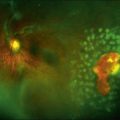
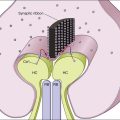
Chapter 41 Hereditary Vitreoretinal Degenerations
Hereditary vitreoretinal degeneration, also known as hereditary vitreoretinopathy, is classically characterized by early-onset cataracts, vitreous anomalies, coarse fibrils and membranes, and retinal detachment. Genetic and clinical advances in the last two decades have enabled a reassessment of the essential criteria that define this group of conditions. More recently, these conditions have been defined as the presence of congenital abnormalities of the vitreous, including severe degeneration or maldevelopment, early-onset progressive cataracts, and an increased predisposition to rhegmatogenous retinal detachment.1 Additional ocular and systemic features may be present, depending on the underlying cause.
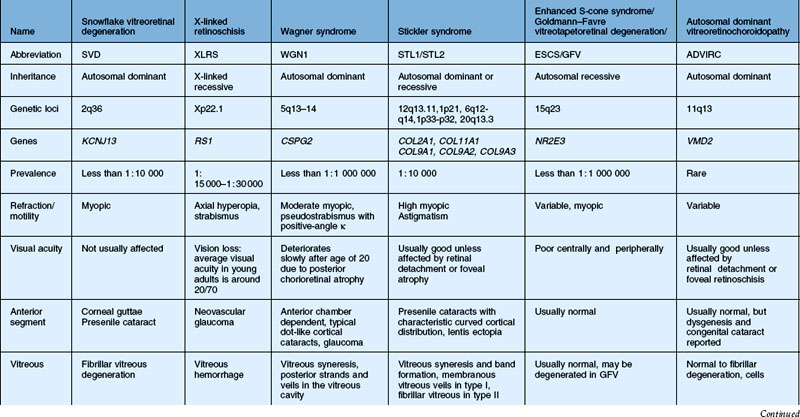
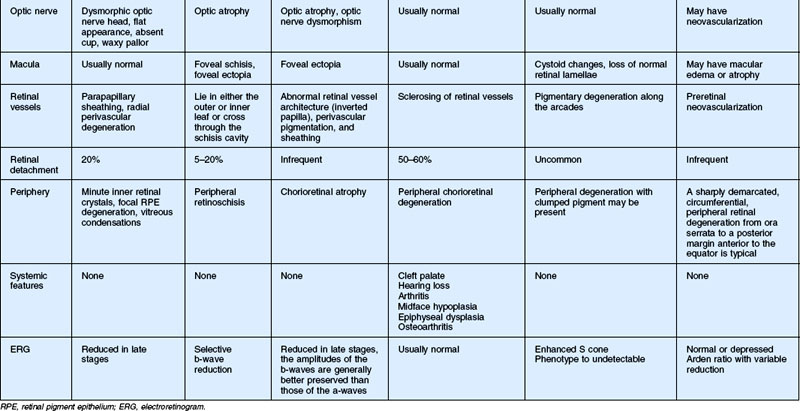
In this chapter, we discuss five main types of vitreoretinopathies: (1) snowflake vitreoretinal degeneration; (2) X-linked juvenile retinoschisis; (3) the chromosome 5q vitreoretinopathies (e.g., Wagner syndrome); (4) chondrodysplasias with vitreoretinal degeneration (e.g., Stickler syndrome); and (5) enhanced S-cone syndrome (ESCS)/Goldmann–Favre vitreotapetoretinal degeneration. Several additional vitreoretinal degenerations are also summarized, including autosomal dominant neovascular inflammatory vitreoretinopathy (ADNIV) and autosomal dominant vitreoretinochoroidopathy (ADVIRC). We review the common features of vitreoretinal degenerations and highlight those features that can be used to distinguish them from each other clinically. Lattice degeneration and familial exudative vitreoretinopathy are not discussed in this chapter. Table 41.1 summarizes the main features of these disorders. Note that a given patient need not have all of the listed features. The pattern of clinical findings, rather than a specific feature, is the best strategy to make the diagnosis.
Snowflake vitreoretinal degeneration
General features
The term “snowflake vitreoretinal degeneration” (SVD) was originally coined by Hirose et al.2 in 1974, who described an American family of European extraction with early-onset cataracts, fibrillar vitreous degeneration, vascular sheathing, peripheral minute crystalline-like deposits, and retinal detachment. The condition received its name from the minute crystalline-like deposits that can be seen by contact lens biomicroscopy in some patients.2 The vitreous degeneration is fibrillar to a variable extent; the bands of condensed vitreous fibrils may obscure fundus details and peripheral condensations of vitreous on the retinal surface can be observed. Radial or circumferential lattice degeneration is not observed. The average spherical equivalent is –2.90 D, indicating moderate myopia. Other distinguishing features include a dysmorphic optic nerve head that appears flat and often without a cup. Peripapillary vascular sheathing and atrophy, as well as waxy pallor, may be present.
Clinical findings
Ocular features
There were 31 individuals in the Hirose family enrolled in the original study in 1974 (13 affected individuals, 14 unaffected individuals, and 4 unaffected spouses). The 13 subjects diagnosed with SVD ranged from 12 to 85 years of age, with clinical features such as early-onset cataract, fibrillar vitreous degeneration, vascular sheathing, and retinal detachment.2 The inheritance pattern was autosomal dominant, and no obligatory carriers of the snowflake trait were found to be normal. After about 20 years, Lee et al.3 restudied 6 of these 13 patients and identified additional clinical features, including corneal guttata (4 out of 5 patients) and optic nerve head dysplasia (the exact number of affected individuals was not recorded). Early-onset cataracts (5 out of 6), fibrillar vitreous degeneration (6 out of 6), and peripheral retinal abnormalities (5 out of 6), including minute crystalline-like deposits called snowflakes (4 out of 6), were common. Compared to other hereditary vitreoretinal degenerations, there was a relatively low rate of retinal detachment, occurring in 1 of the 6 examined family members.3 Orofacial features, early-onset hearing loss, and arthritis that are typical of Stickler syndrome were absent. Thus, the clinical findings were not typical of Stickler syndrome or chromosome 5q retinopathies, suggesting SVD may be a distinct form of vitreoretinal degeneration.
According to the dominant features present on fundoscopic examination, SVD has been classified into four stages: (1) extensive white with pressure; (2) snowflake degeneration; (3) sheathing of retinal vessels and fundus pigmentation; and (4) further pigmentation and disappearance of the peripheral retinal vessels. Hejtmancik et al. classified the clinical features of SVD into subgroups of congenital and progressive abnormalities.4 The congenital abnormalities include optic nerve head dysmorphism with fibrillar degeneration of the vitreous. Progressive ocular features include corneal guttae and peripheral retinal degeneration within which minute crystalline deposits, referred to as snowflakes, might be seen. These characteristics distinguish SVD from other vitreoretinal degenerations.
Using Hejtmancik’s genetic studies, it is evident that although the term “snowflake” has been used in reports of other families, they do not appear to be the same condition according to the clinical criteria and/or genetic evaluation. Moreover, the proportion of patients diagnosed with non-syndromic Stickler syndrome that ultimately share a common genetic basis with SVD patients is currently unknown, thus the real prevalence of SVD to date is difficult to estimate. So far, there is only one family reported in the literature with a case history similar to the classic description of SVD. This family was reported by Pollack and colleagues5,6 and showed an autosomal dominant vitreoretinal degeneration with minute crystalline-like dots, probably similar to the Hirose family. A distinguishing feature of this family, however was the appearance of neovascular tufts in the temporal periphery in 4 of the 9 affected members of the middle generation.6 Two other reports have described subjects with SVD. Robertson et al.7 reported familial clustering of granular deposits 100–200 µm in diameter in 10 patients from four families, which was described as snowflake degeneration of the retina in 1982. The deposits were evenly distributed about the circumference of the eye near the equatorial fundus. Smaller crystalline deposits were observed between the granular deposits. However, findings of vitreoretinal degeneration, such as early-onset cataract, severe vitreous degeneration, and retinal detachment, were not observed. The granular deposits are also not similar to those in patients with COL2A1 mutations causing Stickler syndrome.2,3 Chen and colleagues8 reported a family with vitreoretinal degeneration in 1986, but they were distinguished from the Hirose family by nyctalopia, poor visual acuity, annular scotomas, and attenuated retinal vessels. They may also have shown different-appearing deposits compared to the classical SVD description.2 Unfortunately, these two families have not been subject to genetic analysis.
Molecular genetics of SVD
Jiao et al.9 re-examined the family and localized the mutation to chromosome 2q36. Molecular genetic investigation excluded the known locations of genes causing vitreoretinal degeneration,3 and a novel gene location was subsequently identified. One of the authors (XD) studied the location of Kir7.1 (the protein product of KCNJ13) in the retina, and found that it is bound to the inner limiting membrane and the retinal pigment epithelium, which suggests that the mutation could affect development of the vitreous through alteration of Müller cell function.4 The mutation in KCNJ13 demonstrates that classic vitreoretinal degeneration can arise from gene mutations that are not structural components of the vitreous. Alteration in potassium transport provides a mechanism for the electrophysiological abnormalities seen in these patients, but further study is required for a precise explanation. The condition arises due to a mutation in KCNJ13 disrupting the selective transport of potassium through the channel.4 Thus, SVD can be clinically and genetically confirmed as a unique form of vitreoretinal degeneration.
Visual psychophysics
Kinetic perimetry shows peripheral defects, which are more pronounced in the superior field.10 Flicker perimetry reveals abnormalities undetected by kinetic perimetry and dark adaptation tests show elevated rod thresholds, except during the early stage of the disease.
Differential diagnosis
Stickler syndrome type I
This is the most common form of vitreoretinal degeneration. Mutations leading to haploinsufficiency of the collagen 2A1 (COL2A1) gene cause Stickler syndrome type I (STL1, MIM 108300). These patients have a vitreous degeneration characterized by a unique vitreous appearance vestigial vitreous gel occupying the immediate retrolental space and no discernible gel in the central vitreous cavity. The expression of syndromic features, including hearing loss, facial dysmorphism, and joint pain, exhibits variability both between and within families.11
Stickler syndrome type II
Mutations leading to haploinsufficiency of the collagen 11A1 (COL11A1) gene cause Stickler syndrome type II (STL2, MIM 604841). Unlike COL2A1 disease, these mutations lead to a fibrillar vitreous degeneration with limited and random fibrils throughout the vitreous cavity.12–14 In some cases, severe fibrillar degeneration of the vitreous can also be seen.
