Genetics and Cell Biology
Tumors of the Peripheral Nervous System: Ewing Sarcoma and Primitive Neuroectodermal Tumors
Clinical Description and Pathology
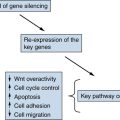
Genetics and Cell Biology
Rhabdomyosarcoma
Clinical Description and Pathology
Genetics and Cell Biology
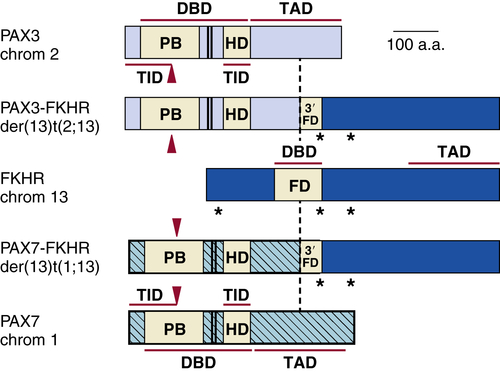
Childhood Sarcomas: Osteosarcoma
Clinical Description and Pathology
Genetics and Cell Biology
Cancer Predisposition Syndromes
Li-Fraumeni Syndrome
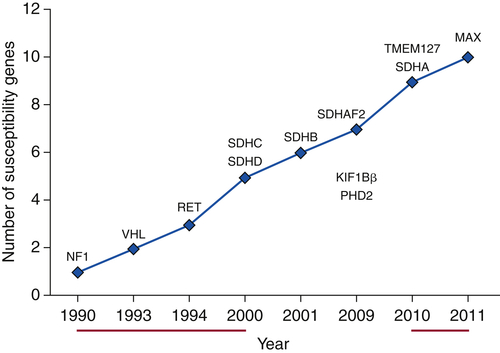
Hereditary Paraganglioma Syndromes
Table 27-2
Hereditary Syndromes Associated with Childhood Neoplasms
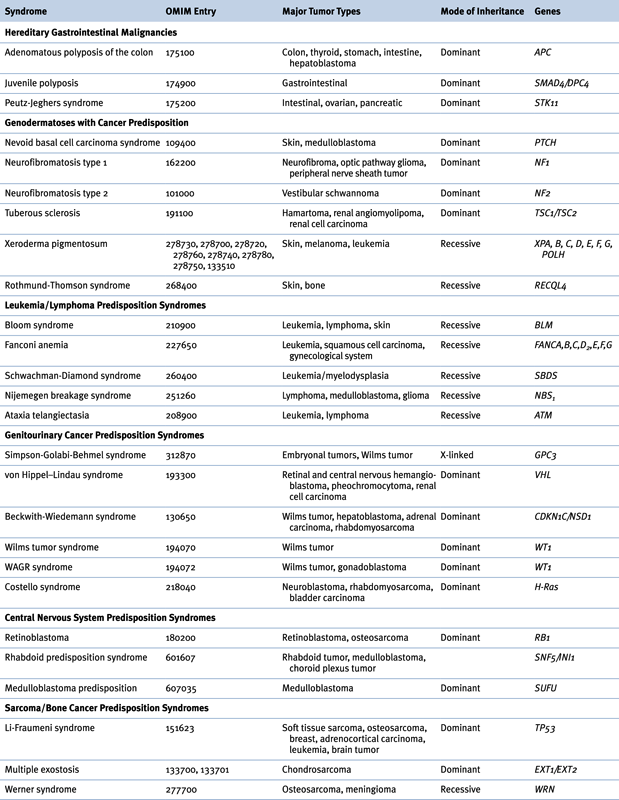
OMIM, Online Mendelian Inheritance in Man.
