Adrenocortical Carcinoma
Epidemiology
Benign adrenal tumors belong to the most common human neoplasias, with a prevalence of >4% in computed tomography (CT) studies1–3 and an even higher prevalence in autopsy series.4 In contrast, adrenocortical carcinoma (ACC) is a rare malignancy with an incidence of only 1 to 2 per million population per year,5,6 leading to 0.2 % of cancer deaths according to U.S. data. However, these data may underestimate the true incidence of ACC. An unusually high incidence of ACC has been found in children in southern Brazil (3.4 to 4.2 per 1 million children versus an estimated worldwide incidence of 0.3 per 1 million children younger than 15 years) related to a founder germline p53 tumor-suppressor gene mutation.7 ACC is more frequent in women than in men (ratio: 1.5). The incidence shows a maximum around the 4th and 5th decade, but the tumor can appear at any age.8
Molecular Pathology
The molecular pathogenesis of adrenocortical tumors is incompletely understood.9,10 Important insights come from hereditary tumor syndromes associated with the development of ACC. In the Li-Fraumeni syndrome,11 the frequency of ACC is up to 4%,12 and 70% of patients with Li-Fraumeni syndrome have germline mutations of the p53 tumor-suppressor gene located at the 7p13 locus.13 A second variant is caused by a heterozygous germline mutation in the hCHK2 gene.14 In children in southern Brazil, a specific germline mutation of p53 encoding an R337H amino acid substitution has been demonstrated. This mutation leads to a pH-sensitive and temperature-dependent alteration in the function of the p53 protein.15 Somatic mutations in the p53 gene have also been found in tumors of patients with sporadic ACC.16 Another hereditary syndrome associated with ACC is the Beckwith-Wiedemann Syndrome (BWS), a congenital overgrowth syndrome characterized by exomphalos, macroglossia, gigantism, and the development of childhood tumors.17 BWS has been mapped to the 11p15.5 region and is associated also with Wilms tumor and hepatoblastoma. Genes located at 11p15 and implicated in the pathogenesis of BWS are insulin-like growth factor 2 (IGF-2), H19, and cyclin-dependent kinase inhibitor 1C (CDKN1C, p57kip2). IGF-2 is maternally imprinted, whereas H19 and p57kip2 are both paternally imprinted. Uniparental paternal isodisomy for this locus associated with IGF-2 overexpression has been observed in BWS. In sporadic ACC, rearrangement at the 11p15 locus, with overexpression of IGF-2, is frequently observed. These are caused either by duplications of the paternal 11p15 allele or by loss of the maternal allele containing the H19 gene. In fact, increased IGF-2 expression is a hallmark of adrenocortical carcinoma and has been consistently described in a variety of studies, including microarray analyses.18–21 These observations indicate that IGF-2 overexpression is of particular importance in the pathogenesis of ACC. Accordingly, inhibition of IGF-2 action by blocking the IGF-1 receptor leads to reduced growth of ACC cells in vitro.22
An important observation relates to phosphodiesterase 11a (PDE11a) and the genetic predisposition to adrenocortical tumors.23 PDE11a inactivating germline mutations are more frequent in patients harboring adrenocortical tumors, including ACC, than in age- and sex-matched controls (odds ratio 3.53), suggesting that PDE11a alterations predispose to adrenocortical tumors.
In sporadic ACC, a variety of somatic mutations have been identified. In both benign and malignant adrenocortical tumors, β-catenin accumulation has been frequently observed, indicating activation of the Wnt-signaling pathway. This is explained in a subset of these tumors by somatic mutations of the β-catenin gene (CTNNB1) which may contribute to tumor progression.24
No activating mutations were found in the ACTH receptor in adrenal tumors.25 In fact, in ACC a loss of heterozygosity of the ACTH receptor, with reduced expression of ACTH receptor mRNA, was observed, supporting the view that ACTH is mainly a differentiating factor for adrenocortical cells and that growth-promoting activities of pro-opiomelanocortin (POMC) may reside in the N-terminus of POMC.26,27
Chromosomal instability has been described in malignant adrenal tumors, indicating defects in the mitogenic machinery.28 Using comparative genomic hybridization analysis, a significantly higher number of changes in ACCs compared to adrenocortical adenomas (mean of 7.6 to 14 changes versus 1.1 to 2 changes) have been demonstrated.29,30 The likelihood of chromosomal changes increased with tumor size. Similarly, loss of heterozygosity (LOH) analysis has found LOHs of 17p13, 11p15, 11q13, 17q22-24, and 2p16 in sporadic ACC.10 It has been shown that the number of somatic aberrations in ACC also predicts prognosis.31
Furthermore, telomere maintenance mechanisms are critical for the malignant phenotype in ACC. It has been demonstrated that telomerase activity is the major mechanism for telomere maintenance, but subsets of ACCs also show evidence of alternative telomere lengthening.32
Clinical Presentation
Most patients with ACC (60%) present with signs and symptoms of adrenal steroid excess. Rapidly progressing Cushing’s syndrome with or without virilization is the most frequent presentation.8,33 Androgen-secreting ACCs in women present with hirsutism and virilization, with male-pattern baldness and oligo/amenorrhea of recent onset. Estrogen-secreting tumors in males lead to gynecomastia and testicular atrophy. Rare aldosterone-producing adrenocortical carcinomas present with severe hypertension and profound hypokalemia (mean serum potassium 2.3 ± 0.08 mmol/L).34 However, low serum potassium is more often the result of excessive cortisol production leading to incomplete renal inactivation by 11-β-dehydrogenase type 2, with consecutive mineralocorticoid excess. Careful search for abnormal adrenal steroid secretion will often reveal increased dehydroepiandrosterone sulfate (DHEAS) concentrations or elevation of androstenedione or 17α-hydroxyprogesterone, thereby confirming the adrenocortical origin of the tumor and defining a marker for follow-up. Using gas chromatography/mass spectroscopy (GC-MS) for sophisticated urinary steroid analysis, hormonal activity can be demonstrated in almost all ACC cases.35 However, owing to low efficiency of intratumoral steroidogenesis or the exclusive secretion of steroid precursors, tumors may appear clinically as hormonally inactive.
