Chapter 81 Viral Infections of the Nervous System
Throughout recorded history, viral infections have caused considerable human suffering [Hughes, 1977]. Approximately 100 species of viruses from 13 different families have been associated directly or indirectly with disorders of the central or peripheral nervous system (Box 81-1) [Johnson, 1998]. Although many viral infections have become infrequent or less severe because of immunization programs or antiviral chemotherapy (Figure 81-1), others remain potential threats to the well-being of children throughout the world. This chapter describes virus-induced neurologic disorders, emphasizing pathogenesis, epidemiology, clinical manifestations, treatment, and prevention.
Box 81-1 Selected Viruses Associated with Human Neurologic Disease
























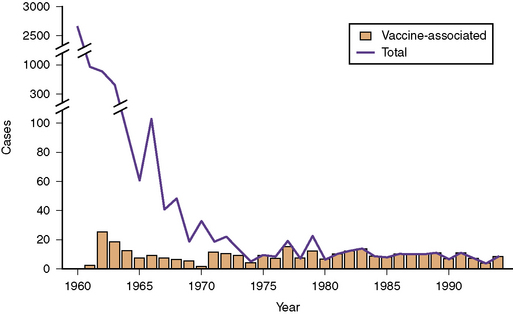
Fig. 81-1 Data for paralytic poliomyelitis in the United States from 1960 to 1994.
(Centers for Disease Control Prevention. Paralytic poliomyelitis – United States, 1980–1994. MMWR Morb Mortal Wkly Rep 1997;46:79).
General Considerations
The pathogenesis of a viral neurologic infection reflects the complex interactions of a viral pathogen and the infected host. Virus-induced neurologic disorders begin with virus entry and replication in the skin, conjunctiva, or mucosal surfaces of the gastrointestinal, respiratory, or genital tracts [Knipe et al., 2007; Johnson, 1998]. Consequently, several host factors, such as the gastric pH, local immune responses, integrity of the skin or mucosal barriers, or enzymes that inactivate viruses, influence whether viral pathogens successfully invade human tissues.
Viruses, among the smallest microorganisms causing human disease, consist of an outer capsid, composed of glycoproteins and lipids, and an inner core, composed of ribonucleic acid (RNA) or deoxyribonucleic acid (DNA) [Knipe et al., 2007]. Virus attachment and penetration of host cells depend on the virus encountering and attaching to receptors located on the surface of host cells. The molecular components of this interaction consist of the viral surface glycoproteins and the host cell surface proteins, such as pre-existing cellular receptors that mediate certain essential cell functions [Knipe et al., 2007; Johnson, 1998]. An example of the virus–host interaction that leads to successful viral replication is the attachment of glycoprotein 120 of the human immunodeficiency virus (HIV) to the CD4 molecule and chemokine receptors of host cells [Knipe et al., 2007].
Viruses enter host cells by endocytosis, release of genomic material, or fusion of the virus envelope with the cell membrane followed by release of viral RNA or DNA. Viruses use host-cell synthetic pathways to uncoat their nucleic acid and to produce virus-encoded nucleic acids and proteins. Replicative strategies vary among viruses [Knipe et al., 2007]. The double-stranded DNA herpesviruses use RNA polymerases in the host-cell nucleus to transcribe viral DNA into viral messenger RNA (mRNA). The RNA of positive-strand RNA viruses serves as the viral mRNA, whereas negative-strand RNA viruses first must be transcribed into an mRNA strand. By contrast, RNA-containing retroviruses, such as HIV, replicate via a DNA intermediary synthesized in the host-cell cytoplasm by a viral-encoded reverse transcriptase [Knipe et al., 2007].
Next, virus-encoded immediate-early and early proteins facilitate the production of additional viral proteins and assembly of mature virus particles, called virions. Viral replication occurs within the nucleus or cytoplasm of the host cell, depending on the viral species, and final assembly of viral particles uses components of existing host-cell structures, such as the nuclear membrane or the Golgi apparatus [Knipe et al., 2007]. Mature virus particles are released from host cells by budding or lysis. Effective replication ultimately releases large quantities of infectious virions into the lymphatics and peripheral circulation of the infected host.
Most viruses reach the central nervous system (CNS) hematogenously and penetrate the blood–brain barrier via the choroid plexus or through the vascular endothelium [Johnson, 1998]. CNS infection depends on the magnitude and duration of viremia, which reflect the efficiency of viral replication at extraneural sites and the ability of the virus to evade host defense mechanisms. CNS invasion, a relatively infrequent event, occurs during infections associated with prolonged viremia or high viral loads.
A few viruses, notably the rabies virus and certain herpesviruses, reach the CNS predominantly through neural routes [Johnson, 1998]. Rabies virus, a negative-strand RNA virus, infects neuromuscular junctions, enters nerve endings, and travels by retrograde axonal transport to the neurons of the spinal cord. The virus then ascends to the brain. Transmission via nerve pathways also mediates the pathogenesis of herpes simplex virus type 1 encephalitis and the mucocutaneous reactivations of herpes simplex virus type 1, herpes simplex virus type 2, and varicella zoster virus.
Viruses induce neurologic signs and symptoms by damaging neural cells directly or by stimulating host-dependent immune responses that perturb neural cell function [Johnson, 1998]. The spectrum and severity of neurologic signs or symptoms depend on several factors, including neurovirulence (the capacity of the virus to cause disease within the nervous system), neurotropism (the propensity of the virus to infect specific cell groups of the CNS), and the nature of the host immune response.
Certain viruses, such as herpes simplex virus type 1, produce lytic infection of neuronal cells and cause hemorrhagic necrosis within the brain, whereas other viruses, such as the La Crosse encephalitis virus, produce minimal cellular damage, despite infection of neural cells. The JC virus, the cause of progressive multifocal leukoencephalopathy, selectively infects oligodendrocytes, causing progressive demyelination [Johnson, 1998]. Some viruses may not infect neural cells directly, but induce immune-mediated processes, such as acute disseminated encephalomyelitis, a condition that accounts for approximately 10–15 percent of acute encephalitis cases in the United States[Johnson, 1996].
Cellular and humoral host immune responses and responses mediated by interferons and inflammatory cytokines (e.g., tumor necrosis factor and the interleukins) have major roles in inhibiting viral infections of the CNS [Zhang et al., 2007a]. The unique roles that specific immune factors have in controlling viral infections in humans continue to constitute an area of intense scientific scrutiny. In animal models, certain Toll and Toll-like receptors, or pattern recognition receptors, participate in the host immune responses to viruses, and in rare instances, human mutations in the genes encoding such receptors have been linked to susceptibility to severe viral infections, such as herpes simplex virus encephalitis [Zhang et al., 2007b]. Immune responses may contribute to the severity of certain diseases, such as herpes simplex virus encephalitis [Conrady et al., 2009], and may provoke certain neurologic disorders, such as Guillain–Barré syndrome, Bell palsy, transverse myelopathy, or acute disseminated encephalomyelitis. The latter disorders can complicate several childhood infections, including relatively benign systemic viral syndromes [Johnson, 1996, 1998].
Clinical Manifestations of Viral Neurologic Disorders
Aseptic Meningitis
Aseptic meningitis, the most frequent CNS disorder associated with viral infections (Box 81-2), produces headache, vomiting, irritability, and neck or back pain. Children appear moderately ill, with photophobia and signs of meningeal irritation, including Kernig’s sign (involuntary spasm of the hamstring muscle provoked by knee extension in a supine patient) or Brudzinski’s sign (flexion of the knees provoked by forced flexion of the neck). In a young infant, signs of meningeal irritation are often absent, and the features of CNS infection, in general, are subtle.



























Encephalitis
Numerous viruses (see Box 81-2) can produce encephalitis in childhood. In addition to the headache, vomiting, and systemic features common in aseptic meningitis, children with viral encephalitis have seizures and altered sensorium, ranging from somnolence to coma [Whitley, 1990; Johnson, 1996]. Fever, a common but not invariable feature, ranges from low-grade to 40°C or higher.
Other Neurologic Disorders
Myelitis, Guillain–Barré syndrome, Bell palsy, acute cerebellar ataxia, myositis, and poliomyelitis-like disorders are additional neurologic conditions that can be associated with antecedent childhood viral infections (Box 81-3). Myelitis and Guillain–Barré syndrome frequently begin with vague sensory phenomena, but flaccid weakness and areflexia become the predominant neurologic signs. Marked sensory dysfunction, such as a sensory loss corresponding to a spinal level, indicates spinal cord involvement, whereas facial paralysis favors Guillain–Barré syndrome. Autonomic nervous system dysfunction also can be a major complication of the latter condition, producing life-threatening cardiac dysrhythmias or blood pressure fluctuations.

























Cerebellar ataxia (see Chapter 67), as summarized by Connolly and associates, can complicate systemic infections with several different viruses [Connolly et al., 1994]. Varicella-associated ataxia typically begins 5–14 days after the onset of the characteristic rash, although occasional cases can appear during the pre-eruptive phase of infection. Virus-induced ataxia and the associated behavioral disturbance peak in severity at onset or within 1–2 weeks, then improve during the ensuing 4–8 weeks.
Polioviruses (usually type 1) could infect the anterior horn cells of the spinal cord and cause asymmetric paralysis of the extremities (infantile paralysis) and often respiratory failure [Johnson, 1998]. Several other viruses, including West Nile virus, enterovirus 71, and other nonpolio enteroviruses, can produce disorders that can mimic classic poliomyelitis [Chen et al., 2001; Huang et al., 1999; Li et al., 2003].
