PMS2, MSH6
BMPR1A
PTEN
Hereditary Colorectal Cancer Syndromes and Molecular Pathways of Colorectal Carcinogenesis
Adenomatous polyposis coli Gene: Gatekeeper in Familial Adenomatous Polyposis and Sporadic Cancer
Familial Adenomatous Polyposis
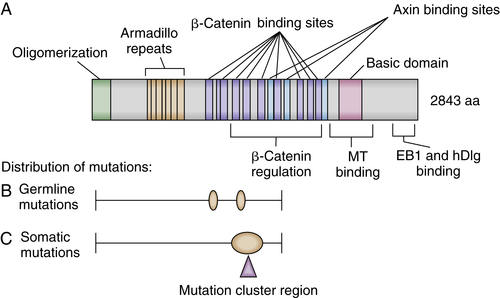
Somatic APC Mutations in Sporadic Tumors
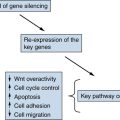
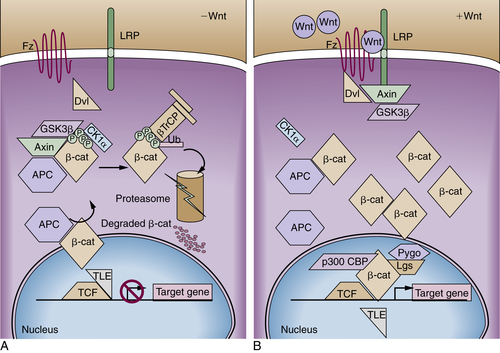
APC Protein Function
Mouse Models of FAP and Genetic and Epigenetic Modifiers
Other Forms of Intestinal Polyposis
DNA Mismatch Repair Deficiency and Lynch Syndrome
Inflammation and Colon Cancer
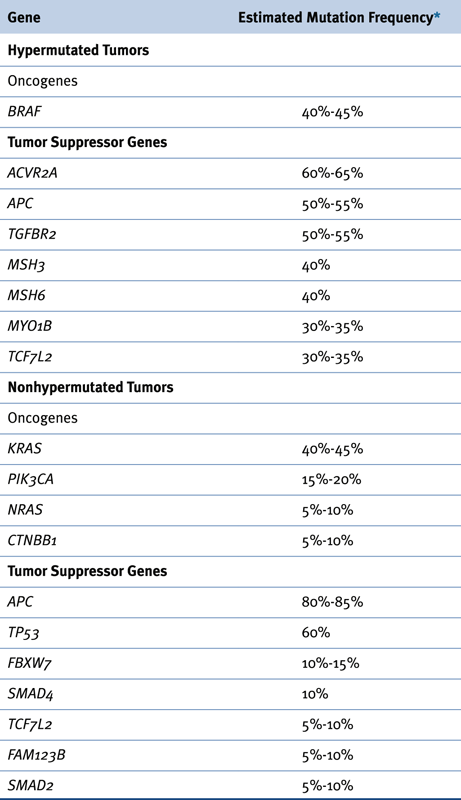
Recurrent Somatic Alterations in Colorectal Cancer
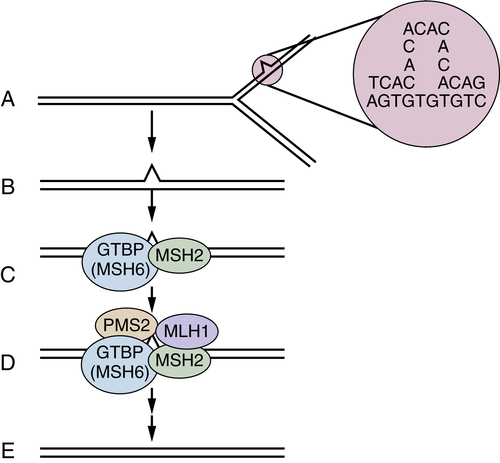
Genetic Instability: Chromosomal Instability versus DNA Mismatch Repair Deficiency
Epigenetic Changes in Colorectal Carcinogenesis: the CpG Island Hypermethylation Phenotype
Altered Gene Expression in Colorectal Cancer
Multistep Genetic Models of Colorectal Tumor Development
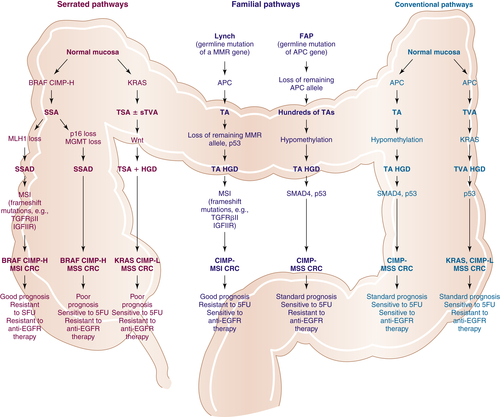
Clinical Applications of Molecular Genetic Insights
Risk Assessment
Early Detection
Prognostic and Predictive Markers
Summary and Future Directions
1. Cancer statistics, 2013 . CA: Cancer J Clin . 2013 ; 63 : 11 – 30 .
2. Colorectal cancer model of health disparities: understanding mortality differences in minority populations . J Clin Oncol . 2006 ; 24 : 2179 – 2187 .
