Chapter 75 Vogt–Koyanagi–Harada Disease
Introduction and historical aspects
Vogt–Koyanagi–Harada (VKH) disease is a bilateral granulomatous uveitis often associated with exudative retinal detachment and with extraocular manifestations, such as pleocytosis in the cerebrospinal fluid and, in some cases, vitiligo, poliosis, alopecia, and dysacusis.1
Poliosis associated with ocular inflammation was first described by Ali-ibn-Isa, an Arab physician who lived in the 1st century AD (cited by Pattison).2 This association was reported by Schenkl in 1873,3 by Hutchinson in 1892,4 and by Vogt in l906.5 Harada described a primary posterior uveitis with exudative retinal detachments in association with cerebrospinal fluid pleocytosis.6
Three years later, in 1929, Koyanagi described six patients with bilateral chronic iridocyclitis, patchy depigmentation of the skin, patchy hair loss, and whitening of the hair, especially the eyelashes.7 This constellation of findings was termed “uveitis with poliosis, vitiligo, alopecia, and dysacusis.”7 Babel in 19328 and Bruno and McPherson in 1945 combined the findings of Vogt, Koyanagi, and Harada and suggested that these processes represent a continuum of the same disease,9 thereafter recognized as Vogt–Koyanagi–Harada syndrome.
When a patient presents with the ocular and the extraocular manifestations, the diagnosis of VKH is made with certainty. However, extraocular manifestations such as dysacusis and cutaneous changes are relatively rare, and the dermatologic changes mainly occur late in the course of the disease.1,10 Because of the variation in clinical presentations of VKH, the American Uveitis Society (AUS) in 1978 recommended the following diagnostic criteria: (1) the absence of any history of ocular trauma or surgery; and (2) the presence of at least three of the following four signs: (a) bilateral chronic iridocyclitis; (b) posterior uveitis, including exudative retinal detachment, forme fruste of exudative retinal detachment, disc hyperemia or edema and “sunset glow” fundus; (c) neurologic signs of tinnitus, neck stiffness, cranial nerve, or central nervous system disorders, or cerebrospinal fluid pleocytosis; and (d) cutaneous findings of alopecia, poliosis, or vitiligo.11
Since VKH manifestations vary depending upon the clinical course, a given patient may not initially present with the features required for the diagnosis of VKH by AUS criteria. Read and Rao recently evaluated the utility of the existing AUS criteria in 71 consecutive patients with VKH who were diagnosed based on the clinical features and the course of the disease, combined with fluorescein angiography with or without utrasonography in selected cases.12 The authors concluded that AUS criteria for diagnosis of VKH may not be adequate. Taking into account the multisystem nature of VKH and allowing for the different ocular findings present in the early and late stages of the disease, the First International Workshop on VKH proposed revised diagnostic criteria to include clinical manifestations at various stages of disease.13 These revised diagnostic criteria are summarized in Box 75.1.
Box 75.1
The revised diagnostic criteria proposed by the First International Workshop on Vogt–Koyanagi–Harada (VKH) disease*
B. Incomplete VKH disease (point 1 and either 2 or 3 must be present)
(Modified from Read RW, Holland GN, Rao NA et al. Revised diagnostic criteria for Vogt–Koyanagi–Harada disease: report of an international committee on nomenclature. Am J Ophthalmol 2001;131:647–52.13)
In the past, constellation of these ocular signs and symptoms warranted the term “syndrome,” but in recent years the entity has been well characterized; thereafter the International Workshop on VKH adopted the term Vogt–Koyanagi–Harada disease.13
Epidemiology
The incidence of VKH is variable. It appears to be more common in Japan, where it accounts for 6.7% of all uveitis referrals.14 In the USA it accounts for 1–4% of all uveitis clinic referrals.
VKH tends to affect more pigmented races, such as Asians, Hispanics, American Indians, and Asian Indians.1,l5 In the USA there appears to be variability in the racial distribution of patients with VKH disease.1,11,16,17 In northern California VKH was seen mainly in Asians (4l%), followed by whites (29%), Hispanics (16%), and blacks (14%).17 In contrast, reports from southern California show that 78% of VKH patients were Hispanic while 3% were white, 10% were Asian, and 6% were black.1 A series reported from the National Institutes of Health (NIH) showed that 50% of VKH patients were white, 35% were black, and 13% were Hispanic.17 However, most of those patients reported in the NIH series had remote American Indian ancestry. Most studies report that women tend to be affected more frequently than men; however, Japanese investigators have not found such a female predilection.15 Most patients are in their second to fifth decades of life, but children may also be affected.1,18
Clinical description
Typical clinical features of VKH include bilateral panuveitis associated with exudative retinal detachment, meningism associated with headache and pleocytosis of cerebrospinal fluid, tinnitus or hearing loss, and cutaneous changes, such as alopecia, poliosis, and vitiligo. However, all of these extraocular features are rarely seen during the initial presentation, and the clinical features vary depending upon the stage of the disease as well as the effect of medical treatment. Presence of ocular and two or more extraocular features is considered as a complete form of VKH disease.13 Incomplete VKH disease includes bilateral typical ocular involvement plus either neurologic/auditory or cutaneous changes, whereas probable VKH disease is composed of just ocular manifestations.13 However, some of these probable VKH patients can develop cutaneous manifestations during the chronic or chronic recurrent stage of the disease.
