Chapter 6 Vision Loss
In children, the complaint of blurred vision or vision loss is often nonspecific and may be difficult to elicit. Although most causes of vision loss in children result from ocular problems, neurologic disorders may have vision loss as a characteristic and early-manifesting feature [Thompson and Kaufman, 2003]. The ability to determine the cause of vision loss frequently aids in the diagnosis of the underlying neurologic disorder, may help determine prognosis, and can be used to monitor treatment efficacy. A thorough understanding of the anatomy of the visual system, the normal stages of visual development in children, and the signs and symptoms of visual dysfunction in children can greatly aid clinicians in evaluating children with vision loss.
Visual Development
Of all the sensory systems, the visual system is perhaps the most immature at birth. It is structurally incapable of processing sensory stimuli to yield maximum visual acuity, and structural changes must occur in the first months of life if normal vision is to be achieved. Postnatal developmental reorganization of the retina takes place during the first several months of life, with intercellular connections forming between the photoreceptors and inner retinal cells [Daw, 1994; Dubowitz et al., 1983, 1986]. Myelination of the optic radiations through the temporal, parietal, and occipital lobes occurs in the first year of life [Barkovich et al., 1992]. The most dramatic structural reorganization occurs in the striate cortex, where cortical cells responsible for the first stages of visual processing require normal focused visual input to develop in the correct orientation and to achieve maximum visual acuity. As described by Hubel and Wiesel [1962], visual deprivation causes abnormal formation of the striate cortical cells and leads to amblyopia. For vision to develop normally, all of the anatomic components of the visual system must be properly formed during development. The sensory neurologic end organ for processing vision is the eye itself. The focusing components of the eye must be developing normally, with clear corneas, crystalline lenses, and optically transparent vitreous media. Properly focused light energy is converted to electrical signals in the photoreceptors that transmit this electrical information through a complex series of interactions through multiple layers of the retina to the ganglion cells, which send projections through the superficial layers of the retina to the optic nerve, finally synapsing in the lateral geniculate nucleus. At the level of the lateral geniculate nucleus, the first levels of cortical processing and organization take place. From the lateral geniculate nucleus, axons project along the optic radiations through the parietal and temporal lobes to synapse in the striate cortex of the occipital lobe. Visual processing occurs in multiple locations in the occipital and temporal lobes in areas labeled V1–V5 [Amedi et al., 2003]. Processing of information from these centers occurs in visual association cortical centers, linking vision with speech, cognition, and more complex, higher cortical functions. A structurally intact neurologic substrate must receive properly focused visual information consistently over the first several years of life if normal, maximal visual acuity is to be achieved [Boothe et al., 1985].
Assessment and Quantification of Visual Acuity
Vision Assessment in Infancy
Although fixation behavior allows the clinician to determine whether the infant can see, it does not quantify visual acuity. Two techniques have been devised for this purpose. They are used clinically and for research purposes, and have allowed estimation of visual acuity in visually immature infants [Droste et al., 1991; Fulton et al., 1981]. The visual-evoked potential is an electrophysiologic test in which visual stimuli are presented to an alert and focused infant, and the cortical response to the visual stimulus is measured in a repeatable and quantifiable fashion [Iinuma et al., 1997] (see Chapter 12). Typically, the infant is placed in front of a computer monitor at a close distance and presented with a visually interesting pattern. The pattern may be an alternating checkerboard, or horizontally or vertically aligned black and white stripes [McCulloch and Taylor, 1992; Sokol et al., 1983]. The size of the stripes or checkerboard is described as cycles per degree or cycles per centimeter, which can be correlated to standard methods of visual or decimal visual acuity. Large targets are initially used to confirm that a cortical signal is recorded by occipitally placed scalp electrodes. The stimulus pattern is then slowly decreased in size until a recordable response can no longer be elicited. The limit of visual acuity is estimated when there is no longer a recordable electrical response to a viewed visual stimulus. The prerequisite for successfully completing this test is a child who is awake and paying attention to the monitor. As a child who looks elsewhere will not give an appropriate response, the examination must be performed quickly by a trained tester who is skilled at attracting the attention of young infants and maintaining their fixation [Good, 2001; Iinuma et al., 1997].
A second test used to quantify visual acuity is the forced-choice preferential looking test (PLT) [Mayer and Fulton, 1985]. Instead of using a cortically recorded electrical response, visual stimuli are presented and the child’s ability to see is determined by the ability to move the eyes toward the visual stimulus. Vertically or horizontally aligned black and white stripes are presented to the child on a test card. One side of the card has the stimulus; the other side of the card has a homogeneous gray background (Figure 6-1A). The infant is directed toward the test card in an apparatus where other visual stimuli are blocked from the infant’s view. When given a choice between a pattern background and a homogeneous background, infants instinctively are interested in the pattern background and make an eye movement or saccade toward the black and white stripes. An observer who is watching through a peephole in the middle of the card records in a masked fashion whether he or she sees the infant make the saccade. When the observer reliably identifies the eye movements, the card is removed, and the procedure repeated with test cards using smaller stimuli to quantify visual acuity. Limitations of this test include the need for an infant who is not irritable, a trained observer, and the appropriate apparatus (see Figure 6-1B) [Lewis and Maurer, 1986].

Vision Assessment in Children
Once a child becomes verbal but is still preliterate, matching tests can be used to quantify visual acuity (Figure 6-2). In the past, the tumbling E has been used; the examiner asks the child to show the direction of the letter E as it is presented in numerous different positions. This test requires some visual spatial integration and visual spatial perception, which some children find difficult, and so may give falsely low estimates of visual acuity. These tests have been replaced by matching games such as HOTV cards and child recognition symbols or Lea symbols that ask a child to match test optotypes that are presented in progressively smaller sizes (see Figure 6-2). These tests have been validated and are consistent with Snellen visual acuity, which is the gold standard for measurement of visual acuity in children and adults. Snellen visual acuity can be recorded in numerous forms using notations such as 20/20, decimal notation, or “logmar” notation, a method of quantification that allows more useful statistical analysis when conducting studies of visual acuity in children and adults.

Assessment of Ocular Motility
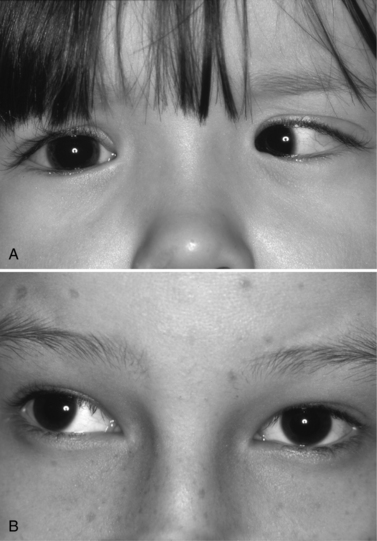
Assessment of ocular motility allows the examiner to assess function of cranial nerves III, IV, and VI (see Chapters 2–4). Ductions and versions can be tested using brightly colored toys and objects. It is important to check the child binocularly first before patching the infant’s eye because patching may be distracting and preclude acquiring useful information. Alignment should be checked using the alternate cover test where fixation is maintained and the visual axis of each eye is occluded alternately. Refixation of the eyes during alternate occlusion may indicate the presence of strabismus such as esotropia, exotropia, or hypertropia (Figure 6-3).
Clinical Features Associated with Vision Loss
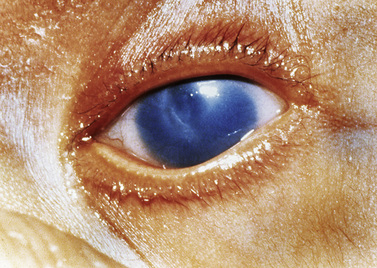
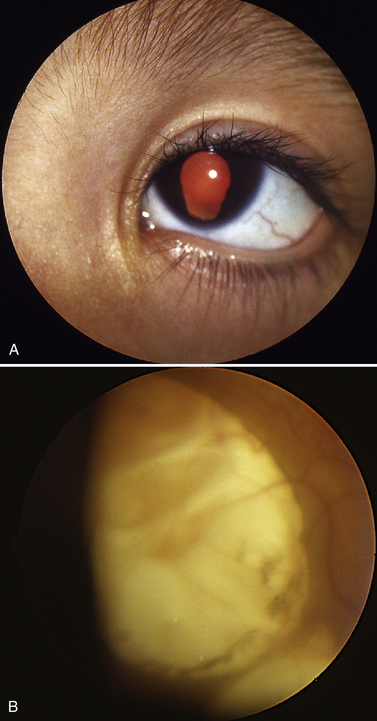
Because symptoms are inconsistently reported, clinicians must be familiar with the physical signs of unilateral vision loss. During the newborn’s physical examination, pediatricians must look for the presence of a red pupillary reflex in each eye. The presence of a white pupil is called leukocoria (i.e., white body), and it is associated with poorly developed vision in one or both affected eyes in the infant (Figure 6-4). The causes of leukocoria are variable, and at a minimum, the condition can cause loss of vision; in more serious situations, leukocoria can be associated with life-threatening conditions such as retinoblastoma. Poor vision in one eye from birth often leads to strabismus that is noticed by the caretaker. Nystagmus is common when there is decreased visual acuity in one or both eyes resulting from a structural anomaly or to a functional deficit preventing visual information from being transmitted from the eye to the cortex [Good, 2001; Hoyt and Fredrick, 1998; Jan and Freeman, 1998]. A vision screening examination failed because of decreased visual acuity in one eye is one of the most common reasons why children are referred to their pediatrician and ophthalmologist. In an infant, bilateral vision loss is manifested by strabismus or nystagmus and visual inattention with poor fixation after 2 months of age. Older children with mild vision loss are usually asymptomatic and their problems are not detected until vision is screened by their pediatrician or family practitioner. Children who have progressive loss of visual acuity exhibit behaviors such as sitting extremely close to the television, being disinterested in distant objects or activities, and having difficulty with tasks that require fine visual acuity.
Associated ocular features of vision loss are important to confirm. Infants with poor vision often develop nystagmus by 2 months of age. They are visually inattentive and do not fix and follow well by this age, and they frequently manifest strabismus or a “wandering eye.” Older children usually do not complain of vision problems but have strabismus. They often close one eye or squint the eye in different lighting conditions, rub their eyes frequently, and occasionally complain of double vision when strabismus occurs suddenly. Children with significant vision loss have disrupted circadian rhythms and disturbed sleep–wake cycles [Leger et al., 1999, 2002; Okawa et al., 1987; Palm et al., 1997]. Other associated neurologic symptoms include headache, nausea, and vomiting. Children with profound vision loss demonstrate stereotyped behaviors such as body rocking, rhythmic head movement, and gazing at their fingers or hands as they are moved rapidly in front of their eyes and face [Fazzi et al., 1999].
