[level-membership-for-opthalmology-category]
Chapter 76 White Spot Syndromes and Related Diseases
Introduction
The white spot syndromes (WSS) are a group of diseases characterized by inflammation and dysfunction of the outer retina, retinal pigment epithelium, choroid, or a combination of these. They often present a diagnostic and therapeutic challenge for clinicians and researchers. The etiologies of WSS remain unknown. This chapter discusses: birdshot chorioretinopathy (BCR), acute posterior multifocal placoid pigment epitheliopathy (APMPPE), serpiginous choroiditis, relentless placoid chorioretinitis, persistent placoid maculopathy, multifocal choroiditis and panuveitis (MFC), punctate inner choroidopathy (PIC), progressive subretinal fibrosis and uveitis syndrome (PSFU), multiple evanescent white dot syndrome (MEWDS), acute zonal occult outer retinopathy (AZOOR), acute idiopathic blind spot enlargement (AIBSE), and acute macular neuroretinopathy (AMN). Many entities are included in the differential diagnosis of these diseases. These include: granulomatous diseases, such as sarcoid, tuberculosis, sympathetic ophthalmia; masquerade syndromes like syphilis and intraocular lymphoma; infectious etiologies including toxoplasmosis, pneumocystis choroidopathy; and other entities such as presumed ocular histoplasmosis, and Behçet disease.1 In addition, in some cases, degenerative processes such as drusen may appear similar to the whitish-yellow lesions of WSS.
An autoimmune etiology has been hypothesized2 and proposed specifically for birdshot chorioretinopathy, acute zonal occult outer retinopathy (AZOOR), and multiple evanescent white dot syndrome (MEWDS).3 As yet, no characteristic pattern of antiretinal antibodies has been found. These entities need to be distinguished from entities with known neoplasias such as cancer-associated retinopathy (CAR) or melanoma-associated retinopathy (MAR). There are similar features including panretinal dysfunction, rapid progression, ERG changes, family history of autoimmune disease, retinal antibody activity on Western blot, and improvement in symptoms with immunosuppression. An increased prevalence of systemic autoimmunity in both patients with WSS and their first- as well as second-degree relatives, was found by Pearlman and colleagues.4 This suggests that this group of diseases occurs in families with inherited immune dysregulation that predisposes to autoimmunity.
It has also been suggested that several of these entities may be “related” or even represent a spectrum of the same process. Multifocal choroiditis (MFC), punctate inner choroiditis (PIC), MEWDS, acute macular neuroretinopathy (AMN), and AZOOR share commonalities and Gass referred to them as the AZOOR complex.5 These include female preponderance, zones of visual field loss that are usually contiguous to the blind spot, photopsias, and ERG changes such as reduced amplitudes. In addition, MFC and APMPPE have also been found in the same patient decades apart.6 Other diseases that have been noted in the same patient include MEWDS and AZOOR as well as MFC and PIC. This overlap may give support to a common underlying genetic predisposition.2
Birdshot chorioretinopathy
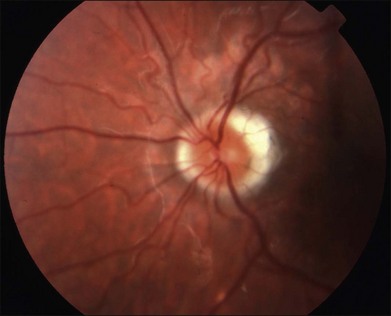
The term birdshot retinochoroidopathy was first used in 1980 by Ryan and Maumenee.7 It was a descriptive term for patients with multiple small, cream-colored fundus findings. These lesions are scattered around the optic disc and radiate to the equator in a “shotgun” pattern. Other terms such as vitiliginous chorioretinitis have been used.8 It has come to be known as birdshot chorioretinopathy (BCR) due to the histopathologic evidence that the primary lesions of the disease are in the choroid.9 It is a bilateral, chronic process with vitritis, retinal vasculitis, and cystoid macular edema (CME). Furthermore, it demonstrates the strongest link between any disease and any HLA class I antigen.10
Clinical course
Clinical symptoms
Patients present with complaints of blurred vision, floaters, and photopsias. Most have vision of 20/40 or better.9 The eye is generally not red or painful. Severe nyctalopia despite normal visual acuity may be a presenting symptom.11 Individuals also may describe an alteration in color vision12 or visual fields.13 Although Gass described a few patients who concurrently had vitiligo of the skin,8 BCR is thought to be a purely ocular disease.
Epidemiology
BCR is a rare chronic posterior uveitis. Shah and colleagues did an extensive review of English literature in 2005 and reported the following epidemiologic characteristics.9 Birdshot chorioretinopathy accounts for 0.6–1.5% of patients referred to tertiary centers for uveitis, or 6–7.9% of patients with posterior uveitis. There is a slight female predominance in the literature, at 54.1%. The mean reported age at the onset of disease is 53 years. It is generally not a disease of children. The oldest reported age at onset was 79 years. Patients are predominantly white; there have only been two reported exceptions. Finally, the HLA-A29 allele (which is present in about 7% of Caucasians) is strongly associated with BCR.14 The presence of this allele has been associated with a risk factor of 50–224 to develop BCR. Ninety percent or more of patients with BCR are HLA-A29-positive. Further sequencing of this allele has uncovered 11 subtypes. The HLA-A29*02 subtype is 20 times more prevalent in Caucasians than the HLA-A29*01 type, and the rest are exceedingly rare.15 HLA-A29*02 subtype is most commonly associated with BCR.
Fundus findings
It is important to recognize that symptoms can precede the onset of the classic fundus findings by several years.16 There is a range of presentations of the lesions associated with BCR. These birdshot lesions can be oval or round in shape, typically about 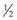 or
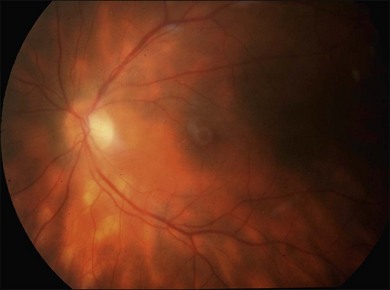
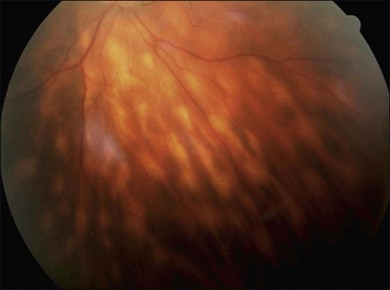
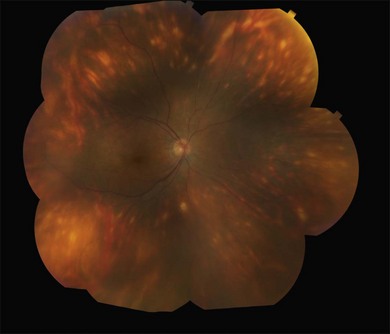
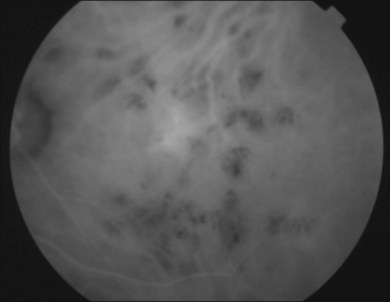
or 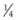 disc diameter in size. They appear deep to the retina (Fig. 76.1). They can be subtle, and asymmetric between eyes. The lesions can be distinct or poorly defined and can occasionally coalesce. They tend to cluster near the nerve and most commonly nasal and inferior to the disc.17,18 There is a pattern of linear radiation away from the nerve to the periphery7 (Fig. 76.2). They may appear to follow choroidal blood vessels peripherally. There is no hyperpigmentation or clumping noted. The retinal pigment epithelium (RPE) and overlying retina appear intact. There has been a report of these choroidal lesions preceding symptom onset in BCR, but this is not the norm.19
disc diameter in size. They appear deep to the retina (Fig. 76.1). They can be subtle, and asymmetric between eyes. The lesions can be distinct or poorly defined and can occasionally coalesce. They tend to cluster near the nerve and most commonly nasal and inferior to the disc.17,18 There is a pattern of linear radiation away from the nerve to the periphery7 (Fig. 76.2). They may appear to follow choroidal blood vessels peripherally. There is no hyperpigmentation or clumping noted. The retinal pigment epithelium (RPE) and overlying retina appear intact. There has been a report of these choroidal lesions preceding symptom onset in BCR, but this is not the norm.19
Other ocular findings
The anterior segment generally has minimal inflammation without prominent keratic precipitates. Fine keratic precipitates have been described in some series.18 Due to the mild nature of anterior segment inflammation, posterior synechiae do not occur. Signs of posterior inflammation include retinal vasculitis, optic disc edema, CME, and epiretinal membrane formation. Rhegmatogenous retinal detachment has been reported.7,20,21 It is unclear if this is related to this disease entity or to an association with uveitis. Other common findings include diffuse narrowing of the retinal arterioles, perivascular nerve fiber layer hemorrhages, and tortuosity of retinal vessels. Choroidal neovascularization (CNV) can occur.10,22,23 It has been postulated that this occurs due to the uveitic component causing CNV rather than ischemic factors.24,25
A consensus document on the diagnostic criteria for BCR was published in 2006.26 Required characteristics include: bilateral disease, presence of at least three peripapillary lesions inferior or nasal to the optic nerve in at least one eye, low-grade anterior segment inflammation (less than or equal to 1+ cells), and low grade vitreous inflammation (less than or equal to 2+ vitreous haze). Supportive findings include: HLA-A29 positive, retinal vasculitis, and CME. Finally, exclusion criteria include: presence of significant keratic precipitates, posterior synechiae, or diagnosis of infectious, neoplastic, or inflammatory disease that may cause multifocal choroidal lesions.
Clinical course and prognosis
The disease is chronic in nature and apparently does not regress. Again, symptoms of blurred vision, color deficiency, contrast sensitivity issues, and visual field problems may be present for years prior to the onset of fundus lesions. Visual acuity may be normal despite these complaints. It remains a poorly understood disease and no consensus on management and treatment has been found. Many patients have a slow decline in vision, despite treatment.9A final visual acuity of 20/40 or better in the best-seeing eye was reported in 75.1% of patients. However, 9.8% of patients were legally blind at follow-up (in the review of literature by Shah and colleagues9). Generally, macular edema was the commonest cause of visual decline in 50.5%.9 Choroidal neovascular membranes developed in 5% of eyes.22,23 These occur near the optic disc and can be bilateral. Finally, optic disc edema leading to atrophy can also affect visual prognosis.
Imaging
Fluorescein angiography
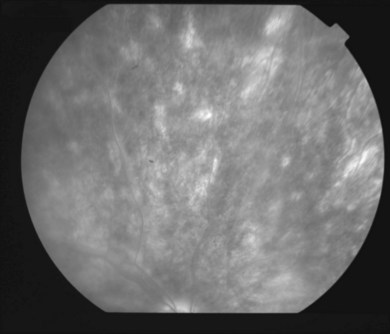
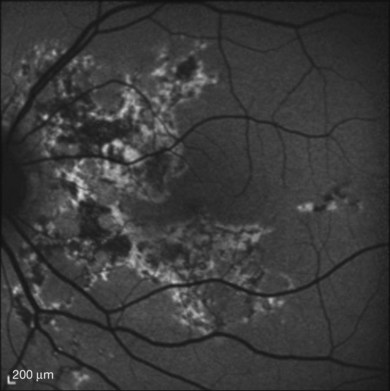
The creamy white spots have variable appearance on fluorescein angiography (FA) and the disease may be more evident clinically than with this mode of imaging (Fig. 76.3). The lesions can hypofluoresce in the early phase and there can be diffuse hyperfluorescence in the late phases (Fig. 76.4). One theory suggests that the lesions are likely in the outer choroid and associated with large choroidal vessels, thus many of the lesions show neither hypofluorescence or hyperfluorescence in any phase. The diffuse hyperfluorescence seen may represent a deep inflammatory focus that accumulates fluorescein.18 Angiographic findings may include increased transit time, leakage from retinal vasculature leading to CME, optic disc hyperfluorescence (Fig. 76.5), and disc or retinal neovascularization.8,27,28
Indocyanine green angiography
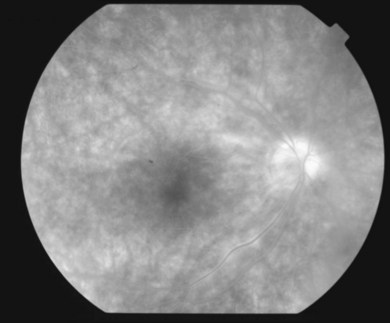
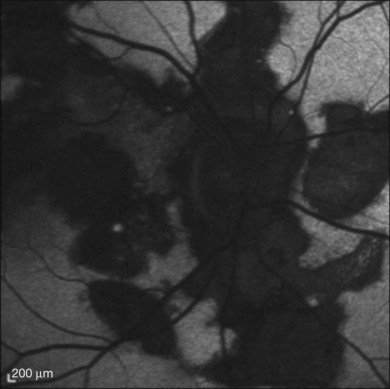
Indocyanine green angiography (ICGA) is an important diagnostic test. In the active disease,28 the birdshot lesions appear hypofluorescent during the intermediate phase of angiography and appear to be bordered by medium-to-large vessels28–30 (Fig. 76.6). The choroidal vessels appear indistinct. Late in the ICGA, there is diffuse choroidal hyperfluorescence. As with all the white spot syndromes, theories to explain this hypofluorescence include choroidal ischemia versus blockage from inflammatory infiltrates. However, in birdshot chorioretinopathy it is agreed upon that the site of the pathology is primarily the choroid. In the acute stages, the inflammatory infiltrates may be denser and block fluorescence. In late stages of the disease, it is thought that the lesions become more atrophic and choroidal vasculature may become more visible. The lesions can become more isofluorescent in this phase of the disease or they may remain hypofluorescent.27,28
Optical coherence tomography
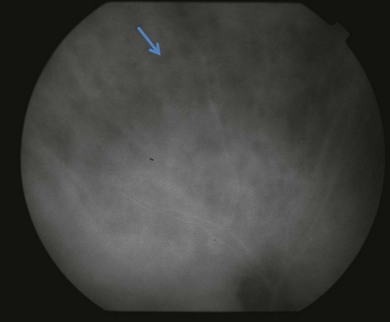
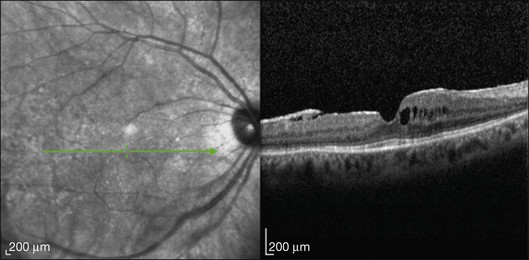
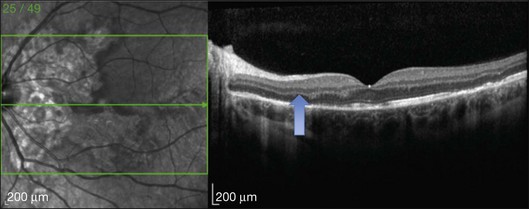
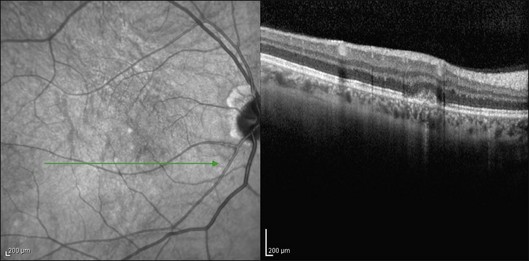
This mode of imaging has primarily been used to follow CME, which is the most common cause of vision loss in BCR31 (Fig. 76.7). Photoreceptor loss is not a prominent finding early on in the course.