The acute uveitic stage
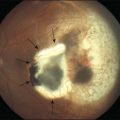
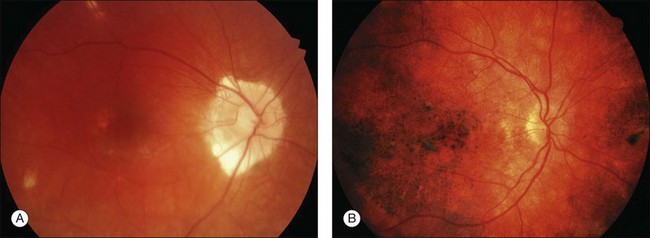
This stage follows the prodromal phase and presents with blurring of vision in both eyes. One eye may be affected first, followed a few days later by the second eye. Despite a delay in symptoms, careful examination will reveal bilateral posterior uveitis. This uveitis consists of thickening of the posterior choroid with elevation of the peripapillary retinochoroidal layer, multiple serous retinal detachments (Fig. 75.1), hyperemia and edema of the optic nerve head.
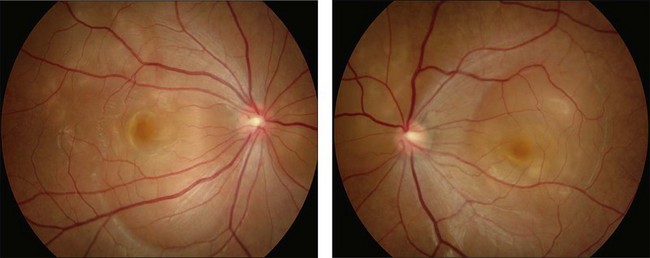
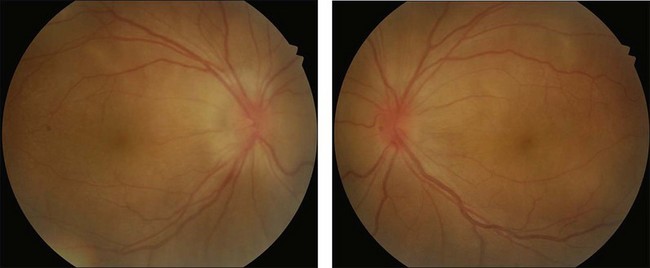
Rarely VKH disease can present with optic disc hyperemia and edema without serous retinal detachments (Fig. 75.2).
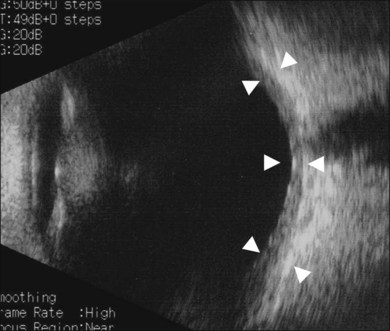
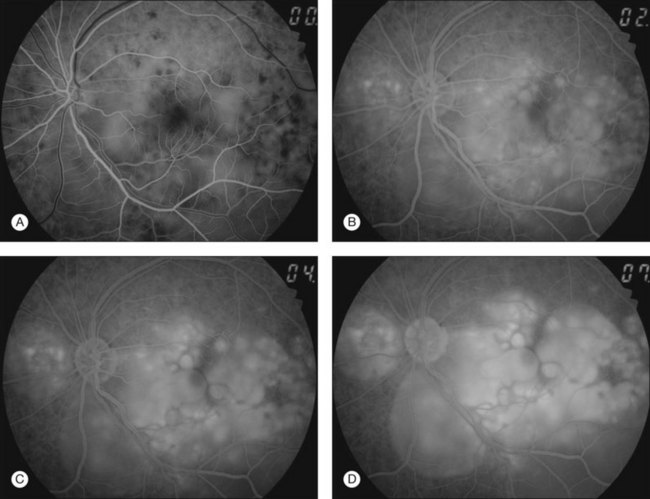
Thickened choroid can be detected by ultrasonography (Fig. 75.3). Alteration in the retinal pigment epithelium (RPE) associated with multifocal choroidal inflammation is easily observed with fluorescein angiography, which reveals hypofluorescent dots at the early phase followed by multiple focal areas of leakage and subretinal fluid accumulation at the late phase (Fig. 75.4).
