Aldosterone
Secretion and Action
The steroid hormone aldosterone first appeared in evolution with the appearance of terrestrial life and the consequent need to conserve sodium and water.1 The primary and best characterized actions of aldosterone are those that stimulate sodium retention in transporting epithelia, particularly the distal nephron, distal colon, and salivary glands.2 At these epithelia, the conservation of sodium is associated with increased secretion of both potassium and hydrogen ions. Aldosterone also has so-called nonclassical actions at the heart, the vasculature, and the central nervous system.
The existence of an adrenal corticoid natriuretic factor, distinct from the other adrenocorticoid steroid hormones, had been suspected for several years before its isolation in 1953.3 Using the toad urinary bladder as a model system, Crabbè4 was the first to show that in vitro aldosterone increases sodium transport. Subsequent studies demonstrated the presence of binding sites for aldosterone in the toad bladder and in other target tissues, particularly the principal cell of the cortical collecting duct of the kidney.5
Feedback Control of Aldosterone Secretion
Both serum sodium concentration and total body sodium are maintained within a narrow range by a complex set of endocrine feedback loops (Fig. 4-1). The most important of these involves the renin-angiotensin system, which responds to volume status. The feedback loops involved with potassium homeostasis operate in parallel and overlap those for sodium.
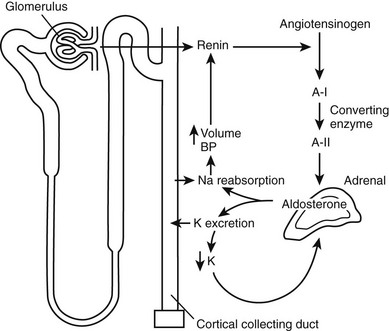
FIGURE 4-1 Interacting feedback loops controlling aldosterone secretion. Volume is regulated through the renin-angiotensin system, and potassium is regulated through direct feedback. A-I, Angiotensin I; A-II, angiotensin II; BP, blood pressure.
Volume status is sensed by the renin-secreting juxtaglomerular cells of the kidney. Where the sodium status (and hence, volume) is low, renin will be secreted. Renin, an aspartyl protease, is synthesised as inactive prorenin that is activated by the action of a protease. Renin release from the juxtaglomerular cells is influenced by a number of factors (Table 4-1), including renal perfusion pressure, the sympathetic nervous system, and prostaglandins (which are stimulatory), dopamine, atrial natriuretic peptide (ANP), and angiotensin II, all of which are inhibitory. Renin acts on angiotensinogen to release the decapeptide angiotensin I, which in turn is subject to further proteolysis by angiotensin-converting enzyme, primarily in the pulmonary vascular bed, to yield the octapeptide angiotensin II. Angiotensin II acts via its specific G protein–coupled receptor in the vasculature as a potent vasoconstrictor (thereby defending plasma volume and blood pressure) and on the adrenocorticoid glomerulosa cells to stimulate aldosterone synthesis.6 The latter response promotes sodium retention with a consequent increase in plasma volume. Aldosterone biosynthesis in the zona glomerulosa of the adrenal cortex is regulated by transcription of the aldosterone synthase gene (CYP11B2). As with other steroidogenic enzymes, steroidogenic factor-1 (SF-1) is required for aldosterone synthase expression. Members of the NR4A family of nuclear receptors have been shown to be regulators of aldosterone synthase gene expression.7 Although angiotensin II is important in the regulation of aldosterone, a response to low-salt or high-potassium diet is also seen in mice in which the angiotensinogen gene has been deleted.8 In these mice, the regulation of aldosterone is directed primarily by serum potassium levels.
Table 4-1
Factor Regulating Renin Release
| Stimulatory | Inhibitory |
| Decreased perfusion pressure | Increased chloride delivery at the macula densa |
| PGl2 | Angiotensin II |
| ACTH | Atrial natriuretic factor |
| Vasopressin | |
| β-Adrenergic stimulation | α-Adrenergic stimulation |
| Dopamine |
Aldosterone secretion is also subject to negative regulation. ANP is a potent inhibitor of aldosterone secretion, consistent with its role to promote natriuresis. Dopamine is a well-characterized inhibitor of aldosterone secretion.9 Other inhibitors have been described, but their physiologic relevance is not clear (Table 4-2).
Table 4-2
Factors Regulating Aldosterone Secretion
| Factor | Stimulatory | Inhibitory |
| Peptides | Angiotensin II | Atrial natriuretic peptide |
| Angiotensin III | Somatostatin | |
| ACTH | ||
| Vasopressin | ||
| Endothelin | ||
| Ions | Plasma potassium | |
| Other | Serotonin | Dopamine |
| Ouabain |
Dietary sodium has a major impact on the state of the renin-angiotensin-aldosterone system (RAS). Sodium deficiency increases adrenal sensitivity to angiotensin II over time; the converse is true of the vasopressor response. Aldosterone-induced sodium retention restores volume status by maintaining the balance between volume and capacity.10
The response of the individual to aldosterone-mediated sodium retention is self-limiting in that after 3 to 4 days, expansion of the extracellular volume plateaus and of sodium secretion returns to control levels. This process is termed escape.11 It should be noted that the kaliuretic effect persists despite the escape of sodium retention. Intrarenal regulators, particularly prostaglandins, are probably the critical mediators of the escape, although other factors (e.g., ANP) may play a role.12,13
A local renin-angiotensin system has been reported to operate in a number of tissues, including the submaxillary glands, gonads, smooth muscle cells, adipose tissue, pituitary, brain, and adrenal cortex. The existence of this system is often determined by the presence of mRNA for renin, angiotensinogen, and angiotensin-converting enzyme (ACE); the relative physiologic importance of these local systems has recently been called into question.14
Potassium Homeostasis
Aldosterone is primarily involved in the chronic regulation of plasma potassium levels.15 Acute regulation involves nonrenal mechanisms such as those mediated by insulin and β-adrenergic agonists. Aldosterone regulates potassium homeostasis through direct effects on transport of epithelia, including its effects on sodium homeostasis. Small fluctuations in plasma potassium influence aldosterone secretion. Although the mechanism of these effects has not been determined, it is known to be independent of angiotensin II levels; however, plasma potassium levels do alter the sensitivity of the adrenal to angiotensin II. The local adrenal RAS has been implicated in the adrenal response to potassium; the circulating system is inhibited by potassium, whereas local adrenal production is increased.
Aldosterone secretion is also subject to regulation by adrenocorticotropic hormone (ACTH); however, aldosterone regulation is normal in patients with hypopituitarism.16
Mineralocorticoid Receptors
High-affinity cytosol and nuclear binding of 3H-aldosterone were first described in classic mineralocorticoid target tissues such as kidney5,17 and parotid18 more than 30 years ago. Spironolactone was shown to block aldosterone binding and action on urinary electrolytes in parallel,19 providing evidence that these sites are physiologic mineralocorticoid receptors (MRs). MR were subsequently cloned from human kidney,20 and the rat homologue was cloned from a hippocampal cDNA library.21 In contrast to glucocorticoid receptors (GRs), which are expressed ubiquitously, MRs have a tissue-specific pattern of expression, with highest levels observed in the distal nephron,22 distal colon,23 and hippocampus.20 Lower levels of expression are observed elsewhere in the gastrointestinal tract; in cardiovascular tissues; and in a range of other tissues, both epithelial and nonepithelial.20,22,23
The human mineralocorticoid receptor is a protein of 984 amino acids and, together with the GR, progesterone receptor (PR), and androgen receptor (AR), forms a distinct subfamily within the steroid/thyroid/retinoid/orphan receptor superfamily.24 This receptor superfamily is defined by a central cysteine-rich DNA-binding domain. At the C terminus is the ligand-binding domain (LBD), which has a highly conserved tertiary structure. The N-terminal domain has little or no homology between receptors. Within the MR/GR/PR/AR subfamily, MR and GR are closely related, with 94% amino acid identity in the central cysteine-rich DNA-binding domain, and 57% identity in the C-terminal ligand-binding domain (Fig. 4-2). The MR and GR, however, are located on different chromosomes (MR on 4q31.225; GR on 5q3126).