Table 27-3
Clinical Criteria for Classic Li-Fraumeni Syndrome, LFS-Like Criteria, and Chompret Criteria

Genetics
Beckwith-Wiedemann Syndrome
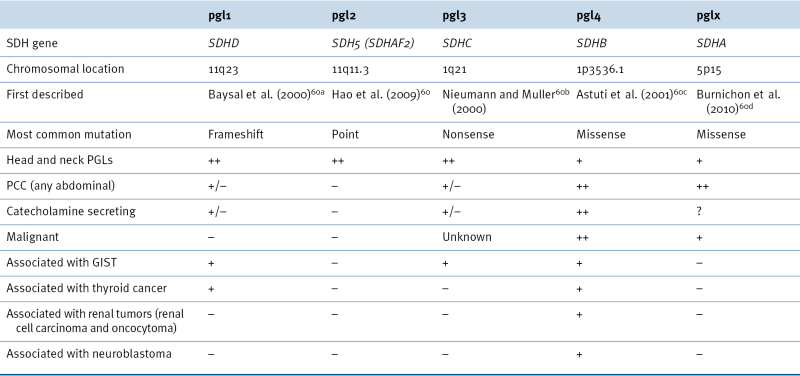
Table 27-4
SDHX Genotype: Phenotype Correlations
Gorlin Syndrome
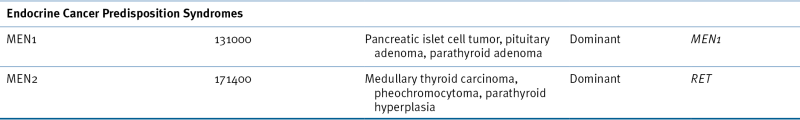
Multiple Endocrine Neoplasia
Table 27-5
Beckwith-Wiedemann Syndrome Genetic and Epigenetic Subgroups
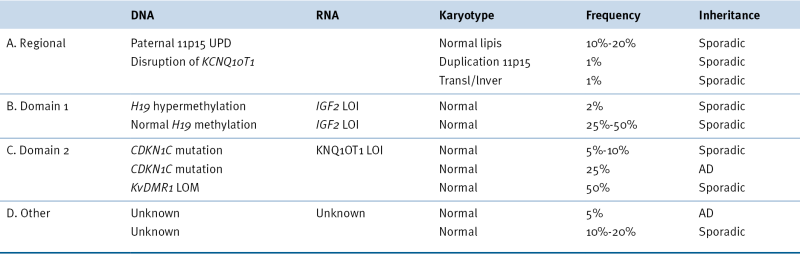
AD, Autosomal dominant; LOI, loss of imprinting; LOM, loss of methylation; UPD, uniparental disomy.
DICER1 Syndrome
Molecular and Clinical Surveillance for Cancer Predisposition in Children
1. Cancer statistics . CA Cancer J Clin . 1998 ; 1998 ( 48 ) : 6 – 29 .
2. Hereditary cancer risk assessment in a pediatric cancer followup clinic . Pediatric Blood Cancer . 2012 ; 58 : 85 – 89 .
3. The incidence of retinoblastoma . Am J Ophthalmol . 1975 ; 80 : 263 – 265 .
4. Mutation and childhood cancer: a probabilistic model for the incidence of retinoblastoma . Proc Natl Acad Sci U S A . 1975 ; 72 : 5116 – 5120 .
5. The practical management of retinoblastoma . Trans Am Ophthalmol Soc . 1969 ; 67 : 462 – 534 .
6. Cloning of the esterase D gene: a polymorphic gene probe closely linked to the retinoblastoma locus on chromosome 13 . Proc Natl Acad Sci U S A . 1986 ; 83 : 6573 – 6577 .
7. Expression of recessive alleles by chromosomal mechanisms in retinoblastoma . Nature . 1983 ; 305 : 779 – 784 .
8. A human DNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma . Nature . 1986 ; 323 : 643 – 646 .
9. Characterisation of retinoblastoma without RB1 mutations: genomic, gene expression, and clinical studies . Lancet Oncol . 2013 pii: S1470-2045(13) 0045–7 .
10. Targeting the p53 pathway in retinoblastoma with subconjunctival Nutlin-3a . Cancer Res . 2011 ; 71 : 4205 – 4213 .
11. Nephrogenic rests, nephroblastomatosis, and the pathogenesis of Wilms tumor . Pediatr Pathol . 1990 ; 10 : 1 – 36 .
12. Chromosomal imbalance in the aniridia-Wilms tumor association: 11p interstitial deletion . Pediatrics . 1978 ; 61 : 604 – 610 .
13. The Denys-Drash syndrome . J Med Genet . 1994 ; 31 : 471 – 477 .
14. Complete and incomplete forms of Beckwith-Wiedemann syndrome: their oncogenic potential . J Pediatr . 1980 ; 96 : 47 – 50 .
15. Genetic linkage of Beckwith-Wiedemann syndrome to 11p15 . Am J Hum Genet . 1989 ; 44 : 720 – 723 .
16. An X chromosome gene, WTX, is commonly mutated in Wilms tumor . Science . 2007 ; 315 : 642 – 645 .
17. A proposed staging system for children with neuroblastoma . Cancer . 1971 ; 27 : 374 – 378 .
18. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour . Nature . 1983 ; 305 : 245 – 248 .