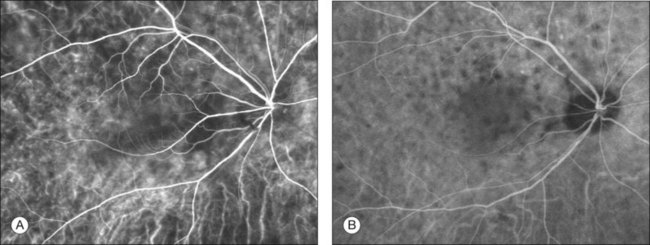
Indocyanine green angiography (ICGA) (Fig. 75.5) could be useful to evaluate choroidal inflammatory changes such as early choroidal stromal vessel hyperfluorescence and leakage, and hypofluorescent dark dots at the level of the choroid.19,20
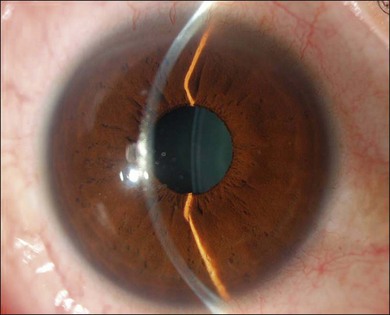
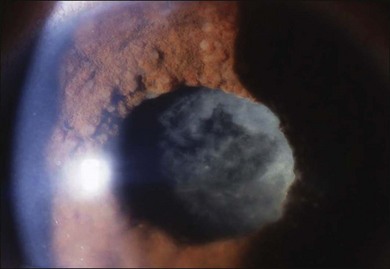
The inflammation eventually becomes diffuse, extending into the anterior segment and revealing the presence of flare and cells in the anterior chamber. Less commonly, mutton-fat keratic precipitates, small nodules on the iris surface and pupillary margin, may be observed;1 however, these anterior inflammatory changes are more common in the recurrent phase. The inflammatory infiltrate in the ciliary body and choroid may cause forward displacement of the lens iris diaphragm (Fig. 75.6), leading to acute angle closure glaucoma or annular choroidal detachment.21,22 These intraocular changes are typically bilateral; rarely, however, the process can be restricted to one eye.23
The chronic uveitic stage
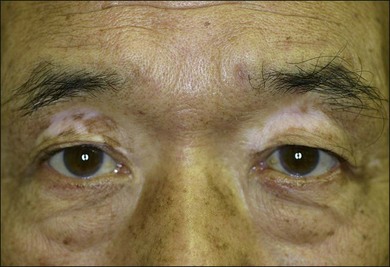
The chronic or convalescent stage occurs several weeks after the acute uveitic stage and is characterized by development of vitiligo (Fig. 75.7), poliosis, and depigmentation of the choroids. Perilimbal vitiligo, also known as Sugiura’s sign (Fig. 75.8), may develop at this stage among the patients who have melanosis at the palisade of Vogt, such as Japanese patients.1,13
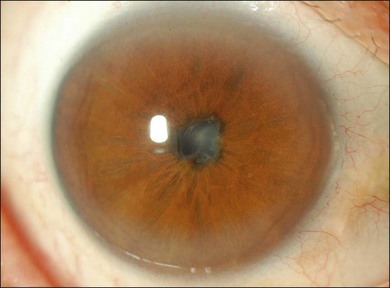
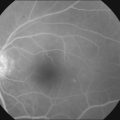
Choroidal depigmentation occurs a few months after the uveitic phase. This leads to the characteristic pale disc with a bright red–orange choroid known as sunset glow fundus (Fig. 75.9). In Hispanics, the sunset glow fundus may show foci of RPE changes in the form of hyperpigmentation or hypopigmentation. The juxtapapillary area may show marked depigmentation. At this stage small, yellow, well-circumscribed areas of chorioretinal atrophy may appear, mainly in the inferior midperiphery of the fundus. This convalescent phase may last for several months.
The chronic recurrent stage
The chronic recurrent stage consists of a smoldering panuveitis with acute episodic exacerbations of granulomatous anterior uveitis. Recurrent posterior uveitis with exudative retinal detachment is uncommon. The anterior uveitis may be resistant to local and systemic corticosteroid therapy. Iris nodules may be seen during this phase (Fig. 75.10). The most visually debilitating complication of the chronic inflammation during this stage appears to be the development of subretinal neovascular membranes.24 Posterior subcapsular cataract, as well as glaucoma, either angle closure or open angle, and posterior synechiae, may also be seen.25,26 Recurrence of the intraocular inflammation may lead to extensive chorioretinal atrophy.
Frequency of distinguishing clinical features
Clinical features of ocular and extraocular manifestations of 180 patients with VKH disease and 967 patients with non-VKH disease analyzed by stepwise logistic regression models are listed in Table 75.1.27 In the acute stage, exudative retinal detachment was most likely to be found, whereas in the chronic stage, sunset glow fundus was most common. Prevalence of sunset glow fundus can be low (67.5%) in patients treated with systemic corticosteroid from the initial acute uveitic stage.28
Table 75.1 Distinguishing clinical features of acute and chronic VKH disease27
| Dependent variable = VKH | Odds ratio estimate (95% CI) | P value |
|---|---|---|
| Acute disease | ||
| Exudative retinal detachment | >999 (48.02, >999) | <0.0001 |
| Alopecia | 81.23 (2.47, >999) | 0.01 |
| Disc hyperemia | 5.28 (1.02, 27.42) | 0.05 |
| Asian | 24.48 (2.38, 251.9) | 0.007 |
| Hispanic | 59.76 (3.77, 948.2) | 0.004 |
| Chronic disease | ||
| Sunset glow fundus | 141.66 (54.65, 367.2) | <0.0001 |
| Vitiligo | 11.73 (3.59, 38.33) | <0.0001 |
| Alopecia | 3.20 (1.40,7.31) | 0.0005 |
| Nummular chorioretinal scars | 2.83 (1.34, 5.98) | 0.01 |
| Vitreous cells | 0.39 (0.18, 0.83) | 0.02 |
| Asian | 3.48 (1.60, 7.60) | 0.002 |
| Hispanic | 13.25 (4.63, 37.88) | 0.0003 |
(Reproduced with permission from Rao NA, Gupta A, Dustin L, et al. Frequency of distinguishing clinical features in Vogt–Koyanagi–Harada disease. Ophthalmology 2010;117:591–9 [Table 8].)