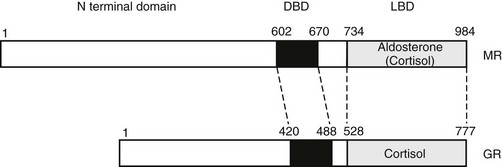
FIGURE 4-2 Domain structure of the mineralocorticoid receptor (MR) and the closely related glucocorticoid receptor (GR) showing the three principal functional domains: the N-terminal domain, the DNA-binding domain (DBD), and the ligand-binding domain (LBD). These domains share less than 15%, 94%, and 57% identity, respectively.
The cysteine residues of the DNA-binding domain complex around two zinc atoms to form two α-helices, one of which lies in the major groove and binds with a common consensus sequence in the DNA, the hormone response element.27 The LBD consists of 11 α-helices, which form a three-layered structure with the ligand-binding pocket buried in the middle.28–30 The N-terminal domain contains a transcription activation function that is relatively unstructured. The N-terminal domains are not conserved between steroid receptors, although in several (including the MR31), a functional interaction has been described between the N-terminal and ligand-binding domains. It is curious that for the MR, this interaction is seen only with aldosterone; cortisol acts as an antagonist.
The unliganded receptor is predominantly cytoplasmic,32 being complexed with the heat shock proteins 70 and 90 and their co-chaperones.33 This configuration maintains the receptor in a transcriptionally inactive high-affinity binding state. The interaction of the ligand-binding domain with this complex is an important determinant of ligand-binding affinity and specificity.34 The mineralocorticoid receptor antagonists, spironolactone and eplerenone, appear to be accommodated into the ligand-binding pocket without distortion,35 suggesting that the mechanism of their antagonism differs from that of the estrogen receptor antagonists such as tamoxifen and raloxifene. These latter compounds exhibit tissue-specific antagonism, in contrast to spironolactone, which is a pure antagonist. Evidence that cortisol/corticosterone may antagonize the actions of aldosterone at the MR in certain tissues suggests that different ligands may induce differing conformations on binding the receptor.31 At a cellular level, these differing conformations result in differential interactions with the transcriptional machinery through the mediators of this signaling, the co-regulatory molecules. In contrast to the other steroid receptors, such interactions are only now being characterized for the MR.36
Very good evidence suggests that not all MRs are physiologic receptors for aldosterone. In brief, cortisol and corticosterone (the physiologic glucocorticoid in the rat) have an affinity for the MR equivalent to that for aldosterone and substantially higher than their affinity for the GR.20 MRs are distributed widely in tissues in which a physiologic effect of aldosterone on Na+ homeostasis is unlikely (e.g., the hippocampus20,37). Given the much higher circulating levels of glucocorticoids than of aldosterone, these sites appear to be high-affinity GRs in such tissues. In nonepithelial tissues, the response of MR to cortisol/corticosterone and aldosterone often is not equivalent,38 leading to speculation about the relationship of epithelial to nonepithelial MR. Most evidence to date would suggest that although the MR gene uses multiple, tissue-specific promoters,39–41 the coding region is unaltered between tissues, with the possible exception of some minor isoforms.42 The explanation for such differences between tissues may lie in the nature of the conformation that the MR adopts after ligand binding.43 Such conformational differences may alter some but not all transactivation functions, such that tissue-specific receptor co-activators or co-repressors36 may mediate different responses in different tissues.
A second question, given the equivalent affinity of MR for glucocorticoids and aldosterone, is that of the mechanism(s) allowing aldosterone occupancy of MR in physiologic mineralocorticoid target tissues. This matter is discussed later in this chapter. Not only do MRs appear unable to distinguish between physiologic mineralocorticoids and glucocorticoids, but evidence suggests an equivalent lack of selectivity at the level of the response elements, where both MR and GR act as transcription factors.20,44 Because no MR-selective response elements have been characterized to date, the evidence for this lack of specificity is indirect but clearly established for epithelial tissues.45 In vitro studies on cultured cortical collecting tubule cells have shown aldosterone, dexamethasone, and the highly specific GR agonist RU28362 to have indistinguishable effects on unidirectional Na+ and K+ fluxes and on the transepithelial potential difference as measured by short-circuit current.46 In vivo studies on adrenalectomized rats given the highly specific GR agonist RU28362 similarly have shown that GR, appropriately activated by ligand, can activate the same genes as the MR2 and can produce a classic mineralocorticoid effect on urinary electrolytes. On the other hand, differences in the action of MR and GR in the same cells can be demonstrated,44,45,47,48 suggesting the possibility of greater complexity in certain circumstances (e.g., when GR but not MR can be shown to interact with other transcription factors such as adaptor protein-1 [AP-1]).47
In addition to its effects on ion flux in classic mineralocorticoid target tissues, aldosterone has been shown to have effects via MR occupancy in a variety of other tissues. Aldosterone elevates blood pressure in the rat when infused into the cerebral ventricles49; this effect clearly results from unprotected MR, because it is blocked by simultaneous infusion of low doses of corticosterone. Thus, corticosterone in the AV3V region acts as an aldosterone antagonist on MR, in contrast with the kidney and other epithelia, where its action is to mimic aldosterone.46,50 An additional difference between epithelial and nonepithelial tissues is that in the former, activation of GR has been shown to mimic that of MR,46,50 whereas in nonepithelial tissues, this clearly is not the case. In the heart, a nonepithelial tissue that expresses MR, studies conducted in vivo show that levels of aldosterone inappropriately high for the Na+ status of the rat produce diffuse perivascular and interstitial fibrosis, an effect that can be antagonized by corticosterone or spironolactone.51 The clinical correlation of these observations is found in two recent large trials, which show a benefit of the addition of a mineralocorticoid antagonist to the conventional regimen in the treatment of individuals with severe cardiac failure, with respect to both morbidity and mortality.52,53
Mice homozygous for inactivating mutations in the MR gene54 (MR knockout, or MRKO) show classical features of aldosterone deficiency—salt-wasting, hyperkalemia, and dehydration—but have marked hyperaldosteronism; these features are also seen in the syndrome of pseudohypoaldosteronism (PHA) (see Chapter 13). MRKO mice are born at the expected frequency from heterozygote matings; untreated, they begin to deteriorate from day 5, and they die between day 8 and day 11; treatment by salt supplementation allows survival and normal growth. Mutations of the MR have been reported in the autosomal dominant form of PHA,55–57 which thus appear to be equivalent to mice heterozygous for the MR gene knockout. In the more severe autosomal recessive form and in many sporadic cases of PHA, mutations of the MR are not observed.56,57
