6 (1.8; 22.8) Gy |
| Desquamation | 11.2 (8.5; 17.6) Gy |
| Lung: pneumonitis ≤90 d after radiotherapy | >8.8 Gy |
| Oral mucosa: mucositis | 8-15 Gy |
| LATE-RESPONDING NORMAL TISSUES | |
| Skin: Telangiectasia | ~2.7 (−0.1; 8.1) Gy |
| Fibrosis | 1.7 (0.6; 3.0) Gy |
| Breast: poor cosmesis | 3.4 (2.3; 4.5) Gy |
| Fibrosis | 3.1 (1.8; 4.4) Gy |
| Lung: pneumonitis >90 days after radiotherapy | 4.0 (2.2; 5.8) Gy |
| Bowel: perforation/stricture | 3.9 (2.5; 5.3) Gy |
| Spinal cord: myelopathy | <3.3 Gy |
| TUMORS | |
| Head and neck: nasopharynx | 16 (−11; 43) Gy |
| Vocal cord | ~13 Gy |
| Tonsil | 7.2 (3.6; ∞) Gy |
| Larynx | 14.5 (4.9; 24) Gy |
| Lung: squamous cell carcinoma | ~50-90 Gy |
| Cervix: squamous cell carcinoma | >13.9 Gy |
| Skin: Squamous cell carcinoma | 8.5 (4.5; 11.3) Gy |
| Melanoma | 0.6 (−1.1; 2.5) Gy |
| Prostate | 1.1 (−3.3; 5.6) Gy |
| Breast (early-stage invasive ductal, lobular, and mixed) | 4.6 (1.1; 8.1) Gy |
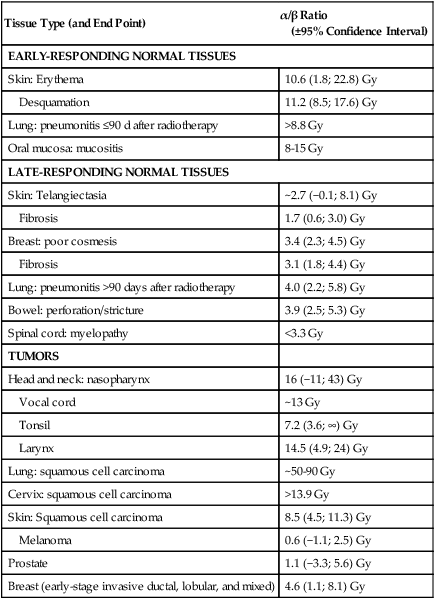
Data from Joiner M, van der Kogel A, editors. Basic clinical radiobiology. 4th ed. London: Hodder Arnold; 2009.
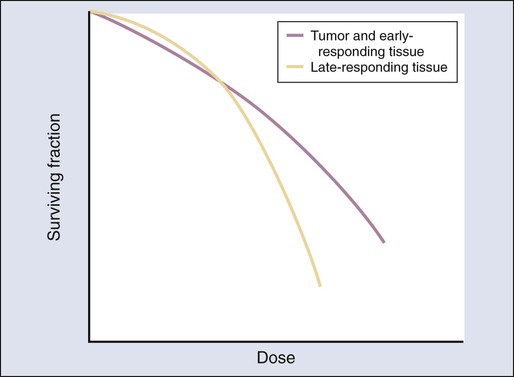
Representative dose-response curve shapes for tissues characterized by low or high α/β ratios are shown in Figure 27-17. The curve with the low α/β ratio has a shallow initial slope at low doses but curves downward rather abruptly as the dose increases. Conversely, the curve with the high α/β ratio has a steeper initial slope and bends more gradually as the dose increases.
In parallel with the fractionation experiments used to determine tissue α/β ratios, other investigators124 quantified the fractionation sensitivity of experimental tumors and normal tissues using a plot of the log of the total dose to achieve a particular tissue end point versus dose per fraction; this is called an isoeffect curve, and this method of plotting tolerance dose data had been in common practice in radiation oncology since the 1940s.125 Plotted in this way, the curves for slowly proliferating or nonproliferating normal tissues such as the kidney and spinal cord, for example, were found to be steeper than those for more rapidly proliferating, early-responding tissues, such as skin and gut epithelium, and most tumors.124,125 Taken together, the low α/β ratio and steep isoeffect curve suggest that late-responding tissues are more sensitive to changes in dose per fraction, thus experiencing greater sparing with decreasing fraction size than their early effects counterparts. This phenomenon is illustrated graphically in Figure 27-18. It was immediately evident that this phenomenon could be exploited for clinical benefit.
