Chapter 48 Autistic Spectrum Disorders
The autistic spectrum disorders (ASD) represent a wide continuum of associated cognitive and neurobehavioral deficits, including deficits in socialization and communication, with restricted and repetitive patterns of behaviors [American Psychiatric Association, 1994, 2000]. The terms autism and autistic spectrum disorders are used interchangeably throughout this chapter and refer to the broader umbrella of pervasive developmental disorders (PDD), as defined by the Fourth Edition of the Diagnostic and Statistical Manual of Mental Disorders [American Psychiatric Association, DSM-IV, 1994; DSM-IV-TR, 2000].
Historical Perspective of the DSM
Although Kanner [1943] first described a syndrome of “autistic disturbances” in 11 children who shared “unique” and previously unreported patterns of behavior, including social remoteness, obsessiveness, stereotypy, and echolalia, the first set of formal diagnostic criteria for this disorder was not formulated until the 1970s [Ritvo and Freeman, 1978; Rutter and Hersov, 1977]. In the DSM-III [American Psychiatric Association, 1980], the term “autism” was included for the first time, and was clearly differentiated from childhood schizophrenia and other psychoses under a new diagnostic umbrella of pervasive developmental disorders; the possible PDD diagnoses included the terms infantile autism (onset before age 30 months) and childhood-onset pervasive developmental disorder (onset after age 30 months), with each further subclassified as full syndrome present or residual state. The DSM-IIIR [American Psychiatric Association, 1987] broadened the spectrum of PDD and narrowed the specific diagnoses to two: autistic disorder and PDD – not otherwise specified (PDD-NOS). The DSM-IV [1994] and DSM-IV-TR [2000] included five possible diagnoses under the PDD umbrella: autistic disorder, Asperger’s disorder, childhood disintegrative disorder, Rett’s syndrome, and PDD-NOS/atypical autism. With an anticipated publication date of 2013, DSM-V [in press] will most likely eliminate the term PDD and instead will use autistic spectrum disorders as the umbrella term, with autistic disorder and atypical autism as the two possible diagnostic categories (Box 48-1).







Clinical Features of ASD
All individuals on the autistic spectrum demonstrate deficits in three core domains: reciprocal social interactions, verbal and nonverbal communication, and restricted and repetitive behaviors or interests [American Psychiatric Association, 1994, 2000]. There is marked variability in the severity of symptoms across patients, and cognitive function can range from profound mental retardation through the superior range on conventional IQ tests. Symptoms and signs are discussed in detail in the DSM-IV, in the monograph edited by Rapin [1996], in the Wing Autistic Disorders Interview Checklist – Revised [Wing, 1996], and in numerous additional publications [Allen, 1988; Allen and Rapin, 1992; Barbaro and Dissanayake, 2009; Filipek et al., 1999; Greenspan et al., 2008; Rapin and Tuchman, 2008; Zwaigenbaum et al., 2009] (Box 48-2).
Box 48-2 DSM-IV/DSM-IV-TR Diagnostic Criteria for 299.00 Autistic Disorder
(From American Psychiatric Association. Diagnostic and statistical manual of mental disorders, 4th edn. Washington, DC: American Psychiatric Association, 1994.)
Qualitative Impairment in (Verbal and Nonverbal) Communication
The communication deficits seen in the autistic spectrum are far more complex than presumed by simple speech delay, and they are similar to the deficits seen in children with developmental language disorders or specific language impairments [Allen and Rapin, 1992]. Expressive language function across the autistic spectrum ranges from complete mutism to verbal fluency, although fluency is often accompanied by many semantic (i.e., word meaning) and verbal pragmatic (i.e., use of language to communicate) errors. Some mute autistic children do not respond to their names, and often, they are initially presumed to be severely hearing-impaired.
Restricted, Repetitive, and Stereotypic Patterns of Behaviors, Interests, and Activities
Some children have obvious stereotypical movements, such as florid hand-clapping or arm-flapping whenever excited or upset, which is pathologic if it occurs after the age of about 18–24 months. Running aimlessly, rocking, spinning, bruxism (teeth grinding), toe-walking, or other odd postures are commonly seen in autistic children. Others may repetitively tap the back of the hand in a less obtrusive manner, or touch or smell items. In higher-functioning youngsters, the stereotypic movements may become “miniaturized” as they get older into more socially acceptable behaviors, such as pill rolling [Bauman, 1992; Rapin, 1996].
Asperger’s Disorder
The validity of Asperger’s disorder as an entity separate from high-functioning (verbal) children with ASD remains controversial [Ariella Ritvo et al., 2008; Frith, 2004; Howlin, 2003; Macintosh and Dissanayake, 2004; Sanders, 2009; Schopler, 1996; Witwer and Lecavalier, 2008; Woodbury-Smith and Volkmar, 2009], and Asperger’s disorder will most likely not be included as a separate entity under the ASD umbrella in DSM-V [in press]. Clinically, the diagnosis of Asperger’s disorder is often inappropriately given as an alternative, more acceptable, “A-word” to high-functioning autistic children [Bishop, 1989]. The similarity and overlap of signs and symptoms of Asperger’s disorder with nonverbal learning disabilities (NLD) additionally expand the spectrum of these developmental disorders [Harnadek and Rourke, 1994; Klin et al., 1995; Rourke, 1989]; a recent report, however, demonstrates a lack of difficulty with spatial- or problem-solving tasks – a main principle in the NLD model – in a small cohort of children with Asperger’s disorder [Ryburn et al., 2009].
In sharp contrast to autistic disorder, DSM-IV-TR Asperger’s criteria state that “there are no clinically significant delays in early language (e.g., single words are used by age 2, communicative phrases by age 3)” [2000, p. 81]. Normal or near-normal cognitive function is also the rule, including self-help skills, “adaptive behavior (other than in social interaction), and curiosity about the environment in childhood” [1994, p. 77]. Although absence of language delay is required for diagnosis, the DSM-IV definition of single words by age 2 and communicative phrases by age 3 is none the less considerably outside the recognized norm for language development [Coplan and Gleason, 1993; Rossetti, 1990; Sanders, 2009; Zimmerman et al., 2002]. Asperger’s criteria for the qualitative impairments in social interaction and restrictive and repetitive patterns of behaviors and activities are identical to those for autistic disorder (for a recent review, see Woodbury-Smith and Volkmar [2009]).
High verbal skills are the rule in Asperger’s disorder, which typically leads to later clinical recognition than with autistic disorder [Volkmar and Cohen, 1991; Woodbury-Smith and Volkmar, 2009]. Despite the DSM-IV definition [1994, 2000], language in Asperger’s disorder is clearly not typical or normal. For example, there usually is pedantic and poorly intoned speech, poor nonverbal pragmatic or communication skills, and intense preoccupation with circumscribed topics, such as the weather or railway timetables [Ghaziuddin and Gerstein, 1996; Klin et al., 1995; Wing, 1981]. Individuals with Asperger’s use fewer personal pronouns, temporal expressions, and referential expressions [Colle et al., 2008]. They often exhibit deficits in the semantics and verbal pragmatics of language, resulting in concrete and literal speech; their answers often miss the point. They also demonstrate deficits in general receptive language [Koning and Magill-Evans, 2001; Noterdaeme et al., 2009; Saalasti et al., 2008] and in prosodic comprehension [Jarvinen-Pasley et al., 2008]. Szatmari et al. [1995] further define this disorder by the complete lack of delayed echolalia, pronoun reversal, or neologisms in language production.
Socially, individuals with Asperger’s disorder are usually unable to form true friendships. Because of their naive, inappropriate, one-sided social interactions and lack of empathy, they may be ridiculed by their peers. Often, they cease their attempts to develop friendships because of the cruel ridicule and then remain extremely socially isolated. Fine and gross motor deficits have been described, including clumsy and uncoordinated movements and odd postures [Jansiewicz et al., 2006; Klin et al., 1995; Nishitani et al., 2004; Rinehart et al., 2006; Wing, 1981]. However, frank motor apraxia is an inconsistent finding [Dziuk et al., 2007; Mostofsky et al., 2006].
Autistic Regression and Childhood Disintegrative Disorder
Approximately 22–35 percent of autistic children initially appear to develop normally until at least 12 months of age, followed by loss of language and/or social skills [Baird et al., 2008; Meilleur and Fombonne, 2009; Rogers, 2004; Tuchman and Rapin, 1997; Wiggins et al., 2009]. Loss of language skills has been found to be specific for ASD [Kurita, 1996; Pickles et al., 2009]. Parents usually report that infants were socially responsive, smiled, waved bye-bye, and said some words, but they then suddenly or gradually stopped speaking and seemed to withdraw. In an on-going surveillance program, Wiggins et al. [2009] found that, not surprisingly, children with a known ASD diagnosis had a higher rate of parentally reported regression than those identified with ASD through the retrospective record review (26 percent vs. 17 percent, respectively). Regression occurred at a median age of 24 months; boys were more likely to demonstrate regression than girls, and at earlier ages. Children who experienced regression were diagnosed with ASD much earlier (mean age 4.2 years) than those without regression (mean age 6.2 years) [Shattuck et al., 2009].
One difficulty hindering a better understanding of autistic regression involves the disentangling of age at onset from age at recognition [Chawarska et al., 2007; Volkmar et al., 1985]. Many children thought by parents to be normal in the first 18 months may indeed show signs or symptoms on retrospective evaluation of home movies and videotapes by as early as 12 months of age [Baranek, 1999; Goldberg et al., 2003; Maestro et al., 2005; Osterling and Dawson, 1994, 1999; Ozonoff et al., 2005; Werner and Dawson, 2005]. Goldberg et al. [2008] found significant concordance between parental report and retrospective analysis of home videotapes of regression only in the language domains.
As recently reviewed by Tuchman [2006, 2009], there is considerable controversy surrounding the relation between autistic regression and epilepsy, with regression associated with an epileptiform electroencephalogram (EEG) approximately 20 percent of the time. Studies report both higher [Hrdlicka, 2008; Kobayashi and Murata, 1998] and lower rates [Baird et al., 2008; Tuchman et al., 1991] of epilepsy in regression. The behavioral phenotypes of autistic regression, Landau–Kleffner syndrome (LKS), and continuous spike-wave during slow-wave sleep (CSWS) overlap considerably, and may represent distinct syndromes based on age of regression, degree and type of regression, and frequency of epilepsy and EEG abnormalities [Tuchman, 2009]. Children with LKS and isolated language regression are more likely to have epileptiform EEGs and seizures than those with an autistic regression [McVicar et al., 2005].
Mitochondrial disorders have recently been reported to be associated with autism, and with regression in particular [Poling et al., 2006]. In a cohort of 25 patients with ASD and definite or probable mitochondrial disease by the Modified Walker and Mitochondrial Disease Criteria [Bernier et al., 2002; Wolf and Smeitink, 2002], Weissman found that 56 percent experienced regression of previously acquired skills; 64 percent of the regressions were multiple, and 43 percent had the regression(s) after 3 years of age [Weissman et al., 2008]. Shoffner et al. [2009] found that 61 percent of children with ASD and mitochondial disease experienced a regression, 71 percent associated with and 29 percent without fever.
By DSM-IV definition, childhood disintegrative disorder (CDD) refers to the rare phenomenon of normal early development until at least age 24 months, followed by the loss of language, social, play, or motor skills which culminate most often in symptoms of autism. Previously called Heller’s syndrome, dementia infantalis, or disintegrative psychosis, CDD usually occurs between 36 and 48 months of age but may occur up to age 10 years [American Psychiatric Association, 1994, 2000]. There is, therefore, much overlap between CDD and autistic regression, which has led to significant controversy [Hendry, 2000; Malhotra and Gupta, 2002]. The category of CDD will most likely be retained in DSM-V [in press] under the umbrella of ASD; however, the diagnostic criteria may be changed to reflect the increased understanding of the phenomenon of regression in autism. The lower age limit of CDD will most likely be increased to age 3 years, with autistic regression occurring prior to age 3 years remaining under autism.
CDD is considered rare, with recent epidemiological data suggesting a prevalence estimate of 2 per 100,000 [Fombonne, 2002b, 2009]. It is usually associated with more severe autistic symptoms than is early-onset autism, including profound loss of cognitive skills resulting in mental retardation. There is a 4:1 male predominance and a mean age of onset of 29 ± 16 months; more than 95 percent demonstrate symptoms of speech loss, social disturbances, stereotyped behaviors, resistance to change, anxiety, and deterioration of self-help skills [Kurita et al., 2004a, b; Mouridsen, 2003; Volkmar and Rutter, 1995]. The risk of epilepsy may be as high as 70 percent [Mouridsen et al., 1999]. Children with CDD after age 3 years are more likely to have seizures than those who regress before age 24 months [Klein et al., 2000; Shinnar et al., 2001; Wilson et al., 2003]. Treatment experience in CDD has been generally limited to anticonvulsant therapy for seizures, although Mordekar et al. [2009] recently reported amelioration of behavior, language, and motor regression after corticosteroid treatment in two children with CDD, seizures, and/or epileptiform EEG patterns.
Pervasive Developmental Disorder – Not Otherwise Specified and Atypical Autism
The diagnosis of atypical autism or PDD-NOS is used when clinically significant autistic symptoms are present involving reciprocal social interactions, verbal or nonverbal communication, or stereotyped behavior, interests, and activities, but criteria are not met for a specific diagnostic category under the umbrella of autistic spectrum or pervasive developmental disorders (e.g., a child who does not meet the required 6 of 12 criteria for the diagnosis of autistic disorder) [American Psychiatric Association, 1994, 2000]. Children whose symptoms are atypical or not as severe are coded under this diagnosis. It should be noted that the DSM-IV definition of PDD-NOS required that a child meet only 1 of the 12 criteria in any of the three core domains; in DSM-IV-TR, the definition was changed to require impairment in the development of reciprocal social interaction and either impairment in verbal and nonverbal communication skills or the presence of stereotyped behavior, interests, and activities. It is expected that this diagnostic category will be eliminated in the DSM-V [in press, Box 48-3].
Box 48-3 Proposed Revision to 299.00 in DSM-V: Autism Spectrum Disorder
Must meet criteria 1, 2, and 3:
Rationale
 New name for category, autism spectrum disorder, which includes autistic disorder (autism), Asperger’s disorder, childhood disintegrative disorder, and pervasive developmental disorder not otherwise specified
New name for category, autism spectrum disorder, which includes autistic disorder (autism), Asperger’s disorder, childhood disintegrative disorder, and pervasive developmental disorder not otherwise specified
 Differentiation of autism spectrum disorder from typical development and other “nonspectrum” disorders is done reliably and with validity, while distinctions among disorders have been found to be inconsistent over time, variable across sites, and often associated with severity, language level, or intelligence, rather than features of the disorder
Differentiation of autism spectrum disorder from typical development and other “nonspectrum” disorders is done reliably and with validity, while distinctions among disorders have been found to be inconsistent over time, variable across sites, and often associated with severity, language level, or intelligence, rather than features of the disorder
 Three domains become two:
Three domains become two:














(American Psychiatric Association. Proposed Revisions to 299.00 Autistic Disorder in DSM-V, 2010. Retrieved 28 February 2010, from http://www.dsm5.org/ProposedRevisions/Pages/proposedrevision.aspx?rid=94#.)