Marshall syndrome
Mutations altering intron–exon splicing of the COL11A1 gene lead to Marshall syndrome (MIM 154780), distinguished from SVD and Stickler syndrome by a more pronounced facial dysmorphism and lower frequency of retinal detachment.15,16 Membranous vitreous veils and radial lattice have also been noted in patients with Marshall syndrome.17
Wagner syndrome
Wagner syndrome (MIM 143200) is caused by noncoding mutations that are thought to affect the splicing of chondroitin sulfate proteoglycan-2 (CSPG2), the gene encoding versican.18,19 These mutations may lead to disease through abnormal ratios of versican isoforms. The distinguishing features of Wagner syndrome are pseudostrabismus, thickened and partially detached posterior hyaloids with an empty vitreous cavity, variable degeneration of the retina and choroid, and the absence of systemic manifestations.20 The absence of nyctalopia, posterior chorioretinal atrophy, and tractional retinal detachment distinguishes Wagner syndrome from other chromosome 5q vitreoretinopathies such as Jansen syndrome and erosive vitreoretinopathy (ERVR).
Goldmann–Favre vitreotapetoretinal degeneration
Patients with Goldmann–Favre vitreotapetoretinal degeneration have nonrecordable ERGs and central and/or peripheral retinoschisis. This is in contrast to patients with snowflake degeneration, who have fairly good ERG responses and absence of central or peripheral retinoschisis.21
Management
Common issues in the management of hereditary vitreoretinal degenerations are described below:
1. Cataract surgery is difficult in these patients due to lack of vitreous support during surgery, and should be performed by experienced surgeons with specific experience with vitrectomized eyes that behave similarly. Microinvasive 23- or 25-gauge infusion cannulae via the pars plana are beneficial for maintaining the intraocular pressure.
2. Glaucoma can occur, usually after cataract surgery, and should be monitored and treated using established methods.
3. Prophylactic cryopexy is performed by some groups, while peripheral laser retinopexy is favored by others. Although a randomized trial has yet to be published comparing the two methods, a recent report using cryopexy is the most comprehensive study to date.22 Ang et al. found that, in patients with type I Stickler syndrome, the prevalence of RD was significantly less in bilateral 360° prophylactic cryotherapy than that in untreated patients, suggesting that prophylactic cryotherapy may be substantially beneficial.22
4. Retinal detachments are also common and should be managed using standard vitrectomy-based approaches, including vitrectomy, artificial posterior vitreous detachment, and release of peripheral traction, perfluorocarbon-mediated retinal reattachment, scleral buckle, laser retinopexy, and tamponade with gas or silicone oil.
The chromosome 5Q retinopathies: wagner syndrome, jansen syndrome, erosive vitreoretinopathy, and related conditions
General features
Wagner syndrome is characterized by an optically empty vitreous with avascular vitreous strands and veils, moderate myopia, presenile cataracts, and retinal degeneration with atrophy.23 Stickler syndrome is also associated with craniofacial abnormalities and a progressive arthropathy.24 Wagner syndrome and Stickler syndrome were once incorrectly considered as one entity: the Wagner–Stickler syndrome.
Wagner syndrome is an autosomal dominant genetic disorder first mapped to chromosome 5q13–14 in 1995. A mutation in the chondroitin sulfate proteoglycan 2 gene (CSPG2), now named VCAN, encoding for the versican protein, was found in 2005.20,25,26 VCAN is the only gene currently associated with Wagner syndrome and ERVR.19,27 Autosomal dominant Stickler syndrome is an inherited progressive disorder of the collagen connective tissues and is associated with the mutation of extracellular matrix collagen genes, such as COL2A1, COL11A, COL11A2, and COL9A1.24,28–30
Jansen syndrome was described as vitreoretinal and lenticular degeneration associated with retinal detachments in the absence of nonocular findings. However, the disease gene for the original Jansen family was demonstrated to be linked to the same region of chromosome 5q1431 where genes for Wagner syndrome and ERVR were located.25
ERVR also displays an autosomal dominant inheritance pattern and shares some clinical features with Wagner syndrome. The critical region of the genetic defect underlying ERVR was found to overlap with the critical region for Wagner syndrome, 5q13-q14.19
Clinical findings
Ocular features
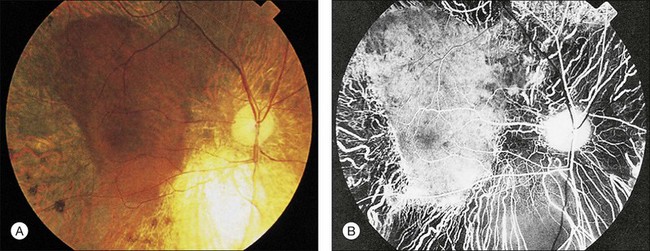
Wagner syndrome is characterized by an optically empty vitreous with equatorial avascular vitreous veils. Additional features include its early-onset, moderate myopia, and typical dot-like cortical cataracts, foveal ectopia, abnormal retinal vessels (inverted papilla), perivascular pigmentation and sheathing, retinal thinning, as well as slowly progressive chorioretinal atrophy. Patients with Wagner syndrome have pseudostrabismus from congenital temporal displacement of the fovea. They also have nyctalopia early in life and final dark adaptation thresholds are elevated in some patients.32 Most patients under the age of 20 have normal vision; however, cataract, retinal detachment, optic atrophy, and chorioretinal atrophy may be progressive and cause visual loss with advancing age20,23,25–27 (Fig. 41.1). In addition to the clinical features of Wagner syndrome, ERVR reveals progressive nyctalopia and visual field constriction. A conspicuous loss of retinal pigment epithelium and choriocapillaries is observed by fluorescein angiography.19 The vitreous findings are marked syneresis with prominent membranes. Rhegmatogenous retinal detachment is usually described as a feature of ERVR, and is less frequent in the original description of Wagner syndrome.33
Visual psychophysics
Nyctalopia can be present early in life in some patients. Vision is usually normal as the pathologic process initially involves the retinal periphery, but severe loss of vision will occur in patients when diffuse cone–rod loss ensues as there is progressive chorioretinal atrophy.1,25
Electrophysiology
Both the ERG and dark adaptation of patients with the chromosome 5q retinopathies appear to be normal early in life but become progressively abnormal throughout the patient’s life. The rod and cone systems are affected to varying degrees but in a family-specific manner. While both a-wave and b-wave amplitudes are reduced, b-wave amplitudes are generally better preserved. Visual field findings can be variable and may include diffuse peripheral loss or partial/complete midperipheral ring scotomas as the chorioretinal atrophy progresses.34
Differential diagnosis
Autosomal dominant vitreoretinopathies
Snowflake vitreoretinal degeneration
SVD is a progressive hereditary eye disorder caused by mutations in KCNJ13. Diagnostic features of SVD consist of fibrillar vitreous degeneration, early-onset cataract, minute crystalline deposits in the neurosensory retina, and retinal detachment.4 However, membranous degeneration of the vitreous with avascular strands and veils is not observed in SVD. Retinal defects typically start in the superficial retinal layers and retinal detachment is uncommon.4,27
Stickler syndrome
Stickler syndrome is genetically distinguished from Wagner syndrome and other chromosome 5q retinopathies. Type I Stickler syndrome is due to COL2A1 mutation and is associated with retrolental membranous vitreous, while type II Stickler syndrome is due to mutations in COL11A1 and is associated with a fibrillar or beaded vitreous phenotype. Both type I and II Stickler syndrome have ocular and systemic manifestations, while type III Stickler syndrome, associated with COL11A2 mutations, has systemic manifestations only. Most, but not all, patients with Stickler syndrome have congenital, nonprogressive, and high-degree myopia. The cataracts may be congenital and nonprogressive, and have an unusual characteristic curved cortical distribution.35 Retinal detachment is much more common in Stickler syndrome (50%) than in chromosome 5q vitreoretinopathies (15%). Systemic abnormalities are present in Stickler syndrome, such as midface hypoplasia, midline cleft of the palate, bifid uvula, sensorineural hearing loss, and skeletal abnormalities.24,27–29 Abnormal dark adaptation associated with alterations in the ERG that is common in chromosome 5q retinopathies has not been described in Stickler syndrome.1,27
Autosomal dominant vitreoretinochoroidopathy
ADVIRC is caused by mutations in VMD2 and also has characteristic retinal and vitreous findings, in particular a peripheral retinal circumferential hyperpigmented band, vitreous fibrillar condensation, punctate white opacities in the retina, breakdown of the blood–retinal barrier, and retinal neovascularization.36
Autosomal recessive vitreoretinopathies
Goldmann–Favre syndrome (GFS) and enhanced S-cone syndrome
GFS and ESCS share common mutations in the NR2E3 gene and are usually associated with night blindness and visual field abnormalities. ERG typically reveals a severe reduction in rod function and a relatively enhanced function of the short-wavelength-sensitive cones.27,37 GFS manifests with progressive vitreous changes, hemeralopia, chorioretinal atrophy, and pigmentary retinal degeneration, later resulting in marked visual field loss, retinoschisis in the periphery and/or macula, presenile cataract, and hyperopia rather than myopia.27,38,39 ESCS lacks the typical marked vitreous changes of GFS.
