[level-membership-for-basic-science-category]
Chapter 19 Musculoskeletal System
Bisphosphonates (BPs)




MOA (Mechanism of Action)
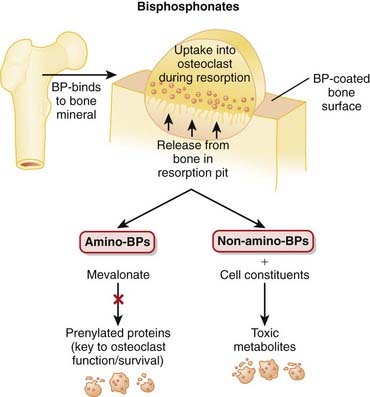
 The structural integrity of bone is determined to a large extent by the balance between the activity of osteoclasts, which break down bone (resorptive), and the activity of osteoblasts, which build bone.
The structural integrity of bone is determined to a large extent by the balance between the activity of osteoclasts, which break down bone (resorptive), and the activity of osteoblasts, which build bone.





Pharmacokinetics







Contraindications


Side Effects
All

Serious
 Esophagitis or esophageal erosion is more commonly seen with the aminobisphosphonates. It may result from a direct irritant effect from tablets lodged in the esophagus or from reflux of gastric acid including the acidic form of the BP. Patients are advised to avoid reclining for at least 30 minutes after taking a BP, reducing the chance of tablet staying in the esophagus or reflux.
Esophagitis or esophageal erosion is more commonly seen with the aminobisphosphonates. It may result from a direct irritant effect from tablets lodged in the esophagus or from reflux of gastric acid including the acidic form of the BP. Patients are advised to avoid reclining for at least 30 minutes after taking a BP, reducing the chance of tablet staying in the esophagus or reflux.

Important Notes






Evidence
Risedronate versus Placebo or Calcium and Vitamin D or Both in Postmenopausal Osteoporosis
 A 2008 Cochrane review (7 trials, N = 14,049 females) found no statistically significant effects for risedronate with respect to primary prevention of vertebral and nonvertebral fractures. For secondary prevention, risedronate demonstrated statistically significant relative risk reductions (RRRs) of vertebral fractures (39%), nonvertebral fractures (20%), and hip fractures (26%). The corresponding absolute risk reductions were small: 5%, 2%, and 1%, respectively. No statistically significant differences were found for adverse events.
A 2008 Cochrane review (7 trials, N = 14,049 females) found no statistically significant effects for risedronate with respect to primary prevention of vertebral and nonvertebral fractures. For secondary prevention, risedronate demonstrated statistically significant relative risk reductions (RRRs) of vertebral fractures (39%), nonvertebral fractures (20%), and hip fractures (26%). The corresponding absolute risk reductions were small: 5%, 2%, and 1%, respectively. No statistically significant differences were found for adverse events.Etidronate versus Placebo and/or Calcium and Vitamin D in Postmenopausal Osteoporosis
 A 2008 Cochrane review (11 trials, N = 1248 females) found no statistically significant effects of etidronate with respect to primary prevention of any fractures. A statistically significant RRR of 47% was found for secondary prevention of vertebral fractures but not for nonvertebral, hip, or wrist fractures. No statistically significant differences were found for adverse events.
A 2008 Cochrane review (11 trials, N = 1248 females) found no statistically significant effects of etidronate with respect to primary prevention of any fractures. A statistically significant RRR of 47% was found for secondary prevention of vertebral fractures but not for nonvertebral, hip, or wrist fractures. No statistically significant differences were found for adverse events.BPs versus Placebo or No Treatment in Myeloma
 A 2002 Cochrane review (11 trials, N = 2183 patients) found that BPs prevented pathologic vertebral fractures (number needed to treat [NNT] = 10) and relieved pain (NNT = 11). BPs did not affect mortality, nonvertebral fractures, or hypercalcemia. No significant adverse events were associated with the BPs.
A 2002 Cochrane review (11 trials, N = 2183 patients) found that BPs prevented pathologic vertebral fractures (number needed to treat [NNT] = 10) and relieved pain (NNT = 11). BPs did not affect mortality, nonvertebral fractures, or hypercalcemia. No significant adverse events were associated with the BPs.