Vision Loss in Infants
Clinical Manifestations
Strabismus is another common complaint in children and infants who have poor visual acuity. Strabismus early in life is not rare. Up to 30 percent of infants manifest intermittent deviations in the first 2 months of life, with exotropia occurring more commonly than esotropia [Nixon et al., 1985]. The deviation is usually intermittent and decreases in frequency over the first few months of life. Any child who has a strabismus lasting longer than a few months should be assessed for an underlying ocular anomaly. Constant deviations require close follow-up and early evaluation, particularly when there is constant exodeviation. Whereas infantile esotropia is readily apparent to the parents and quite frightening for them, this condition usually is not associated with an underlying neurologic abnormality. In contrast, constant exotropia should alert the practitioner that an underlying neurologic abnormality exists. If a constant esotropia is not associated with a sixth nerve palsy, it is usually not associated with neurologic problems.
Infants with visual problems also often demonstrate behavioral mannerisms that help suggest the cause of the vision loss [Brown et al., 1997b; Good and Hoyt, 1989]. Children with retinal dysfunction from congenital dysfunction of the photoreceptors or from retinopathy of prematurity often press their eyes to generate some sort of photic stimulation [Sonksen and Dale, 2002]. Children with cortical visual impairment may demonstrate overlooking behavior, an eccentric fixation, to maximize visual function in the visual fields that are least damaged from the underlying cortical injury. Patients with achromatopsia or congenital glaucoma may be quite photosensitive and demonstrate behaviors to shield their eyes from the light to minimize the dysphoric sensation they receive from visual stimulation.
Differential Diagnosis of Vision Loss in Infants
Structural Anomalies
Retinopathy of prematurity
Retinopathy of prematurity remains a common cause of vision impairment in infants, causing blindness in more than 500 infants each year in the United States. Despite intensive efforts by neonatologists and ophthalmologists to screen and treat the early stages of retinopathy, the disease progresses in some children. These patients develop cicatricial changes in the retina leading to vision impairment [Pierce and Mukai, 1994]. Premature infants who do not develop retinopathy of prematurity are still at risk for cerebral vision impairment due to periventricular leukomalacia [Jacobson et al., 2009].
Congenital cataracts
The causes of congenital cataracts vary throughout the world [Merin and Crawford, 1971]. In developed countries, the most common cause is autosomal-dominant cataracts, for which there is a clear history of congenital cataracts affecting multiple generations. These cataracts usually involve both eyes, with characteristic morphologic features. Infectious cataracts are uncommon in developed countries but are a leading cause of blindness in developing nations [Bale and Murph, 1992]. Immunizations are effective in reducing the incidence of infectious cataracts, but other viral illnesses can cause cataracts, as can other malformations. Metabolic causes of cataracts include galactosemia, with which infants present with poor feeding and failure to thrive, and galactokinase deficiency, with which infants may be quite healthy. These two metabolic disorders can be distinguished by performing enzymatic assays of erythrocytes. Early detection and dietary treatment may prevent the neurologic sequelae of these disorders.
Most developed countries have programs to identify children at risk for cataracts, but screening occasionally fails, and the clinician should be aware of the oil droplet morphology of these refractive types of cataracts. Early recognition and treatment can prevent loss of vision and may result in reversal of early cataracts in patients with galactosemia. Often, cataracts are idiopathic, with no previous family history of cataracts. Other neurometabolic conditions, such as mitochondrial disorders, can be associated with cataracts. Frequently, these are not present at birth but evolve in the first few years [Marcel, 1998; Sher et al., 1979]. For this reason, children with a suspected neurometabolic disorder should be followed to determine whether cataracts develop.
Treatment involves prompt recognition, removal of visually significant cataracts, and visual rehabilitation with intraocular lens implantation, extended-wear soft contact lenses, or aphakic spectacles [Basti et al., 1996].
Corneal opacity
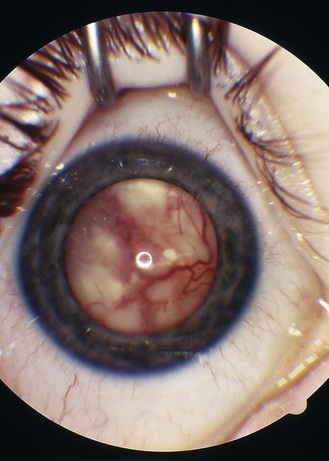
Corneal opacities usually are easily detected by pediatricians or parents at birth when a white spot, or leukoma, is detected within the cornea. These opacities are associated with other structural anomalies, such as microphthalmos, or small eye, and anterior segment dysgenesis, which may also be associated with glaucoma or birth trauma (Figure 6-5). These corneal opacities can cause significant vision impairment and are difficult to correct, because the success rate of corneal transplantation is poor in infants.
Embryologically, the cornea and crystalline lens originate from surface ectoderm, and the iris, uvea, and ciliary body arise from neural crest and mesenchymal cells, with all of the structures forming the anterior segment of the eye. When development is abnormal, the condition is called anterior segment dysgenesis, which can be associated with congenital corneal and lens opacities and with glaucoma, all of which can lead to permanent vision impairment in infants. Patients with anterior segment dysgenesis often have other associated dysmorphic features that indicate the presence of a syndrome or sequence that, if unrecognized, can lead to profound morbidity of the affected child. One example of a form of abnormal anterior segment development is the absence or profound hypoplasia of the iris, a condition known as aniridia. Aniridia is associated with abnormal retinal development leading to nystagmus and vision impairment. It may occur in isolation, or it may occur as a feature of a syndrome characterized by Wilms’ tumor, genitourinary abnormalities, and mental retardation (WAGR) that is associated with deletion on the short arm of chromosome 11 (11p13). Anterior segment anomalies involving the cornea, lens, and iris are known as Peters’ anomaly, and if the child also has mental retardation, the diagnosis of Peters’ syndrome is made. Dysgenesis of the iris and peripheral cornea causing glaucoma is called Rieger’s anomaly, and if associated dental, cardiac, and cerebellar abnormalities are present, the term Rieger’s syndrome is used. Collaboration with a medical geneticist is helpful in identifying other dysmorphic features and determining appropriate genetic testing. In developing countries, leading causes of blindness associated with corneal opacities are vitamin A deficiency and measles [Semba and Bloem, 2004].
Ocular coloboma
Coloboma, or absence of tissue, can affect vision profoundly. If the coloboma involves the iris but not deeper tissues, visual acuity can be normal (Figure 6-6). However, when the coloboma involves the central retina, macula, or the optic nerve, vision can be severely impaired [Apple et al., 1982]. Children with bilateral colobomata are at high risk for underlying neurologic problems [Chestler and France, 1988; Russell-Eggitt et al., 1990]. Any child with bilateral colobomata should be evaluated for chromosomal trisomies and the CHARGE association (i.e., coloboma, heart defects, atresia choanae, retardation of growth and development, genitourinary problems, and ear anomalies). Aicardi’s syndrome should be considered in any female with a seizure disorder and ocular colobomata (Figure 6-7). Patients with Aicardi’s syndrome have ectopic gray matter and other CNS malformations; the disorder is X-linked and lethal for males [Carney et al., 1993; Gloor et al., 1989]. Coloboma of the optic nerve can also be associated with underlying renal disease, known as the papillorenal syndrome; this diagnosis is made by genetic testing for mutations in the PAX6 gene [Alur et al., 2010].
Congenital glaucoma
Congenital glaucoma occurs when the aqueous drainage pathways of the iris form abnormally, leading to increasing intraocular pressure, corneal edema, and optic nerve injury. Most cases of congenital glaucoma are not associated with specific syndromes or underlying neurologic disorders. The exception is Sturge–Weber syndrome, in which vascular anomalies involving the eye lead to increased episcleral venous pressure and glaucoma (see Chapter 40). These patients typically have cerebrovascular malformations, epilepsy, and contralateral hemiplegia.
Retinal dysplasia
Structural anomalies of the retina not associated with retinopathy of prematurity can lead to significant vision impairment. These forms of retinal dysplasia are frequently associated with a variety of neurologic malformations. An example is Walker–Warburg syndrome, in which congenital retinal dysplasia is associated with cerebral structural abnormalities such as hydrocephalus, agyria, and occasionally encephalocele (see Chapters 22–27). Muscle–eye–brain disease is another example of neurologic and retinal dysplasia resulting from abnormal glial development due to defective glycosylation of α-dystroglycan. This results in profound CNS involvement and significant vision loss [Shenoy et al., 2010]. Norrie’s disease is an X-linked condition in which retinal dysplasia is associated with mental retardation and deafness. In this condition, a genetic defect in the gene product norrin leads to abnormal endothelial cell migration and proliferation [Mintz-Hittner et al., 1996].
Optic nerve hypoplasia
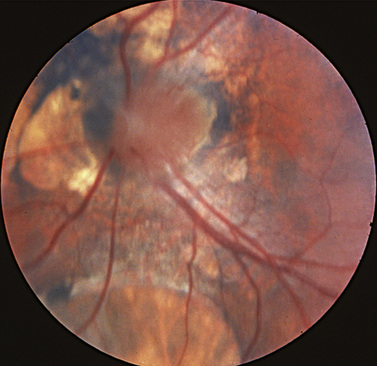
Failure of the optic nerves to form properly leads to a small dysfunctional optic nerve [Siatkowski et al., 1997]. In optic nerve hypoplasia, the nerve is small and its morphology abnormal (Figure 6-8). Frequently, there is a double-ring sign; the scleral canal of the optic nerve is present, but the optic nerve tissues comprise only a small portion of the canal, leading to two distinct rings. Children with optic nerve hypoplasia frequently present with nystagmus. Children with optic nerve hypoplasia may have de Morsier’s syndrome, or septo-optic dysplasia, characterized by midline structural defects of the CNS (e.g., absence of the septum pellucidum, agenesis of the corpus callosum) in addition to neuroendocrine dysfunction (see Chapter 97). All children with optic nerve hypoplasia should undergo neuroimaging, with particular attention to the septum pellucidum, corpus callosum, and pituitary body [Brodsky et al., 1990]. The presence of an ectopic bright spot places the child at higher risk for neuroendocrine dysfunction. Patients suspected of having septo-optic dysplasia should have their growth and endocrine status monitored closely [Siatkowski et al., 1997; Skarf and Hoyt, 1984]. The absence of cerebral developmental anomalies does not mean that endocrine abnormalities will not occur, and children require continued endocrinologic follow-up [Garcia-Filion, 2008a & b]. A child may have normal endocrine function early in life and later develop panhypopituitarism. There have been numerous cases of sudden death associated with septo-optic dysplasia, in which affected children develop a febrile illness that leads to rapid decompensation and death due to adrenal insufficiency [Brodsky et al., 1997]. This complication may occur in children who have or have not received corticosteroid therapy. Parents should be advised concerning these potential risks and treat all illnesses seriously.
The cause of optic nerve hypoplasia is unknown, although there have been numerous case reports of optic nerve hypoplasia occurring in infants exposed prenatally to quinine, LSD, alcohol, and antiepileptic drugs [Lambert et al., 1987]. The condition is usually seen in young mothers and first-born children. Some infants with optic nerve hypoplasia develop moderate visual function, and the clinician should be careful in prognosticating long-term visual function based on the appearance and size of the optic nerve.
