Chapter 28 Mechanisms of Macular Edema and Therapeutic Approaches
 For additional online content visit http://www.expertconsult.com
For additional online content visit http://www.expertconsult.com
Introduction
The breakdown of the blood–retinal barrier (BRB), modulated via different growth factors, results from a disturbance of the integrity of the tight junctions.1 Starling’s law predicts that macular edema will develop if the hydrostatic pressure gradient between capillary and retinal tissue is increased. That can occur, for example, in the presence of elevated blood pressure, or if the osmotic pressure gradient is decreased by excessive protein accumulation in the extracellular space within the retina.2
Macular edema as A result of various disease mechanisms
Causes of macular edema
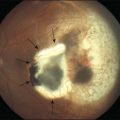
The classic pattern of cystoid macular edema (CME) with the petaloid appearance originating from the fluorescein leakage from perifoveal capillaries may be seen in cases of advanced edema of various origins (Fig. 28.1, panel B online). This includes postsurgical CME as well as CME associated with one of the following conditions: diabetes, vascular occlusion, hypertensive retinopathy, epiretinal membranes, intraocular tumors (e.g., melanoma, choroidal hemangioma), intraocular inflammation (e.g., pars planitis), macroaneurysm, retinitis pigmentosa, choroidal neovascularization, and radiation retinopathy.
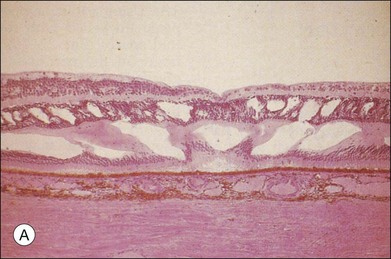
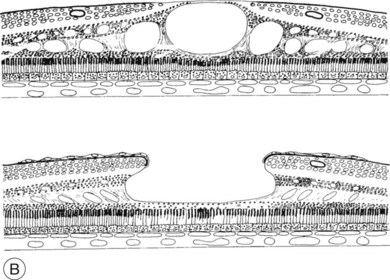
Fig. 28.1, online (B) Schematic drawing of the central retina with large cysts (top), finally resulting in a pseudohole formation (bottom).
Given the heterogeneous etiology of macular edema, its effective treatment depends upon a better understanding of its pathogenesis. In general, formation of macular edema is related to metabolic changes, ischemia, hydrostatic forces, inflammatory and toxic mechanisms, or mechanical forces that occur to various degrees in different conditions (Table 28.1).
Table 28.1 Causes of macular edema in relation to underlying disorders
| Disease group | Disorder | Pathogenesis |
|---|---|---|
| Metabolic alterations | Diabetes | Abnormal glucose metabolism Aldose reductase |
| Retinitis pigmentosa | CME: leakage at the level of RPE | |
| Inherited CME (autosomal-dominant) | Müller cell disease: leakage from perifoveolar capillaries | |
| Ischemia | Vein occlusion Diabetic retinopathy |
Inner BRB (retinal capillary hypoperfusion) |
| Severe hypertensive retinopathy HELLP syndrome Vasculitis, collagenosis |
Outer BRB (ischemic hypoperfusion of the choroid: serous detachment) | |
| Hydrostatic forces | Retinal vascular occlusions Venous occlusion Arterial hypertension Low IOP |
Increased intravascular pressure Failure of the BRB |
| Mechanical forces | Vitreous traction on the macula | Epiretinal membranes with tangential traction Vitreomacular traction syndrome |
| Inflammation | Intermediate uveitis | Mediated by prostaglandins CME is considered an indication for treatment |
| Postoperative CME | Perivascular leukocytic infiltrates | |
| DME | Diabetic leukostasis mediates vascular leakage by endothelial cell apoptosis | |
| Choroidal inflammatory diseases | Vogt–Koyanagi–Harada syndrome Birdshot retinochoroidopathy |
|
| Pharmacotoxic effects | e.g., Epinephrine (in aphakia) Betaxolol Latanoprost |
Mostly via prostaglandins |
CME, cystoid macular edema; RPE, retinal pigment epithelium; BRB, blood–retinal barrier; HELLP syndrome, hemolytic anemia, elevated liver enzymes, and low platelet count; IOP, intraocular pressure; DME, diabetic macular edema.
Molecular and cellular alterations leading to macular edema
Much of the knowledge on the pathophysiology of macular edema has been determined from extensive experimental studies on diabetic retinopathy and diabetic vascular leakage. A variety of techniques measuring accumulation of material from plasma in the neural retina have been investigated to assess permeability. Such accumulation seems diffuse in nature and focal defects have not been reproducibly described in diabetic mice; as well, interpretations of techniques involving tracer accumulation have not been validated in terms of “gold standard” permeability surface area product.3 Interestingly, edema has not been demonstrated in the retina of diabetic mice based on retinal thickness measurements despite the indication of increased permeability.
The BRB consists of the retinal pigment epithelium (RPE) layer (outer BRB), and the vascular endothelium (inner BRB), that prohibit the passage of macromolecules and circulating cells from the vascular compartment to the extracellular compartment and therefore intraretinal space.4 Intracellular edema (or cytotoxic edema) is defined as cellular swelling that occurs without opening of the BRB. Extracellular (or vasogenic) edema is characterized by retinal thickening in association with loss of BRB integrity (Fig. 28.2). While for diabetes and ischemic retinopathies the inner BRB was found to play a dominant role in vascular leakage, the importance of the outer BRB has recently been supported.5 The outer BRB separates the neural retina from the choroidal vasculature, which is responsible for approximately 80% of the blood supply in the eye. The outer BRB-specific leakage of fluorescent macromolecules can be visualized in diabetic and ischemic rodents and substantial leakage of macromolecules through the outer BRB can be detected.
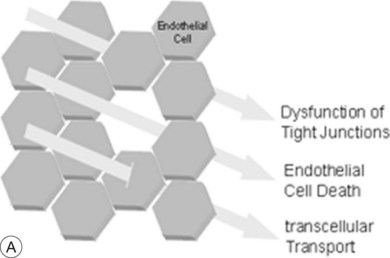
The breakdown of the inner BRB may occur to a variable extent via dysfunction of intercellular junctions, increased transcellular transport, or increased endothelial cell destruction, and result in an increase in vascular permeability (Fig. 28.3B, online).
Cell-to-cell junctions and vascular permeability
Fluid homeostasis and endothelial permeability are mostly regulated by intercellular junctions in the nondiseased retina. Intercellular junctions are complex structures formed by the assembly of a transmembrane and cytoplasmic/cytoskeletal protein components. At least four different types of endothelial junctions have been described: tight junctions, gap junctions, adherence junctions, and syndesmos. Tight junctions are the most apical component of the intercellular cleft (Fig. 28.4, online).
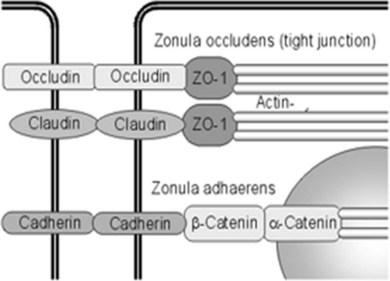
Although the molecular structure of tight junctions generally appears to be similar in all barrier systems, there are some differences between epithelial and endothelial tight junctions, and between tight junctions of peripheral and retinal endothelial cells.6 Expression of selected endothelial cell tight junction genes and particularly that of occludin and claudin-5 is reduced in the diabetic retina.7 In contrast to tight junctions in epithelial systems, structural and functional characteristics of tight junctions in endothelial cells respond promptly to ambient factors. It is likely that inflammatory agents increase permeability by binding to specific receptors that transduce intercellular signals, which in turn cause cytoskeletal reorganization and widening of the interendothelial clefts. For example, tumor necrosis factor-alpha (TNF-α) signals through protein kinase C (PKC)ζ/nuclear factor-kappa B (NF-κB) to alter the tight junction complex and increase retinal endothelial cell permeability.8 Endothelial junctions also regulate leukocyte extravasation. Once leukocytes have adhered to the endothelium, a coordinated opening of interendothelial cell junctions occurs.
Inflammation and vascular permeability
In diabetes, activated leukocytes adhere to the retinal vascular endothelium.9,10 Increased leukostasis is one of the first histologic changes in diabetic retinopathy and occurs prior to any apparent clinical pathology.
Adherent leukocytes play a crucial role in diabetic retinopathy by directly inducing endothelial cell death in capillaries,11 causing vascular obstruction and vascular leakage. Endothelial cell death precedes the formation of acellular capillaries.10 With time, however, acellular capillaries prevail and become widespread. Although the mechanism of this destructive process remains elusive, it is clear that the interaction between the altered leukocytes and the endothelial cells and the subsequent endothelial damage represents a crucial pathogenic step9,11,12 (Fig. 28.5).