Vitamin D Replacement


MOA (Mechanism of Action)

 Vitamin D is an important regulator of calcium and phosphate homeostasis and bone metabolism. It works in conjunction with PTH. The overall effect of vitamin D is to increase serum calcium concentrations. These effects are mediated via the following:
Vitamin D is an important regulator of calcium and phosphate homeostasis and bone metabolism. It works in conjunction with PTH. The overall effect of vitamin D is to increase serum calcium concentrations. These effects are mediated via the following:




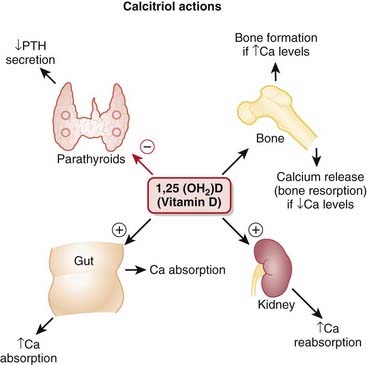







Important Notes
 Vitamin D is synthesized in the skin, liver, and kidney. Vitamin D supplementation is therefore frequently required in patients with renal failure.
Vitamin D is synthesized in the skin, liver, and kidney. Vitamin D supplementation is therefore frequently required in patients with renal failure.


Advanced
 Vitamin D supplements are sometimes used in the treatment of psoriasis, a common skin condition involving rapid turnover and inflammation of the skin. The evidence for this use, however, is not strongly conclusive.
Vitamin D supplements are sometimes used in the treatment of psoriasis, a common skin condition involving rapid turnover and inflammation of the skin. The evidence for this use, however, is not strongly conclusive.
| Common Name | Drug Name | Abbreviation |
|---|---|---|
| Vitamin D2 | Ergocalciferol | D2 |
| 1-Hydroxyvitamin D2 | Doxercalciferol | 1(OH)D2 |
| Vitamin D3 | Cholecalciferol | D3 |
| 25-Hydroxyvitamin D3 | Calcifediol | 25(OH)D3 |
| 1,25-Dihydroxyvitamin D3 | Calcitriol | 1,25(OH)2D3 |
| 24,25-Dihydroxyvitamin D3 | Secalcifediol | 24,25(OH)2D3 |
Evidence

Vitamin D Plus Calcium and Bone Fractures in the Elderly
 The same meta-analysis in 2008 showed that vitamin D plus calcium supplements do reduce hip fractures in the elderly (8 trials, N = 46,658 participants, relative risk [RR] 0.84). Hypercalcemia is significantly more common in people receiving vitamin D or an analogue, with or without calcium (18 trials, N = 11,346 participants, RR 2.35). There is a significant but modest increase in gastrointestinal symptoms (RR 1.04) and a small but significant increase in renal disease (RR 1.16).
The same meta-analysis in 2008 showed that vitamin D plus calcium supplements do reduce hip fractures in the elderly (8 trials, N = 46,658 participants, relative risk [RR] 0.84). Hypercalcemia is significantly more common in people receiving vitamin D or an analogue, with or without calcium (18 trials, N = 11,346 participants, RR 2.35). There is a significant but modest increase in gastrointestinal symptoms (RR 1.04) and a small but significant increase in renal disease (RR 1.16).FYI
 The concept that vitamin D comes from the sun is inaccurate; the inert precursor (7-dehydrocholesterol) is present in the skin, and exposure to ultraviolet light converts it to cholecalciferol, which is then isomerized to vitamin D3. Reduced exposure to sunlight is one cause of vitamin D deficiency.
The concept that vitamin D comes from the sun is inaccurate; the inert precursor (7-dehydrocholesterol) is present in the skin, and exposure to ultraviolet light converts it to cholecalciferol, which is then isomerized to vitamin D3. Reduced exposure to sunlight is one cause of vitamin D deficiency.

