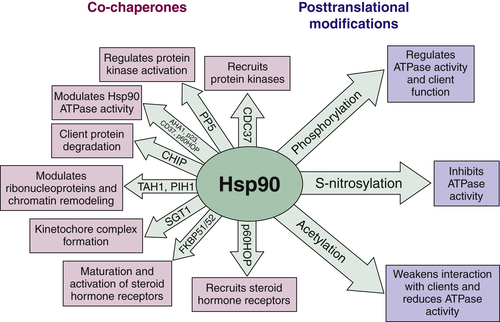
Hsp90 ATPase Activity and Chaperone Function
Hsp90 Inhibitors Target Client Proteins to the Proteasome
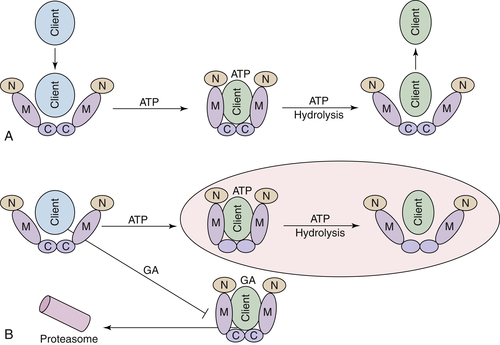
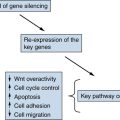
Hsp90 Inhibitors May Prevent Oncogenic Switching
Table 56-1
List of Selected Hsp90 Clients Involved in Cancer
| Cancer | Hsp90 Client | References |
| Anaplastic large cell lymphomas | NMP-ALK | 103, 104 |
| Acute myeloid leukemia | FLT3 | 105, 106 |
| Chronic myelogenous leukemia | BCR-ABL | 107, 108 |
| Human mastocytosis Gastrointestinal stromal tumors |
KIT | 109 |
| Melanomas | B-RAF EGFR |
89, 110–113 |
| Prostate cancer | Androgen receptor | 114–116 |
| Clear cell renal cell carcinoma (ccRCC) Nonhereditary sporadic ccRCC |
HIF1-α | 117, 118 |
| Glioblastoma | VEGF | 119 |
| Leukemia | BCL2 APAF |
120 |
| Small-cell lung cancer | MET | 121–124 |
| Multiple endocrine neoplasia type 2 (MEN2A, MEN2B) Familial medullary thyroid carcinoma (FMTC) Papillary carcinoma of thyroid |
RET | 125–128 |
The Proteasome as an Anticancer Molecular Target
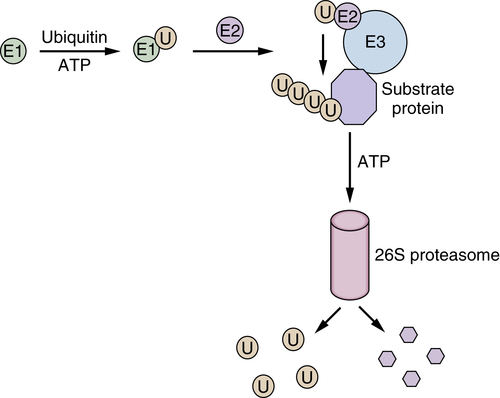
Anticancer Activity of the Proteasome Inhibitor Bortezomib
Second-Generation Proteasome Inhibitors
Combined Inhibition of Hsp90 and the Proteasome
Additional Rationales for Inhibiting the Ubiquitin-Proteasome System in Cancer
Why Are Tumor Cells Uniquely Sensitive to Hsp90 and Proteasome Inhibition?
Conclusion
1. The onset and extent of genomic instability in sporadic colorectal tumor progression . Proc Natl Acad Sci U S A . 1999 ; 96 : 15121 – 15126 .
2. Modelling the molecular circuitry of cancer . Nat Rev Cancer . 2002 ; 2 : 331 – 341 .
3. Insights from pre-clinical studies for new combination treatment regimens with the Bcr-Abl kinase inhibitor imatinib mesylate (Gleevec/Glivec) in chronic myelogenous leukemia: a translational perspective . Leukemia . 2002 ; 16 : 1213 – 1219 .