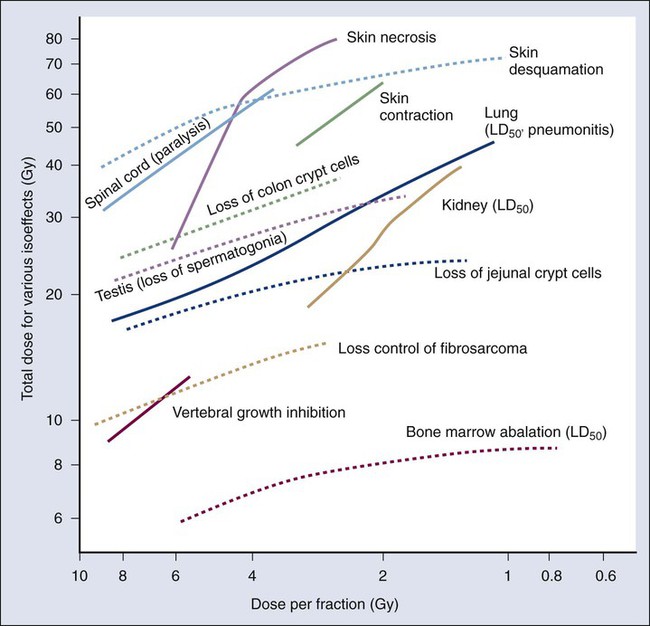
Clinical Application of the LQ Isoeffect Model
The shapes of tissue and tumor isoeffect curves and their calculated α/β ratios have a number of clinical applications. One is to use α/β ratios to generate alternate treatment schedules using different-sized doses per fraction in order to match the probability of causing a tissue injury. Making such a calculation assumes the overall treatment times are similar in both schedules or that the tissue at risk of a complication is relatively insensitive to treatment duration.126 The equation,
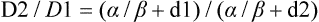
can be used for this purpose, where D1 and d1 are, respectively, the total dose and dose per fraction of one radiotherapy treatment plan and D2 and d2 are, respectively, the total dose and dose per fraction for an alternate treatment plan intended to be biologically equivalent for a particular tissue effect, and with the fractionation sensitivity of that tissue defined by its α/β ratio. One example of the utility of this approach is to express particular time-dose-fractionation schedules in terms of “equivalent total dose if the treatment had been given in 2 Gy fractions” or EQD2,
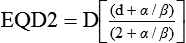
where D is the total dose actually delivered and d is the dose per fraction actually used. When comparing clinical trial data from multiple institutions, one can specify treatment parameters using EQD2 doses rather than the actual doses used, which makes the pooling of data with varying fractionation schedules more manageable, with the caveat that the results will only apply to the tissue of interest.
Another important implication of the steeper isoeffect curves for late-responding tissues compared with those for tumors is that it might be possible to increase the therapeutic ratio by using larger numbers of smaller fractions to a somewhat higher total dose than traditionally used.127 Although such treatments would be expected to exacerbate acute effects in some normal tissues (undesirable, but usually manageable) and in the tumor (desirable), late effects would be spared preferentially (also desirable). The use of multiple fractions per day of smaller than conventional size (less than about 1.8 Gy) but to a somewhat higher total dose, with little or no change in overall treatment time, has been termed hyperfractionation.