Genomic Versus Nongenomic Aldosterone Actions
Considerable interest has been expressed with respect to steroid hormone action in terms of whether all responses are mediated through the classical nuclear receptor with direct regulation of gene expression, or whether other pathways, perhaps involving novel cell membrane receptors, exist58; the evidence for novel receptors is not compelling.59 Clear evidence has been found both in vitro and in vivo for rapid nongenomic signaling. This can involve activation by src kinase of the epidermal growth factor (EGF) receptor with consequent downstream signaling through the mitogen-activated protein (MAP)-kinase pathway; the signaling appears to require only the LBD of the MR.60 McEneaney et al.61 defined rapid effects on protein kinase signaling, Mihailidou et al.62 reported rapid effects in isolated cell patches from cardiomyocytes, and Alzamora et al.63 observed rapid effects in vascular cells. Karst et al.64 found rapid nongenomic effects of corticosterone on glutamate release from the CA1 pyramidal neurons of the hippocampus. This response also involves mitogen-activated protein kinase/extracellular signal–related kinase (MAPK/ERK) signaling.65 In each case, the receptor involved is the classical MR. The relative contribution to the mineralocorticoid response by this signaling has not yet been evaluated, although it is speculated that this rapid response may prime the transcriptional response65 or may alter the dynamic range of the response.65
Specificity-Conferring Enzymes
11β-Hydroxysteroid Dehydrogenase Type 2
Although the affinity of MR in vitro20,37 is equal for aldosterone, corticosterone, and cortisol, in vivo cortisol is excluded from such receptors in the kidney, parotid, and colon (but not in the hippocampus).66 This reflects the activity of the enzyme 11β-hydroxysteroid dehydrogenase (11β-HSD), which is responsible for the interconversion of cortisol and cortisone (in the rat, corticosterone and 11-dehydrocorticosterone). In the kidney, the predominant direction of conversion is cortisol to cortisone, as is shown by the reduced cortisol-to-cortisone ratio in human renal venous blood.67 Initially, rat liver 11β-HSD was purified, cloned, and sequenced,68 as was its human homologue69; it is expressed at high levels in the liver, testis, lung, and renal proximal tubule,70 none of which are physiologic mineralocorticoid target tissues. Subsequently, a second isoform (11β-HSD2, with the “liver” isoform termed 11β-HSD1) was isolated by expression cloning71 and was shown to be the enzyme responsible for the aldosterone selectivity of MR in epithelial aldosterone target tissues.72,73 Unlike 11β-HSD1, it co-localizes with MR32 in renal distal tubular elements, colon, sweat, and salivary glands74; it is also expressed at high abundance in the placenta71 and in select areas (subcommissural organ, ventromedial ventrolateral hypothalamus) of the rat brain.75 It has a low Km (i.e., high affinity) for both corticosterone (≈5 nM) and cortisol (≈50 nM), unlike the micromolar Km of 11β-HSD1; in vivo, it appears operationally unidirectional, acting uniquely as a dehydrogenase, whereas 11β-HSD1 appears to act predominantly as a reductase.72 Deletion of the 11β-HSD2 gene in mice yields a phenotype76 consistent with the clinical syndrome of human apparent mineralocorticoid excess (AME).72,73,77
In AME, 11β-HSD2 activity is congenitally low, indicated by a markedly increased ratio of urinary cortisol to cortisone metabolites; abnormally high intrarenal cortisol levels occupy receptors normally protected by the enzyme, resulting in increased Na+ retention and elevation of blood pressure.72,73,78 Kindred with AME have been examined for the presence of mutations in the gene coding for 11β-HSD2.77,79,80 The mutations are autosomal recessive and thus are commonly seen in the context of consanguinuity, although compound heterozygosity has also been reported.77 Although heterozygotes may show subtle departures from normal in terms of their ratio of urinary cortisol to cortisone metabolites, little evidence indicates a substantial deficit in such individuals (see also Chapter 13).
Licorice ingestion has long been known to cause Na+ retention, hypokalemia, and hypertension; the mechanism of its action was elucidated by elegant studies78 on human volunteers, in whom ingestion of 250 g of licorice a day for 10 days produced a clinical picture equivalent to a mild form of apparent mineralocorticoid excess. When rats were given glycyrrhetinic acid (the active principal of licorice)81 or carbenoxolone (glycyrrhetinic acid hemisuccinate)82 to block 11β-HSD, the normal aldosterone selectivity of epithelial MR was abolished.
Cellular Actions of Aldosterone
As the term mineralocorticoid implies, the classical effects of aldosterone reflect its role in the regulation of electrolyte flux across transporting epithelia. The molecular basis of this regulation, particularly the steps downstream of receptor binding, has yet to be fully established.2,83,84 The more recently defined roles of aldosterone in the central regulation of blood pressure,47 in salt appetite,85 and in the pathogenesis of cardiac fibrosis51,86 are even less well understood in terms of their cellular basis.
Sodium Transport
Aldosterone increases transepithelial sodium flux in a number of target tissues, of which the amphibian bladder, the mammalian distal nephron, and the mammalian distal colon are the best characterized.2,83 The temporal pattern of response comprises a lag period of 30 to 60 minutes, followed by an early phase in which preexisting pumps and channels are activated; this is followed by a so-called late phase.83 The late phase starts 3 to 6 hours after steroid exposure and is characterized by an increase in the number of pumps and channels; with longer exposure, morphologic and functional changes are observed.87 The latent phase is consistent with the concept that the effects of aldosterone are primarily genomic, with genes primarily regulated by the MR as it mediates sodium transport per se, or by modulating components of the transport pathway. In the classic model of aldosterone-induced transport, sodium entry at the apical membrane occurs through an amiloride-sensitive electrogenic sodium channel, and efflux via the sodium pump at the basolateral membrane is energy dependent, with ATP generation needed to drive the efflux process (Fig. 4-3).88
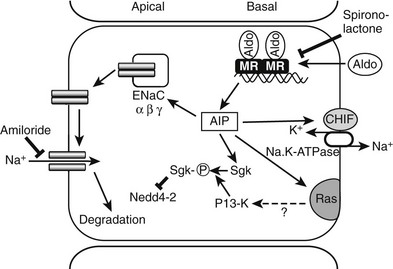
FIGURE 4-3 Aldosterone (Aldo) acts on epithelial cells of the distal nephron and the distal colon via the mineralocorticoid receptor (MR) to induce aldosterone-induced proteins (AIP). ENaC, Epithelial sodium channels; sgk, serum and glucocorticoid-induced kinase; CHIF, channel-inducing factor; PI 3-K, phosphoinositide 3-kinase.