4. Cancer robustness: tumour tactics . Nature . 2003 ; 426 : 125 .
5. Protein misfolding and disease: the case of prion disorders . Cell Mol Life Sci . 2003 ; 60 : 133 – 143 .
6. Chaperone-related immune dysfunction: an emergent property of distorted chaperone networks . Trends Immunol . 2006 ; 27 : 74 – 79 .
7. Navigating the chaperone network: an integrative map of physical and genetic interactions mediated by the hsp90 chaperone . Cell . 2005 ; 120 : 715 – 727 .
8. Inhibition of Hsp90: a new strategy for inhibiting protein kinases . Biochim Biophys Acta . 2004 ; 1697 : 233 – 242 .
9. Hallmarks of cancer: the next generation . Cell . 2011 ; 144 : 646 – 674 .
10. Heat-shock protein 90 inhibitors in antineoplastic therapy: is it all wrapped up? Expert Opin Investig Drugs . 2005 ; 14 : 569 – 589 .
11. Hsp90: the vulnerable chaperone . Drug Discov Today . 2004 ; 9 : 881 – 888 .
12. Combinatorial attack on multistep oncogenesis by inhibiting the Hsp90 molecular chaperone . Cancer Lett . 2004 ; 206 : 149 – 157 .
13. Altered Hsp90 function in cancer: a unique therapeutic opportunity . Mol Cancer Ther . 2004 ; 3 : 1021 – 1030 .
14. Targeting multiple signal transduction pathways through inhibition of Hsp90 . J Mol Med . 2004 ; 82 : 488 – 499 .
15. The Hsp90 chaperone complex as a novel target for cancer therapy . Ann Oncol . 2003 ; 14 : 1169 – 1176 .
16. Heat shock protein 90 as a molecular target for cancer therapeutics . Cancer Cell . 2003 ; 3 : 213 – 217 .
17. Hsp90 as a capacitor for morphological evolution . Nature . 1998 ; 396 : 336 – 342 .
18. Hsp90 as a capacitor of phenotypic variation . Nature . 2002 ; 417 : 618 – 624 .
19. HSP90 and the chaperoning of cancer . Nat Rev Cancer . 2005 ; 5 : 761 – 772 .
20. Identification of aneuploidy-selective antiproliferation compounds . Cell . 2011 ; 144 : 499 – 512 .
21. Geldanamycin, a new antibiotic . J Antibiot (Tokyo) . 1970 ; 23 : 442 – 447 .
22. The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin . Cancer Chemother Pharmacol . 1998 ; 42 : 273 – 279 .
23. Ansamycin inhibitors of Hsp90: nature’s prototype for anti-chaperone therapy . Curr Top Med Chem . 2009 ; 9 : 1386 – 1418 .
24. Targeting the dynamic HSP90 complex in cancer . Nat Rev Cancer . 2010 ; 10 : 537 – 549 .
25. Hsp90 molecular chaperone inhibitors: are we there yet? Clin Cancer Res . 2012 ; 18 : 64 – 76 .
26. Co-chaperone regulation of conformational switching in the Hsp90 ATPase cycle . J Biol Chem . 2004 ; 279 : 51989 – 51998 .
27. Hsp70 and Hsp90—a relay team for protein folding . Rev Physiol Biochem Pharmacol . 2004 ; 151 : 1 – 44 .
28. Structure and functional relationships of Hsp90 . Curr Cancer Drug Targets . 2003 ; 3 : 301 – 323 .
29. Identification of potent water soluble purine-scaffold inhibitors of the heat shock protein 90 . J Med Chem . 2006 ; 49 : 381 – 390 .
30. Evaluation of 8-arylsulfanyl, 8-arylsulfoxyl, and 8-arylsulfonyl adenine derivatives as inhibitors of the heat shock protein 90 . J Med Chem . 2005 ; 48 : 2892 – 2905 .
31. Development of purine-scaffold small molecule inhibitors of Hsp90 . Curr Cancer Drug Targets . 2003 ; 3 : 371 – 376 .
32. Development of a purine-scaffold novel class of Hsp90 binders that inhibit the proliferation of cancer cells and induce the degradation of Her2 tyrosine kinase . Bioorg Med Chem . 2002 ; 10 : 3555 – 3564 .