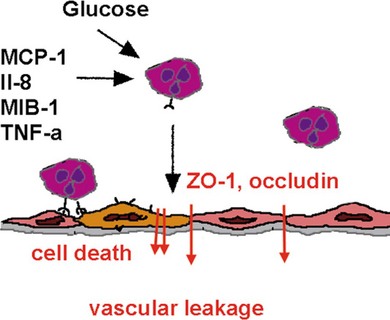
Inflammatory cytokines such as TNF-α decrease the protein and mRNA content of the tight junction proteins zonula occludens (ZO)-1 and claudin-5.8 TNF-α and interleukin-1 beta (IL-1β) are elevated in the vitreous of diabetic patients and in the retina of diabetic rats associated with increased retinal vascular permeability and leukostasis13,14 (Fig. 28.6, online). Furthermore, TNF-α is involved in ischemic vascular changes.15
Growth factors, vasoactive factors, and vascular permeability
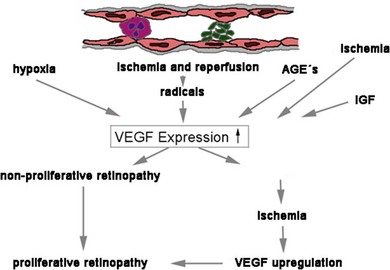
The disruption of endothelial integrity leads to retinal ischemia and vascular endothelial growth factor (VEGF)-mediated iris and retinal neovascularization.9,16,17 VEGF is 50 000 times more potent than histamine in causing vascular permeability.18–20 Previous work has shown that retinal VEGF levels correlate with diabetic BRB breakdown in rodents21 and humans.22 Flt-1(1–3 Ig)Fc, a soluble VEGF receptor, reverses early diabetic BRB breakdown and diabetic leukostasis in a dose-dependent manner.17 Early BRB breakdown localizes, in part, to retinal venules and capillaries of the superficial inner retinal circulation23 and can be sufficiently reduced by VEGF inhibition (Fig. 28.7, online). Although VEGF is only one of the cytokines involved in the pathogenesis of the vascular leakage, it is likely to be one of the most effective therapeutic targets.
On a cellular level, VEGF has been implicated in many different mechanisms, which lead to macular edema. VEGF has, for example, been shown to decrease the proteins responsible for the tightness of the intercellular junctions and induces rapid phosphorylation of the tight junction proteins occludin and ZO-1, resulting in breakdown of the BRB.24 VEGF-induced BRB breakdown appears to be effected via nitric oxide.17 VEGF also increases paracellular transport without altering the solvent drag reflection coefficient.25 Furthermore, VEGF activation of PKC stimulates occludin phosphorylation and contributes to endothelial permeability.26
There are tight connections between inflammation and VEGF expression.17 Recently, Müller-cell-derived VEGF was shown to be essential for diabetes-induced retinal inflammation and vascular leakage.27
Investigations on cell–cell interactions by D’Amore and coworkers demonstrated an inhibitory effect of TGF-β secreted by pericytes on endothelial cell growth. In diabetic retinopathy formation of sorbitol via aldose reductase leads to PKC activation, resulting in a loss of the inhibitory balance28 (Fig. 28.8).
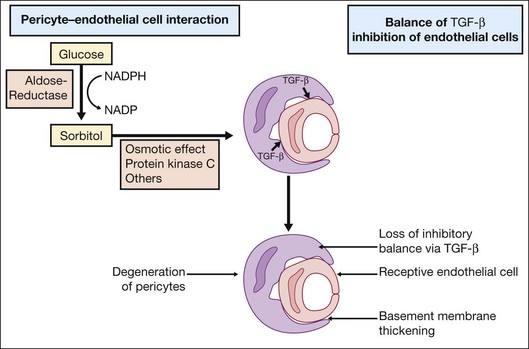
High glucose concentration leads to increased diacylglycerol (DAG) by two pathways: de novo synthesis and through dehydrogenation of phosphatidylcholine. Increased levels of DAG mediate PKC activation. Several studies have shown that a decrease in retinal blood flow occurs with PKC activation. Conversely, inhibition of PKC with LY333531 (Eli Lilly, Indianapolis, IN) normalized decreased retinal blood flow in diabetic rats.29,30
Furthermore, retinal vascular endothelial cells are very sensitive to histamine. Several studies have documented increased vascular histamine synthesis in diabetic rats and humans.31–33 The administration of histamine reduces ZO-1 protein expression and thus correlates with vascular permeability. The H1 receptor stimulates PKC that has been implicated in increased retinal vascular permeability.34 Interestingly, Aiello and coworkers showed that administration of LY333531, a PKC-β isoform-selective inhibitor, does not significantly decrease histamine-induced permeability but instead VEGF-induced permeability. In contrast, administration of nonisoform-selective PKC inhibitors did significantly suppress histamine-induced permeability.35
Furthermore, in vascular endothelial cells, advanced glycation end-products (AGE) may affect the gene expression of ET-1 and modify VEGF expression. The AGE-stimulated increased VEGF expression is dose- and time-dependent and additive to hypoxia.36,37
Endothelial cell death and vascular permeability
Where intraluminal pressure falls below a critical closing pressure the tone of the arteriolar wall cannot be maintained and the downstream capillary bed collapses and endothelial cells become “fibrin-locked.” Endothelial cells deprived of their circulation and nutrition die and only acellular basement membranes persists. A reduction in intraocular pressure may cause macular edema with cystoid degenerative changes and secondary atrophic alterations at the outer retina. Similarly, a reduction in retinal perfusion pressure, often linked to carotid/ophthalmic artery insufficiency, can have similar retinal manifestations and in extreme circumstances there may be retrograde filling of arteries from fellow veins. Stasis of the blood flow in capillaries after venous or arterial occlusions results in rapid apoptosis of endothelial cells.38
Similarly, in diabetes, retinal barrier breakdown is at least in part due to endothelial cell damage and apoptosis. The proapoptotic molecule Fas-ligand (FasL) induces apoptosis in cells that carry its receptor Fas (CD 95).39 There is evidence that FasL is expressed on vascular endothelium where it functions to inhibit leukocyte extravasation. The expression of FasL on vascular endothelial cells might thus prevent detrimental inflammation by inducing apoptosis in leukocytes as they attempt to enter the vessel. In fact, during inflammation and ensuing TNF-α release, the retinal endothelium upregulates several adhesion molecules40 that mediate the adherence of the leukocytes, but also downregulates FasL thus allowing leukocyte survival and migration to active sites of inflammation. In experimental diabetic retinopathy, inhibition of Fas-mediated apoptotic cell death reduces vascular leakage.41 The cumulative endothelial cell death during the course of diabetes plays a causal role in the pathogenesis of the diabetic vascular leakage and maculopathy.
Extracellular matrix alterations and vascular permeability
Degradation of the extracellular matrix affects endothelial cell function at many levels causing endothelial cell lability which is required for cellular invasion and proliferation, or influencing the cellular resistance and therefore the vascular permeability. The degradation and modulation of the extracellular matrix are exerted by matrix metalloproteinases (MMPs), a family of zinc-binding, calcium-dependent enzymes.42 Elevation of MMP-9 and MMP-2 expression has been shown in diabetic neovascular membranes,43,44 although a direct effect of glucose on MMP-9 expression in vascular endothelial cells could not be shown.45 It is probable that MMPs participate at various stages during the course of the BRB dysfunction and breakdown. Their actions include early changes of the endothelial cell resistance with influence on intercellular junction formation and function46 to active participation in endothelial and pericyte cell death47 that occurs late in the course of the disease.
Transcellular transport and vascular permeability
Disruption of the BRB is an early phenomenon in preclinical diabetic retinopathy. Two vascular permeability pathways may be affected, the paracellular pathway involving endothelial cell tight junctions, and the endothelial transcellular pathway mediated by endocytotic vesicles (caveolae). Despite the fact that pinocytic transport is critically involved in the transepithelial fluid exchange, its role in the pathogenesis of increased vascular leakage in diabetes is just emerging.48,49 The importance of the regulation of fluid homeostasis by active cellular transport of nutrients and fluid via pinocytosis is underlined by recent data suggesting a transient induction of the paracellular pathway and prolonged involvement of transcellular endothelial transport mechanisms in the increased permeability of retinal capillaries in diabetes.7
It is currently known that one of the factors involved in the regulation of pinocytic transport is VEGF. VEGF increases vascular permeability not only by disrupting the intercellular tight junctions between the retinal endothelial cells but also by inducing the formation of fenestrations and vesiculovacuolar organelles. The role of VEGF in the disruption of the pinocytic transport that is translated into increased vascular permeability in disease states is still controversial.50 Whereas, in higly permeable blood vessels the number of pinocytotic vesicles at the endothelial luminal membrane transporting plasma immunoglobulin G is significantly increased, no fenestrations or vesicles were found in the endothelial cells of the VEGF-affected eyes when examined by electron microscopy.
Neuronal involvement in the formation of macular edema
Recent research on DME emphasizes the role of neuronal cells in the diabetic retinal damage. The retina consists of a network of neurons and glia (astrocytes, Müller cells, and microglial cells) that comprise approximately 95% of the tissue, with blood vessels representing less than 5% of the retinal mass.51 As the network of retinal neurons and glia is intimately linked, there is no doubt that the neural (photoreceptors, bipolar cells, horizontal cells, amacrine cells, and ganglion cells) and vascular components of the retina are closely associated by metabolic synergy and paracrine communication.52,53 Neuroglial cells are involved in vision, and blood vessels provide nutrients to facilitate the process.54
In the inner retina, metabolic substrates, such as glucose, flow from vascular endothelium to astrocytes to neurons. In the outer retina, substrates reach Müller cells and photoreceptors from the choroid via the RPE.52 Microglia associate intimately with neurons that express molecules, such as CX3CL1 (fractalkine) and CD20, that negatively regulate microglial activation through their respective receptors. As such, perturbation of expression of ligand or receptor during stress would activate microglia to produce proinflammatory cytokines and acquire an activated morphology. Activated microglia produce chemokines such as monocyte chemoattractant protein-1, inducing expression of adhesion molecules, which can promote the leukostasis of neutrophils on endothelium, and potentially inducing the extravasation of inflammatory macrophages.52,54 Induction of glial fibrillary acidic protein (GFAP) is a marker of glial activation and increased expression of this protein occurs in Müller cells from the retinas of diabetic patients, but also after ischemic injury.