Ocular or oculocutaneous albinism
Normal pigment formation is essential for normal ocular development and normal function of the retinal pigment epithelium [Brodsky et al., 1993]. Albinism may involve the eye and skin (oculocutaneous albinism), or only the eye (ocular albinism); both forms are associated with decreased visual acuity. Patients with oculocutaneous albinism are more severely affected, with visual acuity in the 20/200 range, whereas those with ocular albinism have acuity in the range of 20/60 to 20/80. Both conditions manifest with nystagmus early in life. The diagnosis of ocular albinism is made by documenting transillumination defects in the iris during slit-lamp examination. This test can be performed in infants, and it obviates the need for further evaluation. Patients with oculocutaneous albinism should be assessed for systemic disease such as Chediak–Higashi syndrome, which is associated with white blood cell dysfunction and recurrent infections, and Hermansky–Pudlak syndrome, which increases the risk for rheologic abnormalities and clotting disorders [Carden et al., 1998].
Leber’s congenital amaurosis
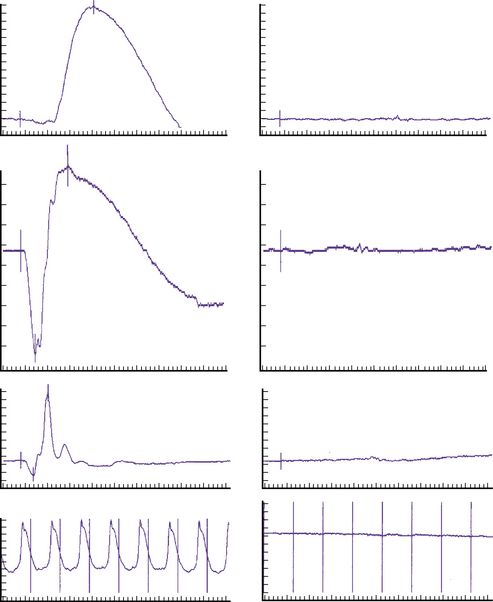
Leber’s congenital amaurosis is a disorder of the photoreceptors and the retinal pigment epithelium in which photoreceptor function is extinguished [Babel et al., 1989; Brecelj and Steirn-Kranjc, 1999; Fulton et al., 1981]. Infants present with large-amplitude, slow-frequency, roving nystagmus. They frequently begin to press on their eyes by 2–3 months of age [Lambert et al., 1997; Sullivan et al., 1994], and they may have a completely normal ophthalmoscopic examination with normal-appearing optic nerve and retina. The diagnosis is established by electroretinography [Weleber, 2002]. In this test, the electrical amplitude of the retina is measured using a contact lens placed on the eye that is stimulated by bright lights to elicit a cone response and dim lights to stimulate a rod response (Figure 6-9). In congenital amaurosis, both rod and cone responses are extinguished [al-Salem, 1997; Heher et al., 1992].
Vision Loss Due to Cortical Visual Impairment
In developed countries, decreased vision caused by cortical or cerebral visual impairment is the leading cause of vision impairment in infants [Blohme and Tornquist, 2000;Gilbert and Awan, 2003; Gilbert et al., 1999; Huo et al., 1999; Mervis et al., 2000]. Damage to the visually immature brain impedes normal visual development and leads to lifelong subnormal vision [Brodsky et al., 2002; Hoyt, 2003]. The most common causes of cortical visual impairment in developed countries are neonatal encephalopathies [Casteels et al., 1997; Flanagan et al., 2003; Lanzi et al., 1998] (see Chapter 17). The second most common cause of cortical visual impairment is periventricular leukomalacia [Afshari et al., 2001; Hoyt and Fredrick, 1998; Huo et al., 1999] (see Chapter 19). Injury and ischemia lead to damage in the periventricular white matter and frequently affect visual development. The location of periventricular leukomalacia determines the form of cortical visual impairment. Lesions involving the anterior visual pathways (e.g., anterior corpus callosum) frequently result in oculomotor anomalies such as apraxia, saccadic paresis, and strabismus. These infants often have difficulty generating saccades and have poor visual fixation. Visual acuity in affected infants may be good but not useful because of the patient’s inability to move the eyes. Damage to the posterior visual pathways (e.g., parieto-occipital white matter, striate cortex) is usually associated with more severe vision loss, poor acuity, and a poor prognosis for recovery [Dutton and Jacobson, 2001; Hoyt and Fredrick, 1998; Jacobson et al., 1998; Sonksen and Dale, 2002].
Clinical features of cortical visual impairment are those of an infant who fails to develop visual fixation behavior after 2–3 months of age [Afshari et al., 2001]. These children are often neurologically impaired and have delayed motor milestones and abnormal findings for the neurologic examination [Levtzion-Korach et al., 2000; Whiting et al., 1985]. Magnetic resonance imaging (MRI) in affected children usually provides evidence of leukoencephalopathy [Casteels et al., 1997]. Infants with cortical visual impairment do not fix or follow, and appear to be visually disinterested in the environment [Dutton and Jacobson, 2001; Good et al., 1994]. Often, the first stimulus of interest to the infant is a bright light or shiny object. Patients often keep their eyes elevated, looking upward to see the light in an overhanging light fixture or a window near the infant’s crib. Infants commonly demonstrate off and on visual behavior, during which there are moments of what appears to be normal visual fixation interspersed with longer periods of visual inattention. Infants respond well to high-contrast targets such as black and white toys and large pattern images. Children with profound cortical visual impairment early in infancy can demonstrate a progressive increase in visual function over several years and may become quite visually proficient [Marcel, 1998]. It is important to refer children with cortical visual impairment for low-vision services that can provide sensory stimulation exercises, which improve the visual performance of infants and provide emotional, social, and educational support for parents [Amedi et al., 2003; Ashmead et al., 1998; Levtzion-Korach et al., 2000; Zihl, 1980].
Structural Cerebral Anomalies Causing Cortical Visual Impairment
Hydrocephalus
Ophthalmic signs of hydrocephalus and increased intracranial pressure include the setting sun sign, which describes the infant’s gaze held in a downward fixed position, with the eyelids retracted and the infant unable to elevate the eyes willfully. Because the cranial sutures are not closed, papilledema usually does not occur early in infancy. After the cranial vault is closed, papilledema can occur as with any child with increased intracranial pressure (see Chapter 77). Children with hydrocephalus require surgical relief of their obstruction by ventriculostomy or ventriculoperitoneal shunt placement. An ophthalmologic examination should be obtained to document the presence of normal optic nerves or the absence of optic atrophy. Many older children with chronic hydrocephalus present with optic atrophy that precludes future useful information about the presence of intracranial pressure because atrophic optic nerves do not swell and cannot reflect increased intracranial pressure. Other ocular signs of increased intracranial pressure include cranial nerve VI paresis, often manifesting as new-onset esotropia. Some children with hydrocephalus are at risk for superior oblique muscle dysfunction (cranial nerve IV) leading to strabismus, in which the esotropia is worse in upward gaze.
Structural brain anomalies
Children with schizencephaly frequently have decreased visual acuity due to damage to the optic radiations and pathways. Contralateral hemianopias and epilepsy are common clinical manifestations. Visual function in children with large schizencephalic clefts, as well as those with porencephaly or hydrocephalus, may improve despite a very abnormal appearance on neuroimaging once the patient is shunted and the cortex re-expands [Summers and MacDonald, 1990].
There are numerous congenital disorders associated with brain malformations, such as the Walker–Warburg syndrome, Dandy–Walker syndrome, and muscle–eye–brain disease, in which structural brain anomalies (see Chapters 22–27) are accompanied by decreased visual function due to striate cortex involvement or associated ocular anomalies such as retinal dysplasia [Liu et al., 2000; Yamamoto et al., 2004].
Vision loss due to epilepsy
Children with epilepsy frequently have poor visual function. When the seizure disorder results from a structural abnormality, there is often concomitant strabismus, nystagmus, and developmental delay. Patients with seizures may develop visual auras before the seizure (see Chapter 54), and functional blindness during the postictal period [Bauer et al., 1991]. Children with frequent seizures throughout the day often have poor visual fixation development. Abnormal electrical activity can interfere with the development of useful vision [Trevathan et al., 1997]. The use of antiepileptic drugs may sedate the child to the point where general development is delayed, and this can affect development of visual acuity [Remler et al., 1990]. Optimizing antiepileptic drug therapy should be encouraged, because visual acuity can markedly improve when seizures are well controlled [Shahar and Barak, 2003]. Certain antiepileptic drugs such as vigabatrin may be associated with specific retinal or ophthalmic abnormalities (see Chapter 59).
Delayed visual maturation
Occasionally, a healthy infant older than 2 months is referred because of failure of development of visual fixation behaviors. Results of ophthalmologic and neurologic examinations may be completely normal. Such infants may have the condition of delayed visual maturation, a diagnosis of exclusion in which visual development is delayed but eventually becomes normal. Often, the onset of visual fixation is dramatic and usually occurs by 6 months of age. Three types of delayed visual maturation have been described [Fielder et al., 1991]. In type I, the child is healthy and has normal vision by 1 year of age. In type II, the child has associated neurologic or systemic disease, and in type III, ocular anomalies occur. Most ophthalmologists reserve the diagnosis of delayed visual maturation for children who have the type I form [Hoyt et al., 1983].
Diagnostic Evaluation of Infants with Poor Vision
An infant who fails to develop visual fixation should first be referred to an ophthalmologist [Kivlin et al., 1990]. Examination by an ophthalmologist most often uncovers the causes of decreased vision, which may be due to any of the congenital structural anomalies described previously. If examination findings are completely normal, the next considerations are a neurologic examination and possibly neuroimaging. Certain neurologic signs and symptoms may suggest a specific neurologic diagnosis [Backhouse et al., 1999]. MRI is preferred to computed tomography (CT) for evaluating structural anomalies. Neurometabolic testing should be performed to exclude reversible and potentially treatable inborn errors of metabolism involving carbohydrate or urea cycle metabolism or mitochondrial disorders [Cooper et al., 2002; Goebel, 1995; Hansen et al., 1979; Santavuori et al., 1993]. These conditions are described in detail in various chapters in this textbook.
In an infant with poor visual function but normal neurologic examination results and normal neuroimaging findings, electroretinography may be used to determine the presence of photoreceptor dysfunction. Such patients usually demonstrate signs of retinal disease, including nystagmus and eye-pressing behavior. An electroretinogram is obtained by placement of a contact lens attached to electrodes on the surface of the cornea. Lights are used to stimulate the retina and the electrical responses are recorded. Depending on the light stimulus, determination can be made whether there is rod-related (affecting night vision) or cone-related dysfunction that affects central vision or color, or both (see Figure 6-9).
Vision Loss in Children
Symptoms and Signs of Vision Loss
Children with vision loss frequently do not complain about vision loss unless it is bilateral. Children often do not use the term blurred vision, but may say “I can’t see,” “things are fuzzy,” or “things are double.” Signs of vision loss are much more helpful. The first sign of vision loss is squinting behavior. When the child squints and closes the eyelids, the pinhole effect helps focus out-of-focus light. This behavior is common in children with refractive errors. Children also rub their eyes in attempt to clear their vision. Those with acute bilateral loss of vision will sit close to the television or become disinterested in activities occurring at a distance. They may hold objects very close to their faces to see them clearly. Children with new-onset strabismus associated with vision loss frequently close one eye to avoid diplopia. They may be sensitive to sunlight and shield their eyes because bright light may markedly decrease their visual acuity, especially when there is associated retinal dysfunction. Children may also tilt their heads when vision is reduced in one eye [Nucci and Rosenbaum, 2002].