33. The identification, synthesis, protein crystal structure and in vitro biochemical evaluation of a new 3,4-diarylpyrazole class of Hsp90 inhibitors . Bioorg Med Chem Lett . 2005 ; 15 : 3338 – 3343 .
34. Novel, potent small-molecule inhibitors of the molecular chaperone Hsp90 discovered through structure-based design . J Med Chem . 2005 ; 48 : 4212 – 4215 .
35. Update on Hsp90 inhibitors in clinical trial . Curr Top Med Chem . 2009 ; 9 : 1479 – 1492 .
36. The Hsp90 complex–a super-chaperone machine as a novel drug target . Biochem Pharmacol . 1998 ; 56 : 675 – 682 .
37. HSP40 binding is the first step in the HSP90 chaperoning pathway for the progesterone receptor . J Biol Chem . 2002 ; 277 : 11873 – 11881 .
38. Stimulation of the weak ATPase activity of human hsp90 by a client protein . J Mol Biol . 2002 ; 315 : 787 – 798 .
39. Regulation of Hsp90 ATPase activity by the co-chaperone Cdc37p/p50cdc37 . J Biol Chem . 2002 ; 277 : 20151 – 20159 .
40. Polypeptide release by Hsp90 involves ATP hydrolysis and is enhanced by the co-chaperone p23 . EMBO J . 2000 ; 19 : 5930 – 5940 .
41. Activation of the ATPase activity of hsp90 by the stress-regulated cochaperone aha1 . Mol Cell . 2002 ; 10 : 1307 – 1318 .
42. Dynamic tyrosine phosphorylation modulates cycling of the HSP90-P50(CDC37)-AHA1 chaperone machine . Mol Cell . 2012 ; 47 : 434 – 443 .
43. Post-translational modifications of Hsp90 and their contributions to chaperone regulation . Biochim Biophys Acta . 2012 ; 1823 : 648 – 655 .
44. Swe1Wee1-dependent tyrosine phosphorylation of Hsp90 regulates distinct facets of chaperone function . Mol Cell . 2010 ; 37 : 333 – 343 .
45. Casein kinase 2 phosphorylation of Hsp90 threonine 22 modulates chaperone function and drug sensitivity . Oncotarget . 2011 ; 2 : 407 – 417 .
46. Hsp90 inhibitors as novel cancer chemotherapeutic agents . Trends Mol Med . 2002 ; 8 ( 4 suppl ) : S55 – S61 .
47. Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu . Proc Natl Acad Sci U S A . 2002 ; 99 : 12847 – 12852 .
48. Regulation of Hsp90 client proteins by a Cullin5-RING E3 ubiquitin ligase . Proc Natl Acad Sci U S A . 2009 ; 106 : 20330 – 20335 .
49. The heat shock protein 90 antagonist geldanamycin alters chaperone association with p210bcr-abl and v-src proteins before their degradation by the proteasome . Cell Growth Differ . 2000 ; 11 : 355 – 360 .
50. Hsp90 phosphorylation, Wee1 and the cell cycle . Cell Cycle . 2010 ; 9 : 2310 – 2316 .
51. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies . Science . 2007 ; 318 : 287 – 290 .
52. HER kinase activation confers resistance to MET tyrosine kinase inhibition in MET oncogene-addicted gastric cancer cells . Mol Cancer Ther . 2008 ; 7 : 3499 – 3508 .
53. Targeting Hsp90 prevents escape of breast cancer cells from tyrosine kinase inhibition . Cell Cycle . 2008 ; 7 : 2936 – 2941 .
54. Cancer cells harboring MET gene amplification activate alternative signaling pathways to escape MET inhibition but remain sensitive to Hsp90 inhibitors . Cell Cycle . 2009 ; 8 : 2050 – 2056 .
55. Hsp90 inhibition overcomes HGF-triggering resistance to EGFR-TKIs in EGFR-mutant lung cancer by decreasing client protein expression and angiogenesis . J Thorac Oncol . 2012 ; 7 : 1078 – 1085 .
56. Functions of the proteasome: From protein degradation and immune surveillance to cancer therapy . Biochem Soc Trans . 2007 ; 35 : 12 – 17 .