Intrauterine and Perinatal Viral Infections
Intrauterine infections with certain viruses (and occasional nonviral pathogens) (see Box 81-3) can seriously damage the developing CNS, causing hydrocephalus, cerebral atrophy, intracranial calcifications, schizencephaly, porencephaly, or hydranencephaly [Bale, 2009]. Linked conceptually by the TORCH (toxoplasmosis, rubella, cytomegalovirus, and herpes simplex virus) acronym, these agents produce similar neonatal symptoms and signs consisting of intrauterine growth retardation, hepatosplenomegaly, jaundice, and petechial or purpuric rash [Bale, 2009]. Neurologic features can include microcephaly, chorioretinitis, macrocrania, sensorineural hearing loss, seizures, hypotonia, and focal deficits. Though the TORCH paradigm reminds clinicians that such pathogens produce overlapping clinical manifestations, some agents are associated with relatively unique or characteristic features. Rubella virus, as an example, is the only agent linked convincingly with congenital heart lesions, and varicella zoster virus is the only agent causing a dermatomal pattern of skin scarring known as a cicatrix. Osteopathy suggests congenital syphilis or congenital rubella syndrome, whereas isolated chorioretinitis can occur with congenital toxoplasmosis or lymphocytic choriomeningitis (LCM) virus infection.
Because congenital infections must begin with maternal infection, maternal immune status is a major determinant in the outcome of maternal infection. The fetuses of rubella-immune women, as indicated by the presence of rubella-virus specific IgG in mothers preconceptually, are protected from congenital rubella syndrome [Dudgeon, 1975; CDC, 2005b]; women who are seropositive for cytomegalovirus pre-conceptually are less likely than seronegative women to give birth to infants with congenital cytomegalovirus disease [Fowler et al., 2003]. The timing of maternal infection also affects the outcome of fetal infection and the pathogenesis of the CNS abnormalities associated with congenital infection. Maternal rubella during the initial 8–12 weeks may lead to ophthalmologic abnormalities and congenital heart disease, while infection during the initial 16 weeks can cause sensorineural hearing loss [Dudgeon, 1975; Hanshaw and Dudgeon, 1978]. By contrast, infections after the 20th week, although producing sensorineural hearing loss in some infants, occur silently and rarely cause neurodevelopmental disabilities. Cytomegalovirus-induced defects of neuronal migration, such as lissencephaly, schizencephaly, polymicrogyria, and cortical dysplasia, suggest that infections during critical gestational periods, especially before the 20th week, can be associated with severe neurodevelopmental abnormalities. Early infections with the parasite Toxoplasma gondii also seem to be associated with more severe outcome [Swisher et al., 1994]. The outcome of LCM virus infections is closely linked to the timing of maternal infection, a conclusion supported by studies of LCM virus infection of experimental animals [Bonthius et al., 2007a]. Congenital varicella syndrome, as an additional example of this phenomenon, rarely complicates maternal varicella zoster virus infections before the 5th week or after the 24th week of gestation [Sauerbrei, 2007].
Diagnosing perinatal infections with certain viruses, such as herpes simplex virus type 1 or 2 or parechovirus, remains challenging because the clinical features can be nonspecific during the initial phase of the neurological disease. Early symptoms of serious viral CNS disease in neonates, including herpes encephalitis, may consist only of poor feeding or fussiness, and the physical examination often reveals few signs localizing to the CNS [Bale and Miner, 2005]. Neonatal infections with parechovirus, a picornavirus with at least 14 types, can be associated with encephalitis and CNS white-matter injury [Verboon-Maciolkek et al., 2008a, b]. This disorder should be suspected in infants with fever, rash, and seizures, features that mimic neonatal infection with other enteroviruses.
Slow Viral Infections
Conventional viral pathogens can occasionally produce chronic disorders of the CNS, historically termed slow viral infections. These include subacute sclerosing panencephalitis, postrubella panencephalitis [Townsend et al., 1975], chronic nonpolio enterovirus infection, and progressive multifocal leukoencephalopathy caused by the JC virus (a polyomavirus named for the initials of the patient from whom the virus initially was isolated). Children with these disorders generally lack systemic signs of infection and have clinical signs restricted to the CNS, such as dementia, spasticity, paralysis, vision loss, and seizures. These slow viral disorders typically cause progressive debility and invariably end in death.
Epidemiology of Viral Infections
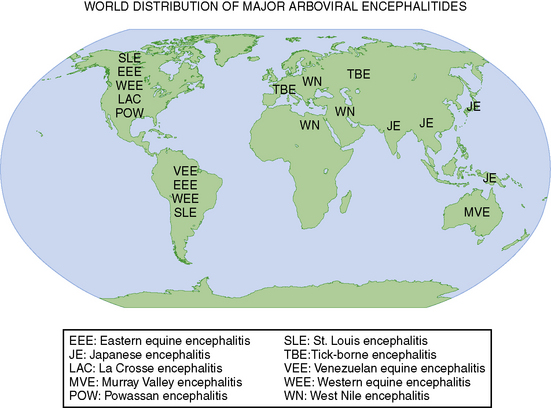
Worldwide epidemiological data indicate that viruses cause neurological disease (meningitis, encephalitis, and others) in many thousands of persons annually. The rates of encephalitis range from 3 cases per 100,000 persons [Johnson, 1996; Nicolosi et al., 1986] to 33 per 100,000 person-years, with rates highest among children [Koskiniemi et al., 1991, 2001]. Japanese encephalitis, a disease of young children, accounts for several thousand cases of encephalitis annually among unimmunized children in the endemic regions of India and eastern Asia [Dhillon and Raina, 2008].
Until the emergence of West Nile virus in the United States in 1999, the Centers for Disease Control and Prevention (CDC) received reports of 200–500 cases of arboviral encephalitis and many cases of aseptic meningitis every year. As the West Nile viral epidemic spread, numerous cases of West Nile virus infection were reported to the CDC. In 2003 alone, the CDC recorded more than 2800 human cases of neuroinvasive disease caused by West Nile virus [CDC, 2004a]. Recent West Nile virus activity in the United States peaked in 2006, when more than 4500 cases of West Nile virus infection were reported to the CDC; approximately 50 percent have been neuroinvasive infections [CDC, 2009d].
Human rabies cases average between 1 per 100,000 and 1 per million inhabitants annually in Africa and Asia, producing several thousand deaths [Dutta, 1999; Fishbein and Robinson, 1993]. By contrast, the United States has fewer than 5 indigenous cases of human rabies per year because of mandatory vaccination of domestic dogs [Blanton et al., 2009; Plotkin, 2000]. Rabies virus persists in many wild and domestic animal reservoirs, including bats, raccoons, skunks, and foxes in the United States, and wild canines or other mammals in many regions. In 2008, the CDC received reports of nearly 7000 cases of animal rabies, including some among domestic cats, dogs, and cattle [Blanton et al., 2009].
Environmentally derived viral pathogens display relatively uniform epidemiologic characteristics. Mosquitoes, ticks, and biting flies serve as the vectors for most human viral diseases. Human disease occurs when vectors are active, typically in spring, summer, and fall in temperate climates, and often displays distinct epidemiological characteristics that correspond to the habitat of the vector (Figure 81-2). Hiking, camping, and outdoor professions that facilitate vector contact enhance the probability of infections with vector-borne diseases [Miller et al., 2009].
By contrast, human-derived viral pathogens require direct contact with the infected host, either by exposure to aerosols that contain infectious viruses or by contact with tissues, blood, or body fluids from infected individuals. Herpes simplex virus type 1 can be acquired by oral–oral or oral–genital contact, whereas varicella zoster virus (the virus causing chickenpox) usually is transmitted by exposure to aerosolized particles that contain infectious virus. Cytomegalovirus can be acquired by direct contact with infected secretions, transfusion of blood products, or transplantation of organs or tissues from cytomegalovirus-seropositive persons [Bale, 1999].
La Crosse virus encephalitis, a frequently recognized arthropod-borne neurologic infection in the United States, illustrates the epidemiologic characteristics of environmentally derived agents. The disorder affects children who reside in the eastern and midwestern United States (Figure 81-3), a prime habitat for the Aedes triseriatus mosquito, a species that breeds preferentially in water that collects in tree trunks (“tree-holes”) and discarded automobile tires. Cases occur from June to early October, reflecting the seasonal activity of the mosquito vector, and frequently involve males 5–15 years old who enter wooded areas to hunt or play.
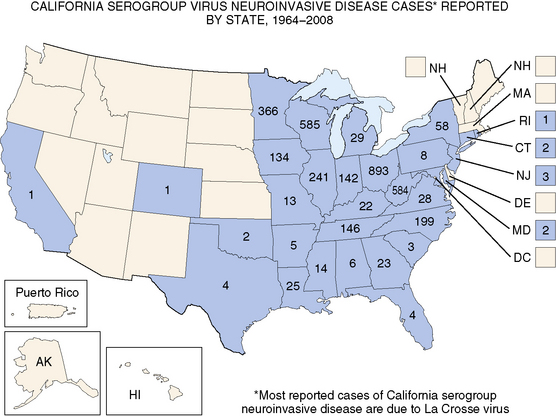
Approximately 70 cases are reported annually.
(Adapted from the Centers for Disease Control and Prevention. Available at: http://www.cdc.gov/ncidod/dvbid/arbor/images/LAC_Map.jpg.)