Epidemiology
The reported prevalence of autism has dramatically increased, and it is now recognized as one of the most common developmental disorders. Most studies come from industrialized countries, but there is increasing awareness of autism and other developmental disabilities in less developed communities around the world. For many years after autism was first described in the 1940s, prevalence was considered to be 2–4 cases per 10,000 children [Wing and Potter, 2002]. Fombonne [2003a] reviewed a total of 32 epidemiological studies published from 1966 through 2001. For the 16 studies published from 1966 to1991, the median prevalence was 4.4 per 10,000; for the 16 studies published from 1992 to 2001, the median was 12.7 per 10,000.
The Centers for Disease Control and Prevention (CDC) examined children in metropolitan Atlanta, Georgia, who were 3–10 years old in 1996, and found a prevalence of children who were diagnosed with ASD of 3.4 per 1000 (CI = 3.0–3.7) [Yeargin-Allsopp et al., 2003]. A 2002 CDC survey of 400,000 children aged 8 years (born in 1994) found a prevalence of 6.6 per 1000 with a wide variation across the 14 states in the study [ADDM, 2007]. Most recently, the CDC reported a prevalence rate of 9.0 per 1000 in 2006 in 307,790 8-year-old children across 11 states [ADDM, 2009]. All of these CDC studies relied on abstraction of health and education records.
Higher numbers have been more recently reported in other studies that relied on active screening and diagnosis of populations of children. The prevalence of ASDs in 55,000 British 8- and 9-year-old children was 11 per 1000 [Baird et al., 2006b], and in a separate study of children ages 5–9 years, cases were documented at a rate of 1 per 100, but the authors thought that not all cases were likely to have been found [Baron-Cohen et al., 2009]. Based on the most recent parent-reported U.S. diagnostic survey, the prevalence for ASD was as high as 11 per 1000 [Kogan et al., 2009]. Definitions used, screening methods, diagnostic criteria, and completeness of sampling varied in these studies; all have methodologic issues affecting prevalence results [Bresnahan et al., 2009; Charman et al., 2009; Hertz-Picciotto and Delwiche, 2009; King and Bearman, 2009; Nassar et al., 2009].
A number of factors contribute to this apparent increase. Diagnostic criteria have evolved and broadened; the concept of autism is now defined as autistic disorder plus the broader autistic spectrum disorders, including Asperger’s syndrome and PDD-NOS; there is now co-diagnosis with known medical disorders such as fragile X syndrome, Tourette’s syndrome (TS) and Down syndrome; and the growing public awareness among parents and teachers has led, in developing countries, to earlier and more accurate diagnoses. The increased availability of services [Nassar et al., 2009] and the ability to diagnose children at younger ages [Parner et al., 2008] may influence the frequency of diagnosis. Children earlier diagnosed as mentally retarded may have met current criteria for autism [King and Bearman, 2009; Nassar et al., 2009; Prior, 2003]. Bishop et al. [2008] found that up to 60 percent of adults previously diagnosed with developmental language disorder would meet more recent criteria for PDD. Case ascertainment methodology is also a factor, because using multiple sources and broad population screening increases the number of cases found. There are little data on prevalence in older populations.
Clearly, a substantial proportion of the increase seen in autism is due to factors such as a combination of better, more population-based studies and changes in the diagnostic criteria and age at diagnosis. However, the increase cannot be solely attributed to known factors and there may, in fact, be a true increase in incidence. It is important for etiologic reasons and for public health and educational planning to ascertain whether the rise in cases is genuine, if it is continuing, and to what degree. The CDC is monitoring the prevalence of autism over time in a number of U.S. sites using consistently applied ascertainment and diagnostic protocols [Croen et al., 2002a; Fombonne, 2003a].
The proportion of children with ASDs who had IQs less than or equal to 70 ranged from about 30–50 percent in the CDC’s Autism and Developmental Disabilities Monitoring Network. A higher proportion of females had cognitive impairment compared to males. The mean male to female ratio is 4:1 or greater for the milder forms, but as severity of cognitive impairment increases, the male to female ratio decreases to 1.3:1 [Yeargin-Allsopp et al., 2003]. The rate of PDD-NOS is approximately 1.5 times that of autistic disorder; the rate of Asperger’s disorder is one-fourth that of autistic disorder. Children with autistic disorder and a measurable IQ of less than 50 are more likely than those who are high-functioning to be female and to have minor physical anomalies, neuroimaging abnormalities, microcephaly, and epilepsy [Nicolson et al., 1999]. Those with specific, known inherited conditions, such as tuberous sclerosis or phenylketonuria, are likely to be more severely cognitively impaired [Rutter et al., 1994].
Risk Factors
Sibling Studies
The risk of ASDs in a sibling has been reported to be 3–8 percent when there is one affected child [Chakrabarti and Fombonne, 2001; Micali et al., 2004]. However, a recent report from Japan by Sumi et al. [2006] found gender differences in the risk for subsequent siblings: general sibling risk was 10 percent, 7.7 percent if the proband was male, and 20.0 percent if the proband was female. The risk is 25 percent if there are already two siblings with ASD [Folstein and Rosen-Sheidley, 2001].
Infant siblings of children with autism have garnered recent research attention as a high-risk group with the hope of identifying the earliest warning signs of ASD [Barbaro and Dissanayake, 2009; Brian et al., 2008; Cassel et al., 2007; Elder et al., 2008; Elsabbagh and Johnson, 2007; Elsabbagh et al., 2009a, b; Goldberg et al., 2005; Ibanez et al., 2008; Iverson and Wozniak, 2007; Landa and Garrett-Mayer, 2006; Landa et al., 2007; Loh et al., 2007; Merin et al., 2007; Mitchell et al., 2006; Sigman et al., 2004; Toth et al., 2007; Zwaigenbaum et al., 2005, 2007; see Rogers [2009] for a recent review]. Age at entry into the studies varies considerably, and one might anticipate that parents of those infants enrolled at later ages might have already recognized warning signs that prompted their participation in the study. This may contribute to the fact that the rate of an eventual diagnosis of ASD also varies highly across the studies, reported as 10 percent for infants enrolled by 5 months of age [Iverson and Wozniak, 2007], 14 percent for those enrolled between 12 and 23 months of age [Yoder et al., 2009], 23 percent for those enrolled between 6 and 12 months of age [Brian et al., 2008], and 62 percent for those enrolled by 18 months of age [Landa and Garrett-Mayer, 2006]. As a result, the true sibling recurrence rate cannot be currently ascertained through the available studies.
Developmental differences in infant siblings who are later diagnosed with an ASD (Sib-ASD) appear to emerge by around 12 months of age, with the developmental gap widening at a decreasing rate over the second year of life [Brian et al., 2008; Rogers, 2009; Stone et al., 2007; Yoder et al., 2009]; to date, studies have not reported significant differences at 6 months of age. Delays in fine and gross motor development have been noted by some [Landa and Garrett-Mayer, 2006] but not all studies [Iverson and Wozniak, 2007; Ozonoff et al., 2008; Toth et al., 2007]. Although stereotypic behaviors are “expected” in infants during the course of motor development [Thelen, 1979], specific atypical and repetitive behaviors (specifically spinning, rotating, rolling, and, most commonly, unusual visual regard of toys) occurred more frequently at 12 months of age [Ozonoff et al., 2008], and Loh et al. [2007] found that arm-waving at 12 and covering of the ears at 18 months of age occurred significantly more often in Sib-ASD.
Delays in verbal and nonverbal communication have been noted in Sib-ASD, beginning only at 12 months of age by almost every research group [Gamliel et al., 2007; Goldberg et al., 2005; Landa and Garrett-Mayer, 2006; Landa et al., 2007; Toth et al., 2007; Yirmiya et al., 2006; Yoder et al., 2009; Zwaigenbaum et al., 2005]. However, no consistent specific deficits have emerged to date across the studies as characteristic of Sib-ASD. Response to name has been explored by several researchers, as well [Brian et al., 2008; Nadig et al., 2007; Yirmiya et al., 2006; Zwaigenbaum et al., 2005], and Sib-ASD responded typically at 6 months, but not at 12 months of age.
Studies of response to joint attention in Sib-ASD have found fewer responses in the second year of life [Cassel et al., 2007; Presmanes et al., 2007; Sullivan et al., 2007], particularly in those situations requiring both head turn and verbal prompt [Presmanes et al., 2007]. Yoder et al. [2009] found that the response to joint attention at 12 months was predictive of degree of social impairment and eventual ASD diagnosis at 3 years of age. Zwaigenbaum et al. [2005] were able to differentiate Sib-ASD infants on imitation of body, oral, and object acts, which was not found in high-risk infants who did not develop ASD [Toth et al., 2007].
Neonatal Intensive Care and Prematurity
Matsuishi et al. [1999] first reported a significantly increased rate of ASD in children born between 1983 and 1987, with a mean gestational age of 35.4 ± 4.6 weeks, requiring neonatal intensive care, and who were followed up between 5 and 8 years of age using DSM-III-R criteria [APA, 1987]; a history of meconium aspiration was significantly more common in those children with ASD than in the comparison groups of children with cerebral palsy and those with typical development. Badawi et al. [2006] also have reported an increased rate of ASD at 5 percent in term neonatal intensive care unit (NICU) survivors of newborn encephalopathy, defined as either seizures alone or any two of the following lasting for longer than 24 hours: abnormal consciousness, difficulty maintaining respiration (of presumed central origin), difficulty feeding (of presumed central origin), and abnormal tone and reflexes [Badawi et al., 1998].
Several recent publications have documented a much higher rate of positive screening for ASD in infants with extreme prematurity using the Modified Checklist for Autism in Toddlers (M-CHAT) [Robins and Dumont-Mathieu, 2006; Robins et al., 2001] and other screening instruments. Limperopoulos et al. [2008] found a 25 percent rate of positive screening for ASD at 18–24 months of age in 91 infants who were less than 1500 g and 31 weeks’ gestation at birth. Kuban et al. [2009] noted a 22 percent rate of positive M-CHAT screens in 988 NICU survivors at 24 months of age who were less than 28 weeks’ gestation at birth and who were followed in the multicenter ELGAN study. Major motor, cognitive, visual, and hearing impairments appeared to account for more than half of the positive M-CHAT screens in this cohort. Even after the toddlers with those impairments were eliminated, 10 percent of children – nearly double the expected rate – screened positive.
In a large Swedish population-based study, Buchmayer et al. [2009] reported that the increased risk of autistic disorders related to preterm birth was mediated primarily by prenatal and neonatal complications that occur more commonly among preterm infants, predominantly pre-eclampsia, but also intracranial hemorrhage, cerebral edema, low Apgar scores, and seizures. Limperopoulos [2009] suggests that the incidence of ASD among survivors of preterm birth is inversely related to gestational age. If so, as survival rates continue to improve in extremely premature infants, the resulting morbidity of ASD may also continue to increase. As noted by Fombonne [2006], it is important that all practitioners have a heightened awareness of these risk factors to screen toddlers and preschoolers with suboptimal perinatal histories systematically.
Other Risk Factors
Risk of ASD is higher with increasing age of mothers, and independently, with increasing age of fathers [Durkin et al., 2008]. In a large population of children born between 1989 and 1994, mothers older than 35 years were three times more likely to have an autistic child than women younger than 20 years [Croen et al., 2002a]. One California study found that, when adjusted for age of the other parent and other covariates, risk of autism increases by up to 40 percent for each 10-year increase in maternal age and by 20–25 percent for each 10-year increase in paternal age [Grether et al., 2009a]. In another study also from California, maternal age was linearly correlated with risk but increased paternal age was a risk factor only in mothers over 30 years old [Shelton et al., 2010]. Some studies found that socioeconomic level does not affect risk [Bhasin and Schendel, 2007; Larsson et al., 2005], but in another study, women with a postgraduate education were twice as likely to have an autistic child as women with less than a high-school education [Croen et al., 2002b]. Risk was also increased in multiple births (RR = 1.7; 95 percent CI = 1.4–2.0) and in black children (RR = 1.6, 95 percent CI = 1.5–1.8) [Croen et al., 2002a].
Advanced parental age and some of the other risk factors for autism that have been suggested may act through increasing risk for de novo mutations. There may also be mutagens in the environment, such as mercury, cadmium, nickel, trichloroethylene, and vinyl chloride. Factors associated with vitamin D deficiency may cause mutations as vitamin D contributes to repair of DNA damage [Kinney et al., 2010]. The number of fetal ultrasounds does not seem to be associated with increased risk [Grether et al., 2009b], and perinatal risk factors associated with fetal distress (other than breech presentation) did not contribute significantly to risk [Bilder et al., 2009].
Pathophysiology and Etiology
Animal Models
No single animal model exists for autism, but several animal models that exhibit some of the major features of autism have provided an opportunity to understand the neural substrates of functional impairments. In the macaque monkey, social behavior is mediated by the amygdala, temporal cortex, orbitofrontal cortex, and superior temporal gyrus [Lord et al., 2000]. In another monkey model (rhesus), bilateral removal of the medial temporal lobes leads to abnormal social behavior during maturation that resolves in adults [Bachevalier, 1994; Machado and Bachevalier, 2003]. Symptoms include abnormal social interaction, absence of facial and body expression, and stereotypic behaviors. Social anxiety and fear have been studied in primates [Amaral, 2002] and in rats [Wolterink et al., 2001], and are related to amygdaloid circuitry.
A behavioral syndrome in Lewis rats with analogies to autism is the result of Borna disease virus infection in neonates [Pletnikov et al., 2003; Weissenbock et al., 2000]. Neonatal infection produces specific behavioral abnormalities, with disturbances in sensorineural development, lower startle responsiveness, and abnormal social play. Proprioceptive systems were abnormal that involved use of hind-limbs and balance. Pathologically, Borna disease virus induces regional neuronal loss in the cerebellum. This model provides some insights into mechanisms of pre- and perinatal infection causing damage to the developing brain, but it does not indicate that Borna disease virus is an etiologic agent in autism.
Other animal models have been developed by knocking out different candidate genes [Lijam et al., 1997], by oxytocin and vasopressin administration [Insel et al., 1999], and by exposing embryos to teratogens such as valproic acid [Ingram et al., 2000]. Behavioral studies have focused on social interaction and memory deficits. Because the neurexin 1-alpha gene has been linked to ASD phenotypes, a knockout mouse model has been developed with analogies to at least one core domain of ASD [Etherton et al., 2009].
Neuropathology
Abnormalities in major cortical and subcortical brain structures have been found through postmortem and magnetic resonance imaging (MRI) studies of autistic subjects. Comprehensive examination of nine autistic postmortem brains was carried out by Kemper and Bauman [1994]. They described three major findings: curtailment of normal development in forebrain neurons, which were smaller and more densely distributed than normal; an apparent congenital decrease in the number of Purkinje cells; and age-related changes in cell size and the number of neurons in the diagonal band of Broca, the cerebellar nuclei, and the inferior olive.