Treatments
Refractive error is corrected by spectacles or contact lenses. Cataract is managed by the phacoemulsification and implantation of an intraocular lens. Retinal breaks without retinal detachment are treated with laser photocoagulation. Vitreoretinal surgery is needed for retinal detachment, vitreoretinal traction involving the macula, or epiretinal membranes involving the macula. Prophylactic cryotherapy has been recently reported to reduce the risk of retinal detachment markedly.1
Chondrodysplasias associated with vitreoretinal degeneration: the stickler syndromes, marshall syndrome, kniest dysplasia, knobloch syndrome, and weissenbacher–zweymuller syndrome
General features
Chondrodysplasias refer to a group of hereditary and systematic disorders that affect skeletal development and growth. These conditions may also feature ocular, central nervous system, or renal abnormalities. The genes involved in these syndromes affect types II, III, V, X, or XI, collagen molecules that are found mainly in cartilage and vitreous and are essential for the normal development of bones and other connective tissue, thus accounting for the symptoms observed clinically. Antenatal diagnosis, including fetal magnetic resonance imaging, computed tomography, ultrasonography, and genetic testing of fetal DNA obtained from aminocentesis or chorionic villus sampling, is important for the proper management of children and counseling of parents.41–43
Stickler syndrome, also known as hereditary progressive arthro-ophthalmopathy, is considered to be the most common chondrodysplasia associated with vitreoretinal degeneration. Types I, II, and III are the three subgroups of Stickler syndrome classified by genetic heterogeneity. Types I and II Stickler syndrome, respectively caused by mutations in the COL2A1 gene encoding type II collagen44 and in the COL11A1 gene encoding type XI collagen,45 can be differentiated successfully by vitreous phenotypes. Mutations in COL2A1 usually result in a congenital membranous vitreous anomaly, while mutations in COL11A1 result in an irregular and beaded vitreous.46,47 Recently, a new subgroup of COL2A1 mutations was found that lead to a hypoplastic vitreous which is either optically empty or contains sparse irregular lamellae.13 Due to the differential splicing, COL2A1 gene transcription products can be divided into two types: collagen IIA with an exon2-encoded length of 69 (rich in amino acid homocysteine) and collagen IIB without the exon2-encoded peptide. Whereas both collagen IIA and IIB loss results in systemic connective tissues disease, collagen IIA loss also leads to ocular abnormalities. It is because collagen IIA primarily exists in the vitreous.48,49 Recent studies have indicated that mutations in any collagen IX genes, like COL9A1, COL9A2, or COL9A3, can cause autosomal recessive Stickler syndrome.50–52 Thus far, the diagnosis of Stickler syndrome has been based on clinical manifestations without consensus on the minimal clinical diagnostic criteria. The anomalous formation of the vitreous gel structure and its corresponding characteristic abnormalities are essential for the diagnosis of types I and II Stickler syndrome. In additional to these clinical manifestations, a detailed family history should be obtained and the diagnosis can be confirmed by genetic analysis. Recently, the COL2A1 mutation in peripheral white blood cells has been used to identify patients with type I Stickler syndrome.51
Marshall syndrome, an autosomal dominant chondrodysplasia, is caused by a splicing mutation of 54-bp exons in the c-terminal region of the COL11A1 gene which is located on the short arm of chromosome 1. Although Marshall syndrome and Stickler syndrome are now considered two distinct diseases, a case of a patient with overlapping phenotypes has been described.53
Knobloch syndrome is a rare and clinically heterogeneous autosomal recessive disorder. Collagen XVIII, which is a basement membrane proteoglycan, distributes in multiple organs of the body and plays an important role in the function and development of the eye, kidney, and nervous system. In most patients with this symptom, null mutations in the COL18A1 gene mapped to the long arm of chromosome 21 are supposed to induce the changes in collagen XVIII.54 There are also case reports of Knobloch syndrome without COL18A1 gene mutations, and thus immunofluorescent histochemistry of skin biopsy samples has proved to be a useful preliminary and complementary test for the diagnosis of Knobloch syndrome.55
Weissenbacher–Zweymuller syndrome, also called Pierre Robin syndrome with fetal chondrodysplasia, is an autosomal recessive disorder characterized by a single-base mutation in the COL11A2 gene, which substitutes glutamate for glycine.56 In the past, it was often misdiagnosed as Stickler syndrome. Recently, investigators have differentiated these two syndromes successfully by prenatal ultrasonography.57
Clinical findings
Extraocular features
Stickler syndrome features a highly variable systemic phenotype, including conductive and sensorineural hearing loss, immunoglobulin deficiency,20 cleft palate, mid facial underdevelopment, mild spondyloepiphyseal dysplasia, and precocious arthritis58 (Fig. 41.2).

Kniest dysplasia has the typical manifestations of short-trunk dwarfism with kyphoscoliosis, enlarged joints with decreased motion, flat midface, cleft palate, and hearing loss.59
Weissenbacher–Zweymuller syndrome is characteristic of midface hypoplasia with a flat nasal bridge, small upturned nasal tip, micrognathia, sensorineural hearing loss, and rhizomelic limb shortening. Radiographic examination may detect dumbbell-shaped femora and humeri, and vertebral coronal clefts in these patients.53 However, some congenital abnormalities, such as dwarfism, developmental delays, and radiological abnormalities, may return to normality at school age, so it is hypothesized to be dysmaturational other than syndromic.60
Ocular features
Types I and II Stickler syndrome have a high risk of ocular complications, including congenital high myopia, cataract, and retinal problems,46 such as vitreous changes, radial perivascular retinal degeneration, and rhegmatogenous retinal detachment61 (Fig. 41.3). These findings may manifest at any age and produce vision loss. Unlike Stickler syndrome, rhegmatogenous retinal detachment is unusual in Marshall syndrome and cataracts may be spontaneously absorbed.
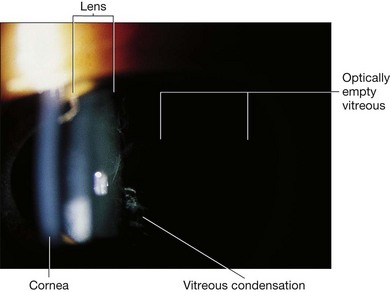
Fig. 41.3 Stickler syndrome. Slit-lamp photograph of the child shown in Figure 41.2 showing congenital vitreous abnormality.
The ocular manifestations of Kniest dysplasia mainly consist of high myopia, optically empty vitreous with retrolental and peripheral vitreous membranes, lattice degeneration, and retinal detachment. Ocular manifestations involved in Knobloch syndrome include high myopia, cataract, vitreoretinal degeneration, and retinal detachment which lead to progressive and irreversible vision loss. Weissenbacher–Zweymuller syndrome mainly shows strabismus and various refractive errors, which should be treated at an early age to prevent amblyopia.62
Differential diagnosis
Wagner syndrome
As noted above, Wagner syndrome is often confused with Stickler syndrome. Key differentiating features are the typical vitreous abnormalities, higher risk of retinal detachment, and systemic findings of Stickler syndrome versus the characteristic nyctalopia, retinal pigmentary changes, and dark adaption problems of Wagner syndrome.20,23
Erosive vitroeretinopathy
ERVR, as described above, is characterized by an “optically empty vitreous” and avascular vitreous strands and veils, mild or occasionally moderate to severe myopia, presenile cataract, night blindness of variable degree associated with progressive chorioretinal atrophy, retinal detachment at advanced stages, and reduced visual acuity. Systemic abnormalities are not observed. Rhegmatogenous retinal detachment occurs more frequently in ERVR than in Wagner syndrome.63
Management
The most common management strategy is symptomatic treatment for complications such as mandibular distraction osteogenesis for pediatric airway management,64 mandibular advancement for malocclusion and micrognathia, correction of refractive errors with spectacles, vitrectomy with silicone oil injection for retinal detachment, and symptomatic treatment for arthropathy.
Prevention of secondary complications and surveillance are essential.65 The rate of retinal detachment has been reported to be as high as 60%,24 so frequent follow-up and potential interventions, including laser therapy, are of great importance in prevention.
Recently, several transgenic mouse models have been developed with mutations in the pro-alpha collagen chain. These animals may serve as useful models for arthro-ophthalmopathies and eventually provide a basis for gene-directed therapy for these conditions.43,48
X-linked retinoschisis
General features
X-linked retinoschisis (XLRS) is a human inherited retinal degenerative disease caused by mutations of the RS1 gene on Xp22.1. Haas66 first described the disease that is now known as retinoschisis in 1898. XLRS was first documented as being X-linked in 1913.67 The term “X-linked retinoschisis,” first used in 1953,68 is now the generally accepted terminology.69–71 XLRS is the most common form of juvenile-onset retinal degeneration in males, with a prevalence of between 1 in 15 000 and 1 in 30 000, and causes vision loss.72,73 XLRS is characterized by schisis of the neural retina, including cystic maculopathy, peripheral schisis, and reduced amplitude of the b-wave on the ERG.74 The common sight-threatening complications of XLRS include retinal detachment, vitreous hemorrhage, and foveal schisis.75 Females who are heterozygous for the RS1 mutation commonly have no clinical symptoms of XLRS.76 To date, no treatment is available to halt the development of schisis in patients with XLRS. Surgical interventions are required for XLRS patients with severe complications, such as retinal detachment and nonclearing vitreous hemorrhage.77
Since the causative gene RS1 was identified in 1997,78 approximately 177 mutations of the RS1 gene responsible for XLRS have been found (http://www.dmd.nl/rs/index.html). RS1 is exclusively expressed in the photoreceptors and retinal bipolar cells,79,80 and encodes a 224-amino-acid homo-oligomeric secretory protein complex – retinoschisin. Retinoschisin can be detected throughout the neural retina layers despite its restricted pattern of gene expression.79–81 A mutant RS1 gene appears to interfere with the secretion and/or octamerization and function of retinoschisin.82–84 Wu et al.82 found that RS1 exists as a novel octamer in which the eight subunits are joined together by Cys59-Cys223 intermolecular disulfide bonds. Each subunit consists of a 157-amino-acid discoidin domain. Within the discoidin domain, one cysteine (Cys83) exists in its reduced state, and two cysteine pairs (Cys63-Cys219 and Cys110-Cys142) form intramolecular disulfide bonds that are important in protein folding. Mutations of RS1 disrupt subunit assembly and then cause XLRS. Although insertions, deletions, and splice site mutations have been described, the mutations that encode the discoidin domain are predominantly missense and clustered in exons 4–6.