Members of the herpesvirus family (herpes simplex virus types 1 and 2; cytomegalovirus; Epstein–Barr virus; varicella zoster virus; and human herpesviruses types 6, 7, and 8) display few seasonal or geographic predilections [Whitley et al., 1982]. When Whitley and colleagues reviewed the epidemiology of herpes simplex virus encephalitis, they observed no unique characteristics with regard to race, sex, age, season, or geographic location [Whitley et al., 1982]. Epidemiologic data indicate, however, that herpes simplex virus type 1 is the most common cause of nonepidemic encephalitis in persons older than 50 years. These data reflect the current opinion that most cases of herpes simplex virus encephalitis in older individuals result from reactivated herpes simplex virus infections rather than primary exposures to the virus.
Infection with HIV illustrates well the effects that human behavior and immune competence can have on the spectrum of disease resulting from a neurotropic viral agent. Early in the epidemic of acquired immunodeficiency syndrome (AIDS) in the United States, intravenous drug use, homosexuality, and sexual contact with persons in these groups increased the probability of HIV infection substantially. When CD4+ cell counts decrease to less than 200/mL in persons infected with HIV, opportunistic infections, including infections resulting from other neurotropic viruses, become more likely. The latter conditions include progressive multifocal encephalopathy and cytomegalovirus retinitis and encephalitis [Jacobson, 1997], which are uncommon disorders in immunocompetent persons.
Diagnostic Evaluation
Cerebrospinal Fluid
The CSF in viral meningitis or encephalitis typically has a lymphocytic pleocytosis (5–500 cells/mL), mildly elevated protein content (usually 50–200 mg/dL), and normal or slightly depressed glucose content (Table 81-1). The CSF profile can vary, however, depending on the specific viral pathogen and the timing of the lumbar puncture with respect to the onset of neurological disease. Many patients (5–15 percent) with herpes simplex virus encephalitis have an entirely normal initial CSF examination [Whitley et al., 1982], and 10 percent or more of children with California encephalitis have normal cerebrospinal fluid parameters [McJunkin et al., 2001].
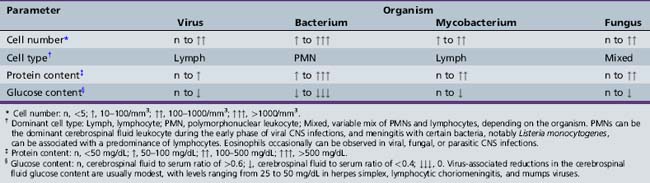
Early in the course of many viral CNS infections, the CSF has a preponderance of neutrophils, thus mimicking bacterial disease, but the CSF profile typically shifts to a lymphocytic pleocytosis over time [Feigin and Shackelford, 1973]. Protein elevations in viral meningitis and encephalitis tend to be modest (often <100 mg/dL), and glucose content in viral diseases, although modestly depressed in some cases (30–50 mg/dL), rarely reaches the low levels (e.g., <20 mg/dL) common in acute bacterial meningitis. None the less, some viral infections, such as eastern equine encephalitis, may have CSF parameters that seem more “bacterial” than “viral” [Przelomski et al., 1988].
Electrodiagnostic Studies
The EEG has an important role in evaluating pediatric patients with suspected viral encephalitis. Although the EEG occasionally can be normal in such patients, infants or children with encephalitis usually have abnormal studies, consisting of slowing or epileptiform discharges that can be diffuse or focal. Because herpes simplex virus encephalitis of children or adults frequently produces focal EEG features [Whitley et al., 1982], consisting of slowing, sharp-wave discharges, or periodic lateralizing epileptiform discharges, the EEG has considerable value in suspected cases of herpes simplex virus encephalitis. Although these abnormalities usually localize to the temporal lobes in children, atypical cases of herpes simplex virus encephalitis with other cortical localizations have been confirmed by MRI and polymerase chain reaction (PCR) detection of herpes simplex virus DNA in the CSF [Schlesinger et al., 1995]. Neonates also may have early EEG abnormalities that alert clinicians to the possibility of herpes simplex virus encephalitis [Mikati et al., 1990; Mizrahi and Tharp, 1982].
The EEG has modest sensitivity and specificity in several other forms of encephalitis. Between 40 and 50 percent of patients with La Crosse encephalitis or eastern equine encephalitis have lateralizing EEG abnormalities that suggest focal brain disease [Hilty et al., 1972; Przelomski et al., 1988]. In many cases, the EEG shows only diffuse slowing of variable severity. In contrast to the abnormalities observed in virus encephalitis, the EEG is usually normal in cases of aseptic meningitis [Pollak et al., 2001].
Neuroimaging
Computed tomography (CT) and MRI have major roles in evaluating infants and children with CNS viral infections [Bale, 1999; Baskin and Hedlund, 2007; Kimberlin, 2004]. Depending on their clinical status, patients with suspected viral encephalitis should undergo a neuroimaging study early in the course of their illnesses. Although large mass lesions, such as tumors or brain abscess, can be detected reliably by CT, especially when obtained with contrast enhancement, MRI has sensitivity that exceeds CT, especially in patients with herpes simplex encephalitis [Baskin and Hedlund, 2007; Domingues et al., 1998; Schroth et al., 1987]. Because of the sensitivity of MRI for the early changes of herpes simplex encephalitis, MRI is the preferred imaging modality in children with suspected viral encephalitis.
After the neonatal period, MRI in herpes simplex encephalitis usually shows T2 prolongation in the medial temporal lobe, orbitofrontal region, or cingulate gyrus, and cortical enhancement in these regions when the paramagnetic agent, gadolinium, is administered intravenously [Baskin and Hedlund, 2007]. Atypical features can be observed, however, including parieto-occipital localization of T2 prolongation and abnormalities of diffusion-weighted images [Leonard et al., 2000; Maschke et al., 2004].
Neuroimaging frequently reveals diffuse or focal brain edema in infants with neonatal herpes simplex encephalitis, a disease that differs pathogenetically from herpes simplex encephalitis of older children in most instances [Bale and Miner, 2005; Kimberlin, 2004]. Neonatal herpes simplex encephalitis usually results from hematogenous transmission of the virus to brain, whereas herpes simplex encephalitis in older individuals usually reflects reactivation and neural transmission of herpes simplex virus.
Several other viral-induced CNS disorders have relatively pathognomonic neuroimaging features. MRI can detect focal abnormalities of the basal ganglia in children with Japanese or Epstein–Barr virus encephalitis [Maschke et al., 2004], and patients with neuroinvasive West Nile virus infections can have imaging abnormalities of the substantia nigra, brainstem, or spinal cord [Maschke et al., 2004]. In acute disseminated encephalomyelitis (ADEM), a condition that mimics virus encephalitis, MRI reveals multifocal areas of T2 prolongation involving the white matter of the cerebrum, cerebellum, and brainstem. The sensitivity of MRI exceeds that of CT in ADEM [Rust, 2000] a condition that often responds to corticosteroid therapy. MRI in other forms of viral encephalitis can be normal; reveal diffuse, nonspecific changes of edema or inflammation; or have focal abnormalities that mimic herpes simplex virus encephalitis [Baskin and Hedlund, 2007]. Children with aseptic meningitis typically have normal brain parenchyma when studied by CT or MRI.
Infants with suspected intrauterine viral infections should be evaluated by CT initially because small intracranial calcifications, a common feature of intrauterine infection, can be detected better by this imaging modality than by MRI or ultrasonography [Bale, 2009; Baskin and Hedlund, 2007]. In addition, most hospitals can perform CT without sedating young infants. These infants may benefit from a subsequent MRI study, however, to identify coexisting brain abnormalities, such as lissencephaly-pachygyria, periventricular leukomalacia, or cortical dysplasia, that are missed or poorly characterized by CT. CT or MRI may reveal hydrocephalus, hydranencephaly, or cystic encephalomalacia in infants with intrauterine or perinatal viral infections.
Microbiologic Evaluation
The diagnostic techniques available to the clinician include serology, cell culture, immunohistology, and molecular methods, such as PCR (Table 81-2). Serology provides indirect evidence of infection because it depends on the host to mount an antibody response specific to the etiologic agent. Certain infections, such as those resulting from Epstein–Barr, lymphocytic choriomeningitis, and West Nile viruses, usually are identified by serologic studies, and others, such as La Crosse, eastern and western equine, St. Louis, and Japanese encephalitis viruses, can be diagnosed rapidly by immunoglobulin M (IgM) antibody capture methods.
Table 81-2 Preferred Diagnostic Methods for Selected Viral Infections of the Nervous System
| Agent | Neonatal Infection | Non-Neonatal Infection |
|---|---|---|
| Herpes simplex virus | Cultures of CSF; swabs of nasopharynx, conjunctivae, and skin lesions; serum and CSF PCR | CSF PCR |
| Cytomegalovirus | Cultures of urine and saliva | Cultures of urine, saliva; serum and CSF PCR |
| Varicella zoster virus | Difficult by any method | Culture of skin vesicle fluid, immunohistology, CSF PCR |
| Epstein–Barr virus | – | Acute and convalescent sera; CSF PCR |
| Human herpesviruses 6, 7, 8 | – | CSF PCR |
| Enteroviruses | Serum and CSF PCRs | Serum and CSF PCRs |
| Arthropod-borne encephalitis viruses | Virus-specific IgM | Virus-specific CSF or serum IgM; CSF PCR; acute and convalescent sera |
| Rabies virus | – | Virus culture, serum IgG after day 15; immunohistology of skin biopsy; CSF PCR |
| Lymphocytic choriomeningitis virus | Serologic studies of infant and mother | Acute and convalescent sera; CSF PCR |
| Human immunodeficiency virus 1 | Plasma PCR | Serologic studies; Western blot; plasma and CSF PCR |
CSF, cerebrospinal fluid; PCR, polymerase chain reaction.