Parathyroid Hormone

MOA (Mechanism of Action)
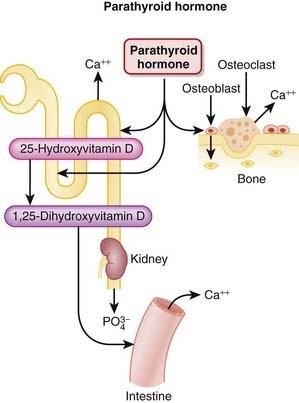
 PTH is released from the parathyroid gland. It regulates calcium and phosphate flux across cell membranes in bone and kidney. The key effects of PTH are as follows:
PTH is released from the parathyroid gland. It regulates calcium and phosphate flux across cell membranes in bone and kidney. The key effects of PTH are as follows:

 The stimulation of osteoclasts increases bone remodeling. PTH increases both bone resorption and formation; however, the net effect of excess PTH is to increase bone resorption (Figure 19-3).
The stimulation of osteoclasts increases bone remodeling. PTH increases both bone resorption and formation; however, the net effect of excess PTH is to increase bone resorption (Figure 19-3).







Contraindications






Side Effects




Important Notes
 The greatest safety concern associated with teriparatide is osteosarcoma (malignant bone cancer). However, this concern is based on observations in rodents exposed to prolonged high doses and on the ability of teriparatide to stimulate osteoblasts. So far, there does not appear to be an elevated risk of osteosarcoma with teriparatide use in humans.
The greatest safety concern associated with teriparatide is osteosarcoma (malignant bone cancer). However, this concern is based on observations in rodents exposed to prolonged high doses and on the ability of teriparatide to stimulate osteoblasts. So far, there does not appear to be an elevated risk of osteosarcoma with teriparatide use in humans.

Evidence
Bisphosphonates or Teriparatide in Postmenopausal Women
 A 2005 systematic review (90 trials) compared all BPs and teriparatide with calcium, calcium plus vitamin D, calcitriol, hormone replacement therapy, exercise, and placebo or no treatment. They found that only teriparatide and risedronate reduced the risk of nonvertebral fracture in women with severe osteoporosis and adequate calcium intake.
A 2005 systematic review (90 trials) compared all BPs and teriparatide with calcium, calcium plus vitamin D, calcitriol, hormone replacement therapy, exercise, and placebo or no treatment. They found that only teriparatide and risedronate reduced the risk of nonvertebral fracture in women with severe osteoporosis and adequate calcium intake.
RANKL Inhibitors











Important Notes
 Osteoporosis can be evaluated through plain x-ray films of bones, but the gold standard is dual-energy x-ray absorptiometry (DXA).
Osteoporosis can be evaluated through plain x-ray films of bones, but the gold standard is dual-energy x-ray absorptiometry (DXA).






Colchicine

MOA (Mechanism of Action)
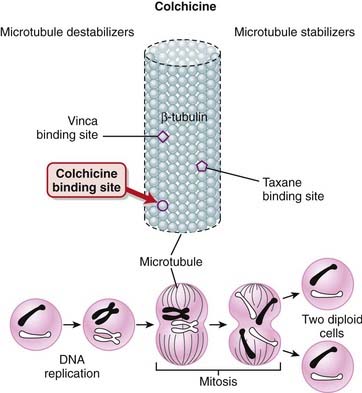
 Most of the pharmacologic effects of colchicine result from colchicine binding to tubulin (Figure 19-4). Tubulin is required for microtubule assembly, and colchicine prevents microtubule assembly. Microtubules are important components of the cytoskeleton and are involved with the following cellular functions:
Most of the pharmacologic effects of colchicine result from colchicine binding to tubulin (Figure 19-4). Tubulin is required for microtubule assembly, and colchicine prevents microtubule assembly. Microtubules are important components of the cytoskeleton and are involved with the following cellular functions:


Pharmacokinetics









Important Notes
 Gout is a common disorder of uric acid metabolism whereby uric acid levels in the blood are elevated, which results in uric acid crystals being deposited into joints, causing inflammation and intense pain. The great toe is the joint most commonly affected.
Gout is a common disorder of uric acid metabolism whereby uric acid levels in the blood are elevated, which results in uric acid crystals being deposited into joints, causing inflammation and intense pain. The great toe is the joint most commonly affected.





Evidence
Acute Gout Pain
 A Cochrane review in 2006 (one RCT, N = 43), compared with placebo, colchicine demonstrated an absolute reduction of 34% for pain scales and a 30% reduction for tenderness, swelling, redness, and pain. The NNT to reduce pain was three. All participants treated with colchicine experienced gastrointestinal side effects (diarrhea and/or vomiting), and the number needed to harm (NNH) with colchicine versus placebo was one. Note: The sample size was very small, and only one study met inclusion criteria in this review.
A Cochrane review in 2006 (one RCT, N = 43), compared with placebo, colchicine demonstrated an absolute reduction of 34% for pain scales and a 30% reduction for tenderness, swelling, redness, and pain. The NNT to reduce pain was three. All participants treated with colchicine experienced gastrointestinal side effects (diarrhea and/or vomiting), and the number needed to harm (NNH) with colchicine versus placebo was one. Note: The sample size was very small, and only one study met inclusion criteria in this review.