Differential Diagnosis of Vision Loss in Children
Amblyopia
Amblyopia is a functional and structural condition wherein an abnormal visual stimulus leads to abnormal development of cortical visual processing cells with smaller cell size and abnormal intercellular connections [Hubel and Wiesel, 1962]. These structural changes are reversible if detected early in life and treated with occlusion therapy. By removing the visual impairment, straightening the eye, or focusing the vision through spectacles, and then patching the unaffected eye to stimulate the immature visual system, amblyopia can be reversed. Whether or not vision can be restored depends on the age of detection. Success in reversing amblyopia lies in the underlying causative factor and the amount of compliance with occlusion therapy. Visual deprivation amblyopia is the most profound and must be reversed within the first few months of age, whereas anisometropic amblyopia can be reversed, even in the second decade of life, with aggressive patching therapy. As a rule, most children with amblyopia should be detected and treated by the age of 6 months for the best visual prognosis.
Ocular Anomalies Causing Vision Loss
Anomalies of the retina
Degenerative diseases of the retina can cause gradual loss of visual acuity and may be extremely difficult to diagnose in young children (Table 6-1). Unlike congenital retinal dysfunction that leads to large-amplitude nystagmus, marked vision impairment, and clear-cut electroretinographic findings, retinal degeneration in older children may be insidious in onset, unaccompanied by significant visual symptoms and with equivocal findings on electroretinography.
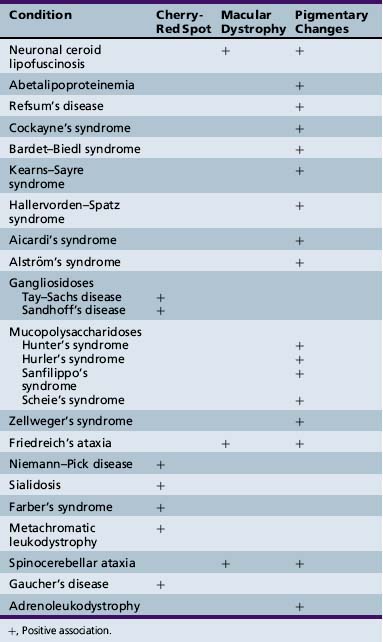
One of the most common causes of visual dysfunction due to retinal dysfunction is Stargardt’s disease [Weleber et al., 1984; Weleber, 2002], also known as fundus flavimaculatus. This disease is a degenerative condition of the retinal pigment epithelium leading to photoreceptor dysfunction [Szlyk et al., 1998]. There is a defect in flipase (ABCA4), with different mutations causing different presentations [Weber, 2003]. Children present with slowly decreasing visual acuity. There are characteristic changes on funduscopic examination, and diagnostic tests such as fluorescein angiography, visual-field testing, and electroretinography can help confirm the diagnosis (Figure 6-10). Mutations of the gene involved in phototransduction have been identified and characterized in some families with this condition [Zack et al., 1999].
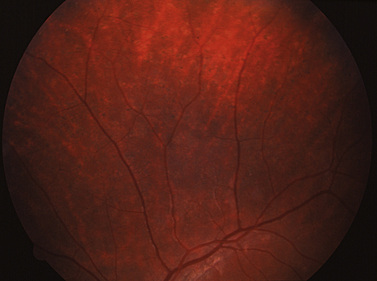
Retinitis pigmentosa
In retinitis pigmentosa, abnormalities of the retinal pigment epithelium can lead to photoreceptor dysfunction and death. Retinitis pigmentosa typically affects rods before affecting cones [Foxman et al., 1985; Heidemann and Beck, 1987]. This process leads to initial symptoms of night blindness and constriction of the peripheral visual field, eventually affecting cones and central visual acuity. A typical “bone spicule” pattern of the retinal pigment epithelium is diagnostic. Electrodiagnostic tests, such as electroretinography, may be helpful. Retinitis pigmentosa has been characterized as involving rods or cones, or both. All modes of inheritance patterns have been described, and there may be variations of phenotypic expression within families. Although some investigators feel that vitamin supplementation may delay disease progression, there have been no studies proving any benefit with any treatment intervention for retinitis pigmentosa in children [Szlyk et al., 1998].
Neurometabolic retinal dysfunction
Several neurometabolic disorders have been associated with retinal dysfunction and secondary vision loss [Collins et al., 1990]. In neuronal ceroid lipofuscinosis, abnormal accumulation of neurotoxic products within the retina leads to cell dysfunction and death [Backhouse et al., 1999; Bohra et al., 2000; Brown et al., 1993; Fulton et al., 1985; Spalton et al., 1980] (see Chapter 41). There are multiple forms of neuronal ceroid lipofuscinosis occurring in different age groups. Characteristic of all forms of neuronal ceroid lipofuscinosis is the development of decreased visual acuity resulting from poor retinal function [Bohra et al., 2000]. Degeneration of the ganglion cell layer results in a typical funduscopic appearance, and ophthalmoscopic examination coupled with electrophysiologic testing can help diagnose these children who present with seizures and progressive dementia [Cotlier, 1971]. Retinal dysfunction occurs in inherited mitochondrial cytopathies such as Kearns–Sayre syndrome, in which a pigmentary retinopathy is associated with decreased visual acuity, external ophthalmoplegia, and cardiac conduction defects. Retinitis is an unusual cause of vision loss in children, but it can occur in patients with cytomegalovirus infection, herpes simplex or zoster infection, cat-scratch disease, and Lyme disease [Clarke et al., 2001; Dreyer et al., 1984; Purdy et al., 2003]. A history of immune suppression or recent systemic illness also can suggest the appropriate diagnosis.
Optic Nerve Disorders
Papilledema
The presence of increased intracranial pressure leads to edema of the optic nerve (i.e., papilledema). The borders of the optic nerve are indistinct and the vessels are swollen; the nerve itself is elevated, with surrounding hemorrhage or exudates (Figure 6-11). Papilledema in children can be caused by obstruction of the ventricular system, craniosynostosis [Fishman et al., 1971; Stavrou et al., 1997], or communicating hydrocephalus. Whereas early papilledema in adults rarely causes visual symptoms, papilledema can be chronic in children, with slow onset and relatively late discovery of disease, and decreased visual acuity can be a presenting complaint. Usually, this does not occur unless the increase in intracranial pressure is rapid and significant in onset or has been of long duration, leading to chronic axonal compression and edema formation in the retina and causing decreased visual acuity or cell death and incipient optic atrophy. For papilledema in children, mandatory neuroimaging should be followed by lumbar puncture [Brodsky and Glasier, 1995]. Papilledema also occurs in children with malignant hypertension, leading to permanent vision loss if not corrected [Browning et al., 2001]. Pseudotumor cerebri or benign intracranial hypertension is not an infrequent cause of papilledema in overweight children [Baker et al., 1985, 1989]. The diagnosis is made after neuroimaging excludes an obstructive lesion and lumbar puncture reveals increased intracranial pressure and no abnormal cytology. Occasionally, pseudotumor cerebri may be associated with sinovenous thrombosis that can be detected with magnetic resonance venography (MRV). Because many drugs can cause benign intracranial hypertension, the treatment is withdrawal of inciting agents such as tetracycline and its derivatives, and vitamin A analogs. Improvement is also seen with substantial weight loss. Visual dysfunction associated with possible secondary ischemic optic neuropathic changes should prompt consideration for optic nerve sheath fenestration or lumbar or ventriculoperitoneal shunting to relieve pressure to prevent permanent loss of visual acuity [Brodsky and Rettele, 1998].
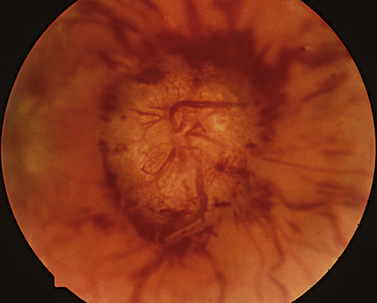
Pseudopapilledema
In pseudopapilledema, the optic nerve appears to be elevated, but there is a lack of edema surrounding the nerve, which is seen with true papilledema (Figure 6-12). Pseudopapilledema can be seen with optic nerve head drusen. Optic nerve drusen are extracellular deposits of material within the nerve fiber layer that cause a lumpy elevation of the optic nerve. Later in childhood, the material develops a glistening calcific appearance and can be easily detected by autofluorescence angiography or by ultrasonography [Bec et al., 1984; Friedman et al., 1977]. In earlier stages of the disease, the bright signal intensity and reflective characteristics are not as evident, making the diagnosis one of exclusion considered only after lumbar puncture and neuroimaging have eliminated more dangerous conditions [Savage et al., 1985]. Pseudopapilledema can also be seen in hyperopia, in which the nerve head may look elevated. The absence of hemorrhages and of blurred disc margins suggests that pseudopapilledema is more likely to be present. Serial examinations and documentation by photography can help differentiate true papilledema from pseudopapilledema [Mustonen, 1983].
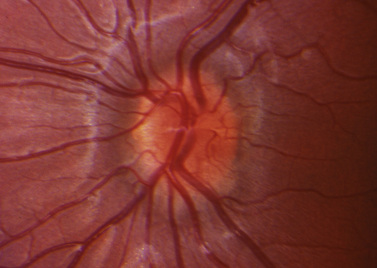
Optic neuritis
Whereas adults with optic neuritis usually present with unilateral disease, bilateral presentation is more common in children [Kennedy and Carroll, 1960]. Children rarely complain of decreased vision in one eye. This phenomenon of failure to detect the unilateral disease may lead to the higher reported incidence of bilaterality and may be artifactual. Optic neuritis in children frequently follows viral illnesses; it is most commonly associated with inflammation and swelling of the optic nerve head, and may be accompanied by vasculitis (Figure 6-13) [Bar et al., 1990; Chrousos et al., 1990; Winterkorn, 1990]. In children, there is usually edema of the optic nerve associated with loss of vision and an afferent pupillary defect, whereas in adults, most cases of optic neuropathy are retrobulbar with no visible changes on ophthalmoscopy [Sato et al., 1998]. Any demyelinating episode may be the first sign of multiple sclerosis, and the risk of a pediatric patient eventually developing multiple sclerosis varies from 7 to 56 percent [Bye et al., 1985]. Children who have white matter changes on neuroimaging have a higher risk of developing multiple sclerosis and require close observation so that use of interferons may be considered [Bonhomme et al., 2009]. A particular form of optic neuritis that occurs more frequently in children than adults is acute disseminated encephalomyelitis (ADEM), as described in Chapter 72 [Brodsky and Beck, 1994]. This can lead to peripheral neurologic changes, central visual loss, and decreased visual acuity. ADEM is usually responsive to corticosteroid or interferon treatment, but relapses can occur, and patients may be followed with MRI and careful observation of vision, assessment of color vision, and examination of the pupils [Sato et al., 1998]. Neuromyelitis optica, or Devic’s disease, is a rare, but debilitating form of optic neuritis that occurs in association with transverse myelitis; diagnosis is made by serologic detection of aquaporin-4 autoimmunity (NMO-IgG), a test which should be performed on all children with optic neuritis [McKeon et al., 2008].