Patients with a nonfunctioning ACC usually present with symptoms of abdominal discomfort (nausea, vomiting, abdominal fullness) or back pain caused by the large tumor mass. In particular, local pain may indicate invasive tumor growth and point to malignancy in a larger adrenal lesion. More frequent and improved abdominal imaging have led to an increasing percentage of ACC being discovered incidentally.36,37 Nonspecific symptoms like fever, weight loss, and loss of appetite are rare. In fact, patients may carry a large tumor burden without much evidence of systemic disease besides signs and symptoms of hormone excess.
Diagnosis
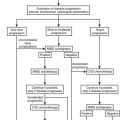
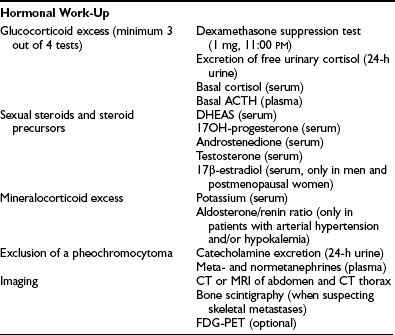
Detailed hormonal evaluation is performed prior to surgery for ACC to identify tumor markers for long-term follow-up and to guide perioperative treatment strategies (e.g., replacement of glucocorticoids after removal of a cortisol-secreting ACC).38 Guidelines for hormonal evaluation in suspected or established ACC have been provided by the ACC Working Group of the European Network for the Study of Adrenal Tumors (ENSAT; www.ensat.org) (Table 11-1). Hormone concentrations are of limited value in predicting malignancy. However, high testosterone in women, or high estradiol in men, or cosecretion of glucocorticoids and sex hormones are indicative of a malignant adrenal lesion. In addition, benign adrenocortical tumors very often show low DHEAS concentrations, whereas highly elevated DHEAS is suggestive of ACC. Similarly, high levels of steroid precursors such as 17α-hydroxyprogesterone or androstenedione are often observed in seemingly hormonally inactive ACCs. Measurement of urinary catecholamine excretion or plasma metanephrines is required prior surgery to exclude pheochromocytoma.
Imaging
Imaging plays a key role in the diagnostic workup of patients with suspected adrenal malignancy39–41 (also discussed in Chapter 10). Both size and appearance of an adrenal mass on CT, magnetic resonance imaging (MRI), and 18F-fluorodeoxyglucose positron emission tomography (FDG-PET) are highly relevant to distinguish between benign and malignant lesions. For an adrenal mass >6 cm, the likelihood of ACC increases dramatically.42 In the German ACC Registry (n = 489), the median tumor size at diagnosis was 11.6 ± 4.7 cm (range 3 to 40 cm). Thus, in many patients, size alone is a strong indicator of malignancy. However, stage I ACC with a diameter of <5 cm has clearly the best prognosis and it is, therefore, important to identify adrenal malignancy as early as possible. Tumors between 3 and 6 cm represent a major diagnostic challenge.
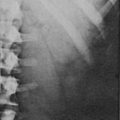
Thin-collimation CT offers high spatial resolution and is fast and widely available. Large size, inhomogeneous appearance, irregular shape, and invasion into surrounding structures indicate malignancy. Frequently, tumor extension into the inferior vena cava, enlarged regional lymph nodes, and other metastases (lung and liver) are already found at presentation of ACC (Fig. 11-1). Measurement of Hounsfield units (HU) in unenhanced CT can often differentiate benign adrenal lesions from malignancy. If the density is <10 HU, diagnosis of adenoma has a sensitivity of 71% and a specificity of 98%, respectively.43 However, lipid-poor benign adenomas are frequent and show unenhanced HU values >10.44 In these cases, delayed postcontrast CT yields high sensitivity and specificity.45,46 Calculating the percentage washout for adrenal masses at 10- to 15-min-delayed enhanced CT, a washout of more than 40% to 50% is highly suggestive of a benign mass, whereas a delayed attenuation of more than 35 HU and a washout of less than 50% suggest malignancy. Delayed enhanced CT is also able to characterize some adrenal masses that cannot be characterized by chemical shift MRI.47
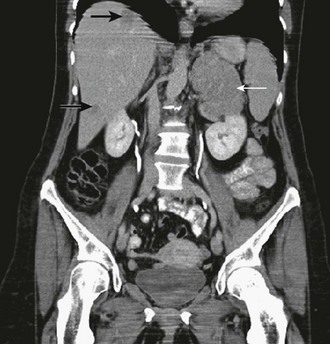
FIGURE 11-1 CT after contrast media of 8.5 cm in homogeneous ACC of the left adrenal gland (white arrow) and liver metastases (black arrows). (Image provided by W. Kenn, Dept. of Radiology, University of Würzburg, Germany.)
Modern MRI of adrenal tumors should include T1- and T2-weighted images plus chemical shift imaging, which consists of in-phase and out-of-phase imaging. Multiplanar MRI is particularly suited to separating adrenal masses from surrounding structures like liver, spleen, pancreas, and kidney. ACCs typically present isointense to liver on T1-weighted images and show an increase in intensity in T2-weighted sequences. Heterogeneity of signals is noted both on T1-weighted and T2-weighted images due to necrosis or hemorrhage. Enhancement after gadolinium is typical, and washout is slow. Modern MRI technology can differentiate benign from malignant adrenal lesions with a sensitivity of 81% to 89% and a specificity between 92% and 99%.41,48,49 However, the optimum MRI method (T1/T2 relaxation time, chemical shift, fast low angle shot, in vivo proton MR spectroscopy, etc.) for diagnosis of ACC remains a matter of controversy.37,50,51
In the past, adrenal scintigraphy with 131I-6β-iodomethyl-norcholesterol (NP59) has been used to characterize adrenal lesions.52 Benign hypersecretory adenomas show enhanced tracer uptake. However, NP 59 scintigraphy is time consuming and associated with a high radiation dose. In contrast, FDG-PET may be highly valuable in patients with suspected ACC. High uptake of 18F-FDG demonstrates increased glucose metabolism, and with few exceptions indicates malignancy. Thus, FDG-PET may be highly valuable for evaluation adrenal masses that are indeterminate by both CT and MRI. However, some benign adenomas or pheochromocytomas also show uptake of FDG.41,50,53 Imaging with FDG-PET has the additional advantage of simultaneously detecting metastases at other sites, but metastatic lesions <1 cm (particularly in the lung) are not easily detected by FDG-PET,54,55 indicating that PET cannot substitute for CT imaging.