Nonsteroidal Antiinflammatory Drugs (NSAIDs)
Description
NSAIDs are antiinflammatory drugs that do not possess a steroidal structure (nonsteroidal).




MOA (Mechanism of Action)
 Cyclooxygenase (COX) is an enzyme that catalyzes the conversion of arachidonic acid to prostaglandin (PG) G (PGG) and PGH. These intermediaries are then converted to a variety of important PGs as well as thromboxane A2 (TXA2).
Cyclooxygenase (COX) is an enzyme that catalyzes the conversion of arachidonic acid to prostaglandin (PG) G (PGG) and PGH. These intermediaries are then converted to a variety of important PGs as well as thromboxane A2 (TXA2).




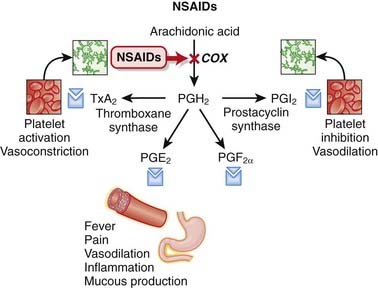









Side Effects
Nonselective




All
 Central nervous system (CNS): Confusion, dizziness, depression, and hallucinations may occur. The mechanism is not confirmed, but COX-2 is the most abundant COX isoform in the CNS, and COX-2 may play a role in neurotransmission. The frequency of CNS side effects appears to be higher with COX-2–selective inhibitors and possibly with indomethacin. These reactions are uncommon.
Central nervous system (CNS): Confusion, dizziness, depression, and hallucinations may occur. The mechanism is not confirmed, but COX-2 is the most abundant COX isoform in the CNS, and COX-2 may play a role in neurotransmission. The frequency of CNS side effects appears to be higher with COX-2–selective inhibitors and possibly with indomethacin. These reactions are uncommon.Important Notes
 Indomethacin is considered to be a particularly potent COX inhibitor with very strong antiinflammatory effects. However, this enhanced efficacy is balanced with an increased severity of adverse effects, particularly gastrointestinal effects. Indomethacin is therefore typically reserved for use in more severe inflammatory conditions rather than everyday analgesia.
Indomethacin is considered to be a particularly potent COX inhibitor with very strong antiinflammatory effects. However, this enhanced efficacy is balanced with an increased severity of adverse effects, particularly gastrointestinal effects. Indomethacin is therefore typically reserved for use in more severe inflammatory conditions rather than everyday analgesia.



Advanced
 A potential role for COX inhibitors in the treatment of cancer has been under investigation for many years. The COX-2 enzyme, which may stimulate cell division, has been selectively targeted in indications such as colon cancer. Inhibition of COX-2 may promote apoptosis, inhibit angiogenesis, and inhibit cell growth.
A potential role for COX inhibitors in the treatment of cancer has been under investigation for many years. The COX-2 enzyme, which may stimulate cell division, has been selectively targeted in indications such as colon cancer. Inhibition of COX-2 may promote apoptosis, inhibit angiogenesis, and inhibit cell growth.Evidence
Alone or in Combination with Opiates for Treatment of Cancer Pain
 A 2005 Cochrane review (42 trials, N = 3084 patients) found that NSAIDs were more effective than placebo for cancer pain. No conclusions could be drawn about the efficacy of NSAIDs relative to one another. The combination of opiates and NSAIDs was slightly and statistically better than either agent alone in 9 of 14 trials and was no different in 4 of 14 trials. The authors noted that the generalizability of the findings was limited by the short term of the studies.
A 2005 Cochrane review (42 trials, N = 3084 patients) found that NSAIDs were more effective than placebo for cancer pain. No conclusions could be drawn about the efficacy of NSAIDs relative to one another. The combination of opiates and NSAIDs was slightly and statistically better than either agent alone in 9 of 14 trials and was no different in 4 of 14 trials. The authors noted that the generalizability of the findings was limited by the short term of the studies.Versus Opiates for Treatment of Acute Renal Colic
 A 2006 Cochrane review (20 trials, N = 1613 patients) compared NSAIDs with opiates for treatment of renal colic. Results of the trials were too heterogeneous to perform a meta-analysis. The authors concluded that both NSAIDs and opiates can significantly relieve pain in acute renal colic. Opiates appeared to cause more adverse effects, particularly vomiting, and particularly pethidine.
A 2006 Cochrane review (20 trials, N = 1613 patients) compared NSAIDs with opiates for treatment of renal colic. Results of the trials were too heterogeneous to perform a meta-analysis. The authors concluded that both NSAIDs and opiates can significantly relieve pain in acute renal colic. Opiates appeared to cause more adverse effects, particularly vomiting, and particularly pethidine.