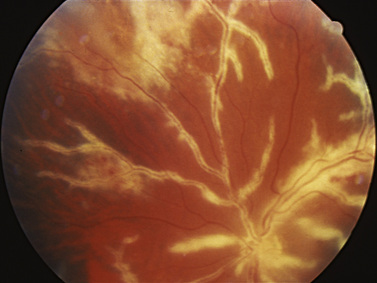
Optic atrophy
In children, optic atrophy is often not diagnosed until both eyes are affected (Table 6-2). The most worrisome cause of optic atrophy is compressive disease of the optic nerve [Liu, 2001; Varma et al., 2003]. Atrophy may occur from increased intracranial pressure due to obstructive intracranial lesions, or from compression of the optic nerve from orbital processes or intrinsic tumors of the optic nerve (e.g., optic nerve gliomas) [Belgaumi et al., 1997; Fletcher et al., 1986]. Children with optic atrophy should undergo neuroimaging to establish a specific diagnosis. Neuroimaging will detect sellar lesions, such as pituitary adenoma or histiocytosis, suprasellar masses such as geminoma, and infiltrative hypothalamic/chiasmal gliomas. It is most difficult to formulate therapeutic approaches for this last condition, as treatment – whether by surgery, radiation, or chemotherapy – is unpredictable in outcome and may be associated with significant morbidity [Opocher et al., 2006]. Children with neurofibromatosis type 1 and optic gliomas require close ophthalmic monitoring, as these tumors may demonstrate spontaneous regression, and treatment should only be considered if there is documented progressive loss of visual acuity or peripheral visual field.
Table 6-2 Neurologic Disease Associated with Vision Loss and Optic Atrophy
| Category | Disease or Syndrome |
|---|---|
| Developmental anomalies | Optic nerve hypoplasia |
| Septo-optic dysplasia or de Morsier’s syndrome | |
| Prenatal or perinatal ischemia | |
| Infarction | |
| Periventricular leukomalacia | |
| White matter disease of prematurity | |
| Cerebral structural anomalies | |
| Hydrocephalus | |
| Schizencephaly | |
| Walker–Warburg syndrome | |
| Encephalocele | |
| Holoprosencephaly | |
| Degenerative disease | Leukodystrophies |
| Adrenoleukodystrophy | |
| Krabbe’s leukodystrophy | |
| Pelizaeus–Merzbacher disease | |
| Alexander’s disease | |
| Canavan’s disease | |
| Leigh’s syndrome | |
| Lysosomal disorders | |
| Gangliosidoses GM1 and GM2 | |
| Mucopolysaccharidoses | |
| Niemann–Pick disease | |
| Ataxias | |
| Friedreich’s ataxia | |
| Charcot–Marie–Tooth disease | |
| Miscellaneous conditions | |
| Neuronal ceroid lipofuscinosis | |
| Wolfram’s syndrome | |
| Zellweger’s syndrome | |
| Inflammatory conditions | Infectious diseases |
| Collagen–vascular disease | |
| Autoimmune disorders | |
| Ischemic conditions | Renal disease |
| Sickle cell disease | |
| Moyamoya disease | |
| Trauma | Direct |
| Indirect | |
| Demyelinating conditions | Optic neuritis |
| Acute disseminated encephalomyelopathy | |
| Multiple sclerosis | |
| Leigh’s disease | |
| Compressive conditions | Pituitary tumor |
| Hypothalamic tumor | |
| Craniopharyngioma | |
| Optic nerve glioma | |
| Optic nerve meningioma | |
| Rhabdomyosarcoma | |
| Papilledema | |
| Idiopathic intracranial hypertension | |
| Craniosynostosis or craniofacial dysostosis | |
| Hereditary conditions | Kjer’s optic atrophy |
| Leber’s congenital amaurosis | |
| Mitochondrial cytopathy | |
| Toxicities | Lead |
| Copper | |
| Streptomycin | |
| Hydroxyquinolones | |
| Methanol | |
| Ethambutol |
When neuroimaging fails to demonstrate compressive lesions, hereditary optic atrophy should be considered [Brown,1990; Eliott et al., 1993; Vinkler et al., 2003]. Kjer’s optic atrophy is transmitted in an autosomal-dominant pattern; it presents with slow onset of visual acuity loss, first in a 20/80 to 20/100 range and then stabilizing in the 20/400 range. Kjer’s optic atrophy has been mapped to OPA1 on chromosome 3 [Egan and Kerrison, 2003]. Wolfram’s disease includes optic atrophy as one of its clinical features (i.e., diabetes insipidus, diabetes mellitus, optic atrophy deafness [DIDMOAD]), and this autosomal-recessive condition maps to WFS1 on chromosome 4p (Egan and Kerrison, 2003). Examination of parents, siblings, and other relatives can be helpful in the diagnosis of hereditary optic neuropathies [Brown et al., 1997a; Kline and Glasier, 1979]. Neurometabolic diseases may cause optic atrophy and are usually diagnosed by the constellation of neurologic and physical findings associated with the disease [Huber, 1994]. Ischemic optic neuropathy has been described in children with underlying renal insufficiency in which the patient develops sudden changes in blood pressure because of illness or blood loss [Bates et al., 1999; Browning et al., 2001; Thomas et al., 2003].
When children sustain damage to the CNS prenatally or perinatally, damage to the occipital cortex and radiations can lead to death of the ganglion cells by trans-synaptic degeneration across the lateral geniculate nucleus [Miller and Newman, 1981]. This damage is most commonly seen in preterm infants with periventricular leukomalacia, in which optic atrophy may be accompanied by extensive optic cupping due to death of the ganglion cell. Neuroimaging demonstrates periventricular leukomalacia or other white matter injury, and it can help confirm the diagnosis when suspected because of premature birth or birth accompanied by hypoxic-ischemic encephalopathy [Uggetti et al., 1997].
Toxic optic neuropathies can occur with exposure or ingestion of heavy metals, disorders of mineral metabolism, and poisoning from methanol or medications such as ethambutol (see Chapter 100). Another group of disorders that can lead to optic atrophy are the mitochondrial encephalopathies (see Chapter 37). Patients with maternally transmitted Leber’s hereditary optic neuropathy present with loss of visual acuity in the second decade of life [Acaroglu et al., 2001; Newman, 1993]. This is associated with characteristic unilateral changes in the optic nerve head and telangiectatic changes in the optic nerve head vessels. Bilateral involvement frequently develops within months. There have been numerous genetic polymorphisms described in patients with Leber’s optic atrophy, and molecular DNA testing is available [Johns et al., 1992, 1993]. Included in this group of disorders is Kearns–Sayre syndrome, mitochondrial encephalomyopathy with lactic acidosis and strokelike syndrome (MELAS) [Rummelt et al., 1993], and myoclonic epilepsy with ragged red fiber disease (MERRF) (see Chapter 37).
Cerebral Vision Impairment
Whereas cerebral vision impairment in infants results from ischemic encephalopathy or white matter disease, impairment in older children usually is traumatic in nature [Billingsley et al., 2002]. An ischemic episode such as a near-drowning, meningitis, or stroke can also cause cortical vision impairment. Toxic cortical blindness can be caused by vincristine, cyclosporine, and tacrolimus. Therapy should be discontinued in children receiving these agents to reverse the symptoms [Jarosz et al., 1997; Schouten et al., 2003]. Visual acuity can be profoundly affected and show slow, progressive improvement over 1–2 years. The diagnosis is made by a combination of history, neuroimaging, and normal ocular examination findings.
Nystagmus in Infancy
Like the visual system, the ocular motor system is immature at birth. Abnormal eye movement such as ocular flutter, bobbing, saccadic intrusions, or saccadic paresis may be transient in the first few weeks of life, but they usually resolve completely. Nystagmus, or oscillation of the eyes in a stereotypic fashion, is rarely seen at birth but usually develops by 2 months of age. There are three primary causes of nystagmus in infancy, each having different visual consequences and health considerations [Lambert et al., 1989].
Nystagmus Caused by Visual Deprivation
Any condition that causes failure of a formed visual image to be perceived by the striate cortex can lead to bilateral nystagmus (Table 6-3). The most common causes are bilateral congenital cataracts, bilateral optic nerve hypoplasia, aniridia, albinism, and retinal anomalies. When the visual acuity is less than 20/200, nystagmus can be large in amplitude and slow in frequency at birth, but it usually becomes more rapid with a lower amplitude later in life. Depending on interventions to clear the visual axis and improve visual acuity, nystagmus may decrease, and visual acuity may be quite good if the condition is detected and treated early [Jan et al., 1990]. If the structural anomaly is in the optic nerve or in the retina, surgical or optical interventions are usually not helpful in significantly improving visual acuity. The presence of an ocular anomaly usually obviates the need for additional neuroimaging and assessment.
Table 6-3 Causes of Bilateral Infantile Vision Impairment Manifesting with Nystagmus by Age 2 Months
| Category | Cause of Impairment |
|---|---|
| Disorders of corneal clarity | Developmental anomalies |
| Peters’ syndrome | |
| Rieger’s syndrome | |
| Sclerocornea | |
| Congenital glaucoma | |
| Forceps birth trauma | |
| Crystalline lens opacity | Congenital cataract |
| Uveal anomalies | Aniridia |
| Oculocutaneous or ocular albinism | |
| Vitreous anomalies | Vitreous hemorrhage |
| Persistent hyperplastic primary vitreous | |
| Retinal anomalies | Leber’s congenital amaurosis |
| Achromatopsia or monochromatopsia | |
| Retinopathy of prematurity | |
| Retinal dysplasia | |
| Chorioretinal scarring | |
| Congenital toxoplasmosis | |
| Chorioretinal coloboma | |
| Optic nerve anomalies | Optic nerve hypoplasia |
| Optic nerve atrophy | |
| Optic nerve coloboma | |
| Morning glory disc |
Congenital Motor Nystagmus
Although congenital motor nystagmus is presumed to arise from a neurologic abnormality of fixation, it is not known whether the molecular defect is located in the eye or in the brain. It may be inherited as an autosomal-dominant, autosomal-recessive, or X-linked disease. Congenital motor nystagmus has been linked to Xq26–q27 [Kerrison et al., 1999].
Transient Episodic Vision Loss in Children
To hear a child complain about occasional abnormal vision is not a rare phenomenon. School-age children frequently complain about blurred vision after prolonged distance or near-visual tasks such as reading, taking tests, or taking notes from the white board. These symptoms are usually brief and resolve after a short period of rest. Complaints of more profound transient and episodic visual loss or blindness should make the clinician consider three causes: intracranial hypertension, migraine, and functional vision loss. Increased intracranial pressure can cause transient obscuration of vision, especially when children change position from supine to standing. They may also complain of positive scotoma and “black-out spells.” Migraines in children do not usually manifest as classic migraines but frequently have an atypical presentation with vision loss, frequent episodes of abdominal pain, and absence of headache [Barlow, 1994; Ehyai and Fenichel, 1978]. Obtaining a family history of migraines and excluding intracranial pathology are important in establishing this frequently missed diagnosis [Hockaday, 1979; Tomsak and Jergens, 1987]. A child may complain about decreased visual acuity but have normal vision without ocular pathology. This form of functional visual loss has a variety of causes and may be difficult to diagnose and detect [Schwartz and Vahgei, 1998]. Children most often complain about bilateral vision loss and initially report visual acuity in the 20/400 range. Their symptoms may be exaggerated, but the children rarely bump into objects when entering a room despite reports of markedly diminished visual acuity. This is a diagnosis of exclusion, and children should be carefully examined, perhaps on repeated occasions, before the diagnosis is made.