Mechanical factors involved in the formation of macular edema
Clinical and anatomic evidence indicates that abnormalities in the structure of the vitreoretinal interface may play an important role in the pathogenesis of DME.55–57 It was suggested that vitreoretinal adhesions in diabetic eyes are stronger than the shear forces of traction from vitreous shrinkage and this in turn may lead to the development of vitreomacular traction and subsequently to macular edema.58 Nevertheless, the risk of developing diffuse macular edema was 3.4-fold lower in the group of eyes with complete posterior vitreous attachment or complete vitreoretinal separation compared to the eyes with vitreomacular adhesion.59
The vitreous humor is a gel-like structure composed mostly of water (99%), hyaluronic acid, and collagen. A structural barrier between the vitreous cavity and the retina is formed by the inner limiting membrane (ILM), which is localized between the innermost layer of the retina and the outer boundary of the vitreous. The ILM shows typical ultrastructural characteristics of a basal lamina, is found in close contact with the foot processes of Müller cells, and contains proteins that are typically found in basal laminae such as collagen type IV and laminin.60 Striated collagen fibrils of the vitreous cortex insert into the inner portion of the ILM,61 which is also known as the hyaloid membrane of the vitreous. Detachment of the posterior hyaloid membrane with aging or pathology results in a condensation of the posterior vitreous surface (membrana hyaloidea posterior). In youth, there is adhesion between the vitreous cortex and the ILM that is stronger than Müller cells themselves and Müller cell foot processes become separated from their main cell body and remain connected to the posterior aspect of the ILM when this is separated from the retinal surface.62
There has been a controversial discussion regarding the embryonic origin of the ILM, which can be demonstrated as early as 4 weeks after gestation in the human eye.63,64 Traditionally, the ILM has been considered to be synthesized by Müller cells. This concept has been challenged by data presented from Sarthy, who investigated the expression of collagen type IV during development of the mouse eye.65 ILM proteins appear to originate largely from lens and ciliary body, although a contribution of retinal glial cells in ILM synthesis cannot be excluded. In support of this are data which show that also other ILM proteins such as perlecan, laminin-1, nidogen, and collagen XVIII are expressed predominantly in lens and ciliary body, but are not detected in the retina.66
Diffuse DME has been found in association with an attached, thickened, and taut posterior hyaloid.67 As immunocytochemical staining for cytokeratin (found in RPE) and GFAP protein (found in astrocytes and Müller cells) demonstrated the existence of cells in the premacular posterior hyaloids, suggesting a possible role for cell infiltration in the development or maintenance of macular edema. It remains to be elucidated whether these cells in the posterior vitreous cause macular edema physiologically rather than mechanically through the production of cytokines.
Treatment of macular edema
Laser treatment
Many studies have demonstrated a beneficial effect of photocoagulation therapy for DME.68–73 The exact mechanism of action of laser photocoagulation-induced resolution of DME is unknown. In short, a laser-induced destruction of oxygen-consuming photoreceptors has been discussed as well as cell death and scarring (involving gliosis and RPE hyperplasia) induced by the temporary rise in tissue temperature. Oxygen that normally diffuses from the choriocapillaris into the outer retina can now diffuse through the laser scar to the inner retina, thus relieving inner retinal hypoxia.74,75 There are contrasting data whether an increased preretinal oxygen partial pressure is involved and allows for microvascular repair in the treated areas.76,77
When studying the diameter of retinal arterioles, venules, and their macular branches before and after macular laser photocoagulation in eyes with DME, the macular arteriolar branches were found to be constricted by 20.2% and the venular branches 13.8%. This was attributed to an improved retinal oxygenation caused by the laser treatment leading to autoregulatory vasoconstriction, improving the DME.78
According to another theory, the beneficial effect of laser photocoagulation is due to an enhanced proliferation of RPE and endothelial cells leading to repair and restoration of the BRB.79 The RPE cells may respond to the injury in several ways: if the lesion is relatively small, the RPE defect can be filled by cell spreading; if the defect is relatively large, the cells can proliferate to resurface the area, and the RPE can produce cytokines (e.g., TGF-β) that antagonize the permeabilizing effects of VEGF.80,81
While focal laser coagulation reduces hypoxic areas and directly occludes leaky microaneurysms, the rationale for grid laser treatment in DME is not yet well established. Potentially, grid laser may have its beneficial effect by thinning the retina, bringing retinal vessels closer to choroidal vessels, permitting the retinal vessels to constrict by autoregulation, thereby decreasing retinal blood flow and consequently decreasing edema formation.82
Potential side-effects are choroidal neovascularization induction (Fig. 28.9) or lack of effect in cases of diffuse edema (Fig. 28.10).
Medical treatment
General aspects of systemic and topical medical therapy
In many cases, macular edema is caused by a generalized health problem such as diabetes, high blood pressure, or inflammatory conditions.83 These generalized diseases need to be treated prior to any other measures. There have been several reports in the literature that such treatments – particularly in diabetes, high blood pressure, and inflammatory diseases – can cure macular edema without any additional specific ocular treatment.84
Modification of the systemic blood flow, including hyperbaric oxygen treatment, is thought to alter blood flow via vasoconstriction, and facilitates the reformation of damaged junctional complexes in the vessel wall. However, despite an improvement in visual acuity in patients with chronic CME after cataract extraction, there was no correlation to macular edema.85 In uveitis-associated CME hyperbaric oxygen had no signifcant effect. Other rheological treatments such as plasma membrane filtration demonstrated good effects in initial studies; however these treatments did not enter large-scale prospective studies.86 Still, there is no evidence that rheological measures have any effect on inflammatory or DME.
Carbonic anhydrase inhibitors and nonsteroidal anti-inflammatory drugs
CA inhibitors have been used clinically for over 20 years in the treatment of macular edema. The initial observation on its therapeutic efficacy was reported in 1988 in a study of 41 patients with CME of various etiologies.87 The rationale of CA inhibitors as a therapeutic agent in the treatment of macular edema is to improve the ability of the RPE to pump fluid out of the retina by modulating the polarized distribution of CA at the level of the RPE.88
In the retina, CA is found in the cytoplasm of red/green cones (albeit not in rods) and especially inside Müller cells. The RPE, however, appears to contain almost exclusively the membrane-bound form of CA.89 The latter appears to regulate and modulate the extracellular pH gradients created by the metabolic activity of cells and may act as a bicarbonate channel.89,90 The CA activity in the RPE shows a clearcut polarized distribution with a large amount of enzyme on the apical surface of the cell, whereas there is less CA activity on the basolateral portion of the cell membrane.91 Further immunohistochemical differentiation has shown the isozyme IV is responsible for apical CA activity in the RPE.89 Intravenous injection of acetazolamide has been shown to decrease the pH in the subretinal space in both chicks and cats.89,92 This acidification was followed immediately by a reduction of the subretinal volume, and it has been postulated that it is the acidification that induces changes in ion and consequent fluid transport through the RPE91 (Fig. 28.11, panel B online).
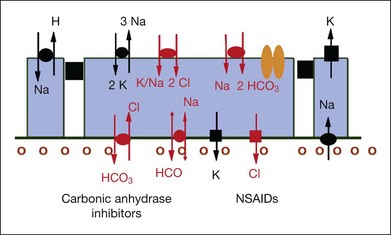
Fig. 28.11, online (B) Membrane-bound carbonic anhydrase (CA IV) 55 kDa. NSAIDs, nonsteroidal anti-inflammatory drugs.
(Adapted with permission from Wolfensberger TJ, Mahieu I, Jarvis-Evans J, et al. Membrane-bound carbonic anhydrase in human retinal pigment epithelium. Invest Ophtholmol Vis Sci 1994;35:3401–7.)
Under normal conditions, roughly 70% of the subretinal fluid is removed by metabolic transport to the choroid. In an in vivo rabbit model, this fluid transport, which is driven to a large extent by active ion transport through the RPE, can be enhanced by acetazolamide93,94 Furthermore, experiments in an animal model of iatrogenically induced retinal detachments showed that the disappearance of fluorescein through the RPE increased by 25% after intravenous injection of acetazolamide.95 The same authors also observed a marked increase in resorption of subretinal fluid at a higher dosage of 50–65 mg/kg body weight. Further studies on the frog RPE demonstrated that active chloride and bicarbonate transport probably occurs at the basal surface, which faces the choroidal blood supply and it was postulated that subretinal fluid absorption occurs at this level.90
Currently, there are no randomized studies available that confirm a beneficial effect of CA inhibitors in the treatment of macular edema. Nonrandomized observations demonstrated improved visual function in patients with postsurgical macular edema, e.g., after cataract surgery or buckling procedures.87,96 The effect lasts only as long as the patient takes the drug (on–off effect, tachyphylaxis).96 The favorable reports that were described at first regarding the application of CA inhibition in patients with macular edema secondary to retinitis pigmentosa are not supported by the long-term observation. With the continuous use of methazolamide a rebound phenomenon is observed.97
Nonsteroidal anti-inflammatory drugs (NSAIDs)
As COX inhibitors block the synthesis and release of prostaglandins, nonsteroidal drugs have been investigated in the prophylaxis and therapy of postsurgical CME. The action is based on the inhibition of the enzyme COX, which in turn inhibits the production of prostaglandins, a degradation product of arachidonic acid in the eye.98 Diclofenac sodium in high doses inhibits the formation of leukotrienes, which amplify cellular infiltration during an inflammatory reaction. Other NSAIDs have been shown to modulate chloride movement, and, as a consequence, fluid movement through the RPE.99 The effect of COX inhibitors on inflammatory aspects of DME was only demonstrated in preclinical studies13 (Fig. 28.12, online).