Cell culture provides direct evidence of infection by detecting viral pathogens in CSF, blood, or other body fluids, although cell culture is gradually been supplanted by molecular methods of detecting viruses, such as PCR. Viruses that lend themselves to detection by cell culture include the herpesviruses (especially cytomegalovirus and certain herpes simplex virus infections), enteroviruses, adenoviruses (a rare cause of CNS infection), HIV, and rabies virus. Cultures of CSF or mucosal surfaces often yield virus in neonatal herpes simplex virus infections [Kimberlin, 2004], but infectious virus can be detected in the CSF in less than 5 percent of herpes simplex virus encephalitis cases in children and adolescents. Consequently, PCR has become the gold standard for evaluating infants and children with suspected herpes simplex virus encephalitis or meningitis, especially as laboratories employ molecular detection methods with high sensitivity and specificity [Mitchell et al., 1997; Whitley and Gnann, 2002]. Specific immunohistologic detection of herpes simplex virus and varicella zoster virus has supplanted the nonspecific and insensitive Tzanck preparation.
PCR allows diagnosis of infections with the herpes simplex viruses, cytomegalovirus, enteroviruses, JC virus, human herpesvirus 6, rabies virus, varicella zoster virus, and HIV-1, and can also be used to detect nonviral agents that cause intrauterine infection, encephalitis, or aseptic meningitis [Huttunen et al., 2009; Read et al., 1997]. PCR detects herpes simplex virus DNA in the CSF in most cases of herpes simplex virus encephalitis or meningitis in infants, children, or adolescents [Domingues et al., 1998; Kimberlin et al., 1996; Lakeman and Whitley, 1995; Mitchell et al., 1997; Read et al., 1997]. The specificity of PCR in herpes simplex virus encephalitis approaches 100 percent, whereas the sensitivity ranges from 75 percent in neonates to 95 percent or greater in children or adolescents [Johnson, 1996; Kimberlin, 2004]. Although PCR has an analytical sensitivity and specificity (i.e., the ability to detect viral DNA consistently in “spiked” CSF samples) that approaches 100 percent for many viruses, the clinical sensitivity of CSF PCR is unknown for many viral CNS infections, given the absence of appropriate gold standards against which to compare PCR. Consequently, clinicians must use sound clinical judgment, especially when making decisions regarding the initiation or discontinuation of antiviral therapy.
Overview of Treatment
Therapy for CNS viral infections must be tailored to illness severity, the suspected agent, and the availability of specific antiviral medications. Numerous nonviral conditions can produce clinical signs that mimic signs of viral CNS infection. Children with life-threatening viral diseases, such as herpes simplex virus encephalitis, require hospitalization in a skilled nursing unit, such as a pediatric intensive care unit, which can provide frequent assessment of the child’s responsiveness and anticipatory monitoring for seizures and increased intracranial pressure. Outcome of viral CNS infections varies according to the viral pathogen, the specific disorder, the child’s age, and availability of specific antiviral therapy (Table 81-3).
Table 81-3 Outcome of Selected Viral Central Nervous System Disorders
| Disorder | Mortality (%) | Morbidity (%) |
|---|---|---|
| HERPES SIMPLEX VIRUS* | ||
| Congenital infection | 10 | 100 |
| Neonatal encephalitis | <10 | 40 |
| Non-neonatal encephalitis | <10 | 40 |
| CYTOMEGALOVIRUS | ||
| Symptomatic congenital | 5 | 95 |
| Encephalitis-AIDS | >50 | High |
| ENTEROVIRUS | ||
| Aseptic meningitis | Negligible | Negligible |
| Encephalitis | Negligible | ≤10 |
| TOGAVIRUSES | ||
| Eastern equine encephalitis | 40 | 50 |
| Western equine encephalitis | 10 | 15 |
| RUBELLA VIRUS | ||
| Symptomatic congenital | 5 | 90 |
| FLAVIVIRUSES | ||
| Japanese encephalitis | 40 | 50 |
| St. Louis encephalitis | 10 | 20 |
| West Nile virus | 5 | 33 |
| BUNYAVIRUSES | ||
| La Crosse encephalitis | Negligible | ≤15 |
| PARAMYXOVIRUSES | ||
| Measles encephalomyelitis | 20 | 50 |
| Subacute measles encephalitis | 80 | 100 |
| SSPE | 100 | – |
| Rabies virus† | Nearly 100 | – |
| LYMPHOCYTIC CHORIOMENINGITIS VIRUS | ||
| Symptomatic congenital | 40 | 80 |
| Aseptic meningitis | Negligible | Negligible |
| RETROVIRUSES‡ | ||
| HTLV-I | Variable | 100 |
| HIV | Variable | Variable |
AIDS, acquired immunodeficiency syndrome; HIV, human immunodeficiency virus; HTLV-I, human T-cell lymphotropic virus type I; SSPE, subacute sclerosing panencephalitis.
* Assumes appropriate management with acyclovir. Rates for untreated cases are higher.
† Survival reported in <10 persons (total) with symptomatic rabies [American Academy of Pediatrics, 2009].
‡ Outcomes related to current management.
Supportive Care
Children or adolescents with optic neuritis, myelitis, inflammatory vasculitis, or acute disseminated encephalomyelitis often benefit from corticosteroid therapy. The successful treatment of optic neuritis in adults serves as a model for the pharmacologic treatment of several inflammatory/immune-mediated conditions in childhood or adolescence. Children can receive 2 weeks of daily corticosteroids, beginning with a short course of high-dose methylprednisolone intravenously (e.g., 15 mg/kg/day divided equally every 6 hours for 3–5 days), followed by prednisone orally (e.g., 1–2 mg/kg/day for 7–10 days). Some authorities have utilized a short (3–5 days only), high-dose corticosteroid regimen, consisting of 15–20 mg/kg/day of methylprednisolone intravenously in equally divided doses every 6 hours [Tenembaum et al., 2007]. Children with postviral Guillain–Barré syndrome may improve after intravenous immunoglobulin [Abd-Allah et al., 1997; Jansen et al., 1992].
Specific Viral Infections
Herpesviruses
Herpes Simplex Virus
Clinical features
Most humans acquire herpes simplex virus type 1 by oral transmission during childhood [Chayavichitsilp et al., 2009; Corey and Spear, 1986]. Primary herpes simplex virus infections are frequently asymptomatic, although some children experience vesicular pharyngitis or gingivostomatitis, a condition associated with fever, diminished oral intake, drooling, and herpetic lesions of the face and mouth. By contrast, herpes simplex virus type 2, usually acquired by sexual contact, causes genital lesions in young adults [Corey and Wald, 2009]. In children, herpes simplex virus-induced neurologic infections can be due to primary or reactivated infections, whereas most herpes simplex virus-induced disease after age 30 years, whether due to herpes simplex virus type 1 or type 2, reflects reactivated infection.
Neonatal herpes simplex virus infection, most often due to herpes simplex virus type 2, can be categorized as (1) skin, eyes, and mouth disease; (2) disseminated disease; or (3) encephalitis, affecting approximately 43 percent, 23 percent, and 34 percent of herpes simplex virus-infected infants, respectively [Corey and Wald, 2009; Kimberlin, 2004; Malm, 2009; Whitley et al., 1988]. Neonatal herpes simplex virus disease usually begins with nonspecific clinical features (poor oral intake, behavioral change, or fever), as early as the first day of life, but more often in the second or third weeks [Kimberlin, 2004; Koskiniemi et al., 1989]. Neonatal herpes simplex encephalitis produces focal or generalized seizures, paralysis, apnea, and lethargy or coma [Bale and Miner, 2005; Kimberlin, 2004; Koskiniemi et al., 1989; Malm, 2009; Whitley et al., 1988]. Herpes simplex encephalitis in neonates can be accompanied by pneumonitis, hepatitis, or disseminated intravascular coagulopathy, but one-third of infants with neonatal herpes simplex virus encephalitis have disease restricted to the CNS [Koskiniemi et al., 1989]. Approximately two-thirds of infants have a vesicular rash. The infection can be rapidly fatal, with multiorgan system failure, or be associated with apnea, impaired bulbar function, and intractable seizures. Approximately 5 percent of infants infected with herpes simplex virus acquire the virus in utero and display a relatively characteristic pattern of physical signs consisting of microcephaly, cataracts, intracranial calcifications (Figure 81-4), intrauterine growth retardation, and vesicular rash or other skin lesions [Grose, 1994; Hutto et al., 1987; Koch et al., 2009].
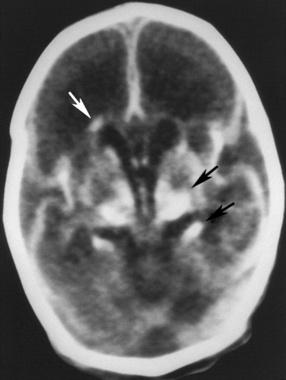
Fig. 81-4 Unenhanced computed tomography scan of an infant with congenital herpes simplex virus infection.
(From Souza IE, Bale JF. The diagnosis of congenital infections: Contemporary strategies. J Child Neurol 1995;10:271.)