The ophthalmologist can use certain techniques to uncover functional visual loss, including placing the child behind the phoropter, and surreptitiously and purposefully blurring the vision using high plus lenses, and then uncovering one eye or the other and monitoring visual acuity as the vision is being cleared. Often, patients with functional vision loss have miraculous improvement of vision when given a pair of glasses with very low refractive power. These children simply want a pair of glasses and are pretending to have poor visual acuity. Other children who are feigning vision loss to seek attention may be more difficult to diagnosis. An inconsistency between near visual acuity and distance visual acuity, the presence of stereovision when tested with polarized lenses, and nonphysiologic visual fields should cause the examiner to suspect functional visual loss [Mewasingh et al., 2002]. Functional visual loss is a diagnosis of exclusion only after the patient has had a thorough ophthalmic examination and more than one measurement of visual acuity. Occasionally, the practitioner must resort to neuroimaging and/or electrophysiologic retinal testing before becoming confident in diagnosing functional vision loss. After the diagnosis is made, supportive reassurance for the child and the family that vision will return and that the child will be able to see usually leads to rapid clearing of the visual “impairment.” Special consideration should be given to children, because there have been numerous cases of sexual abuse occurring in children who present with decreased visual acuity [Kathol et al., 1983].
The vast majority of children with complaints of vision loss have benign refractive conditions that are easily corrected, but the complaint of blurred vision should always be taken seriously by pediatricians and primary care providers, as it can be a harbinger of more serious conditions. In the absence of focal or specific neurologic findings, an examination by an ophthalmologist can often determine the diagnosis and spare the child unnecessary and costly diagnostic interventions, and reserve neurologic consultation for appropriate patients. Knowledge of the common causes of vision loss in children (Table 6-4) will help the neurologist work in a multidisciplinary fashion to assure prompt diagnosis and institution of vision-saving therapies.
Table 6-4 Causes of Vision Loss, Associated Findings, and Diagnostic Recommendations
| Cause of Vision Loss | Associated Findings/Syndrome* | Diagnostic Evaluation |
|---|---|---|
| Leber’s congenital amaurosis | Large-amplitude, slow-frequency, roving nystagmus at 2 months of age; eye pressing (oculodigital sign) | Electroretinogram for photoreceptor function; renal consultation to rule out Senior’s syndrome |
| Ocular albinism/oculocutaneous albinism | Moderate-amplitude nystagmus by 2 months, fair skin, X-linked inheritence; rule out Chediak–Higashi and Hermansky–Pudlak syndromes | Slit-lamp examination for iris transillumination defects; examine mother for signs of retinal hypopigmentation |
| Aniridia | Nystagmus at 2 months; consider WAGR syndrome | Slit-lamp examination shows lack of iris development; obtain genetic consult and urologic consultation to rule out and follow for Wilms’ tumor |
| Achromatopsia | High-frequency, low-amplitude nystagmus due to lack of cone photoreceptor development; photophobia | Electroretinogram for photoreceptor function |
| Optic nerve coloboma | Nystagmus if severe and bilateral, strabismus if unilateral; CHARGE sequence, papillorenal syndrome | MRI of brain to rule out encephalocele and other structural anomalies; genetic and renal consultation to rule out other syndromes |
| Optic nerve hypoplasia | Nystagmus by 2 months if bilateral, strabismus if unilateral; septo-optic dysplasia/Aicardi’s syndrome; hypoglycemic at birth, signs of panhypopituitarism | MRI of brain – look for agenesis of corpus callosum, absence of septum pellucidum, absence or ectopy of pituitary bright spot, gray matter heterotopia in Aicardi’s syndrome |
| Retinopathy of prematurity | History of prematurity, oculodigital sign | Ophthalmic examination |
| Norrie’s syndrome | Nystagmus at 2 months, X-linked, bilateral retinal detachment with cataract | Ophthalmic examination; genetic consultation |
| Congenital cataract | Nystagmus at 2 months if complete cataract, strabismus if unilateral; stigmata of trisomies; metabolic signs if enzymatic or mitochondrial | Ophthalmic examination; if bilateral, consider galatosemia screen, urine amino acids (Lowe’s syndrome), urine for reducing substances, genetic consultation; examine parents to rule out autosomal-dominant cataract; TORCH evaluation |
| Optic neuritis | Vision loss, possible pain with eye movements, afferent pupil defect, focal neurologic findings; ADEM, neuromyelitis optica (Devic’s disease), multiple sclerosis, syphilis, Lyme disease, cat-scratch disease, toxin (nutritional, ethambutol, methanol), Leber’s hereditary optic neuropathy | Ophthalmic examination, MRI, lumbar puncture with opening pressure, serology and monoclonal bands, infectious disease consultation, genetic evaluation if positive family history; test for Leber’s hereditary optic neuropathy |
| Optic atrophy | Vision loss, strabismus, associated focal neurologic findings, endocrine signs/symptoms, deafness, loss of milestones in NCL/neurometabolic disorders/Leigh’s disease; family history | Attention to pre- and perinatal history, neuroimaging, genetic consultation, endocrinologic consultation, assess for mitochondrial dysfunction |
| Cerebral visual impairment | Vision loss, sterotypic features: off/on, saccadic paresis; history of pre-/perinatal ischemia, intraventricular hemorrhage/prematurity, meningitis, cerebral developmental anomaly, cerebral palsy, seizure disorder | Neuroimaging; genetics if no known etiology |
| Refractive errors/amblyopia | Often sudden discovery or during screening examination; family history common; nonfocal neurologic examination; refractive errors correct with pinhole | Ophthalmic examination |
| Papilledema | Vision loss only if chronic or rapid onset, scotoma on change in position, constricted fields, esotropia with abducens paresis, headache, nausea; risk factors for idiopathic intracranial hypertension – obese, female, estrogen use, tetracycline, vitamin A, steroids, growth hormone, cerebral venous thrombosis | Ophthalmic examination – loss of spontaneous venous pulsations and edema of optic nerve, MRI and MRV, lumbar puncture with opening pressure |
* ADEM, acute disseminated encephalomyelitis; CHARGE, coloboma, heart defects, atresia choanae, retardation of growth and development, genitourinary problems, and ear anomalies; NCL, neuronal ceroid lipafuscinosis; TORCH, toxoplasmosis, rubella, cytomegalovirus, herpes simplex virus; WAGR, Wilms’ tumor, genitourinary abnormalities, and mental retardation.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Acaroglu G., Kansu T., Dogulu C.F. Visual recovery patterns in children with Leber’s hereditary optic neuropathy. Int Ophthalmol. 2001;24:349.
Afshari M.A., Afshari N.A., Fulton A.B. Cortical visual impairment in infants and children. Int Ophthalmol Clin. 2001;41:159.
al-Salem M. Leber’s congenital amaurosis in 22 affected members of one family. J Pediatr Ophthalmol Strabismus. 1997;34:254.
Alur R.P., Vijayasarathy C., Brown J.D., et al. Papillorenal syndrome-causing missense mutations in PAX2/Pax2 result in hypomorphic alleles in mouse and human. PLoS Genet. 2010;6(3):e1000870.
Amedi A., Raz N., Pianka P., et al. Early ‘visual’ cortex activation correlates with superior verbal memory performance in the blind. Nat Neurosci. 2003;6:758.
Apple D.J., Rabb M.F., Walsh P.M. Congenital anomalies of the optic disc. Surv Ophthalmol. 1982;27:3.
Ashmead D.H., Wall R.S., Ebinger K.A., et al. Spatial hearing in children with visual disabilities. Perception. 1998;27:105.
Babel J., Klein D., Roth A. Leber’s congenital amaurosis associated with high hyperopia in four sisters. Ophthalmic Paediatr Genet. 1989;10:55.
Backhouse O., Leitch R.J., Thompson D., et al. A case of reversible blindness in maple syrup urine disease. Br J Ophthalmol. 1999;83:250.
Baker R.S., Baumann R.J., Buncic J.R. Idiopathic intracranial hypertension (pseudotumor cerebri) in pediatric patients. Pediatr Neurol. 1989;5:5.
Baker R.S., Carter D., Hendrick E.B., et al. Visual loss in pseudotumor cerebri of childhood. A follow-up study. Arch Ophthalmol. 1985;103:1681.
Bale J.F.Jr, Murph J.R. Congenital infections and the nervous system. Pediatr Clin North Am. 1992;39:669.
Bar S., Segal M., Shapira R., et al. Neuroretinitis associated with cat scratch disease. Am J Ophthalmol. 1990;110:703.
Barkovich A.J., Lyon G., Evrard P. Formation, maturation, and disorders of white matter. Am J Neuroradiol. 1992;13:447.
Barlow C.F. Migraine in the infant and toddler. J Child Neurol. 1994;9:92.
Basti S., Ravishankar U., Gupta S. Results of a prospective evaluation of three methods of management of pediatric cataracts. Ophthalmology. 1996;103:713.
Bates C.M., Baum M., Hunchik M., et al. Acute vision loss in children with autosomal recessive polycystic kidney disease. Am J Kidney Dis. 1999;34:1125.
Bauer J., Schuler P., Feistel H., et al. Blindness as an ictal phenomenon: Investigations with EEG and SPECT in two patients suffering from epilepsy. J Neurol. 1991;238:44.
Bec P., Adam P., Mathis A., et al. Optic nerve head drusen. High-resolution computed tomographic approach. Arch Ophthalmol. 1984;102:680.
Belgaumi A.F., Kauffman W.M., Jenkins J.J., et al. Blindness in children with neuroblastoma. Cancer. 1997;80:1997.
Billingsley R.L., Lang F.F., Slopis J.M., et al. Visual-spatial neglect in a child following sub-cortical tumor resection. Dev Med Child Neurol. 2002;44:191.
Blohme J., Tornqvist K. Visually impaired Swedish children. The 1980 cohort study–a 19-year ophthalmological follow-up. Acta Ophthalmol Scand. 2000;78:553.
Bohra L.I., Weizer J.S., Lee A.G., et al. Vision loss as the presenting sign in juvenile neuronal ceroid lipofuscinosis. J Neuroophthalmol. 2000;20:111.
Bonhomme G.R., Waldman A.T., Balcer L.J., et al. Pediatric optic neuritis: brain MRI abnormalities and risk of multiple sclerosis. Neurology. 2009;72(10):881.
Boothe R.G., Dobson V., Teller D.Y. Postnatal development of vision in human and nonhuman primates. Annu Rev Neurosci. 1985;8:495.