Hypofractionation, that is, the use of one or a few large dose fractions delivered over short periods (as would be the case for stereotactic radiosurgery or intraoperative radiation therapy), is also an option. Indications for the use of hypofractionation would include cases in which the complete ablation of small tumors is the goal, or in the relatively unusual circumstance in which the tumor is suspected of having a low, rather than high, α/β ratio. Prostate cancer and melanoma appear to be tumor types that meet this criterion, although this topic remains controversial, as does the use of hypofractionation in general.130–130
With particularly aggressive tumors that proliferate rapidly, another clinical strategy has been suggested, although it is not explicitly derived from the LQ model. Multiple treatments per day might also be useful to decrease the overall treatment time, thereby allowing less time for repopulation of clonogenic tumor cells.131 Treatment with multiple daily fractions of approximately standard size and number (and to about the same total dose), but in shorter overall times, has been called accelerated fractionation. In practice, however, a combination of accelerated and hyperfractionated treatment is most often used.132
Of course, determining the best treatment schedule for a particular patient is not simply a matter of avoiding a normal tissue complication or delivering a curative dose to the tumor, but rather, how the probabilities of both change when comparing different treatment possibilities. The “biologically effective dose” or BED method,133 another derivative of the LQ model, attempts to address this issue. BEDs are theoretical doses to achieve a particular tissue effect under conditions in which the fractionation pattern consists of an infinite number of infinitely small dose fractions. BEDs would be quite large for tissues characterized by low α/β ratios and steep isoeffect curves (like many late-responding normal tissues) and appreciably smaller for tissues characterized by high α/β ratios and shallower isoeffect curves (like most tumors and early-responding normal tissues). BEDs are not real doses but extrapolations based on the shapes of dose-response curves characterized by particular α/β ratios. For this reason, the units used to describe these theoretical, extrapolated doses are, for example, “Gy3‘s” or “Gy10‘s” rather than “Gy’s,” with the subscripts 3 and 10 referring to the α/β ratios for the tissues at risk. Although two radiotherapy treatment schedules can be compared qualitatively on the basis of their respective Gy3 or Gy10 “doses,” Gy3‘s and Gy10‘s cannot be intercompared.
Even with the BED concept being only semiquantitative at best, its use for treatment planning purposes during the past two decades has changed the radiotherapy standards of care for several tumor types and provided a wealth of clinical data that has allowed a better definition of what is or isn’t tolerable for particular normal tissues, in terms of Gy3‘s or Gy10‘s. Using head and neck cancer as an example, Fowler133 has suggested that for acute mucosal reactions, the maximally tolerated BED is in the range of 59 to 61 Gy10, and for brain necrosis, it is approximately 110 to 115 Gy3.
Radiation Therapy Delivery Approaches
1. Radiation therapy alone. Radiation therapy alone is often used for small tumors for which surgery would be excessive therapy either because of the precise location of the tumor or because of the limitations of surgical resection. Radiation therapy can also be used in this manner when a patient is too frail for a surgical procedure or when a patient refuses surgical intervention. Radiation therapy alone is used much less commonly for locally advanced disease, because it is now often combined with concurrent chemotherapy, and surgical procedures can often be performed even for extensive local-regional disease. Examples of this category include treatment of early stage cancers of the head and neck, prostate cancer of all stages, early-stage cervical cancer, and selected basal cell carcinomas of the skin.
2. Preoperative or postoperative radiation therapy. For some tumors, surgical therapy is required, but surgery alone is likely to leave residual disease either in the tumor bed or in regional lymphatics that are not completely resected. Radiation therapy in this situation will often be given with concurrent chemotherapy for the radiation-sensitizing effects of the chemotherapy. Examples include soft tissue sarcomas, certain cancers of the head and neck, intermediate and high-grade gliomas, and early-stage breast cancers.
3. Primary radiation therapy with concurrent chemotherapy. For many tumors, surgical resection is not an option because of the inability to perform a grossly complete surgical resection, and thus radiation therapy becomes the primary local therapy. Chemotherapy can be added either concurrently as a radiation sensitizer to enhance local control or before or after radiation therapy for control of systemic disease. Examples include all anal cancers and locally advanced tumors of the uterine cervix, esophagus, pancreas, and head and neck.
4. Tri-modality therapy. Often all three primary therapeutic modalities—surgery, radiation therapy, and chemotherapy—are needed for control of local, regional, and systemic disease. The timing of the therapies is dependent on the individual clinical situation. Tri-modality therapy is perhaps the most common approach at the present time. Examples include a large group of tumors, including esophageal, glioblastoma, head and neck, esophagus, pancreas, breast, and rectum tumors.
Radiation Therapy Delivery Techniques
External Beam Fractionated Radiation Therapy
Simulation, Treatment Planning, and Delivery of External Beam Radiotherapy
Poor soft tissue contrast from fluoroscopic imaging means that, on a conventional simulator, treatment parameters such as isocenter and beam boundaries can only be defined relative to visible landmarks, such as bony anatomy. More recently, techniques have been developed to use dedicated computed tomography (CT) scanners for “virtual” simulation, replacing conventional simulators in many radiation oncology departments. Virtual simulation is simulation that is based solely on the CT scan of a patient, and it is made possible by the ability to reproduce any arbitrary radiograph from a CT data set. In virtual simulation, the patient is placed in the orientation and position to be used for treatment delivery. A CT scan is acquired, and, with the patient still positioned on the scanning table, the physician uses the superior soft tissue contrast of the CT scan to select a location for isocenter within the target area (Fig. 27-19). An integrated laser system then moves to indicate the position of the physician-selected isocenter on the patient surface, allowing the external markers or tattoos to be placed for future alignment with the linear accelerator vault laser systems. The superior soft tissue contrast on CT images allows improved tumor localization and improved beam selection compared with fluoroscopy-based conventional simulators.