Fundus autofluorescence
Giuliari and colleagues looked at fundus autofluorescence (FAF) of 18 eyes and found a spectrum of patterns.31 They found that hypoautofluorescent lesions on FAF did not necessarily correspond to clinical birdshot lesions. Commonly (15/18 eyes) more FAF lesions denoting RPE defects were seen than clinical lesions. Koizumi and colleagues looked at 8 patients. They found that some of these hypofluorescent lesions could be placoid in nature and involve the macula.32 In addition, a perivascular (retinal) linear hypofluorescent FAF pattern was noted by both groups of investigators.31,32 It was postulated that this finding may suggest that the retinal vessels may play a role in inflammatory-driven damage of the RPE and serve as a conduit for inflammatory factors. The observation that the clinical choroidal lesions did not always correspond to the FAF defects suggests that the choroid and RPE may be affected independently.32 Furthermore, RPE defects in the macula could also be another cause of vision loss in these individuals. FAF may be valuable in evaluating these patients as lesions may be clinically difficult to see.
Electrophysiology
Priem and coworkers also studied pattern-evoked cortical potentials and found abnormal findings in 53.3% of patients.33
Electroretinogram
Electroretinography (ERG) is an important test for following BCR. Abnormal electroretinograms (ERGs) were reported in 88.8% of patients.9 There appear to be two groups of patients: those who develop abnormal ERG early in the disease process and those that develop abnormal results late in the course.34 Early ERGs may demonstrate supernormal ERG amplitudes which may be related to retinal inflammation. In this stage, there may be a greater decrease in b-wave amplitude versus a-wave amplitude.21,34 This is a negative ERG pattern, and is not pathognomonic to BCR. It suggests that the Müller and bipolar cells are more affected than the photoreceptor–retinal pigment epithelium complex. This has been seen in autoimmune retinopathy. Rod dysfunction may occur before cone dysfunction: the rod b-wave may be affected prior to photopic b-wave and flicker response in most patients. In late phases of the disease, there is a progressive decrease in a- and b-wave amplitudes. ERG findings have been noted to improve with treatment.35 Some authors argue that the reversible nature of the ERG abnormalities suggest a non-ischemic etiology.15 Inflammation of the retinal vasculature could lead to inner retinal dysfunction, while choroidal inflammation could be the cause of altered outer retinal function.
Visual field testing
Visual field abnormalities are present in patients with BCR. These include: peripheral constriction, enlarged blind spot, central or paracentral scotomas, and generalized diminished sensitivity.8,17,18,20,33 It is not certain whether these visual field defects occur due to ganglion cell, optic nerve, or outer retinal dysfunction, but it is unlikely that the birdshot lesions themselves cause blind spots.9 Visual field abnormalities may be reversible with immunosuppression.36
Systemic associations
There are no definitive systemic associations. Shah and colleagues’9 review of the literature revealed hearing loss in some patients.8,37 As mentioned earlier, cutaneous vitiligo associated with BCR has also been reported in a few patients.8,18 In addition, Priem and Oosterhuis reported an increased incidence of vascular disease in their series. Of 102 patients, 16 had hypertension, 5 had coronary artery disease, 2 had a history of cerebrovascular accident, and 2 had a central retinal vein occlusion.18 These numbers are not impressive considering the age of the patients.
Pathogenesis
The pathogenesis of BCR is unknown. Inflammation appears to be a primary feature. The histopathological findings in a few eyes with BCR have been reported. These suggest that the spots may be related to accumulation of lymphocytes in the choroid at multiple levels, occasionally associated with hemorrhage.38 Some of the foci were adjacent to the choroidal vessels. The RPE, ciliary body, and iris were not involved. Some lymphocytes were found around the retinal blood vessels and in the optic disc. The lymphocytes were primarily CD8+T-lymphocytes. Inflammation is strongly associated with HLA-A29, which suggests a genetic predisposition to this disease. However the fact that HLA-A29 is present in 7% of the white population, and BCR is so rare tells us other factors are at work. Other genes are highly suspected. The more recent discovery of HLA class I-specific killer cell immunoglobulin-like receptors (KIR) led to epidemiological studies implicating KIR-HLA gene combinations in disease.39 The combination of KIR and HLA gene variants appears to increase the risk of developing BCR in HLA-A29-positive individuals. The presence of these in the absence of strong inhibition may activate natural killer cells and T cells against intraocular self-antigens thus inciting an autoimmune process. Familial history of autoimmunity is likely.2
Differential diagnosis
Birdshot chorioretinopathy can usually be distinguished from other disorders by history and physical findings. However, entities such as pars planitis, intraocular B-cell lymphoma, syphilitic chorioretinitis, sarcoidosis, sympathetic ophthalmia, and other white spot diseases, especially multifocal choroiditis and panuveitis syndrome, should be considered. Sarcoidosis (see Chapter 78, Sarcoidosis) and BCR may be the most difficult to distinguish from each other.9
Management/treatment
In the review of the literature by Shah and colleagues9 most cases were treated; however, no definitive guidelines for the initiation of treatment were given. This chronic progressive disease may not be sight-threatening in the early course, but macular edema with retinal damage, photoreceptor dysfunction, RPE atrophy, and optic nerve damage are the end results. Corticosteroids have been the mainstay of treatment. Oral, sub-Tenon’s, intraocular, and most recently sustained release fluocinolone acetonide40 have been used. Corticosteroids can reduce CME,7,21,41,42 inflammation,21 and optic disc edema.41 They have also been reported to decrease symptoms such as nyctalopia and issues with contrast sensitivity.11 Systemic steroids carry their own risks. Local steroid injections increase the risk of cataract and cause glaucoma with repeated dosing. Implantation of a fluocinolone acetonide sustained-release device has been shown to eliminate the need for systemic therapy, however it also has a high risk of cataract progression and glaucoma.40
Immunosuppressive therapy
Steroid-sparing agents have been used for long-term management of refractory cases. The three classes of immunosuppressives have been used. Cyclosporine has been used as it inhibits T lymphocytes and prevents S-Ag-induced experimental uveitis.10 Low-dose cyclosporine has proven to have positive visual effect in conjunction with steroids or alone.43 Nephrotoxicity is the primary side-effect and hypertension can be a problem.43 Antimetabolites such as azathioprine, mycophenolate mofetil, and methotrexate have been adjunctive or used in monotherapy. Side-effects of these drugs include bone-marrow suppression and hepatotoxicity.44 The use of the alkylating agents cyclophosphamide and chlorambucil has also been reported. Side-effects such as bone-marrow suppression and development of malignancies must be weighed for this class. Daclizumab, a monoclonal antibody against the alpha-subunit of the IL-2 receptor of T cells, has recently been found to have value in treating BCR.44,45 The long-term efficacy of these agents needs to be determined and weighed against potential side-effects.
Anti-VEGF therapy has been documented to be useful in treating CNV associated with inflammatory chorioretinal disorders.46
Placoid diseases
Acute posterior multifocal placoid pigment epitheliopathy
Acute posterior multifocal placoid pigment epitheliopathy (APMPPE) was first described in 1968 by Gass.47 He presented 3 healthy young female patients who developed the acute onset of bilateral central vision loss associated with multifocal placoid (plate-like) lesions at the level of the outer retina and the RPE, although he had also considered a primary choroiditis. Discussion regarding the site and etiology of the lesions would continue as the disease was further described in later years.48–57
Clinical course
Clinical symptoms
Patients present with rapid onset of central vision loss that may be described as blurred vision, paracentral scotoma, metamorphopsia, “spots” in the vision, and photopsias.58 Initial vision at presentation is 20/25 or worse in about 77% of eyes and 20/40 or worse in 58%.58 Deficits can be unilateral or bilateral (more common 75%).59 If unilateral, the second eye can become involved in a few days or weeks. Headaches, stiff neck, and malaise may accompany these ocular symptoms. A history of an antecedent viral syndrome or recent vaccination may be obtained.
Epidemiology
Males and females are equally affected and generally this occurs in young adults. Typically this presents between age 20 and 50 with the mean age of onset being 26.58,59 Recently, Taich and Johnson describe a syndrome resembling APMPPE in older adults (over age 50).60 These elderly individuals characteristically may have a worse outcome, with moderate or severe vision loss due to the development of geographic atrophy and CNV. These patients resemble the entity persistent placoid maculopathy (see below) and may not have APMPPE.
Fundus findings
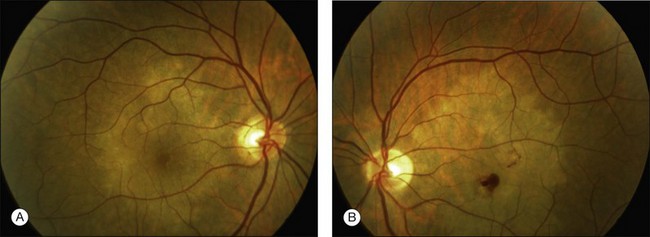
Gass described the presence of multiple round and confluent cream colored, flat lesions with indistinct margins scattered in the posterior pole. Lesions are not found anterior to the equator. The presence of these lesions is typically bilateral (Figs 76.8, 76.9). Fresh lesions can develop over the next few weeks, therefore lesions of differing ages can be visible. The placoid lesions tend to clear centrally initially leaving hypopigmentation. Later there is mild pigment mottling which develops into condensation of the pigment midperipherally, finally an increasing degree of coarse pigment clumping occurs (Fig. 76.10). These lesions can enlarge, but generally do not. In the original article, there was no description of serous macular detachment associated with APMPPE. However, later reports described localized serous retinal detachments over the lesions.52,55,61–65 This feature occasionally makes it difficult to distinguish APMPPE from Harada’s disease,62,66,67 and some have thought that these entities may form a spectrum of disease.68 It is more likely that there is an underlying common pathology that leads to the serous fluid.69 Gass commented on how remarkably the choroid and retina remain relatively intact during the course of the disease. However, Spaide described choroidal infiltrations in the periphery70 of a patient with acute APMPPE. In addition, multiple authors have described an association with retinal vasculitis71,72 and retinal vein occlusion has also been seen.71,72 Other findings can include subhyaloid hemorrhage72 and rare CNV.73 Case reports have also illustrated optic nerve involvement with disc edema.71,74
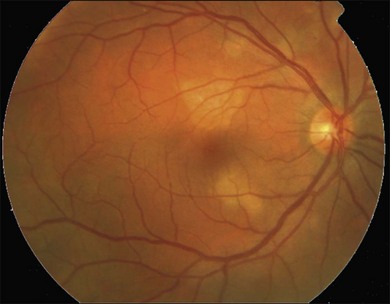
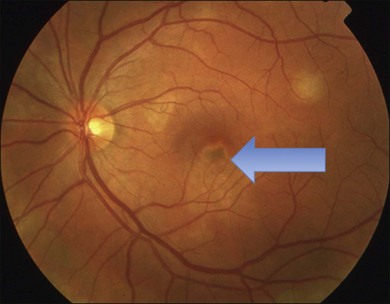
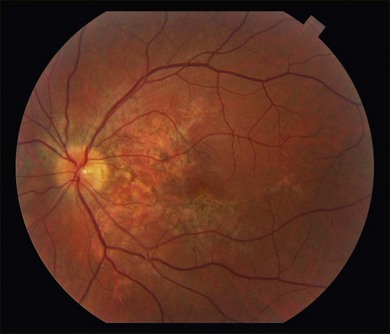
Other ocular findings
Although vitritis is not a significant component of APMPPE, the degree of ocular inflammation varies widely. An anterior uveitis75 and granulomatous anterior uveitis76 have been described. In addition, corneal stromal infiltrates77 have been mentioned.
Clinical course and prognosis
Generally there is improvement of the visual symptoms within 2–4 weeks. APMPPE has a relatively good prognosis when compared with other placoid white spot syndromes. However Fiore and colleagues58 reviewed the literature as well as a cohort from their own institution and showed that approximately 50% of patients have an incomplete recovery and 25% of patients have 20/40 vision or worse. Sixty percent of eyes have residual visual symptoms. Foveal involvement at presentation is an important prognosticator. Eighty-eight percent of eyes without foveal involvement proceed to full visual recovery in contrast to 53% of eyes that presented with foveal involvement. Unfortunately, approximately 70% of eyes present with foveal involvement. Photoreceptor involvement may ultimately limit visual prognosis. Progressive improvement of visual acuity usually follows the resolution of the lesions. Gass initially reported that visual recovery could continue on for months after the lesions resolved, even up to 6 months.47 Most visual recovery occurs within 1 month.49,51,52,55,58,61
Imaging
Fluorescein angiography
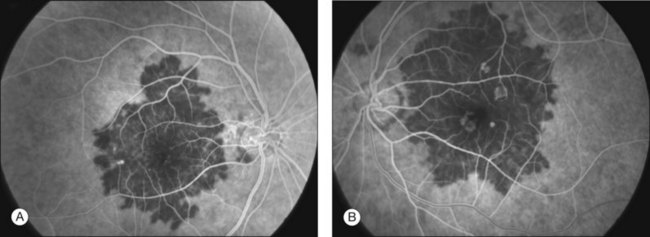
Gass47 described the lesions in the early phase as nonfluorescent (Fig. 76.11) and that the choroidal fluorescence is obstructed. Later in the angiogram there is a progressive, irregular staining of the lesions (Fig. 76.12). Ryan and Maumenee postulated that this could represent localization to the RPE or choriocapillaris rather than implying an infiltrate of the outer choroid.48 Gass also stated that the underlying choroidal circulation appeared intact below the healing lesions.47 However, later ICGA studies suggested that some choroidal hypoperfusion did exist.78,79 This is discussed below. As the process becomes inactive, hyperfluorescence corresponding to window defects in the RPE develops and staining is no longer evident (Fig. 76.13). There is a clear visibility of the large choroidal vessels in areas of confluent atrophy. However, this does not exist in all areas, implying some RPE remains intact.53 Either loss of the choriocapillaris or abnormal circulation in the confluent atrophied areas is suggested. In addition to these FA findings, there is also blocked fluorescence from pigment clumping.
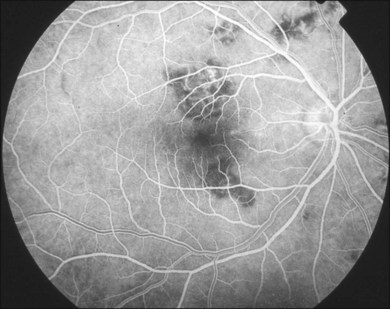
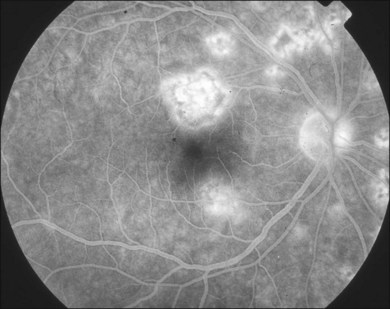
The retinal vessels and optic nerve appeared normal47,53 in original descriptions; however later reports described an association with retinal vasculitis, retinal vein occlusions, and disc edema.
