[level-membership-for-basic-science-category]
Chapter 18 Infectious Diseases
Introduction to Antibiotics





Suggestions
 Know which category the individual drugs belong to, and learn the category as a whole. For example, ceftriaxone is a third-generation cephalosporin. Therefore learn about cephalosporins in general, and then know the general differences among first-, second-, third-, and fourth-generation cephalosporins. In some circumstances there might be one or two extra important bits of information pertaining to individual drugs that are important to learn.
Know which category the individual drugs belong to, and learn the category as a whole. For example, ceftriaxone is a third-generation cephalosporin. Therefore learn about cephalosporins in general, and then know the general differences among first-, second-, third-, and fourth-generation cephalosporins. In some circumstances there might be one or two extra important bits of information pertaining to individual drugs that are important to learn.





Important Considerations
 It is required that you learn the mechanism of action (MOA) of a drug and also the mechanism of resistance, which enables an organism to live in the presence of the drug. This is important because when you are selecting an antibiotic, if your first choice does not work, you will have a better understanding of why it did not work and will be better educated to select a different antibiotic that will have a greater probability of killing the organism. For example, if a penicillin (a β-lactam) was given and the bug is known to produce β-lactamase, a third-generation cephalosporin (another β-lactam) might be a poor next choice.
It is required that you learn the mechanism of action (MOA) of a drug and also the mechanism of resistance, which enables an organism to live in the presence of the drug. This is important because when you are selecting an antibiotic, if your first choice does not work, you will have a better understanding of why it did not work and will be better educated to select a different antibiotic that will have a greater probability of killing the organism. For example, if a penicillin (a β-lactam) was given and the bug is known to produce β-lactamase, a third-generation cephalosporin (another β-lactam) might be a poor next choice. Make sure that the drug you select can get to the tissue in which the infection is located. Special examples include the following:
Make sure that the drug you select can get to the tissue in which the infection is located. Special examples include the following:




Viruses are technically not alive, so antivirals are usually not referred to as antimicrobials.
Advanced Killing Techniques
Knowledge of pharmacokinetics is required to enable a clinician to be really good at knowing how to kill off an infection. Understanding some very important fundamental concepts are required (Figure 18-1).
 Minimum inhibitory concentration (MIC): The MIC of an antimicrobial is the minimum concentration that will inhibit growth of a pathogen. Obviously, it is desirable to have the concentration in the patient’s infected tissues to be greater than the MIC. If drug A has a lower MIC than drug B, then drug A kills the pathogen at a lower concentration of drug and would therefore be better at killing that particular pathogen, assuming all other factors are identical. When the MIC level of a drug for a given pathogen is low, the pathogen is considered sensitive to the drug (i.e., the drug will kill it). When the MIC is a moderate value, the pathogen’s sensitivity to the drug will be intermediate, and when the MIC is high, the pathogen will be resistant to that drug. Laboratory values often report sensitivities as S, I, or R to report sensitive, intermediate, and resistant, respectively.
Minimum inhibitory concentration (MIC): The MIC of an antimicrobial is the minimum concentration that will inhibit growth of a pathogen. Obviously, it is desirable to have the concentration in the patient’s infected tissues to be greater than the MIC. If drug A has a lower MIC than drug B, then drug A kills the pathogen at a lower concentration of drug and would therefore be better at killing that particular pathogen, assuming all other factors are identical. When the MIC level of a drug for a given pathogen is low, the pathogen is considered sensitive to the drug (i.e., the drug will kill it). When the MIC is a moderate value, the pathogen’s sensitivity to the drug will be intermediate, and when the MIC is high, the pathogen will be resistant to that drug. Laboratory values often report sensitivities as S, I, or R to report sensitive, intermediate, and resistant, respectively.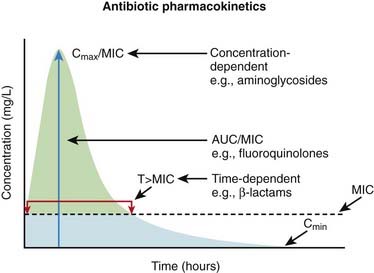
Now, exactly how the concentration of the antimicrobial stays above the MIC in the body is very important and is different for different drugs. The most important concepts are illustrated in Figure 18-1 and include:
 Time dependence is the total length of time that a drug level stays above the MIC. What matters is the total duration that the concentration is above the MIC. These drugs are called time-dependent antimicrobials. β-Lactams (penicillins, cephalosporins, and carbapenems) all fall into this category. Note that it does not matter if the drug level is just barely above the MIC or is 10 times the MIC.
Time dependence is the total length of time that a drug level stays above the MIC. What matters is the total duration that the concentration is above the MIC. These drugs are called time-dependent antimicrobials. β-Lactams (penicillins, cephalosporins, and carbapenems) all fall into this category. Note that it does not matter if the drug level is just barely above the MIC or is 10 times the MIC.
Penicillins




Moa (Mechanism of Action)





Cell Wall Destruction
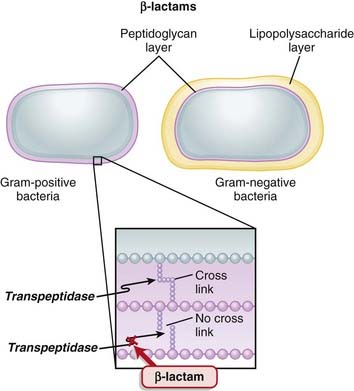
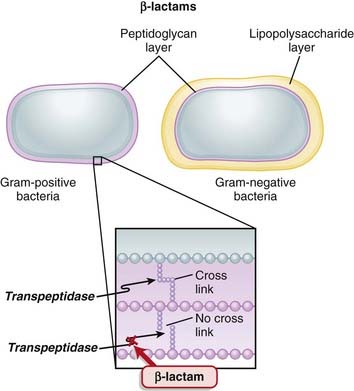
 Transpeptidases are enzymes that cross-link peptidoglycan molecules in bacterial cell walls. Cross-linking these molecules gives strength to the cell wall.
Transpeptidases are enzymes that cross-link peptidoglycan molecules in bacterial cell walls. Cross-linking these molecules gives strength to the cell wall. β-Lactams work by inhibiting transpeptidase, preventing it from forming cross-links. This results in a bacterium with a structurally deficient cell wall, typically leading to bacterial lysis (Figure 18-2).
β-Lactams work by inhibiting transpeptidase, preventing it from forming cross-links. This results in a bacterium with a structurally deficient cell wall, typically leading to bacterial lysis (Figure 18-2).