Epithelial Sodium Channels
Although a variety of amiloride-sensitive sodium channels have been identified in various epithelia,89 only one has been shown clearly to be directly involved in aldosterone-dependent sodium transport. The epithelial sodium channel (ENaC) genes cloned from the rat distal colon encode three homologous subunits (α, β, and γ).90 Each subunit consists of two transmembrane domains with intracellular N and C termini. When the α subunit is expressed in Xenopus oocytes, a weak, amiloride-sensitive sodium flux can be demonstrated; expression of the other subunits alone occurs without activity, and co-expression of all three subunits is required for maximal amiloride-sensitive sodium transport.90 The subunits exhibit a heterotrimeric complex, as is demonstrated by the recent characterization of the tertiary structure of a closely related acid-sensing ion channel.91
The genes for the ENaC subunits are members of the DEG/ENaC superfamily of sodium channels; the degenerins (DEGs) mediate mechanosensory transduction in Caenorhabditis elegans.92 Insights into the structural determinants of function of the ENaC subunits have been derived from a combination of both naturally occurring and engineered mutations.92 Deletion of the αENaC gene in transgenic mice results in early neonatal death caused by defective clearance of lung liquid93; partial restoration of αENaC expression in these mice by transgenesis results in a phenotype very similar to that seen in PHA.94 Mice with the βENaC or the γENaC gene deleted show a less severe pulmonary phenotype but die between 24 and 36 hours with salt wasting and profound hyperkalemia, again reminiscent of PHA95,96; low levels of expression of the βENaC subunit gene in mice in which the βENaC gene has been disrupted also result in a milder phenotype analogous to PHA.97 Autosomal recessive PHA indeed is associated with inactivating mutations of all three of the ENaC subunits98,99 (see also Chapter 13).
The molecular characterization of Liddle’s syndrome100 has also provided important insights into the function of the C-terminal intracellular domain and emphasizes the central role of the channel in amiloride-sensitive sodium transport2,94 (see also Chapter 13). Liddle’s syndrome, or pseudoaldosteronism, has the reciprocal pathophysiology to PHA,100 with apparent aldosterone excess (i.e., hypertension, hypokalemia, and suppressed plasma renin activity) but low aldosterone levels, and no response to spironolactone but a response to amiloride. Several studies have identified mutations in the β– or γENaC gene in various kindred.100,101 The mutations are either nonsense or missense mutations localized to the C terminus; the latter are particularly informative because they identify a key motif, proline-proline-proline-X-tyrosine (PY), which is conserved across the subunits and is disrupted in all cases of Liddle’s syndrome.92,101 ENaC is a relatively short-lived protein that is ubiquinated on residues in the N terminus of the α and γ but not β subunits; the PY motif interacts with Nedd4-2, a ubiquitin protein-ligase, whose role is to target the channels for proteosomal degradation.102 Nedd4-2 is not itself regulated by aldosterone.103
These studies demonstrated the central role of the ENaC in mediating aldosterone-induced epithelial sodium transport. ENaC subunit gene expression is regulated by aldosterone. Both β and γENaC subunit mRNA levels were increased in the colon by sodium depletion104 and by dexamethasone or aldosterone treatment for 6105 or 24 hours106,107; in the kidney, however, levels remained unaltered, although when separate regions of the kidney were distinguished, an increase in αENaC mRNA levels was seen in the inner medulla.108 Cultured medullary collecting duct cells show increased α- but not β- or γENaC mRNA levels after 3 hours of aldosterone exposure; in cultured cortical collecting duct cells, a γENaC subunit response to aldosterone required 24 hours of treatment.109 Although it is clear from the above studies that aldosterone can increase ENaC synthesis (at least in the late phase), an effect in the early phase (i.e., a primary effect) has not been demonstrated. This is in agreement with a number of electrophysiologic88 and biosynthetic studies.110
Studies in amphibian A6 cells111 and rabbit cortical collecting duct cells112 provided the first conclusive evidence for an early phase, direct mediator of aldosterone action in the kidney. Aldosterone treatment in vivo rapidly increased levels of the serine, threonine kinase, serum, and glucocorticoid-regulated kinase (sgk111), with a time course clearly consistent with an effect of aldosterone on transcription.113,114 Sgk directly interacts with Nedd4-2 to block its binding of the ENaC and, as a consequence, slows ENaC degradation.115,116 Regulation of ENaCα subunit gene expression involves sgk-1 through relief of Dotla-Af9–mediated transcriptional repression.117 Nedd4-2 is also regulated by Usp2-45, a deubiquitinylation enzyme that itself is regulated by aldosterone.118 Sgk requires phosphorylation by the phosphatidylinosital 3-kinase (PI 3-kinase) pathway for full activity.119 This may be a point at which the signaling of nuclear receptors and that of membrane-associated receptors such as the insulin receptor are integrated.119 PI 3-kinase may be activated by small monomeric G proteins, including ras. K-ras 2A has been identified as an aldosterone-induced gene in an amphibian-derived renal cell line84; this has been confirmed in the rodent distal colon.120 The glucocorticoid-induced leucine zipper protein (GIL2) is also aldosterone-induced; it acts to repress ERK signaling.121
Na+/K+-ATPase
Active extrusion of Na+ from the cell reflects sodium pump activity in the basolateral cell membrane. Na+/K+-ATPase activity is increased in a number of epithelia by aldosterone. Toad bladder and A6 cells provide evidence that Na+/K+-ATPase α and β subunit gene expression is significantly increased in the early phase of the response to aldosterone,83 although this does not appear to be the case in mammalian systems.122 Na+/K+-ATPase activity is very sensitive to intracellular sodium concentrations, and in isolated cortical tubules the early Na+/K+-ATPase response to aldosterone is blocked by amiloride, suggesting that the increased activity is secondary to sodium influx at the apical membrane.123 In the late phase of the aldosterone response, levels of Na+/K+-ATPase mRNA, protein, and activity are all increased.124 Channel-inducing factor (CHIF) was first identified as a novel corticosteroid-induced gene in the rat distal colon.125 CHIF is a member of the FXYD family of small transmembrane proteins that includes the γ subunit of Na+/K+-ATPase.126 Under experimental conditions, CHIF is upregulated in the distal colon in response to aldosterone127,128; this appears to be a primary transcriptional response.129 It has been shown recently126 that CHIF increases the affinity of Na+/K+-ATPase for sodium. It would appear that the early aldosterone-induced increase in Na+/K+-ATPase activity is mediated by CHIF.
Potassium Transport
Potassium flux occurs in transporting epithelia in response to aldosterone as a result of Na+/K+-ATPase–mediated exchange at the basolateral membrane, with the resulting electrochemical gradient favoring potassium excretion.130 Regulation of potassium homeostasis by aldosterone is independent of its effects on sodium transport, and Na+/K+-ATPase–mediated basolateral membrane exchange of sodium and potassium130 has been described; recent evidence131 suggests that a mediator of potassium transport, the α subunit of a putative K+-ATPase, is regulated by dietary potassium and corticosteroids. Aldosterone and dexamethasone treatment for 2 days upregulates levels of K+-ATPase mRNA in the epithelial cells of both the outer medullary collecting duct and the distal colon.131 Sgk-1 has been reported to increase the channel density of ROMK, a potassium channel, through direct phosphorylation or perhaps through Nedd4-2.132
Hydrogen Ion Transport
As for potassium, aldosterone also has effects on proton excretion over and above a simple cation exchange for sodium across the epithelium to maintain electroneutrality.133 The targets for this effect are carbonic anhydrase–rich cells, particularly the intercalated cells within the outer medullary collecting ducts. Transport across the apical membrane occurs through an H+-ATPase activity134 (which is upregulated by aldosterone), coupled with increased activity of the basolateral Cl−/HCO3− exchanger.135 These effects are largely sodium independent, as demonstrated by the lack of an effect of amiloride. Aldosterone also has an effect on the Na+-H+ antiporter in a range of tissues; in some tissues, the response is very rapid and represents a nongenomic action.