Many studies have been focused on understanding the function and role of retinoschisin in retinal cell adhesion. Recently, retinoschisin has been reported to interact with β2 laminin within the extracellular space and αB crystallin intracellularly as it moves through the secretory pathway.73,85,86 Molday et al.87 have confirmed the co-localization of retinoschisin with Na/K-ATPase and SARM1 in photoreceptors and retinal bipolar cells. Gehrig et al.88 suggested that activation of microglia/glia may trigger the photoreceptor degeneration in retinoschisin-deficient mice and that the Erk1/2-Egr1 pathway may be activated in the pathogenesis of RS. Also, RS1 efficiently and reversely binds galactose-agarose and lactose-agarose, indicating the possibility of interaction between RS1 and glycosylation sites on Na/K ATPase, providing a method for the purification of RS1, which may facilitate further functional studies of RS1.89 There may be other influencing factors such as genetic modifiers or environmental influences, since no correlations are between mutation type and disease severity or progression exists.90,91 Certainly, the roles of these potential molecular interactions in XLRS require further investigation.
Clinical findings
Ocular features
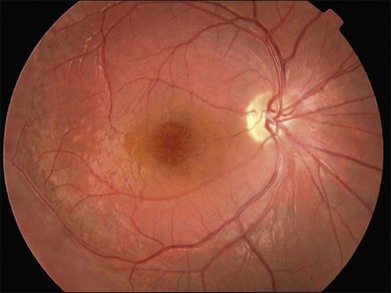
XLRS has variable disease severity, even when caused by the same RS1 mutation.90 Each eye of a patient may have asymmetric progression, but both eyes are invariably involved. Foveal schisis is the characteristic sign of XLRS and is present in 98–100% of cases.92,93 Although macular changes are present in almost all XLRS patients, the typical foveal schisis, seen as a spokewheel pattern of folds radiating out from the fovea (Fig. 41.4), has been found to appear in only about 70% of XLRS patients. Peripheral retinoschisis is often noted in the inferotemporal region and is present in around 50% of patients94 (Fig. 41.5). The splitting occurs in the superficial retinal layers, and so retinal vessels may lie in either the outer or inner leaf or cross from one to the other through the schisis cavity. Breaks occur within the inner layer, varying from small holes to large tears,92 and the fragmentation of the inner leaf can lead to membranous remnants referred to vitreous veils. Other changes, including subretinal linear fibrosis, pigmentation, white retinal flecks, and vascular attenuation or sheathing, often appear in peripheral retina. The common sight-threatening complications of XLRS include traction or rhegmatogenous retinal detachment, dense vitreous hemorrhage, hemorrhage within a large schisis cavity, and intraretinal splitting involving the macula. Other less common complications include neovascular glaucoma,95 vitreoretinal traction with secondary macular dragging,96,97 and optic atrophy.98 For most retinal detachments associated with XLRS, fluid usually accesses the subretinal space through either outer leaf/layer breaks in the areas of peripheral retinoschisis with inner leaf holes, or full-thickness retinal tears following vitreous detachment. Vitreous hemorrhage and retinal detachment are the most serious complications of XLRS. About 5–20% of XLRS patients may progress to retinal detachment,92,93 and up to a third of patients develop vitreous hemorrhage,73,99 which causes severe vision loss.
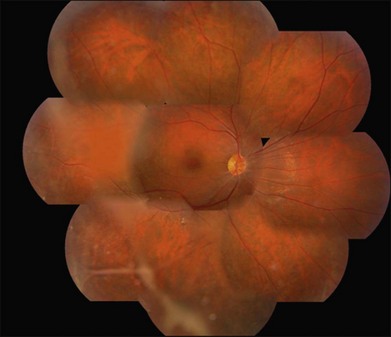
Less commonly, the disorder may present in early infancy with strabismus, nystagmus, axial hyperopia, foveal ectopia, or bilateral very large bullous retinoschisis, often with hemorrhage within the schisis cavity or into the vitreous.99–101
Visual psychophysics
Vision loss is the most common clinical presentation in XLRS patients. Visual acuity may deteriorate during the first and second decades of life, presenting as young as 3 months,77 then remain relatively stable with very slow progression of macular atrophy until the fifth or sixth decade,102 with eventual progression to legal blindness (acuity <20/200) by the sixth or seventh decade. XLRS patients are often detected when having reading difficulties and poor vision at school age. Visual acuity ranges from 20/20 to less than 20/200. The average visual acuity in young adults is around 20/70. Retinal detachment and vitreous hemorrhage may be the cause of a precipitous drop in visual acuity. Defective color vision (red–green dyschromatopsia) can also be present in XLRS patients.103 The visual field shows an absolute scotoma in the field corresponding to the location of the peripheral retinal schisis.
Optical coherence tomography
Optical coherence tomography (OCT) helps to enhance the visualization of macular pathologic features in XLRS (Fig. 41.6).104 The schisis can occur in different layers of the neural retina.75 OCT findings may vary depending on the disease stage.105
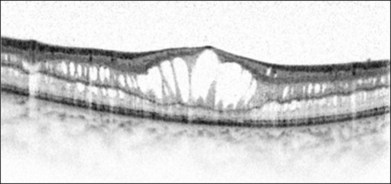
Electrophysiology
ERG is helpful in the diagnosis of XLRS. There is typically reduced b-wave amplitude with a relatively preserved a-wave amplitude, although a few patients may have a relatively preserved b-wave amplitude.94 The alteration of the b : a ratio (“negative” waveform, with a-wave amplitude exceeding the b-wave amplitude) is considered to be an important diagnostic parameter.106 However, not all individuals with XLRS show the classic electronegative ERG, and b-wave amplitudes may not be significantly different from normal. The a-wave can be normal or near-normal in an XLRS patient, whereas it may also be reduced due to the progressive atrophy of the retinal pigment epithelium.107 The severity of ERG abnormalities does not appear to correlate with the mutation type.108 Multifocal ERG can also demonstrate the reduced amplitudes and longer implicit times in the central macula (Fig. 41.7).
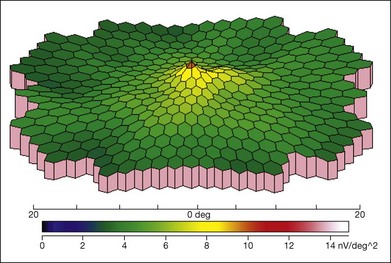
Differential diagnosis
The typical foveal schisis in a male with a reduced b-wave on ERG and a family history consistent with X-linked inheritance makes the diagnosis of XLRS very likely.73 A domelike or slight elevation of a very thin layer of retinal tissue containing blood vessels can be visualized by ophthalmoscopy. Ancillary studies, including OCT and fluorescein angiography, may be useful in supporting the clinical diagnosis when minute foveal schisis is difficult to find by ophthalmoscopy. XLRS patients can be further confirmed with gene diagnosis of the RS1 gene mutation.
Cystoid macular edema is often observed in association with disorders such as retinal vein occlusion, diabetic retinopathy, uveitis, retinitis pigmentosa, dominantly inherited cystoid macular edema, or intraocular surgery (Irvine–Gass syndrome).109 Although angiography is not required for diagnosis of XLRS, the absence of leakage in XLRS may aid in differentiating foveal schisis from other causes of cystoid macular edema characterized by late hyperfluorescence in a petaloid pattern (Fig. 41.8).
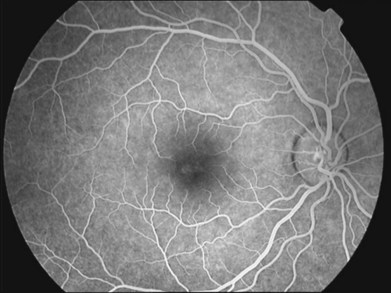
Degenerative retinoschisis is an idiopathic, degenerative splitting of the outer layers of the peripheral retina without ERG abnormalities or RS1 mutations, occurring in an older age group, typically unilaterally.73,110
Goldmann–Favre vitreoretinal degeneration and ESCS caused by mutations in the NR2E3 gene can also lead to foveal schisis, but the severely impaired vision, including marked visual field loss and nyctalopia, pigmentary clumping, absence of vitreous veils, and markedly reduced a-waves and b-waves with altered timing, should help to differentiate this disease from XLRS.39
Treatment
Pharmacological treatment
The successful treatment of schisis cavities with carbonic anhydrase inhibitor has been previously reported.111 Genead et al.112 treated 29 eyes of 15 XLRS patients with topical dorzolamide for 4–41 months and noted some positive effects on visual acuity, cystoid macular lesions, and central foveal thickness. Further studies are required to elucidate the true frequency and completeness of the response to dorzolamide, as well as to evaluate for possible recurrence of foveal cystic change.113 More specific pharmacotherapies may become possible as the pathogenetic mechanisms of XLRS are better understood.
Laser
Laser photocoagulation is often considered as an adjuvant or preventive treatment for XLRS.114,115 However, scatter laser photocoagulation performed in order to flatten peripheral schisis cavities and reduce the likelihood of retinal detachment resulted in retinal detachment in many cases.116 So the administration time and effectiveness of laser treatment for XLRS should be carefully considered.
Surgery
Surgical intervention may be required for XLRS patients with severe complications, such as retinal detachment and vitreous hemorrhage. Currently, the main surgeries include scleral buckle,115 vitrectomy, and perfluorocarbon liquid, perfluorodecalin or sulfur hexafluoride gas tamponade. Vitreous surgery includes core vitrectomy, surgical induction of posterior vitreous detachment, removal of the internal limiting membrane, and gas tamponade.117In addition, Wu et al.118 used autologous plasmin enzyme-assisted vitreoretinal surgery to treat XLRS patients and achieved retinal reattachment in 91% (20/21) of eyes and postoperative visual improvement in 53% (8/15).
Gene therapy
Gene therapy also holds great promise for the treatment of inherited retinal degenerations.119 Gene therapy might be an effective treatment for XLRS patients.120 The RS1 gene delivered intraocularly in RS1-knockout mice was found to restore b-wave amplitude of the treated mice.121,122 XLRS patients may benefit from the replacement of RS1 gene, even at advanced stages, and gene replacement may be a promising treatment for XLRS patients in the near future.123
Retina and/or progenitor cell transplantation
Retinal transplantation or replacement also holds promise as a potential future therapy for XLRS disease in conjunction with the development of new surgical techniques and instrumentation. However, due to the limited source of human tissue and ethical considerations, it is important to find an alternative cell source for retinal replacement therapy. Transplantation of stem cells and/or progenitor cells, including retinal progenitor cells, bone marrow-derived cells, and induced pluripotent cells, may eventually provide an alternative approach to restore vision.124 Compared to human embryonic stem cells, the use of human bone marrow-derived cells and induced pluripotent cells does not have significant ethical issues and may eliminate the risk of immunorejection.