Indocyanine green angiography
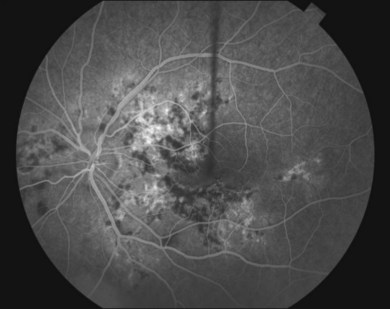
Since ICGA studies focus on the choroidal circulation, its findings in APMPPE have been important in developing theories of pathogenesis.79–84 Acute lesions show early hypofluorescence (Fig. 76.14). In the late phases, these lesions become more defined in shape. These areas are more numerous than the placoid lesions seen clinically. As the disease heals, the hypofluorescence in the late phase becomes less defined and smaller. This lends some support to the theory of choroidal ischemia as an underlying factor in the pathogenesis of APMPPE. It is postulated that there is more hypofluorescence in the acute phase due to the additional presence of swollen outer retina or RPE cells in response to choroidal ischemia (clinically presenting as placoid lesions). As these disappear, there is less blockage and thus less hypofluorescence. Photoreceptor damage may also play a role in this as well, as elucidated by OCT studies and discussed below. Studies show that the hypofluorescence in the late phases can also completely resolve,78 suggesting that choroidal vasculopathy, if present, may be a transient process.
Optical coherence tomography
Many studies have described the OCT findings in APMPPE.67,85–90 Garg and Jampol reported outer retinal abnormalities in APMPPE using time domain technology. A serous retinal detachment had reflective material within the subretinal fluid. It was postulated that this was either proteinaceous material or edematous RPE. There was rapid resolution of the serous detachment and the material disappeared.64
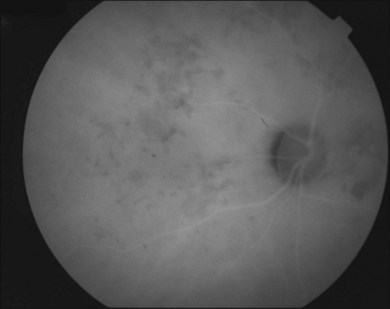
Lofoco and colleagues showed that in the acute phases the OCT revealed a mild hyperreflective area above the RPE in the photoreceptor layer corresponding to the placoid lesions. Later the OCT scan revealed a nodular hyperreflective lesion in the plane of the RPE with mild underlying backscattering. They theorized that the hyperreflective areas may indicate inflammatory tissue and inflammatory cells or the presence of ischemic edema in the outer retinal layers.90 As ultra-high resolution (UHR)-OCT developed, Scheufele and colleagues show disruption of the outer retina early in the disease (Fig. 76.15). RPE disruption occurs as the lesions heal.91 Backscatter of lesions was observed in the acute inflammatory phase in UHR as well. They found photoreceptor atrophy as the lesions began to heal and persistence of it post resolution. They suggested that the backscattering of acute lesions in the outer retina represents inflamed or damaged photoreceptor cell bodies. In addition to illustrating photoreceptor and RPE degeneration, spectral domain OCT has also shown that in some patients with fluid associated with the placoid lesions, that there may actually be accumulation of intraretinal fluid rather than an exudative retinal detachment.64,85
Fundus autofluorescence
FAF imaging is directed at the retinal pigment epithelial layer and is therefore especially useful in APMPPE. Several studies have described FAF findings67,87,89,92,93 (Fig 76.16).
Spaide compared angiographic findings with autofluorescence. He noted that the early hypofluorescence seen on angiogram did not match up with observable changes of the RPE and he suggested this means there are choriocapillaris perfusion defects. In the late phase of the angiogram, there was staining of some of the lesions. These late-staining lesions matched the size and shape of lesions seen in fundus autofluorescence. As the lesions resolved clinically, they became pigmented centrally with a depigmented halo. On autofluorescence, centrally there was intense hyperautofluorescence, and the depigmented halo was hypoautofluorescent, implying atrophy. He postulated there was a centripetal contraction of the placoid lesions that produced this appearance. He notes the autofluorescence changes lagged behind the clinical appearance. In addition, he found that choroidal abnormalities seemed more numerous on fluorescein and indocyanine green angiography. He concluded the RPE abnormalities were a result of the choroidal abnormalities.93
Electrophysiology
Although these functional studies are not essential in the diagnosis of APMPPE, they do emphasize the role of RPE involvement as described by Fishman and colleagues. The electroretinogram is normal to minimally subnormal. However, abnormal light:dark ratios have been documented on EOG, suggesting a diffuse RPE problem. Furthermore, the ERG and EOG abnormalities can normalize, suggesting that this can be a transient RPE problem.53
Systemic associations
APMPPE has been linked to CNS manifestations94–96 including cerebral vasculitis,97–103 meningo-encephalitis104 and stroke.105–109 Cases of APMPPE associated with CN VI palsy110 and transient hearing loss111 have also been cited. Headaches are a common symptom and APMPPE has mimicked migraine with aura.112 Although many patients give a history of viral illness, the symptoms of malaise and headaches may thus be more related to a widespread underlying vasculitis. Cerebrospinal fluid analysis has shown pleocytosis,113 which lends credence to this. The CNS associations are not benign and death, though uncommon, is possible.
Systemic vasculitis114 has been implicated in APMPPE and has been described in a P-ANCA-positive patient.115,116 Other associations with erythema nodosum,49,57 ulcerative colitis,117 thyroiditis,118 nephritis,119,120 and juvenile rheumatoid arthritis121 lead to an underlying immune mediated or inflammatory link. Other associations include granulomatous diseases such as: Wegener’s granulomatosis (granulomatosis with polyangiitis),122 pulmonary TB,123 sarcoidosis.97,124
Pathogenesis
When Gass first described APMPPE, he believed that the abnormalities were primarily at the level of the RPE. He believed that there was blockage of fluorescence due to damaged RPE cells, and these cells stained in the late phases. He stated that the retinal and choroidal circulations looked remarkably intact.47 Van Buskirk and coworkers first described an alternate theory that choroidal perfusion was the primary problem and that the hypofluorescence seen with angiography was due to lack of perfusion of the choriocapillaris.57 ICG studies found that the choroid did appear hypoperfused as described above. However, Fishman and colleagues’ study of ERG and EOG revealed that the ERG and thus the retina remained relatively intact compared to the EOG. Therefore, a diffuse RPE process was implicated in the acute phase of the disease. It also confirmed the transitory nature of this process as the EOG could normalize.53 The fact that the ERG was generally normal, did not favor a primary widespread choriocapillaris defect.
Currently, some believe that there is a vascular insult affecting the choroid that may cause partial choroidal ischemia leading to RPE damage and ultimately affect photoreceptors. Imaging studies support this. In addition, the systemic associations of this disease point to an underlying vasculitis. It is still possible that a primary process involving the outer retina and RPE could secondarily cause choroidal abnormalities on angiography. It appears that there may be a trigger, either inflammatory or infectious in nature, that incites this process. Furthermore, an association with HLA-B7 and HLA-DR2125 has been described and this suggests there are certain individuals who are more susceptible to this process.
Although exact etiology is unknown, there may be an association with viral illness;126 specifically, mumps127 and adenovirus have been mentioned.128,129 In half of the cases presented by Ryan and Maumenee there was an overt viral illness prodrome.48 In addition to viral infections, an association with bacterial infections has also been described. Two case reports implicate Lyme disease.130,131 Another case report described APMPPE post acute bacterial infection with group A streptococcus.132 APMPPE has also been found in cases post various vaccinations such as influenza,133 varicella,134 meningococcal C conjugate,135 and hepatitis B.136 Molecular mimicry has been implicated in the cases of post-vaccination APMPPE.133 Sequence similarities between the introduced antigens and RPE could incite an immune reaction.
A delayed-type hypersensitivity (DTH) reaction may bring all these associations together.82 In this process, there is an activation of sensitized T lymphocytes by various stimuli such as bacteria, viruses, and fungi. Previously primed T cells release lymphokines that then activate macrophages and cytotoxic T cells. Macrophages then give rise to epithelioid cells and giant cells that can lead to granulomas. The systemic associations such as CNS inflammation as mentioned above could be explained in this manner. The fluorescein and ICG findings could also be explained by a choroidal DTH reaction causing choroidal vascular occlusion.
A thrombotic process of the choroid has also been postulated. Elevated anticardiolipin antibodies were found in a patient with acute APMPPE. These levels were normal a year after resolution of the disease.137 The association with retinal vein occlusion supports a thrombotic etiology. Furthermore, elevated anticardiolipin antibodies have been associated with a number of viral infections and in many cases also associated with thrombosis.138
Differential diagnosis
The diagnosis of APMPPE is made clinically based on examination, time course, and imaging characteristics. Other white spot diseases are in the differential diagnosis and should be considered. These include: serpiginous choroidopathy (which should be considered in recurrent, chronic cases), relentless placoid chorioretinitis, which should be thought of in severe, persistent, and recurrent cases, and multifocal evanescent white dot syndrome. Other considerations include: diffuse unilateral subacute neuroretinitis, Harada’s disease,62,66–68 TB, sarcoid, fungal disease, choroidal metastasis or lymphomatous infiltrate,48 and syphilis.139
Management/treatment
Laboratory investigations are usually not necessary. Certain investigations may be useful in questionable cases. Spinal fluid analysis for pleocytosis,114 anticardiolipin antibodies,137 and association with HLA-B7 and HLA-DR2125 are among them.
No therapy has been proven useful in the management of acute APMPPE. A review of the data from the literature on corticosteroids does not provide clear direction.58 However, some make a case for intravenous steroids and immunosuppressive agents to be considered when there is central nervous system involvement.140
Rarely CNV can develop. Bruch’s membrane appears less affected in APMPPE compared to serpiginous choroiditis, and therefore CNV is less prevalent. Anti-VEGF agents have been found to be useful in treating CNV.141,142 Photodynamic therapy has been used as well; however, its use is cautioned as it can exacerbate inflammation and possible choroidal ischemia, which are present in the acute stages of APMPPE.73
Serpiginous choroiditis
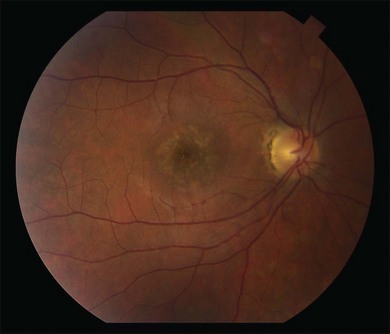
Serpiginous choroiditis (SC) also known as helicoid peripapillary chorioretinal degeneration,143 geographic helicoid peripapillary choroidopathy,144 and geographical choroidopathy145 is a rare disease of unknown etiology. It is usually bilateral, chronic, and progressive. The lesions involve the outer retina, RPE, choriocapillaris, and large choroidal vessels.146 Patients present with acute geographic or serpentine lesions that are gray or gray–yellow (due to disruption of the RPE or outer retina).
Clinical course
Clinical symptoms
Patients are often asymptomatic until the lesions affect the fovea. Blurred vision occurs at this point.147,148 Although this is a bilateral disease, typically the patient becomes symptomatic unilaterally with foveal involvement. Small central or paracentral scotomas may be present. These are absolute in their acute stages, and become relative with healing.148
Epidemiology
SC is a disease of healthy individuals. The onset is usually between the ages of 30 and 70. However, cases of SC have been reported in younger individuals.148 Most reports of SC involve Caucasians; however, SC has also been reported in Asians,149 African Americans,148 and Hispanics.150 The literature reflects a slight predilection for males.146 No family history is elicited. One study done in a Finnish population showed a higher prevalence of HLA-B7 in patients with SC.151
Fundus findings
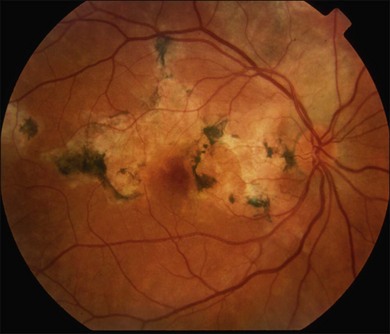
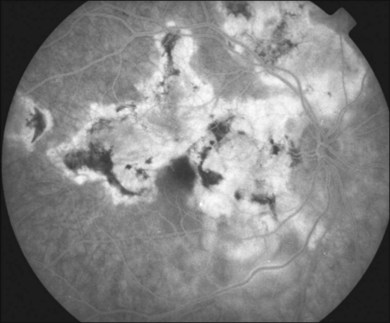
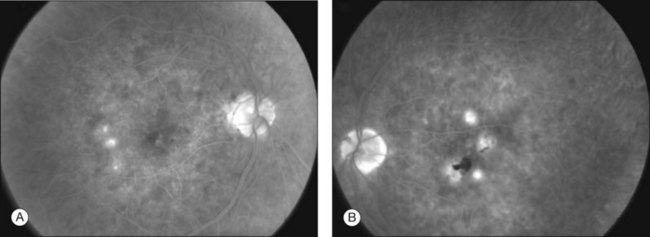
Two variants of SC are seen. The classic appearance, seen in approximately 80% of cases,146 starts with geographic patches of gray or creamy yellow placoid lesions in the peripapillary region. It generally progresses in a centrifugal manner with finger-like or serpentine projections. In one report, a centripetal direction was described.147 The outer retina appears edematous, and serous retinal detachments may be seen.152,153 As these active lesions resolve (with or without) treatment over the next several weeks, extensive RPE and choriocapillaris atrophy occurs (Fig. 76.17). SC has recurrences and therefore it is common to see lesions of different ages. Recurrences can occur at the edge of old scars (Fig. 76.18), but not always. As the disease becomes chronic, chorioretinal atrophy, subretinal fibrosis, and RPE clumping is seen.146 Since most patients become symptomatic only when the fovea becomes involved, about two-thirds present with bilateral chorioretinal scarring.154
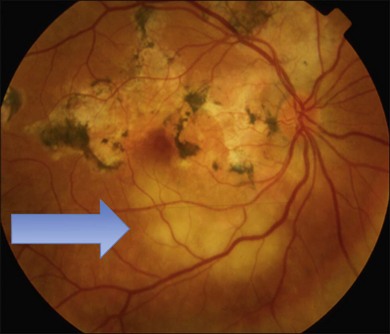
Macular serpiginous choroiditis149,150 is the second variant (Figs 76.19–76.22). There is no difference in the lesions of macular serpiginous and peripapillary diseases except for location.151 There is generally a poorer prognosis for the macular variant as the fovea is affected at the start. Vision loss may also be related to presence of CNV.150 However, Sahu and coworkers, in a more recent report, did not observe CNV in their series.155 Macular SC may be misdiagnosed as macular degeneration, macular dystrophies, or toxoplasmosis.146
Other ocular findings
Although serpiginous choroiditis is not usually associated with a marked inflammatory response, nongranulomatous anterior uveitis156,157 has been seen. More commonly – estimated in about a third of patients – there are fine vitreous cells.153 The eye is white. Additional retinal findings have been described. These include CNV, which affects 13–20% of eyes,158–163 vein occlusions,164,165 retinal vasculitis,165 usually a periphlebitis, CME,166 and bilateral full-thickness macular holes.167 Optic nerve abnormalities such as optic disc edema147 and disc neovascularization162 can be seen at the active stage of serpiginous choroiditis.
Clinical course and prognosis
Serpiginous choroiditis is characterized by multiple recurrences at intervals of months to years. Healing of individual lesions takes place in 2151 to 8 weeks,146 but new lesions appear later.147 Generally acute lesions are seen in one eye at a time.