In older children or adolescents, herpes simplex encephalitis, typically herpes simplex virus type 1, begins nonspecifically with headache, fever, malaise, vomiting, and behavioral changes [Hsieh et al., 2007]. Focal neurologic signs, such as hemiparesis, dysphasia, or visual field defects, appear subsequently in most patients. Seizures affect approximately 40 percent of patients with biopsy-confirmed herpes simplex encephalitis; seizures can be generalized but are more often partial [Whitley et al., 1982]. Focal abnormalities reflect the predilection of herpes simplex virus for infecting frontotemporal brain regions, but other areas of the brain can be affected [Leonard et al., 2000]. The clinical course of herpes simplex encephalitis can be rapidly progressive, with coma and intractable seizures, or indolent, with memory loss and behavioral disturbances.
In addition to causing necrotizing encephalitis, herpes simplex virus has been linked to Bell’s palsy by detecting herpes simplex virus type 1 DNA in facial nerve tissues or in the geniculate ganglia [Burgess et al., 1994]. Herpes simplex virus also has been associated with primary or recurrent aseptic meningitis (Mollaret meningitis) [Bergström et al., 1990]. Common clinical manifestations of herpes simplex virus-associated aseptic meningitis include fever, photophobia, vomiting, and headache, but children with herpes simplex virus-induced meningitis usually lack herpetic lesions of the skin or mucosal surfaces [Rathore et al., 1996]. Although children with herpes simplex virus type 1 meningitis can recover uneventfully without therapy [Rathore et al., 1996], acyclovir therapy should be administered. Herpes simplex virus infection also has been associated with a necrotizing myelopathy in childhood, although it more commonly occurs in adults with reactivated herpes simplex virus infections [Ellstein-Shturman et al., 1976].
Diagnosis
Neonates with herpes simplex virus infections may have elevated serum transaminases, direct hyperbilirubinemia, and laboratory features of disseminated intravascular coagulopathy [Kimberlin, 2004; Koskiniemi et al., 1989]. The CSF in neonatal herpes simplex encephalitis reveals a lymphocytic pleocytosis and elevations of the protein content; the CSF glucose can be normal or mildly low. MRI and CT usually reveal diffuse edema during the acute stage, and later exhibit atrophy, parenchymal calcification, or cystic encephalomalacia [Tien et al., 1993; Vossough et al., 2008]. MRI can reveal a “watershed” distribution of abnormalities [Vossough et al., 2008]. EEGs are frequently abnormal in infants with encephalitis, revealing slow background activity and paroxysmal discharges that occasionally are periodic [Mikati et al., 1990].
Herpes simplex virus infection of the newborn can be confirmed by isolating virus from CSF, skin lesions, or mucosal surfaces of the eye, oropharynx, or rectum, or by detecting herpes simplex virus DNA in CSF or serum by using PCR. In a retrospective analysis of CSF samples accumulated by the National Institutes of Health–Collaborative Antiviral Study Group, herpes simplex virus DNA was detected by PCR in the CSF of 93 percent of the infants with disseminated infection, 76 percent of infants with encephalitis, and only 24 percent of infants with skin-eyes-mouth disease [Kimberlin et al., 1996].
Because older infants, children, and adolescents with herpes simplex virus encephalitis rarely have systemic signs of infection, neurodiagnostic and CSF PCR studies have major roles in establishing the diagnosis [Mitchell et al., 1997; Whitley and Lakeman, 1995; Whitley et al., 1982]. MRI may indicate unilateral or bilateral abnormalities consisting of T2 prolongation in the temporal lobe or insula, and enhancement of the insula, temporal cortex, and cingulate gyrus on T1-weighted, gadolinium-enhanced images [Schroth et al., 1987; Tien et al., 1993]. Focal abnormalities involving other cortical regions, including parietal or occipital lobes, can be observed, however, in atypical cases of herpes simplex encephalitis [Schlesinger et al., 1995]. EEGs, abnormal in approximately 80 percent or more of patients, reveal focal or diffuse slowing and epileptiform discharges that can be periodic and lateralizing.
In National Institutes of Health–Collaborative Antiviral Study Group studies of adults, herpes simplex virus DNA was detected in the CSF of 98 percent of patients with biopsy-proven herpes simplex virus encephalitis, whereas only 6 percent of biopsy-negative patients had herpes simplex virus DNA in CSF [Lakeman and Whitley, 1995]. These results, as well as results from other laboratories, indicate that the sensitivity and specificity of PCR in herpes simplex virus encephalitis of children or adolescents exceed 90–95 percent [Mitchell et al., 1997; Read et al., 1997]. None the less, clinicians must use caution when interpreting the results of CSF PCR studies for herpes simplex virus, especially when negative. Full courses of acyclovir may be necessary when clinical features, especially MRI findings, are compatible with herpes simplex virus encephalitis but CSF PCR studies are negative.
Treatment and outcome
Infants with herpes simplex virus infections should receive acyclovir, 20 mg/kg every 8 hours for 14 days in skin and mucous membrane infections, and 20 mg/kg every 8 hours for 21 days in disseminated infections or encephalitis [Bale and Miner, 2005; James et al., 2009; Kimberlin et al., 2001b; Whitley et al., 1986, 1991a, b]. CSF PCR should be obtained at 21 days in the latter infants, and if the PCR remains positive for herpes simplex virus DNA, acyclovir should be continued for another 7 days. Children with herpes simplex virus encephalitis should be treated with 20 mg/kg every 8 hours for 21 days, and adolescents can receive 1500 mg/m2 for 21 days. Potential side effects include tremulousness, hematuria, and reversible nephropathy. The drug is very well tolerated, although complete blood counts and serum creatinine levels should be monitored during prolonged courses of acyclovir. Although favorable outcome after neonatal or childhood herpes simplex virus infections correlates with the early initiation of acyclovir, sequelae are common, even after appropriate medical management.
All acyclovir-treated infants with skin, eye, and mouth infections with herpes simplex virus survive, and nearly all such infants have no long-term sequelae of their infections, other than the propensity to have recurrent herpetic skin lesions [Kimberlin et al., 2001a]. As many as 30 percent of the infants with disseminated neonatal herpes simplex virus infections die, however, despite appropriate therapy, and 20–30 percent of the survivors have sequelae ranging from mild (speech delay or mild motor delay) to severe (spastic quadriparesis or severe developmental delay). Neonates with encephalitis have less than 10 percent mortality, but approximately 40 percent of the surviving infants have severe sequelae, despite acyclovir therapy [Kimberlin et al., 2001b]. Infants who survive herpes simplex virus encephalitis can have permanent pseudobulbar palsy with mutism and feeding difficulties (Figure 81-5). Infants with congenital herpes simplex virus infections generally have poor prognoses [Grose, 1994; Hutto et al., 1987]. Potential sequelae of herpes simplex virus encephalitis in children and adolescents consist of partial and generalized seizure disorders, developmental delay, cerebral palsy, language and memory dysfunction, behavioral abnormalities, and motor deficits [Hsieh et al., 2007; Kimberlin et al., 2001a, b; Whitley et al., 1991b].
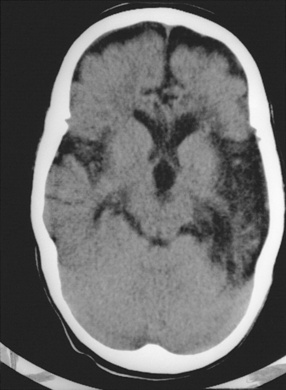
Fig. 81-5 Unenhanced computed tomography scan of a young child who survived herpes simplex virus encephalitis.
Relapse or persistence of symptoms in herpes simplex virus encephalitis, associated with fever, lethargy, headache, and movement disorder, can develop despite appropriate initial management and acyclovir therapy [Barthez-Carpentier et al., 1995; Gutman et al., 1986]. Such children require repeat CSF PCR studies for herpes simplex virus, MRI, repeat treatment with acyclovir, and corticosteroid therapy if imaging results suggest acute disseminated encephalitis. Because of the substantial morbidity associated with neonatal and childhood herpes simplex virus encephalitis, on-going studies are evaluating the role for prolonged suppressive therapy with acyclovir in neonates with herpes simplex virus infections [James et al., 2009; Kimberlin, 2004].
Cytomegalovirus
Clinical features
Infection with cytomegalovirus, a ubiquitous member of the herpesvirus family, has been associated with several childhood neurologic conditions, including intrauterine infection; infantile spasms; Guillain–Barré syndrome; and encephalitis, myelitis, or retinitis in immunosuppressed hosts, especially persons with AIDS [Bale, 1999; McCutchan, 1995]. Approximately 1 percent of infants excrete cytomegalovirus at birth, and the virus is acquired steadily thereafter throughout life. Young children who attend daycare centers, adults who have contact with young children, and persons with multiple sexual partners acquire cytomegalovirus at annual rates of 8–25 percent or more, considerably higher than the background infection rates of 2–5 percent per year [Demmler, 1991; Murph and Bale, 1988]. Children acquire cytomegalovirus by contact with persons who shed cytomegalovirus in urine or saliva, and by transfusion of blood products or transplantation of organs from cytomegalovirus-seropositive donors [Demmler, 1991].
At birth, 0.4–2.5 percent of newborns shed cytomegalovirus in urine or saliva, indicating congenital infection [Demmler, 1991; Malm and Engman, 2007]. Approximately 90 percent of cytomegalovirus-infected infants are asymptomatic at birth. Although sensorineural hearing loss develops subsequently in 7–10 percent of these infants [Fowler et al., 1997], they rarely have other CNS complications. The remaining 10 percent of infants with congenital cytomegalovirus infection have hepatomegaly, splenomegaly, jaundice, petechial or purpuric skin rash, intrauterine growth retardation, microcephaly, or chorioretinitis [Boppana et al., 1997; Demmler, 1991; Malm and Engman, 2007].