Other Tissues
Although the last decade has seen significant advances in our understanding of the molecular responses to aldosterone in epithelial cells, the nature of the response in other tissues is much less well characterized. In the heart, for instance, the acute inflammatory response to mineralocorticoid treatment in the presence of salt loading is likely to involve a distinctively different set of genes.51,86 Responses in the central nervous system also appear to differ.136 The role of aldosterone in other tissues, particularly those of the cerebrovascular and cardiovascular systems, has been a major focus of recent research. In the heart, mineralocorticoid treatment in the presence of salt loading results in an inflammatory response that leads to cardiac fibrosis.34 Several studies have identified aldosterone-induced genes in the heart,137,138 although their full physiologic significance remains to be determined. The same is true in the central nervous system.139,65 It is important to note that in several, but not all of these tissues, the biology may be that of cortisol activation of the MR rather than of aldosterone.
References
1. Vinson, GP, Whitehouse, BJ, Goddard, C, et al. Comparative and evolutionary aspects of aldosterone secretion and zona glomerulosa function. J Endocrinol. 1979;81:5P–24P.
2. Rogerson, FM, Fuller, PJ. Mineralocorticoid action. Steroids. 2000;65:61–73.
3. Simpson, SA, Tait, JF, Wettstein, A, et al. Isolierung eines neuen kristallisierten Hormons aus Nebennieren mit besonders hoher Wirksamkeit auf den Mineral-stoffwechsel. Experientia. 1953;9:333–335.
4. Crabbè, J. Stimulation of active sodium transport across toad bladder with aldosterone in vitro. J Clin Invest. 1961;76:2103–2110.
5. Feldman, D, Funder, JW, Edelman, IS. Subcellular mechanisms in the action of adrenal steroids. Am J Med. 1972;53:545–560.
6. Weir, MR, Dzau, VJ. The renin-angiotensin-aldosterone system: a specific target: hypertension management. Am J Hypertens. 1999;12:205S–213S.
7. Nogueira, EF, Vargas, CA, Otis, M, et al. Angiotensin-II acute regulation of rapid response genes in human, bovine, and rat adrenocortical cells. J Mol Endocrinol. 2007;39:365–374.
8. Okubo, S, Niimura, F, Nishimura, H, et al. Angiotensin-independent mechanism for aldosterone synthesis during chronic extracellular fluid volume depletion. J Clin Invest. 1997;99:855–860.
9. Carey, RM. Physiologic and possible pathophysiologic relevance of dopaminergic mechanisms in the control of aldosterone secretion. In: Mantero F, Biglieri EG, Funder JW, Scoggins BA, eds. The Adrenal and Hypertension. New York: Raven; 1995:55–69.
10. Hollenberg, NK, Chenitz, WR, Adams, DF, et al. Reciprocal influence of salt intake on adrenal glomerulosa and renal vascular responses to angiotensin II in normal man. J Clin Invest. 1974;54:34–42.
11. Knox, F, Burnett, JJ, Kohan, D, et al. Escape from the sodium-retaining effects of mineralocorticoids. Kidney Int. 1980;17:263–276.
12. Zimmerman, RS, Edwards, BS, Schwab, TR, et al. Atrial natriuretic peptide during mineralocorticoid escape in the human. J Clin Endocrinol Metab. 1987;64:624–627.
13. Gaillard, CA, Koomans, HA, Rabelink, TJ, et al. Enhanced natriuretic effect of atrial natriuretic factor during mineralocorticoid escape in humans. Hypertension. 1988;12:450–456.
14. Ye, P, Kenyon, CJ, MacKenzie, SM, et al. The aldosterone synthase (CYP11B2) and 11β-hydroxylase (CYP11B1) genes are not expressed in the rat heart. Endocrinology. 2005;146:5287–5293.
15. Young, DB. Quantitative analysis of aldosterone’s role in potassium regulation. Am J Physiol. 1988;55:F811–F822.
16. Williams, GH, Rose, LI, Dluhy, RG, et al. Aldosterone response to sodium restriction and ACTH stimulation in panhypopituitarism. J Clin Endocrinol Metab. 1971;32:27–35.
17. Rousseau, G, Baxter, JD, Funder, JW, et al. Glucocorticoid and mineralocorticoid receptors for aldosterone. J Steroid Biochem. 1972;3:219–227.
18. Funder, JW, Feldman, D, Edelman, IS. Specific aldosterone binding in rat kidney and parotid. J Steroid Biochem. 1972;3:209–218.
19. Marver, D, Stewart, J, Funder, JW, et al. Renal aldosterone receptors: studies with (3H)aldosterone and the anti-mineralocorticoid (3H)spirolactone (SC-26304). Proc Natl Acad Sci U S A. 1974;71:1431–1435.
20. Arriza, JL, Weinberger, C, Cerelli, G, et al. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science. 1987;237:268–275.
21. Patel, PD, Sherman, TG, Goldman, DJ. Molecular cloning of a mineralocorticoid (type I) receptor complementary DNA for rat hippocampus. Mol Endocrinol. 1989;3:1877–1885.
22. Todd-Turla, MD, Schnermann, J, Fejes-Toth, G, et al. Distribution of mineralocorticoid glucocorticoid receptor mRNA along the nephron. Am J Physiol. 1993;264:F781–F791.
23. Fuller, PJ, Verity, K. Mineralocorticoid receptor gene expression in the gastrointestinal tract: distribution and ontogeny. J Steroid Biochem. 1990;36:263–267.
24. Mangelsdorf, DJ, Thummel, C, Beato, M, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839.
25. Fan, Y-S, Eddy, RL, Byers, LL, et al. The human mineralocorticoid receptor gene (MLR) is located on chromosome 4 at q31.2. Cytogenet Cell Genet. 1989;52:83–84.
26. Theriault, A, Boyd, E, Harrap, SB, et al. Regional chromosomal assignment of the human glucocorticoid receptor gene to 5q31. Hum Genet. 1989;83:289–291.
27. Luisi, BF, Xu, WX, Itwubiwsju, Z, et al. Crystallographic analysis of the interaction of the glucocorticoid receptor with DNA. Nature. 1991;352:497–505.
28. Fagart, J, Huyet, J, Pinon, GM, et al. Crystal structure of a mutant mineralocorticoid receptor responsible for hypertension. Nature Struct Mol Biol. 2005;12:554–555.
29. Bledsoe, R.K., Madauss, KP, Holt, JA, et al. A ligand-mediated hydrogen bond network required for the activation of the mineralocorticoid receptor. J Biol Chem. 2005;280:31283–31293.
30. Li, Y, Suino, K, Daugherty, J, et al. Structural and biochemical mechanisms for the specificity of hormone binding and coactivator assembly by mineralocorticoid receptor. Mol Cell. 2005;19:367–380.
31. Rogerson, FM, Fuller, PJ. Interdomain interactions in the mineralocorticoid receptor. Mol Cell Endocrinol. 2003;200:45–55.