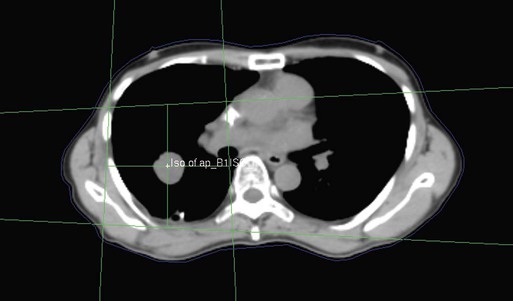
After simulation is completed, the CT scan and other images can be sent to a computer-based planning workstation, where the team of physicians, dosimetrists, and physicists can begin the treatment planning process. Treatment planning includes the identification of target volumes and normal structures requiring dose tracking and the selection and modification of beams to achieve specified dosimetric goals. The contouring process of identifying the boundaries of all treatment and avoidance structures takes place on the CT scan acquired at the time of simulation, but it may be supplemented by other imaging modalities, such as magnetic resonance imaging, positron emission tomography, or ultrasound. Normal structures are typically defined at their anatomic boundaries. The volume to be treated, defined as the target volume inInternational Commission on Radiation Units & Measurements Report 60 (ICRU 60),134 is created by combining three structures: the gross tumor volume (GTV), the clinical tumor volume (CTV), and the planning target volume (PTV). The GTV includes all disease detectable on each imaging modality, including visible primary tumor and involved lymph nodes. The CTV is an expansion of the GTV that includes regions of possible or probable microscopic disease, including both adjacent tissue and draining lymph nodes. The PTV is a further expansion of the CTV to account for anatomic motion and expected variations of patient daily setup. Expansions from GTV to CTV to PTV vary with disease, technique, and patient history, but typical expansion numbers from CTV to PTV are usually on the order of 1 cm or less. Expansion from GTV to CTV is entirely dependent on the biology of the clinical situation. At times, no GTV is present (such as treatment in the postoperative setting), in which case the CTV is based entirely on the assumed high-risk areas for residual disease.
Once target and avoidance structures are identified and defined on the patient images, a specific strategy for dose delivery can be developed by the physicians, physicists, and dosimetrists involved. The essential task of treatment planning is to select, arrange, and characterize a group of radiation beams to deliver a high dose to the tumor while keeping the dose delivered to normal structures under acceptable limits. The treatment plan must be specified in terms of the total dose and the number of fractions in which the dose will be delivered. Individual beams can be customized by specifying the beam energy, orientation, and shape. Beams are typically shaped to match the cross section of the PTV plus some small margin to maximize overlap with the tumor while limiting the amount of normal tissue directly exposed to the radiation beam. This goal can be accomplished by designing a customized block made from an easily cast metal alloy such as Cerrobend, but on modern machines it is typically performed with use of automated beam-shaping devices such as a multileaf collimator (Fig. 27-20). Use of multiple beams overlapping on the tumor site can maximize the dose received by the tumor. The appropriate number, orientation, and relative weighting of beams in a treatment plan will depend on geometric factors, such as tumor location and the proximity of normal structures, as well as clinical factors, such as prior or planned surgery, overall patient health, history of prior irradiation, likelihood of future irradiation, and the effects of concurrent chemotherapy.