Pathology and pathogenesis
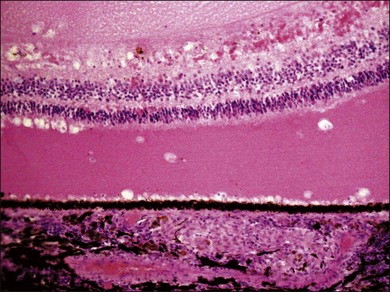
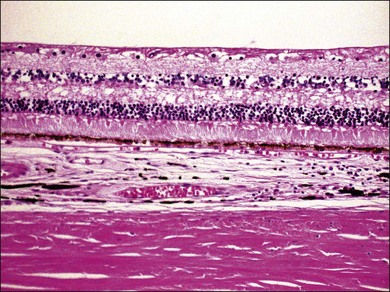
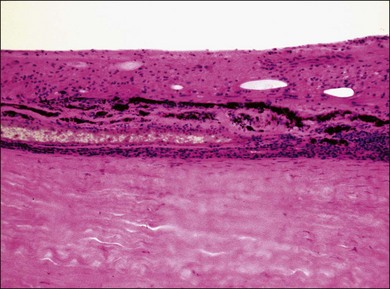
VKH is a non-necrotizing diffuse granulomatous inflammation involving the uvea. Although a granulomatous process is the primary feature of the disease, the histopathologic changes vary depending on the stage of the disease.29 Uvea is thickened by diffuse infiltration of lymphocytes and macrophages, admixed with epithelioid cells and multinucleated giant cells containing melanin granules. The neural retina is detached from the RPE, and the subretinal space contains proteinaceous fluid exudates (Fig. 75.11). Dalen–Fuchs nodules, which represent focal aggregates of epithelioid histiocytes admixed with RPE, are located between Bruch’s membrane and the RPE.29 In the convalescent stage, the choroidal melanocytes decrease in number and disappear (Fig. 75.12), resulting in the sunset glow appearance of the fundus.29 During the chronic stage, the numerous focal yellowish oval or round lesions seen in the inferior peripheral fundus by ophthalmoscopy histologically display a focal loss of RPE cells and the formation of chorioretinal adhesions.29 In the long-standing chronic recurrent stage, the RPE and neural retina show degenerative changes (Fig. 75.13). The RPE may reveal hyperplasia and fibrous metaplasia with or without associated subretinal neovascularization.29
Although the exact cause for the inflammation directed at the melanocytes remains unknown, current evidence suggests that it involves an autoimmune process driven by T lymphocytes against an as-yet unidentified antigen(s) associated with melanocytes.1,30,31
The antigenic peptides to induce autoimmune response in the uveal tract may include tyrosinase or tyrosinase-related proteins.32,33 Experimental animal studies, as well as T-cell clones raised specific to tyrosinase family protein from the peripheral blood of patients with VKH, suggest that autoreactive T cells against tyrosinase and/or tyrosinase-related proteins may play a role in the development of VKH in a genetically susceptible individual.33
Cytokines are known to play an important role in the pathogenesis of VKH disease. Interleukin-21, a member of the IL-2 family exerting pleiotropic effects on the immune system, could be involved in the pathogenesis of VKH disease, possibly by promoting IL-17 secretion.34
CD4+CD25high Treg cells have been shown to be involved in the pathogenesis of autoimmune diseases. Reduction of number or impaired function of CD4+CD25high Treg cells has been reported in patients with VKH.35
There is a strong association with the human leukocyte antigen (HLA) DR4 in Japanese patients with VKH disease, and these patients and individuals from Korea showed predominant alleles of DRB1*0405 and HLA-DRB1 *0410.36 Significant difference in the prevalence of HLA-DRB1 *0405 between the VKH patients and control subjects was also demonstrated in the Middle East.37 However, in other racial groups, such as mixed Hispanic individuals from southern California, either HLA-DR1 or HLA-DR4 was found in 84% of patients with VKH disease.38 Indeed, there was a higher relative risk with HLA-DR1 than HLA-DR4 (4.11 versus 1.96, respectively). Similar HLA-DR1 and HLA-DR4 subtypes were noted in 89% of mixed Mexican patients.39 These studies indicate that specific HLA genes may confer risk for development of VKH disease.
Investigations
Imaging studies
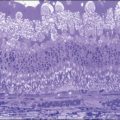
In patients with inadequate pupillary dilation caused by posterior synechiae or dense vitritis that obscures the view of the fundus, ultrasonography may help to establish the diagnosis.40 Ultrasound biomicroscopic examination and other recent ophthalmic viewing system during the uveitic stage may reveal shallow anterior chamber, ciliochoroidal detachment, and thickened ciliary body.