These neuropathologic findings may account for many clinical features of autism. Perinatally acquired lesions in limbic system structures could lead to disruption in memory processes involved in the ability to learn new information. In contrast, striatal and cortical areas involved in habitual memory were spared, potentially relating to the need for sameness, narrow interests, and capacity for rote memory. Disruption of cerebellar function may lead to a number of motor and sensory system deficiencies, including mental imagery, anticipatory planning, and timing and integration of sensory and motor information [Kemper and Bauman, 1998]. Support of brain-tissue banking is critical to continued research into the neuropathologic underpinnings of autism [Pickett, 2001].
Another approach to determining the neuropathologic underpinnings of autism has been suggested by Rodier [2002]. She found that children exposed to thalidomide in the first trimester developed autism at an increased rate, supporting the idea that the brain abnormality originates at the time of closure of the neural tube [Rodier et al., 1996]. This finding was corroborated by postmortem examination of a brain from a subject with autism not exposed to thalidomide, but whose mother was an alcoholic, showing near-complete absence of the facial nucleus and superior olive and narrowing of the brainstem between the trapezoid body and inferior olive. This deficit was reproduced in an HOXA1-knockout mouse model and by exposing rat embryos to valproic acid on the day of neural tube closure [Ingram et al., 2000]. The investigators concluded that central nervous system injuries occurring during or just after neural tube closure can lead to selective loss of neurons derived from the basal plate of the rhombencephalon, and this finding may indicate that the initiating injury in some individuals with autism takes place around the time of neural tube closure.
Additional supporting evidence for this theory comes from a Nova Scotia cohort of 61 autistic children [Rodier et al., 1997]. Forty-two percent of these children with autism had posterior rotation of the external ears, compared with 18 percent of controls. They postulated that this could have been associated with a disruption of otic disc formation in the fourth week of embryonic life and that ear anomalies found in some children with autism could possibly be a marker for initiating events in utero.
An important additional neuropathologic finding described by Casanova et al. [2008, 2002] is the finding of abnormalities in the structure of minicolumns in the brain of autistic individuals. Minicolumns consist of 80–100 neurons, and they are believed to be the smallest unit of functional organization in the cortex. In autistic individuals, the minicolumns were described as more numerous but smaller than those of controls and with less space in between. This structural difference may cause the firing of too many processing units at once and prohibit the units from coherently responding to signals. Over-arousal or under-arousal could easily result. This abnormality could also be responsible for the increased incidence of seizures in individuals with autism.
Neuroimaging
Both structural and functional imaging have contributed to the understanding of autism. Quantitative volume analysis using MRI has provided information that the outer layers of white matter are enlarged in autistic subjects compared with controls. Herbert et al. [2005, 2004] compared 13 subjects with high-functioning autism, 14 subjects with developmental language disorder, and 14 controls. The inner zones of white matter were not different in autistic subjects from those in controls, but the outer zone of white matter was larger than controls in autistic individuals and subjects with developmental language disorder. In the autistic group, frontal lobe enlargement was proportionally greater than other areas, but not in the developmental language disorder sample. These areas myelinate relatively late, beginning in the second half of the first year and continuing into the second year of life and later, which is consistent with the timing of increased head circumference seen in autistic subjects. By the time autistic children are 2–4 years old, 90 percent have above-average brain volume, and 37 percent have developmental macrocephaly, defined as brain volume exceeding 2 standard deviations above the normal mean for age [Courchesne et al., 2001]. This finding suggests an on-going postnatal process that primarily affects interhemispheric and cortical connections.
In other studies, children with ASDs were found to have significantly increased cerebral volumes compared with normal and with developmentally delayed children, and white-matter enlargement was seen [Sparks et al., 2002]. There was greater enlargement of the frontal cortex [Carper et al., 2002] compared with other lobes, and reduced amygdala volume [Aylward et al., 1999]. Juranek et al. [2006] also found a positive correlation between amygdalar volume, particularly the right side, and anxiety levels as measured by the Child Behavior Checklist in a cohort of 42 children with ASD, which supports evidence for a neurobiologic relationship between symptoms of anxiety and depression with amygdalar structure and function.
Courchesne et al. [2003] have found specific neuroanatomic abnormalities, increased cerebellar and cerebral white matter, and cerebral cortical gray matter in 2- to 3-year-old children compared with controls, but not in older children or adolescents with autism. Frontal lobes were most affected with greatest involvement in the dorsolateral and medial prefrontal cortex. A widely debated finding in the MRI literature is reduction in size of one or another subregion of the cerebellar vermis [Courchesne et al., 1994a, b, 1988; Hashimoto et al., 1995]. Other investigations have failed to replicate these findings [Filipek, 1995; Hardan et al., 2001; Piven and Arndt, 1995].
Increasing evidence from functional MRI studies (fMRI) points to changes in activation and synchronization of cortical networks. Functional connectivity is lowered, leading to deficits in language, social cognition, motor planning, and perception [Levy et al., 2009]. Autistic children have profound deficits in face-recognition tasks [Klin et al., 1999]. Technologies involving the use of eye-tracking devices and fMRI have yielded important information regarding the pathways for face recognition in autism. The fusiform face area (on the lateral aspect of the fusiform gyrus) is hypoactive as seen by fMRI, and the degree of hypoactivation strongly correlates with the degree of disability [Critchley et al., 2000; Kanwisher et al., 1997; Pierce et al., 2001; Schultz et al., 2000]. Hubl et al. [2003] saw lower blood oxygen-level dependent signals in ten autistic subjects during face processing than in controls, and they observed higher activation in the medial occipital gyrus, an area related to processing objects. Schultz et al. [2000] also found lower activation in autistic subjects in the right fusiform gyrus, but they identified higher activity in the right inferior temporal gyrus. Mechanisms that underlie learning of novel movement patterns are different in children with autism compared to controls, and may lead to impaired skill development [Gidley Larson and Mostofsky, 2008].
Serotonin
Elevated blood levels of serotonin (5-hydroxytryptamine – 5HT) in autistic subjects, first reported in 1961 [Schain and Freedman, 1961], are caused by an elevation within circulating platelets, an increase in the serotonin transporter concentration, and a decrease in 5HT2 receptor binding [Cook and Leventhal, 1996]. Elevated concentrations of a low-molecular-weight peptide that increases the uptake of 5HT into platelets [Pedersen et al., 1999] was demonstrated in autistic subjects. Elevation of platelet serotonin levels has been well studied and generally replicated.
It has been hypothesized that, if siblings of children with autism have higher platelet 5HT levels, it may indicate a higher familial risk [Piven et al., 1991b]. A familial pattern of hyperserotonemia was confirmed by Leboyer et al. [1999] in a sample of 62 autistic subjects and 122 first-degree relatives. Levels of whole-blood 5HT did not change with age in subjects with autism but did change in controls. There does not seem to be a correlation between 5HT levels and specific defined phenotypes [Kuperman et al., 1987]. However, Leboyer et al. [1999] suggested that the level of 5HT in blood could be considered an intermediate or surrogate phenotype that could be correlated with DNA polymorphisms in the 5HT transporter gene.
Serotonergic disturbances, including defects in 5HT transporter expression and decreases in plasma tryptophan, may play a role in the pathophysiology of autism [Croonenberghs et al., 2000; Marazziti et al., 2000]. 5HT has an important role in central nervous system development, affecting social behavior, sleep, aggression, anxiety, and affective regulation, and it plays a role in the modulation of synaptogenesis [Chugani, 2002]. It may have a role in the development of neuropathologic abnormalities in the hippocampus, amygdala, and cerebellum [Bauman and Kemper, 1994] and in thalamocortical connections. Depletion of 5HT in critical developmental periods may also cause a decrease in growth of dendritic spines [Yan et al., 1997]. Using positron emission tomography (PET) scans, Chugani et al. [1997] found alterations of 5HT synthesis in the frontal and thalamic pathways that are important for language production and sensory integration.
About one-half of the patients with Smith–Lemli–Opitz syndrome, a malformation syndrome with mental retardation that is caused by an inborn error in cholesterol biosynthesis [Opitz et al., 2002], exhibit autistic behavior. A mouse model of the Smith–Lemli–Opitz syndrome was created by eliminating DHCR7, which encodes 7-dehydrocholesterol reductase, the terminal enzyme required for cholesterol biosynthesis [Waage-Baudet et al., 2003]. These knockout mice have a 300 percent increase in 5HT immunoreactivity in hindbrains compared with control mice, and 5HT-immunoreactive cells are present in unusual locations and represent an increase in the total number of 5HT neurons and fibers. These observations provide a basis for the autistic phenotype seen in Smith–Lemli–Opitz syndrome, and they support the focus of therapeutic interventions aimed at modulation of the serotonergic system.
Vaccines
Multiple studies concluded that there was no increase in risk of autistic disorder with exposure to the measles-mumps-rubella vaccine [Chen et al., 2004; Dales et al., 2001; Fombonne and Chakrabarti, 2001; Kaye et al., 2001; Madsen and Vestergaard, 2004; Taylor et al., 1999]; see monograph by Offit [2008]. Additional data derived from passive surveillance systems also consistently failed to detect an association [Patja et al., 2000; Peltola et al., 1998; Plesner et al., 2000]. There has been no evidence that the incidence of regressive autism has increased after administration of the measles-mumps-rubella vaccine. The measles-mumps-rubella vaccine has been given at a constant rate since 1979 in the United States, 1982 in Finland, and 1998 in the United Kingdom and Denmark, but rates of autism have steadily risen [Klein and Diehl, 2004]. The Immunization Safety Review Committee of the Institute of Medicine of the National Academies (IOM) [Stratton et al., 2001] concluded that there was consistent evidence of no causal association between administration of the measles-mumps-rubella vaccine and autism.
However, the IOM report did not think there was sufficient evidence to accept or reject a causal association between exposure to thimerosal and autism [McCormick, 2003]. Exposure to thimerosal is a concern because mercury is a known neurotoxin, and there are claims of abnormal levels of mercury in autistic children who respond to chelation therapy. There are no studies involving low doses, and the pharmacokinetics of ethyl mercury in human infants is unknown. Almost all studies available regarding mercury toxicity are related to exposure to methyl rather than ethyl mercury, and the clinical picture of mercury poisoning from any dose, duration, or age of exposure is different from that of autism [Nelson and Bauman, 2003]. Analysis of data from California, Sweden, and Denmark [Stehr-Green et al., 2003] found that, although thimerosal exposure from vaccines was eliminated in Sweden and Denmark by the early 1990s, the incidence of autism was accelerated. This finding was confirmed by a retrospective cohort study [Verstraeten et al., 2003].
A claim has been made that persistent measles virus found in the gastrointestinal tract of ten children with autism and bowel symptoms was causally related to their autism [Wakefield et al., 1998]. Wakefield’s hypothesis was that measles-mumps-rubella vaccine is a risk factor for children with regressive autism. However, there was no clear clinical description of the subjects, there were no controls, and the history and timing of regression was determined years later retrospectively. Most of the other authors have since rescinded the conclusions of this study [Murch et al., 2004], the paper has been officially retracted by the Lancet [Dyer, 2010; Godlee et al., 2011]. Taylor et al. [1999] found 6.6 percent of children with autism had regression and bowel symptoms, but the onset of symptoms was not associated with the measles-mumps-rubella vaccine. Another prospective report [Fombonne and Chakrabarti, 2001] did not find any association in a large, prospective sample. Further strong evidence against the association of autism with persistent measles virus in the gastrointestinal tract or measles-mumps-rubella exposure came from a case control study by Hornig et al. [2008]. Gastrointestinal tissue samples from children with autism, 90 percent of whom showed behavioral regression, and from controls, was examined for presence of measles virus RNA in a blinded study, with results consistent over three laboratory sites [Hornig et al., 2008]. There were no differences between case and control groups in the presence of measles virus RNA, and gastrointestinal symptom and autism onset were unrelated to timing of the measles-mumps-rubella vaccine.
Diagnostic Evaluation and Screening
ASDs can be reliably diagnosed in children as young as 2 years [Lord, 1995; Moore and Goodson, 2003], and early intervention is beneficial [Lord, 1995; National Research Council, 2001]. However, the average age of diagnosis is reported as 3–6 years [Howlin and Moore, 1997; Mandell et al., 2002], and it has remained fairly stable over the past decade. There is evidence that age of diagnosis varies as a function of ethnicity and socioeconomic status [Mandell et al., 2002]. Parents often report concerns about their child’s social and communication skills, and they seek medical assistance long before a formal diagnosis is obtained.
The American Academy of Pediatrics and the CDC launched an awareness campaign to sensitize pediatricians and parents to the early symptoms of autism, to encourage caregivers to listen to parents’ concerns, and to encourage parents to be assertive in making sure that their concerns are addressed. The campaign includes evaluating strategies for screening and developing resources and tools for physicians. The CDC website offers downloadable educational information and screening instruments: http://www.cdc.gov/ncbddd/autism/hcp-screening.html.
The American Academy of Pediatrics Council of Children with Disabilities recently published a set of guidelines [Johnson and Myers, 2007; Myers and Johnson, 2007] on the identification and management, respectively, of children with ASD. Risk factors that should call attention to the possibility of ASD include having a sibling with ASD, or when there is parental, other caregiver, or pediatrician concern.
Screening and Diagnostic Instruments
The Child Neurology Society and American Academy of Neurology published a “Practice Parameter for the Screening and Diagnosis of Autism” [Filipek et al., 1999, 2000a]. A two-level approach is recommended. Level 1 involves developmental screening as part of routine well-child care, followed by an ASD-specific screen for children identified with developmental delay. Level 2 involves formal diagnostic procedures to determine whether the child has ASD. This level of diagnostic evaluation is conducted by an experienced clinician, and it uses information gleaned from the medical history, neurologic evaluation, and developmental testing to determine the child’s developmental profile.
Instruments for Autistic Spectrum Disorders Screening
“Red flags” for language development can be easily recognized and should engender prompt referral (Box 48-4). Screening instruments for ASDs are in various stages of development. Presented here are those that have reported good psychometric properties (although they all need further development and psychometric evaluation) and that are available for use by clinicians. These instruments are most sensitive when they follow general developmental screening and are applied when a child has already been identified with delayed or atypical development. Some of these instruments are also being validated for use in the general population as a first-step screening, which would be desirable in many settings. However, administration of ASD-specific screening tools after screening for and identification of atypical development is the method with the most sensitivity (i.e., probability of correctly identifying a child with ASD) and specificity (i.e., proportion of non-ASD cases correctly identified).