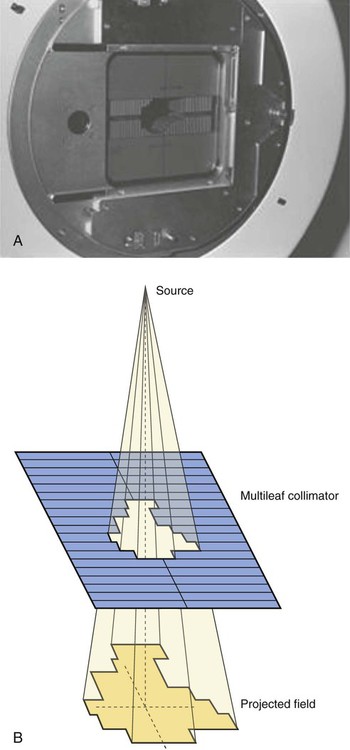
The dosimetry of a sample treatment plan must be evaluated for likely treatment efficacy of the tumor and the possibility of toxicity to normal structures. Dosimetry can be evaluated by examining isodose lines and dose-volume histograms. Isodose lines are a 3D representation of dose levels superimposed on the patient image and indicate regions of high and low dose on any user-selected plane. As shown in Figure 27-21, isodose lines correlate dose with anatomic location and can indicate regions of insufficient or excessive dose and suggest beam arrangements and weightings that can better meet the dosimetric goals for the plan. Aggregate dose to contoured structures are expressed as dose-volume histograms, which display the percentage or absolute volume of the structure receiving a specified dose or lower (Fig. 27-22). Most dose constraints applied to treatment planning are expressed in terms of dose-volume histograms. For example, a typical constraint imposed on treating lung lesions is to limit the total volume of lung receiving 20 Gy or more to 30%.135 Determining appropriate dose constraints for normal structures from clinical data is critical; compilations of so-called “tolerance doses” for different normal tissue effects are generated periodically as clinical data accrues and have most recently been aggregated in the Quantitative Analyses of Normal Tissue Effects in the Clinic (QUANTEC) publications.136
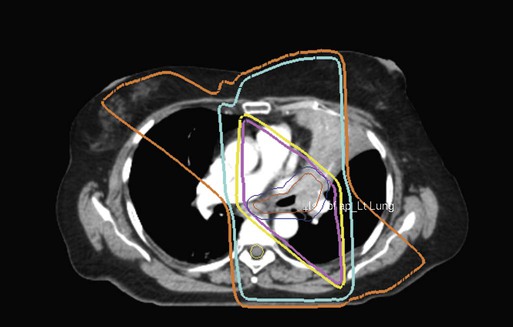
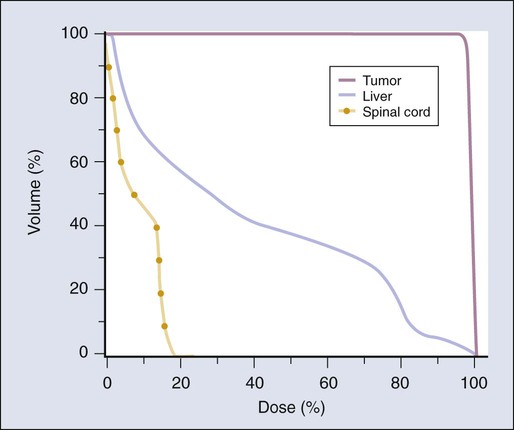
Intensity-Modulated Radiation Therapy
In conventional radiotherapy, treatment plans are designed to deliver a roughly homogeneous dose distribution to the tumor region. This goal is most easily accomplished by covering the target with overlapping homogeneous beams. For this reason, most modern linacs are designed to deliver just such flat dose distributions. Attaining a homogeneous dose distribution across a target can be complicated by a variety of factors. For example, geometric concerns such as beam obliquity or irregular patient surfaces commonly lead to uneven dose distributions across a given beam depth. In other cases, the presence of a sensitive normal structure in the vicinity of the target region may make it desirable that the photon intensity be lessened across all or part of a given beam. Varying the intensity of multiple radiation beams in a manner that improves the overall homogeneity of the dose to the target and helps to decrease the dose to normal tissue is referred to as intensity-modulated radiation therapy (IMRT).137
The simplest form of intensity modulation, wedge filters, has been in use in radiotherapy since the 1960s. A wedge is an angled piece of high-density material that preferentially attenuates a photon beam on its thick end, resulting in a dose gradient across the beam. Wedges are often used to flatten the dose distribution for beams striking sloped surfaces, as illustrated in Figure 27-23. More recently, complex intensity distributions have been created by combining multiple beamlets of varying intensity into a single beam having an intensity profile customized to meet the needs of a specific patient (Fig. 27-24). The delivery of multiple beamlets for IMRT is made practical by the development of the multileaf collimator, which allows each beamlet to be formed and delivered in sequence without requiring the intervention of a radiation technologist. The widespread implementation of multileaf collimators has allowed IMRT to grow from an experimental procedure practiced at a small number of academic centers in the 1990s to a standard technique applied in clinics all around the world.

