Table 48-1
Dose Escalation/De-escalation Decisions Associated with Toxicity Outcomes at a Given Dose for a Popular Version of the 3 + 3 Design
| No. of Patients with Dose-Limiting Toxicity | Decision |
| 0/3 | Escalate one level |
| 1/3 | Treat 3 more at same level |
| 1/3 + 0/3∗ | Escalate one level |
| 1/3 + 1/3∗ | Stop and choose previous dose as the MTD |
| 1/3 + (2/3 or 3/3)∗ | Stop and choose previous dose as the MTD |
| 2/3 or 3/3 | Stop and choose previous dose as the MTD |
∗ Note that those rows with number of toxicities equal to 1/3 + t/3 (for t = 0, . . . , 3) corresponds to situations in which one toxicity is observed in the first cohort of 3 patients enrolled at the current dose and t toxicities are observed in the second cohort of patients enrolled at that dose.
Modifications to the Traditional Design
Cytotoxic versus Targeted Design
Pharmacokinetically Guided Dose-Escalation Method (PGDE)
Continual Reassessment Method (CRM)
Statistical Considerations of Phase I Studies
Pharmacodynamic Markers in Phase I Studies: Tissue Analysis
Overview of Pharmacodynamic Markers in Tissues
Quantitative Analysis of Pharmacodynamic Markers in Tissues
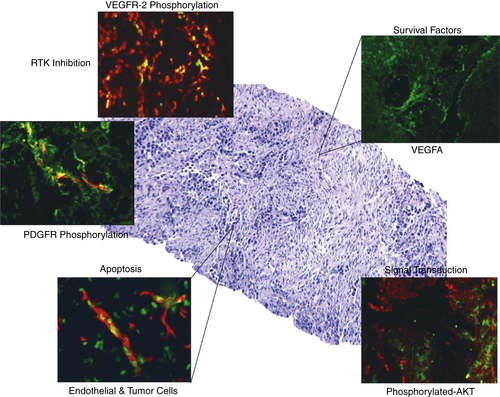
Pharmacodynamic Analysis of Receptor Tyrosine Kinase Targeted Therapies
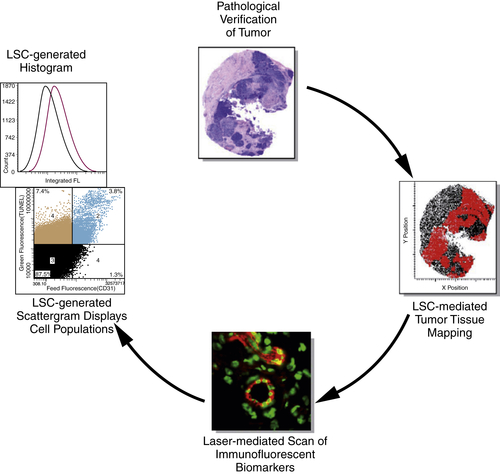
Table 48-2
Recent Successful Phase I Trials
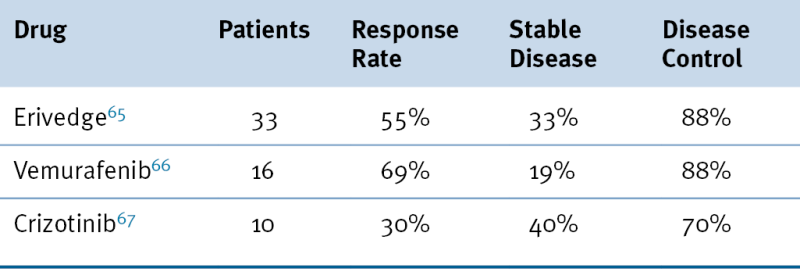
Gefitinib
Pharmacodynamic Analysis of Signal Transduction Inhibitors and Other Targets
Vemurafenib
Recent Therapeutic Successes with Phase I Trials
Challenges and Perspectives
Imaging Techniques in Phase I Studies
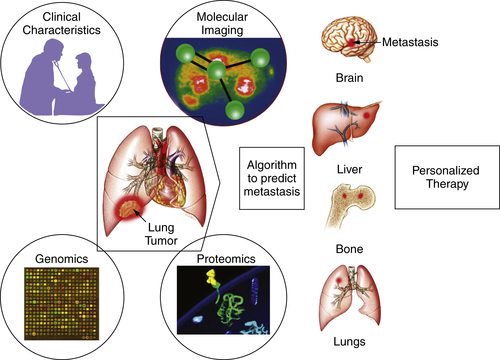
Conclusion
1. Risks and benefits of phase 1 oncology trials, 1991 through 2002 . N Engl J Med . 2005 ; 352 : 895 – 904 .
2. Hospice benefits and phase I cancer trials . Ann Intern Med . 2004 ; 140 : 70 – 71 author reply 71 .
3. For phase I studies, ethical and practical concerns abound . J Natl Cancer Inst . 2004 ; 96 : 1354 – 1355 .
4. Ethics of phase 1 oncology studies: reexamining the arguments and data . JAMA . 2003 ; 290 : 1075 – 1082 .
5. Radiosynthesis—a vital role supporting drug development? Drug Discov World . 2004 ; 5 : 59 – 62 .
6. Molecular imaging in drug discovery and development . Nat Rev . 2003 ; 2 : 123 – 131 .