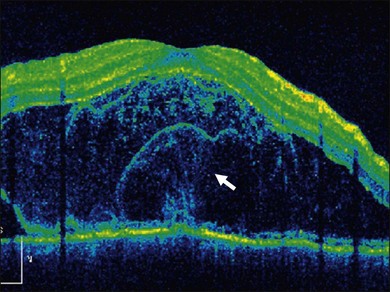
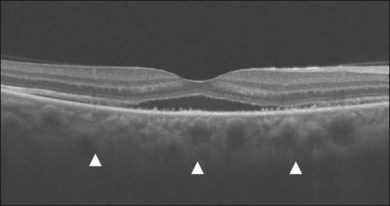
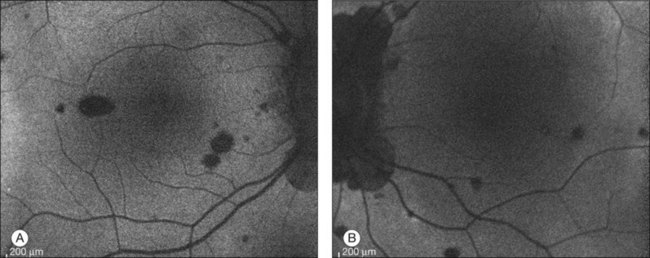
In addition to angiography and ultrasonography, optical coherence tomography (OCT) and scanning laser ophthalmoscopy (SLO) have been found useful for substantiating the diagnosis. Serous retinal detachment can be clearly observed by OCT (Fig. 75.14). Thickened choroid can be demonstrated by OCT with enhanced depth imaging (Fig. 75.15). OCT has been used not only for diagnosis but for monitoring resolution of the serous retinal detachment41,42 and choroidal thickness43 with corticosteroid therapy. In the chronic stage, changes of the RPE can be clearly depicted as decreased fundus autofluorescence44 (Fig. 75.16).
Lumbar puncture
Although lumbar puncture is not necessary for the diagnosis of VKH disease with typical ocular and extraocular manifestations, this procedure is a useful adjunctive test in cases with atypical features. Ohno et al. found that more than 80% of patients with VKH disease had cerebrospinal fluid pleocytosis, consisting mostly of lymphocytes.17 In their study, the pleocytosis was present in 80% of patients within 1 week and in 97% of patients within 3 weeks of the onset of uveitis. The cerebrospinal fluid pleocytosis, however, is transient and resolves within 8 weeks even in patients who develop recurrences of intraocular inflammation. Cytologic analysis may reveal melanin-containing histiocyte in patients with pleocytosis.45
Differential diagnosis
The differential diagnosis of VKH disease includes sympathetic ophthalmia, uveal effusion syndrome, posterior scleritis, acute posterior multifocal placoid pigment epitheliopathy (APMPPE), and sarcoidosis.1 Sympathetic ophthalmia can present with bilateral panuveitis associated with retinal detachment and meningism. However, a history of penetrating ocular injury is the rule in this disorder. Extraocular manifestations, such as dysacusis, vitiligo, poliosis, and alopecia, can occur in sympathetic ophthalmia, but they are rare.46
Treatment
Although data from randomized trials are lacking, early and aggressive use of systemic corticosteroids followed by slow tapering over 3–6 months is the acceptable treatment of choice to suppress the intraocular inflammation and to prevent the development of complications related to the ocular inflammation.1 Such treatment may prevent progression of the disease to the chronic recurrent stage and may also reduce the incidence and/or severity of extraocular manifestations. If the ocular inflammation relapses after tapering of systemic corticosteroids, the relapse may reflect too-rapid tapering of the corticosteroids. Such recurrences become increasingly steroid-resistant, and cytotoxic or immunosuppressive agents are usually required to control the inflammation. Patients with inflammatory cell infiltration in the anterior chamber require topical corticosteroids and cycloplegics to reduce ciliary spasm and prevent posterior synechiae formation.
High-dose oral corticosteroids, 80–100 mg per day of prednisone or 200 mg of intravenous methylprednisolone for 3 days, followed by oral administration of high-dose corticosteroids with a slow taper, are the mainstay of therapy for VKH disease.47,48 The use of intravenous corticosteroids, up to 1 g/day for 3 days, followed by a slow taper has been recommended by some. However, Sasamoto et al. found a similar beneficial effect of decreased intraocular inflammation with either 1 g or 200 mg per day of intravenous corticosteroids.48 Of course, as is always the case when corticosteroids are prescribed, careful attention to possible risks and side-effects is warranted.
Although the initial episode of uveitis can be managed successfully in the majority of cases with intravenous and/or oral corticosteroids, recurrences do not respond as well to systemic corticosteroid treatment.1 Such patients may show some initial response to subtenon injections of triamcinolone acetonide, but they usually require immunosuppressive or cytotoxic agents, such as cyclosporine, azathioprine, cyclophosphamide, chlorambucil, mycophenolate mofetil (CellCept), and tacrolimus (FK506). Cyclosporine 5 mg/kg per day is generally preferred when the intraocular inflammation is corticosteroid-resistant or when the patient experiences intolerable side-effects from the long-term use of corticosteroids. Recently, a biologic agent, anti-TNF-α antibody, has been reported in the management of VKH disease.49
Administration of the immunosuppressives, cytotoxic agents, and biologics requires a careful pretreatment evaluation and careful subsequent evaluations during the follow-up examinations for any side-effects associated with the therapy. Various immunosuppressive and cytotoxic agents used in the treatment of VKH disease are summarized in Box 75.2.