Retinal nuclear receptor (NR2E3)-related diseases: enhanced S-cone syndrome and goldmann–favre vitreotapetoretinal degeneration
General features
NR2E3 (retinal nuclear receptor subfamily 2, group E, member 3), formerly called photoreceptor-specific nuclear receptor (PNR), is a transcription factor of the nuclear hormone receptor superfamily whose expression is uniquely restricted to photoreceptors. Its physiological activity is essential for proper rod and cone development and maintenance. Mutations in NR2E3 may suppress cone proliferation during retinal development125 and lead to an autosomal recessive retinal degeneration of variable severity, encompassing GFS,126,127 ESCS,128 and clumped pigmentary retinal degeneration.39,129
Goldmann–Favre vitreotapetoretinal degeneration (also called Favre microfibrillar vitreoretinal degeneration), a rare condition that affects the retina, vitreous body, and crystalline lens, was first described in 1958.130,131 The characteristic features are early-onset nyctalopia, fibrillar vitreous degeneration, foveal cysts, peripheral retinoschisis, and retinal degeneration with clumped pigment, and an unusual ERG.132 An estimated 0.5% of patients with retinitis pigmentosa have clumps of pigment in the mid-peripheral fundus, referred to as “clumped pigmentation.” To et al. reviewed the clinical and pathological findings in patients with clumped pigmentation and found that they had signs and symptoms of NR2E3 disease. Histopathologic study showed that the clinically distinct areas of clumped pigment are due to excessive accumulation of melanin granules in retinal pigment epithelial cells.129
ESCS was described in 1990 and named after the enhanced sensitivity of the S-cone system128,133 due to an increase in the blue cone population with associated variable degeneration of the rod and red and green cone photoreceptors. Clinical features include early-onset night blindness, cystic maculopathy, and peripheral retinal degeneration characterized by mild visual field loss. The enhanced S-cone ERG shows no response to dim light in the dark-adapted state, but a large, slow response to bright light which persists with light adaptation.128
Many psychophysical,134 histological,129,135,136 and animal studies137,138 have demonstrated that mutations in NR2E3 lead to an absolute increase in S cones at the expense of M and L cones, decreased rod development, and retinal disorganization and degeneration. Severe loss of rod sensitivity is evident throughout the retina, along with the characteristic enhanced S-cone ERG.
Clinical findings
Ocular features
Patients with GFS have progressive loss of vision similar to retinitis pigmentosa, which is caused by retinoschisis, cataract, and pigmentary chorioretinal degeneration. It is the macular retinoschisis that most often accounts for the poor central vision and central scotoma in this disorder (Fig. 41.9). The presenting symptom is frequently night blindness, which is often detected in the first decade of life.132,139 The intraretinal pigmentation is described as spots of pigment, rather than the bone spicule pattern seen in typical retinitis pigmentosa. The retinoschisis in some patients affects both the central and peripheral retina, and resembles juvenile XLRS. Macular lesions may be isolated from or continuous with the peripheral area of schisis. The peripheral retinoschisis frequently shows oval holes in the inner layer and causes absolute field defects in the peripheral visual field.
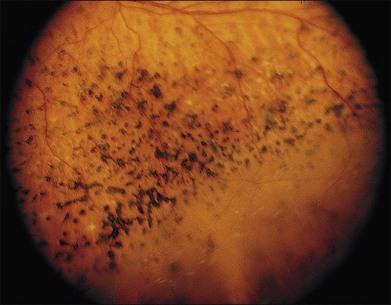
Fig. 41.9 Goldmann–Favre vitreotapetoretinal degeneration, illustrating pigment spots and retinoschisis.
(Courtesy of Gerald A. Fishman, MD.)
The most striking vitreous change is liquefaction, which converts a large portion of the vitreous body into an optically empty space, somewhat similar to that found in Wagner syndrome.139 The space may contain fine fibrous strands and is surrounded by semiliquefied gel containing loose, plated membranes. These membranes have no visible edge and vary in density from one part of the vitreous body to another. The outer layer of the posterior cortex is condensed, resembling a preretinal membrane. When the posterior cortex is detached, it shows depressions that appear to be molded to retinal vessels. The cortex usually adheres to the retinoschisis and to areas with chorioretinal pigmentary proliferation.
Electrophysiology
The characteristic ERG shows an undetectable rod-specific response, similar photopic and scotopic responses to a standard single flash, and a 30-Hz lower-amplitude photopic a-wave response. High variability is common and at least partially related to the severity of retinal degeneration.132 As discussed earlier, the response from the S-cone system accounts for much of the waveform under both scotopic and photopic conditions. The ERG may become undetectable over time.
In addition, ERGs of patients with either ESCS or GFS have greater amplitudes to short-wavelength (e.g., blue) light flashes than to intensity-matched, long-wavelength (e.g., orange) light flashes.135,140,141 Although rods and S cones are both maximally sensitive to blue light and either might hypothetically mediate this hypersensitivity, the similar ERG amplitudes under scotopic and photopic conditions to single flashes of bright white light indicate that it is mediated predominantly by S cones.141–143 Evidence from the shape of the ERG a-wave and from psychophysical studies of color sensitivity indicates that the affected retinas have an overabundance of S-cone photoreceptors at birth, a reduced number of L and M cones, and few, if any, functional rod photoreceptors.143 The relative abundance of S-cone photoreceptors persists even late in the disease, when visual function is severely reduced, as shown by histopathological examination at autopsy of the eyes of a patient with ESCS.135
Management
There is no satisfactory treatment for this condition. Standard vitreoretinal approaches are used for rhegmatogenous retinal detachment and prophylactic treatment of retinal tears using laser photocoagulation should be performed. As discussed in the section on congenital XLRS, prophylactic treatment of breaks in the outer layer of the retinoschisis is generally not recommended. Laser photocoagulation has been used to treat elevated macular retinoschisis.144
Other vitreoretinal degenerations and vitreoretinopathies
Autosomal dominant vitreoretinochoroidopathy
Kaufman and colleagues described a unique condition with 360° of peripheral chorioretinal atrophy between the ora serrata and a very distinct posterior border near the equator.145 This feature is not seen in any other disorder to our knowledge. Cataract, moderate fibrillar vitreous degeneration with pigmented cells, cystoid macular edema, neovascularization of the disc, punctate white opacities on the surface of the retina, and retinal detachment may be observed.146–148 The electro-oculogram may be abnormal,149 but the ERG is typically normal and nyctalopia is absent.
Autosomal recessive inherited vitreoretinal dystrophy
Sarra and colleagues reported a family with 4 of 8 siblings affected with early-onset high myopia, vitreous liquefaction, macular staphyloma with chorioretinal atrophy, diffuse peripheral atrophy of the retinal pigment epithelium, and early cataract. There was also a peripheral veil in one subject, but no extraocular manifestations.150 Monophasic dark adaptation was observed and the ERG showed a severe rod–cone retinal degeneration.150 Linkage analysis identified chromosome 22q13 with a 2-point lod score of 2.18 as the likely locus.
Hereditary neovascular vitreoretinopathies
These conditions are characterized by hereditary peripheral retinal neovascularization with vitreoretinal traction. We discuss here hereditary conditions without primary vitreal degeneration, unaccompanied by systemic clinical manifestations, and incontinentia pigmenti, sickle-cell retinopathy, and other peripheral proliferative retinopathies that have been reviewed previously.151
Autosomal dominant neovascular inflammatory vitreoretinopathy
ADNIV is an apparently rare condition characterized by cataract, cystoid macular edema, peripheral retinal scarring and pigmentation, peripheral arteriolar closure, and neovascularization of the peripheral retina at the ora serrata.152 Young adults are asymptomatic, but have vitreous cell and selective b-wave loss on the ERG. Neovascularization may result in tractional retinal detachment. About half of patients will develop rubeosis or neovascular glaucoma by age 60 or older. The gene was localized to chromosome 11q13.153 Vitreous bands and sheets are not observed and the vitreous was not optically empty, enabling differentiation from classical vitreoretinal degenerations such as Stickler, Wagner, and SVD. The peripheral retinal vessels are initially normal in ADNIV, and dragging of the macular vessels as seen in familial exudative vitreoretinopathy is absent.
Dominantly inherited peripheral retinal neovascularization
Gitter and colleagues described a family with 7 of 15 members affected with early cataract, uveitis, prominence of the vitreous base, lattice degeneration, and severe peripheral retinal neovascularization leading to vitreous hemorrhage and retinal detachment. The syndrome appears similar to ADNIV, but after reviewing photographs of the ADNIV family, the condition was deemed to be distinct.154
1 Edwards AO. Clinical features of the congenital vitreoretinopathies. Eye (Lond). 2008;22:1233–1242.
2 Hirose T, Lee KY, Schepens CL. Snowflake degeneration in hereditary vitreoretinal degeneration. Am J Ophthalmol. 1974;77:143–153.
3 Lee MM, Ritter R, 3rd., Hirose T, et al. Snowflake vitreoretinal degeneration: follow-up of the original family. Ophthalmology. 2003;110:2418–2426.
4 Hejtmancik JF, Jiao X, Li A, et al. Mutations in KCNJ13 cause autosomal-dominant snowflake vitreoretinal degeneration. Am J Hum Genet. 2008;82:174–180.
5 Gheiler M, Pollack A, Uchenik D, et al. Hereditary snowflake vitreoretinal degeneration. Birth Defects Orig Artic Ser. 1982;18:577–580.
6 Pollack A, Uchenik D, Chemke J, et al. Prophylactic laser photocoagulation in hereditary snowflake vitreoretinal degeneration. A family report. Arch Ophthalmol. 1983;101:1536–1539.