Calculating the proper intensity distributions for multiple intersecting beams is too complex a task for human hand calculations. Rather than having the physician and physicist create a treatment plan by manually selecting beam orientations and field shapes, IMRT fields are designed by computer algorithms called optimizers. The treatment planners specify the hoped-for dose distribution, expressed as dose goals and toxicity limits, and the optimizer uses mathematical techniques to alter the intensity profile of each beam until the dose goals are met to the greatest extent possible. The first optimizers only considered purely dosimetric goals, but recent efforts have been made to include biological end point models in optimization functions, such as effective uniform dose, tumor control probability, and normal tissue complication probability. IMRT optimization has been used in a variety of clinical circumstances and has been shown to have clinical benefits for treatments in many anatomic sites, such as treatments for prostate,138 lung,139 and head and neck cancers.140
Once a treatment plan has been completed, approved, peer reviewed, and passed through a rigorous quality assurance program, the patient is ready to begin treatment. On each treatment day the patient must be positioned in an orientation identical to that used during simulation, and thus the position within the patient designated as the isocenter must be placed in the room position matching the center of rotation of the linear accelerator. Rough alignment can be accomplished by aligning the tattoos from the simulation step with in-room lasers in the accelerator vault. For more precise alignment of the patient, image-guided radiation therapy (IGRT) techniques can be used. Global patient positioning can be refined using video imaging of the patient surface141 or two-dimensional imaging of bony anatomy (MV portal films or kV radiographs). Techniques to image soft tissue structures to improve patient alignment include the use of in-room CT scanners, linac-mounted cone-beam CT scans,142 implantable fiducial markers detectable by radiograph, radiofrequency signal,143 or ultrasound.144 Adjustments to patient position based on these techniques can be made prior to radiation delivery. To ensure that the patient moves as little as possible between positioning and the delivery of radiation, several forms of patient immobilization may be used.
Brachytherapy
Simulation, Treatment Planning, and Delivery of Brachytherapy
The short range of radiation from most brachytherapy sources confines the majority of the dose to the immediate vicinity of the sources themselves, allowing highly conformal dose distributions through the careful placement of sources within or near the target volume. This arrangement allows a degree of sparing of nearby normal structures that may be superior to that achievable with use of external beam radiation therapy. Because of the high degree of conformity involved, brachytherapy is most effective for relatively small, well-localized tumors. Most radionuclides used for radiation therapy use γ-ray photon emission as the primary source of therapeutic energy. Early brachytherapy procedures used radium or radon sources, but these sources have been almost completely replaced by safer, artificially produced radionuclides such as cesium-137, iridium-192, iodine-125, and palladium-103 (Table 27-1). Brachytherapy sources are generally enclosed in multiple layers of inert material. The inert layers prevent leakage of the radioactive material and, for photon sources, absorb unwanted decay products such as α-particles and electrons. Brachytherapy doses can either be delivered over a short period (temporary implants) or gradually over the lifetime of the seeds (permanent implants).
Brachytherapy treatments are generally delivered using either interstitial or intracavitary techniques. In interstitial brachytherapy, radioactive seeds are surgically placed within the tumor volume. The seed placement can be permanent or temporary. Permanent seeds are deposited in the tumor volume using needles. Temporary seeds are placed and removed from the tumor volume through implanted catheters. A common permanent interstitial procedure is treatment for early-stage prostate cancer, in which approximately 100 125I seeds, each the size of a grain of rice, are placed in the prostate. Temporary interstitial procedures typically use fewer seeds, with MammoSite therapy for partial breast irradiation using a single 192Ir seed applied through a catheter.145
Stereotactic Radiosurgery
Stereotactic Radiosurgery and Stereotactic Body Radiotherapy Physics
1. well-circumscribed tumors (minimal or no CTV expansion);
2. minimal treatment margin around the lesion (minimal PTV expansion);
3. rapid dose falloff away from the target area; and
4. very precise (1 to 5 mm) targeting and localization of the tumor.