None of the imaging methods mentioned can reliably differentiate ACC from a metastasis of other origin or a pheochromocytoma. In this context, a new method for adrenal imaging is promising: 11C-metomidate PET.56,57 Metomidate specifically binds to adrenal 11β-hydroxylase and aldosterone synthase, so uptake indicates the adrenocortical origin of an adrenal lesion. A potentially more widely available tracer is 123I-iodometomidate for SPECT imaging.58
Prior to surgery, high-resolution CT of chest and abdomen should be performed to detect frequent lung and liver metastases. A bone scan is only required if the patient complains of bone pain (see Table 11-1). Fine-needle biopsy of suspected ACC is rarely justified and is associated with the risk of needle-track metastasis. In our view, a biopsy should only be performed if the tumor cannot be removed surgically, and medical therapy needs to be based on clear pathologic evidence.
Histopathology
The pathologic diagnosis of ACC may be difficult because of the lack of clear-cut morphologic criteria,59 and in all cases, it is recommended to involve a specialized pathologist.
Weight and size are important criteria for malignancy. Most adenomas have a weight of <50 g, whereas most carcinomas weigh >100 g. The likelihood of an ACC increases steeply with a diameter of more than 6 cm.60
The differential diagnosis of carcinomas and adenomas is based largely on morphologic features. Different diagnostic scores61–63 have been introduced for diagnosis of malignancy. The Weiss system is most widely used and combines nine morphologic parameters, which include three parameters related to structure (description of cytoplasm, diffuse architecture, necrosis), three parameters related to cytology (atypia, atypical mitotic figures, mitotic count), and three related to invasion (veins, sinusoids, and capsule). Further morphologic parameters of importance are broad fibrous bands and hemorrhage. It has been shown that the mitotic index also has prognostic importance.64,65 In addition, periadrenal tissue infiltration and vena cava or renal vein invasion carry prognostic significance.66 Careful assessment of the resection status (R0, R1, R2) is also of great importance in that it may define further treatment strategies. For the same reason, violation of the capsule must be reported.
Important information is also provided by immunohistochemistry. Here, Ki-67 expression can be used both for differentiating benign from malignant tumors and for prognosis in ACC. A cutoff value between adenomas and ACCs varying from 1.5% to 4% has been reported. In patients with ACC, a Ki-67 labeling index of >10% was associated with poor survival in the German ACC registry. Other markers like Melan A, D11, inhibin α, and SF-1 are helpful to define the adrenocortical origin of the tumor, whereas ACC is typically negative for chromogranin A and S100.59,60,67,68 A number of new markers (LOH at 17p13, IGF-2 overexpression, cyclin E, matrix metalloproteinase-2 [mmp-2], telomerase activity, topoisomerase IIα, and N-cadherin) have been used to separate benign from malignant adrenal lesions. However, currently none of these markers has gained widespread acceptance.
Staging
In 2004, for the first time, a staging system was published by the Union International Contre Cancer (UICC) and the World Health Organization69 (Table 11-2). Stages I and II describe localized tumors 5 cm or smaller and larger than 5 cm, respectively. Locally invasive tumors or tumors with regional lymph node metastases are classified as stage III, and stage IV consists of tumors invading adjacent organs or presenting with distant metastases. Unfortunately, this staging system, which is largely based on the Macfarlane classification as modified by Sullivan, showed limited prognostic power.66 Thus, a revised TNM classification was proposed by ENSAT (see Table 11-2). In this staging system, stage III is defined by tumor infiltration in surrounding tissue or in adjacent organs, thrombus in vena cava/renal vein, or positive lymph nodes, whereas stage IV is defined by the presence of distant metastasis. The ENSAT staging system provides a powerful prognostic tool, predicting both disease-free and disease-specific survival in patients with ACC (Fig. 11-2).
Table 11-2
Staging Systems for Adrenocortical Carcinoma Proposed by the UICC 2004 and ENSAT 200866,69
| Stage | UICC/WHO 2004 | ENSAT 2008 |
| I | T1, N0, M0 | T1, N0, M0 |
| II | T2, N0, M0 | T2, N0, M0 |
| III | T1-2, N1, M0 T3, N0, M0 |
T1-2, N1, M0 T3-4, N0-1, M0 |
| IV | T1-4, N0-1, M1 T3, N1, M0 T4, N0-1, M0 |
T1-4, N0-1, M1 |
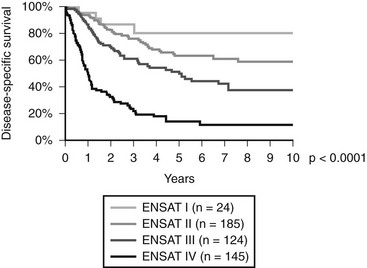
FIGURE 11-2 Disease-specific survival according to tumor stage (ENSAT Classification; see Table 11-2). (Data derived from the German ACC Registry, September 2008.)