Cytomegalovirus has been linked to infantile spasms, usually in infants with intrauterine infection. Immunocompetent children or adolescents with cytomegalovirus infections can have a mild, self-limited encephalitis [Studahl et al., 1992] or Guillain–Barré syndrome. The latter disorder begins with paresthesias and causes weakness that usually begins in the lower extremities. Affected children can have bilateral facial paresis, respiratory failure, or cardiac dysrhythmias. Symptoms of a cytomegalovirus-induced mononucleosis syndrome [Horwitz et al., 1986] – malaise, low-grade fever, or headache – may precede or accompany the onset of Guillain–Barré syndrome.
Patients immunocompromised by AIDS, malignancy, transplantation, or immunosuppressive therapy are at risk for cytomegalovirus-induced retinitis, encephalitis, myelopathy, and Guillain–Barré syndrome-like disorders. Given the demographics of AIDS, most reported patients with cytomegalovirus retinitis, encephalitis, or myelitis are adults between the ages of 20 and 50 years [Holland et al., 1994; Jacobson, 1997]. Immunocompromised infants or children, especially those with HIV infection, are at risk for retinitis, colitis, pneumonitis, and CNS complications of cytomegalovirus infections [Darin et al., 1994].
Diagnosis
The diagnosis of intrauterine cytomegalovirus infection can be established by assaying urine or saliva using the shell vial cell culture method. PCR can detect cytomegalovirus DNA in urine, saliva, or serum, but the shell vial assay is sensitive and cost-effective [Demmler et al., 1988]. Cytomegalovirus DNA can also be detected by PCR analysis of dried blood spots obtained from newborn infants, but the sensitivity of this detection method does not exceed that of urine or saliva assay [Atkinson et al., 2009]. Common, nonspecific laboratory features of congenital cytomegalovirus infections include thrombocytopenia, direct hyperbilirubinemia, and elevated serum transaminases [Demmler, 1991].
CT scans reveal intracranial calcifications (Figure 81-6) in 25–50 percent of symptomatic infants [Boppana et al., 1997; Pass et al., 1980]. Hydranencephaly, atrophy, lissencephaly, schizencephaly, and cerebellar hypoplasia also can accompany congenital cytomegalovirus infections [de Vries et al., 2004; Iannetti et al., 1998], and white-matter abnormalities are commonly seen when congenitally infected infants undergo brain MRI [van der Knapp et al., 2004]. Approximately 50 percent of symptomatic infants have abnormal auditory-evoked responses, indicating sensorineural hearing loss [Boppana et al., 1997; Pass et al., 1980; Rivera et al., 2002].
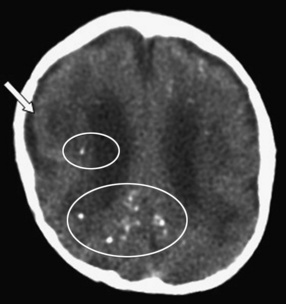
Infection in older infants or children can be established by isolating cytomegalovirus from urine, saliva, or circulating leukocytes; by detecting cytomegalovirus-specific antigens in circulating leukocytes; or by detecting cytomegalovirus DNA in CSF, serum, or urine [Clifford et al., 1993]. Some patients with cytomegalovirus infection have elevated serum transaminases or hematologic abnormalities, such as leukopenia, anemia, or thrombocytopenia. CT or MRI may reveal abnormalities of the brain or spinal cord consisting of demyelination or inflammation involving cortical or periventricular regions [Miller et al., 1997].
Treatment and outcome
Infants who survive symptomatic intrauterine cytomegalovirus infections often have permanent neurodevelopmental sequelae consisting of mental retardation, seizure disorders, behavioral disturbances, visual impairment, and sensorineural hearing loss [Boppana et al., 1997; Malm and Engman, 2007; Pass et al., 1980]. Because hearing loss can be progressive, even after silent, intrauterine cytomegalovirus infection, infants with known congenital cytomegalovirus infection require serial audiometry at frequent intervals during infancy and childhood [Fowler et al., 1997]. Infants with abnormal CT scans are more likely to have adverse neurodevelopmental outcomes [Boppana et al., 1997; Noyola et al., 2001].
Although intrauterine cytomegalovirus infection cannot currently be prevented by vaccination, recent studies suggest that a vaccine based on glycoprotein B, a major immunogenic protein of human cytomegalovirus, can reduce the risk of maternal infection [Pass et al., 2009]. Ganciclovir, an antiviral drug closely related to acyclovir, provides some benefit to infants with congenital cytomegalovirus disease. A controlled trial of ganciclovir indicated that prolonged courses of ganciclovir, 10–12 mg/kg/day for 42 days, can improve hearing outcomes [Kimberlin et al., 2003]. Based on these encouraging data, ganciclovir should be considered for infants with congenital cytomegalovirus disease, given the high risk of sensorineural hearing loss in such infants. Bone marrow suppression and line infections can be potential adverse effects of prolonged intravenous therapy. Studies of oral therapy, using the prodrug valganciclovir, are on-going.
Immunocompromised children with invasive cytomegalovirus disease, especially retinitis, should receive ganciclovir, beginning with 5 mg/kg intravenously twice daily for approximately 14 days. Long-term maintenance therapy, using 5–10 mg/kg/day, is required in persons with cytomegalovirus retinitis and HIV infection [Jacobson, 1997]. Complete blood counts and renal function should be monitored frequently. Outcome of acquired cytomegalovirus infection depends greatly on the immunocompetence of the infected host. Patients who are immunosuppressed by AIDS or transplantation have increased risks of death during systemic or CNS cytomegalovirus infections. By contrast, most children with normal immune systems generally recover from acquired cytomegalovirus infections without lasting sequelae. The prognosis of cytomegalovirus-induced Guillain–Barré syndrome in immunocompetent children is comparable with that of Guillain–Barré syndrome resulting from other causes.
Varicella Zoster Virus
Clinical features
Varicella zoster virus, the cause of chickenpox (varicella) and shingles (zoster), has been associated with several neurologic conditions in children, including acute ataxia, myelitis, stroke, postherpetic neuralgia, Bell’s palsy, Ramsay Hunt syndrome, aseptic meningitis, encephalitis, congenital varicella syndrome, and Reye’s syndrome [Gilden, 2004; Weller, 1983]. These conditions can affect immunocompetent or immunocompromised hosts, although immunosuppressed hosts, such as patients with HIV/AIDS or hematologic malignancies, have greater risks of severe, invasive varicella zoster virus infections. Certain conditions, such as varicella-induced ataxia or Reye’s syndrome (rarely seen, currently), occur almost exclusively in immunocompetent children.
Chickenpox, the illness associated with primary varicella zoster virus infection, peaks among children between the ages of 5 and 9 years and occurs more often in winter months. The virus spreads by aerosolization and respiratory transmission; children are contagious during the interval encompassing 2 days before and 5 days after the onset of the chickenpox rash. The incubation period averages 14 days. After primary infection, the virus enters a latent stage in sensory ganglia and can reactivate, causing zoster or shingles [Gilden, 2004]. Consequently, zoster more commonly affects the elderly or persons with immunocompromising conditions. Zoster can occur, however, in otherwise healthy children or adolescents [Guess et al., 1985]. Less than 0.1 percent of otherwise healthy children with chickenpox experience neurologic complications. Acute cerebellar ataxia, the most common complication, begins approximately 10 days after the onset of the rash, although ataxia preceding the rash or after varicella vaccination has been reported [Sunaga et al., 1995]. Truncal ataxia, dysmetria, vomiting, and irritability are common clinical features. The ataxia, often maximum at onset, frequently inhibits independent ambulation, and nystagmus can be present [Connolly et al., 1994]. The precise pathogenesis of ataxia after varicella, although presumed to be immune-mediated, has not been fully elucidated.
Other varicella zoster virus-related neurologic complications are uncommon. Reye’s syndrome, a now-rare disorder characterized by pernicious vomiting, coma, and increased intracranial pressure, can be prevented by avoiding aspirin therapy in children or adolescents with chickenpox or influenza. Varicella encephalitis, another rare complication of either varicella or zoster, usually appears 3–7 days after the onset of the rash and produces headache, fever, seizures, coma, or paralysis [Gilden, 2004; Hausler et al., 2002]. Myelitis causes urinary retention or incontinence, paraparesis, and sensory abnormalities [Rosenfeld et al., 1993]. Ramsay Hunt syndrome, an uncommon complication of zoster in childhood, consists of acute facial palsy, pain, unilateral hearing loss, and occasionally other cranial palsies in association with zoster of the ear or neck [Grose et al., 2002]. Children infrequently have postherpetic neuralgia.
Stroke [Askalan et al., 2001; Miravet et al., 2007; Sebire et al., 1999] or, rarely, herpes zoster ophthalmicus [Gilden, 2004; Leis and Butler, 1987] has been reported in children after varicella. Usually occurring within 4–8 weeks of varicella or zoster infection, these disorders produce focal deficits, headache, lethargy or somnolence, and occasionally seizures. The pathogenesis of stroke in these pediatric cases appears to reflect vasculopathy mediated by varicella zoster virus, as occurs in herpes zoster ophthalmicus and delayed infarction, a potential complication of varicella zoster virus reactivation in adults [Hilt et al., 1983]. The intrauterine varicella syndrome affects approximately 2–3 percent of the offspring of women who have chickenpox during their first or second trimesters of pregnancy [Alkalay et al., 1987; Brunnell, 1992; Grose, 1994]. The characteristics of this disorder include intrauterine growth retardation, cicatricial skin lesions in a dermatomal distribution, eye abnormalities (chorioretinitis, cataracts, optic atrophy), limb hypoplasia, and neurologic abnormalities (hydrocephalus, microcephaly, seizures, or Horner’s syndrome) [Alkalay et al., 1987; Grose, 1994].