57. Drug discovery in the ubiquitin-proteasome system . Nat Rev Drug Discov . 2006 ; 5 : 596 – 613 .
58. Protein degradation and human diseases: the ubiquitin connection . Ann Intern Med . 2006 ; 145 676-207 .
59. Bortezomib as an antitumor agent . Curr Pharm Biotech . 2006 ; 7 : 441 – 448 .
60. Proteasome inhibitors in multiple myeloma: ten years later . Blood . 2012 ; 120 : 947 – 959 .
61. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer . Cancer Res . 2010 ; 70 : 1970 – 1980 .
62. Investigational agent MLN9708, an oral proteasome inhibitor, in patients (pts) with relapsed and/or refractory multiple myeloma (MM): results from the expansion cohorts of a phase 1 dose-escalation study . ASH Annual Meeting Abstracts . 2011 ; 118 : 301 .
63. CEP-18770: A novel, orally active proteasome inhibitor with a tumor-selective pharmacologic profile competitive with bortezomib . Blood . 2008 ; 111 : 2765 – 2775 .
64. Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome . Cancer Res . 2007 ; 67 : 6383 – 6391 .
65. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma . Blood . 2007 ; 110 : 3281 – 3290 .
66. Emerging role of carfilzomib in treatment of relapsed and refractory lymphoid neoplasms and multiple myeloma . Core Evid . 2011 ; 6 : 43 – 57 .
67. A novel orally active proteasome inhibitor ONX 0912 triggers in vitro and in vivo cytotoxicity in multiple myeloma . Blood . 2010 ; 116 : 4906 – 4915 .
68. Phase 1 Clinical Evaluation of Twice-Weekly Marizomib (NPI-0052), a Novel Proteasome Inhibitor, in Patients with Relapsed/Refractory Multiple Myeloma (MM) . ASH Annual Meeting Abstracts . 2011 ; 118 : 302 .
69. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from bortezomib . Cancer Cell . 2005 ; 8 : 407 – 419 .
70. Proteasome structure, function, and lessons learned from beta-lactone inhibitors . Curr Top Med Chem . 2011 ; 11 : 2850 – 2878 .
71. Polyubiquitination and proteasomal degradation of the p185c-erbB-2 receptor protein-tyrosine kinase induced by geldanamycin . J Biol Chem . 1996 ; 271 : 22796 – 22801 .
72. Pharmacologic shifting of a balance between protein refolding and degradation mediated by Hsp90 . Proc Natl Acad Sci U S A . 1996 ; 93 : 14536 – 14541 .
73. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function . J Biol Chem . 2002 ; 277 : 39858 – 39866 .
74. Aggresome formation in liver cells in response to different toxic mechanisms: role of the ubiquitin-proteasome pathway and the frameshift mutant of ubiquitin . Exp Mol Pathol . 2001 ; 71 : 241 – 246 .
75. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation . Mol Biol Cell . 2001 ; 12 : 1393 – 1407 .
76. Molecular sequelae of proteasome inhibition in human multiple myeloma cells . Proc Natl Acad Sci U S A . 2002 ; 99 : 14374 – 14379 .
77. Tanespimycin with bortezomib: activity in relapsed/refractory patients with multiple myeloma . Br J Haematol . 2010 ; 150 : 428 – 437 .
78. Tanespimycin monotherapy in relapsed multiple myeloma: results of a phase 1 dose-escalation study . Br J Haematol . 2010 ; 150 : 438 – 445 .
79. Tanespimycin and bortezomib combination treatment in patients with relapsed or relapsed and refractory multiple myeloma: results of a phase 1/2 study . Br J Haematol . 2011 ; 153 : 729 – 740 .
80. Inhibition of heat shock protein 90 (HSP90) as a therapeutic strategy for the treatment of myeloma and other cancers . Br J Haematol . 2011 ; 152 : 367 – 379 .
81. Bortezomib-resistant myeloma cell lines: a role for mutated PSMB5 in preventing the accumulation of unfolded proteins and fatal ER stress . Leukemia . 2010 ; 24 : 1506 – 1512 .