7 Robertson DM, Link TP, Rostvold JA. Snowflake degeneration of the retina. Ophthalmology. 1982;89:1513–1517.
8 Chen CJ, Everett TK, Marascalco D. Snowflake degeneration: an independent entity or a variant of retinitis pigmentosa? South Med J. 1986;79:1216–1223.
9 Jiao X, Ritter R, 3rd., Hejtmancik JF, et al. Genetic linkage of snowflake vitreoretinal degeneration to chromosome 2q36. Invest Ophthalmol Vis Sci. 2004;45:4498–4503.
10 Hirose T, Wolf E, Schepens CL. Retinal functions in snowflake degeneration. Ann Ophthalmol. 1980;12:1135–1146.
11 Aylward B, daCruz L, Ezra E, et al. Stickler syndrome. Ophthalmology. 2008;115:1636–1637. author reply 7–8
12 Ang A, Ung T, Puvanachandra N, et al. Vitreous phenotype: a key diagnostic sign in Stickler syndrome types 1 and 2 complicated by double heterozygosity. Am J Med Genet A. 2007;143:604–607.
13 Richards AJ, McNinch A, Martin H, et al. Stickler syndrome and the vitreous phenotype: mutations in COL2A1 and COL11A1. Hum Mutat. 2010;31:E1461–E1471.
14 Richards AJ, Yates JR, Williams R, et al. A family with Stickler syndrome type 2 has a mutation in the COL11A1 gene resulting in the substitution of glycine 97 by valine in alpha 1 (XI) collagen. Hum Mol Genet. 1996;5:1339–1343.
15 Annunen S, Korkko J, Czarny M, et al. Splicing mutations of 54-bp exons in the COL11A1 gene cause Marshall syndrome, but other mutations cause overlapping Marshall/Stickler phenotypes. Am J Hum Genet. 1999;65:974–983.
16 Shanske AL, Bogdanow A, Shprintzen RJ, et al. The Marshall syndrome: report of a new family and review of the literature. Am J Med Genet. 1997;70:52–57.
17 Brubaker JW, Mohney BG, Pulido JS, et al. Vitreous veils and radial lattice in Marshall syndrome. Ophthalmic Genet. 2008;29:184–185.
18 Kloeckener-Gruissem B, Bartholdi D, Abdou MT, et al. Identification of the genetic defect in the original Wagner syndrome family. Mol Vis. 2006;12:350–355.
19 Mukhopadhyay A, Nikopoulos K, Maugeri A, et al. Erosive vitreoretinopathy and Wagner disease are caused by intronic mutations in CSPG2/Versican that result in an imbalance of splice variants. Invest Ophthalmol Vis Sci. 2006;47:3565–3572.
20 Meredith SP, Richards AJ, Flanagan DW, et al. Clinical characterisation and molecular analysis of Wagner syndrome. Br J Ophthalmol. 2007;91:655–659.
21 Batioglu F. Goldmann–Favre vitreoretinal degeneration. Eur J Ophthalmol. 2003;13:307–310.
22 Ang A, Poulson AV, Goodburn SF, et al. Retinal detachment and prophylaxis in type 1 Stickler syndrome. Ophthalmology. 2008;115:164–168.
23 Ronan SM, Tran-Viet KN, Burner EL, et al. Mutational hot spot potential of a novel base pair mutation of the CSPG2 gene in a family with Wagner syndrome. Arch Ophthalmol. 2009;127:1511–1519.
24 Carroll C, Papaioannou D, Rees A, et al. The clinical effectiveness and safety of prophylactic retinal interventions to reduce the risk of retinal detachment and subsequent vision loss in adults and children with Stickler syndrome: a systematic review. Health Technol Assess. 2011;15:iii–xiv. 1–62
25 Brown DM, Graemiger RA, Hergersberg M, et al. Genetic linkage of Wagner disease and erosive vitreoretinopathy to chromosome 5q13–14. Arch Ophthalmol. 1995;113:671–675.
26 Black GC, Perveen R, Wiszniewski W, et al. A novel hereditary developmental vitreoretinopathy with multiple ocular abnormalities localizing to a 5-cM region of chromosome 5q13-q14. Ophthalmology. 1999;106:2074–2081.
27 Kloeckener-Gruissem B, Amstutz C. VCAN-related vitreoretinopathy. In: Pagon RA, Bird TD, Dolan CR, et al. GeneReviews (internet). Seattle, WA: University of Washington, 2009.
28 Fryer AE, Upadhyaya M, Littler M, et al. Exclusion of COL2A1 as a candidate gene in a family with Wagner–Stickler syndrome. J Med Genet. 1990;27:91–93.
29 Snead MP, Yates JR. Clinical and Molecular genetics of Stickler syndrome. J Med Genet. 1999;36:353–359.
30 McLeod D, Black GC, Bishop PN. Vitreous phenotype: genotype correlation in Stickler syndrome. Graefes Arch Clin Exp Ophthalmol. 2002;240:63–65. author reply 6
31 Perveen R, Hart-Holden N, Dixon MJ, et al. Refined genetic and physical localization of the Wagner disease (WGN1) locus and the genes CRTL1 and CSPG2 to a 2- to 2.5-cM region of chromosome 5q14.3. Genomics. 1999;57:219–226.
32 Maumenee IH, Stoll HU, Mets MB. The Wagner syndrome versus hereditary arthroophthalmopathy. Trans Am Ophthalmol Soc. 1982;80:349–365.
33 Go SL, Maugeri A, Mulder JJ, et al. Autosomal dominant rhegmatogenous retinal detachment associated with an Arg453Ter mutation in the COL2A1 gene. Invest Ophthalmol Vis Sci. 2003;44:4035–4043.
34 Graemiger RA, Niemeyer G, Schneeberger SA, et al. Wagner vitreoretinal degeneration. Follow-up of the original pedigree. Ophthalmology. 1995;102:1830–1839.
35 Seery CM, Pruett RC, Liberfarb RM, et al. Distinctive cataract in the Stickler syndrome. Am J Ophthalmol. 1990;110:143–148.
36 Yardley J, Leroy BP, Hart-Holden N, et al. Mutations of VMD2 splicing regulators cause nanophthalmos and autosomal dominant vitreoretinochoroidopathy (ADVIRC). Invest Ophthalmol Vis Sci. 2004;45:3683–3689.
37 Audo I, Michaelides M, Robson AG, et al. Phenotypic variation in enhanced S-cone syndrome. Invest Ophthalmol Vis Sci. 2008;49:2082–2093.
38 Chavala SH, Sari A, Lewis H, et al. An Arg311Gln NR2E3 mutation in a family with classic Goldmann–Favre syndrome. Br J Ophthalmol. 2005;89:1065–1066.
39 Sharon D, Sandberg MA, Caruso RC, et al. Shared mutations in NR2E3 in enhanced S-cone syndrome, Goldmann–Favre syndrome, and many cases of clumped pigmentary retinal degeneration. Arch Ophthalmol. 2003;121:1316–1323.
40 Menzel O, Bekkeheien RC, Reymond A, et al. Knobloch syndrome: novel mutations in COL18A1, evidence for genetic heterogeneity, and a functionally impaired polymorphism in endostatin. Hum Mutat. 2004;23:77–84.
41 Cui YX, Xia XY, Bu Y, et al. Rapid molecular prenatal diagnosis of spondyloepiphyseal dysplasia congenita by PCR-SSP assay. Genet Test. 2008;12:533–536.
42 Wada R, Sawai H, Nishimura G, et al. Prenatal diagnosis of Kniest dysplasia with three-dimensional helical computed tomography. J Matern Fetal Neonatal Med. 2011;24:1181–1184.
43 Yazici Z, Kline-Fath BM, Laor T, et al. Fetal MR imaging of Kniest dysplasia. Pediatr Radiol. 2010;40:348–352.
44 Hoornaert KP, Vereecke I, Dewinter C, et al. Stickler syndrome caused by COL2A1 mutations: genotype–phenotype correlation in a series of 100 patients. Eur J Hum Genet. 2010;18:872–880.
45 Majava M, Hoornaert KP, Bartholdi D, et al. A report on 10 new patients with heterozygous mutations in the COL11A1 gene and a review of genotype-phenotype correlations in type XI collagenopathies. Am J Med Genet A. 2007;143:258–264.
46 Vu CD, Brown J, Jr., Korkko J, et al. Posterior chorioretinal atrophy and vitreous phenotype in a family with Stickler syndrome from a mutation in the COL2A1 gene. Ophthalmology. 2003;110:70–77.
47 Donoso LA, Edwards AO, Frost AT, et al. Identification of a stop codon mutation in exon 2 of the collagen 2A1 gene in a large stickler syndrome family. Am J Ophthalmol. 2002;134:720–727.
48 Donoso LA, Edwards AO, Frost AT, et al. Clinical variability of Stickler syndrome: role of exon 2 of the collagen COL2A1 gene. Surv Ophthalmol. 2003;48:191–203.
49 McAlinden A, Majava M, Bishop PN, et al. Missense and nonsense mutations in the alternatively-spliced exon 2 of COL2A1 cause the ocular variant of Stickler syndrome. Hum Mutat. 2008;29:83–90.
50 Baker S, Booth C, Fillman C, et al. A loss of function mutation in the COL9A2 gene causes autosomal recessive Stickler syndrome. Am J Med Genet A. 2011;155:1668–1672.
51 Nikopoulos K, Schrauwen I, Simon M, et al. Autosomal recessive Stickler syndrome in two families caused by mutations in the COL9A1 gene. Invest Ophthalmol Vis Sci. 2011;52:4774–4779.
52 Yaguchi H, Ikeda T, Osada H, et al. Identification of the COL2A1 mutation in patients with type I Stickler syndrome using RNA from freshly isolated peripheral white blood cells. Genet Test Mol Biomarkers. 2011;15:231–237.
53 Al Kaissi A, Ganger R, Klaushofer K, et al. Significant ophthalmoarthropathy associated with ectodermal dysplasia in a child with Marshall–Stickler overlap: a case report. Cases J. 2008;1:270.
54 Passos-Bueno MR, Suzuki OT, Armelin-Correa LM, et al. Mutations in collagen 18A1 and their relevance to the human phenotype. An Acad Bras Cienc. 2006;78:123–131.