Therapy (Fig. 11-3)
Today most patients are diagnosed in stages I to III (see Fig. 11-2). In these stages, complete removal is the single most important therapeutic measure and offers the best chance for cure. All available data indicate that surgery should be performed by a specialized surgeon aware of the pitfalls of surgery for ACC (violation of the capsule, spilling). An R0 resection (resection margins microscopically disease free) is of utmost importance for long-term prognosis. Often surgery needs to be extensive, with en bloc resection of invaded organs. Spillage during surgery is paramount to an upgrade to stage IV66 and poor prognosis. The presence of a thrombus is compatible with complete resection but occasionally necessitates cardiac bypass technique.70–72 The use of laparoscopic adrenalectomy for ACC is debated. A higher risk of tumor spillage and local recurrences has been reported.73–75 However, these reports suffer from a referral bias, and inferiority of laparoscopic surgery for ACC has never been demonstrated. While most experts still favor open surgery for ACC, an analysis from the German ACC registry suggests that the long-term outcome in terms of disease-free survival and overall survival is not significantly different between the use of laparoscopic adrenalectomy or open surgery in tumors <10 cm. Probably more important than this technical aspect is the expert status of the surgeon.
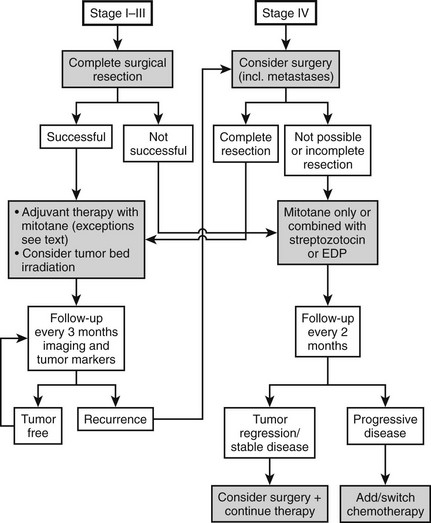
Radiation Therapy
The high rate of local relapse after surgery with curative intent suggests that adjuvant tumor-bed irradiation may have therapeutic potential by preventing local relapse, which often precedes metastatic tumor spread. This view is supported by a small study indicating that adjuvant radiotherapy of the tumor bed reduces the rate of local recurrences.76 Although more data are needed to better define the role of radiation therapy in an adjuvant setting, it may be justified to use adjuvant radiotherapy in selected patients. We recommend radiation therapy of the tumor bed in patients with histologically incomplete (R1) resection. In patients with macroscopically visible residual tumor, a second surgical approach by an expert surgeon should be considered first. Another group of patients that may benefit from adjuvant radiotherapy are patients with advanced locoregional disease (stage III), no evidence of distant metastases, and no evidence of residual disease after surgery. A standard fractionation scheme with single doses of 1.8 to 2.0 Gy on 5 days per week for 5 to 6 weeks is recommended. Total doses should not fall below a minimum of 40 Gy and ideally should reach 50 to 60 Gy.
Medical Therapy: Mitotane
Mitotane (1,1 dichloro-2[o-chlorophenyl]-2-[p-chloro-phenyl]ethane, o,p′-DDD) is an adrenolytic compound with specific adrenocortical activity that was introduced in the treatment of ACC more than 40 years ago by Bergenstal et al.77 Its precise mechanism of action is not fully understood.78 Its therapeutic activity depends on metabolic activation in adrenocortical cells. Mitotane is hydroxylated in the mitochondria and transformed into an acylchloride. The activated metabolites may directly bind to macromolecules, thereby inhibiting their activity and inducing oxidative damage through production of free radicals. Impairment of adrenal steroidogenesis has been reported, and evidence for inhibition of 11β-hydroxylase activity has been demonstrated.
The clinical efficacy of mitotane has long been disputed, and only in 2004 was it approved for advanced ACC by the European Medicine Agency (EMEA). However, there is clear evidence that mitotane can induce significant tumor regression in about 25% of cases with metastasized ACC and achieve control of hypercortisolism in the majority of patients.78 Complete responses have been described, and long-term survival is seen in the occasional patient. The use of mitotane in an adjuvant setting has been a matter of debate. However, a large retrospective analysis has indicated that adjuvant mitotane holds great potential to prolong disease-free survival and overall survival in ACC.79 Different from many previous studies, in this investigation, selection bias was largely avoided.
Mitotane is given as tablets (Lysodren; HRA Pharma Paris, Bristol-Myers Squibb New York). Monitoring mitotane blood concentrations is essential for providing optimal efficacy and reducing toxicity. Treatment aims at mitotane concentrations between 14 and 20 mg/L for antitumor response. Higher concentrations may lead to unacceptable toxicity, whereas a response is unlikely at a concentration <14 mg/L. Thus, mitotane has a narrow therapeutic window. However, some patients may respond to lower mitotane concentrations. Owing to the long half-life of mitotane, target levels are often achieved only after weeks to months.80 This time can be shortened by using high-dose regimens in the early phase of therapy81: treatment is initiated with 1.5 g/day and rapidly increased to 5 to 6 g/day until target concentrations have been reached. Most side effects are related to mitotane plasma concentrations, but gastrointestinal side effects like diarrhea seem to be more related to the daily dose. During long-term treatment, the dose of mitotane can usually be reduced, and many patients need only 2 to 3 g/day during long-term therapy. Adverse effects of mitotane treatment are manifold and common.78 The most troubling side effects involve the central nervous system and include ataxia, confusion, tiredness, and dizziness. In addition, dose-limited gastrointestinal side effects such as nausea, vomiting, anorexia, and diarrhea may occur. Treatment with 5-hydroxytryptamine blockers and loperamide, respectively, may be useful. Because of the long half-life of mitotane, blood levels increase slowly, and adverse effects usually become apparent over time, even if the dose has remained unchanged. In case of significant side effects, drug treatment is interrupted for days or a week, and treatment is restarted with a lower dose. Ingestion of mitotane together with a lipid-rich drink or meal may enhance resorption and increase drug concentrations. Owing to its adrenolytic activity, mitotane treatment induces adrenal insufficiency. Furthermore, mitotane also increases the metabolic clearance of glucocorticoids and increases the concentration of cortisol-binding globulin (CBG).82 High-dose glucocorticoid replacement (50 to 80 mg hydrocortisone daily) is needed. Insufficient glucocorticoid replacement enhances mitotane intolerance. Aldosterone secretion is less often affected, since mitotane primarily acts on the zona fasciculata and the zona reticularis. However, aldosterone secretion should be monitored, and replacement with fludrocortisone may become necessary with long-term use of mitotane. In addition, mitotane increases sex hormone–binding globulin82 and often leads to low free-testosterone concentrations in males. With long-term therapy, free–thyroid hormone concentrations decline in the presence of low to low-normal TSH, suggesting secondary hypothyroidism. Mitotane’s estrogenic activity may facilitate the development of gynecomastia.