19. Decreased expression of N-myc precedes retinoic acid-induced morphological differentiation of human neuroblastoma . Nature . 1985 ; 313 : 404 – 406 .
20. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children’s Cancer Group . N Engl J Med . 1999 ; 341 : 1165 – 1173 .
21. Telomere length is a prognostic factor in neuroblastoma . Cancer . 2006 ; 107 : 1391 – 1399 .
22. Selective targeting of cancer stem cells by telomerase inhibition . Clin Cancer Res . 2011 ; 17 : 111 – 121 .
23. Germline mutations of the paired-like homeobox 2B (PHOX2B) gene in neuroblastoma . Am J Human Genet . 2004 ; 74 : 761 – 764 .
24. Identification of ALK as a major neuroblastoma susceptibility gene . Nature . 2008 ; 455 : 930 – 935 .
25. The genetic landscape of high-risk neuroblastoma . Nat Genet . 2013 ; 45 : 279 – 284 .
26. Cytogenetic characterization of selected small round cell tumors of childhood . Cancer Genet Cytogenet . 1986 ; 21 : 185 – 208 .
27. The IGF-1 receptor gene promoter is a molecular target for the Ewing sarcoma-Wilms tumor 1 fusion protein . J Biol Chem . 1996 ; 271 : 19304 – 19309 .
28. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma . Nat Med . 2009 ; 15 : 750 – 756 .
29. Insulin-like growth factor 1 receptor as a therapeutic target in Ewing sarcoma: lack of consistent upregulation or recurrent mutation and a review of the clinical trial literature . Sarcoma . 2013 doi: 10.1155/2013/450478 .
30. Does expression of different EWS chimeric transcripts define clinically distinct risk groups of Ewing tumor patients? J Clin Oncol . 1996 ; 14 : 1245 – 1251 .
31. A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion . Nat Genet . 2012 ; 44 : 461 – 466 .
32. Rhabdomyosarcoma and the undifferentiated sarcomas . In: Pizzo P.P. , ed. Principles and Practice of Pediatric Oncology . 4th ed . New York, NY : Lippincott Williams & Wilkins ; 2006 .
33. Consistent chromosomal translocation in alveolar rhabdomyosarcoma . Cancer Genet Cytogenet . 1986 ; 19 : 361 – 362 .
34. Chromosomal translocation t(1;13)(p36;q14) in a case of rhabdomyosarcoma . Genes Chromosome Cancer . 1991 ; 3 : 483 – 484 .
35. Fusion gene-negative alveolar rhabdomyosarcoma is clinically and molecularly indistinguishable from embryonal rhabdomyosarcoma . J Clin Oncol . 2010 ; 28 : 2151 – 2158 .
36. Chromosomal localization of the human rhabdomyosarcoma locus by mitotic recombination mapping . Nature . 1987 ; 329 : 645 – 647 .
37. A model for embryonal rhabdomyosarcoma tumorigenesis that involves genome imprinting . Proc Natl Acad Sci U S A . 1989 ; 86 : 7480 – 7484 .
38. Costellow syndrome: a Ras/mitogen activated protein kinase pathway syndrome (rasopathy) resulting from HRAS germline mutations . Genet Med . 2012 ; 14 : 285 – 292 .
39. Recurrent chromosome aberrations in cancer . Mutat Res . 2000 ; 462 : 247 – 253 .
40. Recurrent focal copy-number changes and loss of heterozygosity implicate two noncoding RNAs and one tumor suppressor gene at chromosome 3q13.31 in osteosarcoma . Cancer Res . 2010 ; 70 : 160 – 171 .
41. Allelotype analysis in osteosarcomas: frequent allele loss on 3q, 13q, 17p, and 18q . Cancer Res . 1992 ; 52 : 2419 – 2423 .
42. Stephens PJ, Greenman CD, Fu B, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 144:27-40.
43. Fas expression inversely correlates with metastatic potential in osteosarcoma cells . Oncol Rep . 2002 ; 9 : 823 – 827 .
44. Cyclin E1 is amplified and overexpressed in osteosarcoma . J Med Design . 2011 ; 13 : 289 – 296 .
45. Rhabdomyosarcoma in children: epidemiologic study and identification of a familial cancer syndrome . J Natl Cancer Inst . 1969 ; 43 : 1365 – 1373 .