82. New molecular and biological mechanism of antitumor activities of KW-2478, a novel nonansamycin heat shock protein 90 inhibitor, in multiple myeloma cells . Clin Cancer Res . 2010 ; 16 : 2792 – 2802 .
83. The anti-myeloma activity of a novel purine scaffold HSP90 inhibitor PU-H71 is via inhibition of both HSP90A and HSP90B1 . J Hematol Oncol . 2010 ; 3 : 40 .
84. Simultaneous inhibition of hsp 90 and the proteasome promotes protein ubiquitination, causes endoplasmic reticulum-derived cytosolic vacuolization, and enhances antitumor activity . Mol Cancer Ther . 2004 ; 3 : 551 – 566 .
85. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors . Nat Med . 2004 ; 10 : 1321 – 1328 .
86. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2 . Science . 2004 ; 303 : 844 – 848 .
87. Gene therapy for human small-cell lung carcinoma by inactivation of Skp-2 with virally mediated RNA interference . Gene Ther . 2005 ; 12 : 95 – 100 .
88. Novel roles of Skp2 E3 ligase in cellular senescence, cancer progression, and metastasis . Chin J Cancer . 2012 ; 31 : 169 – 177 .
89. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino,17-demethoxygeldanamycin in patients with advanced malignancies . J Clin Oncol . 2005 ; 23 : 4152 – 4161 .
90. Pharmacokinetics and pharmacodynamics of 17-demethoxy 17-[[(2-dimethylamino)ethyl]amino]geldanamycin (17DMAG, NSC 707545) in C.B-17 SCID mice bearing MDA-MB-231 human breast cancer xenografts . Cancer Chemother Pharmacol . 2005 ; 55 : 21 – 32 .
91. Rational design of shepherdin, a novel anticancer agent . Cancer Cell . 2005 ; 7 : 457 – 468 .
92. Targeting wide-range oncogenic transformation via PU24FCl, a specific inhibitor of tumor Hsp90 . Chem Biol . 2004 ; 11 : 787 – 797 .
93. Physiologically-based pharmacokinetics and molecular pharmacodynamics of 17-(allylamino)-17-demethoxygeldanamycin and its active metabolite in tumor-bearing mice . J Pharmacokinet Pharmacodyn . 2003 ; 30 : 185 – 219 .
94. 17AAG: Low target binding affinity and potent cell activity-finding an explanation . Mol Cancer Ther . 2003 ; 2 : 123 – 129 .
95. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors . Nature . 2003 ; 425 : 407 – 410 .
96. Affinity-based proteomics reveal cancer-specific networks coordinated by Hsp90 . Nat Chem Biol . 2011 ; 7 : 818 – 826 .
97. DT-Diaphorase expression and tumor cell sensitivity to 17-allylamino, 17-demethoxygeldanamycin, an inhibitor of heat shock protein 90 . J Natl Cancer Inst . 1999 ; 91 : 1940 – 1949 .
98. Formation of 17-allylamino-demethoxygeldanamycin (17-AAG) hydroquinone by NAD(P)H:quinone oxidoreductase 1: role of 17-AAG hydroquinone in heat shock protein 90 inhibition . Cancer Res . 2005 ; 65 : 10006 – 10015 .
99. NAD(P)H:quinone oxidoreductase1 (DT-diaphorase) expression in normal and tumor tissues . Cancer Metastasis Rev . 1993 ; 12 : 103 – 117 .
100. Discovery and development of Hsp90 inhibitors: a promising pathway for cancer therapy . Curr Opin Chem Biol . 2010 ; 14 : 412 – 420 .
101. Retaspimycin hydrochloride (IPI-504): a novel heat shock protein inhibitor as an anticancer agent . Expert Opin Investig Drugs . 2009 ; 18 : 1375 – 1383 .
102. Importance of the different proteolytic sites of the proteasome and the efficacy of inhibitors varies with the protein substrate . J Biol Chem . 2006 ; 281 : 8582 – 8590 .
103. Characterization of the transforming activity of p80, a hyperphosphorylated protein in a Ki-1 lymphoma cell line with chromosomal translocation t(2;5) . Proc Natl Acad Sci U S A . 1996 ; 93 : 4181 – 4186 .