3. Disparities in colorectal cancer in African-Americans vs whites: before and after diagnosis . World J Gastroenterol . 2009 ; 15 : 3734 – 3743 .
4. Hereditary and familial colon cancer . Gastroenterology . 2010 ; 138 : 2044 – 2058 .
5. Familial colon cancer syndromes: an update of a rapidly evolving field . Curr Gastroenterol Rep . 2012 ; 14 : 428 – 438 .
6. Primary prevention of colorectal cancer . Gastroenterology . 2010 ; 138 : 2029 – 2043 .
7. Colon cancer: a civilization disorder . Dig Dis . 2011 ; 29 : 222 – 228 .
8. Colon cancer screening in 2005: status and challenges . Gastroenterology . 2005 ; 128 : 1685 – 1695 .
9. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features . Histopathology . 2007 ; 50 : 113 – 130 .
10. The serrated pathway to colorectal carcinoma: current concepts and challenges . Histopathology . 2013 ; 62 : 367 – 386 .
11. Natural history of colorectal polyps and the effect of polypectomy on occurrence of subsequent cancer . Int J Cancer . 1990 ; 46 : 159 – 164 .
12. Guidelines for colonoscopy surveillance after polypectomy: a consensus update by the US Multi-Society Task Force on Colorectal Cancer and the American Cancer Society . Gastroenterology . 2006 ; 130 : 1885 .
13. Risk of proximal colon neoplasia with distal hyperplastic polyps: a meta-analysis . Arch Int Med . 2005 ; 165 : 382 – 390 .
14. Risk of proximal colorectal neoplasia among asymptomatic patients with distal hyperplastic polyps . Am J Med . 2005 ; 118 : 1113 – 1119 .
15. Progression of flat low-grade dysplasia to advanced neoplasia in patients with ulcerative colitis . Gastroenterology . 2003 ; 125 : 1311 – 1319 .
16. Nonpolypoid (flat and depressed) colorectal neoplasms . Gastroenterology . 2006 ; 130 : 566 – 576 .
17. The consensus coding sequences of human breast and colorectal cancers . Science . 2006 ; 314 : 268 – 274 .
18. Molecular genetics of colorectal cancer . Annu Rev Pathol Mech Dis . 2011 ; 6 : 479 – 507 .
19. . Comprehensive molecular characterization of human colon and rectal cancer . Nature . 2012 ; 487 : 330 – 337 .
20. Recurrent R-spondin fusions in colon cancer . Nature . 2012 ; 488 : 660 – 664 .
21. Mutations in the APC gene and their implications for protein structure and function . Curr Opin Genet Dev . 1995 ; 5 : 66 – 71 .
22. The molecular basis of Turcot’s syndrome . N Engl J Med . 1995 ; 332 : 839 – 847 .
23. Severe Gardner syndrome in families with mutations restricted to a specific region of the APC gene . Am J Hum Genet . 1995 ; 57 : 1151 – 1158 .
24. Correlation between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): a review of the literature . Crit Rev Oncol Hematol . 2007 ; 61 : 153 – 161 .
25. Prevention and management of duodenal polyps in familial adenomatous polyposis . Gut . 2005 ; 54 : 1034 – 1043 .
26. Surveillance and management of upper gastrointestinal disease in familial adenomatous polyposis . Fam Cancer . 2006 ; 5 : 263 – 273 .
27. Familial colorectal cancer in Ashkenazim due to a hypermutable tract in APC . Nat Genet . 1997 ; 17 : 79 – 83 .
28. Lessons from hereditary colorectal cancer . Cell . 1996 ; 87 : 159 – 170 .
29. Molecular determinants of dysplasia in colorectal lesions . Cancer Res . 1994 ; 54 : 5523 – 5526 .
30. The adenomatous polyposis coli protein: the Achilles heel of the gut epithelium . Annu Rev Cell Dev Biol . 2004 ; 20 : 337 – 366 .
31. Wnt/β-catenin signaling and disease . Cell . 2012 ; 149 : 1192 – 1205 .
32. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC . Science . 1997 ; 275 : 1787 – 1790 .
33. Canonical Wnt suppressor, Axin2, promotes colon carcinoma oncogenic activity . Proc Natl Acad Sci U S A . 2012 ; 109 : 11312 – 11317 .
34. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse . Science . 1990 ; 247 : 322 – 324 .