The Checklist for Autism in Toddlers (CHAT) [Baird et al., 2000, 2001] was developed in Great Britain, and it is most useful in screening children 18–24 months old. It was designed for use in the general population and to be administered in the home by a medical caregiver. The CHAT is intended to be cost-effective and sensitive to early signs of ASD. Parents respond to nine items asking about their child’s social orienting behaviors, such as pretend play, joint attention, and pointing. The clinician completes five items after observing the child. Unfortunately, when administered in the general population, the sensitivity has been disappointing, with high rates of false-negative results. The developers recommend a screen-rescreen procedure (at 18 and again at 19 months), which increases specificity to within acceptable limits [Charman et al., 2002, 2007].
Scambler et al. [2001] conducted a small study in which they demonstrated better sensitivity (65 percent) and specificity (100 percent) when using the CHAT to distinguish children with autism from among a group already identified with developmental problems. They improved on this further by developing their own criteria (i.e., Denver Criteria) for defining risk status. The CHAT worked best with children whose developmental level was within the 18- to 24-month range, despite them being older in chronologic age. The sample size was small, and the Denver Criteria were developed ad hoc. Although the CHAT looks promising for this type of use, more validation is needed. Development of this tool is on-going, and there are attempts at modifications to improve sensitivity and specificity within the general population.
The Modified Checklist for Autism in Toddlers (M-CHAT) [Robins and Dumont-Mathieu, 2006; Robins et al., 2001] relies only on parent report. There is a 23-item questionnaire completed by a parent. If results of the checklist indicate possible risk for autism, there is a follow-up parent interview. The use of this follow-up interview increases the sensitivity and specificity of the instrument, especially when used in a general pediatric population that has not already been screened for developmental disorders [Kleinman et al., 2008].
The Screening Tool for Autism in Two-Year-Olds (STAT) [Stone et al., 2000, 2008] is composed of interactive items administered by a clinician to children 24–35 months old. It is designed to identify children with autism disorders (not the full range of ASDs) from a population of children identified with developmental delays. The STAT quantifies behaviors in four domains: play, requesting, directing attention, and motor imitation. The instrument requires training in administration and scoring, and it takes about 20 minutes to complete. Strong psychometric properties are reported, although they are derived from small samples, and there is need for further development (e.g., evaluation of usefulness with the full range of ASDs) [Stone et al., 2004]. Preliminary results also suggest that the tool is useful in identifying autism in at-risk toddlers down to 14 months of age [Stone et al., 2008]. The investigators suggest that the qualitative information obtained during the screening can be used to inform intervention goals for individual children. In some settings, this may outweigh the inconvenience of the need for training and the relatively longer administration time.
The screening instruments previously described are designed for early diagnosis. Unfortunately, not all children are diagnosed early, and a neurology practice is likely to encounter older children referred for possible autism. The Social Communication Questionnaire [Berument et al., 1999; Rutter et al., 2003a] was developed to identify older, higher-functioning individuals with ASD. It is a 40-item, parent-completed questionnaire for children 4 years old and older. In a large sample of children referred for possible ASD diagnoses, the instrument performed well when differentiating ASDs from other diagnoses. It also demonstrated good sensitivity and specificity in identifying ASD in school-age children with special learning needs [Charman et al., 2007].
Instruments for ASD Diagnosis
The Childhood Autism Rating Scale (CARS) is a clinician-rated diagnostic instrument for use with children older than 2 years [Chlebowski et al., 2010]. It should be administered by a clinician who is experienced with ASDs. The clinician should view a training videotape before using this rating scale. It takes 30–45 minutes to administer. The CARS is widely recognized and used in clinical settings. Psychometric properties are good to excellent, although in some of these reports, the sample population is not well described. Because the CARS was developed before publication of the DSM-IV [American Psychiatric Association, 1994] and ICD-10 [World Health Organization, 1992], it is less helpful in distinguishing between subgroups within the broader category of ASDs.
The Autism Diagnostic Interview – Revised (ADI-R) [Lord et al., 1994; Rutter et al., 2003b], a structured parent interview, and the Autism Diagnostic Observation Schedule (ADOS) [Lord et al., 1994; Luyster et al., 2009] are considered the gold standards for the diagnosis of autism. These instruments differentiate between autism disorder and PDD-NOS with definitive thresholds. They were developed for use in research, but both have become commercially available for clinical use. The procedures are time-consuming and costly, and they require extensive training. As a result, they are most likely to be used in research or specialty clinics for ASDs and are not widely used in general clinical practice.
The ADOS is a semistructured, observational assessment that includes investigator-directed activities to evaluate communication, reciprocal social interaction, play, stereotypic behavior, restricted interests, and other abnormal behaviors. The procedure takes about 30–45 minutes to complete. Five modules are available, including a new research module for toddlers under 30 months of age. Each of the other four modules corresponds to a developmental level, so the instrument can be used with individuals of all ages and all levels of functional impairment. The ADOS is an observation of current behavior, and it must be combined with a clinical interview to obtain historical information. An algorithm can be used to convert ADOS scores into a severity metric, which facilitates comparison of scores across modules, making it useful to track changes in functioning over time [Gotham et al., 2008].
The Neurologic Evaluation in Autism
Neurologic Examination
Most investigators report that a small proportion of autistic children have frank macrocephaly or megalencephaly. The distribution of head size is clearly shifted upward, with the mean approximately at the 75th percentile [Bailey et al., 1993; Bolton et al., 1994; Courchesne, 2004; Davidovitch et al., 1996; Lainhart, 2003; Lainhart et al., 2006; Woodhouse et al., 1996]. Some investigators have observed that a large head circumference correlates with higher IQ and tends to be a familial trait [Filipek et al., 1992; Miles et al., 2000]. The large head circumference is not usually present at birth, but it may appear in early to middle childhood with increased rates of growth [Lainhart, 1997; Mason-Brothers et al., 1990; Mraz et al., 2009]. Head circumference is generally normal by adolescence and adulthood [Aylward et al., 2002], as is postmortem brain weight by adulthood [Bauman and Kemper, 1997]. Microcephaly is more common in autistic subjects than in controls, and it is associated with abnormal physical morphology, medical disorders, lower IQ, and seizures [Fombonne et al., 1999; Miles et al., 2000].
Abnormalities in the neurologic examination may include hypotonia, which was observed in about 25 percent of 176 autistic children and in 33 percent of 110 nonautistic, developmentally delayed children. Spasticity was found in less than 5 percent of either group (exclusionary criteria for this sample included the presence of lateralizing gross motor findings) [Rapin, 1996]. Motor apraxia was identified in almost 30 percent of autistic children with normal cognitive function; in 75 percent of retarded, autistic children; and in 56 percent of a nonautistic, retarded control group [Dziuk et al., 2007; Mari et al., 2003; Ming et al., 2007; Mostofsky et al., 2006; Rapin, 1996; Smith and Bryson, 2007; Vernazza-Martin et al., 2005; Williams et al., 2004]. Characteristic motor abnormalities include motor stereotypies, which occurred in more than 40 percent of autistic children but in only 13 percent of the nonautistic control group [Rapin, 1996]. Hand or finger mannerisms, body rocking, and unusual posturing are reported in 37–95 percent of individuals, and they often manifest during the preschool years [Lord, 1995; Rapin, 1996; Rogers et al., 2003]. There may be considerable overlap in the repertoire of stereotypic movements that autistic children and normal or mentally retarded children display, but they are more prevalent in children with autism especially, most particularly those who are low-functioning [Singer, 2009].
Clinical Testing
Evaluation of Hearing
Many children diagnosed with autism are first described by parents as acting “as if deaf.” However, most children with autism have normal hearing function. Rosenhall et al. [1999] performed audiologic evaluations on 199 children and adolescents with autism and found that pronounced to profound bilateral hearing loss or deafness was present in 3.5 percent of all cases, a prevalence greater than that for the general population but similar to the rate for individuals with mental retardation. However, the rate of hearing loss in this study was equivalent across all levels of cognitive functioning. In contrast, hyperacusis was commonly found; it affected almost 20 percent of the autistic sample. Tharpe et al. [2006] found that, as might be expected, behavioral responses of children with autism were elevated and less reliable, relative to those of typically developing children. In addition, approximately half of the children with autism demonstrated abnormal behavioral responses to tones (but not to speech sounds), despite having normal to near-normal hearing sensitivity as determined by other audiometric measures. Audiologic evaluation or brainstem auditory-evoked potential testing should be performed in all children with autism so that, if indicated, appropriate referrals can be made for aural habilitation [American Speech-Language-Hearing Association, 1991; Filipek et al., 1999, 2000a; Johnson and Myers, 2007; Myers and Johnson, 2007].
Lead Level
Children with developmental delay who spend an extended period in the oral-motor stage of play (when everything “goes into their mouths”) are at increased risk for lead toxicity, especially in certain environments [Filipek, 2005]. The prevalence of pica in this group can result in high rates of substantial and often recurrent exposure to lead and other metals [Shannon and Graef, 1997]. All children with developmental delay or who are at risk for autism should have a periodic lead screen until the pica disappears [Centers for Disease Control and Prevention, 1997; Shannon and Graef, 1997].
Electroencephalography
There is currently insufficient evidence to recommend for or against the use of routine screening EEGs in autistic patients. A recent review found that epileptiform EEG abnormalities were present in 10.3–72.4 percent of patients and subclinical abnormalities in 6.1–31 percent [Kagan-Kushnir et al., 2005]. The authors concluded that further research is needed about the significance of the EEG abnormalities in the absence of clinical epilepsy (see section on epilepsy p. 14).
Metabolic Testing
The reported co-occurrence of autistic-like symptoms in individuals with inborn errors of metabolism has led to consideration of screening tests as part of the assessment of patients with severe developmental impairment [Steffenburg, 1991]. Although the percentage of children with autism who prove to have an identifiable metabolic disorder has traditionally been considered to be less than 5 percent [Dykens and Volkmar, 1997; Rutter, 1997; Rutter et al., 1994], the recent recognition of the potential association between autism and mitochondrial disorders is bringing this figure into question. Most biochemical analyses are useful only as research tools in the on-going effort to understand the biology of autism, but metabolic testing clearly is indicated by a history of lethargy, cyclic vomiting, early seizures, dysmorphic or coarse features, mental retardation, questionable newborn screening, or birth outside of the United States [Filipek, 2005]. Although on-going research studies may produce evidence to the contrary, at present, selective metabolic testing should continue to be initiated only in the presence of suggestive clinical and physical findings [Curry et al., 1997; Filipek et al., 1999, 2000b].
Neuroimaging Studies
Routine neuroimaging to evaluate a child with autism and macrocephaly is not warranted unless there is evidence of lateralizing signs on neurologic examination. A review of neuroimaging reports for children with autism found a very low prevalence of focal lesions or other abnormalities, none of which turned out consistently to be more than coincidental findings. PET and single-photon emission computed tomography (SPECT) are used only as research tools and are not indicated in the diagnostic evaluation of autism [Filipek, 2005; Filipek et al., 1999, 2000a].
Tests of Unproven Value
Unsupported claims have led a number of parents to subject their children to various tests in the hope of finding a treatable cause of autism. There is inadequate evidence to support routine clinical testing of individuals with autism for trace elements in the hair [Shearer et al., 1982; Wecker et al., 1985], celiac antibodies [Pavone et al., 1997], allergies (particularly food allergies caused by gluten, casein, and Candida and other molds) [Lucarelli et al., 1995], immunologic or neurochemical abnormalities [Cook et al., 1993; Singh et al., 1997; Yuwiler et al., 1992], micronutrients such as vitamin levels [Findling et al., 1997; LaPerchia, 1987; Tolbert et al., 1993], intestinal permeability [D’Eufemia et al., 1996], stool analysis, urinary peptides, thyroid function [Hashimoto et al., 1991], or erythrocyte glutathione peroxidase [Michelson, 1998].
Coexistent Medical Conditions
Feeding Disturbances and Gastrointestinal Problems
Feeding habits and food preferences of children with autism typically are unconventional. Bowers [2002] performed an audit of referrals of autistic children to a dietetic service over a 3-month period and found that, despite selective food preferences in 46 percent, the majority of children had intakes that met or exceeded dietary reference values.
Although there have been reports of gastrointestinal complaints in children with autism dating back more than 30 years [Goodwin et al., 1971; Walker-Smith and Andrews, 1972], the gastrointestinal tract only recently has become a significant focus of study [Filipek, 2005]. In one survey of 500 parents of autistic children, almost 50 percent reported loose stools or frequent diarrhea [Lightdale et al., 2001b]. Other studies found that 19–24 percent of children with autism reported gastrointestinal symptoms, with constipation identified in 9 percent [Fombonne and Chakrabarti, 2001], and diarrhea in 17 percent [Molloy and Manning-Courtney, 2003].
In contrast, others have found that the frequency of gastrointestinal symptoms in ASD is not as common as the literature from gastroenterology clinics might suggest [Black et al., 2002; DeFelice et al., 2003; Kuddo and Nelson, 2003; Mouridsen et al., 2009; Peltola et al., 1998; Taylor et al., 2002]. The recent consensus panel [Buie et al., 2010] reviewed the available literature and concluded that evidence-based recommendations for diagnostic evaluation and management of gastrointestinal problems in children with ASD were not yet available, but that individuals with ASD should receive the same standard of care in the diagnostic work-up and treatment of gastrointestinal concerns, as should occur for patients without ASD. Providers should be aware that problem behavior in patients with ASD may be the primary or sole symptom of underlying medical conditions, including gastrointestinal disorders.
In addition, the consensus panel found that available research data do not support the use of a casein-free diet, a gluten-free diet, or combined gluten-free/casein-free (GFCF) diet as a primary treatment for individuals with ASD. To date, only one small randomized, double-blind crossover study has been published. Similar results were found in both the GFCF diet and non-diet-treated groups [Elder, 2008].
Sleep Disturbances
Sleep disturbances, and particularly abnormalities in sleep-wake cycles, have been a recognized feature of autism for over 25 years (see Didde and Sigafoos [2001]; Ivanenko and Johnson [2008]; Richdale [1999]; and Stores and Wiggs [1998] for reviews). The majority of children with autism have sleep problems, often severe, and usually involving extreme sleep latencies, lengthy nighttime awakenings, shortened night sleep and early morning awakenings [Glickman, 2009; Honomichl et al., 2002; Johnson et al., 2009; Johnson and Malow, 2008; Patzold et al., 1998; Richdale and Schreck, 2009; Schreck and Mulick, 2000; Souders et al., 2009]. Children with autism also have more unusual and obligatory bedtime routines: e.g., requiring that parents hold them, lie down with them, or sit beside their bed; that all family members go to bed at the same time; or that curtains or bedclothes be positioned in a certain way. If these routines are not performed exactly, the result is usually a tantrum or other angry outburst. As might be expected, only the autistic children always followed their bedtime routine [Patzold et al., 1998], and the presence of sleep problems was significantly associated with parental stress [Richdale et al., 2000]. Schreck et al. [2004] recently suggested that both the quantity and quality of sleep per night predicted overall autism scores, as measured by the Gilliam Autism Rating Scale (GARS) [Gilliam, 1995], social skills deficits, and stereotypic behaviors. It is unclear whether sleep disorders in children with autism cause daytime maladaptive behaviors, simply allow them to continue, or actually worsen pre-existing problems [Wiggs and Stores, 1996].