7. The progress and promise of molecular imaging probes in oncologic drug development . Clin Cancer Res . 2005 ; 11 : 7967 – 7985 .
8. Cytotoxic anticancer agents and renal impairment study: the challenge remains . J Clin Oncol . 2006 ; 24 : 533 – 536 .
9. Design, conduct, and interpretation of organ impairment studies in oncology patients . J Clin Oncol . 2006 ; 24 : 3509 – 3510 author reply 3510-3501 .
10. P-5331: a phase I pharmacokinetic (PK) study of STI571 in patients (pts) with advanced malignancies and varying degrees of liver dysfunction (LD) . Proc Am Soc Clin Oncol . 2003 ; 502 : 22 abstr .
11. P-5340: a phase I pharmacokinetic study of STI-571 in patients (pts) with advanced malignancies and varying degrees of renal dysfunction . Proc Am Soc Clin Oncol . 2003 : 22 abstr 503 .
12. Pharmacology of oxaliplatin in solid tumor patients with hepatic dysfunction: a preliminary report of the National Cancer Institute Organ Dysfunction Working Group . Semin Oncol . 2003 ; 30 ( 4 suppl 15 ) : 14 – 19 .
13. Dose-escalating and pharmacological study of oxaliplatin in adult cancer patients with impaired renal function: a National Cancer Institute Organ Dysfunction Working Group Study . J Clin Oncol . 2003 ; 21 : 2664 – 2672 .
14. Administration of oxaliplatin to patients with renal dysfunction: a preliminary report of the national cancer institute organ dysfunction working group . Semin Oncol . 2003 ; 30 ( 4 suppl 15 ) : 20 – 25 .
15. Pharmacokinetics and safety of bortezomib in patients with advanced malignancies and varying degrees of liver dysfunction: phase I NCI Organ Dysfunction Working Group Study NCI-6432 . Clin Cancer Res . 2012 ; 18 : 2954 – 2963 .
16. The use of electrocardiograms in clinical trials: a public discussion of the proposed ICH E14 regulatory guidance April 11-12, 2005, Bethesda, MD, USA . Expert Opin Drug Saf . 2005 ; 4 : 795 – 799 .
17. Phase I evaluation of AZD2171, a highly potent and selective inhibitor of VEGFR signaling, in combination with selected chemotherapy regimens in patients with advanced solid tumors . J Clin Oncol 2006 ASCO Annu Meeting Proc Part I . 2006 ; 24 : 3034 (June 20 Supplement) .
18. Accelerated titration designs for phase I clinical trials in oncology . J Natl Cancer Inst . 1997 ; 89 : 1138 – 1147 .
19. Potential roles for preclinical pharmacology in phase I clinical trials . Cancer Treatment Rep . 1986 ; 70 : 73 – 80 .