Diagnosis
Children or adolescents with neurologic conditions associated with varicella zoster virus have CSF abnormalities consisting of a lymphocytic pleocytosis and elevated protein content. Approximately 50 percent of children with ataxia after varicella have a lymphocytic pleocytosis, although the total leukocyte count and protein content rarely exceed 100 cells/mm3 and 40 mg/dL, respectively [Connolly et al., 1994]. Varicella zoster virus infection is suspected when characteristic skin lesions are present and confirmed by detecting infectious virus in skin lesions or varicella zoster virus-specific antibodies or varicella zoster virus DNA in CSF using PCR [Gilden, 2004; Kido et al., 1991]. Some adults and, presumably, some children or adolescents with varicella zoster virus-related neurologic complications lack cutaneous manifestations of varicella zoster virus [Gilden, 2004]. This is especially true when remote complications of varicella zoster virus, such as childhood stroke, occur [Askalan et al., 2001].
The results of neuroimaging studies vary, depending on the disorder and immune status of the child. Children with ataxia or encephalitis after varicella often have normal imaging studies, although MRI occasionally can detect edema or white-matter lesions compatible with demyelination. Occasionally, features suggesting acute disseminated encephalitis are observed. MRI indicates focal cerebral or cerebellar lesions in children with stroke [Leis and Butler, 1987; Sunaga et al., 1995], and hydrocephalus, cortical atrophy, hydranencephaly, or intracranial calcifications in infants with congenital varicella zoster virus embryopathy [Alkalay et al., 1987; Grose, 1994].
Treatment and outcome
Children with varicella zoster virus-related ataxia recover without antiviral or corticosteroid therapy [Connolly et al., 1994], indicating that conservative management of these children is warranted. Virtually all children with varicella-induced ataxia recover completely within 8 weeks of onset, although behavioral disturbances can persist longer. Children or adolescents with encephalitis or myelitis should receive acyclovir, 1500 mg/m2/day in three equally divided doses for at least 10 days, especially if immunocompromised by HIV/AIDS, solid organ or stem-cell transplantation, or chemotherapy [Gilden, 2004]. Children with Ramsay Hunt syndrome also may benefit from acyclovir.
Corticosteroid therapy (e.g., intravenous methylprednisolone) should be considered in children or adolescents with myelitis, acute disseminated encephalitis, or stroke secondary to varicella zoster virus vasculitis. Outcome of varicella-related stroke in children seems to be more favorable than the outcome in adults. Infants with intrauterine infections have variable outcomes, with high rates of permanent neurodevelopmental sequelae [Grose, 1994]. Varicella can be prevented by vaccination, suggesting that certain neurologic complications also may be preventable.
Epstein–Barr Virus
Clinical features
Epstein–Barr virus, the cause of infectious mononucleosis, has been associated with several neurologic disorders of children and adults, including encephalitis, Guillain–Barré syndrome, optic neuritis, Bell palsy, acute ataxia, chronic daily headaches, acute chorea, and “Alice in Wonderland” syndrome [Connelly and DeWitt, 1994; Doja et al., 2006; Volpi, 2004]. Epstein–Barr virus, transmitted by contact with oral secretions, commonly infects children before the age of 5 years, children and adolescents after age 10 years, and young adults. Most primary infections are asymptomatic, although infected individuals can experience infectious mononucleosis, an illness associated with fever, malaise, headache, sore throat, lymphadenopathy, splenomegaly, and rash [Sumaya and Ench, 1985a].
Epstein–Barr virus encephalitis accounts for 5 percent of cases of acute encephalitis and produces fever, headache, altered consciousness, and seizures, including acute status epilepticus [Connelly and DeWitt, 1994; Doja et al., 2006; Domachowske et al., 1996]. In the National Institutes of Health–Collaborative Antiviral Study Group studies, Epstein–Barr virus was the most common infectious agent mimicking herpes simplex virus encephalitis [Whitley et al., 1989]. “Alice in Wonderland” syndrome, another cerebral manifestation of Epstein–Barr virus infection, causes personality changes and illusions of distorted size, shape, or distance (metamorphopsia) [Liaw and Shen, 1991].
Other neurologic conditions attributed to Epstein–Barr virus, such as optic neuritis [Frey, 1973], Guillain–Barré syndrome, acute hemiplegia, and acute ataxia [Connolly et al., 1994], lack unique, distinguishing features that allow the clinical diagnosis of Epstein–Barr virus infection. The association of Epstein–Barr virus with these disorders often has been established retrospectively by serologic studies and exclusion of other potential causative factors. The lymphoproliferative syndrome associated with Epstein–Barr virus in transplant patients may cause seizures or altered sensorium as a consequence of intracranial mass lesions or lymphomatous meningitis [Boyle et al., 1997].
Diagnosis, treatment, and outcome
Children with Epstein–Barr virus infection frequently have elevated serum transaminases and hematologic abnormalities, including thrombocytopenia, leukopenia, and an atypical lymphocytosis [Sumaya and Ench, 1985a]. A positive heterophil response supports recent Epstein–Barr virus infection, although children younger than 4 years frequently have false-negative results [Sumaya and Ench, 1985b]. The diagnosis of Epstein–Barr virus infection can be established specifically by a serologic panel that assays Epstein–Barr virus-specific antibody responses to viral capsid, early, and nuclear antigens [Sumaya, 1985]. Acute infection is suggested by the presence of viral capsid antigen IgM or IgG and the absence of Epstein–Barr nuclear antigen IgG [Sumaya and Ench, 1985b]. Epstein–Barr virus infection of the CNS also can be diagnosed by detecting Epstein–Barr virus DNA in CSF [Landgren et al., 1994; Volpi, 2004].
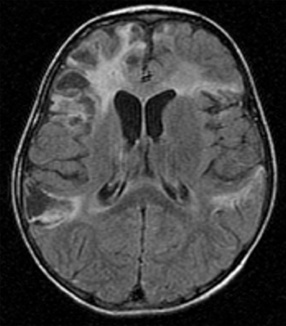
EEG may demonstrate slowing or epileptiform discharges during acute Epstein–Barr virus encephalitis or in the “Alice in Wonderland” syndrome. Neuroimaging studies occasionally can be abnormal (Figure 81-7) [Bale, 1999; Domachowske et al., 1996; Baskin and Hedlund, 2007]. Current antiviral medications generally have minimal effect on CNS infections due to Epstein–Barr virus, although ganciclovir has been used with success during systemic and CNS Epstein–Barr virus infections [Adams et al., 2006]. Although most children with Epstein–Barr virus encephalitis recover completely with supportive care, severe CNS damage can occur and may have permanent neurodevelopmental sequelae (Figure 81-8) [Domachowske et al., 1996].
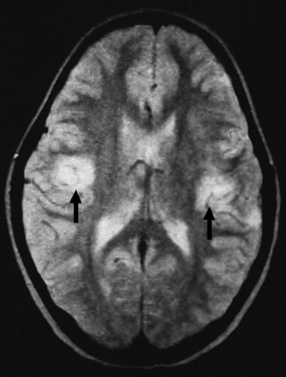
Human Herpesviruses Types 6, 7, and 8
Human herpesviruses type 6 and type 7, discovered in 1986 and 1990, cause roseola (exanthem subitum) in young children, and either virus has been associated with neurologic complications in children or adults [Leach et al., 1992; Torigoe et al., 1996]. These viruses commonly infect infants and toddlers, such that virtually all children have serologic evidence of infection by 5 years of age. Although primary infections are frequently asymptomatic, human herpesvirus type 6 and human herpesvirus type 7 produce roseola, an acute childhood illness with high fever (often >40°C), lymphadenopathy, and an erythematous rash that characteristically appears after the fever. By causing high fever at a vulnerable age, these viruses account for a substantial proportion of febrile seizures of childhood [Barone et al., 1995; Hall et al., 1994].
In addition to inducing febrile seizures, human herpesvirus type 6 and human herpesvirus type 7 have been linked with acute encephalopathy or encephalitis in young children, and myelitis or encephalitis in adults [Barone et al., 1995; Gewurz et al., 2008; Hall et al., 1994; Leach et al., 1992; Suga et al., 1993; Torigoe et al., 1996]. Young children with symptomatic infections have high fevers and seizures that are usually generalized. Transient or permanent hemiparesis and coma are additional clinical features. The diagnosis can be established serologically or by PCR [Yoshikawa et al., 2000].
Human herpesvirus type 8, a herpesvirus that was linked to Kaposi’s sarcoma in 1994, is acquired after adolescence in the United States and Europe, an epidemiologic feature that suggests sexual transmission [Chang et al., 1994; Martin et al., 1998]. Nonsexual modes contribute to the transmission of human herpesvirus type 8 in endemic regions of the developing world, however [Andreoni et al., 1999]. Although neurologic complications of human herpesvirus type 8 are rare, human herpesvirus type 6 and human herpesvirus type 7, as well as human herpesvirus type 8, can be opportunistic CNS pathogens in persons with impaired cell-mediated immunity [Volpi, 2004]. The role of these viruses in other neurological conditions, such as multiple sclerosis and CNS neoplasms, continues to be an area of scientific investigation [Crawford et al., 2009; Dewhurst 2004].