Despite the plethora of adverse effects of mitotane, most patients can be managed with acceptable toxicity in the long term.80,83 This is particularly important in an adjuvant setting, because treatment must be given for a minimum of 2 years.
Cytotoxic Chemotherapy
Experience with cytotoxic chemotherapy in ACC is still limited.38,84 In general, it has been shown that a combination of mitotane with cytotoxic agents is superior to treatment with cytotoxic agents alone.78 The best results in terms of response rate have been reported for a combination of mitotane with etoposide, doxorubicin, and cisplatin.85 According to WHO criteria, the overall response rate in 72 patients was 49%, including 5 patients with a complete response. A response rate of 36% was reported for a combination of mitotane and streptozotocin.86 This protocol is more easily administered and may have less toxicity. At the time of this writing, these two cytotoxic regimens (Table 11-3) are directly compared in the first ever phase III trial in ACC (www.firm-act.org). Results will be available in 2011 and will provide a reference cytotoxic drug regimen against which future treatments will be compared. A variety of other protocols have been used in small series of patients.87 From this experience, it can be concluded that platinum-based therapies are in general more successful than cytotoxic regimens without platinum compounds. For other tumors, it has been shown that expression of ERCC1 plays a key role in the resistance to platinum compounds. In a retrospective series, it has been demonstrated that patients with low or absent expression of ERCC1 in their tumor respond better to treatment with a platinum-based cytotoxic chemotherapy than patients who have tumors with high expression of ERCC1.88 This observation may be a first step towards an individualized therapy of patients with ACC.
Table 11-3
Recommended First-Line Cytotoxic Drug Regimens
Etoposide, doxorubicin, and cisplatin (EDP) plus mitotane (EDP/M) (adapted from ref. 85) every 28 days:
Day 1: 40 mg/m2 D
Day 2: 100 mg/m2 E
Days 3 + 4: 100 mg/m2 E + 40 mg/m2 P plus oral mitotane aiming at blood levels between 14 and 20 mg/L
Streptozotocin plus mitotane (Sz/M) (ref. 86):
Induction: days 1-5: 1 g Sz/day
Afterwards: 2 g/d Sz every 21 days plus oral mitotane aiming at blood levels between 14 and 20 mg/L
Targeted Therapies
Current success with cytotoxic chemotherapy in ACC is not very satisfactory, with less than 50% of tumors responding to treatment and most of them for only a limited period of time. Thus, new treatment options are of great interest, and the advent of targeted therapies has led to the first small trials incorporating such compounds89 (www.clinicaltrials.gov). However, combinations of erlotinib plus gemcitabine exhibited only limited efficacy as salvage therapy for patients with advanced ACC.90 Similarly, in a series from M.D. Anderson in Houston, no response to the EGF receptor antagonist gefitinib was seen in 19 patients with advanced ACC.91 Furthermore, in 9 patients we saw no response to a combination of bevacizumab plus capecitabine given as salvage treatment, also suggesting that this combination has no relevant activity in advanced ACC. Because ACCs express high levels of IGF-2, which acts via the IGF-1 receptor, blockade of the IGF-1 receptor has been suggested as a promising treatment target in ACC.20–22 Trials have been initiated, but no results have been published.
Another group of compounds that holds promise for the treatment of solid tumors are multi-tyrosine kinase inhibitors (e.g., sunitinib, sorafenib). Again, the available evidence is still very limited. Occasional tumor responses have been reported for the antiangiogenic compound thalidomide.92 Thus, an intensive search for improved treatment protocols has been initiated, and it is expected that within the next decade, major changes in the treatment of advanced ACC will take place.
References
1. Grumbach, MM, Biller, BM, Braunstein, GD, et al. Management of the clinically inapparent adrenal mass (“incidentaloma”). Ann Intern Med. 2003;138(5):424–429.
2. Bovio, S, Cataldi, A, Reimondo, G, et al. Prevalence of adrenal incidentaloma in a contemporary computerized tomography series. J Endocrinol Invest. 2006;29(4):298–302.
3. Song, JH, Chaudhry, FS, Mayo-Smith, WW. The incidental adrenal mass on CT: prevalence of adrenal disease in 1,049 consecutive adrenal masses in patients with no known malignancy. AJR Am J Roentgenol. 2008;190(5):1163–1168.
4. Abecassis, M, McLoughlin, MJ, Langer, B, et al. Serendipitous adrenal masses: prevalence, significance, and management. Am J Surg. 1985;149(6):783–788.
5. National-Cancer-Institute. Third national cancer survey: incidence data. DHEW Publ No (NIH). 1975;75-787(NCI monograph):41.
6. Kebebew, E, Reiff, E, Duh, QY, et al. Extent of Disease at Presentation and Outcome for Adrenocortical Carcinoma: Have We Made Progress? World J Surg. 2006;30:872–878.
7. Ribeiro, RC, Sandrini, F, Figueiredo, B, et al. An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc Natl Acad Sci U S A. 2001;98(16):9330–9335.
8. Koschker, AC, Fassnacht, M, Hahner, S, et al. Adrenocortical carcinoma—improving patient care by establishing new structures. Exp Clin Endocrinol Diabetes. 2006;114(2):45–51.
9. Barlaskar, FM, Hammer, GD. The molecular genetics of adrenocortical carcinoma. Rev Endocr Metab Disord. 2007;8(4):343–348.
10. Soon, PS, McDonald, KL, Robinson, BG, et al. Molecular markers and the pathogenesis of adrenocortical cancer. Oncologist. 2008;13(5):548–561.