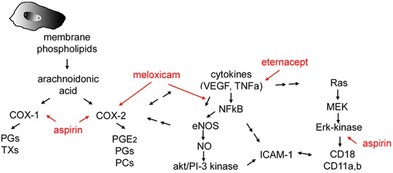
Thus, NSAIDS target the inflammatory mediators that are responsible for the edema formation and, although they may not be an optimal standalone treatment, they can be used as steroid-sparing agents. Topical NSAIDs have become the mainstay in the treatment of inflammatory CME.100 The clinical efficacy of topical NSAIDs has been shown to be of value both in the prevention101–103 and in the treatment104,105 of inflammatory CME, particularly when related to cataract surgery. Two double-masked, placebo-controlled studies in which corticosteroids were not used demonstrated that ketorolac 0.5% ophthalmic solution, administered for up to 3 months, improves vision in some patients with chronic CME after cataract surgery.106,107 A meta-analysis of the results from several different randomized controlled trials suggests that NSAIDs are beneficial as a medical prophylaxis for aphakic and pseudophakic CME and as medical treatment for chronic CME.108
There may be several explanations for why NSAIDs cannot improve vision in DME, such as chronic edema, inflammation, and ischemia that induce permanent structural alterations. Although effects on diabetic vascular leakage were achieved in preclinical studies,13 there is so far no clinical evidence for an effect of NSAIDs in DME.
Corticosteroids
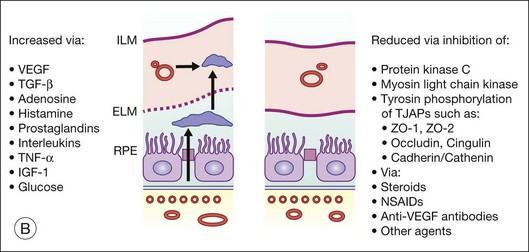
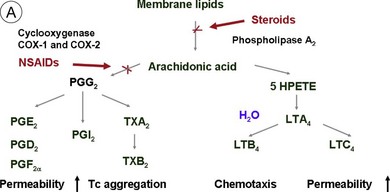
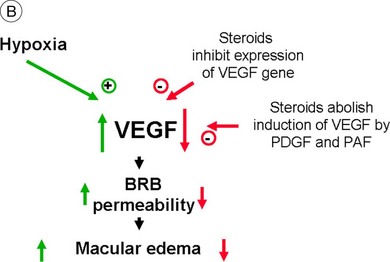
Steroids are currently regaining attention by the growing use of intravitreal triamcinolone. Corticosteroids block the release of arachidonic acid from cell membranes by inhibiting the enzyme COX and thus reduce the synthesis of prostaglandins, but they also have a multitude of other anti-inflammatory effects by acting, among others, on IL-1 and by reducing vascular permeability109 (Fig. 28.13A, online). Their additive anti-inflammatory effect to NSAIDs has been shown to be useful in the treatment of various postoperative inflammatory conditions. One potential mode of action is the increased resorption of fluid through the RPE, although the exact mechanism of this is not as yet clear. Steroids specifically stabilize endothelial tight junctions and increase their numbers.110,111 Another action of steroids is the downregulation of the production of the VEGF, which, in turn, renders the BRB tighter (Fig. 28.13B, online). Steroids also downregulate VEGF production,112 specifically in the retina,113,114 and this explains the clinical observation that the application of steroids both intravitreally and into the subtenon space can reduce macular edema considerably. Furthermore, steroids have been shown to prevent the induction of VEGF production by platelet-activating factor and platelet-derived growth factor.115 Triamcinolone also inhibits IL-6- and VEGF-induced angiogenesis downstream of the IL-6 and VEGF receptors.116 Leukocyte adhesion plays an important role in macular edema – particularly so in diabetic maculopathy.11,12 The endothelial damage resulting from this leukocyte adherence to vessel walls is mediated by nitric oxide, adhesion molecules, and other inflammatory mediators.17,117 Subtenon triamcinolone inhibits leukocyte–endothelium interactions in the retina and downregulates adhesion molecules of retinal vascular endothelium118 and thus decreases the retinal thickness.
The Müller cells represent a further site of action of steroids.119 Macular edema is thought to be partly linked to the downregulation of the Müller cell protein Kir4.1. The resultant increase in intracellular K+ leads to the uptake of proteins and osmotic swelling of the Müller cells via aquaporin 4 channels. The administration of triamcinolone reduces the production of VEGF, arachidonic acid, and prostaglandins, allowing the reactivation of fluid clearance by Müller cells via endogenous adenosine and an increase in TWIK-related acid-sensitive potassium (TASK) channels. These processes lead to an efflux of potassium, thus correcting the downregulation of the Kir4.1 protein.119
Corticosteroids have different potency levels depending on their chemical composition120 and the newer synthetically produced compounds show an up to 25-fold increase in activity, as compared to cortisone. These new agents, such as triamcinolone, dexamethasone, and fluocinolone acetonide, have fluor at the 9α position, which increases corticosteroid receptor binding. Routes of clinical administrations are manifold, including topical, periocular, intraocular, oral, and intravenous routes. Subtenon injections of corticosteroids are widely used in patients with asymmetric or unilateral uveitis. The advantages of the periocular injections are high concentrations of corticosteroids in the posterior eye, and reduction of the adverse effects compared to systemic administration. Intraocular levels of corticosteroids are identical between subtenon and retrobulbar administration.121 For oral administration, the initial high dose (1–1.5 mg/kg) is subsequently decreased according to clinical effect.122
Recent publications suggest that the intravitreal application of triamcinolone seems to be a promising therapeutic method for macular edema that fails to respond to conventional treatment.123,124 Martidis et al. published a prospective, noncomparative, interventional case series to determine if intravitreal injection of triamcinolone acetonide is safe and effective in treating DME unresponsive to prior laser photocoagulation.124 Most recent data of a randomized clinical trial demonstrated that, over a 2-year period, focal/grid photocoagulation is more effective and has fewer side-effects than intravitreal triamcinolone, supporting that focal/grid photocoagulation currently should be the benchmark against which other treatments are compared in clinical trials of DME.125 In comparison to VEGF inhibitors triamcinolone has a lesser long-term effect. Cataract and secondary glaucoma formation have to be taken into consideration.
Similar studies were performed for patients with uveitic macular edema, central vein occlusion, and CME after cataract surgery.123,124 In most published reports, complications do not appear to be prohibitive.
Reviewing the published data on intravitreal injections of triamcinolone acetonide, the therapeutic window seems very wide. The dose range of intravitreally injected triamcinolone acetonide varies from 2 mg125 to 4 mg124 and even 25 mg in a single report.126 Interestingly, reaccumulation of fluid in cystoid spaces occurs between 6 weeks and 3 months after injection, and this does not seem to be dose-dependent. Repeated injections at intervals ranging from 10 weeks127 to more than 6 months show a variable treatment response.
Jaffe and coworkers128,129 constructed a fluocinolone acetonide drug delivery device that releases fluocinolone acetonide in a linear manner over an extended period. A clinical phase III study by Bausch & Lomb investigated the efficacy of 0.5 mg (slow-release) fluocinolone acetonide in 80 patients with diffuse DME. Patients receiving the implant showed a statistically significant regression of retinal thickness after 6 months in comparison to the control group.
The safety and efficacy of dexamethasone intravitreal implant (DEX implant; Ozurdex, Allergan, Irvine, CA) compared with sham in eyes with vision loss due to macular edema associated with branch retinal vein occlusion or central retinal vein occlusion as well as posterior uveitis was assessed. The device was found to reduce the risk of vision loss in these conditions, while the vitreoretinal pharmacokinetic profiles were, in contrast to triamcinolone, similar between nonvitrectomized and vitrectomized eyes.130–132
Antiangiogenic treatment
Anti-VEGF agents
As discussed above, there are several reports which support the notion that corticosteroids act as indirect anti-VEGF agents. However, more recently, directly acting anti-VEGF agents have come to the forefront as promising treatment options for macular edema of different origins.133–135 Ranibizumab (Lucentis) and bevacizumab (Avastin) are antibodies with high affinity for VEGF, which bind to all VEGF isoforms.
The intravitreal injection of bevacizumab potentially reduces not only VEGF but also stromal cell-derived factor 1α. This suggests that intravitreal bevacizumab may influence intraocular mediators other than VEGF.136
Clinical studies have proven the effect of ranibizumab in several forms of macular edema; it is approved for the treatment of DME and macular edema after vein occlusion in the USA and Europe (Fig. 28.14). Other VEGF inhibitors such as VEGF trap or small-molecule inhibitors are currently being investigated in clinical trials. The VEGF aptamer pegaptanib (modified RNA oligonucleotide, which binds and inactivates VEGF165 only) has been shown in animal models to restore the BRB in diabetic retinopathy.137 Similar to the developments in AMD, lesser injections and slow-release systems are in demand as well as better knowledge about the combination of VEGF inhibitors with systemic treatment of the underlying disease.
Other medical treatments
Steroid-sparing immunosuppressive drugs are frequently used as additional second-line agents, particularly in patients with severe intraocular inflammation and CME.1 The rationale for these treatments relies on the inhibition of several different proinflammatory cytokines, which are specifically involved in causing macular edema by breaking down the BRB in intraocular inflammatory disorders.