32. Odermatt, A, Arnold, P, Frey, FJ. The intracellular localization of the mineralocorticoid receptor is regulated by 11beta-hydroxysteroid dehydrogenase type 2. J Biol Chem. 2001;276:28484–28492.
33. Bruner, KL, Derfoul, A, Robertson, NM, et al. The unliganded mineralocorticoid receptor is associated with heat shock proteins 70 and 90 and the immunophilin FKBP-52. Recept Signal Transduct. 1997;7:85–98.
34. Fuller, PJ, Young, MJ. Mechanisms of mineralocorticoid action. Hypertension. 2005;46:1227–1235.
35. Rogerson, FM, Yao, YZ, Smith, BJ, et al. Determinants of spironolactone binding specificity in the mineralocorticoid receptor. J Mol Endocrinol. 2003;31:573–582.
36. Viengchareun, S, Le Menuet, D, Martinerie, L, et al. The mineralocorticoid receptor: insights into its molecular and (patho)physiological biology. Nucl Recept Signal. 2007;5:1–16.
37. Krozowski, ZS, Funder, JW. Renal mineralocorticoid receptors and hippocampal corticosterone binding species have identical intrinsic steroid specificity. Proc Natl Acad Sci U S A. 1983;80:6056–6060.
38. de Kloet, ER, Vreugdenhil, E, Oitzl, MS, et al. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19:269–301.
39. Kwak, SP, Patel, PD, Thompson, RC, et al. 5′-heterogeneity of the mineralocorticoid receptors messenger ribonucleic acid: differential expression and regulation of splice variants within the rat hippocampus. Endocrinology. 1993;133:2344–2350.
40. Zennaro, M-C, Keightley, MC, Kotelevtsev, Y, et al. Human mineralocorticoid receptor genomic structure and identification of expressed isoforms. J Biol Chem. 1995;270:21016–21020.
41. Zennaro, M-C, Le Menuet, D, Lombes, M. Characterization of the human mineralocorticoid receptor gene 5′-regulatory region: evidence for differential hormonal regulation of two alternative promoters via non-classical mechanisms. Mol Endocrinol. 1996;10:1549–1560.
42. Zennaro, MC, Souque, A, Viengchareun, S, et al. A new human MR splice variant is a ligand-independent transactivator modulating corticosteroid action. Mol Endocrinol. 2001;15:1586–1598.
43. Couette, B, Fagart, J, Jalaguier, S, et al. Ligand-induced conformational change in the human mineralocorticoid receptor occurs within its hetero-oligomeric structure. Biochem J. 1996;315:421–427.
44. Farman, N, Rafestin-Oblin, ME. Multiple aspects of mineralocorticoid selectivity. Am J Physiol Renal Physiol. 2001;280:F181–F192.
45. Lim-Tio, SS, Keightley, M-C, Fuller, PJ. Determination of specificity of transactivation by the mineralocorticoid or glucocorticoid receptors. Endocrinology. 1997;138:2537–3543.
46. Naray-Fejes-Toth, A, Fejes-Toth, G. Glucocorticoid receptors mediate mineralocorticoid-like effects in cultured collecting duct cells. Am J Physiol. 1990;259:F672–F678.
47. Pearce, D, Yamamoto, KR. Mineralocorticoid and glucocorticoid receptor activities are distinguished by nonreceptor factors at a composite response element. Science. 1993;259:1661–1665.
48. Lim-Tio, SS, Fuller, PJ. Intracellular signaling pathways confer specificity of transactivation by mineralocorticoid and glucocorticoid receptors. Endocrinology. 1998;139:1653–1661.
49. Gomez-Sanchez, EP, Venkataraman, MT, Thwaites, D, et al. Intracerebroventricular infusion of corticosterone antagonizes ICV-aldosterone hypertension. Am J Physiol. 1990;258:E649–E653.
50. Funder, JW, Pearce, P, Myles, K, et al. Apparent mineralocorticoid excess, pseudohypoaldosteronism and urinary electrolyte excretion: towards a redefinition of “mineralocorticoid” action. FASEB J. 1990;4:3234–3238.
51. Young, MJ, Funder, JW. Mineralocorticoid receptors and pathophysiological roles for aldosterone in the cardiovascular system. J Hypertens. 2002;20:1465–1468.
52. Pitt, B, Zannad, F, Remme, WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341:709–717.
53. Pitt, B, Remme, W, Zannad, F, et al. Eplerenone post-acute myocardial infarction heart failure efficacy and survival study investigators. N Engl J Med. 2003;348:1309–1321.
54. Berger, S, Bleich, M, Schmid, W, et al. Mineralocorticoid receptor knockout mice: pathophysiology of Na+ metabolism. Proc Natl Acad Sci U S A. 1998;95:9424–9429.
55. Geller, DS, Rodriguez-Soriano, J, Boado, AV, et al. Mutations in the mineralocorticoid receptor gene cause autosomal dominant pseudohypoaldosteronism type 1. Nat Genet. 1998;19:279–281.
56. Pujo, L, Fagart, J, Gary, F, et al. Mineralocorticoid receptor mutations are the principal cause of renal type 1 pseudohypoaldosteronism. Hum Mutat. 2007;28:33–40.
57. Geller, DS, Zhang, J, Zennaro, MC, et al. Autosomal dominant pseudohypoaldosteronism type 1: mechanisms, evidence for neonatal lethality, and phenotypic expression in adults. J Am Soc Nephrol. 2006;16:1429–1436.
58. Losel, RM, Feuring, M, Falkenstein, E, et al. Nongenomic effects of aldosterone: cellular aspects and clinical implications. Steroids. 2002;67:493–498.
59. Funder, JW. The nongenomic actions of aldosterone. Endocr Rev. 2005;26:313–321.
60. Grossmann, C, Freudinger, R, Mildenbergr, S, et al. EF-domains are sufficient for nongenomic mineralocorticoid receptor actions. J Biol Chem. 2008;283:7109–7116.
61. McEneaney, V, Harvey, BJ, Thomas, W. Aldosterone regulates rapid trafficking of ENaC subunits in renal cortical collecting duct cells via protein kinase D activation. Mol Endocrinol. 2008;22:881–882.
62. Mihailidou, AS, Mardini, M, Funder, JW. Rapid, nongenomic effects of aldosterone in the heart mediated by epsilon protein kinase C. Endocrinology. 2003;145:773–780.
63. Alzamora, R, Marusic, ET, Gonzalez, M, et al. Nongenomic effect of aldosterone on Na+,K+-adenosine triphosphatase in arterial vessels. Endocrinology. 2003;144:1266–1272.
64. Karst, H, Berger, S, Turiault, M, et al. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glucamate transmission by corticosterone. Proc Natl Acad Sci U S A. 2005;102:19204–19207.
65. Joels, M, Karst, H, DeRijk, R, et al. The coming out of the brain mineralocorticoid receptor. Trends Neurosci. 2008;31:1–7.
66. Sheppard, K, Funder, JW. Type I receptors in parotid, colon and pituitary are aldosterone-selective in vivo. Am J Physiol. 1987;253:E467–E471.
67. Walker, BR, Campbell, JC, Fraser, R, et al. Mineralocorticoid excess and inhibition of 11β-hydroxysteroid dehydrogenase in patients with ectopic ACTH syndrome. Clin Endocrinol. 1992;37:483–489.