Mechanisms of Resistance
 Gram-negative bacteria, as described previously, inherently have an outer protective lipopolysaccharide layer that guards the peptidoglycan layer from attack by some β-lactams.
Gram-negative bacteria, as described previously, inherently have an outer protective lipopolysaccharide layer that guards the peptidoglycan layer from attack by some β-lactams. The main mechanisms of resistance to β-lactams include the following:
The main mechanisms of resistance to β-lactams include the following:
 Production of β-lactamase, which enzymatically destroys the four-carbon β-lactam ring
Production of β-lactamase, which enzymatically destroys the four-carbon β-lactam ring
Pharmacokinetics
 Eighty percent of penicillin is cleared by the kidneys within 4 hours. Therefore it is very quickly eliminated from the body, which is an undesirable characteristic if the goal is to expose the bacterial infection to prolonged concentrations of the drug.
Eighty percent of penicillin is cleared by the kidneys within 4 hours. Therefore it is very quickly eliminated from the body, which is an undesirable characteristic if the goal is to expose the bacterial infection to prolonged concentrations of the drug.











Important Notes


FYI
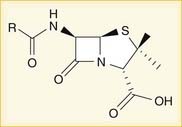
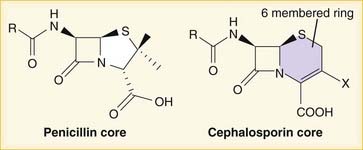
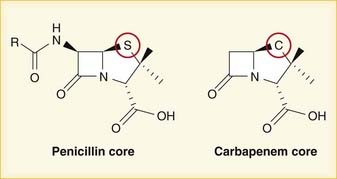
 A lactam is a chemical ring. A β-lactam is a four-molecule ring (three carbons and one nitrogen) and is the nucleus of the penicillin molecule. The penicillin nucleus is shown in Figure 18-3. Modifications to the five-membered ring are the basis on which cephalosporins and carbapenems (two other β-lactam antibiotics) were derived.
A lactam is a chemical ring. A β-lactam is a four-molecule ring (three carbons and one nitrogen) and is the nucleus of the penicillin molecule. The penicillin nucleus is shown in Figure 18-3. Modifications to the five-membered ring are the basis on which cephalosporins and carbapenems (two other β-lactam antibiotics) were derived.








Cephalosporins
Prototypes and common drugs


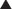

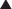

Moa (Mechanism of Action)





Cell Wall Destruction




Mechanisms of Resistance
 The main mechanisms of resistance to β-lactams include the following:
The main mechanisms of resistance to β-lactams include the following:
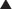
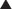 Production of β-lactamase, which enzymatically destroys the four-carbon β-lactam ring
Production of β-lactamase, which enzymatically destroys the four-carbon β-lactam ring










Side Effects



Important Notes

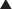
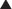

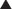
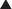

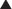

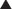
FYI
 Note how the four-membered ring (the β-lactam) is the same as in penicillins; however, the other ring is six-membered, whereas in penicillins it is five-membered. The diagram shows the core nucleus of cephalosporins. There are currently four generations of cephalosporins (Figure 18-4).
Note how the four-membered ring (the β-lactam) is the same as in penicillins; however, the other ring is six-membered, whereas in penicillins it is five-membered. The diagram shows the core nucleus of cephalosporins. There are currently four generations of cephalosporins (Figure 18-4).


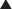
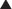

Carbapenems


Moa (Mechanism of Action)





Cell Wall Destruction


Mechanisms of Resistance
 The main mechanisms of resistance to β-lactams include the following:
The main mechanisms of resistance to β-lactams include the following:
 Production of β-lactamase, which enzymatically destroys the four-carbon β-lactam ring
Production of β-lactamase, which enzymatically destroys the four-carbon β-lactam ring















Important Notes
 MRSA is an important resistant strain of S. aureus. It is not sensitive to carbapenems. It is treated with vancomycin.
MRSA is an important resistant strain of S. aureus. It is not sensitive to carbapenems. It is treated with vancomycin. VRE is another important resistant strain. About 50% of VRE infections are sensitive to carbapenems.
VRE is another important resistant strain. About 50% of VRE infections are sensitive to carbapenems.



FYI
 Carbapenems differ structurally from penicillins in that the five-membered ring that is attached to the β-lactam ring contains a carbon atom instead of a sulfur atom. The name carbapenem comes from carbon and penicillin. Figure 18-6 shows the core structure of a carbapenem but does not show the side chains.
Carbapenems differ structurally from penicillins in that the five-membered ring that is attached to the β-lactam ring contains a carbon atom instead of a sulfur atom. The name carbapenem comes from carbon and penicillin. Figure 18-6 shows the core structure of a carbapenem but does not show the side chains.
Glycopeptides


Moa (Mechanism of Action)
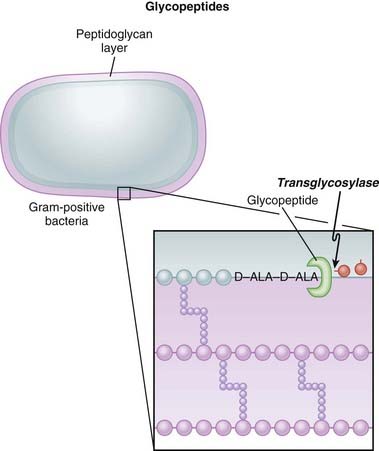
 Glycopeptides inhibit cell wall synthesis by attaching to the end of the peptidoglycan precursor units (a short four– or five–amino acid sequence called the d-alanyl-d-alanine [D-ALA-D-ALA] terminus) that are required to be laid down into the matrix. This step is catalyzed by transglycosylase. Because the glycopeptide binds to the end of the precursor, the precursor is not released from the carrier and thus peptidoglycan synthesis stops (Figure 18-7).
Glycopeptides inhibit cell wall synthesis by attaching to the end of the peptidoglycan precursor units (a short four– or five–amino acid sequence called the d-alanyl-d-alanine [D-ALA-D-ALA] terminus) that are required to be laid down into the matrix. This step is catalyzed by transglycosylase. Because the glycopeptide binds to the end of the precursor, the precursor is not released from the carrier and thus peptidoglycan synthesis stops (Figure 18-7).





Pharmacokinetics











Side Effects
 Flushing: When vancomycin is administered quickly, the blood pressure can fall because of histamine release. Therefore vancomycin must be administered slowly (usually over 1 hour). The flushing caused by histamine release has been called red man syndrome and is not an allergy but a predictable response to rapidly administered vancomycin.
Flushing: When vancomycin is administered quickly, the blood pressure can fall because of histamine release. Therefore vancomycin must be administered slowly (usually over 1 hour). The flushing caused by histamine release has been called red man syndrome and is not an allergy but a predictable response to rapidly administered vancomycin.


Important Notes
 Although vancomycin can kill S. aureus organisms that are resistant to cloxacillin (or methicillin [i.e., MRSA]), cloxacillin has a lower MIC than vancomycin for strains that are β-lactam susceptible. Therefore cloxacillin is a better killer of S. aureus when there is no resistance. Vancomycin is not a stronger antibiotic. It is simply a different but paradoxically slightly weaker one that avoids β-lactam resistance.
Although vancomycin can kill S. aureus organisms that are resistant to cloxacillin (or methicillin [i.e., MRSA]), cloxacillin has a lower MIC than vancomycin for strains that are β-lactam susceptible. Therefore cloxacillin is a better killer of S. aureus when there is no resistance. Vancomycin is not a stronger antibiotic. It is simply a different but paradoxically slightly weaker one that avoids β-lactam resistance.