Visual loss is correlated with proximity to the fovea. There can be some incomplete recovery, but due to the recurrences, 75% of patients develop central visual loss in one or both eyes. Final visual acuity is less than 20/200 in up to 25% of eyes regardless of treatment.168
Imaging
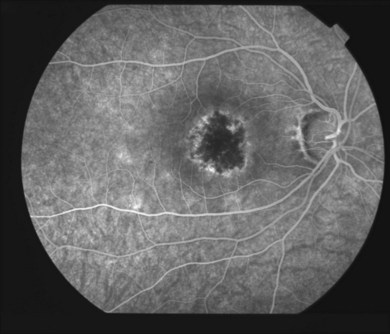
Fluorescein angiography
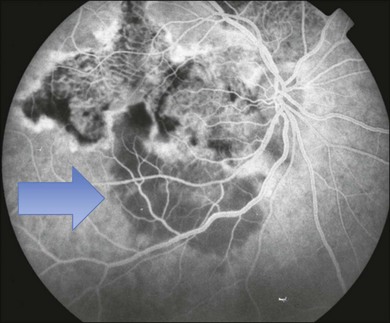
In the acute SC, the lesions show hypofluorescence during the early phase of the study144 (Fig. 76.23). This is likely due to a combination of blockage by swollen outer retina and RPE and nonperfusion of the choriocapillaris. The borders of the lesion may be hyperfluorescent, representing intact choriocapillaris. As the study progresses, the previous hypofluorescent lesions become variably hyperfluorescent and this intensifies over time representing staining of the acute lesions (Fig. 76.24). Retinal vascular staining may be seen near active lesions. Pigmentary changes then develop and there is often pronounced atrophy. Angiography at this stage shows mottled hyperfluorescence with increased fluorescence in the late phases representing leakage of dye from the damaged choriocapillaris at the periphery of the lesion. CNV can occur and appears as late leakage, usually at the edge of a scar.
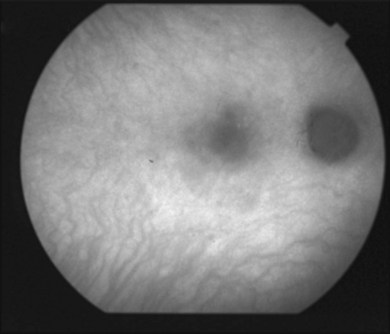
Indocyanine green angiography
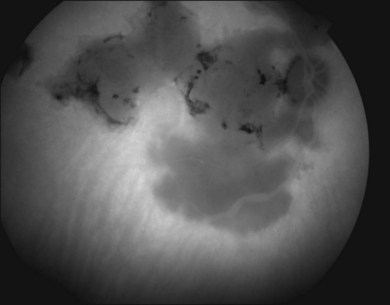
ICGA169–172 is useful in evaluating SC. Giovannini and colleagues describe how ICGA allows a greater understanding of the disease. In particular, ICGA allowed: (1) better staging of SC, revealing choroidal alterations when there was no clinical or FA evidence; (2) better identification of the active lesions, which appear to be larger at the choroidal level in comparison with the corresponding retinal lesions; and (3) persistence of choroidal activity even when the signs of retinal activity had disappeared.171 The ICG pattern is characterized by hypofluorescent areas persisting from early to late phases (Fig. 76.25). The hypofluorescence has been reported to be less pronounced in later phases, which may represent delayed perfusion rather than nonperfusion.173 The hypofluorescent lesions appear more extensive on ICGA in comparison to clinical examination and fluorescein angiography.
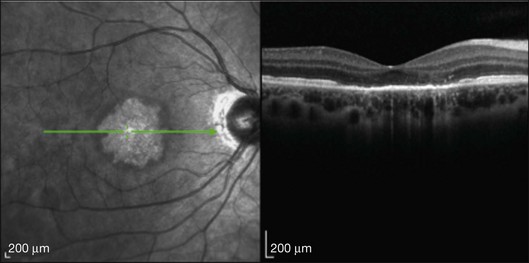
Optical coherence tomography
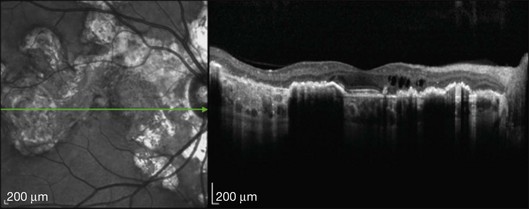
OCT findings have been described recently.174–178 Spectral domain OCT illustrates retinal atrophy with disruption of the photoreceptor layers in affected areas. There is thinning of the RPE and intraretinal fluid including cystic changes can be seen.174 There is increased reflectance of the choroid and the deeper retinal layers. Disruption of the photoreceptor inner/outer segments is seen in active and inactive lesions175 (Fig. 76.26).
Fundus autofluorescence
As SC involves the RPE, FAF175,178,179 images are helpful in demarcating this disease. New lesions appear hyperautofluorescent and old lesions are hypoautofluorescent (Fig. 76.27). Recurrences are characterized by hyperautofluorescent lesions at the borders of old hypoautofluorescent areas.179 Hyperautofluorescence was detected 2–5 days after the appearance of acute lesions.178 Piccolino and colleagues compared FAF to ICGA and OCT and found that the acute hyperautofluorescent areas were less extensive than the perfusion defect delineated by ICGA. When compared to OCT, there was increased reflectance of the photoreceptor layer corresponding to the area of hyperautofluorescence. As the disease progressed, the hyperautofluorescent area became hypoautofluorescent and the photoreceptor changes persisted.
In developing countries, especially India, individuals with evidence of tuberculosis may have serpiginous-like lesions. (See differential diagnosis section below and Chapter 82, Mycobacterial infections). FAF appears to distinguish the lesions of SC from TB-related disease. TB-related diseases have a stippled hypoautofluorescence corresponding to lesions, which is not as homogenous as SC.179
Electrophysiology
Results of ERG are normal,148,153 and EOG is abnormal only when there is extensive disease.153
Perimetry
Goldmann visual field examination shows that scotomas corresponding to an area of activity are not permanent.148 Scotomas corresponding to fundus lesions are present, but are not absolute and usually are denser centrally and less peripherally. More recently microperimetry180 studies confirm this finding.
Systemic associations
Reports have described SC in patients with various systemic diseases: Crohn’s disease,181 SLE,182 celiac disease,183 and extrapyramidal dystonia.184 These are likely coincidental associations. Systemic vasculitides can cause choroidal ischemic syndromes, which may resemble SC.
Pathogenesis
The pathogenesis of serpiginous choroiditis remains unknown. Postulated etiologies have included autoimmune, infectious, vascular, and degenerative.146
The inflammatory nature of the disease is supported by histopathology with inflammatory lymphocytic infiltrate in the choroid (more prominent at the margin of the lesion).161,185 Clinically, the presence of anterior uveitis, vitritis, and retinal vasculitis also points to an inflammatory etiology. Like APMPPE, the report of an association with HLA- B7 suggests some individuals are more susceptible to this process.151 Broekhuyse found a sensitization to S-antigen in SC patients.186 Acute SC lesions respond to steroids and immune modulating agents consistent with an immune-mediated process.
A microbial origin of SC has been suggested, but the evidence is not convincing. Herpes virus has been found in aqueous humor187 and VZV and HSV DNA were detected in the aqueous of a subset of patients with serpiginous choroiditis, suggesting that these viruses may be involved. Conversely, Akpek and Chan described the lack of herpes virus DNA in choroidal tissues at autopsy of a patient with SC.185 No viral DNA was amplified, including HSV, CMV, EBV, VZV, or HHV-8. Furthermore, there are reports of recurrences while on antiviral therapy.168 Pisa and colleagues studied 5 patients with SC to assess the presence of fungal infection by the presence of antibodies in human serum samples.188 Antibodies against Candida were apparent in 4 of the 5 patients.
Vasculopathy, either primary or secondary, has also been proposed.146,173,189 The clinical presence of phlebitis and branch retinal vein occlusions supports this etiology. King and colleagues noted elevated factor VIII in association with SC.190 This could lead to an occlusive vascular phenomenon due to vascular endothelial injury as seen in diseases such as scleroderma, Raynaud disease, polymyalgia rheumatica, and temporal arteritis. Fluorescein and ICGA also suggest vascular involvement. Hayreh’s early work showed that closure of the cilioretinal vessels could produce lesions that look something like SC.191 However, the lack of systemic vascular disease in SC does not support vasculopathy.
Finally, a degenerative etiology has been proposed. The nature of SC’s chronic and progressive nature and later onset in life lends some support to this.146 One case showed an association with unilateral extrapyramidal dystonia,184 which could be supportive or coincidental. The presence of inflammation and lack of familial inheritance, the sporadic nature of SC, and occasional recovery of vision are not supportive of a degenerative condition.
Differential diagnosis
Tuberculous serpiginous choroiditis (see Chapter 82, Mycobacterial infections) can resemble idiopathic serpiginous choroiditis. TB-related serpiginous choroiditis is associated with more vitritis compared to serpiginous choroiditis. In addition, patients with TB choroiditis have more multifocal lesions involving the periphery. SC has larger lesions compared to TB and is more likely to have lesions extending from the optic nerve head.192 Fluorescein angiography does not distinguish between these two entities. Fundus autofluorescence may. Hypofluorescence corresponding to RPE loss in SC is more homogeneous compared to the variegated pattern and stippled hyperautofluorescence of tuberculous disease.178
Other diagnostic considerations include: sarcoidosis,193 systemic non-Hodgkin lymphoma,194 antiphospholipid antibody syndrome,195 toxoplasmosis,196 syphilis, and posterior scleritis.197
With macular SC, one must think about choroidal ischemia in the differential diagnosis.150 This can occur with systemic vascular diseases such as systemic lupus erythematosus, toxemia of pregnancy, disseminated intravascular coagulation, thrombotic thrombocytopenic purpura, and malignant hypertension as an etiology for macular vision loss.
Management/treatment
Although the diagnosis is suspected by clinical history, examination, and imaging characteristics, a preliminary workup could include TB skin test, chest X-ray, quanterferon gold test if TB is strongly suggested,198 ACE, VDRL, fluorescent treponemal antibody absorption test, toxoplasma titers, and a viral screen. Abrez and colleagues suggest an aqueous tap if anterior chamber cells are noted with an evaluation by PCR for viral etiology.157
As serpiginous choroiditis is a rare disease, controlled trials comparing treatments are not available. Much of what we know comes from small case series.199 Due to the relapsing and progressive nature of SC, treatment should be aimed at both the acute inflammatory episode as well as preventing recurrences.
Steroids have been a mainstay of treatment and multiple routes of administration have been described. Oral prednisone,152 sub-Tenon’s triamcinolone, intravenous pulse methylprednisolone therapy,200 intravitreal triamcinolone acetonide,201–204 and intravitreous fluocinolone acetonide implant.205 Systemic prednisone therapy of 60–80 mg/day is a commonly prescribed therapeutic regimen. Aggressive corticosteroid therapy is useful in treating acute attacks, but since the steroids are tapered, is not useful in preventing recurrences. Relapses are common when tapering the medication. A case report described using an intravitreous fluocinolone acetonide implant which resulted in ongoing control of the disease for 14 months postoperative follow-up.205 This delivery route avoids the side-effects of systemic steroids. However, cataract and glaucoma from this route of administration are often seen.
Corticosteroids can be used alone, or in combination with other immunosuppressive therapy.206,207 Antibiotics (cyclosporine),208,209 antimetabolites (azathioprine),210 alkylating agents (cyclophosphamide and chlorambucil)211,212 have been used. Cyclosporine alone has been used with mixed results. Triple-agent immunosuppression with cyclosporine, azathioprine, and prednisone was described by Hooper and Kaplan.213 They reported 5 patients with bilateral SC in whom visual recovery was promoted by this regimen. The therapy was given for 8 weeks prior to tapering. Two patients relapsed. Another study reported 4 patients maintained on a low dose of triple therapy for 12–69 months. Three of 4 patients did achieve remission off the drugs.206 Alkylating agents have shown some promise, but side-effects including bone-marrow suppression, nausea, fatigue, as well as one case of development of transitional epithelial carcinoma of the bladder, did occur.211 For this reason, these agents should probably be reserved for sight-threatening disease that has failed conventional immunosuppressive therapy.146
Other more recent therapies have included infliximab214 and interferon-alpha-2a.215 They appear promising, but the risk of systemic side-effects should be weighed. Patients receiving systemic management for SC are often jointly managed with a rheumatologist, internist, or oncologist.
Treatment is also aimed at managing complications of SC. These include CNV and CME. Reports have described the use of anti-VEGF agents for inflammatory chorioretinal disorders.216–219 CNV in SC appears to be responsive to this treatment. Photodynamic therapy220,221 has also been used with some success, however the clinician should be mindful that choroidal ischemia and inflammation is inherent in SC and could be exacerbated by this treatment. For extrafoveal CNV, thermal photocoagulation is possible.
Acetazolamide222 may be useful in the management of CME related to SC. It is thought that CME develops due to RPE dysfunction. After 2 weeks of treatment, there was complete resolution of CME with improvement of visual acuity in one case report.
Follow-up can include Amsler grid use by the patient to monitor for relapses and foveal involvement.
Relentless placoid chorioretinitis
Relentless placoid chorioretinitis (RPC) was described in 2000 by Jones and colleagues.223 They reported 6 patients resembling both APMPPE and serpiginous choroiditis, but with an atypical time course and retinal distribution. The term ampiginous has been used to describe similar patients.224 It is likely that some previous descriptions of multifocal serpiginous225 and recurrent APMPPE represent this entity.226
Clinical course
Clinical symptoms
The commonest complaint is sudden painless blurring.227 Patients also describe metamorphopsia, floaters, or can be asymptomatic.
Epidemiology
Patients were aged 17–51 in the report by Jones and colleagues.223 There was no gender predilection. In a series by Jyotirmay and colleagues, a male preponderance was found with a mean age of 34.227 The patients have no consistent medical disorder or viral prodrome.
Fundus findings
In most cases, patients have bilateral posterior creamy-white lesions at the level of the RPE (Fig. 76.28). The lesions (in almost all cases) tend to be smaller than those of APMPPE (approximately 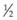 disc area)227 (Fig. 76.29). Lesions can be active in both eyes simultaneously. As they heal, pigmented chorioretinal atrophy develops. Lesions can persist and grow. A hallmark of this disease is the eventual presence of numerous (>50 to hundreds) lesions with involvement anterior and posterior to the equator. This is in contrast to APMPPE which is limited to the posterior pole. One study found that lesions more commonly appear in the periphery first and in the posterior pole later.227 The fovea is commonly involved.
disc area)227 (Fig. 76.29). Lesions can be active in both eyes simultaneously. As they heal, pigmented chorioretinal atrophy develops. Lesions can persist and grow. A hallmark of this disease is the eventual presence of numerous (>50 to hundreds) lesions with involvement anterior and posterior to the equator. This is in contrast to APMPPE which is limited to the posterior pole. One study found that lesions more commonly appear in the periphery first and in the posterior pole later.227 The fovea is commonly involved.
Other findings have included a mild vitritis, occasional subretinal fluid, and disc swelling.223 In their study of 26 eyes in 16 patients, Jyotirmay and colleagues found subretinal fibrosis, as well as epiretinal membranes as rare findings.227 Interestingly, CNV was not described in either case series.222,227,228
Other ocular findings
Jones et al. described a patient with herpetic keratitis and corneal infiltrates in their series. Iritis with keratic precipitates as well as episcleritis can also occur.223,228
Clinical course and prognosis
Permanent vision loss is usually mild. However, central vision was affected in all untreated cases. Vision dropped as much as 6 lines in the acute stage in Jones et al.’s study.223 Some patients showed improvement in vision. Patients who received prolonged systemic steroids seemed to have decreased activity and improved visual outcome. Only 2 out of 6 patients had a final vision worse than 20/40 in the original series. Jyotirmay and colleagues report a favorable visual outcome in over 96% of their patients.227
Imaging
Optical coherence tomography
There has been only one report. During the active stage of the disease with foveal lesions, the OCT showed subfoveal fluid.229 In addition, a pigment epithelial detachment with hyperreflectivity of the inner and outer retinal layers was present. Further study is warranted.