Uricosurics


MOA (Mechanism of Action)
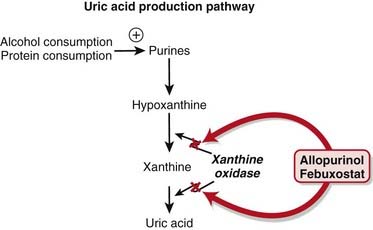
 Gout is a condition whereby uric acid crystals precipitate in joints and the crystals induce an intense inflammatory reaction within the synovial space, leading to severe pain. Uric acid is an organic acid and a byproduct of purine metabolism.
Gout is a condition whereby uric acid crystals precipitate in joints and the crystals induce an intense inflammatory reaction within the synovial space, leading to severe pain. Uric acid is an organic acid and a byproduct of purine metabolism.

 In the proximal tubule, a transporter that exchanges urate for either an organic or an inorganic anion is the channel responsible for uric acid reabsorption; uricosuric drugs compete with urate for this brush-border transporter, thereby inhibiting its reabsorption. Probenecid is completely reabsorbed by the proximal tubule (Figure 19-6).
In the proximal tubule, a transporter that exchanges urate for either an organic or an inorganic anion is the channel responsible for uric acid reabsorption; uricosuric drugs compete with urate for this brush-border transporter, thereby inhibiting its reabsorption. Probenecid is completely reabsorbed by the proximal tubule (Figure 19-6).


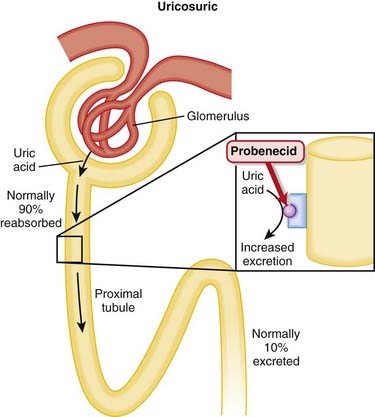
Pharmacokinetics









Important Notes
 Alcohol and high protein intake will increase uric acid levels in the blood. Therapy for gout must include dietary modification.
Alcohol and high protein intake will increase uric acid levels in the blood. Therapy for gout must include dietary modification.








Xanthine Oxidase Inhibitors


MOA (Mechanism of Action)
 Allopurinol is a purine analogue of hypoxanthine and is a substrate for, and inhibitor of, the enzyme xanthine oxidase.
Allopurinol is a purine analogue of hypoxanthine and is a substrate for, and inhibitor of, the enzyme xanthine oxidase.






Pharmacokinetics
 Allopurinol increases the half-life of probenecid and enhances its uricosuric effect, whereas probenecid increases the clearance of oxypurinol, thereby increasing dose requirements of allopurinol. The clinical relevance of this interaction is that both drugs are used to treat gout and therefore are potentially coadministered.
Allopurinol increases the half-life of probenecid and enhances its uricosuric effect, whereas probenecid increases the clearance of oxypurinol, thereby increasing dose requirements of allopurinol. The clinical relevance of this interaction is that both drugs are used to treat gout and therefore are potentially coadministered.
Indications