Fluoroquinolones


MOA (Mechanism of Action)
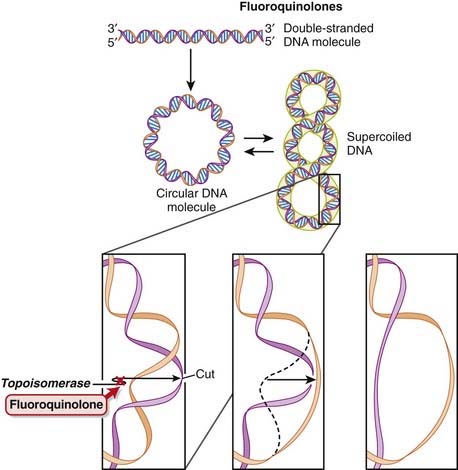
 DNA is normally supercoiled. Supercoiled DNA is under too much tension to be separated, so an extra step is required before replication and transcription can occur. DNA gyrase relaxes supercoiled DNA by cutting it, allowing rotation to occur, and then reattaching it.
DNA is normally supercoiled. Supercoiled DNA is under too much tension to be separated, so an extra step is required before replication and transcription can occur. DNA gyrase relaxes supercoiled DNA by cutting it, allowing rotation to occur, and then reattaching it.





Pharmacokinetics
 Fluoroquinolones can enter human cells easily and therefore are often used to treat intracellular pathogens.
Fluoroquinolones can enter human cells easily and therefore are often used to treat intracellular pathogens.











Important Notes




FYI
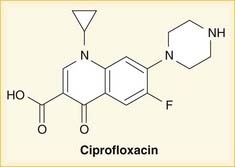
 As the name suggests, fluoroquinolones possess a fluorine ion. They all contain two six-membered rings. Ciprofloxacin is shown in the diagram (Figure 18-9). Earlier generations of fluoroquinolones were not fluorinated and were simply called quinolones. They are infrequently used now.
As the name suggests, fluoroquinolones possess a fluorine ion. They all contain two six-membered rings. Ciprofloxacin is shown in the diagram (Figure 18-9). Earlier generations of fluoroquinolones were not fluorinated and were simply called quinolones. They are infrequently used now.


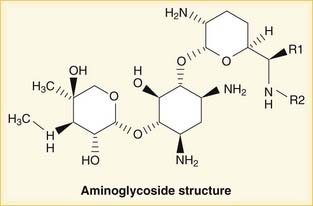
Aminoglycosides


MOA (Mechanism of Action)
 The production of proteins from mRNA is called translation; this step requires ribosomes, transfer RNA (tRNA), and messenger RNA (mRNA) (Figure 18-10).
The production of proteins from mRNA is called translation; this step requires ribosomes, transfer RNA (tRNA), and messenger RNA (mRNA) (Figure 18-10).
 The ribosomes are different sizes:
The ribosomes are different sizes:

 There are two theories about how aminoglycosides work:
There are two theories about how aminoglycosides work:


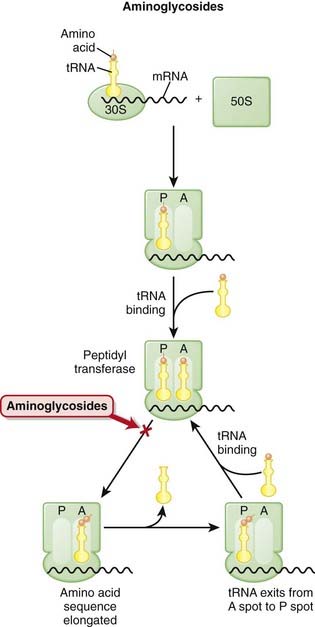


Pharmacokinetics
 Penetration of biologic membranes is poor because of the drug’s polar structure. Therefore all aminoglycosides are poorly absorbed in the GI tract and are not administered orally, and intracellular concentrations are usually low.
Penetration of biologic membranes is poor because of the drug’s polar structure. Therefore all aminoglycosides are poorly absorbed in the GI tract and are not administered orally, and intracellular concentrations are usually low.












Important Notes
 Serum levels of aminoglycosides must be monitored. Peak and trough levels help determine required doses.
Serum levels of aminoglycosides must be monitored. Peak and trough levels help determine required doses.

Advanced
 Neuromuscular blockade occurs because of a reduction in acetylcholine release. These drugs do not paralyze patients, but under anesthesia in patients who are already receiving a neuromuscular blocking drug (such as rocuronium or vecuronium), the duration of blockade can be longer than normal. Other conditions in which neuromuscular blockade could become problematic are:
Neuromuscular blockade occurs because of a reduction in acetylcholine release. These drugs do not paralyze patients, but under anesthesia in patients who are already receiving a neuromuscular blocking drug (such as rocuronium or vecuronium), the duration of blockade can be longer than normal. Other conditions in which neuromuscular blockade could become problematic are:

FYI
 Nomenclature:
Nomenclature:
 The suffix –mycin is for drugs that are derived from Streptomyces, a genus of bacteria commonly found in soil. Streptomycin is named after Streptomyces, but it is no longer commonly used.
The suffix –mycin is for drugs that are derived from Streptomyces, a genus of bacteria commonly found in soil. Streptomycin is named after Streptomyces, but it is no longer commonly used.







Lincosamides


MOA (Mechanism of Action)
 The production of proteins from mRNA is called translation; this step requires ribosomes, tRNA, and mRNA.
The production of proteins from mRNA is called translation; this step requires ribosomes, tRNA, and mRNA. mRNA contains the sequencing code based on DNA; tRNA contains single amino acids and binds to a three-base sequence of mRNA. Ribosomes are like “assembly line” processing machinery, bringing the mRNA and tRNA together to assemble sequences of amino acids (Figure 18-12).
mRNA contains the sequencing code based on DNA; tRNA contains single amino acids and binds to a three-base sequence of mRNA. Ribosomes are like “assembly line” processing machinery, bringing the mRNA and tRNA together to assemble sequences of amino acids (Figure 18-12). The ribosomes are different sizes:
The ribosomes are different sizes:




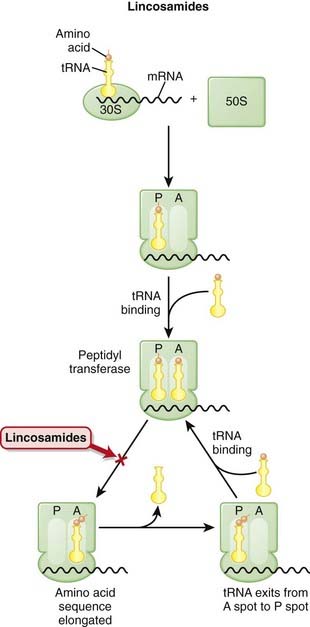
Mechanisms of Resistance
 Mutation of the ribosomal receptor site or modification of the receptor by a methylase enzyme results in decreased binding to the 50S subunit.
Mutation of the ribosomal receptor site or modification of the receptor by a methylase enzyme results in decreased binding to the 50S subunit.