Apart from the well-known agents such as VEGF, prostaglandins, and leukotrienes, these cytokines also include insulin-like growth factor 1, IL-6, stromal cell-derived factor 1, and hepatocyte growth factor. Particularly elevated levels of intraocular VEGF and IL-6 have been correlated with the severity of uveitic macular edema138 and treatments directed specifically against these factors have been proposed.
Promising results have also been reported using interferon α2139 as a treatment for long-standing refractory CME in uveitis. In addition, a beneficial effect of interferon on inflammatory CME was noted in a retrospective study of patients with multiple sclerosis-associated intermediate uveitis.140 Others reported comparable efficacy of cyclosporine A to prednisolone in the treatment of macular edema in patients with endogenous uveitis.141 Anti-TNF therapy has also been demonstrated as a promising therapy for uveitic macular edema.142 Somatostatin analogs such as octreotide may also be effective in the treatment of CME by blocking the local and systemic production of growth hormone, insulin-like growth factor, and VEGF.143 Treatment with octreotide resulted in marked improvement, or even complete resolution, of CME in uveitic patients.144 Similarly, calcium dobesilate prevents the BRB breakdown induced by diabetes, by restoring tight junction protein levels and organization and decreasing leukocyte adhesion to retinal vessels. The protective effects of calcium dobesilate likely involve the inhibition of p38 mitogen-activated protein kinase and NF-κB activation, possibly through the inhibition of oxidative/nitrosative stress.
Surgical approaches
There is clinical evidence that traction forces at the vitreoretinal interface may play an important role in the pathogenesis of macular edema. Several authors have studied vitrectomy for persistent macular edema and have suggested that release of the tractional forces at the vitreomacular interface may improve resolution of the macular edema and restore visual acuity (Fig. 28.15).
Although pars plana vitrectomy may be considered as a very simple surgical procedure, its manifold effects on a cellular level are becoming more and more understood.2
Tractional origin of macular edema and surgical aspects
The initial rationale for using vitrectomy in cases of macular edema was entirely structural, i.e., aimed at the removal of vitreous traction on the macula.55,145 The effect of traction on retinal structures becomes more understandable using Newton’s third law: to any action there is always an equal reaction in the opposite direction. The force of vitreoretinal traction will thus be met by an equal and opposite force in the retina, resulting in the retinal tissues being pulled apart. Eventually this results in the lowering of the tissue pressure within the retina, which in turn increases the difference between the hydrostatic pressure in the blood vessels and the tissue and contributes thus to edema formation (Starling’s law). Releasing the traction will increase tissue pressure and lower the hydrostatic pressure gradient, reducing the water flux from blood vessels into retinal tissue.2
Vitreoretinal traction associated with macular edema has been identified in diabetic retinopathy, following complicated cataract surgery (Irvine–Gass syndrome) and in several other disease entities. The removal of such traction by vitreoretinal surgery has been found to be beneficial.55,145,146 Peeling of the ILM of the retina ensures complete release of tractional forces, removes a potential diffusional barrier, and inhibits reproliferation of fibrous astrocytes.147
The rational for vitrectomy (removal of the hyaloid) plus peeling of the ILM is the postulated improvement of fluid diffusion from the retina to the vitreous cavity and is explained in more detail in Chapter 118, CME and vitreomacular traction.
This is particularly true for macular edema of vascular origin, such as diabetes or retinal vein occlusion. The beneficial effect of vitrectomy is thought to be based, at least in part, on two mechanisms. First, it has been found, for example, that oxygen transport between the anterior and posterior segments of the eye is increased in the vitrectomized lentectomized eyes.148,149 Others have shown that pharmacologic vitreolysis also improves oxygen diffusion within the vitreous cavity.150 This means that following vitrectomy and/or posterior vitreous detachment, the transport of molecules to and from the retina is increased.
Second, it has been shown that several growth factors such as VEGF, IL-6, platelet-derived growth factor, and others are secreted in large amounts into the vitreous during proliferative vasculopathies such as diabetic retinopathy or retinal vein occlusion151,152 and it is conceivable that a complete vitrectomy will remove this excess of growth factors mechanically with the desired effect of a restitution of the BRB. The rapid clearance of VEGF and other cytokines may thus help to prevent macular edema and retinal neovascularization in ischemic retinopathies, such as diabetic retinopathy and retinal vein occlusions. Vitreous clearance of growth factors may indeed have the same effect as the presence of, for example, VEGF antibodies in the vitreous cavity.2,153,154
Discussion and conclusion
 Bonus images for this chapter can be found online at http://www.expertconsult.com
Bonus images for this chapter can be found online at http://www.expertconsult.com
Fig. 28.1(B) Schematic drawing of the central retina with large cysts (top), finally resulting in a pseudohole formation (bottom).
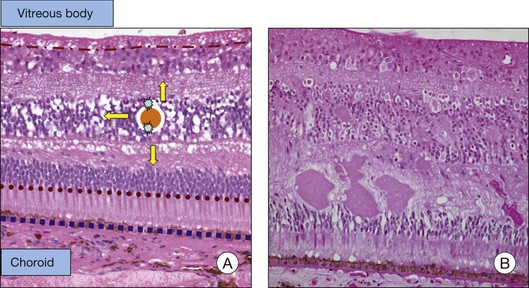
Fig. 28.3(A) In general, water outflow through vessels is possible via three major routes: paracellular via dysfunction of tight junctions, transcellular via increased transport, e.g., mediated via growth factors, and finally directly via endothelial gaps after cell death.
Fig. 28.4 Intercellular junctions in endothelial cells. Endothelial cells are connected and communicate with each other by tight junctions and adherens junctions. Tight junctions resemble a major part of the inner blood–retinal barrier. They are built by different proteins, including occludin, ZO-1, and the claudin family.
Fig. 28.6 Inflammatory mediators are involved in leukocyte–endothelial interaction that, via reduction of tight junction protein expression and induction of apoptosis, results in vascular leakage. MCP-1, monocyte chemoattractant protein; IL-8, interleukin-8; MIB-1, macrophage-inhibitory factor; TNF-α, tumor necrosis factor-alpha; ZO-1, zona occludens-1.
Fig. 28.7 Vascular endothelial growth factor (VEGF) is the key mediator of vascular damage and finally proliferation in the diabetic retina. AGE, advanced glycation end-products; IGF, insulin-like growth factor.
Fig. 28.11(B) Membrane-bound carbonic anhydrase (CA IV) 55 kDa. NSAIDs, nonsteroidal anti-inflammatory drugs. (Adapted with permission from Wolfensberger TJ, Mahieu I, Jarvis-Evans J, et al. Membrane-bound carbonic anhydrase in human retinal pigment epithelium. Invest Ophtholmol Vis Sci 1994;35:3401–7.)
Fig. 28.12 Nonsteroidal anti-inflammatory drugs (NSAIDs). Interaction of NSAID with vascular endothelial growth factor (VEGF) and inflammatory cytokines and their signaling pathways. COX-1, cyclooxygenase 1; PGs, prostaglandins; TXs, thromboxanes; TNF-α, tumor necrosis factor-α; NF-κB, nuclear factor-kappa B; eNOS, endothelial nitric oxide synthase; NO, nitric oxide; ICAM-1, intercellular adhesion molecule 1; MEK, miogen-activated protein kinase.
Fig. 28.13(A) Action of steroids on arachidonic acid. (B) Action of steroids on growth factor release. COX, cyclooxygenase; NSAIDs, nonsteroidal anti-inflammatory drugs; PG, prostaglandin; TX, thromboxane; LT, leukotriene; VEGF, vascular endothelial growth factor; PDGF, platelet-derived growth factor; PAF, platelet-activating factor; BRB, blood–retinal barrier.
1 Tranos PG, Wickremasinghe SS, Stangos NT, et al. Macular edema. Surv Ophthalmol. 2004;49:470–490.
2 Stefansson E. Physiology of vitreous surgery. Graefes Arch Clin Exp Ophthalmol. 2009;247:147–163.
3 Kern TS, Tang J, Berkowitz BA. Validation of structural and functional lesions of diabetic retinopathy in mice. Mol Vis. 2010;16:2121–2131.
4 Antcliff RJ, Marshall J. The pathogenesis of edema in diabetic maculopathy. Semin Ophthalmol. 1999;14:223–232.
5 Xu HZ, Le YZ. Significance of outer blood–retina barrier breakdown in diabetes and ischemia. Invest Ophthalmol Vis Sci. 2011;52:2160–2164.
6 Schulze C, Firth JA. Immunohistochemical localization of adherens junction components in blood–brain barrier microvessels of the rat. J Cell Sci. 1993;104:773–782.
7 Klaassen I, Hughes JM, Vogels IM, et al. Altered expression of genes related to blood–retina barrier disruption in streptozotocin-induced diabetes. Exp Eye Res. 2009;89:4–15.
8 Aveleira CA, Lin CM, Abcouwer SF, et al. TNF-α signals through PKCζ/NF-κB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes. 2010;59:2872–2882.
9 Miyamoto K, Khosrof S, Bursell S-E, et al. Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc Natl Acad Sci U S A. 1999;96:10836–10841.
10 Schröder S, Palinski W, Schmidt-Schönbein GW. Activated monocytes and granulocytes, capillary nonperfusion, and neovascularization in diabetic retinopathy. Am J Pathol. 1991;139:81–100.