Electrophysiology
A decrease in the electrooculogram and electroretinogram results (scotopic, photopic, and flicker) has been reported.223 Other reports do not show this finding.229
Systemic associations
In Jones and colleagues’ case series, one patient had Hashimoto thyroiditis and aseptic meningitis. Two other patients presented with nonspecific upper respiratory tract infections.223 Jyotirmay et al.’s series reported 2 patients with Hashimoto thyoroiditis, and one patient with type 1 diabetes mellitus.227 There has been a recent case report of RPC associated with central nervous system lesions in a 20-year-old male.231
Pathogenesis
Choroidal ischemia either primary or secondary due to an inflammatory etiology is a consideration, as it is in APMPPE and SC. The presence of RPC in a patient with CNS lesions may indicate a small-vessel vasculitis.231
Differential diagnosis
APMPPE and serpiginous choroiditis are the main considerations (Table 76.1). Time course as well as the number and location of the lesions distinguish these diseases. There may be some resemblance to other white spot diseases including: multifocal choroiditis and birdshot chorioretinopathy. Other processes such as Harada’s disease, neoplastic infiltration of the choroid, infections such as syphilis, and granulomatous diseases such as tuberculosis and sarcoidosis can also be considered.223
Management/treatment
The best regimen is as yet unknown. Jones et al. described decreased activity and improved vision in patients who were treated with prolonged steroids versus patients who were untreated or treated less aggressively.223 Antiviral therapy was not effective. Immunosuppression with cyclosporine was also described, but recurrence was noted when it was tapered. Mycophenolate mofetil with prednisone was used in one patient who also had CNS lesions.232 Jyotirmay and colleagues used steroids (including sub-Tenon’s) in combinations with azathioprine, or cyclophosphamide.
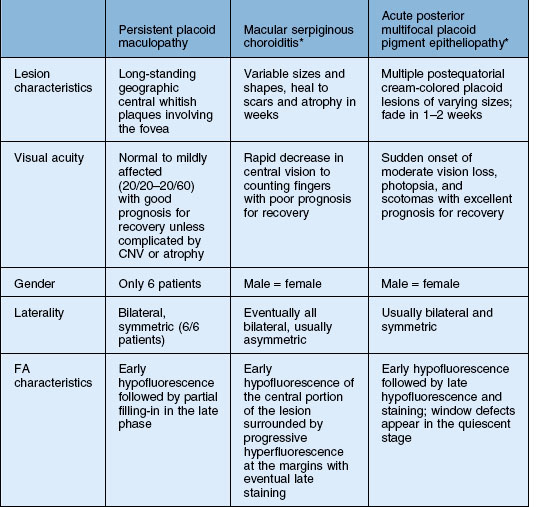
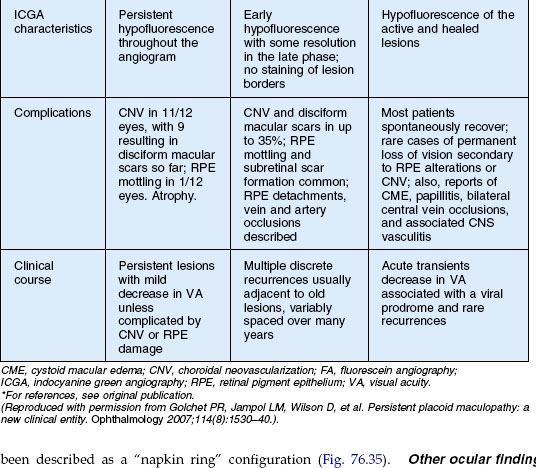
Persistent placoid maculopathy
Persistent placoid maculopathy (PPM) was recently described by Golchet and colleagues. It superficially resembles macular serpiginous choroiditis but differs in its clinical course and visual prognosis.233 CNV is a common feature of PPM, and usually the major cause of visual loss.
Clinical course
Clinical symptoms
Patients present with gradual, painless, decreased vision more commonly in one eye. Photopsias and decline in color vision can be seen.233
Epidemiology
In the original description there were 6 patients. Five patients were men and one was a woman. The range of ages was 50–68. All patients were Caucasian. There were no consistent systemic medical problems.233
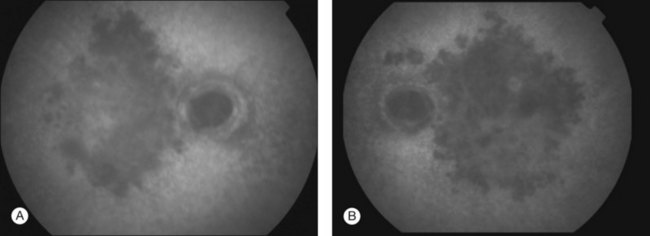
Fundus findings
Patients have bilateral symmetric whitish plaque-like lesions at the level of the outer retina and RPE (Fig. 76.30). These lesions are centered in the fovea and not contiguous with the optic disc. There is a jigsaw pattern to margins of the lesions. During the course of the disease, there is very gradual fading of the whitish lesions in all patients.233 This can take months to years. In addition to the central lesions, one patient had small white lesions nasal and superior to the disc at the time of presentation. Another patient developed a small lesion nasal to the disc in the course of follow-up. The central lesion grew in size in one of 6 patients in Golchet’s series. RPE mottling and pigment clumping without CNV developed in 2 eyes. In all other eyes, no increase in pigmentation, atrophy, or scarring occurred until CNV developed.
Clinical course and prognosis
This macular disorder superficially resembles macular serpiginous. Vision is minimally affected, however, until the development of CNV or atrophy. The largest decrease in vision in eyes with no CNV was 4 Snellen lines to 20/60 in Golchet et al.’s series. This patient improved to 20/25 with corticosteroids.233 Only one eye without CNV had a poor visual outcome. Nine of 12 eyes developed CNV and disciform scar formation.
Imaging
Fluorescein angiography
FA showed early hypofluorescence with partial filling in the late phase. No leakage or staining was seen, unless CNV was present (Figs 76.31, 76.32).
Indocyanine green angiography
ICGA revealed persistent hypofluorescence throughout the angiogram (Fig. 76.33). The large choroidal vessels in the affected areas were seen. In one case, the hypofluorescence showed partial resolution on follow-up.233
Systemic associations
Three patients had vascular disease, including cardiac disease, myocardial infarction, hypertension, transient ischemic attack, and coronary artery disease. Two patients had type 2 diabetes. One patient had colon carcinoma and thrombocytopenia. Hyperthyroidism and cutaneous pemphigus were also reported by Golchet et al.233
Pathogenesis
There is similar speculation about pathology as with other placoid white spot entities. Choroidal hypoperfusion,235 blockage by swollen RPE cells and inflammatory lesions in the outer retina, or a combination of these have been suggested to explain the hypofluorescence on ICG.235 However, extensive choroidal hypoperfusion would not be expected in patients who maintain good vision.
Differential diagnosis
In addition to macular serpiginous choroiditis, other considerations include APMPPE, relentless placoid chorioretinitis, and syphilitic posterior placoid chorioretinitis233 (Table 76.2). The visual outcome, number of lesions, and common association with CNV of this process distinguishes itself from the other white spot entities. The fluorescein angiogram in syphilis shows late staining unlike PPM, as well as clinical signs of inflammation due to the infectious etiology (see Chapter 84, Spirochetal infections).
Management/treatment
Oral or periocular corticosteroids have been used with subsequent improvement in vision. The role of steroids requires further investigation. Anti-VEGF agents have been used to treat CNV, with good results in one report.236,237
Multifocal choroiditis and panuveitis, punctate inner choroidopathy, and progressive subretinal fibrosis and uveitis syndrome
Multifocal choroiditis and panuveitis (MFC), punctate inner choroidopathy (PIC), and progressive subretinal fibrosis and uveitis syndrome (PSFU) are conditions characterized by inflammation at the level of the RPE and outer retina. It does not appear to be a true choroiditis. Chorioretinal scars are left by the inflammation. The etiologies and pathogenesis of these entities are unclear. Jampol and Wiredu238 have classified these entities as the same disease as their ophthalmosopic appearance and clinical courses are similar. MFC and PIC will be presented first, with a common differential and treatment. This section will end with a brief discussion of the rare PSFU.
Multifocal choroiditis and panuveitis
MFC was first described in 2 young patients in 1973 by Nozik and Dorsch.239 They reported a chorioretinopathy that resembled presumed ocular histoplasmosis (POHS) but was associated with a bilateral anterior uveitis (see Chapter 70, Ocular histoplasmosis). Later, Dreyer and Gass240 coined the term multifocal choroiditis and panuveitis, describing 28 patients with uveitis and lesions at the level of the RPE and choriocapillaris. Deutsch and Tessler241 reported a series of 28 patients in 1984 with what they termed inflammatory pseudohistoplasmosis. Their series included patients with concurrent systemic diseases such as tuberculosis, sarcoidosis, and syphilis. Morgan and Schatz,242 2 years later, described 11 patients with a recurrent idiopathic multifocal choroiditis.
Clinical course
Clinical symptoms
Patients present with decreased central vision, photopsias, floaters, metamorphopsia, paracentral or temporal scotomas, ocular discomfort, and photophobia. Photopsias often correspond to lesions in the peripapillary area and indicate activity of the disease. Visual acuity can range from 20/20 to light perception. More than half present with vision less than 20/100 in the worse eye.240,242
Epidemiology
MFC occurs predominantly in Caucasian,243 myopic women between the second and sixth decades of life.240,242 Most affected patients are in their 30s. In addition, they have never lived in areas that are endemic for histoplasmosis.
Fundus findings
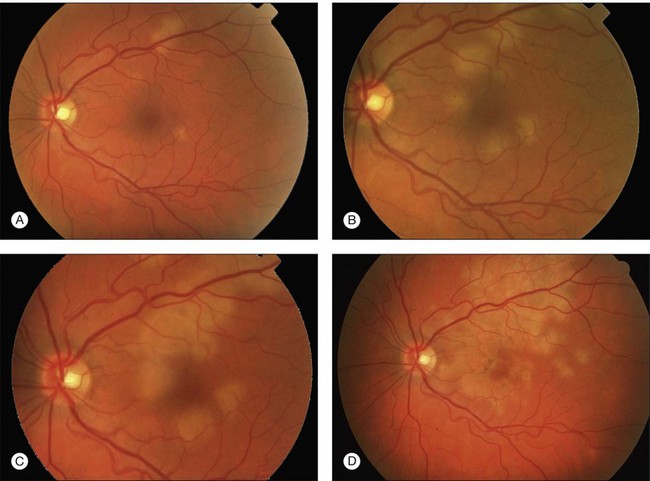
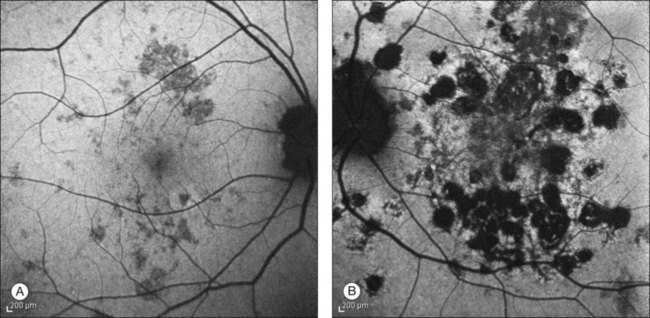
In the acute phase of MFC, yellow round or oval lesions, ranging in number from one to scores, are seen in the outer retina and RPE. They range in size from 50 to 1000 µm. They occur in the posterior pole, peripapillary region, and midperiphery. The nasal retina often shows clustering.244 The lesions can also be arranged in linear scars parallel to the ora in the periphery245 (Fig. 76.34). Previously, this linear appearance was thought to be pathognomonic for POHS.246,247 Active lesions can be associated with subretinal fluid. As inflammation resolves, the lesions become atrophic with a variable amount of pigment (“punched out” appearance). They can also enlarge in size. There can be bridging subretinal scars between the disciform or atrophic scars.248 Cantrill and Folk described 5 patients who developed sharply angulated subretinal scars as the lesions healed.249 They noted broad scar formation in the macula as the lesions coalesced that could be related to serous and hemorrhagic macular detachment. During active phases, exudative retinal detachment may overlie areas of activity.
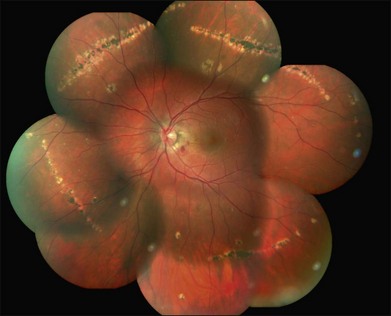
Fig. 76.34 Linear scars parallel to the ora are seen in the periphery.
(Courtesy of Medical University of Innsbruck, Austria.)
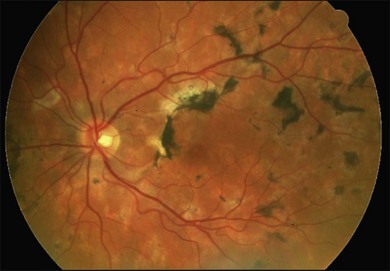
In recurrent episodes, new chorioretinal lesions may develop. Optic disc edema can be seen and atrophy may occur. Optic disc neovascularization can very rarely be seen.241 The peripapillary region may have a characteristic subretinal fibrosis250 that has been described as a “napkin ring” configuration (Fig. 76.35). Retinal vessels may be narrowed. Periphlebitis may develop. Cystoid macular edema occurs in 10–20% of individuals. CNV develops in 25–30% of cases and can be the presenting sign.240,242,243
Other ocular findings
MFC may have a mild to moderate anterior uveitis. Nongranulomatous keratic precipitates and posterior synechiae can be present. Iris abnormalities, such as neovascularization or abnormal avascular areas on fluorescein angiography, have been reported.251 Vitritis, if present, is usually mild or moderate. Findings may be asymmetric.
Clinical course and prognosis
MFC waxes and wanes, but vision loss can occur. Most patients tend to have recurrent episodes that can involve the central or peripheral vision. These can resolve spontaneously or with the aid of immunosuppressants. Some patients retain excellent vision, but have persistent photopsias. About 25% of patients have a more chronic course that involves more inflammation, or CNV. Recurrent inflammation may cause CME, vitreous haze, and swelling around old scars (Fig. 76.36). New lesions that are not contiguous with old scars can develop. CNV is the most common cause of visual loss in MFC and can be seen with either inactive macular scars or recurrent inflammation. The long-term visual prognosis of MFC is variable. Brown and colleagues244 followed 41 patients (68 eyes) for a mean of 39 months. They found that 66% of eyes ended up with 20/40 vision or better. Only 21% of eyes ended up 20/200 or worse. The majority of the cases with poor vision were due to CNV and less commonly CME. Foveal atrophy and uveitic neovascular glaucoma were other causes in their series. About one-third of patients developed CNV. Thorne and colleagues252 studied 66 patients (122 eyes) and found that 55% of patients presented with vision worse than 20/50 and 38% presented with vision worse than 20/200. CNV was found in 22% of patients at presentation and was the most common cause of vision loss. Presence of epiretinal membrane and CME were other causes of vision loss. Use of immunosuppressive therapy was associated with an 83% reduction in the risk of posterior pole complications, including new or recurrent CNV.