Pharmacokinetics










Side Effects
 Pseudomembranous colitis (aka C. difficile colitis or “C diff” colitis) may occur. Bacterial flora of the colon changes with antibiotic administration. Some antibiotics (clindamycin is the worst offender) wipe out the normal flora and enable pathologic flora to grow, resulting in inflammation of the colon and diarrhea. This is a major problem and requires a second antibiotic (usually metronidazole or vancomycin) to be used to treat the diarrhea, which can be moderate to severe in intensity. C. difficile infections can sometimes be very difficult to eradicate.
Pseudomembranous colitis (aka C. difficile colitis or “C diff” colitis) may occur. Bacterial flora of the colon changes with antibiotic administration. Some antibiotics (clindamycin is the worst offender) wipe out the normal flora and enable pathologic flora to grow, resulting in inflammation of the colon and diarrhea. This is a major problem and requires a second antibiotic (usually metronidazole or vancomycin) to be used to treat the diarrhea, which can be moderate to severe in intensity. C. difficile infections can sometimes be very difficult to eradicate.Important Notes





FYI
 The S in 50S and 23S refers to Svedberg units. A Svedberg unit is a measurement of time and equals 10-13 seconds. It is used to describe speed of sedimentation during centrifuging. Combining two molecules (30S + 50S) and predicting the combined Svedberg units are not simply an additive process because the surface area changes. Thus in the bacterial ribosome, 30S + 50S = 70S (and not 80).
The S in 50S and 23S refers to Svedberg units. A Svedberg unit is a measurement of time and equals 10-13 seconds. It is used to describe speed of sedimentation during centrifuging. Combining two molecules (30S + 50S) and predicting the combined Svedberg units are not simply an additive process because the surface area changes. Thus in the bacterial ribosome, 30S + 50S = 70S (and not 80).Tetracyclines


MOA (Mechanism of Action)
 The production of proteins from mRNA is called translation; this step requires ribosomes, transfer RNA (tRNA), and messenger RNA (mRNA) (Figure 18-13).
The production of proteins from mRNA is called translation; this step requires ribosomes, transfer RNA (tRNA), and messenger RNA (mRNA) (Figure 18-13).
 The ribosomes are different sizes:
The ribosomes are different sizes:






Mechanisms of Resistance
 There are three commonly recognized mechanisms by which tetracycline resistance is conferred to organisms:
There are three commonly recognized mechanisms by which tetracycline resistance is conferred to organisms:
Pharmacokinetics
 Tetracyclines are bound and inactivated by divalent cations such as calcium and magnesium, and co-ingestion of these agents (e.g., in the form of calcium supplements or antacids) interferes with the effectiveness of tetracyclines. Therefore, tetracyclines should be taken on an empty stomach.
Tetracyclines are bound and inactivated by divalent cations such as calcium and magnesium, and co-ingestion of these agents (e.g., in the form of calcium supplements or antacids) interferes with the effectiveness of tetracyclines. Therefore, tetracyclines should be taken on an empty stomach.





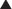



Side Effects







Important Notes




Advanced
 Porphyria cutanea tarda is a form of porphyria that affects the skin. Porphyria is a group of diseases caused by enzyme deficiencies resulting in accumulation of porphyrins, which are precursors to heme. Tetracycline can cause a rare form of phototoxicity that mimics porphyria and is called pseudoporphyria (but does not involve porphyrins).
Porphyria cutanea tarda is a form of porphyria that affects the skin. Porphyria is a group of diseases caused by enzyme deficiencies resulting in accumulation of porphyrins, which are precursors to heme. Tetracycline can cause a rare form of phototoxicity that mimics porphyria and is called pseudoporphyria (but does not involve porphyrins).









Macrolides


MOA (Mechanism OF Action)
 The production of proteins from mRNA is called translation; this step requires ribosomes, transfer RNA (tRNA), and messenger RNA (mRNA) (Figure 18-14).
The production of proteins from mRNA is called translation; this step requires ribosomes, transfer RNA (tRNA), and messenger RNA (mRNA) (Figure 18-14).
 The ribosomes are different sizes:
The ribosomes are different sizes:









Pharmacokinetics
 Half-lives of erythromycin, clarithromycin, and azithromycin are 1.5, 6, and 68 hours, and administration is four times daily, twice daily, and once daily, respectively. At high doses, clarithromycin is sometimes administered once a day.
Half-lives of erythromycin, clarithromycin, and azithromycin are 1.5, 6, and 68 hours, and administration is four times daily, twice daily, and once daily, respectively. At high doses, clarithromycin is sometimes administered once a day.









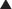

Indications
 In addition to antibacterial activity, erythromycin specifically is used to enhance GI motility. Through direct stimulation of the GI tract, this mechanism is responsible both for the enhanced motility that is of benefit in patients who have dysmotility and also, unfortunately, for the GI intolerance (side effects) in patients who have normal motility.
In addition to antibacterial activity, erythromycin specifically is used to enhance GI motility. Through direct stimulation of the GI tract, this mechanism is responsible both for the enhanced motility that is of benefit in patients who have dysmotility and also, unfortunately, for the GI intolerance (side effects) in patients who have normal motility.





Important Notes








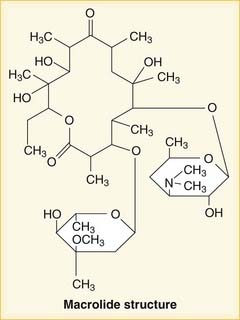
Oxazolidinones

MOA (Mechanism of Action)
 The production of proteins from mRNA is called translation; this step requires ribosomes, transfer RNA (tRNA), and messenger RNA (mRNA) (Figure 18-16).
The production of proteins from mRNA is called translation; this step requires ribosomes, transfer RNA (tRNA), and messenger RNA (mRNA) (Figure 18-16).
 The ribosomes are different sizes:
The ribosomes are different sizes:











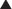







Important Notes
 Linezolids were approved in 2000, and linezolid resistance was first reported in 2001. New drugs often have low rates of resistance, but over time, increased evolutionary pressure from exposure of the drug to bacteria results in increased levels of resistance.
Linezolids were approved in 2000, and linezolid resistance was first reported in 2001. New drugs often have low rates of resistance, but over time, increased evolutionary pressure from exposure of the drug to bacteria results in increased levels of resistance.


Sulfonamides and Other Folate Synthesis Inhibitors

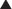


MOA (Mechanism of Action)
 Most bacteria must synthesize their own folate for survival; they cannot use external sources. In contrast, humans do not synthesize folate but rather obtain it through the diet.
Most bacteria must synthesize their own folate for survival; they cannot use external sources. In contrast, humans do not synthesize folate but rather obtain it through the diet.







Pharmacokinetics
 Metabolism of sulfonamides is via the liver and excretion by the kidney. In patients with advanced renal dysfunction, doses should be reduced.
Metabolism of sulfonamides is via the liver and excretion by the kidney. In patients with advanced renal dysfunction, doses should be reduced.