Box 75.2
Immunosuppressive/cytotoxic agents used in the treatment of Vogt–Koyanagi–Harada disease
• Oral prednisone 100–200 mg initially, followed by gradual taper over 3–6 months
• Pulse dose of methylprednisolone 1 g/day for 3 days, followed by gradual tapering of oral prednisone over 3–6 months
• Intravenous methylprednisolone 100–200 mg/day for 3 days, followed by gradual tapering of oral prednisone over 3–6 months
• FK506 0.1–0.15 mg/kg per day
• Azathioprine 1–2.5 mg/kg per day
• Mycophenolate mofetil 1–3 g/day
• Cyclophosphamide 1–2 mg/kg per day
• Chlorambucil 0.1 mg/kg per day; dose adjusted every 3 weeks to a maximum of 18 mg/day
Complications and management
A retrospective analysis of the records of 101 patients with VKH disease followed at the Doheny Eye Institute revealed the development of at least one complication in 51% of eyes.50 Cataract occurred in 42%, glaucoma in 27%, choroidal neovascularization in 11% (Fig. 75.17), and subretinal fibrosis in 6% (Fig. 75.18). The patients who developed these complications had a significantly longer median duration of disease and significantly more recurrences than did those patients who developed no complications. Moreover, eyes possessing a better visual acuity at presentation had better visual acuity at final follow-up, and patients who developed VKH at a more advanced age had a worse visual acuity.50
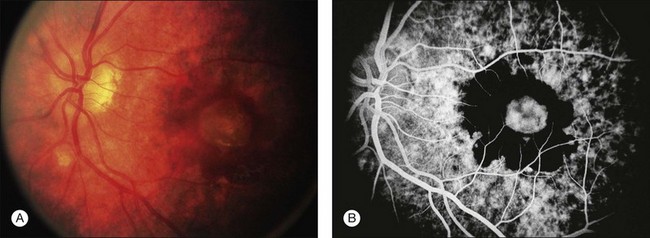
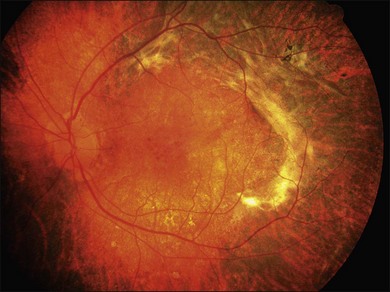
Fig. 75.18 Subretinal fibrosis in a patient with chronic recurrent stage of Vogt–Koyanagi–Harada disease.
There is general agreement that cataract surgery should be delayed until the intraocular inflammation has subsided, at which time safe cataract extraction with posterior-chamber intraocular lens implantation can be successfully accomplished. Occasionally, patients with significant vitreous opacities and debris may require a combined procedure of pars plana vitrectomy and lensectomy.24
Angle closure due to peripheral anterior synechiae and posterior synechiae may cause glaucoma.25 Acute angle closure with elevated intraocular pressure has been reported as a presenting sign of VKH disease. Although sustained elevated intraocular pressure can be controlled by medical therapy alone, most patients require surgical intervention in the form of iridectomy, trabeculectomy with 5-fluorouracil or mitomycin C and tube-shunt surgery.
Chronic recurrent anterior uveitis and fundus pigmentary disturbances seem to predispose patients to the development of choroidal neovascularization24 (Fig. 75.18). These subretinal membranes present with white gliotic raised masses, which may be associated with subretinal hemorrhage. ICG angiography is useful for detecting the membranes and photocoagulation may help with the management. Photodynamic therapy with verteporfin for subfoveal choroidal neovascularization has been attempted with some success.51 Intravitreal injection of an anti-vascular endothelial growth factor (VEGF) may be an option to treat CNV in eyes with VKH syndrome.52
Prognosis
In general, those VKH patients who are treated with initial high-dose systemic corticosteroids followed by gradual tapering will usually have a fair visual prognosis; nearly two-thirds of these patients retain 20/40 or better visual acuity.1,48 On average, most patients require treatment for 6 months. The complications of chronic recurrent VKH disease include cataract, glaucoma, choroidal neovascularization, subretinal fibrosis, and optic atrophy.1,50,53
1 Moorthy RS, lnomata H, Rao NA. Vogt–Koyanagi–Harada syndrome. Surv Ophthalmol. 1995;39:265–292.
2 Pattison EM. Uveo-meningoencephalitic syndrome (Vogt–Koyanagi–Harada). Arch Neurol. 1965;12:197–205.
3 Schenkl A. Ein Fall von plötzlich aufgetretener Poliosis circumscripta der Wimpern. Arch Dermatol Syph. 1873;5:137–139.