68. Agarwal, AK, Monder, C, Eckstein, B, et al. Cloning and expression of rat cDNA encoding corticosteroid 11β-dehydrogenase. J Biol Chem. 1989;264:18939–18943.
69. Tannin, GM, Agarwal, AK, Monder, C, et al. The human gene for 11β-hydroxysteroid dehydrogenase: Structure, tissue distribution, and chromosomal localization. J Biol Chem. 1991;266:16653–16658.
70. Monder, C, Lakshmi, V. Corticosteroid 11β-dehydrogenase of rat tissues: immunological studies. Endocrinology. 1990;126:2435–2443.
71. Albiston, AL, Obeyesekere, VR, Smith, RE, et al. Cloning and tissue distribution of the human 11 beta-hydroxysteroid dehydrogenase type 2 enzyme. Mol Cell Endocrinol. 1994;105:R11–R17.
72. Quinkler, M, Stewart, PM. Hypertension and the cortisol-cortisone shuttle. J Clin Endocrinol Metab. 2003;88:2384–2392.
73. White, PC, Mune, T, Agarwal, AK. 11β-Hydroxysteroid dehydrogenase and the syndrome of apparent mineralocorticoid excess. Endocr Rev. 1997;18:135–156.
74. Smith, RE, Li, KXZ, Andrews, RK, et al. Immunohistochemical and molecular characterization of the rat 11 beta-hydroxysteroid dehydrogenase type II enzyme. Endocrinology. 1997;138:540–547.
75. Roland, BL, Li, KX, Funder, JW. Hybridization histochemical localization of 11 beta-hydroxysteroid dehydrogenase type 2 in rat brain. Endocrinology. 1995;136:4697–4700.
76. Kotelevtsev, Y, Brown, RW, Fleming, S, et al. Hypertension in mice lacking 11β-hydroxysteroid dehydrogenase type 2. J Clin Invest. 1999;103:683–689.
77. Wilson, RC, Nimkarn, S, New, MI. Apparent mineralocorticoid excess. Trends Endocrinol Metab. 2001;12:104–111.
78. Stewart, PM, Valentino, R, Wallace, AM, et al. Mineralocorticoid activity of licorice: 11β-hydroxysteroid dehydrogenase activity comes of age. Lancet. 1987;2:821–824.
79. Wilson, RC, Krozowski, ZS, Obeyesekere, VR, et al. A mutation in the HSD11B2 gene in a family with apparent mineralocorticoid excess. J Clin Endocrinol Metab. 1995;80:2263–2266.
80. Mune, T, Rogerson, FM, Nikkila, H, et al. Human hypertension caused by mutations in the kidney isozyme of 11 beta-hydroxysteroid dehydrogenase. Nat Genet. 1995;10:394–399.
81. Edwards, CRW, Stewart, PM, Burt, D, et al. Localization of 11β-hydroxysteroid dehydrogenase: tissue-specific protector of the mineralocorticoid receptor. Lancet. 1988;2:986–989.
82. Funder, JW, Pearce, PT, Smith, R, et al. Mineralocorticoid action: target-tissue specificity is enzyme, not receptor-mediated. Science. 1988;242:583–585.
83. Verrey, F. Early aldosterone action: toward filling the gap between transcription and transport. Am J Physiol. 1999;277:F319–F327.
84. Stockand, JD. New ideas about aldosterone signalling in epithelia. Am J Physiol. 2002;282:F559–F576.
85. McEwen, BS, Lambdin, LT, Rainbow, TC, et al. Aldosterone effects on salt appetite in adrenalectomized rats. Neuroendocrinology. 1986;43:38–43.
86. Rocha, R, Funder, JW. The pathophysiology of aldosterone in the cardiovascular system. Ann N Y Acad Sci. 2002;970:89–100.
87. Wade, JB, Stanton, BA, Field, MJ, et al. Morphological and physiological responses to aldosterone: time course and sodium dependence. Am J Physiol. 1990;259:F88–F94.
88. Garty, H, Palmer, LG. Epithelial sodium channels: function, structure and regulation. Physiol Rev. 1997;77:359–396.
89. Benos, DJ, Awayda, MS, Ismailov, II, et al. Structure and function of amiloride-sensitive Na+ channels. J Membr Biol. 1995;143:1–18.
90. Canessa, CM, Schild, L, Buell, G, et al. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature. 1994;367:463–467.
91. Jasti, J, Furukawa, H, Gonzales, EB, et al. Structure of acid-sensing ion channel 1 at 1.9A resolution and low pH. Nature. 2007;449:316–323.
92. Barbry, P, Hofman, P. Molecular biology of Na+ absorption. Am J Physiol. 1997;273:G571–G585.
93. Hummler, E, Barker, P, Gatzy, J, et al. Early death due to defective neonatal lung liquid clearance in αENaC-deficient mice. Nat Genet. 1996;12:325–328.
94. Gründer, S, Rossier, BC. A reappraisal of aldosterone effects on the kidney: new insights provided by epithelial sodium channel cloning. Curr Opin Nephrol Hypertens. 1997;6:35–39.
95. Barker, PM, Nguyen, MS, Gatzy, JT, et al. Role of gamma ENaC subunit in lung liquid clearance and electrolyte balance in newborn mice. Insights into perinatal adaptation and pseudohypoaldosteronism. J Clin Invest. 1998;102:1634–1640.
96. McDonald, FJ, Yang, B, Hrstka, RF, et al. Disruption of the beta subunit of the epithelial Na+ channel in mice: hyperkalemia and neonatal death associated with a pseudohypoaldosteronism phenotype. Proc Natl Acad Sci U S A. 1999;96:1727–1731.
97. Pradervand, S, Barker, PM, Wang, Q, et al. Salt restriction induces pseudohypoaldosteronism type 1 in mice expressing low levels of the beta-subunit of the amiloride-sensitive epithelial sodium channel. Proc Natl Acad Sci U S A. 1999;96:1732–1737.
98. Chang, SS, Grunder, S, Hanukoglu, A, et al. Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nat Genet. 1996;12:248–253.
99. Strautnieks, SS, Thompson, RJ, Gardiner, RM, et al. A novel splice-site mutation in the γ-subunit of the epithelial sodium channel gene in three pseudohypoaldosteronism type 1 families. Nat Genet. 1996;13:248–250.
100. Shimkets, RA, Warnock, DG, Bositis, CM, et al. Liddle’s syndrome: Heritable human hypertension caused by mutations in the β subunit of the epithelial sodium channel. Cell. 1994;79:407–414.
101. Lifton, RP, Gharavi, AG, Geller, DS. Molecular mechanisms of human hypertension. Cell. 2001;104:545–556.
102. Staub, O, Gautschi, I, Ishikawa, T, et al. Regulation of stability and function of the epithelial Na+ channel ENaC by ubiquitination. EMBO J. 1997;16:6325–6336.
103. Rogerson, FM, Brennan, FE, Fuller, PJ. Dissecting mineralocorticoid receptor structure and function. J Steroid Biochem Mol Biol. 2003;85:389–396.
104. Lingueglia, E, Renard, S, Waldmann, R, et al. Different homologous subunits of the amiloride-sensitive Na+ channel are differently regulated by aldosterone. J Biol Chem. 1994;269:13736–13739.