Imaging
Fluorescein angiography
In the acute phase, the clinical lesions appear hypofluorescent on FA. Late in the angiogram, the lesions stain242 (Figs 76.37, 76.38). CNV may be present in the peripapillary or macular areas. In the healed phase, the lesions become atrophic and show a window defect on angiography. A recent study of CNV in MFC revealed that most CNV is classic and clearly demonstrated unless blocked by subretinal blood.253 The researchers further compared this with OCT (both stratus and spectralis), and found that CNV could be missed with this mode of imaging. They recommended FA be used in cases with suspected neovascularization.
Indocyanine green angiography
ICGA shows hypofluorescent round spots that may be far more numerous than seen on clinical examination and fluorescein angiography254–256 (Fig. 76.39).
Optical coherence tomography
In the active phase, stratus OCT findings show an RPE irregularity corresponding to a clinical lesion. This RPE irregularity resolves with treatment.257 In our experience with spectral domain OCT, the lesion disrupts the photoreceptor layer with potential normalization with treatment (Fig. 76.40). Retinal thinning may occur. Vance and colleagues describe SD-OCT imaging of acute lesions that showed drusen-like material between the RPE and Bruch’s membrane as well as a localized choroidal hyperreflectivity below the material.258 These lesions disappear with treatment.
Fundus autofluorescence
Hyperautofluorescence was observed in the areas of fresh lesions. This resolved when immunosuppression was instituted. In the majority of patients, punctate hypoautofluorescent spots were seen on FAF corresponding to areas of chorioretinal atrophy (inactive lesions)259 (Fig. 76.41).
Electrophysiology testing
Dreyer and Gass240 reported ERGs on 29 eyes of 16 patients. They observed normal or borderline results in 41% of patients, moderately reduced results in 17%, and severely reduced results in 21% of eyes. Oh and colleagues used multifocal ERG (mfERG) to study pre- and post-treatment results; however, due to the sensitivity of this test, it becomes almost extinguished with active MFC.260 Electrophysiologic tests are not used commonly in the management of MFC, as with other white spot diseases such as birdshot chorioretinopathy.
Visual field testing
Blind spot enlargement as a manifestation of multifocal choroiditis was described in 1991.261 Enlarged blind spot in the setting of a normal optic nerve has been seen in acute idiopathic blind spot enlargement (AIBSE) and in patients with multiple evanescent white dot syndrome (MEWDS). The enlargement is thought to be due to peripapillary retinal dysfunction rather than an optic nerve problem. This is supported by the fact that color vision and pupillary responses have been reported to be normal in these patients. The enlarged blind spot can resolve with inactivity of disease. Photoreceptor outer segment abnormalities may be a cause of the blind spot enlargement.262 Visual field testing may also demonstrate scotomas related to areas of chorioretinal scarring or serous detachments related to active lesions. Visual fields are an important way to monitor MFC.
Systemic associations
An association between MFC and Epstein–Barr Virus (EBV) has been suggested.263 It was postulated that EBV triggers an immune response that starts a persistent intraocular inflammatory response. Immunoglobulin M antibodies directed against the viral capsid antigen or the Epstein–Barr early antigen were present in patients with MFC in one series with none in controls.263,264 However, this could not be reproduced.265 Some patients with MFC may also have or develop sarcoidosis. These individuals are often not distinguishable from those who have idiopathic MFC.
Punctate inner choroidopathy
PIC was first described in 1984.266 Watzke reported 10 myopic women with blurred vision, photopsias, or paracentral scotomas with small yellow–white lesions of the inner choroid and RPE. These lesions were associated with overlying subretinal fluid. Acute lesions healed to atrophic scars and developed more pigmentation with time. Choroidal neovascular membranes developed in more than half of these individuals.267
Clinical course
Clinical symptoms
In a survey of 77 people with PIC conducted by Gerstenblith and colleagues,268 scotomas were the presenting complaint in 91% of patients, followed by blurred vision (86%), photopsias (73%), floaters (69%), metamorphopsia (65%), and decreased peripheral vision (26%). Most (85%) presented with unilateral symptoms.
Epidemiology
PIC is a disease of young, relatively healthy, myopic women. An estimation of a national incidence is approximately 100–200 cases per year.269 This is probably an underestimation. Ninety percent of patients are women. Eighty-five percent of patients are myopic, and 97% are Caucasian.268 The mean age at presentation is 30 years (range 15–55). Patel and colleagues270 described the presentation and outcome of PIC patients at a tertiary referral center and found similar trends. Mean refraction is -4.6 diopters.271
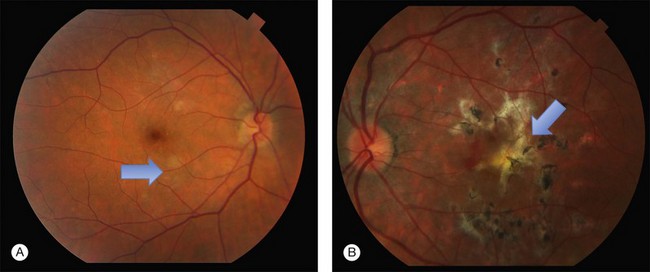
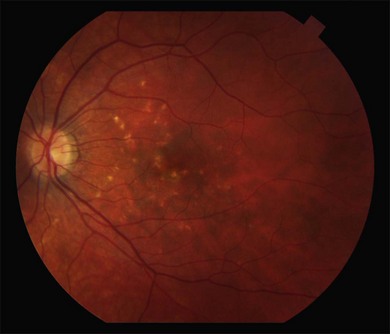
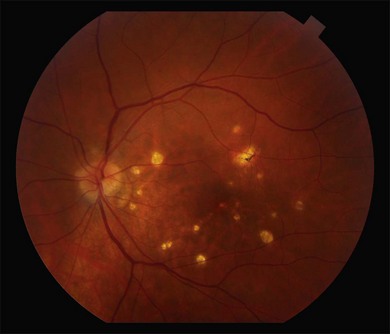
Fundus findings
Multiple gray or yellow, small round lesions are seen scattered throughout the posterior pole.268 (Fig. 76.42). Occasionally there is a linear pattern. The lesions are at the level of the outer retina, RPE, and inner choroid and a neurosensory detachment may be present. With time, these spots evolve into atrophic chorioretinal scars. After 2–3 years, they become more distinct, pigment, and appear similar to the punched out lesions of POHS (Fig. 76.43). Some lesions disappear without sequelae. CNV commonly develops.
Other ocular findings
Generally, PIC is not associated with anterior or posterior segment inflammation.
Clinical course and prognosis
Brown and colleagues reported that 88% of 16 patients had bilateral disease, despite its most commonly being a unilateral complaint.244 Patients generally become asymptomatic after one month. The scars continue to become atrophic and pigmented over the next 2–3 years. These scars appear to involve the RPE and choriocapillaris. Recurrences are common. Brown and colleagues followed patients for a mean length of 51 months.244 They found that 77% of eyes had a final vision of 20/40 or better. Twenty-three percent had a vision of 20/50 or worse. Twenty percent had a vision of 20/200 or worse. The primary cause of this poor vision was CNV. Approximately 40% of eyes developed CNV and it generally appeared within one year of presentation. Smaller areas of CNV (<100 µm) were more likely to spontaneously resolve as compared with larger areas (>200 µm). Essex and colleagues271 in a larger study recently reported the clinical features and outcome of 136 PIC patients (271 eyes) with a mean follow-up of 6.2 years. They found 63 fellow eyes that were normal at presentation. Of these, 56 (88%) remained unchanged. Only 3 (5%) developed PIC lesions and 4 (6%) developed CNV. In eyes with PIC lesions, 49 of 74 eyes (66%) remained unchanged. New PIC lesions developed in 9 eyes (12%) and CNV developed in 16 (22%).
Imaging
Fluorescein angiography
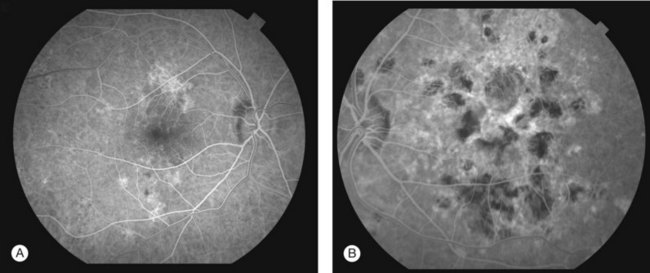
PIC lesions can appear hyperfluorescent in the arterial phase or may appear as blocked fluorescence (Fig. 76.44A). In later phases, the lesions stain266 (Fig. 76.44B). More lesions are seen on FA than are clinically visible. Tiny punctuate hyperfluorescent lesions may be seen scattered in the posterior pole. In later stages of disease, as the RPE atrophies, window defects are seen. Leakage of fluorescein into the subretinal space is also possible.
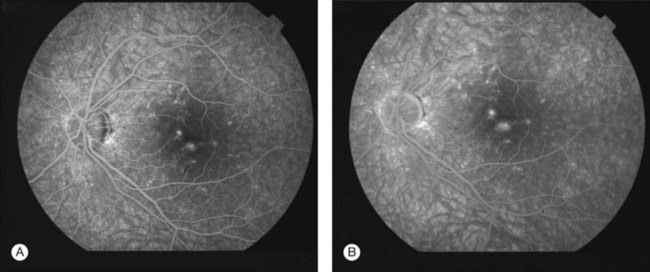
Olsen and colleagues described CNV in six patients with PIC.272 They found that a fibrotic response followed the development of CVN and there was often a dumbbell-shaped pattern of subretinal fibrosis.
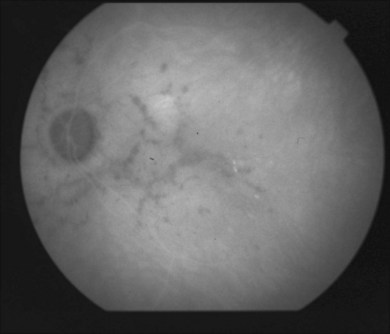
Indocyanine green angiography
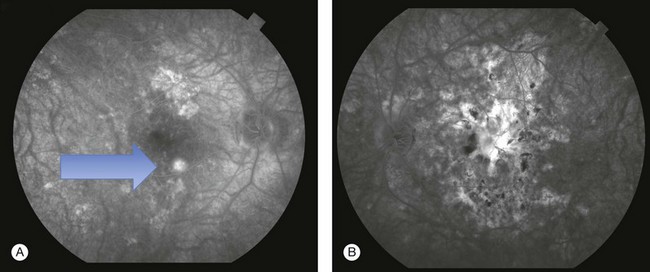
As with other white spot diseases, the PIC lesions hypofluoresce in the early, middle, and late phases of ICGA273,274 (Fig. 76.45). Many more lesions are seen on ICGA than are clinically visible or seen in FA.275 Tiffin and colleagues described choroidal vascular abnormalities.274 In addition to the hypofluorescent lesions that, in some cases, corresponded to clinically seen lesions, larger choroidal vessels were seen crossing these areas. Several choroidal vessels also showed hyperfluorescence of the vessel wall. They postulated focal choroidal ischemia in the areas of hypofluorescence and possibly a vasculitis in the area of hyperfluorescence of the vessel walls.
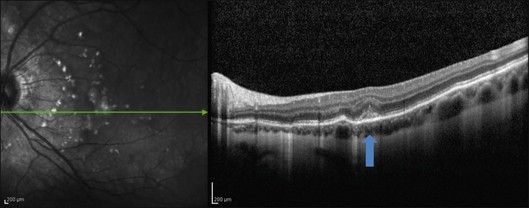
Optical coherence tomography
Stepien and Carroll276 described spectral domain OCT findings in a patient with PIC with recurrent activity. In addition to seeing fluid associated with CNV, outer retinal irregularity was described. A homogenous thickening was seen overlying chorioretinal lesions (Fig. 76.46). This resolved with treatment. The pattern we see with SD-OCT is similar to MFC. There is sub-RPE solid looking material that pushes up the RPE and may extend into the retina.
Electrophysiology
Reddy and colleagues found that 7 out of 16 patients with PIC had normal ERGs.277 Three of the 7 patients had some asymmetry in b-wave amplitudes between the two eyes which correlated to the number of lesions in each eye.
Visual field testing
Reddy and colleagues also studied visual fields in 22 eyes of patients with PIC.277 Ten eyes (45%) had normal visual fields. Nine eyes (41%) had enlargement of the blind spot, many of which extended toward the macula. Three eyes (14%) had central/paracentral scotomas. They postulated that this is related to the peripapillary clustering of lesions. The visual fields improved without treatment in most patients at follow-up. Photoreceptor outer segment abnormalities are the cause of the blind spot enlargement.262
Pathogenesis
The cause of MFC and PIC is not understood. It is uncertain if they are separate entities or on a spectrum of a single disease. They typically affect young, otherwise healthy women. There are yellow lesions at the level of the outer retina as seen by OCT and RPE, as well as evidence of some choroidal involvement. The lesions of MFC and PIC eventually atrophy and look similar to the punched out lesions of POHS. CNV can develop, leading to bridging scars and subretinal fibrosis. Patients can also develop blind spots and scotomas that can improve with or without treatment. These field defects can be associated with abnormalities of the outer retina as seen on ERG. MFC has been reported to occur in patients with other white spot syndromes such as APMPPE and MEWDS.6,278 Becker and Jampol proposed a common genetic hypothesis for autoimmune involvement in these inflammatory processes.2 The interplay of genetics, immune dysregulation, and environmental triggers may play a role in the different presentations of these clinical entities. Atan and colleagues279 recently studied MFC and PIC and found similarities in the association with a IL-10 haplotype, and negative associations with a TNF haplotype. These genetic loci are known to be associated with noninfectious uveitis and autoimmunity. They state that definitive proof will necessitate genomewide sequence analysis. However, their data support the idea that genetic factors have a strong effect on clinical phenotype.
Differential diagnosis
Neoplastic masquerading diseases such as intraocular lymphoma can be considered as well.
Differentiation of MFC and POHS
Both entities may show punched out chorioretinal scars, peripapillary scarring, and CNV. Classically, the teaching is that the difference between these two entities is the presence of inflammation with MFC and absence with POHS. However, MFC can be present without signs of inflammation. Parnell and colleagues248 differentiated MFC from POHS based on clinical features, morphology of photographed fundus lesions, and fluorescein angiography. Clinically, MFC patients may develop photopsias and visual field defects. Clinically, distinctive features of MFC include multiple small white lesions clustered in the macula, nasal retina, or equator, a mixture of acute and inactive lesions, progressive growth of lesions, and subretinal fibrous metaplasia with bridging tissue between scars. Other changes include hyperplastic changes around the nerve (“napkin ring”), myopic-appearing nerve, sheathing of vessels or narrowed vessels, and inflammation seen as CME, vitritis, or disc edema (see Chapter 70, Ocular histoplasmosis.)
Differentiation of MFC and PIC
Although we believe that these two entities are on the same spectrum of a single disease, there are a few studies that have proposed distinct features. Kedhar and colleagues243 studied 66 patients (122 eyes) with MFC and 13 patients (22 eyes) with PIC. They found that the median range of age at presentation was less in PIC patients at 29 years compared with MFC at 45 years. Patients with MFC presented with a higher frequency of structural complications such as cataract, CME and ERM while PIC patients did not have these. They found that PIC patients were more likely to develop CNV (76.9%) versus those with MFC (27.7%). MFC patients were more likely to have complications from intraocular inflammation and bilateral visual impairment of 20/50 or worse. These differences are likely secondary to more widespread inflammatory disease with MFC.