Hering et al. [1999] compared the results of parental questionnaires with electronic movement activity recordings (actigraphy) in three groups of children: group 1 consisted of autistic children whose parents reported sleep difficulties; group 2, autistic children whose parents did not report sleep difficulties; and group 3, typically developing children. The initial questionnaires showed that 50 percent of children in group 1 had sleep disorders versus only 20 percent in group 2 and none in group 3. When sleep was quantified using actigraphy, there were no differences in patterns of sleep between groups 1 and 2, except for more early morning awakening in group 1. These findings support the need for objective study methodologies in these samples. Diomedi et al. [1999] compared polysomnograph parameters in adult autistic individuals, who demonstrated a significant reduction of rapid-eye-movement (REM) sleep, increased interspersed wakefulness, and increased number of awakenings with reduction of sleep efficiency, relative to normal controls (see Harvey and Kennedy [2002] for a comprehensive review of polysomnography in autism and other developmental disabilities).
Epilepsy
Epilepsy occurs frequently in children with autism; approximately one-third will develop epilepsy by adulthood, and all seizure types occur [Volkmar and Nelson, 1990]. There is a bimodal distribution of age of onset, with peaks occurring at younger than 5 years and during adolescence [Spence and Schneider, 2009; Tuchman and Rapin, 2002], and with the rate of epilepsy increased among those with mental retardation or underlying medical conditions. The presence of cerebral palsy or focal motor findings also increases risk. Tuchman et al. [1991] found a prevalence of epilepsy of 14 percent among 314 autistic children, but many were younger than adolescent age; in autistic children with normal or near-normal cognitive function, the risk of epilepsy was less than 10 percent by age 10 years. The rate was 27 percent in the presence of severe mental retardation and 67 percent for those with severe mental retardation and a motor deficit. In a subgroup of 160 children with ASD, normal intelligence, no associated cause, and no family history, the cumulative probability of epilepsy was 6 percent, similar to a rate of 8 percent for children with developmental language disorders. The risk of epilepsy in children with Asperger’s disorder and PDD-NOS was less than 10 percent.
Spence and Schneider [2009] recently reviewed the role of epilepsy and epileptiform EEGs in ASD. They found that the rate of epilepsy, even in idiopathic cases of autism with normal IQ, was indeed significantly higher (13–17 percent) [Canitano, 2007; Canitano et al., 2005; Hara, 2007] than the risk in the general population (1–2 percent). Reports have also focused on the presence of subclinical epileptiform abnormalities [Akshoomoff et al., 2007; Baird et al., 2006a; Canitano, 2007; Chez et al., 2006; Gabis et al., 2005; Giannotti et al., 2008; Giovanardi Rossi et al., 2000; Hara, 2007; Hrdlicka et al., 2004; Hughes and Melyn, 2005; Kim et al., 2006; Rossi et al., 1995; Spence and Schneider, 2009]. Kim et al. [2006] found epileptiform abnormalities in the absence of electrographic and clinical seizures in 19 of 32 children referred for prolonged video EEG monitoring for possible seizure activity. Chez et al. [2006] noted epileptiform EEG abnormalities during sleep in 61 percent of almost 900 children with ASD without a history of epilepsy. Other studies, however, report much lower rates [Canitano, 2007; Gabis et al., 2005; Hara, 2007; Parmeggiani et al., 2007], which may be due to differences in sample characteristics and the use of routine EEGs versus prolonged video monitoring. The high rates of isolated epileptiform abnormalities represent a possible objective physiologic finding in ASD, and the treatment of these discharges in the absence of documented seizures is controversial, pending future well-designed longitudinal studies [Spence and Schneider, 2009].
Autism may follow infantile spasms [Riikonen and Amnell, 1981]. Chugani et al. [1996] found that, among children with infantile spasms, those with bitemporal glucose hypometabolism were likely to manifest autism with severe language impairment and mental retardation. In a recent large population-based study, infantile spasms predicted high risk for ASD, but this was, to a large extent, explained by the association of ASD with the symptomatic origin of the seizures [Saemundsen et al., 2008].
Congenital Blindness
The comorbidity of autism and congenital blindness has received relatively meager attention in the autism research literature. Autistic symptomatology has been anecdotally associated with congenital blindness (CB) for decades; in some studies up to 30 percent of children with CB were also described as being autistic (see reviews by Cass [1998]; Hobson and Bishop [2003]; Hobson et al. [1999]. Rogers and Newhart-Larson [1989] reported a diagnosis of autism in all five boys studied with Leber’s congenital amaurosis. Ek et al. [1998] found that 56 percent of premature babies with retinopathy of prematurity (ROP) had both autistic disorder and mental retardation, and, of those, one-third had coexistent cerebral palsy; in comparison, only 14 percent of those with hereditary retinal disease had autistic disorder. Janson [1993] postulated that, in blind children with ROP, a behavior pattern of unresponsivity and stereotypic object manipulation emerges between 12 and 30 months to distinguish autistic and nonautistic children with CB. Msall et al. [2004] followed children with ROP at ages 5 and 8 years, and found that 23 percent had epilepsy, 39 percent cerebral palsy, and 44 percent learning disabilities. Of the children with no or minimal light perception or totally detached retinas bilaterally, 9 percent were autistic, as compared with only 1 percent of those with more favorable visual status.
Cass et al. [1994] reported that, of an entire sample of over 600 congenitally blind children of differing etiologies, only 17 percent demonstrated no evidence of additional disabilities and were developing normally at age 16 months when first studied. Subsequently, 31 percent had a regression in their development occurring between 16 and 27 months of age; children who regressed tended to have disorders of central nervous system/optic nerve/retina, while children who did not regress had a purely optical cause for their blindness (e.g., congenital cataracts or glaucoma). The more “central” pathophysiology of the blindness in the regression cohort was subsequently confirmed by neuroimaging studies; the children with developmental regression had more central nervous system lesions than those who did not regress [Waugh et al., 1998].
Brown et al. [1997] reported that almost half of their sample with CB met criteria for autism, and that, even in CB without autism, there were significantly more “autistic features” than seen in matched sighted children. Brown et al. [1997] and Hobson et al. [1999] compared congenitally blind (of various etiologies) and sighted autistic children and noted remarkably similar clinical features. The authors’ clinical impressions were that blind autistic children were less severely impaired than sighted autistic children; none was abnormal in listening response, but most were markedly abnormal in body and object use. Mukaddes et al. [2007] examined 257 blind children for autism, and identified 30 (12 percent) who met criteria for DSM-IV Autistic Disorder. Differentiating factors between those CB children with and without autism included severity of blindness, cerebral palsy, and intellectual level, with greater neurological impairments and more severe blindness in those with CB and autism.
Known Genetic and Other Conditions associated with Autism
Known single-gene defects and diagnosed medical conditions account for about 10 percent of cases of autism [Chakrabarti and Fombonne, 2001; Fombonne, 2002a], and there may be subtle but qualitative differences in the symptoms of ASD in specific syndromes. Bolton et al. [1991] and Rutter et al. [1993] found that 8.1 percent of 151 individuals with autism had specific associated medical conditions, including fragile X syndrome, bilateral deafness, cerebral palsy, multiple congenital abnormalities, and identified chromosomal anomalies. About 3.8 percent had other medical concerns that were less likely to be associated with etiologic factors. The possibility of finding any associated medical condition rises with increasing degrees of mental retardation – approaching 50 percent among persons at the severe and profound levels of cognitive dysfunction [Scott, 1994]. In a survey of multiple studies, the fraction of patients with autism who have an associated medical condition of potential etiologic significance was 0–17 percent, with a mean of 6 percent [Fombonne, 2003a, b]. Defined mutations, genetic syndromes, and metabolic diseases account for up to 20 percent of autistic patients [Benvenuto et al., 2009a, b].
These studies investigated the prevalence of medical disorders in populations of individuals with autism. It is important to differentiate that perspective from the prevalence of an autistic phenotype in a population of individuals with the given disorder. Tuberous sclerosis complex (TSC) and fragile X syndrome (FraX) are prime examples to illustrate this concept. In a population of individuals with autism, only a few will have a diagnosis of TSC or FraX; however, in a population of individuals with TSC or FraX, over 50 percent will have a diagnosis of autism. It is therefore important to recognize the autistic phenotype in associated genetic-metabolic and syndromic conditions to optimize intervention plans for these children and adults [Moss and Howlin, 2009].
Fragile X Syndrome
Fragile X syndrome is second only to Down syndrome in terms of a known chromosomal cause of mental retardation. Between 21 and 50 percent of boys with fragile X syndrome are on the autistic spectrum [Moss and Howlin, 2009], and 0–6 percent of autism populations have fragile X syndrome (median of six studies 0.75 percent) [Fombonne et al., 1997b]. A milder presentation of autistic symptoms is more common, and symptoms tend most frequently towards social anxiety, extreme shyness, and gaze avoidance [Roberts et al., 2007]. Summarizing data across 40 studies, Fisch [1992] found virtually identical pooled proportions of fragile X syndrome in autistic males and in mentally retarded males. These studies suggested that autism and fragile X syndrome may occur together, but the prevalence of these cases is much lower than originally thought, and that fragile X syndrome is not a major etiologic factor in autism.
Tuberous Sclerosis Complex
TSC has been strongly associated with autism and identified more frequently in individuals with mental retardation and most commonly in those with epilepsy [Gutierrez et al., 1998; Kandt, 2003]. Rates of ASD in TSC most consistently range between 24 and 60 percent [Moss and Howlin, 2009]. The incidence of autistic individuals with TSC complex has been estimated to be between 0.4 and 4 percent in epidemiologic studies [Chudley et al., 1998; Fombonne et al., 1997b; Smalley, 1998]. This rate increases to 8–14 percent for autistic individuals with epilepsy [Gillberg, 1991].
PET studies of children with TSC and autism have revealed changes in the deep cerebellar nuclei and caudate nuclei that were not seen in nonautistic TSC subjects [Asano et al., 2001], but the presence of cortical tubers in the temporal lobes did not increase the risk for autism.
15q Syndrome
A chromosomal abnormality that occurs in 1–4 percent of autistic individuals involves the proximal long arm of chromosome 15q11–q13 [Browne et al., 1997; Schroer et al., 1998], which is the most frequent of the currently identifiable chromosomal disorders associated with autism. The clinical phenotype is highly variable, ranging from profound psychomotor retardation to normal nonverbal cognitive scores [Filipek et al., 2000c]. The duplication is usually maternally inherited, and involves the area roughly corresponding to the Prader–Willi/Angelman critical region (PWACR) of approximately 4 million base pairs. The additional genetic material may be interstitial, producing a trisomy of 15q11–q13, which may or may not be inverted, or may be a separate marker chromosome, producing a tetrasomy of this region. Over 100 individuals with autism and this chromosomal anomaly have been reported in the literature to date (see Schanen [2006] for a recent review). In addition to MECP2 deletions, Longo et al. [2004] also found 15q11–q13 duplications in 3 out of 63 (4.7 percent) patients with Rett’s syndrome.
Chromosome 22q11 Deletion Syndrome/Velocardiofacial Syndrome
Shprintzen et al. [1981] first described velocardiofacial syndrome (VCFS), which is characterized by cleft palate, cardiac malformations (usually a ventricular septal defect), typical facies (tubular nose, narrow palpebral fissures, and retruded jaw), learning disabilities and/or mental retardation, microcephaly, short stature, central nervous system vascular malformations, and seizures [Coppola et al., 2001; Perez and Sullivan, 2002; Roubertie et al., 2001]. It is now known to be caused by a microdeletion in the TBX1 gene on chromosome 22q11.2, and its prevalence is estimated at 1 in 4000 [Bassett and Chow, 1999]. Gothelf et al. [2001, 2004] reported that 16–25 percent will develop psychotic disorder by adolescence; the prevalence of schizophrenia in VCFS is 25 times that of the general population, and up to 40 percent meet criteria for attention-deficit hyperactivity disorder (ADHD), and 33 percent for obsessive-compulsive disorder. Kozma [1998] was the first to report comorbid autism in VCFS, with associated severe mental retardation. Subsequent studies found that 20–30 percent of VCFS subjects met criteria for autistic disorder, and over 50 percent for an ASD; over 50 percent had mental retardation [Chudley et al., 1998; Fine et al., 2005; Kates et al., 2007; Niklasson et al., 2001, 2002; Roubertie et al., 2001; Vorstman et al., 2006].
Autism has also been associated with neurofibromatosis [Fombonne et al., 1997b; Gillberg and Forsell, 1984; Marui et al., 2004; Mouridsen et al., 1992; Williams and Hersh, 1998], Smith–Lemli–Opitz syndrome [Bukelis et al., 2007; Cohen et al., 2005; Martin et al., 2001; Sikora et al., 2006; Tierney et al., 2006], cerebral folate deficiency [Moretti et al., 2005, 2008; Ramaekers et al., 2007], and biotinidase deficiency [Zaffanello et al., 2003], as well as the classic example of phenylketonuria [Baieli et al., 2003; Chen and Hsiao, 1989; Miladi et al., 1992].
Mitochondrial Disorders
Coleman and Blass [1985] first reported an association of lactic acidosis with autism over 20 years ago, which was corroborated by Laszlo et al. [1994]. Lombard [1998] postulated a mitochondrial etiology for autism, based on, among other things, his unpublished anecdotal observations of carnitine deficiency. Functional neuroimaging methodologies have also related autism and deficient energy metabolism in the brain [Chugani et al., 1999; Levitt et al., 2003; Minshew et al., 1993].
Clark-Taylor and Clark-Taylor [2004] reported a child with autism who also had an abnormal acyl-carnitine profile with elevations of unsaturated fatty-acid metabolites C14:1 and C14:2 and ammonia, and alterations of tricarboxylic acid cycle energy production. Filipek et al. [2003] first reported evidence of mitochondrial dysfunction in two autistic children with inverted duplications of chromosome 15q11–q13. They also found that free and total carnitine and pyruvate were significantly reduced, while ammonia, lactate, and alanine levels were considerably elevated in 100 asymptomatic autistic children in a clinic population, suggestive of mild mitochondrial dysfunction [Filipek et al., 2004]. In a population-based study, Oliveria et al. [2005] found hyperlacticacidemia in 20 percent, with a definite mitochondrial respiratory chain disorder identified in 7.2 percent of 120 children with ASD.
Filiano et al. [2002] reported a group of 12 children presenting with hypotonia, intractable epilepsy, autism, and developmental delay (HEADD syndrome), who demonstrated reduced levels in specific mitochondrial respiratory activities encoded by mitochondrial DNA, with a majority also showing mitochondrial structural abnormalities. Several additional reports have appeared in the literature recently, describing specific mitochondrial DNA abnormalities associated with ASD [Graf et al., 2000; Kent et al., 2008; Pons et al., 2004; Smith et al., 2009]. Weissman et al. [2008], in a record review of 25 children with ASD and mitochondrial disorders, found clinical abnormalities uncommon in idiopathic autism, including constitutional symptoms, especially excessive fatigability, significant non-neurologic medical histories, marked delay in early gross motor milestones, and unusual patterns of regression.