20. Modified Fibonacci search . J Clin Oncol . 2003 ; 21 : 3177 .
21. Mouse to man: statistical problems in bringing a drug to clinical trial . Proceedings of the Fifth Berkeley Symposium on Mathematical Statistics and Probability . ; vol IV Berkeley : University of California Press ; 1967 : 855–866 .
22. The superiority of the time-to-event continual reassessment method to the rolling six design in pediatric oncology Phase I trials . Clin Trials . 2011 ; 8 : 361 – 369 .
23. Shortening the timeline of pediatric phase I trials: the rolling six design . J Clin Oncol . 2008 ; 26 : 190 – 195 .
24. Design and analysis of phase I clinical trials . Biometrics . 1989 ; 45 : 925 – 937 .
25. Phase 1 clinical trial design . In: Budman D.R. , Calvert A.H. , Rowinsky E.K. , eds. Handbook of Anticancer Drug Development . Baltimore, MD : Lippincott Williams & Wilkins ; 2003 : 297 – 308 .
26. Phase I clinical trial of XK469 in patients with chemo-refractory solid tumors . J Clin Oncol (Meeting Abstracts) . 2004 ; 22 ( 14 suppl ) : 2020 .
27. A phase I clinical trial of spicamycin derivative KRN5500 (NSC 650426) using a phase I accelerated titration “2B” design . Invest New Drugs . 2003 ; 21 : 63 – 74 .
28. Phase I clinical trial of BMS-247550, a derivative of epothilone B, using accelerated titration 2B design . Clin Cancer Res . 2005 ; 11 : 6233 – 6239 .
29. Phase I pharmacokinetic study of the novel antitumor agent SR233377 . Clin Cancer Res . 2000 ; 6 : 3088 – 3094 .
30. Phase I clinical trial of 5-fluoro-pyrimidinone (5FP), an oral prodrug of 5-fluorouracil (5FU) . Invest New Drugs . 2002 ; 20 : 63 – 71 .
31. Phase I and II trials of novel anti-cancer agents: endpoints, efficacy and existentialism. The Michel Clavel Lecture, held at the 10th NCI-EORTC Conference on New Drugs in Cancer Therapy, Amsterdam, 16-19 June 1998 . Ann Oncol . 1998 ; 9 : 1047 – 1052 .
32. Dose escalation trial designs based on a molecularly targeted endpoint . Stat Med . 2005 ; 24 : 2171 – 2181 .
33. Nontoxicity endpoints in phase I trial designs for targeted, non-cytotoxic agents . J Natl Cancer Inst . 2004 ; 96 : 977 – 978 .
34. An adaptive dose-finding design incorporating both toxicity and efficacy . Stat Med . 2006 ; 25 : 2365 – 2383 .
35. Pharmacologically guided phase I clinical trials based upon preclinical drug development . J Natl Cancer Inst . 1990 ; 82 : 1321 – 1326 .
36. Innovations in phase 1 trial design: where do we go next? Clin Cancer Res . 2000 ; 6 : 3801 – 3802 .
37. Prediction of the maximal tolerated dose (MTD) and therapeutic effect of anticancer drugs in humans: integration of pharmacokinetics with pharmacodynamics and toxicodynamics . Cancer Treatment Rev . 1995 ; 21 : 133 – 157 .
38. A phase I and pharmacologic evaluation of the DNA intercalator CI-958 in patients with advanced solid tumors . Clin Cancer Res . 2000 ; 6 : 3885 – 3894 .
39. Continual reassessment method: a practical design for phase 1 clinical trials in cancer . Biometrics . 1990 ; 46 : 33 – 48 .
40. The continual reassessment method for dose-finding studies: a tutorial . Clin Trials (Lond) . 2006 ; 3 : 57 – 71 .
41. Improved up-and-down designs for phase I trials . Stat Med . 2003 ; 22 : 69 – 82 .
42. Competing designs for phase I clinical trials: a review . Stat Med . 2002 ; 21 : 2757 – 2770 .
43. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer . N Engl J Med . 2005 ; 353 : 1659 – 1672 .