4 Hutchinson J. A case of blanched eyelashes. Arch Surg. 1892;4:357.
5 Vogt A. Frühzeitiges Ergrauen der Zilien und Bemerkungen über den sogenannten plötzlichen Eintritt dieser Veränderung. Klim Monatsbl Augenheilkd. 1906;44:228–242.
6 Harada E. Beitrag zur klinischen Kenntnis von nichteitriger Choroiditis (choroiditis diffusa acuta). Acta Soc Ophthalmol Jpn. 1926;30:356–378.
7 Koyanagi Y. Dysakusis, Alopecia und Poliosis bei schwerter Uveitis nichttraumatischen Ursprungs. Klin Monatsbl Augenheilkd. 1929;82:l94–211.
8 Babel J. Syndrome de Vogt–Koyanagi (Uveite bilaterale, poliosis, alopecie, vitiligo et dysacousie). Schweiz Med Wochenschr NR. 1932;4:1136–1140.
9 Bruno MG, McPherson SD, Jr. Harada’s disease. Am J 0phthalmol. 1949;32:513–522.
10 Beniz J, Forster DJ, Lean JS, et al. Variations in clinical features of the Vogt–Koyanagi–Harada syndrome. Retina. 1991;11:275–280.
11 Snyder DA, Tessler HA. Vogt–Koyanagi–Harada syndrome. Am J Ophthalmol. 1980;90:69–75.
12 Read RW, Rao NA. Utility of existing Vogt–Koyanagi–Harada syndrome diagnostic criteria at initial evaluation of the individual patient: a retrospective analysis. Ocul Immunol Inflamm. 2000;8:227–234.
13 Read RW, Holland GN, Rao NA, et al. Revised diagnostic criteria for Vogt–Koyanagi–Harada disease: report of an international committee on nomenclature. Am J Ophthalmol. 2001;131:647–652.
14 Goto H, Mochizuki M, Yamaki S, et al. Epidemiological survey of intraocular inflammation in Japan. Jpn J Ophthalmol. 2007;51:41–44.
15 Shimizu K. Harada’s, Behçet’s, Vogt–Koyanagi syndromes: are they clinical entities? Trans Am Acad Ophthalmol Otolaryngol. 1973;77:281–290.
16 Nussenblatt RB. Clinical studies of Vogt–Koyanagi–Harada’s disease at the National Eye Institute, NIH, USA. Jpn J Ophthalmol. 1988;32:330–333.
17 Ohno S, Char DH, Kimura SJ, et al. Vogt–Koyanagi–Harada syndrome. Am J Opthalmlol. 1977;83:735–740.
18 Forster DJ, Green RL, Rao NA. Unilateral manifestation of the Vogt–Koyanagi–Harada syndrome in a 7-year-old child. Am J Ophthalmol. 1991;111:380–382.
19 Herbort CP, Mantovani A, Bouchenaki N. Indocyanine green angiography in Vogt–Koyanagi–Harada disease: angiographic signs and utility in patient follow-up. Int Ophthalmol. 2007;27:173–182.
20 Miyanaga M, Kawaguchi T, Miyata K, et al. Indocyanine green angiography findings in initial acute pretreatment Vogt–Koyanagi–Harada disease in Japanese patients. Jpn J Ophthalmol. 2010;54:377–382.
21 Kawano Y, Tawara A, Nishioka Y, et al. Ultrasound biomicroscopic analysis of transient shallow anterior chamber in Vogt–Koyanagi–Harada syndrome. Am J Ophthalmol. 1996;121:720–723.
22 Yamamoto N, Naito K. Annular choroidal detachment in patients with Vogt–Koyanagi–Harada disease. Graefes Arch Clin Exp Opthalmol. 2004;242:355–358.
23 Usui Y, Goto H, Sakai J, et al. Presumed Vogt–Koyanagi–Harada disease with unilateral ocular involvement: report of three cases. Graefes Arch Clin Exp Ophthalmol. 2009;247:1127–1132.
24 Moorthy RS, Chong LP, Smith RE, et al. Subretinal neovascular membranes in Vogt–Koyanagi–Harada syndrome. Am J Ophthalmol. 1993;116:164–170.
25 Forster DJ, Rao NA, Hill RA, et al. Incidence and management of glaucoma in Vogt–Koyanagi–Harada syndrome. Ophthalmology. 1993;100:613–618.
26 Moorthy RS, Rajeev B, Smith RE, et al. Incidence and management of cataracts in Vogt–Koyanagi–Harada syndrome. Am J Ophthalmol. 1999;118:197–204.
27 Rao NA, Gupta A, Dustin L, et al. Frequency of distinguishing clinical features in Vogt–Koyanagi–Harada disease. Ophthalmology. 2010;117:591–599.
28 Keino H, Goto H, Usui M. Sunset glow fundus in Vogt–Koyanagi–Harada disease with or without chronic ocular inflammation. Graefes Arch Clin Exp Ophthalmol. 2002;240:878–882.
29 Inomata H, Rao NA. Depigmented atrophic lesions in sunset glow fundi of Vogt–Koyanagi–Harada disease. Am J Ophthalmol. 2001;131:607–614.
30 Norose K, Yano A. Melanoma specific Th1 cytotoxic T lymphocyte lines in Vogt–Koyanagi–Harada disease. Br J Ophthalmol. 1996;80:1002–1008.