11. Li, FP, Fraumeni, JF, Jr. Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med. 1969;71(4):747–752.
12. Hisada, M, Garber, JE, Fung, CY, et al. Multiple primary cancers in families with Li-Fraumeni syndrome. J Natl Cancer Inst. 1998;90(8):606–611.
13. Varley, JM, McGown, G, Thorncroft, M, et al. Are there low-penetrance TP53 Alleles? Evidence from childhood adrenocortical tumors. Am J Hum Genet. 1999;65(4):995–1006.
14. Bell, DW, Varley, JM, Szydlo, TE, et al. Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science. 1999;286(5449):2528–2531.
15. DiGiammarino, EL, Lee, AS, Cadwell, C, et al. A novel mechanism of tumorigenesis involving pH-dependent destabilization of a mutant p53 tetramer. Nat Struct Biol. 2002;9(1):12–16.
16. Reincke, M, Karl, M, Travis, WH, et al. p53 mutations in human adrenocortical neoplasms: immunohistochemical and molecular studies. J Clin Endocrinol Metab. 1994;78(3):790–794.
17. Maher, ER, Reik, W. Beckwith-Wiedemann syndrome: imprinting in clusters revisited. J Clin Invest. 2000;105(3):247–252.
18. Gicquel, C, Raffin-Sanson, ML, Gaston, V, et al. Structural and functional abnormalities at 11p15 are associated with the malignant phenotype in sporadic adrenocortical tumors: study on a series of 82 tumors. J Clin Endocrinol Metab. 1997;82(8):2559–2565.
19. Gicquel, C, Bertagna, X, Gaston, V, et al. Molecular markers and long-term recurrences in a large cohort of patients with sporadic adrenocortical tumors. Cancer Res. 2001;61(18):6762–6767.
20. Giordano, TJ, Thomas, DG, Kuick, R, et al. Distinct transcriptional profiles of adrenocortical tumors uncovered by DNA microarray analysis. Am J Pathol. 2003;162(2):521–531.
21. de Fraipont, F, El Atifi, M, Cherradi, N, et al. Gene expression profiling of human adrenocortical tumors using complementary deoxyribonucleic acid microarrays identifies several candidate genes as markers of malignancy. J Clin Endocrinol Metab. 2005;90(3):1819–1829.
22. Almeida, MQ, Fragoso, MC, Lotfi, CF, et al. Expression of IGF-II and its receptor in pediatric and adult adrenocortical tumors. J Clin Endocrinol Metab. 2008.
23. Libe, R, Fratticci, A, Coste, J, et al. Phosphodiesterase 11A (PDE11A) and genetic predisposition to adrenocortical tumors. Clin Cancer Res. 2008;14(12):4016–4024.
24. Gaujoux, S, Tissier, F, Groussin, L, et al. Wnt/ss-catenin and cAMP/PKA signaling pathways alterations and somatic ss-catenin gene mutations in the progression of adrenocortical tumors. J Clin Endocrinol Metab. 2008.
25. Latronico, AC, Reincke, M, Mendonca, BB, et al. No evidence for oncogenic mutations in the adrenocorticotropin receptor gene in human adrenocortical neoplasms. J Clin Endocrinol Metab. 1995;80(3):875–877.
26. Beuschlein, F, Fassnacht, M, Klink, A, et al. ACTH-receptor expression, regulation and role in adrenocortical formation. Eur J Endocrinol. 2001;144(3):199–206.
27. Fassnacht, M, Hahner, S, Hansen, IA, et al. N-terminal pro-opiomelanocortin acts as a mitogen in adrenocortical cells and decreases adrenal steroidogenesis. J Clin Endocrinol Metab. 2003;88(5):2171–2179.
28. Dohna, M, Reincke, M, Mincheva, A, et al. Adrenocortical carcinoma is characterized by a high frequency of chromosomal gains and high-level amplifications. Genes Chromosomes Cancer. 2000;28(2):145–152.
29. Sidhu, S, Marsh, DJ, Theodosopoulos, G, et al. Comparative genomic hybridization analysis of adrenocortical tumors. J Clin Endocrinol Metab. 2002;87(7):3467–3474.
30. Zhao, J, Speel, EJ, Muletta-Feurer, S, et al. Analysis of genomic alterations in sporadic adrenocortical lesions. Gain of chromosome 17 is an early event in adrenocortical tumorigenesis. Am J Pathol. 1999;155(4):1039–1045.
31. Stephan, EA, Chung, TH, Grant, CS, et al. Adrenocortical carcinoma survival rates correlated to genomic copy number variants. Mol Cancer Ther. 2008;7(2):425–431.
32. Else, T, Giordano, TJ, Hammer, GD. Evaluation of telomere length maintenance mechanisms in adrenocortical carcinoma. J Clin Endocrinol Metab. 2008;93(4):1442–1449.
33. Abiven, G, Coste, J, Groussin, L, et al. Clinical and biological features in the prognosis of adrenocortical cancer: poor outcome of cortisol-secreting tumors in a series of 202 consecutive patients. J Clin Endocrinol Metab. 2006;91(7):2650–2655.
34. Seccia, TM, Fassina, A, Nussdorfer, GG, et al. Aldosterone-producing adrenocortical carcinoma: an unusual cause of Conn’s syndrome with an ominous clinical course. Endocr Relat Cancer. 2005;12(1):149–159.
35. Arlt W, Hahner S, Libe R, et al: Steroid profiling in the diagnosis and monitoring of adrenocortical cancer—results of the EURINE ACC Study of the European Network for the Study of Adrenal Tumors (ENSAT). Abstracts of the 90th annual meeting of the Endocrine Society (San Francisco):OR40-2, 2008.
36. Mantero, F, Terzolo, M, Arnaldi, G, et al. A survey on adrenal incidentaloma in Italy. Study Group on Adrenal Tumors of the Italian Society of Endocrinology. J Clin Endocrinol Metab. 2000;85(2):637–644.