Side Effects
 Precipitation in urine: Sulfa molecules might precipitate at neutral or acidic pH, leading to crystalluria (crystals in the urine), and possibly hematuria (blood in urine secondary to reaction from crystals).
Precipitation in urine: Sulfa molecules might precipitate at neutral or acidic pH, leading to crystalluria (crystals in the urine), and possibly hematuria (blood in urine secondary to reaction from crystals).


Rare and Serious
 Stevens-Johnson syndrome is a rare but very serious (life-threatening) hypersensitivity reaction involving skin and mucous membranes.
Stevens-Johnson syndrome is a rare but very serious (life-threatening) hypersensitivity reaction involving skin and mucous membranes.

Important Notes

 Common clinical infections treated by sulfonamides include the following:
Common clinical infections treated by sulfonamides include the following:










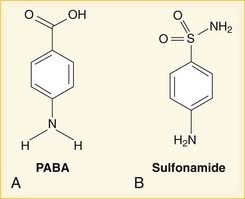
Metronidazole

MOA (Mechanism of Action)
 Metronidazole is a prodrug. The nitrogen group must be reduced (addition of electron) before the chemical obtains its antiinfective function. It is reduced by a nitroreductase enzyme called a ferredoxin (an iron- and sulfur-containing enzyme). The extra nitrogen side chain is reduced in this reaction.
Metronidazole is a prodrug. The nitrogen group must be reduced (addition of electron) before the chemical obtains its antiinfective function. It is reduced by a nitroreductase enzyme called a ferredoxin (an iron- and sulfur-containing enzyme). The extra nitrogen side chain is reduced in this reaction.

Mechanisms of Resistance
 Some bacteria are facultatively anaerobic (can live with or without oxygen) or are microaerophiles (can live in the presence of very little oxygen). In these bacteria, the genes required to produce ferredoxin can be switched off; this would result in metronidazole not becoming reduced, and therefore it would remain in the inactive prodrug state.
Some bacteria are facultatively anaerobic (can live with or without oxygen) or are microaerophiles (can live in the presence of very little oxygen). In these bacteria, the genes required to produce ferredoxin can be switched off; this would result in metronidazole not becoming reduced, and therefore it would remain in the inactive prodrug state.Pharmacokinetics





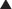


Contraindications







FYI
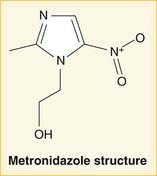
 Metronidazole contains the five-member imidazole ring (three carbons + two nitrogen atoms), but there is a nitrogen side chain; it is therefore a nitroimidazole. It is in a drug category all on its own (Figure 18-19).
Metronidazole contains the five-member imidazole ring (three carbons + two nitrogen atoms), but there is a nitrogen side chain; it is therefore a nitroimidazole. It is in a drug category all on its own (Figure 18-19).


Introduction to Antimycobacterials
The main anti-TB drugs and their abbreviations include the following:
Mutation Rate




Standard Regimen
 Treatment involves an induction phase (high dose, more drugs) followed by a maintenance phase (fewer drugs).
Treatment involves an induction phase (high dose, more drugs) followed by a maintenance phase (fewer drugs).
Ethambutol

MOA (Mechanism of Action) (Figure 18-20)



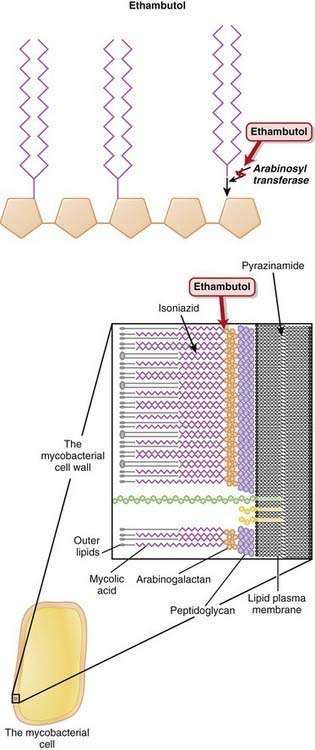





Side Effects

 Neurologic:
Neurologic:
 Optic neuritis: <1% occurrence. Inflammation of the optic nerve results in decreased visual acuity (blurred vision). Recovery usually occurs after discontinuation of the drug. This is a dose-related effect and occurs more commonly with higher doses. One theory for the MOA of ocular toxicity is related to its zinc chelating effect, but the mechanism has not been fully established.
Optic neuritis: <1% occurrence. Inflammation of the optic nerve results in decreased visual acuity (blurred vision). Recovery usually occurs after discontinuation of the drug. This is a dose-related effect and occurs more commonly with higher doses. One theory for the MOA of ocular toxicity is related to its zinc chelating effect, but the mechanism has not been fully established.






Isoniazid
Description
Isoniazid is a first-line anti-TB drug. It is used in combination with other anti-TB drugs.

MOA (Mechanism of Action) (Figure 18-21)



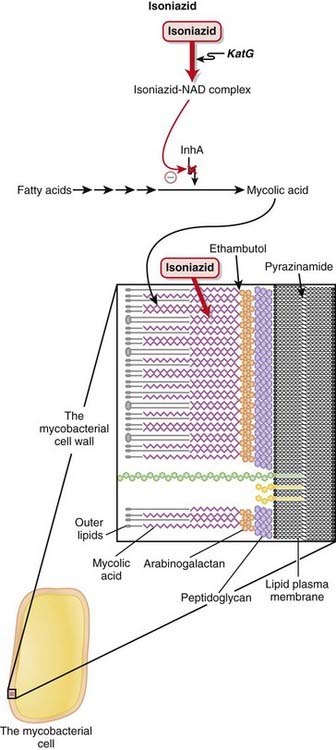

Pharmacokinetics
 Isoniazid is distributed in serum, in cerebrospinal fluid (CSF), and within caseous granulomas (which often surround TB). Caseous means having a cheeselike consistency.
Isoniazid is distributed in serum, in cerebrospinal fluid (CSF), and within caseous granulomas (which often surround TB). Caseous means having a cheeselike consistency.

Side Effects
 Hepatitis, which can be initially asymptomatic and detected only by hepatic enzyme elevation. Significant enzyme elevation might mandate that isoniazid be discontinued in that patient. Note that the term hepatitis here does not refer to viral hepatitis, but rather a noninfectious inflammation of the liver that is directly drug induced.
Hepatitis, which can be initially asymptomatic and detected only by hepatic enzyme elevation. Significant enzyme elevation might mandate that isoniazid be discontinued in that patient. Note that the term hepatitis here does not refer to viral hepatitis, but rather a noninfectious inflammation of the liver that is directly drug induced.


Important Notes
 Baseline measurements of hepatic enzymes are required because of the risk of hepatitis. Repeat measurements as follows:
Baseline measurements of hepatic enzymes are required because of the risk of hepatitis. Repeat measurements as follows:







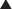
Pyrazinamide

MOA (Mechanism of Action) (Figure 18-22)




Indications














Rifamycins


MOA (Mechanism of Action) (Figure 18-23)
 RNA polymerase reads DNA to produce an mRNA copy of the genetic code. Without RNA, protein synthesis cannot occur.
RNA polymerase reads DNA to produce an mRNA copy of the genetic code. Without RNA, protein synthesis cannot occur.