104. Nucleophosmin-anaplastic lymphoma kinase (NPM-ALK), a novel Hsp90-client tyrosine kinase: down-regulation of NPM-ALK expression and tyrosine phosphorylation in ALK+ CD30+ lymphoma cells by the Hsp90 antagonist 17-allylamino,17-demethoxygeldanamycin . Cancer Res . 2002 ; 62 : 1559 – 1566 .
105. FLT3 tyrosine kinase as a target molecule for selective antileukemia therapy . Cancer Chemother Pharmacol . 2001 ; 48 ( suppl 1 ) : S27 – S30 .
106. Selective apoptosis of tandemly duplicated FLT3-transformed leukemia cells by Hsp90 inhibitors . Leukemia . 2002 ; 16 : 1535 – 1540 .
107. Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study . Blood . 2002 ; 99 : 3530 – 3539 .
108. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia . Cancer Cell . 2002 ; 2 : 117 – 125 .
109. 17-Allylamino-17-demethoxygeldanamycin (17-AAG) is effective in down-regulating mutated, constitutively activated KIT protein in human mast cells . Blood . 2004 ; 103 : 1078 – 1084 .
110. V600E B-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors . Proc Natl Acad Sci U S A . 2006 ; 103 : 57 – 62 .
111. Activated B-RAF is an Hsp90 client protein that is targeted by the anticancer drug 17-allylamino-17-demethoxygeldanamycin . Cancer Res . 2005 ; 65 : 10686 – 10691 .
112. Epidermal growth factor receptors harboring kinase domain mutations associate with the heat shock protein 90 chaperone and are destabilized following exposure to geldanamycins . Cancer Res . 2005 ; 65 : 6401 – 6408 .
113. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status . Nature . 2002 ; 418 : 934 .
114. Effect of geldanamycin on androgen receptor function and stability . Cell Stress Chaperones . 2002 ; 7 : 55 – 64 .
115. Mechanism of antiandrogen action: Key role of Hsp90 in conformational change and transcriptional activity of the androgen receptor . Biochemistry . 2002 ; 41 : 11824 – 11831 .
116. 17-allylamino-17-demthoxygeldanamycin induces the degradation of androgen receptor and HER-2/neu and inhibits the growth of prostate cancer xenografts . Clin Cancer Res . 2002 : 986 – 993 .
117. Hsp90 regulates a von Hippel Lindau-independent hypoxia-inducible factor-1 alpha-degradative pathway . J Biol Chem . 2002 ; 277 : 29936 – 29944 .
118. Geldanamycin induces degradation of hypoxia-inducible factor 1a protein via the proteasome pathway in prostate cancer cells . Cancer Res . 2002 ; 62 : 2478 – 2482 .
119. Geldanamycin inhibits migration of glioma cells in vitro: A potential role for hypoxia-inducible factor (HIF-1alpha) in glioma cell invasion . J Cell Physiol . 2003 ; 196 : 394 – 402 .
120. VEGF promotes survival of leukemic cells by Hsp90-mediated induction of Bcl-2 expression and apoptosis inhibition . Blood . 2002 ; 99 : 2532 – 2540 .
121. Modulation of the c-Met/hepatocyte growth factor pathway in small cell lung cancer . Clin Cancer Res . 2002 ; 8 : 620 – 627 .
122. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene . Cancer Cell . 2003 ; 3 : 347 – 361 .
123. Hepatocyte growth factor signalling stimulates hypoxia inducible factor-1 (HIF-1) activity in HepG2 hepatoma cells . Carcinogenesis . 2001 ; 22 : 1363 – 1371 .
124. Out of air is not out of action . Nature . 2003 ; 423 : 593 – 595 .
125. Molecular mechanisms of RET activation in human cancer . Ann N Y Acad Sci . 2002 ; 963 : 116 – 121 .
126. The RET proto-oncogene in human cancers . Oncogene . 2000 ; 19 : 5590 – 5597 .
127. RET and neuroendocrine tumors . Cancer Lett . 2004 ; 204 : 197 – 211 .
128. Inhibition of medullary thyroid carcinoma cell proliferation and RET phosphorylation by tyrosine kinase inhibitors . Surgery . 2002 ; 132 : 960 – 966 discussion 966–967 .