37. Mansmann, G, Lau, J, Balk, E, et al. The clinically inapparent adrenal mass: update in diagnosis and management. Endocr Rev. 2004;25(2):309–340.
38. Allolio, B, Fassnacht, M. Clinical review: adrenocortical carcinoma: clinical update. J Clin Endocrinol Metab. 2006;91(6):2027–2037.
39. Fassnacht, M, Kenn, W, Allolio, B. Adrenal tumors: how to establish malignancy? J Endocrinol Invest. 2004;27(4):387–399.
40. Ilias, I, Sahdev, A, Reznek, RH, et al. The optimal imaging of adrenal tumors: a comparison of different methods. Endocr Relat Cancer. 2007;14(3):587–599.
41. Heinz-Peer, G, Memarsadeghi, M, Niederle, B. Imaging of adrenal masses. Curr Opin Urol. 2007;17(1):32–38.
42. Ross, NS, Aron, DC. Hormonal evaluation of the patient with an incidentally discovered adrenal mass. N Engl J Med. 1990;323(20):1401–1405.
43. Boland, GW, Lee, MJ, Gazelle, GS, et al. Characterization of adrenal masses using unenhanced CT: an analysis of the CT literature. Am J Roentgenol. 1998;171(1):201–204.
44. Hamrahian, AH, Ioachimescu, AG, Remer, EM, et al. Clinical utility of noncontrast computed tomography attenuation value (Hounsfield units) to differentiate adrenal adenomas/hyperplasias from nonadenomas: Cleveland Clinic experience. J Clin Endocrinol Metab. 2005;90(2):871–877.
45. Caoili, EM, Korobkin, M, Francis, IR, et al. Adrenal masses: characterization with combined unenhanced and delayed enhanced CT. Radiology. 2002;222(3):629–633.
46. Szolar, DH, Korobkin, M, Reittner, P, et al. Adrenocortical carcinomas and adrenal pheochromocytomas: mass and enhancement loss evaluation at delayed contrast-enhanced CT. Radiology. 2005;234(2):479–485.
47. Park, BK, Kim, CK, Kim, B, et al. Comparison of delayed enhanced CT and chemical shift MR for evaluating hyperattenuating incidental adrenal masses. Radiology. 2007;243(3):760–765.
48. Korobkin, M, Lombardi, TJ, Aisen, AM, et al. Characterization of adrenal masses with chemical shift and gadolinium-enhanced MR imaging. Radiology. 1995;197(2):411–418.
49. Honigschnabl, S, Gallo, S, Niederle, B, et al. How accurate is MR imaging in characterisation of adrenal masses: update of a long-term study. Eur J Radiol. 2002;41(2):113–122.
50. Al-Hawary, MM, Francis, IR, Korobkin, M. Non-invasive evaluation of the incidentally detected indeterminate adrenal mass. Best Pract Res Clin Endocrinol Metab. 2005;19(2):277–292.
51. Faria, JF, Goldman, SM, Szejnfeld, J, et al. Adrenal masses: characterization with in vivo proton MR spectroscopy—initial experience. Radiology. 2007;245(3):788–797.
52. Gross, MD, Shapiro, B, Francis, IR, et al. Scintigraphic evaluation of clinically silent adrenal masses. J Nucl Med. 1994;35(7):1145–1152.
53. Caoili, EM, Korobkin, M, Brown, RK, et al. Differentiating adrenal adenomas from nonadenomas using (18)F-FDG PET/CT: quantitative and qualitative evaluation. Acad Radiol. 2007;14(4):468–475.
54. Mackie, GC, Shulkin, BL, Ribeiro, RC, et al. Use of [18F]fluorodeoxyglucose positron emission tomography in evaluating locally recurrent and metastatic adrenocortical carcinoma. J Clin Endocrinol Metab. 2006;91(7):2665–2671.
55. Leboulleux, S, Dromain, C, Bonniaud, G, et al. Diagnostic and prognostic value of 18-fluorodeoxyglucose positron emission tomography in adrenocortical carcinoma: a prospective comparison with computed tomography. J Clin Endocrinol Metab. 2006;91(3):920–925.
56. Khan, TS, Sundin, A, Juhlin, C, et al. 11C-metomidate PET imaging of adrenocortical cancer. Eur J Nucl Med Mol Imaging. 2003;30(3):403–410.
57. Hennings, J, Lindhe, O, Bergstrom, M, et al. [11C]metomidate positron emission tomography of adrenocortical tumors in correlation with histopathological findings. J Clin Endocrinol Metab. 2006;91(4):1410–1414.
58. Hahner, S, Stuermer, A, Kreissl, M, et al. [123 I]Iodometomidate for molecular imaging of adrenocortical cytochrome P450 family 11B enzymes. J Clin Endocrinol Metab. 2008;93(6):2358–2365.
59. Volante, M, Buttigliero, C, Greco, E, et al. Pathological and molecular features of adrenocortical carcinoma: an update. J Clin Pathol. 2008;61(7):787–793.
60. Saeger, W. Histopathological classification of adrenal tumors. Eur J Clin Invest. 2000;30(Suppl 3):58–62.
61. Weiss, LM, Medeiros, LJ, Vickery, AL, Jr. Pathologic features of prognostic significance in adrenocortical carcinoma. Am J Surg Pathol. 1989;13(3):202–206.
62. Hough, AJ, Hollifield, JW, Page, DL, et al. Prognostic factors in adrenal cortical tumors. A mathematical analysis of clinical and morphologic data. Am J Clin Pathol. 1979;72(3):390–399.
63. van Slooten, H, Schaberg, A, Smeenk, D, et al. Morphologic characteristics of benign and malignant adrenocortical tumors. Cancer. 1985;55(4):766–773.
64. Stojadinovic, A, Ghossein, RA, Hoos, A, et al. Adrenocortical carcinoma: clinical, morphologic, and molecular characterization. J Clin Oncol. 2002;20(4):941–950.