105. Stokes, JB, Sigmund, RD. Regulation of rENaC mRNA by dietary NaC1 and steroids: organ, tissue, and steroid heterogeneity. Am J Physiol. 1998;274:C1699–C1707.
106. Renard, S, Voilley, N, Bassilana, F, et al. Localization and regulation by steroids of the α, β and γ subunits of the amiloride-sensitive Na+ channel in colon, lung and kidney. Pflugers Arch. 1995;430:299–307.
107. Asher, C, Wald, H, Rossier, BC, et al. Aldosterone-induced increase in the abundance of Na+ channel subunits. Am J Physiol. 1996;271:C605–C611.
108. Volk, KA, Sigmund, RD, Snyder, PM, et al. rENaC is the predominant Na+ channel in the apical membrane of the rat renal inner medullary collecting duct. J Clin Invest. 1995;96:2748–2757.
109. Denault, DL, Fejes-Tòth, G, Naray-Fejes-Tòth, A. Aldosterone regulation of sodium channel γ-subunit mRNA in cortical collecting duct cells. Am J Physiol. 1996;271:C423–C428.
110. May, A, Puoti, A, Gaeggeler, H-P, et al. Early effect of aldosterone on the rate of synthesis of the epithelial sodium channel α subunit in A6 renal cells. J Am Soc Nephrol. 1997;8:1813–1822.
111. Chen, SY, Bhargava, A, Mastroberadino, L, et al. Epithelial sodium channel regulated by aldosterone-induced protein sgk. Proc Natl Acad Sci U S A. 1999;96:2514–2519.
112. Naray-Fejes-Toth, A, Canessa, C, Cleaveland, ES, et al. sgk is an aldosterone-induced kinase in the renal collecting duct. J Biol Chem. 1999;274:16973–16978.
113. Brennan, FE, Fuller, PJ. Rapid up-regulation of serum and glucocorticoid-regulated kinase (sgk) gene expression by corticosteroids in vivo. Mol Cell Endocrinol. 2000;30:129–136.
114. Bhargava, A, Fullerton, MJ, Myles, K, et al. The serum- and glucocorticoid-induced kinase is a physiological mediator of aldosterone action. Endocrinology. 2001;142:1587–1594.
115. Debonneville, C, Flores, SY, Kamynina, E, et al. Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na+ channel cell surface expression. EMBO J. 2001;20:7052–7059.
116. Snyder, PM, Olson, DR, Thomas, BC. Serum and glucocorticoid-regulated kinase modulates Nedd4-2-mediated inhibition of the epithelial Na+ channel. J Biol Chem. 2002;277:5–8.
117. Zhang, W, Xia, X, Reisenauer, MR, et al. Aldosterone-induced Sgk1 relieves Dot1a-Af9-mediated transcriptional repression of epithelial Na+ channel α. J Clin Invest. 2007;117:773–783.
118. Fakitsas, P, Adam, G, Daidié, D, et al. Early aldosterone-induced gene product regulates the epithelial sodium channel by deubiquitylation. J Am Soc Nephrol. 2007;18:1084–1092.
119. Wang, J, Barbry, P, Maiyar, AC, et al. SGK integrates insulin and mineralocorticoid regulation of epithelial sodium transport. Am J Physiol. 2001;280:F303–F313.
120. Brennan, FE, Fuller, PJ. Mammalian K-ras2 is a corticosteroid-induced gene in vivo. Endocrinology. 2006;147:2809–2816.
121. Soundarajan, R, Wang, J, Melters, D, et al. Differential activities of glucocorticoid-induced leucine zipper protein isoforms. J Biol Chem. 2007;282:36303–36313.
122. Fuller, PJ, Verity, K. Colonic sodium-potassium adenosine triphosphate subunit gene expression: ontogeny and regulation by adrenocortical steroids. Endocrinology. 1990;127:32–38.
123. Petty, KJ, Kokko, JP, Marver, D. Secondary effect of aldosterone on Na-K,ATPase activity in the rabbit cortical collecting tubule. J Clin Invest. 1981;68:1514–1521.
124. Sansom, SC, O’Neil, RG. Mineralocorticoid regulation of apical cell membrane Na+ and K+ transport of the cortical collecting duct. Am J Physiol. 1985;248:F858–F868.
125. Attali, B, Latter, H, Rachamim, N, et al. A corticosteroid-induced gene expressing an “IsK-like” K+ channel activity in Xenopus oocytes. Proc Natl Acad Sci U S A. 1995;92:6092–6096.
126. Beguin, P, Crambert, G, Guennoun, S, et al. CHIF, a member of the FXYD protein family, is a regulator of Na,K-ATPase distinct from the gamma-subunit. EMBO J. 2001;20:3993–4002.
127. Wald, H, Goldstein, O, Asher, C, et al. Aldosterone induction and epithelial distribution of CHIF. Am J Physiol. 1996;271:F322–F329.
128. Brennan, FE, Fuller, PJ. Acute regulation by corticosteroids of CHIF mRNA in the distal colon. Endocrinology. 1999;140:1213–1218.
129. Brennan, FE, Fuller, PJ. Transcriptional control by corticosteroids of CHIF gene expression in the rat distal colon. Clin Exp Physiol Pharmacol. 1999;26:489–491.
130. Young, DB. Quantitative analysis of aldosterone’s role in potassium regulation. Am J Physiol. 1988;255:F811–F822.
131. Jaisser, F, Escoubet, B, Coutry, N, et al. Differential regulation of putative K+-ATPase by low-K+ diet and corticosteroids in rat distal colon and kidney. Am J Physiol. 1996;270:C679–C687.
132. Yoo D, Kim BY, Campo C, et al: Cell surface expression of the ROMK (Kir 1.1) channel is regulated by the aldosterone-induced kinase, sgk-1, and protein kinase A, J Biol Chem 278:23066–23075.
133. Stone, DK, Seldin, DW, Kokko, JP, et al. Mineralocorticoid modulation of rabbit medullary collecting duct acidification. J Clin Invest. 1983;72:77–83.
134. Kuwahara, M, Sasaki, S, Marumo, F. Mineralocorticoids and acidosis regulate H+/HCO3 transport of intercalated cells. J Clin Invest. 1992;89:1388–1394.
135. Hays, TR. Mineralocorticoid modulation of apical and basolateralmembrane H+/OH-/HCO3 transport processes in the rabbit inner stripe of outer medullary collecting duct. J Clin Invest. 1992;90:1–8.
136. Obradovic, D, Tirard, M, Nemethy, ZS, et al. DAXX, FLASH, and FAF-1 modulate mineralocorticoid and glucocorticoid receptor-mediated transcription in hippocampal cells: toward a basis for the opposite actions elicited by two nuclear receptors? Mol Pharmacol. 2004;65:761–769.
137. Turchin, A, Guo, CZ, Adler, GK, et al. Effect of acute aldosterone administration on gene expression profile in the heart. Endocrinology. 2006;147:3183–3189.
138. Fejes Tóth, G, Náray-Fejes-Tóth, A. Early aldosterone-regulated genes in cardiomyocytes: clues to cardiac remodeling? Endocrinology. 2007;148:1502–1510.
139. Rigsby, CS, Cannady, WE, Dorrance, AM. Aldosterone: good guy or bad guy in cerebrovascular disease? Trends Endocrinol Metab. 2005;16:401–406.