44. Phase II, randomized trial comparing bevacizumab plus fluorouracil (FU)/leucovorin (LV) with FU/LV alone in patients with metastatic colorectal cancer . J Clin Oncol . 2003 ; 21 : 60 – 65 .
45. Strategies for optimizing combinations of molecularly targeted anticancer agents . Nat Rev . 2006 ; 5 : 649 – 659 .
46. Surrogate markers in antiangiogenesis clinical trials . Br J Cancer . 2003 ; 89 : 8 – 14 .
47. Sequential tumor biopsies in early phase clinical trials of anticancer agents for pharmacodynamic evaluation . Clin Cancer Res . 2001 ; 7 : 2971 – 2976 .
48. What brown cannot do for you . Nat Biotechnol . 2006 ; 24 : 914 – 916 .
49. Pharmacodynamic analysis of target inhibition and endothelial cell death in tumors treated with the vascular endothelial growth factor receptor antagonists SU5416 or SU6668 . Clin Cancer Res . 2005 ; 11 ( 2 Pt 1 ) : 678 – 689 .
50. Quantitative analysis of biomarkers defines an optimal biological dose for recombinant human endostatin in primary human tumors . Clin Cancer Res . 2004 ; 10 ( 1 Pt 1 ) : 33 – 42 .
51. Development of biologic markers of response and assessment of antiangiogenic activity in a clinical trial of human recombinant endostatin . J Clin Oncol . 2002 ; 20 : 3804 – 3814 .
52. Antiangiogenic therapy targeting the tyrosine kinase receptor for vascular endothelial growth factor receptor inhibits the growth of colon cancer liver metastasis and induces tumor and endothelial cell apoptosis . Cancer Res . 1999 ; 59 : 5412 – 5416 .
53. Automated quantitative analysis of activator protein-2alpha subcellular expression in melanoma tissue microarrays correlates with survival prediction . Cancer Res . 2005 ; 65 : 11185 – 11192 .
54. Immunohistochemistry and quantitative analysis of protein expression . Arch Pathol Lab Med . 2006 ; 130 : 1026 – 1030 .
55. A phase I surrogate endpoint study of SU6668 in patients with solid tumors . Invest New Drugs . 2004 ; 22 : 459 – 466 .
56. Automated quantification of apoptosis after neoadjuvant chemotherapy for breast cancer: early assessment predicts clinical response . Clin Cancer Res . 2003 ; 9 : 955 – 960 .
57. Pharmacodynamic analysis of target receptor tyrosine kinase activity and apoptosis in GIST tumors responding to therapy with SU11248 . Proc Am Soc Clin Oncol . 2005 : 23 Abstr 3006 .
58. Epidermal growth factor receptor targeting in cancer . Semin Oncol . 2006 ; 33 : 369 – 385 .
59. Structural analysis of receptor tyrosine kinases . Prog Biophys Mol Biol . 1999 ; 71 : 343 – 358 .
60. Vascular endothelial growth factor effect on endothelial cell proliferation, migration, and platelet-activating factor synthesis is Flk-1-dependent . J Biol Chem . 1999 ; 274 : 31047 – 31054 .
61. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer (The IDEAL 1 Trial) [corrected] . J Clin Oncol . 2003 ; 21 : 2237 – 2246 .
62. Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer: a randomized trial . JAMA . 2003 ; 290 : 2149 – 2158 .
63. Gefitinib (ZD1839) combined with weekly epirubicin in patients with metastatic breast cancer: a phase I study with biological correlate . Ann Oncol . 2005 ; 16 : 1867 – 1873 .
64. Pharmacodynamic studies of the epidermal growth factor receptor inhibitor ZD1839 in skin from cancer patients: histopathologic and molecular consequences of receptor inhibition . J Clin Oncol . 2002 ; 20 : 110 – 124 .
65. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma . N Engl J Med . 2009 ; 361 : 1164 – 1172 .