11 Joussen AM, Murata T, Tsujikawa A, et al. Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol. 2001;158:147–152.
12 Joussen AM, Poulaki V, Le ML, et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004;18:1450–1452.
13 Joussen AM, Poulaki V, Mitsiades N, et al. Potential use of nonsteroidal anti-inflammatory drugs for prevention of diabetic vascular changes: aspirin prevents diabetic leakage and leukocyte adhesion through inhibition of TNF-α. FASEB J. 2002;16:438–440.
14 Joussen AM, Doehmen S, Le ML, et al. TNF-alpha mediated apoptosis plays an important role in the development of early diabetic retinopathy and long-term histopathological alterations. Mol Vis. 2009;15:1418–1428.
15 Kociok N, Radetzky S, Krohne TU, et al. Pathological but not physiological retinal neovascularization is altered in TNF-Rp55-receptor-deficient mice. Invest Ophthalmol Vis Sci. 2006;47:5057–5065.
16 Mizutani M, Kern TS, Lorenzi M. Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J Clin Invest. 1996;97:2883–2890.
17 Joussen AM, Qin W, Poulaki V, et al. Endogenous VEGF induces retinal ICAM-1 and eNOS expression and initiates early diabetic retinal leukostasis. Am J Pathol. 2002;160:501–509.
18 Senger DR, Van De Water L, Brown LF, et al. Vascular permeability factor (VPF, VEGF) in tumor biology. Cancer Metastas Rev. 1993;12:303–324.
19 Keck PJ, Hauser SD, Krivi G, et al. Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science. 1989;246:1309–1312.
20 Ferrara N, Houck K, Jakeman L, et al. Molecular and biological properties of the vascular endothelial growth factor family of proteins. Endocr Rev. 1992;13:18–32.
21 Engerman RL, Kern TS. Retinopathy and tissue hexose in drug-treated animals. Arch Ophthalmol. 1998;116:543–544.
22 Amin RH, Frank RN, Kennedy A, et al. Vascular endothelial growth factor is present in glial cells of the retina and optic nerve of human subjects with nonproliferative diabetic retinopathy. Invest Ophthalmol Vis Sci. 1999;38:36–47.
23 Qaum T, Xu Q, Joussen AM, et al. Early diabetic blood–retinal barrier breakdown is VEGF-dependent. Invest Ophthalmol Vis Sci. 2001;42:2408–2413.
24 Antonetti DA, Barber AJ, Khin S, et al. Vascular permeability in experimental diabetes is associated with reduced endothelial occludin content: vascular endothelial growth factor decreases occludin in retinal endothelial cells. Penn State Retina Research Group. Diabetes. 1998;47:1953–1959.
25 DeMaio L, Antonetti DA, Scaduto RC, Jr., et al. VEGF increases paracellular transport without altering the solvent-drag reflection coefficient. Microvasc Res. 2004;68:295–302.
26 Harhaj NS, Felinski EA, Wolpert EB, et al. VEGF activation of protein kinase C stimulates occludin phosphorylation and contributes to endothelial permeability. Invest Ophthalmol Vis Sci. 2006;47:5106–5115.
27 Wang J, Xu X, Elliott MH, et al. Müller cell-derived VEGF is essential for diabetes-induced retinal inflammation and vascular leakage. Diabetes. 2010;59:2297–2305.
28 Antonelli-Orlidge A, Saunders KB, Smith SR, et al. An activated form of transforming growth factor beta is produced by cocultures of endothelial cells and pericytes. Proc Natl Acad Sci U S A. 1989;86:4544–4548.
29 Ishii H, Jirousek MR, Koya D, et al. Amelioration of vascular dysfunction in diabetic rats by an oral PKC beta inhibitor. Science. 1996;272:728–731.
30 Whiteside C, Dlugosz J. Mesangial cell protein kinase C isozyme activation in diabetic milieu. Am J Physiol Renal Physiol. 2002;282:F975–F980.
31 Orlidge A, Hollis TM. Aortic endothelial and smooth muscle histamine metabolism in experimental diabetes. Atherosclerosis. 1982;2:142–150.
32 Gilbert RE, Kelly DJ, Cox AJ, et al. Angiotensin converting enzyme inhibition reduces retinal overexpression of vascular endothelial growth factor and hyperpermiability in experimental diabetes. Diabetologia. 2000;43:1360–1367.
33 Gill DS, Barradas MA, Fonseca VA, et al. Plasma histamine concentrations are elevated in patients with diabetes mellitus and peripheral vascular disease. Metabolism. 1989;38:243–247.
34 Gardner T. Histamine, ZO-1 and increased blood retinal permeability in diabetic retinopathy. Trans Am Ophthalmol Soc. 1995;93:583–621.
35 Aiello LP, Bursell S-E, Clermont A, et al. Vascular endothelial growth factor-induced retinal permeability is mediated by protein kinase C in vivo and suppressed by an orally effective beta isoform selective inhibitor. Diabetes. 1997;46:1473–1480.
36 Vlassara H. Recent progress in advanced glycation end products and diabetic complications. Diabetes. 1997;46:S19–S25.
37 Lu M, Kuroki M, Amano S, et al. Advanced glycation end products increase retinal vascular endothelial growth factor expression. J Clin Invest. 1998;101:1219–1224.
38 Donati G, Kapetanios A, Dubois-Dauphin M, et al. Caspase-related apoptosis in chronic ischaemic microangiopathy following experimental vein occlusion in mini-pigs. Acta Ophthalmol. 2008;86:302–306.
39 Cardier JE, Schulte T, Kammer H, et al. Fas (CD95-Apo-1) antigen expression and function in nurine liver endothelial cells: implications for the regulation of apoptosis in liver endothelial cells. FASEB J. 1999;13:1950–1960.
40 Walsh K, Sata M. Is extravasation a Fas-regulated process? Mol Med Today. 1999;5:61–67.
41 Joussen AM, Poulaki V, Mitsiades N, et al. Suppression of Fas-FasL-induced endothelial cell apoptosis prevents diabetic blood–retinal barrier breakdown in a model of streptozotocin-induced diabetes. FASEB J. 2003;17:76–78.
42 Matrisian LM. The matrix-degrading metalloproteinases. Bio Assays. 1992;14:455–463.
43 Das A, McGuire PG, Eriqat C, et al. Human diabetic neovascular membranes contain high levels of urokinase and metalloproteinase enzymes. Invest Ophthalmol Vis Sci. 1999;40:809–813.
44 Salzmann J, Limb GA, Khaw PT, et al. Matrix metalloproteinases and their natural inhibitors in fibrovascular membranes of proliferative diabetic retinopathy. Br J Ophthalmol. 2000;84:1091–1096.
45 Grant MB, Caballero S, Tarnuzzer RT, et al. Matrix metalloproteinases expression in human retinal microvascular cells. Diabetes. 1998;47:1311–1317.
46 Fernandez-Patron C, Zouki C, Whittal R, et al. Matrix metalloproteinases regulate neutrophil-endothelial cell adhesion through generation of endothelin-1. FASEB J. 2001;15:2230–2240.
47 Behzadian MA, Wang XL, Windsor LJ, et al. TGF-beta increases retinal endothelial cell permeability by increasing MMP-9: possible role of glial cells in endothelial barrier function. Invest Ophthalmol Vis Sci. 2001;42:853–859.
48 Abrass CK. Measurement of the rates of basal pinocytosis of horseradish peroxidase and internalization of heat-aggregated IgG by macrophages from normal and streptozotocin-induced diabetic rats. Immunology. 1998;65:411–415.
49 Fitzgerald ME, Caldwell RB. The retinal microvasculature of spontaneously diabetic BB rats: structure and luminal surface properties. Microvasc Res. 1990;39:15–27.
50 Hofman P, Blauwegers HG, Tolentino MJ, et al. VEGF-A induced hyperpermeability of blood–retinal barrier endothelium in vivo is predominantly associated with pinocytotic vesicular transport and not with formation of fenestrations. Vascular endothelial growth factor-A. Curr Eye Res. 2000;21:637–645.
51 Gardner TW, Abcouwer SF, Barber AJ, et al. An integrated approach to diabetic retinopathy research. Arch Ophthalmol. 2011;129:230–235.
52 Antonetti DA, Barber AJ, Bronson SK, et al. Diabetic retinopathy: seeing beyond glucose-induced microvascular disease. Diabetes. 2006;55:2401–2411.
53 Masland RH. The fundamental plan of the retina. Nat Neurosci. 2001;4:877–886.
54 Müther PS, Semkova I, Schmidt K, et al. Conditions of retinal glial and inflammatory cell activation after irradiation in a GFP-chimeric mouse model. Invest Ophthalmol Vis Sci. 2010;51:4831–4839.
55 Lewis H, Abrams GW, Blumenkranz MS, et al. Vitrectomy for diabetic macular traction and edema associated with posterior hyaloidal traction. Ophthalmology. 1992;99:753–759.
56 Harbour JW, Smiddy WE, Flynn HW, Jr., et al. Vitrectomy for diabetic macular edema associated with a thickened and taut posterior hyaloid membrane. Am J Ophthalmol. 1996;121:405–413.
57 Tachi N, Ogino N. Vitrectomy for diffuse macular edema in cases of diabetic retinopathy. Am J Ophthalmol. 1996;8:258–260.
58 Tagawa H, McMeel JW, Furukawa H, et al. Role of the vitreous in diabetic retinopathy. I. Vitreous changes in diabetic retinopathy and in physiologic aging. Ophthalmology. 1986;93:596–601.
59 Lopes de Faria JM, Jalkh AE, Trempe CL, et al. Diabetic macular edema: risk factors and concomitants. Acta Ophthalmol Scand. 1999;77:170–175.