31 Sugita S, Sagawa K, Mochizuki M, et al. Melanocyte lysis by cytotoxic T lymphocytes recognizing the MART-1 melanoma antigen in HLA-A2 patients with Vogt–Koyanagi–Harada disease. Int Immunol. 1996;8:799–803.
32 Gocho K, Kondo I, Yamaki K. Identificaiton of autoreactive T cells in Vogt–Koyanagi–Harada disease. Invest Ophthalmol Vis Sic. 2001;42:2004–2009.
33 Hayakawa K, Ishikawa M, Yamaki K. Ultrastructural changes in rat eyes with experimental Vogt–Koyanagi–Harada disease. Jpn J Ophthalmol. 2004;48:222–227.
34 Li F, Yang P, Liu X, et al. Upregulation of interleukin 21 and promotion of interleukin 17 production in chronic or recurrent Vogt–Koyanagi–Harada disease. Arch Ophthalmol. 2010;128:1449–1454.
35 Chen L, Yang P, Zhou H, et al. Diminished frequency and function of CD4+CD25high regulatory T cells associated with active uveitis in Vogt–Koyanagi–Harada syndrome. Invest Ophthalmol Vis Sci. 2008:49–3475.
36 Shindo Y, Ohno S, Yamamoto T, et al. Complete association of the HLA-DRB1 04 and DQ1 04 alleles with Vogt–Koyanagi–Harada’s disease. Hum Immunol. 1994;39:169–176.
37 Iqniebi A, Gaafar A, Sheereen A, et al. HLA-DRB1among patients with Vogt–Koyanagi–Harada disease in Saudi Arabia. Mol Vis. 2009;15:1876–1880.
38 Weisz JM, Holland GN, Roer LN, et al. Association between Vogt–Koyanagi–Harada syndrome and HLA-DR1 and DR4 in Hispanic patients living in Southern California. Ophthalmology. 1995;102:1012–1015.
39 Arellanes-Garcia L, Bautista N, More P, et al. HLA-DR is strongly associated with Vogt–Koyanagi–Harada disease in Mexican Mestizo patients. Ocul Immunol Inflamm. 1998;6:93–100.
40 Forster DJ, Cano MR, Green RL, et al. Echographic features of the Vogt–Koyanagi–Harada syndrome. Arch Ophthalmol. 1990;108:1421–1426.
41 Yamaguchi Y, Otani T, Kishi S. Tomographic features of serous retinal detachment with multilobular dye pooling in acute Vogt–Koyanagi–Harada disease. Am J Ophthalmol. 2007;144:260–265.
42 Ishihara K, Hangai M, Kita M, et al. Acute Vogt–Koyanagi–Harada disease in enhanced spectral-domain optical coherence tomography. Ophthalmology. 2009;116:1799–1807.
43 Fong AH, Li KK, Wong D. Choroidal evaluation using enhanced depth imaging spectral domain optical coherence tomography in Vogt–Koyanagi–Harada disease. Retina. 2011;31:502–599.
44 Vasconcelos-Santos DV, Sohn EH, Sadda S, et al. Retinal pigment epithelial changes in chronic Vogt–Koyanagi–Harada disease: fundus autofluorescence and spectral domain-optical coherence tomography findings. Retina.. 2010;30:33–41.
45 Tsai JH, Sukavatcharin S, Rao NA. Utility of lumbar puncture in diagnosis of Vogt–Koyanagi–Harada disease. Int Ophthalmol. 2007;27:189–194.
46 Rao NA, Marak GE. Sympathetic ophthalmia simulating Vogt–Koyanagi–Harada’s disease: a clinico-pathologic study of four cases. Jpn J Ophthalmol. 1983;27:506–511.
47 Rubsamen PE, Gass JDM. Vogt–Koyanagi–Harada syndrome. Clinical course, therapy, and long-term visual outcome. Arch Ophthalmol. 1991;109:682–687.
48 Sasamoto Y, Ohno S, Matsuda H. Studies on corticosteroid therapy in Vogt–Koyanagi–Harada disease. Ophthalmologica. 1990;201:162–167.
49 Wang Y, Gaudio PA. Infliximab therapy for 2 patients with Vogt–Koyanagi–Harada syndrome. Ocul Immuno Inflamm. 2008;16:167–171.
50 Read RW, Rechodouni A, Butani N, et al. Complications and prognostic factors in Vogt–Koyanagi–Harada disease. Am J Ophthalmol. 2001;131:599–606.
51 Nowilaty SR, Bouhaimed M. Photodynamic Therapy Study Group. Photodynamic therapy for subfoveal choroidal neovascularisation in Vogt–Koyanagi–Harada disease. Br J Ophthalmol. 2006;90:982–986.
52 Wu L, Evans T, Saravia M, et al. Intravitreal bevacizumab for choroidal neovascularization secondary to Vogt–Koyanagi–Harada syndrome. Jpn J Ophthalmol. 2009;53:57–60.
53 Kuo IC, Rechdouni A, Rao NA, et al. Subretinal fibrosis in a patient with Vogt–Koyanagi–Harada syndrome. Ophthalmology. 2000;107:1721–1728.