35. Secretory phospholipase Pla2g2a confers resistance to intestinal tumorigenesis . Nat Genet . 1997 ; 17 : 88 – 91 .
36. Suppression of intestinal neoplasia by DNA hypomethylation . Cell . 1995 ; 81 : 197 – 205 .
37. Suppression of intestinal neoplasia by deletion of Dnmt3b . Mol Cell Biol . 2006 ; 26 : 2976 – 2983 .
38. Suppression of intestinal polyposis in Apcdelta716 knockout mice by inhibition of cyclo-oxygenase 2 (COX-2) . Cell . 1996 ; 87 : 803 – 809 .
39. Prevention of colorectal cancer through the use of COX-2 selective inhibitors . Cancer Chemother Pharmacol . 2004 ; 54 : S50 – S56 .
40. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas . Nat Genet . 2013 ; 45 : 136 – 144 .
41. Hereditary mixed polyposis syndrome is caused by a 40-kb upstream duplication that leads to increased and ectopic expression of the BMP antagonist GREM1 . Nat Genet . 2012 ; 44 : 699 – 703 .
42. Heredity with respect to carcinoma . Arch Int Med . 1913 ; 12 : 546 .
43. Hereditary factors in cancer: study of two large Midwestern kindreds . Arch Int Med . 1966 ; 117 : 206 – 212 .
44. The tumor spectrum in the Lynch syndrome . Fam Cancer . 2005 ; 4 : 245 – 248 .
45. Genetic mapping of a locus predisposing to human colorectal cancer . Science . 1993 ; 260 : 810 – 812 .
46. Genetic mapping of a second locus predisposing to hereditary non-polyposis colon cancer . Nat Genet . 1993 ; 5 : 279 – 282 .
47. Microsatellite instability in the management of colorectal cancer . Expert Rev Gastroenterol Hepatol . 2011 ; 5 : 385 – 399 .
48. Intestinal inflammation and cancer . Gastroenterology . 2011 ; 140 : 1807 – 1816 .
49. Inflammatory bowel disease therapies and cancer risk: where are we and where are we going . Gut . 2012 ; 61 : 476 – 483 .
50. Inflammation and colorectal cancer: colitis-associated neoplasia . Semin Immunopathol . 2013 ; 35 : 229 – 244 .
51. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses . Nat Med . 2009 ; 15 : 1016 – 1022 .
52. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth . Nature . 2012 ; 491 : 254 – 258 .
53. Genetic instability in human cancers . Nature . 1998 ; 396 : 643 – 649 .
54. The chromosomal instability pathway in colon cancer . Gastroenterology . 2010 ; 138 : 2059 – 2072 .
55. Replication stress links structural and numerical cancer chromosome instability . Nature . 2013 ; 494 : 492 – 496 .
56. Functions of DNA methylation: islands, start sites, gene bodies and beyond . Nat Rev Genet . 2012 ; 13 : 484 – 492 .
57. Epigenetics of colorectal cancer . Gastroenterology . 2012 ; 143 : 1442 – 1460 .
58. Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types . Hum Mutat . 2012 ; 33 : 100 – 103 .
59. mRNA/microRNA gene expression profile in microsatellite unstable colorectal cancer . Mol Cancer . 2007 ; 6 : 54 .
60. Modulation of microRNA processing by p53 . Nature . 2009 ; 460 : 529 – 533 .
61. MicroRNA expression profiles classify human cancers . Nature . 2005 ; 435 : 834 – 838 .
62. Genome regulation by long noncoding RNAs . Annu Rev Biochem . 2012 ; 81 : 145 – 166 .
63. Vogelstein. A genetic model for colorectal tumorigenesis . Cell . 1990 ; 61 : 759 – 767 .
64. Fecal DNA versus fecal occult blood for colorectal-cancer screening in an average-risk population . N Engl J Med . 2004 ; 351 : 2704 – 2714 .
65. Next-generation stool DNA test accurately detects colorectal cancer and large adenomas . Gastroenterology . 2012 ; 142 : 248 – 256 .
66. DNA methylation patterns in blood of patients with colorectal cancer and adenomatous polyps . Int J Cancer . 2012 ; 131 : 1153 – 1157 .
67. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer . N Engl J Med . 2000 ; 342 : 69 – 77 .
68. Use of 5-fluorouracil and survival in patients with microsatellite-unstable colorectal cancer . Gastroenterology . 2004 ; 126 : 394 – 401 .
69. EGFR and KRAS in colorectal cancer . Adv Clin Chem . 2010 ; 51 : 71 – 119 .
70. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers . Nature . 2012 ; 486 : 537 – 540 .
71. , et al. The serrated pathway to colorectal cancer: current concepts and challenges . Histopathology . 2013 ; 62 : 367 – 386 .