60 Bron AJ, Tripathi RC, Tripathi BJ. The inner limiting membrane. In: Wolff’s Anatomy of the eye. London: Chapman & Hall; 1997. Ch. 14, p. 488
61 Hogan MJ, Alvaroda JA, Weddell JE. Retina. In: Histology of the human eye. Philadelphia: W.B. Saunders; 1971:393–522.
62 Sebag J. Age-related differences in the human vitreoretinal interface. Arch Ophthalmol. 1991;109:966–971.
63 Rhodes RH. A light microscopic study of the developing human neural retina. Am J Anat. 1979;154:195–209.
64 Spira AW, Hollenberg MJ. Human retinal development: ultrastructure of the inner retinal layers. Dev Biol. 1973;31:1–21.
65 Sarthy V. Collagen IV mRNA expression during development of the mouse retina: an in situ hybridization study. Invest Ophthalmol Vis Sci. 1993;34:145–152.
66 Halfter W, Dong S, Schurer B, et al. Embryonic synthesis of the inner limiting membrane and vitreous body. Invest Ophthalmol Vis Sci. 2005;46:2202–2209.
67 Jumper JM, Embabi SN, Toth CA, et al. Electron immunocytochemical analysis of posterior hyaloid associated with diabetic macular edema. Retina. 2000;20:63–68.
68 Blankenship GW. Diabetic macular edema and argon laser photocoagulation: a prospective randomized study. Ophthalmology. 1979;86:69–76.
69 Early Treatment Diabetic Retinopathy Study research group. Photocoagulation for diabetic macular edema. Early Treatment Diabetic Retinopathy Study report number 1. Arch Ophthalmol. 1985;103:1796–1806.
70 Early Treatment Diabetic Retinopathy Study research group. Treatment techniques and clinical guidelines for photocoagulation of diabetic macular edema. Early Treatment Diabetic Retinopathy Study report number 2. Ophthalmology. 1987;94:761–774.
71 Early Treatment Diabetic Retinopathy Study research group. Techniques for scatter and local photocoagulation treatment of diabetic retinopathy: Early Treatment Diabetic Retinopathy Study report no. 3. Int Ophthalmol Clin. 1987;27:254–264.
72 Early Treatment Diabetic Retinopathy Study research group. Photocoagulation for diabetic macular edema: Early Treatment Diabetic Retinopathy Study report no. 4. Int Ophthalmol Clin. 1987;27:265–272.
73 Early Treatment Diabetic Retinopathy Study research group. Early photocoagulation for diabetic retinopathy. ETDRS report number 9. Ophthalmology. 1991;98:766–785.
74 Weiter JJ, Zuckerman R. The influence of the photoreceptor–RPE complex on the inner retina. An explanation for the beneficial effects of photocoagulation. Ophthalmology. 1980;87:1133–1139.
75 Bresnick GH. Diabetic maculopathy. A critical review highlighting diffuse macular edema. Ophthalmology. 1983;90:1301–1317.
76 Molnar I, Poitry S, Tsacopoulos M, et al. Effect of laser photocoagulation on oxygenation of the retina in miniature pigs. Invest Ophthalmol Vis Sci. 1985;26:1410–1414.
77 Perry DD, Risco JM. Choroidal microvascular repair after argon photocoagulation. Am J Ophthalmol. 1982;93:787–793.
78 Gottfredsdottir MS, Stefansson E, Jonasson F, et al. Retinal vasoconstriction after laser treatment for diabetic macular edema. Am J Ophthalmol. 1993;115:64–67.
79 Wallow IH, Sponsel WE, Stevens TS. Clinicopathologic correlation of diode laser burns in monkeys. Arch Ophthalmol. 1991;109:648–653.
80 Glaser BM, Campochiaro PA, Davis JL, Jr., et al. Retinal pigment epithelial cells release inhibitors of neovascularization. Ophthalmology. 1987;94:780–784.
81 Boulton ME, Xiao M, Khaki A. Changes in growth factor expression in pig eyes following scatter laser photocoagulation. Invest Ophthalmol Vis Sci. 1995;36(suppl.):95.
82 Wilson D, Finkelstein D, Quingley H, et al. Macular grid photocoagulation: an experimental animal study on the primate retina. Arch Ophthalmol. 1988;106:100–105.
83 Gardner TW, Gabbay RA. Diabetes and obesity: a challenge for every ophthalmologist. Arch Ophthalmol. 2009;127:328–329.
84 Liew G, Mitchell P, Wong TY. Systemic management of diabetic retinopathy. Br Med J. 2009;338:b441.
85 Pfoff DS, Thom SR. Preliminary report on the effect of hyperbaric oxygen on cystoid macular edema. J Cataract Refract Surg. 1987;13:136–140.
86 Widder RA, Brunner R, Walter P, et al. Improvement of visual acuity in patients suffering from diabetic retinopathy after membrane differential filtration: a pilot study. Transfus Sci. 1999;21:201–206.
87 Cox SN, Hay E, Bird AC. Treatment of chronic macular edema with acetazolamide. Arch Ophthalmol. 1988;106:1190–1195.
88 Marmor MF, Maak T. Enhancement of retinal adhesion and subretinal fluid absorbtion by acetazolamide. Invest Ophthalmol Vis Sci. 1982;23:121–124.
89 Wolfensberger TJ, Dmitriev AV, Govardovskii VI. Inhibition of membranebound carbonic anhydrase decreases subretinal pH and volume. Doc Ophthalmol. 1999;97:261–271.
90 Miller SS, Steinberg RH. Active transport of ions across frog retinal pigment epithelium. Exp Eye Res. 1977;25:235.
91 Wolfensberger TJ, Gregor ZJ. Macular edema – rationale for therapy. Dev Ophthalmol. 2010;47:49–58.
92 Yamamoto F, Steinberg RH. Effects of intravenous acetazolamide on retinal pH in the cat. Exp Eye Res. 1992;54:711–718.
93 Wolfensberger TJ, Chiang RK, Takeuchi A, et al. Inhibition of membrane-bound carbonic anhydrase enhances subretinal fluid absorption and retinal adhesiveness. Graefes Arch Clin Exp Ophthalmol. 2000;238:76–80.
94 Marmor MF, Negi A. Pharmacologic modification of subretinal fluid absorption in the rabbit eye. Arch Ophthalmol. 1986;104:1674–1677.
95 Tsuboi S, Pederson JE. Experimental retinal detachment. 10. Effect of acetazolamide on vitreous fluorescein disappearance. Arch Ophthalmol. 1985;103:1557–1558.
96 Weene LE. Cystoid macular edema after scleral buckling responsive to acetazolamide. Ann Ophthalmol. 1992;24:423–424.
97 Fishman GA, Gilbert LD, Anderson RJ, et al. Effects of methazolamidine on chronic macular edema in patients with retinitis pigmentosa. Ophthalmology. 1994;101:687–693.
98 Colin J. The role of NSAIDs in the management of postoperative ophthalmic inflammation. Drugs. 2007;67:1291–1308.
99 Bialek S, Quong JN, Yu K, et al. Nonsteroidal anti-inflammatory drugs alter chloride and fluid transport in bovine retinal pigment epithelium. Am J Physiol. 1996;270:C1175–C1189.
100 Wolfensberger TJ, Herbort CP. Treatment of cystoid macular edema with non-steroidal anti-inflammatory drugs and corticosteroids. Doc Ophthalmol. 1999;97:381–386.
101 Flach AJ, Stegman RC, Graham J, et al. Prophylaxis of aphakic cystoid macular edema without corticosteroids. A paired-comparison, placebo-controlled double-masked study. Ophthalmology. 1990;97:1253–1258.
102 Almeida DR, Johnson D, Hollands H, et al. Effect of prophylactic nonsteroidal antiinflammatory drugs on cystoid macular edema assessed using optical coherence tomography quantification of total macular volume after cataract surgery. J Cataract Refract Surg. 2008;34:64–69.
103 DeCroos FC, Afshari NA. Perioperative antibiotics and anti-inflammatory agents in cataract surgery. Curr Opin Ophthalmol. 2008;19:22–26.
104 Nelson ML, Martidis A. Managing cystoid macular edema after cataract surgery. Curr Opin Ophthalmol. 2003;14:39–43.
105 Sivaprasad S, Bunce C, Patel N. Non-steroidal anti-inflammatory agents for treating cystoid macular oedema following cataract surgery. Cochrane Database Syst Rev. (1):2005. CD004239
106 Flach AJ, Dolan BJ, Irvine AR. Effectiveness of ketorolac tromethamine 0.5% ophthalmic solution for chronic aphakic and pseudophakic cystoid macular edema. Am J Ophthalmol. 1987;103:479–486.
107 Flach AJ, Jampol LM, Weinberg D, et al. Improvement in visual acuity in chronic aphakic and pseudophakic cystoid macular edema after treatment with topical 0.5% ketorolac tromethamine. Am J Ophthalmol. 1991;112:514–519.
108 Rossetti L, Chaudhuri J, Dickersin K. Medical prophylaxis and treatment of cystoid macular edema after cataract surgery. The results of a meta-analysis. Ophthalmology. 1998;105:397–405.
109 Nehme A, Edelman J. Dexamethasone inhibits high glucose-, TNF-α-, and IL-1β-induced secretion of inflammatory and angiogenic mediators from retinal microvascular pericytes. Invest Ophthalmol Vis Sci. 2008;49:2030–2038.