 Rifampin is highly lipophilic. This is extremely important because it confers the property that the drug can easily cross lipophilic membranes:
Rifampin is highly lipophilic. This is extremely important because it confers the property that the drug can easily cross lipophilic membranes:
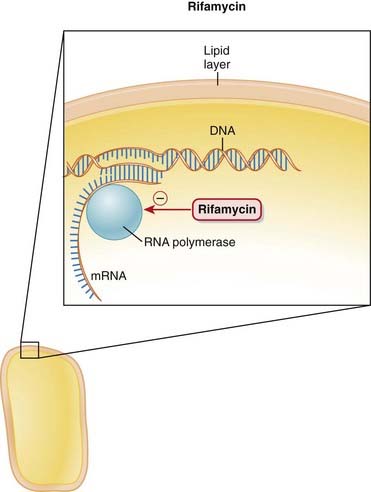


Pharmacokinetics
 Rifamycins are primarily metabolized by the liver and eliminated in the bile. Does adjustments are not required in liver or renal dysfunction.
Rifamycins are primarily metabolized by the liver and eliminated in the bile. Does adjustments are not required in liver or renal dysfunction.








Side Effects
Common and Mild
 Discoloration of secretions: Because of its lipophilic nature, it enters secretions readily and colors them orange or red. Such secretions include tears, urine, sweat, stool, sputum, and saliva. Contact lenses can be stained. Patients should be warned about this so that they do not become unnecessarily alarmed when it happens.
Discoloration of secretions: Because of its lipophilic nature, it enters secretions readily and colors them orange or red. Such secretions include tears, urine, sweat, stool, sputum, and saliva. Contact lenses can be stained. Patients should be warned about this so that they do not become unnecessarily alarmed when it happens.



Important Notes
 Staphylococcal infections (S. epidermidis and S. aureus) are commonly associated with biofilm production. Because of the lipophilic nature of rifampin, it absorbs into and crosses biofilms and can be more effective against these infections.
Staphylococcal infections (S. epidermidis and S. aureus) are commonly associated with biofilm production. Because of the lipophilic nature of rifampin, it absorbs into and crosses biofilms and can be more effective against these infections.

Advanced
 Many guidelines recommend rifampin as a second drug to be used in combination for some staphylococcal and streptococcal infections. However, a meta-analysis found that only orthopedic infections (bone, joint, or joint hardware) with Staphylococcus showed a significant improvement in cure rates with combination treatment.
Many guidelines recommend rifampin as a second drug to be used in combination for some staphylococcal and streptococcal infections. However, a meta-analysis found that only orthopedic infections (bone, joint, or joint hardware) with Staphylococcus showed a significant improvement in cure rates with combination treatment.




CCR5 Antagonists

MOA (Mechanism of Action)











Side Effects
 Antiretrovirals are typically administered in combination regimens, which can make it unclear how each individual drug contributes to side effects.
Antiretrovirals are typically administered in combination regimens, which can make it unclear how each individual drug contributes to side effects.


Important Notes
 In clinical trials, virologic failure typically occurred in patients exhibiting a switch in HIV-1 from CCR5 to CXCR4 or dual-mixed. However, once patients had discontinued maraviroc, they typically reverted back to CCR5.
In clinical trials, virologic failure typically occurred in patients exhibiting a switch in HIV-1 from CCR5 to CXCR4 or dual-mixed. However, once patients had discontinued maraviroc, they typically reverted back to CCR5.






Fusion Inhibitors


MOA (Mechanism of Action) (Figure 18-25)
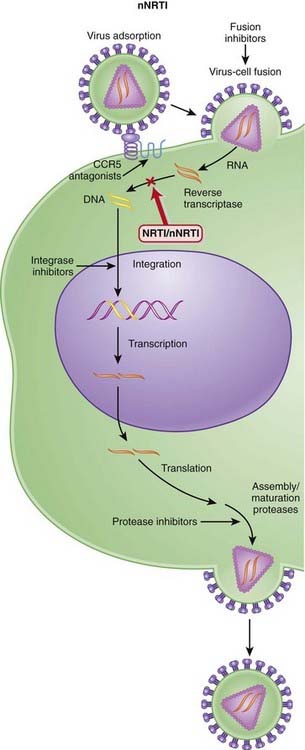
 Several steps are involved in the replication of HIV. The virus must fuse to the host cell, uncoat, enter and be transcribed by reverse transcriptase, become incorporated into the host genome, and be transcribed to viral RNA, which is then translated into polyproteins.
Several steps are involved in the replication of HIV. The virus must fuse to the host cell, uncoat, enter and be transcribed by reverse transcriptase, become incorporated into the host genome, and be transcribed to viral RNA, which is then translated into polyproteins.
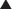
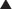

Indications





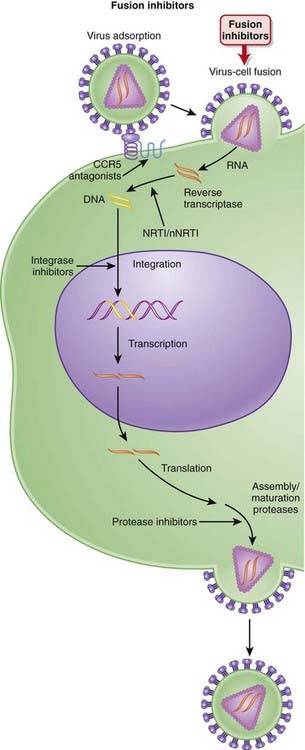
Integrase Inhibitors

MOA (Mechanism of Action)
 Several steps are involved in the replication of HIV. The virus must fuse to the host cell, uncoat, enter and be transcribed by reverse transcriptase, become incorporated into the host genome, and be transcribed to viral RNA, which is then translated into polyproteins (Figure 18-26).
Several steps are involved in the replication of HIV. The virus must fuse to the host cell, uncoat, enter and be transcribed by reverse transcriptase, become incorporated into the host genome, and be transcribed to viral RNA, which is then translated into polyproteins (Figure 18-26).


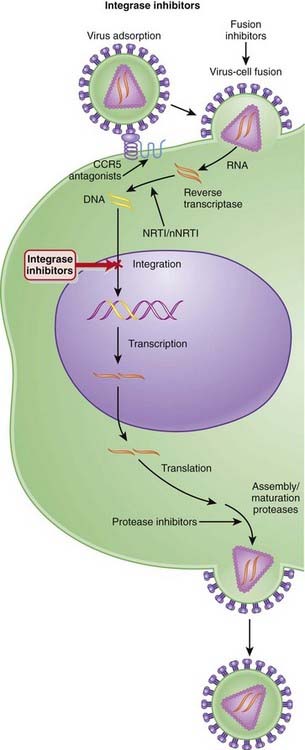


Pharmacokinetics
 The main route of elimination for raltegravir is hepatic; however, dose adjustments are not required in patients with mild to moderate hepatic impairment. Adjustments in severe hepatic impairment have not been studied. Because renal elimination plays a relatively minor role, no adjustments are required in patients with renal impairment.
The main route of elimination for raltegravir is hepatic; however, dose adjustments are not required in patients with mild to moderate hepatic impairment. Adjustments in severe hepatic impairment have not been studied. Because renal elimination plays a relatively minor role, no adjustments are required in patients with renal impairment.