Stereotactic radiosurgery (SRS) refers to a single-fraction delivery of a high dose to an intracranial target. The term “radiosurgery” was coined in 1951 by a neurosurgeon named Lars Leskel,146 who developed the original techniques for SRS in the 1940s using orthovoltage x-rays. Since that time, patients undergoing SRS have been treated with heavy charged particles, megavoltage x-rays, and γ-rays from radioactive sources. During the following decades, the use of SRS has been documented in more than 4000 publications,147 demonstrating the effectiveness of the technique in treating primary and metastatic brain lesions, as well as nonmalignant functional disorders such as arteriovenous malformations and trigeminal neuralgias. The single-fraction doses in SRS are sufficient to kill any tissue, cancerous or otherwise, and thus great care must be taken to produce a high-dose region that is tightly conformal to the target area and to ensure that the dose is delivered to the correct location in the patient. Highly conformal dose distributions can be created using 100 to 200 overlapping beams (Fig. 27-25, A) or by delivering dose continuously as a linear accelerator moves around the patient in one or more arcs (Fig. 27-25, B). Positioning the patient correctly relative to the beams requires much stricter immobilization than is typically used for standard radiotherapy. Traditionally, patients would be placed in a rigid stereotactic head frame, which would then be positioned relative to the radiation beams by affixing the frame to either the treatment table or a room-mounted pedestal. The frame, which is affixed to the patient’s skull prior to patient imaging, provides a known offset between radiation isocenter and the tumor location, allowing dose delivery with an accuracy of approximately 1 mm. Newer SRS immobilization techniques have been able to approach the accuracy of stereotactic frames without requiring fixation to the patient’s skull, having the advantages of being less invasive and more reproducible if fractionation is warranted. Examples of these techniques include vacuum-based frame fixation to the hard palate and IGRT-intensive frameless radiosurgery. SRS can be delivered to multiple tumor sites in the brain during a single treatment and is generally limited to well-defined lesions 2 cm or smaller.
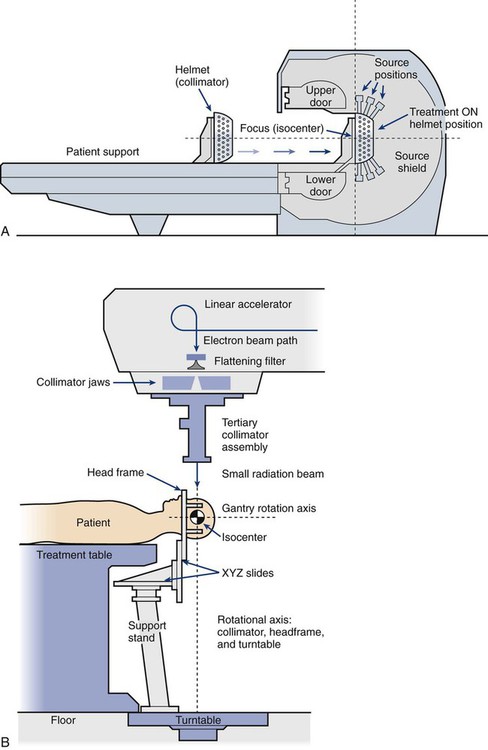
Stereotactic body radiotherapy (SBRT) is hypofractionated treatment applied to extracranial targets, such as in the abdominal, pelvic, and spinal regions. SBRT has a much shorter history than SRS, with the first clinical outcomes for SBRT reported in 1995.148 Early SBRT studies focused on the treatment of lung and liver lesions, but more recent efforts have included studies of spine, prostate, kidney, pancreas, and gynecologic cancers. The delayed development of SBRT relative to SRS is largely due to increased difficulties in target localization. For example, whereas intracranial lesions can be expected to maintain a constant position relative to the patient’s skull, soft tissue lesions in the body can move relative to bony anatomy or any external markers used for patient setup. Minimizing this uncertainty requires the use of in-room imaging devices to ensure accurate beam delivery. Soft-tissue imaging may be facilitated by implanting radiographically visible fiducial markers in the region of the tumor. The extensive use of in-room imaging (IGRT) has largely replaced the use of stereotactic body frames in SBRT. Lesions in the thoracic or abdominal cavities may also move during beam delivery as a result of respiratory motion. Techniques for minimizing the effects of respiratory motion include:
Intraoperative Radiation Therapy
In oncologic surgery, it is not always possible to remove all cancerous tissue during the procedure. Presumed microscopic infiltration of cancer cells into the tumor bed and/or portions of the tumor left unresected because of the excessive risk of morbidity can be treated in the operating room using short-range radiation. Intraoperative radiation therapy (IORT) was attempted as early as 1909149 but did not become a mature treatment modality until the 1980s. In IORT, a large single dose of radiation is delivered to an exposed tumor or tumor bed with the intention of improving the probability of local control. Performing the irradiation during the surgical procedure allows sensitive normal structures to be surgically moved out of the beam path, minimizing the dose received by healthy tissue.