55 Suzuki O, Kague E, Bagatini K, et al. Novel pathogenic mutations and skin biopsy analysis in Knobloch syndrome. Mol Vis. 2009;15:801–809.
56 Pihlajamaa T, Prockop DJ, Faber J, et al. Heterozygous glycine substitution in the COL11A2 gene in the original patient with the Weissenbacher–Zweymuller syndrome demonstrates its identity with heterozygous OSMED (nonocular Stickler syndrome). Am J Med Genet. 1998;80:115–120.
57 Pacella E, Malvasi A, Tinelli A, et al. Stickler syndrome in Pierre-Robin sequence prenatal ultrasonographic diagnosis and postnatal therapy: two cases report. Eur Rev Med Pharmacol Sci. 2010;14:1051–1054.
58 Couchouron T, Masson C. Early-onset progressive osteoarthritis with hereditary progressive ophthalmopathy or Stickler syndrome. Joint Bone Spine. 2011;78:45–49.
59 Spranger J, Winterpacht A, Zabel B. Kniest dysplasia: Dr. W. Kniest, his patient, the molecular defect. Am J Med Genet. 1997;69:79–84.
60 Galil A, Carmi R, Goldstein E, et al. Weissenbacher–Zweymuller syndrome: long-term follow-up of growth and psychomotor development. Dev Med Child Neurol. 1991;33:1104–1109.
61 Watanabe H, Kohzaki K, Kubo H, et al. [Stickler syndrome with rhegmatogenous retinal detachment.]. Nippon Ganka Gakkai Zasshi. 2010;114:454–458.
62 Rabinowitz R, Gradstein L, Galil A, et al. The ocular manifestations of Weissenbacher–Zweymuller syndrome. Eye (Lond). 2004;18:1258–1263.
63 Parma ES, Korkko J, Hagler WS, et al. Radial perivascular retinal degeneration: a key to the clinical diagnosis of an ocular variant of Stickler syndrome with minimal or no systemic manifestations. Am J Ophthalmol. 2002;134:728–734.
64 Miloro M. Mandibular distraction osteogenesis for pediatric airway management. J Oral Maxillofac Surg. 2010;68:1512–1523.
65 Ihanamäki T, Metsäranta M, Rintala M, et al. Ocular abnormalities in transgenic mice harboring mutations in the type II collagen gene. Eur J Ophthalmol. 1996;6:427–435.
66 Haas J. Ueber das Zusammenvorkommen von Veranderungen der Retina und Choroidea. Arch Augenheilkd. 1898;37:343–348.
67 Pagenstecher H. Uebereine unter dem Bildeder Natzhauterblosung verlaufende, erbiche Erkankung der Retina. Graefes Arch Clin Exp Ophthalmol. 1913;86:457–462.
68 Jager G. A hereditary retinal disease. Trans Ophthalmol Soc UK. 1953;73:617–619.
69 Kim SY, Ko HS, Yu YS, et al. Molecular genetic characteristics of X-linked retinoschisis in Koreans. Mol Vis. 2009;15:833–843.
70 Lamey T, Laurin S, Chelva E, et al. Genotypic analysis of X-linked retinoschisis in Western Australia. Adv Exp Med Biol. 2010;664:283–291.
71 Teixeira C, Rocha-Sousa A, Trump D, et al. Identification of XLRS1 gene mutation (608C > T) in a Portuguese family with juvenile retinoschisis. Eur J Ophthalmol. 2005;15:638–640.
72 Biswas S, Funnell CL, Gray J, et al. Nidek MP-1 microperimetry and Fourier domain optical coherence tomography (FD-OCT) in X linked retinoschisis. Br J Ophthalmol. 2010;94:949–950.
73 Sikkink SK, Biswas S, Parry NR, et al. X-linked retinoschisis: an update. J Med Genet. 2007;44:225–232.
74 Garcia-Arumi J, Corcostegui IA, Navarro R, et al. Vitreoretinal surgery without schisis cavity excision for the management of juvenile X linked retinoschisis. Br J Ophthalmol. 2008;92:1558–1560.
75 Lesch B, Szabo V, Kanya M, et al. Clinical and genetic findings in Hungarian patients with X-linked juvenile retinoschisis. Mol Vis. 2008;14:2321–2332.
76 Vainio-Mattila B, Eriksson AW, Forsius H. X-chromosomal recessive retinoschisis in the region of Pori. An ophthalmo-genetical analysis of 103 cases. Acta Ophthalmol (Copenh). 1969;47:1135–1148.
77 Riveiro-Alvarez R, Trujillo-Tiebas MJ, Gimenez-Pardo A, et al. Correlation of genetic and clinical findings in Spanish patients with X-linked juvenile retinoschisis. Invest Ophthalmol Vis Sci. 2009;50:4342–4350.
78 Sauer CG, Gehrig A, Warneke-Wittstock R, et al. Positional cloning of the gene associated with X-linked juvenile retinoschisis. Nat Genet. 1997;17:164–170.
79 Grayson C, Reid SN, Ellis JA, et al. Retinoschisin, the X-linked retinoschisis protein, is a secreted photoreceptor protein, and is expressed and released by Weri-Rb1 cells. Hum Mol Genet. 2000;9:1873–1879.
80 Molday LL, Hicks D, Sauer CG, et al. Expression of X-linked retinoschisis protein RS1 in photoreceptor and bipolar cells. Invest Ophthalmol Vis Sci. 2001;42:816–825.
81 Reid SN, Yamashita C, Farber DB. Retinoschisin, a photoreceptor-secreted protein, and its interaction with bipolar and Müller cells. J Neurosci. 2003;23:6030–6040.
82 Wu WW, Wong JP, Kast J, et al. RS1, a discoidin domain-containing retinal cell adhesion protein associated with X-linked retinoschisis, exists as a novel disulfide-linked octamer. J Biol Chem. 2005;280:10721–10730.
83 Wang T, Zhou A, Waters CT, et al. Molecular pathology of X linked retinoschisis: mutations interfere with retinoschisin secretion and oligomerisation. Br J Ophthalmol. 2006;90:81–86.
84 Fraternali F, Cavallo L, Musco G. Effects of pathological mutations on the stability of a conserved amino acid triad in retinoschisin. FEBS Lett. 2003;544:21–26.
85 Steiner-Champliaud MF, Sahel J, Hicks D. Retinoschisin forms a multi-molecular complex with extracellular matrix and cytoplasmic proteins: interactions with beta2 laminin and alphaB-crystallin. Mol Vis. 2006;12:892–901.
86 Horwitz J. Alpha-crystallin can function as a molecular chaperone. Proc Natl Acad Sci U S A. 1992;89:10449–10453.
87 Molday LL, Wu WW, Molday RS. Retinoschisin (RS1), the protein encoded by the X-linked retinoschisis gene, is anchored to the surface of retinal photoreceptor and bipolar cells through its interactions with a Na/K ATPase-SARM1 complex. J Biol Chem. 2007;282:32792–32801.
88 Gehrig A, Langmann T, Horling F, et al. Genome-wide expression profiling of the retinoschisin-deficient retina in early postnatal mouse development. Invest Ophthalmol Vis Sci. 2007;48:891–900.
89 Dyka FM, Wu WW, Pfeifer TA, et al. Characterization and purification of the discoidin domain-containing protein retinoschisin and its interaction with galactose. Biochemistry. 2008;47:9098–9106.
90 Pimenides D, George ND, Yates JR, et al. X-linked retinoschisis: clinical phenotype and RS1 genotype in 86 UK patients. J Med Genet. 2005;42:e35.
91 Iannaccone A, Mura M, Dyka FM, et al. An unusual X-linked retinoschisis phenotype and biochemical characterization of the W112C RS1 mutation. Vision Res. 2006;46:3845–3852.
92 Kellner U, Brummer S, Foerster MH, et al. X-linked congenital retinoschisis. Graefes Arch Clin Exp Ophthalmol. 1990;228:432–437.
93 Mitamura Y, Miyanishi K, Shizukawa N, et al. A case of X-linked retinoschisis diagnosed in an infant. Retina. 2003;23:731–732.
94 Peachey NS, Fishman GA, Derlacki DJ, et al. Psychophysical and electroretinographic findings in X-linked juvenile retinoschisis. Arch Ophthalmol. 1987;105:513–516.
95 Ando A, Takahashi K, Sho K, Matsushima M, et al. Histopathological findings of X-linked retinoschisis with neovascular glaucoma. Graefes Arch Clin Exp Ophthalmol. 2000;238:1–7.
96 Greven CM, Moreno RJ, Tasman W. Unusual manifestations of X-linked retinoschisis. Trans Am Ophthalmol Soc. 1990;88:211–225. discussion 26–8
97 Tasman W, Greven C, Moreno R. Nasal retinal dragging in X-linked retinoschisis. Graefes Arch Clin Exp Ophthalmol. 1991;229:319–322.
98 Sorsby A, Klein M, Gann JH, et al. Unusual retinal detachment possibly sex-linked. Br J Ophthalmol. 1951;35:1–10.
99 George ND, Yates JR, Moore AT. Clinical features in affected males with X-linked retinoschisis. Arch Ophthalmol. 1996;114:274–280.
100 George ND, Yates JR, Bradshaw K, et al. Infantile presentation of X linked retinoschisis. Br J Ophthalmol. 1995;79:653–657.
101 Garg SJ, Lee HC, Grand MG. Bilateral macular detachments in X-linked retinoschisis. Arch Ophthalmol. 2006;124:1053–1055.
102 Apushkin MA, Fishman GA, Rajagopalan AS. Fundus findings and longitudinal study of visual acuity loss in patients with X-linked retinoschisis. Retina. 2005;25:612–618.
103 McKibbin M, Booth AP, George ND. Foveal ectopia in X-linked retinoschisis. Retina. 2001;21:361–366.
104 Muscat S, Fahad B, Parks S, et al. Optical coherence tomography and multifocal electroretinography of X-linked juvenile retinoschisis. Eye (Lond). 2001;15:796–799.