110 Romero IA, Radewicz K, Jubin E, et al. Changes in cytoskeletal and tight junctional proteins correlate with decreased permeability induced by dexamethasone in cultured rat brain endothelial cells. Neurosci Lett. 2003;334:112–116.
111 Antonetti DA, Wolpert EB, DeMaio L, et al. Hydrocortisone decreases retinal endothelial cell water and solute flux coincident with increased content and decreased phosphorylation of occludin. J Neurochem. 2002;80:667–677.
112 Edelman JL, Lutz D, Castro MR. Corticosteroids inhibit VEGF-induced vascular leakage in a rabbit model of blood–retinal and blood–aqueous barrier breakdown. Exp Eye Res. 2005;80:249–258.
113 Wang K, Wang Y, Gao L, et al. Dexamethasone inhibits leukocyte accumulation and vascular permeability in retina of streptozotocin-induced diabetic rats via reducing vascular endothelial growth factor and intercellular adhesion molecule-1 expression. Biol Pharm Bull. 2008;31:1541–1546.
114 Zhang X, Bao S, Lai D, et al. Intravitreal triamcinolone acetonide inhibits breakdown of the blood–retinal barrier through differential regulation of VEGF-A and its receptors in early diabetic rat retinas. Diabetes. 2008;57:1026–1033.
115 Nauck M, Roth M, Tamm M, et al. Induction of vascular endothelial growth factor by platelet-activating factor and platelet-derived growth factor is downregulated by corticosteroids. Am J Respir Cell Mol Biol. 1997;16:398–406.
116 Ebrahem Q, Minamoto A, Hoppe G, et al. Triamcinolone acetonide inhibits IL-6- and VEGF-induced angiogenesis downstream of the IL-6 and VEGF receptors. Invest Ophthalmol Vis Sci. 2006;47:4935–4941.
117 Leal EC, Manivannan A, Hosoya K, et al. Inducible nitric oxide synthase isoform is a key mediator of leukostasis and blood-retinal barrier breakdown in diabetic retinopathy. Invest Ophthalmol Vis Sci. 2007;48:5257–5265.
118 Mizuno S, Nishiwaki A, Morita H, et al. Effects of periocular administration of triamcinolone acetonide on leukocyte–endothelium interactions in the ischemic retina. Invest Ophthalmol Vis Sci. 2007;48:2831–2836.
119 Reichenbach A, Wurm A, Pannicke T, et al. Muller cells as players in retinal degeneration and edema. Graefes Arch Clin Exp Ophthalmol. 2007;245:627–636.
120 Haynes RC, Jr., Murad F. Adrenocorticotropic hormone: adrenocortical steroids and their synthetic analogs: inhibitors of adrenocortical steroid biosynthesis. In: Gilman AG, Goodman LS, Rall TW, et al. Goodman and Gilman’s The pharmacological basis of therapeutics. New York: Macmillan; 1985:1459–1489.
121 Thach AB, Dugel PU, Flindall RJ, et al. A comparison of retrobulbar versus subtenon’s corticosteroid therapy for cystoid macular edema refractory to topical medications. Ophthalmology. 1997;104:2003–2008.
122 Freeman G. Cystoid macular oedema in uveitis: an unsolved problem. Eye. 2001;15:12–17.
123 Jonas JB, Sofker A. Intraocular injection of crystalline cortisone as adjunctive treatment of diabetic macular edema. Am J Ophthalmol. 2001;132:425–427.
124 Martidis A, Duker JS, Greenberg PB, et al. Intravitreal triamcinolone for refractory diabetic macular edema. Ophthalmology. 2002;109:920–927.
125 Diabetic Retinopathy Clinical Research Network. A randomized trial comparing intravitreal triamcinolone acetonide and focal/grid photocoagulation for diabetic macular edema. Ophthalmology. 2008;115:1447–1449. 1449
126 Jonas JB, Kreissig I, Degenring RF. Intravitreal triamcinolone acetonide as treatment of macular edema in central retinal vein occlusion. Graefes Arch Clin Exp Ophthalmol. 2002;240:782–783.
127 Jonas JB, Kreissig I, Sofker A, et al. Intravitreal injection of triamcinolone for diffuse diabetic macular edema. Arch Ophthalmol. 2003;121:57–61.
128 Jaffe GJ, Yang CH, Guo H, et al. Safety and pharmacokinetics of an intraocular fluocinolone acetonide sustained delivery device. Invest Ophthalmol Vis Sci. 2000;41:3569–3575.
129 Jaffe GJ, Ben-Nun J, Guo H, et al. Fluocinolone acetonide sustained drug delivery device to treat severe uveitis. Ophthalmology. 2000;107:2024–2033.
130 Haller JA, Bandello F, Belfort R, Jr., et al. Randomized, sham-controlled trial of dexamethasone intravitreal implant in patients with macular edema due to retinal vein occlusion. Ophthalmology. 2010;117:1134–1146. e3
131 Lowder C, Belfort R, Jr., Lightman S, et al. Dexamethasone intravitreal implant for noninfectious intermediate or posterior uveitis. Arch Ophthalmol. 2011;129:545–553.
132 Boyer DS, Faber D, Gupta S, et al. Dexametasone intravitreal implant for treatment of diabetic macular edema in vitrectomized patients. Retina. 2011;31:915–923.
133 Cordero Coma M, Sobrin L, Onal S, et al. Intravitreal bevacizumab for treatment of uveitic macular edema. Ophthalmology. 2007;114:1574–1579.
134 Mason JO, 3rd., Albert MA, Jr., Vail R. Intravitreal bevacizumab (Avastin) for refractory pseudophakic cystoid macular edema. Retina. 2006;26:356–357.
135 Spaide RF, Chang LK, Klancnik JM, et al. Prospective study of intravitreal ranibizumab as a treatment for decreased visual acuity secondary to central retinal vein occlusion. Am J Ophthalmol. 2009;147:298–306.
136 Arimura N, Otsuka H, Yamakiri K, et al. Vitreous mediators after intravitreal bevacizumab or triamcinolone acetonide in eyes with proliferative diabetic retinopathy. Ophthalmology. 2009;116:921–926.
137 Starita C, Patel M, Katz B, et al. Vascular endothelial growth factor and the potential therapeutic use of pegaptanib (macugen) in diabetic retinopathy. Dev Ophthalmol. 2007;39:122–148.
138 van Kooij B, Rothova A, Rijkers GT, et al. Distinct cytokine and chemokine profiles in the aqueous of patients with uveitis and cystoid macular edema. Am J Ophthalmol. 2006;142:192–194.
139 Deuter CM, Koetter I, Guenaydin I, et al. Interferon alfa-2a: a new treatment option for long lasting refractory cystoid macular edema in uveitis? A pilot study. Retina. 2006;26:786–791.
140 Becker MD, Heiligenhaus A, Hudde T, et al. Interferon as a treatment for uveitis associated with multiple sclerosis. Br J Ophthalmol. 2005;89:1254–1257.
141 Nussenblatt RB, Palestine AG, Chan CC, et al. Randomized, double-masked study of cyclosporine compared to prednisolone in the treatment of endogenous uveitis. Am J Ophthalmol. 1991;112:138–146.
142 Theodossiadis PG, Markomichelakis NN, Sfikakis PP. Tumor necrosis factor antagonists: preliminary evidence for an emerging approach in the treatment of ocular inflammation. Retina. 2007;27:399–413.
143 Rothova A. Inflammatory cystoid macular edema. Curr Opin Ophthalmol. 2007;18:487–492.
144 Kafkala C, Choi JY, Choopong P, et al. Octreotide as a treatment for uveitic cystoid macular edema. Arch Ophthalmol. 2006;124:1353–1355.
145 Fung WE. Vitrectomy for chronic aphakic cystoid macular edema. Results of a national, collaborative, prospective, randomized investigation. Ophthalmology. 1985;92:1102–1111.
146 Margherio RR, Trese MT, Margherio AR, et al. Surgical management of vitreomacular traction syndromes. Ophthalmology. 1989;96:1437–1445.
147 Gandorfer A, Messmer EM, Ulbig MW, et al. Resolution of diabetic macular edema after surgical removal of the posterior hyaloid and the inner limiting membrane. Retina. 2000;20:126–133.
148 Stefansson E, Novack RL, Hatchell DL. Vitrectomy prevents retinal hypoxia in branch retinal vein occlusion. Invest Ophthalmol Vis Sci. 1990;31:284–289.
149 Holekamp NM, Shui YB, Beebe DC. Vitrectomy surgery increases oxygen exposure to the lens: a possible mechanism for nuclear cataract formation. Am J Ophthalmol. 2005;139:302–310.
150 Giblin FJ, Quiram PA, Leverenz VR, et al. Enzyme induced posterior vitreous detachment in the rat produces increased lens nuclear pO2 levels. Exp Eye Res. 2009;88:286–292.
151 Noma H, Funatsu H, Mimura T, et al. Vitreous levels of interleukin-6 and vascular endothelial growth factor in macular edema with central retinal vein occlusion. Ophthalmology. 2009;116:87–93.
152 Praidou A, Klangas I, Papakonstantinou E, et al. Vitreous and serum levels of platelet-derived growth factor and their correlation in patients with proliferative diabetic retinopathy. Curr Eye Res. 2009;34:152–161.
153 Stefansson E. The therapeutic effects of retinal laser treatment and vitrectomy. A theory based on oxygen and vascular physiology. Acta Ophthalmol Scand. 2001;79:435–440.
154 Stefansson E. Ocular oxygenation and the treatment of diabetic retinopathy. Surv Ophthalmol. 2006;4:364–380.