Specialized Radiotherapy Techniques and Facilities
Investigators have great interest in the use of specialized radiation modalities that require different machines that produce either protons, neutrons, or other high-energy particles. Although treatment with photon beams is the most common form of radiotherapy, particle beam therapy also has many clinically useful properties. As discussed elsewhere in this chapter, light charged particles such as electrons lose their energy gradually, depositing dose approximately evenly as the beam passes through tissue. This process continues until all energy is expended, at which point the dose delivery stops abruptly, as illustrated in Figure 27-3. Thus particle beams can be used to treat a target while delivering almost no dose to anatomic structures beyond the target region. Therapeutic electrons in clinical use are limited to treatment depths of 5 to 6 cm, limiting their applicability to shallow lesions. These high-energy electrons can scatter at large angles, causing the electron beam to spread as it passes through tissue. This effect blurs the edges of the dose distribution, making precision dosimetry difficult.
1. heavier particles scatter at smaller angles, allowing less blurring and more precise dose delivery;
2. heavier particles typically have a higher LET, leading to greater biological effectiveness than an equivalent energy deposition from standard photon or electron therapy; and
3. for particles such as protons, the distal end of the dose distribution is quite sharp, allowing much more accuracy in avoiding irradiation of normal tissues deep to the tumor.
Protons
Proton therapy is the most common type of heavy particle therapy. Proton beams are produced by accelerating ionized hydrogen in a cyclotron or synchrotron to energies in excess of 100 MeV. These devices are considerably larger and more expensive than conventional clinical accelerators, and thus they tend to be built as stand-alone facilities, although research into smaller scale proton devices is ongoing. Unlike electrons that lose energy roughly evenly across the range of therapeutic energies, protons lose their energy at an increasing rate as the beams loses energy with depth. This effect culminates in a region of rapid dose deposition near the depth of maximum penetration, called the Bragg peak (Fig. 27-26). The depth of the Bragg peak increases with beam energy, allowing careful energy selection to ensure that the highest dose is delivered to the tumor volume, with lower doses upstream of the target and negligible dose downstream of the target. The width of the Bragg peak is virtually always narrower than the region to be targeted, requiring a range of proton energies to “paint” high, uniform doses across the target. This widened dose distribution, known as a spread out Bragg peak, can be created either by varying the proton beam energy in a synchrotron or by using a spinning modulator of varying thickness to selectively vary the beam energy, shown in Figure 27-26.
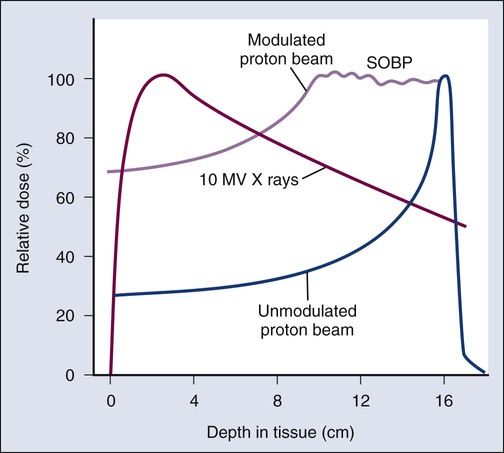
The largest clinical experience with the use of proton radiotherapy has been for the treatment of prostate cancer. However, although their use in this site has a theoretical advantage, that advantage has never been demonstrated in clinical trials.150 Protons are very appealing for the treatment of certain pediatric tumors, because the effects of unwanted irradiation of normal tissues can be severe. In this context, protons have been used extensively in the treatment of pediatric central nervous system tumors.151 Studies exploring the use of protons for therapy of lung cancer and tumors at other anatomic sites are ongoing. Because the cost of a proton irradiator (and its specialized facility) is far greater than for traditional x-ray and electron linacs, it is incumbent upon persons using protons for radiotherapy to demonstrate that the extra expense translates into better tumor control rates and/or fewer normal tissue complications. The lack of such level I evidence to date has made many persons concerned about the rapid proliferation of proton facilities.
Heavy Ions
Investigators have had a long-standing interest in using other heavy charged particles, such as accelerated carbon or neon ions, for therapeutic purposes, and that interest continues to the present day. These particles have the potential to combine both the biological advantages of neutrons and the dose distribution advantage of protons. Machines to produce these particles and the associated operating costs are very expensive, however, which has substantially limited their development and use to only a handful of facilities worldwide.152,153