66. Inhibition of mutated, activated BRAF in metastatic melanoma . N Engl J Med . 2010 ; 363 : 809 – 819 .
67. Clinical activity observed in a phase I dose escalation trial of an oral c-met and ALK inhibitor, PF-02341066 . ASCO Meeting Abstracts . 2009 ; 27 ( 15S ) : 3509 .
68. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma . Nature . 2010 ; 467 : 596 – 599 .
69. Phase I study of intravenous vascular endothelial growth factor trap, aflibercept, in patients with advanced solid tumors . J Clin Oncol . 2010 ; 28 : 207 – 214 .
70. Phase 1 study of aflibercept administered subcutaneously to patients with advanced solid tumors . Clin Cancer Res . 2010 ; 16 : 358 – 366 .
71. Improved survival with vemurafenib in melanoma with BRAF V600E mutation . N Engl J Med . 2011 ; 364 : 2507 – 2516 .
72. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer . N Engl J Med . 2010 ; 363 : 1693 – 1703 .
73. Multigene reverse transcription-PCR profiling of circulating tumor cells in hormone-refractory prostate cancer . Clin Chem . 2004 ; 50 : 826 – 835 .
74. Gordon. Phase I trial of recombinant humanized monoclonal anti-vascular endothelial growth factor (anti-VEGF MAB) in patients (PTS) with metastatic cancer (Meeting abstract) 1998.
75. . Biomarkers and surrogate endpoints: preferred definitions and conceptual framework . Clin Pharmacol Ther . 2001 ; 69 : 89 – 95 .
76. Clinical biomarkers in drug discovery and development . Nat Rev . 2003 ; 2 : 566 – 580 .
77. Functional imaging in phase I studies: decorations or decision making? J Clin Oncol . 2003 ; 21 : 2807 – 2809 .
78. Positron emission tomography microdosing: a new concept with application in tracer and early clinical drug development . Eur J Clin Pharmacol . 2003 ; 59 : 357 – 366 .
79. MR in oncology drug development . NMR Biomed . 2006 ; 19 : 681 – 689 .
80. Progress and promise of FDG-PET imaging for cancer patient management and oncologic drug development . Clin Cancer Res . 2005 ; 11 : 2785 – 2808 .
81. FDG uptake and glucose transporter subtype expressions in experimental tumor and inflammation models . J Nucl Med . 2001 ; 42 : 1551 – 1555 .
82. Biological correlates of FDG uptake in non-small cell lung cancer . Lung Cancer . 2007 ; 55 : 79 – 87 .
83. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors . N Engl J Med . 2002 ; 347 : 472 – 480 .
84. Early prediction of response to sunitinib after imatinib failure by 18F-fluorodeoxyglucose positron emission tomography in patients with gastrointestinal stromal tumor . J Clin Oncol . 2009 ; 27 : 439 – 445 .
85. New guidelines to evaluate the response to treatment in solid tumors . National Cancer Institute of the United States, National Cancer Institute of Canada European Organization for Research and Treatment of Cancer . J Natl Cancer Inst . 2000 ; 92 : 205 – 216 .
86. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) . Eur J Cancer . 2009 ; 45 : 228 – 247 .
87. Evaluation of the response to treatment of solid tumours—a consensus statement of the International Cancer Imaging Society . Br J Cancer . 2004 ; 90 : 2256 – 2260 .
88. Criticism of tumor response criteria raises trial design questions . J Natl Cancer Inst . 2006 ; 98 : 232 – 234 .
89. Response assessment classification in patients with advanced renal cell carcinoma treated on clinical trials . Cancer . 2003 ; 98 : 1611 – 1619 .
90. The effect of measuring error on the results of therapeutic trials in advanced cancer . Cancer . 1976 ; 38 : 388 – 394 .
91. Measuring response in a post-RECIST world: from black and white to shades of grey . Nat Rev Cancer . 2006 ; 6 : 409 – 414 .
92. An alternative model for the evaluation of antitumor activity . Cancer Clin Trials . 1981 ; 4 : 451 – 457 .
93. Cancer clinical trial outcomes: any progress in tumour-size assessment? Eur J Cancer . 2009 ; 45 : 225 – 227 .