Side Effects






Important Notes








[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 19 Musculoskeletal System
Bisphosphonates (BPs)
MOA (Mechanism of Action)
The structural integrity of bone is determined to a large extent by the balance between the activity of osteoclasts, which break down bone (resorptive), and the activity of osteoblasts, which build bone.Pharmacokinetics
Contraindications
Side Effects
All
Serious
Esophagitis or esophageal erosion is more commonly seen with the aminobisphosphonates. It may result from a direct irritant effect from tablets lodged in the esophagus or from reflux of gastric acid including the acidic form of the BP. Patients are advised to avoid reclining for at least 30 minutes after taking a BP, reducing the chance of tablet staying in the esophagus or reflux.Important Notes
Evidence
Risedronate versus Placebo or Calcium and Vitamin D or Both in Postmenopausal Osteoporosis
A 2008 Cochrane review (7 trials, N = 14,049 females) found no statistically significant effects for risedronate with respect to primary prevention of vertebral and nonvertebral fractures. For secondary prevention, risedronate demonstrated statistically significant relative risk reductions (RRRs) of vertebral fractures (39%), nonvertebral fractures (20%), and hip fractures (26%). The corresponding absolute risk reductions were small: 5%, 2%, and 1%, respectively. No statistically significant differences were found for adverse events.Etidronate versus Placebo and/or Calcium and Vitamin D in Postmenopausal Osteoporosis
A 2008 Cochrane review (11 trials, N = 1248 females) found no statistically significant effects of etidronate with respect to primary prevention of any fractures. A statistically significant RRR of 47% was found for secondary prevention of vertebral fractures but not for nonvertebral, hip, or wrist fractures. No statistically significant differences were found for adverse events.BPs versus Placebo or No Treatment in Myeloma
A 2002 Cochrane review (11 trials, N = 2183 patients) found that BPs prevented pathologic vertebral fractures (number needed to treat [NNT] = 10) and relieved pain (NNT = 11). BPs did not affect mortality, nonvertebral fractures, or hypercalcemia. No significant adverse events were associated with the BPs.Vitamin D Replacement
MOA (Mechanism of Action)
Vitamin D is an important regulator of calcium and phosphate homeostasis and bone metabolism. It works in conjunction with PTH. The overall effect of vitamin D is to increase serum calcium concentrations. These effects are mediated via the following:
Important Notes
Vitamin D is synthesized in the skin, liver, and kidney. Vitamin D supplementation is therefore frequently required in patients with renal failure.Advanced
Vitamin D supplements are sometimes used in the treatment of psoriasis, a common skin condition involving rapid turnover and inflammation of the skin. The evidence for this use, however, is not strongly conclusive.| Common Name | Drug Name | Abbreviation |
|---|---|---|
| Vitamin D2 | Ergocalciferol | D2 |
| 1-Hydroxyvitamin D2 | Doxercalciferol | 1(OH)D2 |
| Vitamin D3 | Cholecalciferol | D3 |
| 25-Hydroxyvitamin D3 | Calcifediol | 25(OH)D3 |
| 1,25-Dihydroxyvitamin D3 | Calcitriol | 1,25(OH)2D3 |
| 24,25-Dihydroxyvitamin D3 | Secalcifediol | 24,25(OH)2D3 |
Evidence
Vitamin D Plus Calcium and Bone Fractures in the Elderly
The same meta-analysis in 2008 showed that vitamin D plus calcium supplements do reduce hip fractures in the elderly (8 trials, N = 46,658 participants, relative risk [RR] 0.84). Hypercalcemia is significantly more common in people receiving vitamin D or an analogue, with or without calcium (18 trials, N = 11,346 participants, RR 2.35). There is a significant but modest increase in gastrointestinal symptoms (RR 1.04) and a small but significant increase in renal disease (RR 1.16).FYI
The concept that vitamin D comes from the sun is inaccurate; the inert precursor (7-dehydrocholesterol) is present in the skin, and exposure to ultraviolet light converts it to cholecalciferol, which is then isomerized to vitamin D3. Reduced exposure to sunlight is one cause of vitamin D deficiency.Parathyroid Hormone
MOA (Mechanism of Action)
PTH is released from the parathyroid gland. It regulates calcium and phosphate flux across cell membranes in bone and kidney. The key effects of PTH are as follows:
The stimulation of osteoclasts increases bone remodeling. PTH increases both bone resorption and formation; however, the net effect of excess PTH is to increase bone resorption (Figure 19-3).Buy Membership for Basic Science Category to continue reading. Learn more here
[/not-level-membership-for-basic-science-category]