Knowledge of mitochondrial function and dysfunction is presently expanding exponentially and concurrently with knowledge of the neurobiology and genetics of autism; further research will elucidate the validity and extent of mitochondrial dysfunction in individuals with autism [Lerman-Sagie et al., 2004; Zecavati and Spence, 2009].
Down Syndrome
Down syndrome is the most common chromosomal cause of mental retardation, occurring in approximately 1 in 1000 live births [Bell et al., 2003; Iliyasu et al., 2002]. Although once considered implausible, the comorbidity of autism and Down syndrome is not rare [Bregman and Volkmar, 1988; Ghaziuddin, 1997, 2000; Ghosh et al., 2008; Howlin et al., 1995; Lowenthal et al., 2007; Molloy et al., 2008, 2009; Wakabayashi, 1979; Wing and Gould, 1979]. In fact, Down’s original phenotypic description [1887, pp. 6–7] describes the autistic phenotype. In epidemiological studies, the prevalence of Down syndrome in individuals with autism ranges from 0 to 16.7 percent (see Fombonne et al. [2003] for a review). Some studies that screened samples with Down syndrome for autism found relatively low rates of autism, ranging from 1.0 to 2.2 percent; other series have reported that as many as 39 percent of subjects with Down syndrome also meet criteria for autism [Capone et al., 2005; Ghaziuddin et al., 1992; Gillberg et al., 1986; Kent et al., 1999; Lowenthal et al., 2007; Lund, 1988; Starr et al., 2005; Turk and Graham, 1997] (see Moss and Howlin [2009] for a recent review).
Howlin et al. [1995] eloquently championed the importance of recognizing autism in children with Down syndrome. Although autism diagnoses are typically made in the preschool years, they noted later ages of autistic diagnoses in all cases reported in the literature (range from 7 years to adulthood). This singular diagnostic view creates unnecessary stress for families, and prevents them from using supports and interventions available to families with an autistic child. Reasons for the lack of recognition of autistic signs in Down syndrome are unclear. The stereotyped personality of individuals with Down syndrome is outgoing, affectionate, easy-going, placid, cheerful, highly social, and verbal. Yet, children with comorbid Down syndrome and autism are very different from other children with Down syndrome, demonstrating classic deficits in sociability, immediate and delayed echolalia, poor developmental progress in communication skills, motor stereotypies and ritualistic behaviors or interests, and adaptive behaviors. Even though autism may not be common in Down syndrome, it should be considered in the range of diagnostic possibilities for all individuals with this syndrome.
Williams–Beuren Syndrome
Williams–Beuren syndrome (WBS) is a rare disorder first described over 40 years ago [Beuren et al., 1962; Williams et al., 1961], and caused by a microdeletion on chromosome 7q11.23 that includes the gene for elastin [OMIMTM, 2000]. The association between WBS and autism has not been widely studied, and there are only few cases of comorbidity formally reported in the literature [Feinstein and Singh, 2007; Gillberg and Rasmussen, 1994; Herguner and Mukaddes, 2006; Klein-Tasman et al., 2009; Reiss et al., 1985].
WBS and autism have traditionally been thought to show opposing patterns of cognitive strength and weakness. By definition, individuals with autism often have poor verbal and nonverbal communication skills. In contrast, despite significant early language delay, many individuals with WBS have been described as showing relative sparing of expressive language and linguistic functioning, including high-level syntax and semantics [Bellugi et al., 2001], story-telling and narrative enrichment strategies involving affective prosody and a sense of drama [Reilly et al., 1990], and a reliance on stereotypic, adult phrases [Udwin and Yule, 1990]. Recently however, investigators have more specifically characterized the atypical language development in WMS for example, referential language precedes referential pointing, the opposite of what is seen in normal development. Noted that referential language precedes referential pointing in WBS, the opposite of what is seen in typical language development. Toddlers with WBS also do not spontaneously use the pointing gesture in free-play situations. Laing et al. [2002] reported that, despite superficially “good social skills,” children with WBS were deficient at both initiating and responding to triadic interactions (e.g., child–interlocutor–object), which are essential for joint attention and referential uses of language. Laws and Bishop [2004] demonstrated that children with WBS indeed have difficulties with social relationships and a semantic-pragmatic language disorder (described by some as “loquaciousness”), particularly with inappropriate initiation of conversation and the use of stereotyped conversation; they also have a restricted range of interests, specialized factual knowledge and usual vocabulary. The authors suggested that, “Far from representing the polar opposite of autism, as suggested by some researchers, WBS could seem to share many of the characteristics of autistic disorder” [2004, p. 45].
Prader–Willi and Angelman’s syndromes
Described as “sister imprinting disorders” [Cassidy et al., 2000], Angelman’s syndrome (AS) and Prader–Willi syndrome (PWS) are each the result of either a deletion or uniparental disomy (UPD) in the PW-AS critical region of chromosome 15 (see Clayton-Smith and Laan [2003] for a review). AS, coined the “happy puppet syndrome” [Bower and Jeavons, 1967], presents with severe motor and intellectual retardation, ataxia, hypotonia, epilepsy, absence of speech, and unusual “happy” facies, and has been associated with ASD in recent years [Bonati et al., 2007; Moss and Howlin, 2009; Pelc et al., 2008; Peters et al., 2004; Pickler and Elias, 2009; Steffenburg et al., 1996; Trillingsgaard and Stergaard, 2004; Veltman et al., 2005; Zafeiriou et al., 2007]. Thompson and Bolton [2003] reported one case of Angelman’s syndrome and paternal UPD, and discussed the milder AS symptomatology associated with UPD, including a lack of autistic features.
Genomic imprinting is a form of epigenetic modification in which allele silencing is specific to the parent of origin. This occurs in Angleman’s syndrome, caused by molecular abnormalities that include deletion of a maternally derived copy of the 15q11–q13 chromosomal region. Children with Angleman’s syndrome have many overlapping features of autism [Peters et al., 2004]. Two types of imprinting defects may cause Angleman’s syndrome: inherited deletions of the imprinting center, which are primarily genetic but also have secondary epigenetic effects, and imprinting defects that have no identifiable DNA sequence abnormality [Jiang et al., 2004].
PWS is characterized by obesity, muscular hypotonia, mental retardation, short stature, hypogonadotropic hypogonadism, and small hands and feet. It appears that PWS results from UPD or deletion of the paternal copies of the imprinted small nuclear ribonucleoprotein polypeptide N (SNRPN) and necdin genes, and possibly others as well. Several recent reports note an association between PWS and ASD [Descheemaeker et al., 2002, 2006; Dimitropoulos and Schultz, 2007; Feinstein and Singh, 2007; Greaves et al., 2006; Milner et al., 2005; Veltman et al., 2005]. Veltman et al. [2004] found that maternal UPD cases of PWS would be more likely to exhibit ASD than would cases with deletions in the PWACR. Therefore, the extent of the associations of AS and PWS with autism remains unclear to date, particularly the differential effects of UPD, as compared with deletions of the responsible genes.
Genetic Studies
Although Bettelheim [1967] espoused the theory that autism was caused by parental behavior (the “refrigerator mother”), it has become clear that autism is a complex genetic disorder. Twin studies provide strong evidence that autism is genetic, involving multiple genes and variable expression. The concordance rate for autistic disorder in monozygotic twins has been reported as 36–90 percent [Bailey et al., 1995; Folstein and Rutter, 1977; Ritvo et al., 1989; Steffenburg et al., 1989], compared with 0–23 percent in dizygotic twins [Bailey et al., 1995]. However, when twin pairs are evaluated for a broader autistic phenotype that includes communication and social disorders and stereotypic behaviors, the concordance rate is 92 percent for monozygotic twins and 10 percent for dizygotic twin pairs.
In families of children with autism, characteristics known as the broader autism phenotype are more common than in controls [Fombonne et al., 1997a; Pickles et al., 2000]. These characteristics include difficulty with communication and language, including delayed onset [Folstein et al., 1999], social reticence or phobias, and preference for routine and difficulty with change [Pickles et al., 2000; Piven et al., 1991a] or obsessive-compulsive traits [Hollander et al., 2003]. These traits are not usually associated with difficulties in function [Folstein and Rosen-Sheidley, 2001]. Milder impairments in social and communicative skills also occur in siblings and relatives of probands more often than in controls [Bailey et al., 1998; Szatmari et al., 2000]. Relatives are more likely to have traits within the broader autism phenotype if a proband is higher- rather than lower-functioning [Nicolson and Szatmari, 2003], and if there are multiple probands with autism [Spence, 2004].
It was formerly felt that there were multiple genes that, in various combinations, confer small to moderate effects on the autism phenotype. Using entire-genome screens in families with more than one affected member, autism or autistic traits has been linked to various shared genetic markers that are regions of interest inherited by affected persons more frequently than would be expected by chance. Multiple collaborative groups have facilitated the identification of regions of interest. One consortium recently identified a common genetic variant associated with autism on chromosome 5p14.4 [de Vries, 2009]. Many of these identified markers have not been verified in other populations, and it has been difficult to find robust common variants [Levy et al., 2009]. Using new whole-genome DNA microarray technologies, or high-density screening platforms, progress has been made in identifying structural abnormalities, from microscopic to submicroscopic: e.g., copy number variations (CNVs), microdeletions, and rearrangements. Environmental factors may act to influence the risk for de novo mutations. There may be many ASD cases attributable to multiple interacting genes interacting with environmental factors, but the more recent thinking is that there are also multiple rare variants or CNVs, most commonly deletions, that have a primary effect on producing the autism phenotype. These may be inherited or may arise de novo. CNVs may account for up to 10 percent of nonfamilial cases of ASD, and about 2 percent of familial cases [Sebat et al., 2007]. Deletion variations are seen more frequently in sporadic than in familial cases of ASD. Chromosome microarray testing (CMA) has been reported to have the highest detection rate for patients with ASD, even though many of the copy number variants seen do not have known significance [Shen et al., 2010].
ASD candidate genes have been identified using whole-genome approaches. Candidate genes are selected for study, often through known genetic disorders or animal models, because they are known to affect developmental processes that may be involved in the pathogenesis of autism. Candidate genes that interfere with synaptic maturation or function have emerged as a plausible reason for the aberrant structural and functional connectivity seen in ASDs [Levy et al., 2009].
Studies of serotonin transporter genes have yielded conflicting results [Betancur et al., 2002; Kim et al., 2002; Klauck et al., 1997; Maestrini et al., 1999; Persico et al., 2000; Tordjman et al., 2001; Yirmiya et al., 2001]. Glutamate transporter genes were found to be upregulated in postmortem studies [Purcell et al., 2001], and the glutamate receptor-6 gene (GRIK2, formerly designated GLUR6) was expressed in brain regions involving learning and memory [Jamain et al., 2002]. Another candidate gene involves oxytocin. Oxytocin levels affect social behavior, and two genome-wide screens have found linkage to a locus containing the oxytocin receptor gene [Auranen et al., 2002; Shao et al., 2002].
Many of the genes identified to date involve cell adhesion pathways [Betancur et al., 2009], including Shank 3, a synaptic protein that regulates synaptic scaffolding organization, along with neuroligins [Durand et al., 2006]. Neuroligins are important in excitatory synapses and synaptogenesis; defects may affect cognitive development and communication. De novo structural chromosome variations are seen in cases both with and without intellectual disability [Geschwind, 2009]. A region of interest on chromosome 7q has been identified that is associated with developmental language disorders; because a core feature of autism is a communication disorder, it is possible that autism and severe language impairment could share a gene in this region [Ashley-Koch et al., 1999; Badner and Gershon, 2002; Folstein and Rosen-Sheidley, 2001; IMGSAC, 1998, 2001]. The 7q region also contains a gene called RELN that codes for a protein thought to help neurons migrate to their proper location.
Epigenetics
Exogenous factors can modify the control of gene expression. Epigenetics refers to the stable and heritable or potentially heritable changes in gene expression that do not involve a change in DNA sequence [Jiang et al., 2004]. Epigenetic mechanisms involve a signal or stimulus that changes gene expression and may help to explain the onset of symptoms in autism after a period of apparently normal development; it may also play a role in how the environment affects phenotypic expression. Chromosomes originating from one parent may have an abnormal epigenotype that leads to modulation of DNA methylation, which causes gene silencing [Bjornsson et al., 2004a, b].
Genetic Counseling
If one child in a family has ASD, the risk for subsequent siblings is elevated [Chakrabarti and Fombonne, 2001; Micali et al., 2004], and is higher if the child with autism is a girl [Sumi et al., 2006]. Rates of mental retardation without autism are not increased in families with ASDs, suggesting that the mental retardation is part of the autistic disorder, not a separate condition. There is no prenatal test to identify autism, but it is important to look for physical or dysmorphic features in the identified autistic child that may suggest a diagnosable genetic condition such as fragile X syndrome [Miles and Hillman, 2000] or tuberous sclerosis complex.
Cytogenetic methods of karyotyping and molecular analyses for fragile X, and the implications of a cytogenetic or molecular diagnosis for other family members, justify their routine inclusion in the diagnostic evaluation of a child with autism [Abdul-Rahman and Hudgins, 2006; Battaglia and Carey, 2006; Challman et al., 2003; Curry et al., 1997; Filipek, 2005; Herman et al., 2007; Marshall et al., 2008]. Current data do not support extensive clinical genetic testing of all children with ASD [Johnson and Myers, 2007]. However, fluorescent in situ hybridization (FISH) studies targeted for specific deletions or duplications – such as is seen in the 15q syndrome, for example – may prove useful in individual cases. Subtelomeric FISH screening in ASD has failed to identify any abnormalities in a total of 225 subjects [Battaglia and Bonaglia, 2006; Keller et al., 2003; Wassink et al., 2007].
Even if the karyotyping appears normal, a microarray comparative genomic hybridization test (aCGH) is advised. aCGH studies identify CNVs, microdeletions, and microduplications that otherwise would go undetected [Christian et al., 2008; Miller et al., 2010]. As such techniques become more available, they should be extended to children without dysmorphic features, as microdeletions and microduplications are common among those with no dysmorphic features as well [Lintas and Persico, 2009]. When there are implications of a cytogenetic or molecular diagnosis for other family members, especially when another pregnancy may be a possibility, their inclusion in the diagnostic evaluation of a child with autism is recommended [American College of Medical Genetics: Policy Statement, 1994]. Prenatal genetic testing is not recommended, because almost all of the recognizable genetic or genomic abnormalities responsible for ASD are not detectable, with the exception of very rare major chromosomal rearrangements.
Even if no genetic condition is revealed, genetic counseling should include a discussion of the risks of recurrence of ASDs in a subsequent child and the lack of ability to diagnose this prenatally [Folstein and Rosen-Sheidley, 2001; Sumi et al., 2006]. The goal of counseling is to provide as much information as possible but to leave decision-making and interpretation of the genetic information to the families.