Side Effects








Nucleoside Reverse Transcriptase Inhibitors (NRTIs)



MOA (Mechanism of Action)
 Several steps are involved in the replication of HIV. The virus must fuse to the host cell, uncoat, enter and be transcribed by reverse transcriptase, become incorporated into the host genome, and be transcribed to viral RNA, which is then translated into polyproteins.
Several steps are involved in the replication of HIV. The virus must fuse to the host cell, uncoat, enter and be transcribed by reverse transcriptase, become incorporated into the host genome, and be transcribed to viral RNA, which is then translated into polyproteins.




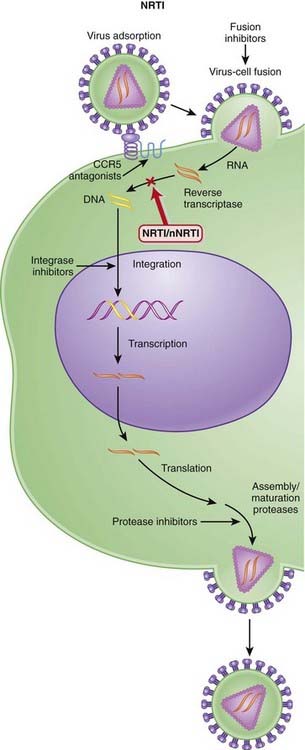


Pharmacokinetics






Side Effects
 Myalgia: One of the most common and earliest side effects associated with NRTIs, myalgia was also the first indication of the mitochondrial toxicity that has become associated with these agents. The mitochondrial toxicity is believed to be caused by the inhibition of DNA polymerase by the NRTI.
Myalgia: One of the most common and earliest side effects associated with NRTIs, myalgia was also the first indication of the mitochondrial toxicity that has become associated with these agents. The mitochondrial toxicity is believed to be caused by the inhibition of DNA polymerase by the NRTI.

Serious









Important Notes
 Lamivudine, emtricitabine, and tenofovir are also active against hepatitis B virus. This presents an opportunity to treat two indications (HIV and hepatitis B) with one drug, but also means that care must be taken when withdrawing these agents from hepatitis B co-infected patients. These patients may experience a rebound and worsening of hepatitis B.
Lamivudine, emtricitabine, and tenofovir are also active against hepatitis B virus. This presents an opportunity to treat two indications (HIV and hepatitis B) with one drug, but also means that care must be taken when withdrawing these agents from hepatitis B co-infected patients. These patients may experience a rebound and worsening of hepatitis B.

Advanced

Drug Interactions
 Didanosine is easily degraded in an acidic environment and is therefore formulated with buffers such as calcium carbonate and magnesium hydroxide. Agents that bind to divalent cations (e.g., calcium and magnesium) should be administered separately from didanosine. Examples of these agents include fluoroquinolones such as ciprofloxacin. The same is true for agents whose dissolution is pH dependent, such as itraconazole.
Didanosine is easily degraded in an acidic environment and is therefore formulated with buffers such as calcium carbonate and magnesium hydroxide. Agents that bind to divalent cations (e.g., calcium and magnesium) should be administered separately from didanosine. Examples of these agents include fluoroquinolones such as ciprofloxacin. The same is true for agents whose dissolution is pH dependent, such as itraconazole.
FYI

 Individual antiretrovirals are commonly referred to by three letter or digit acronyms. Aside from the fact that the abbreviation of drug names can lead to dispensing errors, the acronyms typically reflect the chemical rather than the generic name of the drug and are thus very confusing. See examples in Table 18-1.
Individual antiretrovirals are commonly referred to by three letter or digit acronyms. Aside from the fact that the abbreviation of drug names can lead to dispensing errors, the acronyms typically reflect the chemical rather than the generic name of the drug and are thus very confusing. See examples in Table 18-1.TABLE 18-1 Origins of the Abbreviations for Various Antiretrovirals
| Generic Name | Chemical Name | Abbreviation |
|---|---|---|
| Zidovudine | Azidothymidine | AZT |
| Lamivudine | 3-Thiacytidine | 3TC |
| Emtricitabine | Fluorothiacytidine | FTC |
| Didanosine | Dideoxyinosine | ddI |
Nonnucleoside Reverse Transcriptase Inhibitors (nNRTIs)
Description
nNRTIs, used in the treatment of HIV infection, inhibit HIV by preventing its transcription.



MOA (Mechanism of Action)
 Several steps are involved in the replication of HIV. The virus must fuse to the host cell, uncoat, enter and be transcribed by reverse transcriptase, become incorporated into the host genome, and be transcribed to viral RNA, which is then translated into polyproteins (Figure 18-28).
Several steps are involved in the replication of HIV. The virus must fuse to the host cell, uncoat, enter and be transcribed by reverse transcriptase, become incorporated into the host genome, and be transcribed to viral RNA, which is then translated into polyproteins (Figure 18-28).










Side Effects
 Rash: Macular or papular rash, often pruritic and also self-limiting, may occur with continued drug administration. In a minority of individuals, rash can progress to more serious Stevens-Johnson syndrome.
Rash: Macular or papular rash, often pruritic and also self-limiting, may occur with continued drug administration. In a minority of individuals, rash can progress to more serious Stevens-Johnson syndrome.


Important Notes
 The nNRTIs do not inhibit DNA polymerase and therefore do not exhibit the same mitochondrial toxicities as the NRTIs. Inhibition of DNA polymerase in mitochondria leads to depletion of mitochondrial DNA and subsequent depletion of mitochondrial RNA and peptides involved in oxidative phosphorylation. This leads to mitochondrial dysfunction, and this is believed to be the cause of several of the important toxicities associated with the NRTIs.
The nNRTIs do not inhibit DNA polymerase and therefore do not exhibit the same mitochondrial toxicities as the NRTIs. Inhibition of DNA polymerase in mitochondria leads to depletion of mitochondrial DNA and subsequent depletion of mitochondrial RNA and peptides involved in oxidative phosphorylation. This leads to mitochondrial dysfunction, and this is believed to be the cause of several of the important toxicities associated with the NRTIs.
Combination Therapy