Brecelj J., Steirn-Kranjc B. ERG and VEP follow-up study in children with Leber’s congenital amaurosis. Eye. 1999;13(Pt 1):47.
Brodsky M.C. Morning glory disc anomaly or optic disc coloboma? Arch Ophthalmol. 1994;112:153.
Brodsky M.C., Beck R.W. The changing role of MR imaging in the evaluation of acute optic neuritis. Radiology. 1994;192:22.
Brodsky M.C., Conte F.A., Taylor D., et al. Sudden death in septo-optic dysplasia. Report of 5 cases. Arch Ophthalmol. 1997;115:66.
Brodsky M.C., Fray K.J., Glasier C.M. Perinatal cortical and subcortical visual loss: Mechanisms of injury and associated ophthalmologic signs. Ophthalmology. 2002;109:85.
Brodsky M.C., Glasier C.M. Magnetic resonance visualization of the swollen optic disc in papilledema. J Neuroophthalmol. 1995;15:122.
Brodsky M.C., Glasier C.M., Creel D.J. Magnetic resonance imaging of the visual pathways in human albinos. J Pediatr Ophthalmol Strabismus. 1993;30:382.
Brodsky M.C., Glasier C.M., Pollock S.C., et al. Optic nerve hypoplasia. Identification by magnetic resonance imaging. Arch Ophthalmol. 1990;108:1562.
Brodsky M.C., Rettele G.A. Protracted postsurgical blindness with visual recovery following optic nerve sheath fenestration. Arch Ophthalmol. 1998;116:107.
Brown A.M. Development of visual sensitivity to light and color vision in human infants: A critical review. Vision Res. 1990;30:1159.
Brown F.R.3rd, Voigt R., Singh A.K., et al. Peroxisomal disorders. Neurodevelopmental and biochemical aspects. Am J Dis Child. 1993;147:617.
Brown J.Jr, Fingert J.H., Taylor C.M., et al. Clinical and genetic analysis of a family affected with dominant optic atrophy (OPA1). Arch Ophthalmol. 1997;115:95.
Brown R., Hobson R.P., Lee A., et al. Are there “autistic-like” features in congenitally blind children? J Child Psychol Psychiatry. 1997;38:693.
Browning A.C., Mengher L.S., Gregson R.M., et al. Visual outcome of malignant hypertension in young people. Arch Dis Child. 2001;85:401.
Bye A.M., Kendall B., Wilson J. Multiple sclerosis in childhood: A new look. Dev Med Child Neurol. 1985;27:215.
Carden S.M., Boissy R.E., Schoettker P.J., et al. Albinism: Modern molecular diagnosis. Br J Ophthalmol. 1998;82:189.
Carney S.H., Brodsky M.C., Good W.V., et al. Aicardi syndrome: More than meets the eye. Surv Ophthalmol. 1993;37:419.
Casteels I., Demaerel P., Spileers W., et al. Cortical visual impairment following perinatal hypoxia: Clinicoradiologic correlation using magnetic resonance imaging. J Pediatr Ophthalmol Strabismus. 1997;34:297.
Chestler R.J., France T.D. Ocular findings in CHARGE syndrome. Six case reports and a review. Ophthalmology. 1988;95:1613.
Chrousos G.A., Drack A.V., Young M., et al. Neuroretinitis in cat scratch disease. J Clin Neuroophthalmol. 1990;10:92.
Clarke W.N., Vomiero G., Leonard B.C. Bilateral simultaneous retinal arteriolar obstruction in a child with hemoglobin SS sickle cell disease. J AAPOS. 2001;5:126.
Collins M.L., Traboulsi E.I., Maumenee I.H. Optic nerve head swelling and optic atrophy in the systemic mucopolysaccharidoses. Ophthalmology. 1990;97:1445.
Cooper L.L., Hansen R.M., Darras B.T., et al. Rod photoreceptor function in children with mitochondrial disorders. Arch Ophthalmol. 2002;120:1055.
Cotlier E. Tay-Sachs’ retina. Deficiency of acetyl hexosaminidase A. Arch Ophthalmol. 1971;86:352.
Daw N.W. Mechanisms of plasticity in the visual cortex. The Friedenwald Lecture. Invest Ophthalmol Vis Sci. 1994;35:4168.
Dreyer R.F., Hopen G., Gass J.D., et al. Leber’s idiopathic stellate neuroretinitis. Arch Ophthalmol. 1984;102:1140.
Droste P.J., Archer S.M., Helveston E.M. Measurement of low vision in children and infants. Ophthalmology. 1991;98:1513.
Dubowitz L.M., Mushin J., De Vries L., et al. Visual function in the newborn infant: Is it cortically mediated? Lancet. 1986;1:1139.
Dubowitz L.M., Mushin J., Morante A., et al. The maturation of visual acuity in neurologically normal and abnormal newborn infants. Behav Brain Res. 1983;10:39.
Dutton G.N., Jacobson L.K. Cerebral visual impairment in children. Semin Neonatol. 2001;6:477.
Ehyai A., Fenichel G.M. The natural history of acute confusional migraine. Arch Neurol. 1978;35:368.
Egan R.A., Kerrison J.B. Survey of genetic neuro-ophthalmic disorders. Ophthalmol Clin North Am. 2003;16:595.
Eliott D., Traboulsi E.I., Maumenee I.H. Visual prognosis in autosomal dominant optic atrophy (Kjer type). Am J Ophthalmol. 1993;115:360.
Fazzi E., Lanners J., Danova S., et al. Stereotyped behaviours in blind children. Brain Dev. 1999;21:522.
Fielder A.R., Mayer D.L., Fulton A.B. Delayed visual maturation. Lancet. 1991;337:1350.
Fishman M.A., Hogan G.R., Dodge P.R. The concurrence of hydrocephalus and craniosynostosis. J Neurosurg. 1971;34:621.
Flanagan N.M., Jackson A.J., Hill A.E. Visual impairment in childhood: Insights from a community-based survey. Child Care Health Dev. 2003;29:493.
Fletcher W.A., Imes R.K., Hoyt W.F. Chiasmal gliomas: Appearance and long-term changes demonstrated by computerized tomography. J Neurosurg. 1986;65:154.
Foxman S.G., Heckenlively J.R., Bateman J.B., et al. Classification of congenital and early onset retinitis pigmentosa. Arch Ophthalmol. 1985;103:1502.
Friedman A.H., Beckerman B., Gold D.H., et al. Drusen of the optic disc. Surv Ophthalmol. 1977;21:373.
Fulton A.B., Hansen R.M., Harris S.J. Retinal degenerations and brain abnormalities in infants and young children. Doc Ophthalmol. 1985;60:133.
Fulton A.B., Hansen R.M., Manning K.A. Measuring visual acuity in infants. Surv Ophthalmol. 1981;25:325.
Garcia-Filion P., Epport K., Nelson M., et al. Neuroradiographic, endocrinologic, and ophthalmic correlates of adverse developmental outcomes in children with optic nerve hypoplasia: a prospective study. Pediatrics. 2008;121(3):e653.
Garcia-Filion P., Fink C., Geffner M.E., et al. Optic nerve hypoplasia in North America: a re-appraisal of perinatal risk factors. Acta Ophthalmol (Copenh). 2008;24:547.
Gilbert C., Awan H. Blindness in children. BMJ. 2003;327:760.
Gilbert C.E., Anderton L., Dandona L., et al. Prevalence of visual impairment in children: A review of available data. Ophthalmic Epidemiol. 1999;6:73.
Gloor P., Pulido J.S., Judisch G.F. Magnetic resonance imaging and fundus findings in a patient with Aicardi’s syndrome. Arch Ophthalmol. 1989;107:922.
Goebel H.H. The neuronal ceroid-lipofuscinoses. J Child Neurol. 1995;10:424.
Good W.V. Development of a quantitative method to measure vision in children with chronic cortical visual impairment. Trans Am Ophthalmol Soc. 2001;99:253.
Good W.V., Hoyt C.S. Behavioral correlates of poor vision in children. Int Ophthalmol Clin. 1989;29:57.
Good W.V., Jan J.E., DeSa L., et al. Cortical visual impairment in children. Surv Ophthalmol. 1994;38:351.
Hansen E., Bachen N.I., Flage T. Refsum’s disease. Eye manifestations in a patient treated with low phytol low phytanic acid diet. Acta Ophthalmol (Copenh). 1979;57:899.
Heher K.L., Traboulsi E.I., Maumenee I.H. The natural history of Leber’s congenital amaurosis. Age-related findings in 35 patients. Ophthalmology. 1992;99:241.
Heidemann D.G., Beck R.W. Retinitis pigmentosa. A mimic of neurologic disease. Surv Ophthalmol. 1987;32:45.
Hockaday J.M. Basilar migraine in childhood. Dev Med Child Neurol. 1979;21:455.
Hoyt C.S. Visual function in the brain-damaged child. Eye. 2003;17:369.
Hoyt C.S., Fredrick D.R. Cortically visually impaired children: A need for more study. Br J Ophthalmol. 1998;82:1225.
Hoyt C.S., Jastrzebski G., Marg E. Delayed visual maturation in infancy. Br J Ophthalmol. 1983;67:127.
Hubel D.N., Wiesel T.N. Receptive fields, binocular interactions and functional architecture in the cats visual cortex. J Physiol. 1962;160:106.
Huber A. Genetic diseases of vision. Curr Opin Neurol. 1994;7:65.
Huo R., Burden S.K., Hoyt C.S., et al. Chronic cortical visual impairment in children: Aetiology, prognosis, and associated neurological deficits. Br J Ophthalmol. 1999;83:670.
Iinuma K., Lombroso C.T., Matsumiya Y. Prognostic value of visual evoked potentials (VEP) in infants with visual inattentiveness. Electroencephalogr Clin Neurophysiol. 1997;104:165.
Jacobson L., Lundin S., Flodmark O., et al. Periventricular leukomalacia causes visual impairment in preterm children. A study on the aetiologies of visual impairment in a population-based group of preterm children born 1989-95 in the county of Varmland, Sweden. Acta Ophthalmol Scand. 1998;76:593.
Jacobson L., Hård A.L., Horemuzova E., et al. Visual impairment is common in children born before 25 gestational weeks–boys are more vulnerable than girls. Acta Paediatr. 2009;98(2):261.
Jan J.E., Freeman R.D. Who is a visually impaired child? Dev Med Child Neurol. 1998;40:65.
Jan J.E., Groenveld M., Connolly M.B. Head shaking by visually impaired children: A voluntary neurovisual adaptation which can be confused with spasmus nutans. Dev Med Child Neurol. 1990;32:1061.
Jarosz J.M., Howlett D.C., Cox T.C., et al. Cyclosporine-related reversible posterior leukoencephalopathy: MRI. Neuroradiology. 1997;39:711.
Johns D.R., Smith K.H., Miller N.R. Leber’s hereditary optic neuropathy. Clinical manifestations of the 3460 mutation. Arch Ophthalmol. 1992;110:1577.
Johns D.R., Smith K.H., Savino P.J., et al. Leber’s hereditary optic neuropathy. Clinical manifestations of the 15257 mutation. Ophthalmology. 1993;100:981.