Pharmacologic Therapy
Dosing should start with low amounts and be slowly escalated with careful attention to possible side effects, the most common of which is activation, defined as overactivity, agitation, or emotional lability. Weight gain and metabolic changes are common side effects of neuroleptics [Correll et al., 2009], and the long-term consequences are of particular concern in children, as they may require treatment for an extended time. Target symptoms should be clearly defined, and new drugs should be tried for a sufficient length of time to determine their usefulness. One caution to keep in mind is that sedation in response to pharmacotherapy may be mistaken for a positive response [Volkmar and Pauls, 2003]. It is important to work with teachers and families to monitor a therapy’s effectiveness and side effects. Drug treatment should be one facet of a comprehensive, multidisciplinary treatment approach that includes structured special educational techniques, language or communication interventions, behavior modification, and parent training.
Neuroleptic Agents
Neuroleptics that block dopamine receptors, such as haloperidol, thioridazine, and trifluoperazine, were used until the development of drugs that were more effective in blocking serotonin receptors. Haloperidol decreased motor stereotypies, hyperactivity, withdrawal, and negativism in children with autism [Anderson et al., 1989]. Common side effects are sedation and weight gain [Campbell et al., 1997].
The newer atypical neuroleptics include risperidone, clozapine, olanzapine, and quetiapine. They modulate dopamine (D2) and serotonin (5HT2) receptors [Buitelaar and Willemsen-Swinkels, 2000]. Risperidone and aripiprazole have been approved by the Federal Drug Administration (FDA) for the treatment of irritabilty (including aggression, self-injurious behavior, temper tantrums, and mood swings) in school-age children and adolescents with autistic disorder. However, these agents have been associated with weight gain, prolactin increases, and sedation in some patients.
In an 8-week multisite, double-blind, randomized trial of risperidone in 101 children with ASD [McCracken et al., 2002], a relatively low dose of 0.5–3.5 mg/d improved irritability, hyperactivity, and stereotypies. Treatment effects were maintained over 16 weeks of treatment, and discontinuation of the medication resulted in return of behavioral symptoms [Aman et al., 2005]. Side effects included weight gain that averaged 6 pounds over 8 weeks, and mild to moderate fatigue in about half of the children. There was a very low incidence of acute extrapyramidal symptoms. These results have been replicated in children [Hellings et al., 2006; Shea et al., 2004] and adults [Hellings et al., 2006]. Luby et al. [2006] conducted a small trial of risperidone with 24 younger children (ages 2.5–6) with ASD. There was a modest but statistically significant improvement on a global autism rating scale compared to the placebo group after 6 months. Significantly greater weight gain and elevated prolactin levels were observed in the treatment group. A double-blind, placebo-controlled trial with 31 autistic adults [McDougle et al., 1998] similarly found decreased irritability, repetitive behavior, and overall behavioral symptoms in the group treated with risperidone.
An 8-week double-blind, randomized, placebo-controlled trial with aripiprazole in 218 children and adolescents with autistic disorder compared three doses of the drug (5, 10, or 15 mg/d) to placebo [Marcus et al., 2009]. All doses demonstrated improvement in irritability, although only the 5 mg/day group reached statistical significance, possibly due to a high placebo response (35 percent). Improvement was reported within 2 weeks, at a point at which all participants were taking the lowest dose. About 10 percent of the participants withdrew from the study because of adverse effects; the most common side effect leading to discontinuation was sedation. Extrapyramidal symptoms were reported in 23.1 percent of the treatment group, compared to 11.8 percent in the placebo group, and more subjects in the treatment groups received medication to treat these symptoms. Aripiprazole was associated with a higher incidence of weight gain than placebo, although no participants discontinued treatment for this reason.
Other atypical neuroleptics studied with similar results include olanzapine and ziprasidone, although they were assessed only in open trials [Malone et al., 2001] or with very small number of participants [Hollander et al., 2006b].
Concern about side effects has led to some comparisons studies. Miral et al. [2008] compared haloperidol and risperidone in 30 children and adolescents with autistic disorder. Both agents were effective in reducing tantrums, aggression, and self-injury. Risperidone was slightly superior to haloperidol in improving disruptive behavior. There was a significant worsening of extrapyramidal symptoms reported in the group treated with haloperidol. Similar levels of weight gain occurred in each group, and there was a greater increase in prolactin in individuals taking risperidone. Remington et al. [2001] compared haloperidol to the serotonin reuptake inhibitor, clomipramine, in 36 children and adults (ages 10–36 years) with ASD. Haloperidol was superior to clomiprimine in reducing irritability and hyperactivity. Neither medication was superior to placebo in reducing stereotypies or inappropriate speech. There was a higher dropout rate in the clomipramine group due to side effects, particularly behavioral activation.
Opiate Antagonists
Several published trials used naltrexone, a long-acting opiate antagonist that can be taken orally. The hypothesis for benefit is that autism is associated with hypersecretion of brain opioids, including beta-endorphins, and that many symptoms of autism are similar to those induced by opiate administration, such as decreased socialization, repetitive stereotypic movements, and motor hyperactivity [Feldman et al., 1999]. Naltrexone in doses of 1.0 mg/kg daily in 23 autistic children decreased restlessness and hyperactivity [Campbell et al., 1993] in a parallel study design. Side effects were mild gastrointestinal symptoms, appetite decrease, and drowsiness. In a randomized, double-blind, crossover design, naltrexone was associated with modest improvement of behavior in 11 of 24 children, but no improvement in learning occurred [Kolmen et al., 1995]. In a separate, similarly designed study that looked specifically at communication skills, no benefit was seen [Feldman et al., 1999]. Studies by Willemsen-Swinkels and colleagues [Willemsen-Swinkels et al., 1995a, b, 1996, 1999] also failed to prove any benefit, and in some children, stereotypic behaviors were increased.
Serotonin Reuptake Inhibitors
Symptoms causing major disruption in autism, such as anxiety and repetitive and ritualized behaviors, can impair learning. Because of the efficacy of serotonin reuptake inhibitors (SRI; e.g., clomipramine, fluoxetine, sertraline, fluvoxamine, and paroxetine) on anxiety and obsessive-compulsive symptoms, and the finding of serotonin system abnormalities in individuals with autism [Chandana et al., 2005], there has been considerable interest in treating disruptive behaviors in autism with these agents. Results of open-label and observational studies have been mixed but encouraging for the reduction of ritualistic behavior, anxiety, and aggression, as well as behavioral rigidity, obsessive-compulsive disorder symptoms, and stereotypies [e.g., Brasic et al., 1994; Cook and Leventhal, 1996; Cook et al., 1992; DeLong et al., 2002; Fatemi et al., 1998; Hollander et al., 2000; Martin et al., 2003; Owley et al., 2005; Sanchez et al., 1995, 1996; Steingard et al., 1997]. However, results of recent double-blind, placebo-controlled, randomized trials have been mixed, and suggest that efficacy of SRIs may be moderated by age, with better responsivity in adults than in children. Additionally, the evidence for possible developmentally sensitive altered regulation of serotonin synthesis in autistic children provides a rationale for giving serotonergic drugs to very young autistic children in order to improve synaptic plasticity during periods of brain development. Controlled clinical trials enrolling very young children are difficult to undertake, but at least one is in process.
A randomized, double-blind, placebo-controlled trial enrolling adults with autism found improvement in one-half of the fluvoxamine-treated patients compared with placebo, with reduction of repetitive thoughts and behavior, maladaptive behavior, language, social relatedness, and aggression [McDougle et al., 1996]. There were few side effects at a mean dose of 270 mg/day. A crossover trial of six adults using the selective SRI fluoxetine demonstrated significant improvement in obsessive behaviors and anxiety, and PET scans demonstrated fluoxetine-elevated metabolic rates in the right frontal lobes [Buchsbaum et al., 2001].
Results of trials in children, however, have been less encouraging. Low-dose liquid fluoxetine (mean dose of 10 mg/day) was superior to placebo on a measure of repetitive behaviors, but not on a measure of clinician-rated global improvement, in 39 children and adolescents with autism in a randomized, crossover trial [Hollander et al., 2005]. Agitation resulted in a dose reduction in 16 percent of the subjects in the fluoxetine group, compared with 5 percent in the placebo group, but this difference was not statistically significant.
A large double-blind, randomized, placebo-controlled trial of citalopram in children and adolescents with ASD has failed to find the predicted effects on repetitive behaviors. A multisite study with 149 children with ASD (ages 5–17 years) and high levels of repetitive behavior evaluated the efficacy of citalopram in reducing those behaviors [King et al., 2009]. In 12 weeks of treatment with a flexible dose schedule, citalopram did not separate from placebo in the primary outcome measures of repetitive behavior and global improvement. The mean citalopram dose was 16.5 (SD = 6.5) mg/d. Citalopram levels and high parent-reported compliance to treatment suggest that this was an adequate trial and the doses were similar to those previously reported to be effective in open trials. Adverse events were significantly more likely to occur in the citalopram-treated group. The most frequently reported side effects were activation, stereotypy, diarrhea, insomnia, and dry skin or pruritus. Two subjects treated with citalopram had seizures. Of note, there was a high placebo response rate (34 percent), which underscores the need for placebo-controlled clinical trials.
In summary, SRIs, selective SRIs, and other medications affecting serotonin and dopamine levels can reduce specific symptoms of autism, such as ritualized behaviors, stereotypies, rigid behaviors, aggression, and anxiety in adults. Large multisite clinical trials with children found that SRIs did not reduce repetitive behavior, or other disruptive behaviors, in children with ASD. While SRIs appear to be generally safe, children may be particularly sensitive to the behavioral activation of these drugs [Buitelaar and Willemsen-Swinkels, 2000]. Seizures have emerged under SRI treatment in clinical trials in a few instances, although this is a seizure-prone population and it is not clear whether the medications were causal. Additionally, there has been concern about reports of an association of fluoxetine and possibly other selective SRIs with suicidal ideation in depressed children, and the FDA has recommended that children on these medications should be very carefully monitored.
Stimulants and Drugs to Treat Hyperactivity
Hyperactivity is an important target symptom that can be potentially improved with psychostimulant medication [Handen et al., 2000; Quintana et al., 1995]. The Research Units on Pediatric Psychopharmacology (RUPP) Autism Network [Posey et al., 2007; Research Units on Pediatric Psychopharmacology Autism Network, 2005a] conducted a randomized, double-blind, placebo-controlled trial of methylphenidate in children with ASDs and high levels of hyperactivity and/or impulsiveness. Doses at the 0.25 and 0.5 mg/kg level were effective in reducing hyperactivity and impulsivity, but less effective in reducing inattention, at 4 weeks and after 8 weeks’ continuation. The response rate was about 35 percent compared to typical response rates of around 70 percent in non-PDD children with ADHD. About 18 percent of subjects withdrew due to side effects, primarily irritability. Other common side effects included decreased appetite and trouble falling asleep. Some children responded best to lower doses of methylphenidate. Observational data was available on a subset of 33 study participants, suggesting that the benefits of methylphenidate extended to some aspects of social interactions, self-regulation, and affect [Jahromi et al., 2009].
Atomoxetine is a nonstimulant medication for ADHD that inhibits the presynaptic norepinephrine transporter. In a placebo-controlled, double-blind crossover study with 16 children with ASD and ADHD symptoms, atomoxetine was more effective than placebo in reducing hyperactivity. The response rate was similar to that reported for methylphenidate in children with ASD, and the mean highest dose was 44.2 (SD = 21.9) mg/d. Side effects, including gastrointestinal symptoms, fatigue, and racing heart were common but transient [Arnold et al., 2006]. Only one participant terminated the study due to intolerance of side effects.
Two small placebo-controlled trials found that clonidine, an adrenergic receptor agonist, had some effect in decreasing irritability and hyperactivity [Fankhauser et al., 1992; Jaselskis et al., 1992] in children and adults with autistic disorder.
There is little evidence for any beneficial effect on hyperactivity using the selective SRIs. Naltrexone, however, has improved hyperactivity [Campbell et al., 1993; Kolmen et al., 1995; Willemsen-Swinkels et al., 1995a, b, 1996, 1999] in small randomized, controlled trials. In clinical trials testing the efficacy for atypical antipsychotics for severe behavior disturbance, hyperactivity was decreased with risperidone [Hellings et al., 2006; McCracken et al., 2002; Research Units on Pediatric Psychopharmacology Autism Network, 2005b; Shea et al., 2004].
Antiepileptic Drugs
Several antiepileptic drugs have been used for behavioral manifestations of autism, particularly for treating intense rapid mood shifts. In open-label studies, levetiracetam [Rugino and Samsock, 2002] and divalproex sodium [Hollander et al., 2001] appeared to be well tolerated and to improve repetitive behavior, impulsivity, and mood stability. A retrospective study of topiramate in children and adolescents with ASDs suggested that the drug reduced misconduct, hyperactivity, and inattention in 8 of 15 patients. Two randomized, placebo-controlled trials, however, have had mixed results. Lamotrigine was not better than placebo on several parent-report and clinician ratings of disruptive behavior and autism symptoms in a double-blind, randomized, controlled trial of 28 children [Belsito et al., 2001]. Children in both groups showed improvement. In a small placebo-controlled, double-blind trial of levetiracetam in 8- to 17-year-olds (n = 20), there was no difference from placebo on measures of behavioral disturbance, repetitive behavior, and autism symptoms [Wasserman et al., 2006]. In a randomized, double-blind, controlled trial with 13 individuals (mean age = 9 years), divalproex sodium was better than placebo in decreasing repetitive behavior [Hollander et al., 2006a]. Patients with the most robust response had repetitive behaviors of the compulsive type, as opposed to stereotypies. This small trial hypothesized a specific target (repetitive behavior), as opposed to global improvement, and the participants demonstrated high levels of that behavior at baseline. The results need replication in a larger trial.
It has been hypothesized that the mechanism of benefit from these medications might be their effect on subclinical seizures, which have been reported to occur in this population. However, there is no evidence and no controlled trials investigating whether treatment with anticonvulsants in children with autism who have epileptiform discharges but no clinical seizures might improve behavioral outcomes in children with autism [Spence and Schneider, 2009].
Cholinesterase Inhibitors
Acetylcholine (ACh) plays a significant role in attention and memory performance. Acetylcholinesterase inhibitors (AChE) slow the breakdown of ACh. This is the presumed mechanism by which cholinesterase inhibitors, such as donepezil, slow decline in memory, attention, and learning in Alzheimer’s disease. Animal studies have suggested that administration of these agents early in development may also enhance learning in a prospective way. Because postmortem studies have found abnormalities of the cholinergic system and its nicotinic receptors in the brains of individuals with autism [Lee et al., 2002; Perry et al., 2001], there is growing interest in the potential benefit of these drugs in ameliorating neurodevelopmental disorders [Yoo et al., 2007].