[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 18 Infectious Diseases
Introduction to Antibiotics
Suggestions
Know which category the individual drugs belong to, and learn the category as a whole. For example, ceftriaxone is a third-generation cephalosporin. Therefore learn about cephalosporins in general, and then know the general differences among first-, second-, third-, and fourth-generation cephalosporins. In some circumstances there might be one or two extra important bits of information pertaining to individual drugs that are important to learn.Important Considerations
It is required that you learn the mechanism of action (MOA) of a drug and also the mechanism of resistance, which enables an organism to live in the presence of the drug. This is important because when you are selecting an antibiotic, if your first choice does not work, you will have a better understanding of why it did not work and will be better educated to select a different antibiotic that will have a greater probability of killing the organism. For example, if a penicillin (a β-lactam) was given and the bug is known to produce β-lactamase, a third-generation cephalosporin (another β-lactam) might be a poor next choice. Make sure that the drug you select can get to the tissue in which the infection is located. Special examples include the following:
Viruses are technically not alive, so antivirals are usually not referred to as antimicrobials.
Advanced Killing Techniques
Knowledge of pharmacokinetics is required to enable a clinician to be really good at knowing how to kill off an infection. Understanding some very important fundamental concepts are required (Figure 18-1).
Minimum inhibitory concentration (MIC): The MIC of an antimicrobial is the minimum concentration that will inhibit growth of a pathogen. Obviously, it is desirable to have the concentration in the patient’s infected tissues to be greater than the MIC. If drug A has a lower MIC than drug B, then drug A kills the pathogen at a lower concentration of drug and would therefore be better at killing that particular pathogen, assuming all other factors are identical. When the MIC level of a drug for a given pathogen is low, the pathogen is considered sensitive to the drug (i.e., the drug will kill it). When the MIC is a moderate value, the pathogen’s sensitivity to the drug will be intermediate, and when the MIC is high, the pathogen will be resistant to that drug. Laboratory values often report sensitivities as S, I, or R to report sensitive, intermediate, and resistant, respectively.Now, exactly how the concentration of the antimicrobial stays above the MIC in the body is very important and is different for different drugs. The most important concepts are illustrated in Figure 18-1 and include:
Time dependence is the total length of time that a drug level stays above the MIC. What matters is the total duration that the concentration is above the MIC. These drugs are called time-dependent antimicrobials. β-Lactams (penicillins, cephalosporins, and carbapenems) all fall into this category. Note that it does not matter if the drug level is just barely above the MIC or is 10 times the MIC.Penicillins
Moa (Mechanism of Action)
Cell Wall Destruction
Transpeptidases are enzymes that cross-link peptidoglycan molecules in bacterial cell walls. Cross-linking these molecules gives strength to the cell wall. β-Lactams work by inhibiting transpeptidase, preventing it from forming cross-links. This results in a bacterium with a structurally deficient cell wall, typically leading to bacterial lysis (Figure 18-2).Mechanisms of Resistance
Gram-negative bacteria, as described previously, inherently have an outer protective lipopolysaccharide layer that guards the peptidoglycan layer from attack by some β-lactams. The main mechanisms of resistance to β-lactams include the following:
Production of β-lactamase, which enzymatically destroys the four-carbon β-lactam ring
Pharmacokinetics
Eighty percent of penicillin is cleared by the kidneys within 4 hours. Therefore it is very quickly eliminated from the body, which is an undesirable characteristic if the goal is to expose the bacterial infection to prolonged concentrations of the drug.Important Notes
FYI
A lactam is a chemical ring. A β-lactam is a four-molecule ring (three carbons and one nitrogen) and is the nucleus of the penicillin molecule. The penicillin nucleus is shown in Figure 18-3. Modifications to the five-membered ring are the basis on which cephalosporins and carbapenems (two other β-lactam antibiotics) were derived.Cephalosporins
Prototypes and common drugs
Moa (Mechanism of Action)
Cell Wall Destruction
Mechanisms of Resistance
The main mechanisms of resistance to β-lactams include the following:
Production of β-lactamase, which enzymatically destroys the four-carbon β-lactam ring
Side Effects
Important Notes
FYI
Note how the four-membered ring (the β-lactam) is the same as in penicillins; however, the other ring is six-membered, whereas in penicillins it is five-membered. The diagram shows the core nucleus of cephalosporins. There are currently four generations of cephalosporins (Figure 18-4).Carbapenems
Moa (Mechanism of Action)
Cell Wall Destruction
Mechanisms of Resistance
The main mechanisms of resistance to β-lactams include the following:
Production of β-lactamase, which enzymatically destroys the four-carbon β-lactam ring
Important Notes
MRSA is an important resistant strain of S. aureus. It is not sensitive to carbapenems. It is treated with vancomycin. VRE is another important resistant strain. About 50% of VRE infections are sensitive to carbapenems.FYI
Carbapenems differ structurally from penicillins in that the five-membered ring that is attached to the β-lactam ring contains a carbon atom instead of a sulfur atom. The name carbapenem comes from carbon and penicillin. Figure 18-6 shows the core structure of a carbapenem but does not show the side chains.Glycopeptides
Moa (Mechanism of Action)
Glycopeptides inhibit cell wall synthesis by attaching to the end of the peptidoglycan precursor units (a short four– or five–amino acid sequence called the d-alanyl-d-alanine [D-ALA-D-ALA] terminus) that are required to be laid down into the matrix. This step is catalyzed by transglycosylase. Because the glycopeptide binds to the end of the precursor, the precursor is not released from the carrier and thus peptidoglycan synthesis stops (Figure 18-7).Pharmacokinetics
Side Effects
Flushing: When vancomycin is administered quickly, the blood pressure can fall because of histamine release. Therefore vancomycin must be administered slowly (usually over 1 hour). The flushing caused by histamine release has been called red man syndrome and is not an allergy but a predictable response to rapidly administered vancomycin.Important Notes
Although vancomycin can kill S. aureus organisms that are resistant to cloxacillin (or methicillin [i.e., MRSA]), cloxacillin has a lower MIC than vancomycin for strains that are β-lactam susceptible. Therefore cloxacillin is a better killer of S. aureus when there is no resistance. Vancomycin is not a stronger antibiotic. It is simply a different but paradoxically slightly weaker one that avoids β-lactam resistance.Fluoroquinolones
MOA (Mechanism of Action)
DNA is normally supercoiled. Supercoiled DNA is under too much tension to be separated, so an extra step is required before replication and transcription can occur. DNA gyrase relaxes supercoiled DNA by cutting it, allowing rotation to occur, and then reattaching it.Pharmacokinetics
Fluoroquinolones can enter human cells easily and therefore are often used to treat intracellular pathogens.Important Notes
FYI
As the name suggests, fluoroquinolones possess a fluorine ion. They all contain two six-membered rings. Ciprofloxacin is shown in the diagram (Figure 18-9). Earlier generations of fluoroquinolones were not fluorinated and were simply called quinolones. They are infrequently used now.Aminoglycosides
MOA (Mechanism of Action)
The production of proteins from mRNA is called translation; this step requires ribosomes, transfer RNA (tRNA), and messenger RNA (mRNA) (Figure 18-10). The ribosomes are different sizes:
There are two theories about how aminoglycosides work:
Pharmacokinetics
Penetration of biologic membranes is poor because of the drug’s polar structure. Therefore all aminoglycosides are poorly absorbed in the GI tract and are not administered orally, and intracellular concentrations are usually low.
[/not-level-membership-for-basic-science-category]