Shimada and colleagues280 looked at 14 eyes of 14 patients who were diagnosed with MFC (8 eyes) or PIC (6 eyes) that underwent surgical excision of a choroidal neovascular membrane. They found that CNV obtained from both types of patients expressed VEGF and CD68. In 3 of 8 MFC eyes, intraocular inflammatory findings were found clinically. In these eyes, immunohistochemistry showed that the CNV was infiltrated with CD20-positive B lymphocytes. No B lymphocytes were found in the PIC patients, or the 5 MFC patients without clinical inflammation. Therefore, the amount of inflammation in MFC may explain the differences.
Management/treatment
Treatment is aimed at inflammation and its sequelae (CME and CNV). Multifocal choroiditis and panuveitis may be associated with significant anterior and posterior inflammation. This can be managed with topical, periocular, intraocular, and systemic corticosteroids. Steroid-sparing agents are used when steroids are not tolerated or recurrence is frequent. Although PIC is not associated with significant visible inflammation, the lesions do respond to immunosuppressives. CNV can be managed with thermal laser if extrafoveal, photodynamic therapy,281 steroids and/or anti-VEGF therapy. PDT can be in conjunction with systemic or local steroids as the etiology of the CNV is in part inflammatory in these entities and PDT can incite inflammation. This has been found to be effective.282 Amer and Lois267 in their extensive review of literature on PIC suggest that PDT may best achieve closure of already formed neovascularization, while immunosuppressive agents may be most effective in the early stages of development of CNV. Anti-VEGF therapy has been found to be promising in several studies of CNV caused by MFC283–286 and PIC.282,287,288 An additional complication in these patients due to the demographics (young females) is concomitant pregnancy. Steroid therapy is possible during pregnancy; other agents may affect fertility, however, and teratogenicity is unclear in humans.
Progressive subretinal fibrosis and uveitis syndrome
PSFU is a rare entity that was first described by Palestine and colleagues in 1984.289 Three young women with intraocular inflammation and fibrotic subretinal lesions that progressively enlarged and coalesced were characterized. The patients also developed macular edema. Several authors have described progressive subretinal fibrosis in association with multifocal choroiditis.240,249,290 This entity is also known as diffuse subretinal fibrosis syndrome.
Clinical course
Clinical symptoms
Patients, in the acute phase, present with unilateral decreased vision,291 floaters, possibly photopsias, scotomas, and metamorphopsia.
Epidemiology
Kaiser and Gragoudas291 reviewed the literature and found that patients are predominantly young, healthy, and myopic. Ages generally ranged from 20 to 40 years old. However, there may be a bimodal curve, as Gass described 3 elderly individuals (mean age 71) with biopsy-proved PSFU.292
Fundus findings
Numerous small (100–500 µm) yellow spots are seen at the level of the choriocapillaris, RPE, and deep retina.291 There is clustering in the posterior pole, but there can be extension to the midperiphery. The lesions can be associated with hazy yellow subretinal fluid. There is rapid development of subretinal scarring.
Other ocular findings
Anterior chamber reaction and vitritis is usually mild. Optic disc edema has been reported.293 Other signs of inflammation such as posterior synechiae, keratic precipitates, and iris atrophy have been reported.291
Clinical course and prognosis
PSFU usually presents asymmetrically but is usually bilateral. Within a few days to weeks after development of the multifocal lesions, fluid starts to accumulate. Over the next several months, subretinal fibrosis develops. This further coalesces and forms sheets of fibrotic tissue under the retina. This is associated with marked decrease in vision. In Brown and colleagues’ study, 7 of 10 eyes of patients with PSFU had vision worse than 20/200 after 2 years.244 Only 2 of 10 eyes had vision of 20/40 or better. CNV and subretinal hemorrhages develop. The second eye can develop lesions within months of the first eye.291
Imaging
Fluorescein angiography
The lesions associated with PSFU are characterized by early hyperfluorescence followed by late leakage in the acute phase.249,277,294 As fibrosis develops, there is staining in the late phases. CNV may be seen on FA but is uncommon and may be difficult to assess due to the fibrotic component.
Systemic associations
Case reports of PSFU in association with ectodermal dysplasia295 and ulcerative colitis296 have appeared. This may point to a genetic or inflammatory etiology, but due to the rarity of this disease, no conclusions can be reached.
Pathogenesis
Histopathology from a patient with PSFU showed a severely gliotic retina and thick subretinal fibrotic tissue. There was granulomatous lymphocytic infiltration in the choroid. The subretinal tissue was derived from retinal pigment epithelial cells. There was a predominance of B cells and plasma cells.294
Management/treatment
Treatment is based on early diagnosis and aggressive management. Once severe subretinal fibrosis develops in the macula, treatment is of little benefit. High-dose systemic or intravitreal steroids have been used. However, these have generally been of little visual benefit.249,289 Other immunosuppressives such as cyclosporine and azathioprine have been used with better results244 and more recently a case report using infliximab improved vision.297 Due to the rarity of this entity, however, a description of best treatment is not possible. Chronic therapy may be needed, and systemic glucocorticoids may be helpful in limiting damage to the fellow eye.
[/level-membership-for-opthalmology-category][not-level-membership-for-opthalmology-category]
Chapter 76 White Spot Syndromes and Related Diseases
Introduction
The white spot syndromes (WSS) are a group of diseases characterized by inflammation and dysfunction of the outer retina, retinal pigment epithelium, choroid, or a combination of these. They often present a diagnostic and therapeutic challenge for clinicians and researchers. The etiologies of WSS remain unknown. This chapter discusses: birdshot chorioretinopathy (BCR), acute posterior multifocal placoid pigment epitheliopathy (APMPPE), serpiginous choroiditis, relentless placoid chorioretinitis, persistent placoid maculopathy, multifocal choroiditis and panuveitis (MFC), punctate inner choroidopathy (PIC), progressive subretinal fibrosis and uveitis syndrome (PSFU), multiple evanescent white dot syndrome (MEWDS), acute zonal occult outer retinopathy (AZOOR), acute idiopathic blind spot enlargement (AIBSE), and acute macular neuroretinopathy (AMN). Many entities are included in the differential diagnosis of these diseases. These include: granulomatous diseases, such as sarcoid, tuberculosis, sympathetic ophthalmia; masquerade syndromes like syphilis and intraocular lymphoma; infectious etiologies including toxoplasmosis, pneumocystis choroidopathy; and other entities such as presumed ocular histoplasmosis, and Behçet disease.1 In addition, in some cases, degenerative processes such as drusen may appear similar to the whitish-yellow lesions of WSS.
An autoimmune etiology has been hypothesized2 and proposed specifically for birdshot chorioretinopathy, acute zonal occult outer retinopathy (AZOOR), and multiple evanescent white dot syndrome (MEWDS).3 As yet, no characteristic pattern of antiretinal antibodies has been found. These entities need to be distinguished from entities with known neoplasias such as cancer-associated retinopathy (CAR) or melanoma-associated retinopathy (MAR). There are similar features including panretinal dysfunction, rapid progression, ERG changes, family history of autoimmune disease, retinal antibody activity on Western blot, and improvement in symptoms with immunosuppression. An increased prevalence of systemic autoimmunity in both patients with WSS and their first- as well as second-degree relatives, was found by Pearlman and colleagues.4 This suggests that this group of diseases occurs in families with inherited immune dysregulation that predisposes to autoimmunity.
It has also been suggested that several of these entities may be “related” or even represent a spectrum of the same process. Multifocal choroiditis (MFC), punctate inner choroiditis (PIC), MEWDS, acute macular neuroretinopathy (AMN), and AZOOR share commonalities and Gass referred to them as the AZOOR complex.5 These include female preponderance, zones of visual field loss that are usually contiguous to the blind spot, photopsias, and ERG changes such as reduced amplitudes. In addition, MFC and APMPPE have also been found in the same patient decades apart.6 Other diseases that have been noted in the same patient include MEWDS and AZOOR as well as MFC and PIC. This overlap may give support to a common underlying genetic predisposition.2
Birdshot chorioretinopathy
The term birdshot retinochoroidopathy was first used in 1980 by Ryan and Maumenee.7 It was a descriptive term for patients with multiple small, cream-colored fundus findings. These lesions are scattered around the optic disc and radiate to the equator in a “shotgun” pattern. Other terms such as vitiliginous chorioretinitis have been used.8 It has come to be known as birdshot chorioretinopathy (BCR) due to the histopathologic evidence that the primary lesions of the disease are in the choroid.9 It is a bilateral, chronic process with vitritis, retinal vasculitis, and cystoid macular edema (CME). Furthermore, it demonstrates the strongest link between any disease and any HLA class I antigen.10
Clinical course
Clinical symptoms
Patients present with complaints of blurred vision, floaters, and photopsias. Most have vision of 20/40 or better.9 The eye is generally not red or painful. Severe nyctalopia despite normal visual acuity may be a presenting symptom.11 Individuals also may describe an alteration in color vision12 or visual fields.13 Although Gass described a few patients who concurrently had vitiligo of the skin,8 BCR is thought to be a purely ocular disease.
Epidemiology
BCR is a rare chronic posterior uveitis. Shah and colleagues did an extensive review of English literature in 2005 and reported the following epidemiologic characteristics.9 Birdshot chorioretinopathy accounts for 0.6–1.5% of patients referred to tertiary centers for uveitis, or 6–7.9% of patients with posterior uveitis. There is a slight female predominance in the literature, at 54.1%. The mean reported age at the onset of disease is 53 years. It is generally not a disease of children. The oldest reported age at onset was 79 years. Patients are predominantly white; there have only been two reported exceptions. Finally, the HLA-A29 allele (which is present in about 7% of Caucasians) is strongly associated with BCR.14 The presence of this allele has been associated with a risk factor of 50–224 to develop BCR. Ninety percent or more of patients with BCR are HLA-A29-positive. Further sequencing of this allele has uncovered 11 subtypes. The HLA-A29*02 subtype is 20 times more prevalent in Caucasians than the HLA-A29*01 type, and the rest are exceedingly rare.15 HLA-A29*02 subtype is most commonly associated with BCR.
Fundus findings
It is important to recognize that symptoms can precede the onset of the classic fundus findings by several years.16 There is a range of presentations of the lesions associated with BCR. These birdshot lesions can be oval or round in shape, typically about or disc diameter in size. They appear deep to the retina (Fig. 76.1). They can be subtle, and asymmetric between eyes. The lesions can be distinct or poorly defined and can occasionally coalesce. They tend to cluster near the nerve and most commonly nasal and inferior to the disc.17,18 There is a pattern of linear radiation away from the nerve to the periphery7 (Fig. 76.2). They may appear to follow choroidal blood vessels peripherally. There is no hyperpigmentation or clumping noted. The retinal pigment epithelium (RPE) and overlying retina appear intact. There has been a report of these choroidal lesions preceding symptom onset in BCR, but this is not the norm.19
Other ocular findings
The anterior segment generally has minimal inflammation without prominent keratic precipitates. Fine keratic precipitates have been described in some series.18 Due to the mild nature of anterior segment inflammation, posterior synechiae do not occur. Signs of posterior inflammation include retinal vasculitis, optic disc edema, CME, and epiretinal membrane formation. Rhegmatogenous retinal detachment has been reported.7,20,21 It is unclear if this is related to this disease entity or to an association with uveitis. Other common findings include diffuse narrowing of the retinal arterioles, perivascular nerve fiber layer hemorrhages, and tortuosity of retinal vessels. Choroidal neovascularization (CNV) can occur.10,22,23 It has been postulated that this occurs due to the uveitic component causing CNV rather than ischemic factors.24,25
A consensus document on the diagnostic criteria for BCR was published in 2006.26 Required characteristics include: bilateral disease, presence of at least three peripapillary lesions inferior or nasal to the optic nerve in at least one eye, low-grade anterior segment inflammation (less than or equal to 1+ cells), and low grade vitreous inflammation (less than or equal to 2+ vitreous haze). Supportive findings include: HLA-A29 positive, retinal vasculitis, and CME. Finally, exclusion criteria include: presence of significant keratic precipitates, posterior synechiae, or diagnosis of infectious, neoplastic, or inflammatory disease that may cause multifocal choroidal lesions.
Clinical course and prognosis
The disease is chronic in nature and apparently does not regress. Again, symptoms of blurred vision, color deficiency, contrast sensitivity issues, and visual field problems may be present for years prior to the onset of fundus lesions. Visual acuity may be normal despite these complaints. It remains a poorly understood disease and no consensus on management and treatment has been found. Many patients have a slow decline in vision, despite treatment.9A final visual acuity of 20/40 or better in the best-seeing eye was reported in 75.1% of patients. However, 9.8% of patients were legally blind at follow-up (in the review of literature by Shah and colleagues9). Generally, macular edema was the commonest cause of visual decline in 50.5%.9 Choroidal neovascular membranes developed in 5% of eyes.22,23 These occur near the optic disc and can be bilateral. Finally, optic disc edema leading to atrophy can also affect visual prognosis.
Imaging
Fluorescein angiography
The creamy white spots have variable appearance on fluorescein angiography (FA) and the disease may be more evident clinically than with this mode of imaging (Fig. 76.3). The lesions can hypofluoresce in the early phase and there can be diffuse hyperfluorescence in the late phases (Fig. 76.4). One theory suggests that the lesions are likely in the outer choroid and associated with large choroidal vessels, thus many of the lesions show neither hypofluorescence or hyperfluorescence in any phase. The diffuse hyperfluorescence seen may represent a deep inflammatory focus that accumulates fluorescein.18 Angiographic findings may include increased transit time, leakage from retinal vasculature leading to CME, optic disc hyperfluorescence (Fig. 76.5), and disc or retinal neovascularization.8,27,28
Indocyanine green angiography
Indocyanine green angiography (ICGA) is an important diagnostic test. In the active disease,28 the birdshot lesions appear hypofluorescent during the intermediate phase of angiography and appear to be bordered by medium-to-large vessels28–30 (Fig. 76.6). The choroidal vessels appear indistinct. Late in the ICGA, there is diffuse choroidal hyperfluorescence. As with all the white spot syndromes, theories to explain this hypofluorescence include choroidal ischemia versus blockage from inflammatory infiltrates. However, in birdshot chorioretinopathy it is agreed upon that the site of the pathology is primarily the choroid. In the acute stages, the inflammatory infiltrates may be denser and block fluorescence. In late stages of the disease, it is thought that the lesions become more atrophic and choroidal vasculature may become more visible. The lesions can become more isofluorescent in this phase of the disease or they may remain hypofluorescent.27,28
Optical coherence tomography
This mode of imaging has primarily been used to follow CME, which is the most common cause of vision loss in BCR31 (Fig. 76.7). Photoreceptor loss is not a prominent finding early on in the course.
Fundus autofluorescence
Giuliari and colleagues looked at fundus autofluorescence (FAF) of 18 eyes and found a spectrum of patterns.31 They found that hypoautofluorescent lesions on FAF did not necessarily correspond to clinical birdshot lesions. Commonly (15/18 eyes) more FAF lesions denoting RPE defects were seen than clinical lesions. Koizumi and colleagues looked at 8 patients. They found that some of these hypofluorescent lesions could be placoid in nature and involve the macula.32 In addition, a perivascular (retinal) linear hypofluorescent FAF pattern was noted by both groups of investigators.31,32 It was postulated that this finding may suggest that the retinal vessels may play a role in inflammatory-driven damage of the RPE and serve as a conduit for inflammatory factors. The observation that the clinical choroidal lesions did not always correspond to the FAF defects suggests that the choroid and RPE may be affected independently.32 Furthermore, RPE defects in the macula could also be another cause of vision loss in these individuals. FAF may be valuable in evaluating these patients as lesions may be clinically difficult to see.