105 Yu J, Ni Y, Keane PA, et al. Foveomacular schisis in juvenile X-linked retinoschisis: an optical coherence tomography study. Am J Ophthalmol. 2010;149:973–978. e2
106 Tanimoto N, Usui T, Takagi M, et al. Electroretinographic findings in three family members with X-linked juvenile retinoschisis associated with a novel Pro192Thr mutation of the XLRS1 gene. Jpn J Ophthalmol. 2002;46:568–576.
107 Miyake Y, Shiroyama N, Ota I, et al. Focal macular electroretinogram in X-linked congenital retinoschisis. Invest Ophthalmol Vis Sci. 1993;34:512–515.
108 Bradshaw K, George N, Moore A, et al. Mutations of the XLRS1 gene cause abnormalities of photoreceptor as well as inner retinal responses of the ERG. Doc Ophthalmol. 1999;98:153–173.
109 Deutman AF, Pinckers AJ, Aan de Kerk AL. Dominantly inherited cystoid macular edema. Am J Ophthalmol. 1976;82:540–548.
110 Gehrig A, White K, Lorenz B, et al. Assessment of RS1 in X-linked juvenile retinoschisis and sporadic senile retinoschisis. Clin Genet. 1999;55:461–465.
111 Iannaccone A, Fung KH, Eyestone ME, et al. Treatment of adult-onset acute macular retinoschisis in enhanced s-cone syndrome with oral acetazolamide. Am J Ophthalmol. 2009;147:307–312. e2
112 Genead MA, Fishman GA, Walia S. Efficacy of sustained topical dorzolamide therapy for cystic macular lesions in patients with X-linked retinoschisis. Arch Ophthalmol. 2010;128:190–197.
113 Bastos AL, Freitas Bde P, Villas Boas O, et al. Use of topical dorzolamide for patients with X-linked juvenile retinoschisis: case report. Arq Bras Oftalmol. 2008;71:286–290.
114 Gopal L, Shanmugam MP, Battu RR, et al. Congenital retinoschisis: successful collapse with photocoagulation. Ind J Ophthalmol. 2001;49:265–266.
115 Avitabile T, Ortisi E, Scott IU, et al. Scleral buckle for progressive symptomatic retinal detachment complicating retinoschisis versus primary rhegmatogenous retinal detachment. Can J Ophthalmol. 2010;45:161–165.
116 Tantri A, Vrabec TR, Cu-Unjieng A, et al. X-linked retinoschisis: a clinical and molecular genetic review. Surv Ophthalmol. 2004;49:214–230.
117 Ikeda F, Iida T, Kishi S. Resolution of retinoschisis after vitreous surgery in X-linked retinoschisis. Ophthalmology. 2008;115:718–722. e1
118 Wu WC, Drenser KA, Capone A, et al. Plasmin enzyme-assisted vitreoretinal surgery in congenital X-linked retinoschisis: surgical techniques based on a new classification system. Retina. 2007;27:1079–1085.
119 Simonelli F, Maguire AM, Testa F, et al. Gene therapy for Leber’s congenital amaurosis is safe and effective through 1.5 years after vector administration. Mol Ther. 2010 Ma;18(3):643–650.
120 Dyka FM, Molday RS. Coexpression and interaction of wild-type and missense RS1 mutants associated with X-linked retinoschisis: its relevance to gene therapy. Invest Ophthalmol Vis Sci. 2007;48:2491–2497.
121 Zeng Y, Takada Y, Kjellstrom S, et al. RS-1 gene delivery to an adult Rs1h knockout mouse model restores ERG b-wave with reversal of the electronegative waveform of X-linked retinoschisis. Invest Ophthalmol Vis Sci. 2004;45:3279–3285.
122 Min SH, Molday LL, Seeliger MW, et al. Prolonged recovery of retinal structure/function after gene therapy in an Rs1h-deficient mouse model of x-linked juvenile retinoschisis. Mol Ther. 2005;12:644–651.
123 Janssen A, Min SH, Molday LL, et al. Effect of late-stage therapy on disease progression in AAV-mediated rescue of photoreceptor cells in the retinoschisin-deficient mouse. Mol Ther. 2008;16:1010–1017.
124 Ballios BG, Cooke MJ, van der Kooy D, et al. A hydrogel-based stem cell delivery system to treat retinal degenerative diseases. Biomaterials. 2010;31:2555–2564.
125 Yanagi Y, Takezawa S, Kato S. Distinct functions of photoreceptor cell-specific nuclear receptor, thyroid hormone receptor beta2 and CRX in one photoreceptor development. Invest Ophthalmol Vis Sci. 2002;43:3489–3494.
126 Schorderet DF, Escher P. NR2E3 mutations in enhanced S-cone sensitivity syndrome (ESCS), Goldmann–Favre syndrome (GFS), clumped pigmentary retinal degeneration (CPRD), and retinitis pigmentosa (RP). Hum Mutat. 2009;30:1475–1485.
127 Brydak-Godowska J, Makowiec-Tabernacka M. [Goldmann–Favre syndrome – case report.]. Klin Oczna. 2009;111:346–347.
128 Marmor MF, Jacobson SG, Foerster MH, et al. Diagnostic clinical findings of a new syndrome with night blindness, maculopathy, and enhanced S cone sensitivity. Am J Ophthalmol. 1990;110:124–134.
129 To KW, Adamian M, Jakobiec FA, et al. Clinical and histopathologic findings in clumped pigmentary retinal degeneration. Arch Ophthalmol. 1996;114:950–955.
130 Favre M. [Two cases of hyaloid-retinal degeneration.]. Ophthalmologica. 1958;135:604–609.
131 Francois J, de Rouck A, Cambie E. [Goldmann–Favre vitreo-tapeto-retinal degeneration.]. Ophthalmologica. 1974;168:81–96.
132 Fishman GA, Jampol LM, Goldberg MF. Diagnostic features of the Favre–Goldmann syndrome. Br J Ophthalmol. 1976;60:345–353.
133 Marmor MF. A teenager with nightblindness and cystic maculopathy: enhanced S cone syndrome (Goldmann–Favre syndrome). Doc Ophthalmol. 2006;113:213–215.
134 Hood DC, Cideciyan AV, Roman AJ, et al. Enhanced S cone syndrome: evidence for an abnormally large number of S cones. Vision Res. 1995;35:1473–1481.
135 Milam AH, Rose L, Cideciyan AV, et al. The nuclear receptor NR2E3 plays a role in human retinal photoreceptor differentiation and degeneration. Proc Natl Acad Sci U S A. 2002;99:473–478.
136 Peyman GA, Fishman GA, Sanders DR, et al. Histopathology of Goldmann–Favre syndrome obtained by full-thickness eye-wall biopsy. Ann Ophthalmol. 1977;9:479–484.
137 Haider NB, Naggert JK, Nishina PM. Excess cone cell proliferation due to lack of a functional NR2E3 causes retinal dysplasia and degeneration in rd7/rd7 mice. Hum Mol Genet. 2001;10:1619–1626.
138 Mitton KP, Swain PK, Khanna H, et al. Interaction of retinal bZIP transcription factor NRL with Flt3-interacting zinc-finger protein Fiz1: possible role of Fiz1 as a transcriptional repressor. Hum Mol Genet. 2003;12:365–373.
139 Feiler-Ofry V, Adam A, Regenbogen L, et al. Hereditary vitreoretinal degeneration and night blindness. Am J Ophthalmol. 1969;67:553–558.
140 Jacobson SG, Marmor MF, Kemp CM, et al. SWS (blue) cone hypersensitivity in a newly identified retinal degeneration. Invest Ophthalmol Vis Sci. 1990;31:827–838.
141 Jacobson SG, Roman AJ, Roman MI, et al. Relatively enhanced S cone function in the Goldmann–Favre syndrome. Am J Ophthalmol. 1991;111:446–453.
142 Fishman GA, Peachey NS. Rod–cone dystrophy associated with a rod system electroretinogram obtained under photopic conditions. Ophthalmology. 1989;96:913–918.
143 Greenstein VC, Zaidi Q, Hood DC, et al. The enhanced S cone syndrome: an analysis of receptoral and post-receptoral changes. Vision Res. 1996;36:3711–3722.
144 Khairallah M, Ladjimi A, Ben Yahia S, et al. Elevated macular retinoschisis associated with Goldmann–Favre syndrome successfully treated with grid laser photocoagulation. Retina. 2002;22:234–237.
145 Kaufman SJ, Goldberg MF, Orth DH, et al. Autosomal dominant vitreoretinochoroidopathy. Arch Ophthalmol. 1982;100:272–278.
146 Lafaut BA, Loeys B, Leroy BP, et al. Clinical and electrophysiological findings in autosomal dominant vitreoretinochoroidopathy: report of a new pedigree. Graefes Arch Clin Exp Ophthalmol. 2001;239:575–582.
147 Roider J, Fritsch E, Hoerauf H, et al. Autosomal dominant vitreoretinochoroidopathy. Retina. 1997;17:294–299.
148 Blair NP, Goldberg MF, Fishman GA, et al. Autosomal dominant vitreoretinochoroidopathy (ADVIRC). Br J Ophthalmol. 1984;68:2–9.
149 Han DP, Lewandowski MF. Electro-oculography in autosomal dominant vitreoretinochoroidopathy. Arch Ophthalmol. 1992;110:1563–1567.
150 Sarra GM, Weigell-Weber M, Kotzot D, et al. Clinical description and exclusion of candidate genes in a novel autosomal recessively inherited vitreoretinal dystrophy. Arch Ophthalmol. 2003;121:1109–1116.
151 Jampol LM, Ebroon DA, Goldbaum MH. Peripheral proliferative retinopathies: an update on angiogenesis, etiologies and management. Surv Ophthalmol. 1994;38:519–540.
152 Bennett SR, Folk JC, Kimura AE, et al. Autosomal dominant neovascular inflammatory vitreoretinopathy. Ophthalmology. 1990;97:1125–1135. discussion 35–6
153 Stone EM, Kimura AE, Folk JC, et al. Genetic linkage of autosomal dominant neovascular inflammatory vitreoretinopathy to chromosome 11q13. Hum Mol Genet. 1992;1:685–689.
154 Gitter KA, Rothschild H, Waltman DD, et al. Dominantly inherited peripheral retinal neovascularization. Arch Ophthalmol. 1978;96:1601–1605.