46. 2009 version of the Chompret criteria for Li-Fraumeni syndrome . J Clin Oncol . 2009 ; 27 : e108 – e109 .
47. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms . Science . 1990 ; 250 : 1233 – 1238 .
48. Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome . Nature . 1990 ; 348 : 747 – 749 .
49. Impact of the MDM2 SNP 309 and p53 Arg72Pro polymorphism on age of onset in Li-Fraumeni syndrome . J Med Genet . 2006 ; 43 : 531 – 533 .
50. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations . Cell . 2012 ; 148 : 59 – 71 .
51. Excessive genomic DNA copy number variation in the Li-Fraumeni cancer predisposition syndrome . Proc Natl Acad Sci U S A . 2008 ; 32 : 11264 – 11269 .
52. TP53 PIN3 and SNP 309 polymorphisms as genetic modifiers in the Li-Fraumeni syndrome: impact on age at first diagnosis . J Med Genet . 2009 ; 46 : 766 – 772 .
53. F18-fluorodeoxyglucose-positron emission tomography/computed tomography screening in Li-Fraumeni syndrome . JAMA . 2008 ; 299 : 1315 – 1319 .
54. Imaging and biochemical surveillance in TP53 mutation carriers with Li-Fraumeni syndrome: a prospective observational study . Lancet Oncol . 2011 ; 12 : 559 – 567 .
55. An update on the genetics of paraganglioma, pheochromocytoma and associated hereditary syndromes . Hormon Metab Res . 2012 ; 44 : 328 – 333 .
56. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes . J Clin Endocrinol Metab . 2006 ; 91 : 827 – 836 .
57. Pheochromocytoma and paraganglioma: progress on all fronts . Endocr Pathol . 2012 ; 23 : 1 – 3 .
58. Treatment with sunitinib for patients with progressive metastatic pheochromocytomas and sympathetic paragangliomas . J Clin Endocrinol Metab . 2012 ; 97 : 4040 – 4050 .
59. SDH mutations in tumorigenesis and inherited endocrine tumors: lesson from the pheochromocytoma-paraganglioma syndromes . J Intern Med . 2009 ; 266 : 19 – 42 .
60. SDH5, a gene required for flavination of succinate dehydrogenase is mutated in paraganglioma . Science . 2009 ; 325 : 1139 – 1142 .
60a. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma . Science . 2000 ; 287 ( 5454 ) : 848 – 851 .
60b. Mutations in SDHC cause autosomal dominant paraganglioma, type 3 . Nature Genet . 2000 ; 26 ( 3 ) : 268 – 270 .
60c. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma . Am J Hum Genet . 2001 ; 69 ( 1 ) : 49 – 54 .
60d. SDHA is a tumor suppressor gene causing paraganglioma . Hum Mol Genet . 2010 ; 19 ( 15 ) : 3011 – 3020 .
61. Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma . Clin Chem . 2011 ; 57 : 411 – 420 .
62. Imaging work-up for screening of paraganglioma and pheochromocytoma in SDHx mutation carriers: a multicenter prospective study from the PGL. EVA Investigators . J Clin Endocrinol Metab . 2013 ; 98 : E162 – E173 .
63. Parental imprinting of human chromosome region 11p15.3-pter involved in the Beckwith-Wiedemann syndrome and various human neoplasia . Eur J Hum Genet . 1994 ; 2 : 3 – 23 .
64. Imprinting mutations in the Beckwith-Wiedemann syndrome suggested by altered imprinting pattern in the IGF2-H19 domain . Hum Mol Genet . 1995 ; 4 : 2379 – 2385 .
65. Nevoid basal-cell carcinoma syndrome . Medicine (Baltimore) . 1987 ; 66 : 98 – 113 .
66. The gene for the naevoid basal cell carcinoma syndrome acts as a tumour-suppressor gene in medulloblastoma . Br J Cancer . 1997 ; 76 : 141 – 145 .
67. Positional cloning of the gene for multiple endocrine neoplasia-type 1 . Science . 1997 ; 276 : 404 – 407 .
68. Exploring the endocrine manifestations of DICER1 mutations . Trends Mol Med . 2012 ; 18 : 503 – 505 .
69. miRNA processing and human cancer: DICER1 cuts the mustard . Sci Transl Med . 2011 ; 3 111ps46 .
70. Statement of the American Society of Clinical Oncology: genetic testing for cancer susceptibility. Adopted on February 20, 1996 . J Clin Oncol . 1996 ; 14 : 1730 – 1736 .