Electrophysiology
Priem and coworkers also studied pattern-evoked cortical potentials and found abnormal findings in 53.3% of patients.33
Electroretinogram
Electroretinography (ERG) is an important test for following BCR. Abnormal electroretinograms (ERGs) were reported in 88.8% of patients.9 There appear to be two groups of patients: those who develop abnormal ERG early in the disease process and those that develop abnormal results late in the course.34 Early ERGs may demonstrate supernormal ERG amplitudes which may be related to retinal inflammation. In this stage, there may be a greater decrease in b-wave amplitude versus a-wave amplitude.21,34 This is a negative ERG pattern, and is not pathognomonic to BCR. It suggests that the Müller and bipolar cells are more affected than the photoreceptor–retinal pigment epithelium complex. This has been seen in autoimmune retinopathy. Rod dysfunction may occur before cone dysfunction: the rod b-wave may be affected prior to photopic b-wave and flicker response in most patients. In late phases of the disease, there is a progressive decrease in a- and b-wave amplitudes. ERG findings have been noted to improve with treatment.35 Some authors argue that the reversible nature of the ERG abnormalities suggest a non-ischemic etiology.15 Inflammation of the retinal vasculature could lead to inner retinal dysfunction, while choroidal inflammation could be the cause of altered outer retinal function.
Visual field testing
Visual field abnormalities are present in patients with BCR. These include: peripheral constriction, enlarged blind spot, central or paracentral scotomas, and generalized diminished sensitivity.8,17,18,20,33 It is not certain whether these visual field defects occur due to ganglion cell, optic nerve, or outer retinal dysfunction, but it is unlikely that the birdshot lesions themselves cause blind spots.9 Visual field abnormalities may be reversible with immunosuppression.36
Systemic associations
There are no definitive systemic associations. Shah and colleagues’9 review of the literature revealed hearing loss in some patients.8,37 As mentioned earlier, cutaneous vitiligo associated with BCR has also been reported in a few patients.8,18 In addition, Priem and Oosterhuis reported an increased incidence of vascular disease in their series. Of 102 patients, 16 had hypertension, 5 had coronary artery disease, 2 had a history of cerebrovascular accident, and 2 had a central retinal vein occlusion.18 These numbers are not impressive considering the age of the patients.
Pathogenesis
The pathogenesis of BCR is unknown. Inflammation appears to be a primary feature. The histopathological findings in a few eyes with BCR have been reported. These suggest that the spots may be related to accumulation of lymphocytes in the choroid at multiple levels, occasionally associated with hemorrhage.38 Some of the foci were adjacent to the choroidal vessels. The RPE, ciliary body, and iris were not involved. Some lymphocytes were found around the retinal blood vessels and in the optic disc. The lymphocytes were primarily CD8+T-lymphocytes. Inflammation is strongly associated with HLA-A29, which suggests a genetic predisposition to this disease. However the fact that HLA-A29 is present in 7% of the white population, and BCR is so rare tells us other factors are at work. Other genes are highly suspected. The more recent discovery of HLA class I-specific killer cell immunoglobulin-like receptors (KIR) led to epidemiological studies implicating KIR-HLA gene combinations in disease.39 The combination of KIR and HLA gene variants appears to increase the risk of developing BCR in HLA-A29-positive individuals. The presence of these in the absence of strong inhibition may activate natural killer cells and T cells against intraocular self-antigens thus inciting an autoimmune process. Familial history of autoimmunity is likely.2
Differential diagnosis
Birdshot chorioretinopathy can usually be distinguished from other disorders by history and physical findings. However, entities such as pars planitis, intraocular B-cell lymphoma, syphilitic chorioretinitis, sarcoidosis, sympathetic ophthalmia, and other white spot diseases, especially multifocal choroiditis and panuveitis syndrome, should be considered. Sarcoidosis (see Chapter 78, Sarcoidosis) and BCR may be the most difficult to distinguish from each other.9
Management/treatment
In the review of the literature by Shah and colleagues9 most cases were treated; however, no definitive guidelines for the initiation of treatment were given. This chronic progressive disease may not be sight-threatening in the early course, but macular edema with retinal damage, photoreceptor dysfunction, RPE atrophy, and optic nerve damage are the end results. Corticosteroids have been the mainstay of treatment. Oral, sub-Tenon’s, intraocular, and most recently sustained release fluocinolone acetonide40 have been used. Corticosteroids can reduce CME,7,21,41,42 inflammation,21 and optic disc edema.41 They have also been reported to decrease symptoms such as nyctalopia and issues with contrast sensitivity.11 Systemic steroids carry their own risks. Local steroid injections increase the risk of cataract and cause glaucoma with repeated dosing. Implantation of a fluocinolone acetonide sustained-release device has been shown to eliminate the need for systemic therapy, however it also has a high risk of cataract progression and glaucoma.40
Immunosuppressive therapy
Steroid-sparing agents have been used for long-term management of refractory cases. The three classes of immunosuppressives have been used. Cyclosporine has been used as it inhibits T lymphocytes and prevents S-Ag-induced experimental uveitis.10 Low-dose cyclosporine has proven to have positive visual effect in conjunction with steroids or alone.43 Nephrotoxicity is the primary side-effect and hypertension can be a problem.43 Antimetabolites such as azathioprine, mycophenolate mofetil, and methotrexate have been adjunctive or used in monotherapy. Side-effects of these drugs include bone-marrow suppression and hepatotoxicity.44 The use of the alkylating agents cyclophosphamide and chlorambucil has also been reported. Side-effects such as bone-marrow suppression and development of malignancies must be weighed for this class. Daclizumab, a monoclonal antibody against the alpha-subunit of the IL-2 receptor of T cells, has recently been found to have value in treating BCR.44,45 The long-term efficacy of these agents needs to be determined and weighed against potential side-effects.
Anti-VEGF therapy has been documented to be useful in treating CNV associated with inflammatory chorioretinal disorders.46
Placoid diseases
Acute posterior multifocal placoid pigment epitheliopathy
Acute posterior multifocal placoid pigment epitheliopathy (APMPPE) was first described in 1968 by Gass.47 He presented 3 healthy young female patients who developed the acute onset of bilateral central vision loss associated with multifocal placoid (plate-like) lesions at the level of the outer retina and the RPE, although he had also considered a primary choroiditis. Discussion regarding the site and etiology of the lesions would continue as the disease was further described in later years.48–57
Clinical course
Clinical symptoms
Patients present with rapid onset of central vision loss that may be described as blurred vision, paracentral scotoma, metamorphopsia, “spots” in the vision, and photopsias.58 Initial vision at presentation is 20/25 or worse in about 77% of eyes and 20/40 or worse in 58%.58 Deficits can be unilateral or bilateral (more common 75%).59 If unilateral, the second eye can become involved in a few days or weeks. Headaches, stiff neck, and malaise may accompany these ocular symptoms. A history of an antecedent viral syndrome or recent vaccination may be obtained.
Epidemiology
Males and females are equally affected and generally this occurs in young adults. Typically this presents between age 20 and 50 with the mean age of onset being 26.58,59 Recently, Taich and Johnson describe a syndrome resembling APMPPE in older adults (over age 50).60 These elderly individuals characteristically may have a worse outcome, with moderate or severe vision loss due to the development of geographic atrophy and CNV. These patients resemble the entity persistent placoid maculopathy (see below) and may not have APMPPE.
Fundus findings
Gass described the presence of multiple round and confluent cream colored, flat lesions with indistinct margins scattered in the posterior pole. Lesions are not found anterior to the equator. The presence of these lesions is typically bilateral (Figs 76.8, 76.9). Fresh lesions can develop over the next few weeks, therefore lesions of differing ages can be visible. The placoid lesions tend to clear centrally initially leaving hypopigmentation. Later there is mild pigment mottling which develops into condensation of the pigment midperipherally, finally an increasing degree of coarse pigment clumping occurs (Fig. 76.10). These lesions can enlarge, but generally do not. In the original article, there was no description of serous macular detachment associated with APMPPE. However, later reports described localized serous retinal detachments over the lesions.52,55,61–65 This feature occasionally makes it difficult to distinguish APMPPE from Harada’s disease,62,66,67 and some have thought that these entities may form a spectrum of disease.68 It is more likely that there is an underlying common pathology that leads to the serous fluid.69 Gass commented on how remarkably the choroid and retina remain relatively intact during the course of the disease. However, Spaide described choroidal infiltrations in the periphery70 of a patient with acute APMPPE. In addition, multiple authors have described an association with retinal vasculitis71,72 and retinal vein occlusion has also been seen.71,72 Other findings can include subhyaloid hemorrhage72 and rare CNV.73 Case reports have also illustrated optic nerve involvement with disc edema.71,74
Other ocular findings
Although vitritis is not a significant component of APMPPE, the degree of ocular inflammation varies widely. An anterior uveitis75 and granulomatous anterior uveitis76 have been described. In addition, corneal stromal infiltrates77 have been mentioned.
Clinical course and prognosis
Generally there is improvement of the visual symptoms within 2–4 weeks. APMPPE has a relatively good prognosis when compared with other placoid white spot syndromes. However Fiore and colleagues58 reviewed the literature as well as a cohort from their own institution and showed that approximately 50% of patients have an incomplete recovery and 25% of patients have 20/40 vision or worse. Sixty percent of eyes have residual visual symptoms. Foveal involvement at presentation is an important prognosticator. Eighty-eight percent of eyes without foveal involvement proceed to full visual recovery in contrast to 53% of eyes that presented with foveal involvement. Unfortunately, approximately 70% of eyes present with foveal involvement. Photoreceptor involvement may ultimately limit visual prognosis. Progressive improvement of visual acuity usually follows the resolution of the lesions. Gass initially reported that visual recovery could continue on for months after the lesions resolved, even up to 6 months.47 Most visual recovery occurs within 1 month.49,51,52,55,58,61
Imaging
Fluorescein angiography
Gass47 described the lesions in the early phase as nonfluorescent (Fig. 76.11) and that the choroidal fluorescence is obstructed. Later in the angiogram there is a progressive, irregular staining of the lesions (Fig. 76.12). Ryan and Maumenee postulated that this could represent localization to the RPE or choriocapillaris rather than implying an infiltrate of the outer choroid.48 Gass also stated that the underlying choroidal circulation appeared intact below the healing lesions.47 However, later ICGA studies suggested that some choroidal hypoperfusion did exist.78,79 This is discussed below. As the process becomes inactive, hyperfluorescence corresponding to window defects in the RPE develops and staining is no longer evident (Fig. 76.13). There is a clear visibility of the large choroidal vessels in areas of confluent atrophy. However, this does not exist in all areas, implying some RPE remains intact.53 Either loss of the choriocapillaris or abnormal circulation in the confluent atrophied areas is suggested. In addition to these FA findings, there is also blocked fluorescence from pigment clumping.
The retinal vessels and optic nerve appeared normal47,53 in original descriptions; however later reports described an association with retinal vasculitis, retinal vein occlusions, and disc edema.
Indocyanine green angiography
Since ICGA studies focus on the choroidal circulation, its findings in APMPPE have been important in developing theories of pathogenesis.79–84 Acute lesions show early hypofluorescence (Fig. 76.14). In the late phases, these lesions become more defined in shape. These areas are more numerous than the placoid lesions seen clinically. As the disease heals, the hypofluorescence in the late phase becomes less defined and smaller. This lends some support to the theory of choroidal ischemia as an underlying factor in the pathogenesis of APMPPE. It is postulated that there is more hypofluorescence in the acute phase due to the additional presence of swollen outer retina or RPE cells in response to choroidal ischemia (clinically presenting as placoid lesions). As these disappear, there is less blockage and thus less hypofluorescence. Photoreceptor damage may also play a role in this as well, as elucidated by OCT studies and discussed below. Studies show that the hypofluorescence in the late phases can also completely resolve,78 suggesting that choroidal vasculopathy, if present, may be a transient process.
Optical coherence tomography
Many studies have described the OCT findings in APMPPE.67,85–90 Garg and Jampol reported outer retinal abnormalities in APMPPE using time domain technology. A serous retinal detachment had reflective material within the subretinal fluid. It was postulated that this was either proteinaceous material or edematous RPE. There was rapid resolution of the serous detachment and the material disappeared.64
Lofoco and colleagues showed that in the acute phases the OCT revealed a mild hyperreflective area above the RPE in the photoreceptor layer corresponding to the placoid lesions. Later the OCT scan revealed a nodular hyperreflective lesion in the plane of the RPE with mild underlying backscattering. They theorized that the hyperreflective areas may indicate inflammatory tissue and inflammatory cells or the presence of ischemic edema in the outer retinal layers.90 As ultra-high resolution (UHR)-OCT developed, Scheufele and colleagues show disruption of the outer retina early in the disease (Fig. 76.15). RPE disruption occurs as the lesions heal.91 Backscatter of lesions was observed in the acute inflammatory phase in UHR as well. They found photoreceptor atrophy as the lesions began to heal and persistence of it post resolution. They suggested that the backscattering of acute lesions in the outer retina represents inflamed or damaged photoreceptor cell bodies. In addition to illustrating photoreceptor and RPE degeneration, spectral domain OCT has also shown that in some patients with fluid associated with the placoid lesions, that there may actually be accumulation of intraretinal fluid rather than an exudative retinal detachment.64,85
Fundus autofluorescence
FAF imaging is directed at the retinal pigment epithelial layer and is therefore especially useful in APMPPE. Several studies have described FAF findings67,87,89,92,93 (Fig 76.16).
Spaide compared angiographic findings with autofluorescence. He noted that the early hypofluorescence seen on angiogram did not match up with observable changes of the RPE and he suggested this means there are choriocapillaris perfusion defects. In the late phase of the angiogram, there was staining of some of the lesions. These late-staining lesions matched the size and shape of lesions seen in fundus autofluorescence. As the lesions resolved clinically, they became pigmented centrally with a depigmented halo. On autofluorescence, centrally there was intense hyperautofluorescence, and the depigmented halo was hypoautofluorescent, implying atrophy. He postulated there was a centripetal contraction of the placoid lesions that produced this appearance. He notes the autofluorescence changes lagged behind the clinical appearance. In addition, he found that choroidal abnormalities seemed more numerous on fluorescein and indocyanine green angiography. He concluded the RPE abnormalities were a result of the choroidal abnormalities.93
Electrophysiology
Although these functional studies are not essential in the diagnosis of APMPPE, they do emphasize the role of RPE involvement as described by Fishman and colleagues. The electroretinogram is normal to minimally subnormal. However, abnormal light:dark ratios have been documented on EOG, suggesting a diffuse RPE problem. Furthermore, the ERG and EOG abnormalities can normalize, suggesting that this can be a transient RPE problem.53
Systemic associations
APMPPE has been linked to CNS manifestations94–96 including cerebral vasculitis,97–103 meningo-encephalitis104 and stroke.105–109 Cases of APMPPE associated with CN VI palsy110 and transient hearing loss111 have also been cited. Headaches are a common symptom and APMPPE has mimicked migraine with aura.112
[/not-level-membership-for-opthalmology-category]