Preliminary data provides some support for pursuing larger trials. A retrospective examination of effects of donepezil in eight children with autism suggested improvements in irritability and hyperactivity, but memory and attention were not measured [Hardan and Handen, 2002]. A randomized, controlled 6-week trial was conducted with 43 children with ASDs, examining the effects of donepezil (2.5 mg/d) [Chez et al., 2003]. The investigators concluded that the drug improved language and reduced overall autistic features. However, the statistical analyses pooled blinded and nonblinded data, leaving the results susceptible to confounders such as placebo effects. A prospective, open-label study with 13 children and adolescents with autism treated for 12 weeks with galantamine found reductions in irritability and social withdrawal [Nicolson et al., 2006]. The efficacy of cholinesterase inhibitors is yet to be demonstrated in rigorously designed and replicated trials.
In summary, a number of medications, particularly atypical neuroleptics, psychostimulants, and SRIs, can help decrease specific symptoms associated with autism, such as behavioral outbursts, stereotypic or compulsive behaviors, aggression, anxiety, inattention, and oppositional behavior (Table 48-1; also see chapter 49). Reduction in these behaviors can improve quality of life and promote better opportunities for learning. However, there must be careful attention to matching the medication with the targeted behavior, and to possible developmental differences in treatment response. Side effects are common, and must be monitored closely and weighed carefully against the benefits of the drugs. Unfortunately, there is currently no pharmacologic treatment that has been demonstrated to address effectively the deficits in language, cognition, and social understanding that are core features of autism.
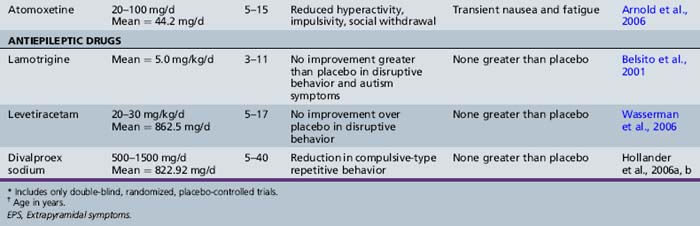
Complementary and Alternative Medicine
A significant number of families seek complementary or alternative medicine treatments, few of which have been studied in well-designed trials [Levy and Hyman, 2008]. One survey [Levy and Hyman, 2003] found that 32 percent of 284 children at a Pennsylvania regional autism center were using complementary and alternative medicine. The investigators suggested that it is important to respect the parents’ belief if the complementary medicine is not toxic, but if the treatment is potentially harmful, negotiating a safer replacement practice should be attempted. Examples of the more commonly used treatments include nutritional supplements, melatonin, hormones, glutein- and caseine-free diet, immunoglobulins, secretin, and chelation therapy.
A review [Nye and Brice, 2005] of vitamin B6 and magnesium concluded that the few studies available were inconclusive and samples sizes were too small for an adequate test of efficacy. A double-blind, placebo-controlled study of pyridoxine and magnesium in 10 patients found no benefit but no significant side effects [Findling et al., 1997].
Dimethylglycine, a nutritional supplement closely related to the inhibitory transmitter glycine, has been proposed [Rimland, 1990] as helpful for autistic children and adults, but it was ineffective in two double-blind, placebo-controlled trials [Bolman and Richmond, 1999; Kern et al., 2001].
Plasma fatty acid levels have been found to be decreased in children with autism when compared with typical controls [Sliwinski et al., 2006; Vancassel et al., 2001], leading to interest in supplementation with omega-3 fatty acids. One small, randomized, double-blind, placebo-controlled pilot study with 13 children reported a nonsignificant trend toward improvement in hyperactivity after 6 weeks of treatment [Amminger et al., 2007]. Mild gastrointestinal side effects were noted.
Melatonin therapy has been used to treat the sleep disturbances in ASD, based on low plasma levels or urinary excretion of melatonin [Kulman et al., 2000; Mulder et al., 2010]. Melke et al. [2008] reported mutations and polymorphisms in the acetylserotonin methyltransferase (ASMT) gene, which encodes the last enzyme of melatonin synthesis, which result in dramatic decreases in ASMT transcripts in blood cell lines. Anecdotal and open-label studies of melatonin therapy have suggested significant improvements in sleep architecture in children with ASD [Andersen et al., 2008; Giannotti et al., 2006; Levy and Hyman, 2008]. Three recent double-blind, placebo-controlled crossover studies were performed in a total of 70 children with ASD using regular (5 mg in Garstang and Wallis [2006]; 3 mg in Wirojanan et al. [2009]) or 5 mg controlled-release melatonin [Wasdell et al., 2008]. Up to 94 percent of children derived significant benefits in sleep without evidence of significant side effects. Larger long-term, double-blind, placebo-controlled crossover studies will be needed to document the efficacy of melatonin across the behavioral subtypes of ASD.
Another popular treatment for ASDs is a gluten-free, casein-free diet. A small randomized, double-blind crossover study was conducted with 15 children with autism on a 12-week GFCF diet [Elder, 2008]. There were no statistically significant differences in autistic symptomatology or urinary peptide levels when participants were on the GFCF diet. The study did, however, demonstrate the feasibility of such a study and the need for an adequately powered trial.
Immunoglobulin has been administered intravenously in open trials [DelGiudice-Asch et al., 1999; Plioplys, 1998] and not found to be useful. A double-blind, placebo-controlled trial of oral human immunoglobulin was conducted with 125 children and adolescents who had autism and persistent gastrointestinal symptoms [Handen et al., 2009]. There was no significant benefit on gastrointestinal symptoms, measures of autistic symptomatology, or behavior disturbances.
Reported dramatic improvement after the administration of secretin as part of endoscopy in three autistic children [Horvath et al., 1998] led to widespread use by parents. Subsequent blinded, randomized trials did not substantiate its efficacy [Carey et al., 2002; Chez et al., 2000; Coniglio et al., 2001; Corbett et al., 2001; Dunn-Geier et al., 2000; Esch and Carr, 2004; Kaminska et al., 2002; Kern et al., 2002, 2004; Levy et al., 2003; Lightdale et al., 2001a; Molloy et al., 2002; Owley et al., 1999; Ratliff-Schaub et al., 2005; Sponheim et al., 2002; Sturmey, 2005; Unis et al., 2002; Williams et al., 2005]. These trials included single- and repeated-dose protocols.
Proponents of chelation therapy suggest that mercury and other heavy metals may be poorly eliminated by children with autism and that it interferes with neurodevelopment via modulation of immune function and other biochemical systems. Despite the lack of scientific evidence of a link between exposure to mercury or other heavy metals and autism, chelation is widely used. One trial [Adams et al., 2009a, b] administered dimercaptosuccinic acid (DMSA) to 69 children with ASD. Those who had a high urinary excretion of toxic metals (n = 49) were then randomized to either DMSA or placebo for an additional six administrations. Some participants in both groups demonstrated improvement of measures of autism symptoms and disruptive behavior, but there were no significant differences between the two groups, and no comparative group that did not receive any treatment. There were no serious adverse effects reported, although a significant increase in excretion of potassium and chromium was noted. No placebo-controlled studies have examined the safety or efficacy of chelation for treating autism, and deaths resulting from hypocalcemia have been reported from the inappropriate use of a chelator, edetate disodium (EDTA) [Brown et al., 2006], including one boy with autism.
It is challenging to perform clinical trials enrolling children with autism, but without well-designed, blinded studies, safe and effective therapies cannot be determined. Problems include standardization of diagnoses, heterogeneity of target problem behaviors, and lack of cooperation of subjects. Many outcome measures are available, including global measures (Clinical Global Impression of Severity) and others targeted to specific symptoms. Whenever feasible, parents should be encouraged to participate in clinical trials to make progress in validating new pharmacologic and behavioral therapies. Information about on-going trials and their locations can be found at www.clinicaltrials.gov.
Educational and Behavioral Interventions
The core deficits associated with autism affect all aspects of the individual’s life, necessitating a comprehensive approach to intervention [Lord and Bailey, 2002; Wetherby et al., 1997]. A primary source of intervention for most children with ASDs is through the educational system. The Individual with Disabilities Education Act (IDEA) ensures a “free and appropriate” public education to “children” between the ages of 3 and 21 who have been diagnosed with learning disabilities. This act specifically covers autistic disorder, but whether the full range of ASDs is covered depends on the particular state’s definition of disabilities. The quality and extent of services that are provided vary from one community to another, even within a particular school district.
Numerous comprehensive early intervention programs for young children with ASDs have been developed and described [Dawson and Osterling, 1997; National Research Council, 2001; Rogers and Vismara, 2008; Seida et al., 2009]. The National Research Council [2001] provides descriptions of ten model programs considered representative of well-described, comprehensive treatment programs with at least some empiric support. Given the great heterogeneity in the population with ASDs, intervention programs must be tailored to the needs of the individual.
The National Research Council [2001] report describes the common elements of successful early intervention programs and makes several recommendations:







Methods based on applied behavior analysis for teaching skills and facilitating more appropriate and adaptive behaviors have been extensively tested for their effectiveness in children and adults with autism and other developmental disabilities [Dunlap and Fox, 1999; Lovaas, 1987; McEachin et al., 1993; Sheinkopf and Siegel, 1998; Smith et al., 2000]. In the most rigorously designed studies of intensive early intervention programs based on applied behavior analysis (ABA), effectiveness is demonstrated at the group level, but response is variable. At the group level, baseline IQ and language skills predict better response to treatment. However, prediction at the individual level is not yet established [Rogers and Vismara, 2008].
Parents are most likely to learn ABA techniques when enrolling their children in a comprehensive treatment program. An alternative to enrollment in a comprehensive school-based program is home-based ABA. In this type of program, parents hire an expert to train the parent and paraprofessionals, who administer the treatment in shifts. As these comprehensive treatment programs have evolved, there have been trends toward teaching parents to implement the programs, toward using the behavior-management techniques in settings that are more naturalistic and during typical activities, and toward developing goals based on the child’s unique developmental profile [Ingersoll et al., 2001]. There is also a trend, at least when applied to programs for children and adults with developmental disabilities, away from emphasizing the consequences of a behavior and toward an emphasis on understanding the triggers of a behavior, proactively providing cues or rehearsal of the appropriate behavior, making changes in the environment to avoid or minimize those triggers, and teaching more adaptive or appropriate responses when the triggers cannot be avoided.
Some comprehensive treatment programs derive strategies from a developmental theoretical framework. For instance, the Denver Model [Rogers et al., 2000] emphasizes the need to establish interpersonal relationships as a foundation to achieving other developmental milestones. Although most of these programs have not been extensively evaluated using rigorous scientific trials, there are theoretical reasons and some preliminary scientific evidence that they can be useful for many children with ASDs [National Research Council, 2001; Rogers, 1998]. An adaptation of the Denver Model for toddlers (18–30 months old), called the Early Start Denver Model (ESDM), was compared to usual community care in a randomized, controlled trial [Dawson et al., 2010]. The ESDM intervention is a parent-delivered treatment applied in the home for 20 hours per week. Intervention began at a mean of 23 months of age. Techniques taught to parents incorporate the developmental techniques from the Denver Model and applied behavior analysis. In this study, children in the ESDM model showed significantly greater improvements in IQ, language, and adaptive skills than the community-treated children with ASD. The gains were greater after 2 years than after 1 year, suggesting benefits from on-going intervention.
Many well-established programs combine elements of behavioral and developmental orientations [Marcus et al., 2000; McGee et al., 2000]. Some have specifically evaluated the effectiveness of the parent-training components of their programs. Parent-training models that are promising based on evidence provided by their developers include the Learning Experiences Alternative Program for Preschoolers and Their Parents [Strain and Cordisco, 2000], the Denver Model [Rogers et al., 2000], the Individualized Support Program at the University of South Florida [Dunlap and Fox, 1999], the Pivotal Response Training Model [Koegel et al., 1999], and the Douglas Developmental Center Program [Harris et al., 2000]. Whatever the theoretical underpinning, well-established and effective programs always include an emphasis on parent–child relationships and overall family support [Dawson and Osterling, 1997]. There is evidence that parents can learn to use these methods and that doing so helps them feel better in general, and more satisfied and confident in their parenting role [Koegel et al., 1996; Ozonoff and Cathcart, 1998; Schreibman, 1997, 2005; Sofronoff and Farbotko, 2002]. One study [Aman et al., 2009] demonstrated that adding a parent training program to medication management for severe behavior disturbances in ASD was more beneficial than medication intervention alone, and allowed for maintenance on a lower dose of the medication.
There is much less evidence when considering intervention programs or treatment options for older children, adolescents, and adults with ASDs. Core deficits in social understanding and social relationships are concerns throughout the life span, and social skills training (SST) is often a component of a treatment plan. Group-based SST programs show promise, but have not been rigorously evaluated [Williams White et al., 2007]. The inclusion of nonautistic peers to assist with SST may be important, but again, rigorous studies are needed [Chan et al., 2009]. A small randomized, controlled trial tested the efficacy of the Program for the Education and Enrichment of Relational Skills (PEERS) for adolescents with ASD [Laugeson et al., 2009]. This group-based intervention integrates parents into the program to help with generalization of skills into the home and community. Results of this study demonstrated benefits in social skills and increased frequency of peer socialization.
The PEERS program described above was originally developed for children with ADHD, and adapted for use with teens with ASD. Similarly, cognitive behavior therapy (CBT) for children with anxiety disorder has been adapted to treat anxiety in high-functioning ASD. A randomized, controlled trial of CBT for anxiety in 7–11-year-old children with ASD demonstrated benefit in reducing anxiety symptoms [Wood et al., 2009]. These studies suggest that adapting evidence-based interventions for specific symptom domains and comorbid symptoms in ASD is a reasonable strategy, but there remains a great need for rigorously designed research to demonstrate efficacy.
Resources for Families
An enormous amount of information is available to families on the Internet; they often need specific counseling regarding evaluation of treatments that are espoused without adequate scientific study. The American Academy of Pediatrics has a useful website (http://www.aap.org/healthtopics/autism.cfm). The National Institutes of Health website (www.nih.gov) can be searched for current research, as can those of specific institutes, including the National Institute of Neurological Disorders and Stroke (www.ninds.nih.gov), National Institute of Child Health and Human Development (www.nichd.nih.gov), and the National Institute of Mental Health (www.nimh.nih.gov). The CDC’s National Center for Birth Defects and Developmental Disabilities has a website (http://www.cdc.gov/ncbddd/autism/index.html) devoted to providing evidence-based information on ASD and its treatment, including links to resources. The National Dissemination Center for Children with Disabilities (www.nichcy.org) has information on special education laws and on educational practices.
Another effort, the First Signs program, is developing methods to inform physicians about the importance of early identification of autism, and it provides resources, including screening tools and referral guidelines. The First Signs website (http://www.firstsigns.org) includes recommendations and information about obtaining autism screening instruments. Educational tools for families can be found on the Exploring Autism website (http://www.exploringautism.com). An organization for autism research has a useful parent’s guide to understanding research on autism (www.researchautism.org). For practitioners, the AAN/CNS practice parameter for evaluation of children with autism and the detailed background paper can be found on the For OC Kids website: (http://www.childneurologysociety.org/resources/practiceparameters).
 The complete list of references for this chapter is available online at
The complete list of references for this chapter is available online at 