Kathol R.G., Cox T.A., Corbett J.J., et al. Functional visual loss: II. Psychiatric aspects in 42 patients followed for 4 years. Psychol Med. 1983;13:315.
Kennedy C., Carroll F.D. Optic neuritis in children. Arch Ophthalmol. 1960;63:747.
Kerrison J.B., Vagefi M.R., Barmada M.M., et al. Congenital motor nystagmus linked to Xq26-q27. Am J Hum Genet. 1999;64:600.
Kivlin J.D., Bodnar A., Ralston C.W., et al. The visually inattentive preterm infant. J Pediatr Ophthalmol Strabismus. 1990;27:190.
Kline L.B., Glasier J.S. Dominant optic atrophy. The clinical profile. Arch Ophthalmol. 1979;97:1680.
Lambert S.R., Hoyt C.S., Narahara M.H. Optic nerve hypoplasia. Surv Ophthalmol. 1987;32:1.
Lambert S.R., Kriss A., Taylor D. Vision in patients with Leber congenital amaurosis. Arch Ophthalmol. 1997;115:293.
Lambert S.R., Taylor D., Kriss A. The infant with nystagmus, normal appearing fundi, but an abnormal ERG. Surv Ophthalmol. 1989;34:173.
Lanzi G., Fazzi E., Uggetti C., et al. Cerebral visual impairment in periventricular leukomalacia. Neuropediatrics. 1998;29:145.
Leger D., Guilleminault C., Santos C., et al. Sleep/wake cycles in the dark: Sleep recorded by polysomnography in 26 totally blind subjects compared to controls. Clin Neurophysiol. 2002;113:1607.
Leger D., Prevot E., Philip P., et al. Sleep disorders in children with blindness. Ann Neurol. 1999;46:648.
Levtzion-Korach O., Tennenbaum A., Schnitzer R., et al. Early motor development of blind children. J Paediatr Child Health. 2000;36:226.
Lewis T.L., Maurer D. Preferential looking as a measure of visual resolution in infants and toddlers: A comparison of psychophysical methods. Child Dev. 1986;57:1062.
Liu G.T., Hunter J., Miki A., et al. Functional MRI in children with congenital structural abnormalities of the occipital cortex. Neuropediatrics. 2000;31:13.
Liu G.T. Visual loss in childhood. Surv Ophthalmol. 2001;46:35.
Marcel A.J. Blindsight and shape perception: Deficit of visual consciousness or of visual function? Brain. 1998;121(Pt 8):1565.
Mayer D.L., Fulton A.B. Preferential looking grating acuities of infants at risk of amblyopia. Trans Ophthalmol Soc U K. 1985;104(Pt 8):903.
McCulloch D.L., Taylor M.J. Cortical blindness in children: Utility of flash VEPs. Pediatr Neurol. 1992;8:156.
McKeon A., Lennon V.A., Lotze T., et al. CNS aquaporin-4 autoimmunity in children. Neurology. 2008;71(2):93.
Merin S., Crawford J.S. The etiology of congenital cataracts. A survey of 386 cases. Can J Ophthalmol. 1971;6:178.
Mervis C.A., Yeargin-Allsopp M., Winter S., et al. Aetiology of childhood vision impairment, metropolitan Atlanta, 1991-93. Paediatr Perinat Epidemiol. 2000;14:70.
Mewasingh L.D., Kornreich C., Christiaens F., et al. Pediatric phantom vision (Charles Bonnet) syndrome. Pediatr Neurol. 2002;26:143.
Miller N.R., Newman S.A. Transsynaptic degeneration. Arch Ophthalmol. 1981;99:1654.
Mintz-Hittner H.A., Ferrell R.E., Sims K.B., et al. Peripheral retinopathy in offspring of carriers of Norrie disease gene mutations. Possible transplacental effect of abnormal Norrin. Ophthalmology. 1996;103:2128.
Mustonen E. Pseudopapilloedema with and without verified optic disc drusen. A clinical analysis I. Acta Ophthalmol (Copenh). 1983;61:1037.
Newman N.J. Leber’s hereditary optic neuropathy. New genetic considerations. Arch Neurol. 1993;50:540.
Nixon R.B., Helveston E.M., Miller K., et al. Incidence of strabismus in neonates. Am J Ophthalmol. 1985;100:798.
Nucci P., Rosenbaum A. Acquired anomalous head posture following loss of vision in one eye. Acta Ophthalmol Scand. 2002;80:109.
Opocher E., Kremer L.C., Da Dalt L., et al. Prognostic factors for progression of childhood optic pathway glioma: a systematic review. Eur J Cancer. 2006;42(12):1807.
Okawa M., Nanami T., Wada S., et al. Four congenitally blind children with circadian sleep-wake rhythm disorder. Sleep. 1987;10:101.
Palm L., Blennow G., Wetterberg L. Long-term melatonin treatment in blind children and young adults with circadian sleep-wake disturbances. Dev Med Child Neurol. 1997;39:319.
Pierce E.A., Mukai S. Controversies in the management of retinopathy of prematurity. Int Ophthalmol Clin. 1994;34:121.
Purdy K.W., Heckenlively J.R., Church J.A., et al. Progressive outer retinal necrosis caused by varicella-zoster virus in children with acquired immunodeficiency syndrome. Pediatr Infect Dis J. 2003;22:384.
Remler B.F., Leigh R.J., Osorio I., et al. The characteristics and mechanisms of visual disturbance associated with anticonvulsant therapy. Neurology. 1990;40:791.
Rummelt V., Folberg R., Ionasescu V., et al. Ocular pathology of MELAS syndrome with mitochondrial DNA nucleotide 3243 point mutation. Ophthalmology. 1993;100:1757.
Russell-Eggitt I.M., Blake K.D., Taylor D.S., et al. The eye in the CHARGE association. Br J Ophthalmol. 1990;74:421.
Santavuori P., Vanhanen S.L., Sainio K., et al. Infantile neuronal ceroid-lipofuscinosis (INCL): Diagnostic criteria. J Inherit Metab Dis. 1993;16:227.
Sato K., Adachi-Usami E., Mizota A. Follow-up studies on pattern reversal visually evoked cortical potentials in a 2-year-old child with optic neuritis. J Neuroophthalmol. 1998;18:21.
Savage G.L., Centaro A., Enoch J.M., et al. Drusen of the optic nerve head–an important model. Ophthalmology. 1985;92:793.
Schouten D., de Graaf S.S., Verrips A. Transient cortical blindness following vincristine therapy. Med Pediatr Oncol. 2003;41:470.
Schwartz T.L., Vahgei L. Charles Bonnet syndrome in children. J AAPOS. 1998;2:310.
Semba R.D., Bloem M.W. Measles blindness. Surv Ophthalmol. 2004;49:243.
Shahar E., Barak S. Favorable outcome of epileptic blindness in children. J Child Neurol. 2003;18:12.
Shenoy A.M., Markowitz J.A., Bonnemann C.G., et al. Muscle-Eye-Brain disease. J Clin Neuromuscul Dis. 2010;11(3):124.
Sher N.A., Letson R.D., Desnick R.J. The ocular manifestations in Fabry’s disease. Arch Ophthalmol. 1979;97:671.
Siatkowski R.M., Sanchez J.C., Andrade R., et al. The clinical, neuroradiographic, and endocrinologic profile of patients with bilateral optic nerve hypoplasia. Ophthalmology. 1997;104:493.
Skarf B., Hoyt C.S. Optic nerve hypoplasia in children. Association with anomalies of the endocrine and CNS. Arch Ophthalmol. 1984;102:62.
Sokol S., Hansen V.C., Moskowitz A., et al. Evoked potential and preferential looking estimates of visual acuity in pediatric patients. Ophthalmology. 1983;90:552.
Sonksen P.M., Dale N. Visual impairment in infancy: Impact on neurodevelopmental and neurobiological processes. Dev Med Child Neurol. 2002;44:782.
Spalton D.J., Taylor D.S., Sanders M.D. Juvenile Batten’s disease: An ophthalmological assessment of 26 patients. Br J Ophthalmol. 1980;64:726.
Stavrou P., Sgouros S., Willshaw H.E., et al. Visual failure caused by raised intracranial pressure in craniosynostosis. Childs Nerv Syst. 1997;13:64.
Sullivan T.J., Heathcote J.G., Brazel S.M., et al. The ocular pathology in Leber’s congenital amaurosis. Aust N Z J Ophthalmol. 1994;22:25.
Summers C.G., MacDonald J.T. Vision despite tomographic absence of the occipital cortex. Surv Ophthalmol. 1990;35:188.
Szlyk J.P., Fishman G.A., Grover S., et al. Difficulty in performing everyday activities in patients with juvenile macular dystrophies: Comparison with patients with retinitis pigmentosa. Br J Ophthalmol. 1998;82:1372.
Thomas W.J., Sahney S., Siegel L.M. Acute visual loss in a child with autosomal recessive polycystic kidney disease: Case report and review of the literature. J AAPOS. 2003;7:217.
Thompson L., Kaufman L.M. The visually impaired child. Pediatr Clin North Am. 2003;50:225.
Tomsak R.L., Jergens P.B. Benign recurrent transient monocular blindness: A possible variant of acephalgic migraine. Headache. 1987;27:66.
Trevathan E., Murphy C.C., Yeargin-Allsopp M. Prevalence and descriptive epidemiology of Lennox-Gastaut syndrome among Atlanta children. Epilepsia. 1997;38:1283.
Uggetti C., Egitto M.G., Fazzi E., et al. Transsynaptic degeneration of lateral geniculate bodies in blind children: in vivo MR demonstration. Am J Neuroradiol. 1997;18:233.
Varma D., George N., Livingston J., et al. Acute visual loss as an early manifestation of metastatic neuroblastoma. Eye. 2003;17:250.
Vinkler C., Lev D., Kalish H., et al. Familial optic atrophy with white matter changes. Am J Med Genet. 2003;121A:263.
Weber R.G. Treatment of retinal and choroidal degenerations and dystrophies: Current status and prospects for gene based therapy. Ophthalmol Clin North Am. 2003;16:583.
Weleber R.G. Infantile and childhood retinal blindness: A molecular perspective. The Franceschetti Lecture. Ophthalmic Genet. 2002;23:71.
Weleber R.G., Tongue A.C., Kennaway N.G., et al. Ophthalmic manifestations of infantile phytanic acid storage disease. Arch Ophthalmol. 1984;102:1317.
Whiting S., Jan J.E., Wong P.K., et al. Permanent cortical visual impairment in children. Dev Med Child Neurol. 1985;27:730.
Winterkorn J.M. Lyme disease: Neurologic and ophthalmic manifestations. Surv Ophthalmol. 1990;35:191.
Yamamoto T., Kato Y., Karita M., et al. Expression of genes related to muscular dystrophy with lissencephaly. Pediatr Neurol. 2004;31:183.
Zack D.J., Dean M., Molday R.S., et al. What can we learn about age-related macular degeneration from other retinal diseases? Mol Vis. 1999;5:30.
Zihl J. “Blindsight”: Improvement of visually guided eye movements by systematic practice in patients with cerebral blindness. Neuropsychologia. 1980;18:71.