65. Assie, G, Antoni, G, Tissier, F, et al. Prognostic parameters of metastatic adrenocortical carcinoma. J Clin Endocrinol Metab. 2007;92(1):148–154.
66. Fassnacht, M, Johanssen, S, Quinkler, M, et al. Limited prognostic value of the 2004 UICC staging classification for adrenocortical carcinoma: proposal for a revised TNM classification. Cancer. 2008.
67. Saeger, W, Fassnacht, M, Chita, R, et al. High diagnostic accuracy of adrenal core biopsy: results of the German and Austrian adrenal network multicenter trial in 220 consecutive patients. Hum Pathol. 2003;34(2):180–186.
68. Sasano, H, Suzuki, T, Moriya, T. Recent advances in histopathology and immunohistochemistry of adrenocortical carcinoma. Endocr Pathol. 2006;17(4):345–354.
69. DeLellis RA, Lloyd RV, Heitz PU, et al: World Health Organization Classification of Tumors. Pathology and Genetics of Tumors of Endocrine Organs. IARC Press, Lyons, France, 136, 2004.
70. Mingoli, A, Nardacchione, F, Sgarzini, G, et al. Inferior vena cava involvement by a left side adrenocortical carcinoma: operative and prognostic considerations. Anticancer Res. 1996;16(5B):3197–3200.
71. Hedican, SP, Marshall, FF. Adrenocortical carcinoma with intracaval extension. J Urol. 1997;158(6):2056–2061.
72. Dackiw, AP, Lee, JE, Gagel, RF, et al. Adrenal cortical carcinoma. World J Surg. 2001;25(7):914–926.
73. Gonzalez, RJ, Shapiro, S, Sarlis, N, et al. Laparoscopic resection of adrenal cortical carcinoma: a cautionary note. Surgery. 2005;138(6):1078–1085.
74. Kebebew, E, Siperstein, AE, Clark, OH, et al. Results of laparoscopic adrenalectomy for suspected and unsuspected malignant adrenal neoplasms. Arch Surg. 2002;137(8):948–951.
75. Schlamp, A, Hallfeldt, K, Mueller-Lisse, U, et al. Recurrent adrenocortical carcinoma after laparoscopic resection. Nat Clin Pract Endocrinol Metab. 2007;3(2):191–195.
76. Fassnacht, M, Hahner, S, Polat, B, et al. Efficacy of adjuvant radiotherapy of the bed on local recurrence of adrenocortical carcinoma. J Clin Endocrinol Metab. 2006;91(11):4501–4504.
77. Bergenstal, D, Lipsett, M, Moy, R, et al. Regression of adrenal cancer and suppression of adrenal function in men by o,p′-DDD. Transactions of American Physicians. 1959;72:341.
78. Hahner, S, Fassnacht, M. Mitotane for adrenocortical carcinoma treatment. Curr Opin Investig Drugs. 2005;6(4):386–394.
79. Terzolo, M, Angeli, A, Fassnacht, M, et al. Adjuvant mitotane treatment in patients with adrenocortical carcinoma. N Engl J Med. 2007;356(23):372–380.
80. Terzolo, M, Pia, A, Berruti, A, et al. Low-dose monitored mitotane treatment achieves the therapeutic range with manageable side effects in patients with adrenocortical cancer. J Clin Endocrinol Metab. 2000;85(6):2234–2238.
81. Faggiano, A, Leboulleux, S, Young, J, et al. Rapidly progressing high o,p′DDD doses shorten the time required to reach the therapeutic threshold with an acceptable tolerance: preliminary results. Clin Endocrinol (Oxf). 2006;64(1):110–113.
82. Nader, N, Raverot, G, Emptoz-Bonneton, A, et al. Mitotane has an estrogenic effect on sex hormone-binding globulin and corticosteroid-binding globulin in humans. J Clin Endocrinol Metab. 2006;91(6):2165–2170.
83. Dickstein, G, Shechner, C, Arad, E, et al. Is there a role for low doses of mitotane (o,p′-DDD) as adjuvant therapy in adrenocortical carcinoma? J Clin Endocrinol Metab. 1998;83(9):3100–3103.
84. Fareau, GG, Lopez, A, Stava, C, et al. Systemic chemotherapy for adrenocortical carcinoma: comparative responses to conventional first-line therapies. Anticancer Drugs. 2008;19(6):637–644.
85. Berruti, A, Terzolo, M, Sperone, P, et al. Etoposide, doxorubicin and cisplatin plus mitotane in the treatment of advanced adrenocortical carcinoma: a large prospective phase II trial. Endocr Relat Cancer. 2005;12(3):657–666.
86. Khan, TS, Imam, H, Juhlin, C, et al. Streptozocin and o,p′DDD in the treatment of adrenocortical cancer patients: long-term survival in its adjuvant use. Ann Oncol. 2000;11(10):1281–1287.
87. Allolio, B, Hahner, S, Weismann, D, et al. Management of adrenocortical carcinoma. Clin Endocrinol (Oxf). 2004;60:273–287.
88. Ronchi CL, Sbiera S, Adam P, et al: ERCC1 expression in adrenocortical cancer: relationship with baseline characteristics and response to platinum-based chemotherapy, Endocr Relet Cancer, in press, 2009.
89. Kirschner, LS. Emerging treatment strategies for adrenocortical carcinoma: a new hope. J Clin Endocrinol Metab. 2005;91:14–21.
90. Quinkler, M, Hahner, S, Wortmann, S, et al. Treatment of advanced adrenocortical carcinoma with erlotinib plus gemcitabine. J Clin Endocrinol Metab. 2008;93(6):2057–2062.
91. Samnotra V, Vassilopoulou-Sellin R, Fojo AT: A phase II trial of gefitinib monotherapy in patients with unresectable adrenocortical carcinoma (ACC), Proc Ann Soc Clin Oncol:abstract no 15527, 2007.
92. Chacon, R, Tossen, G, Loria, FS, et al. CASE 2. Response in a patient with metastatic adrenal cortical carcinoma with thalidomide. J Clin Oncol. 2005;23(7):1579–1580.