Adenoviruses
The adenoviruses, important causes of mild upper respiratory illnesses, pneumonia, and keratoconjunctivitis, have been associated occasionally with encephalitis or aseptic meningitis [Chuang et al., 2003]. Acquired by contact with infected respiratory or conjunctival secretions, childhood adenoviral infections produce low-grade fever, cough, coryza, and conjunctivitis. Adenovirus infections usually remit spontaneously. Infrequently, they can cause coma, seizures, and meningeal signs. The diagnosis can be established by isolating adenoviruses from CSF, respiratory secretions, or urine. Treatment consists of supportive care. Although fatal cases have been described, most children recover uneventfully [Chuang et al., 2003].
Picornaviruses
Clinical features
Members of the picornavirus family, especially the enteroviruses, are important potential causes of neurologic infections worldwide. Although paralytic poliomyelitis has been eliminated from nearly all regions of the world, nonpolio enteroviruses continue to be associated with a spectrum of neurologic disease that includes aseptic meningitis, acute ataxia, acute hemiplegia, opsoclonus-myoclonus, encephalitis, Guillain–Barré syndrome, and poliolike paralysis [Alexander et al., 1994; Chen et al., 2001; Huang et al., 1999; Rorabaugh et al., 1993; Sawyer, 2002]. The last case of wild poliomyelitis in the United States occurred in 1979 (see Figure 81-1), and the World Health Organization has certified that the Americas, Europe, and the western Pacific regions are free of polio [CDC, 1994a, 2004b]. The numbers of wild poliovirus infections have declined dramatically from approximately 350,000 cases worldwide in 1988, the year in which the World Health Organization established the global eradication initiative, to approximately 1700 in 2008 [CDC, 2009a]. Eradicating poliomyelitis from the four remaining endemic locations (Afghanistan, India, Nigeria, and Pakistan) remains the objective of the World Health Organization [CDC, 2009a]. Before the use of the inactivated poliovirus vaccine for the complete childhood immunization series, vaccine-related poliomyelitis occurred occasionally in the United States [CDC, 1997b]; vaccine-associated poliomyelitis continues to be observed in countries relying upon the live oral polio vaccine [CDC, 2009b].
Spread by the fecal–oral route, the nonpolio enteroviruses, coxsackieviruses, and echoviruses produce several distinct systemic syndromes, including pharyngitis, herpangina, gastroenteritis, hand-foot-and-mouth syndrome, neonatal sepsis, and neurologic disorders. Data from the CDC indicate that approximately 10 million or more cases of nonpolio enteroviral infections occur annually in the United States [CDC, 2000]. More than 10,000 cases of nonpolio enterovirus aseptic meningitis are reported to the CDC annually [Sawyer, 2002]; outbreaks of aseptic meningitis appear sporadically in association with several echovirus serotypes, including 9, 13, 18, and 30 [CDC, 2006].
Aseptic meningitis, the most common neurologic manifestation of nonpolio enteroviral infection, appears epidemically during the late summer and early fall, although cases can be observed throughout the year. Affected children have fever, irritability, headache, photophobia, nausea, vomiting, meningeal signs, and other signs of enteroviral disease, including diarrhea, abdominal discomfort, and a maculopapular or petechial rash [Rorabaugh et al., 1993; Sawyer, 2002]. Signs of aseptic meningitis in a young infant may be remarkably subtle, consisting only of fussiness or poor oral intake. Fever is an inconsistent feature.
Occasionally, children with nonpolio enteroviral infections have an acute encephalitic presentation with fever, seizures, somnolence, coma, or focal deficits [Fowlkes et al., 2008]. Such children may have focal clinical, EEG, or neuroimaging abnormalities that resemble herpes simplex encephalitis. Children with acute opsoclonus-myoclonus, a syndrome more commonly associated with occult neural crest tumors, have involuntary, chaotic eye movements and random myoclonus of the extremities. Enterovirus 71, in addition to being associated with encephalitis and aseptic meningitis, causes an acute neurologic disorder with unilateral or bilateral flaccid paralysis that can resemble poliomyelitis or Guillain–Barré syndrome [Alexander et al., 1994; Chen et al., 2001; Huang et al., 1999]. Several other nonpolio enteroviruses have been linked to acute poliomyelitis-like syndromes.
Children with poliomyelitis have a biphasic illness; the initial, nonspecific phase consists of fever, malaise, vomiting, or diarrhea; the second, neurologic phase consists of aseptic meningitis or paralytic disease, usually more severe in the lower extremities [CDC, 1997b]. Mild or inapparent poliovirus infections greatly exceed the number of paralytic cases. Although the pathogenesis of their condition remains uncertain, persons with paralytic poliomyelitis may experience muscle pain, weakness, and fatigue after many years, a condition that has been labeled postpolio syndrome [Windebank et al., 1991].
Infants with nonpolio enteroviral infections can have a disseminated illness that resembles bacterial sepsis or herpes simplex virus infection. Clinical features of this condition include fever, respiratory distress, seizures, lethargy, shock, or disseminated intravascular coagulopathy; cardiomyopathy can complicate neonatal enteroviral infections, especially those due to coxsackieviruses, and be fatal in some infants [Verma et al., 2009]. During community enteroviral outbreaks, numerous young infants acquire enteroviruses, usually asymptomatically. Children with agammaglobulinemia can have a persistent, often fatal, neurologic condition caused by chronic nonpolio enterovirus infection of the CNS [Mease et al., 1981; Wilfert et al., 1977]. Patients with this rare disorder exhibit a dermatomyositis-like disorder and neurologic signs, which may include seizures, mental status changes, hemiparesis, and headache.
Diagnosis
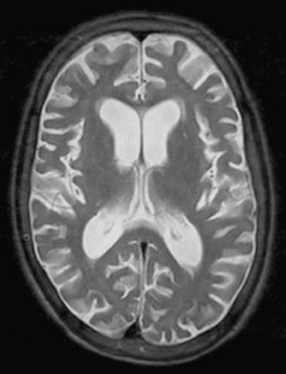
Infants with disseminated enteroviral infections can have leukocytosis, elevated serum transaminases, and thrombocytopenia, whereas infants or children with aseptic meningitis have mild CSF changes, consisting of a lymphocytic pleocytosis and protein elevation [Negrini et al., 2000], and, occasionally, laboratory abnormalities consistent with the syndrome of inappropriate antidiuretic hormone secretion. The diagnosis of enteroviral infection can be established by isolating enteroviruses from CSF or stool or throat swabs, or by detecting enteroviral RNA in stool, serum, or CSF by using reverse transcription PCR [Hamilton et al., 1999; Sawyer, 2002; Schlesinger et al., 1994]. Imaging studies are normal in nonpolio enterovirus aseptic meningitis. Children with progressive nonpolio enterovirus infections have progressive cortical atrophy (Figure 81-9).
Treatment and outcome
In most instances, children with enteroviral CNS infections require supportive care only. Children with severe nonpolio enteroviral infections have been treated with pleconaril, a novel antiviral drug with some efficacy against several RNA viruses, although controlled trials suggest that pleconaril therapy is associated with only modest shortening of enteroviral disease-related symptoms [Abzug et al., 2003; Desmond et al., 2006; Sawyer, 2002]. Children with agammaglobulinemia and persistent echovirus infections may improve during immunoglobulin replacement therapy [Mease et al., 1981]. Infants with enteroviral aseptic meningitis recover uneventfully [Bergman et al., 1987], whereas the outcome in infants with myocarditis or children with encephalitis or enterovirus 71 infections can be less favorable [Sawyer, 2002]. Persistent enterovirus infections in children with agammaglobulinemia are often fatal, despite therapy with intravenous immunoglobulin and the available antiviral drugs.
Togaviruses
Eastern Equine Encephalitis Virus
Although having the lowest incidence of the major North American arthropod-borne encephalitides, with an average of only six human cases reported to the CDC annually during the 44-year interval from 1964 to 2008, eastern equine encephalitis has the highest mortality rate, ranging from 30 to 50 percent during these decades [CDC, 2009c; Deresiewicz et al., 1997; Przelomski et al., 1988]. Cases among equines and humans, usually adults, appear during the summer months in states bordering the Atlantic Ocean and the Gulf of Mexico. Indigenous cases have been observed as far west as Indiana, Michigan, and Wisconsin.
The disorder usually begins abruptly, with high fever, lethargy, vomiting, and convulsions, although some patients have a prodromal phase lasting several days, characterized by fever, headache, malaise, and myalgias. Signs are usually diffuse, but patients may have focal findings, raising consideration of herpes simplex virus encephalitis. EEGs usually exhibit diffuse slowing, and neuroimaging studies can be normal or indicate diffuse edema or focal lesions involving the thalami or basal ganglia [Deresiewicz et al., 1997; Przelomski et al., 1988]. The diagnosis can be confirmed by detecting eastern equine encephalitis virus-specific IgM in serum or CSF, and by detecting eastern equine encephalitis virus RNA using reverse transcription PCR. Treatment consists of supportive care.
Western Equine Encephalitis Virus
Although the western equine encephalitis virus can be detected in bird and mosquito reservoirs throughout the Americas, most human cases occur in the western United States or Canada, peaking in July and August [Calisher, 1994]. The incidence of western equine encephalitis varies considerably from year to year; however, epidemics appear at approximately 10-year intervals, with the largest affecting more than 3000 persons in 1941 [CDC, 1987]. The CDC received reports of nearly 300 cases in 1975 and 37 cases in 1987, the largest outbreak in recent years, but only a total of 639 confirmed cases of western equine encephalitis have been reported since 1964 [CDC, 2009e]. Residing in rural areas or engaging in outdoor occupations is a risk factor associated with acquiring western equine encephalitis.
The clinical features of western equine encephalitis consist of fever, headache, somnolence, stiff neck, nausea, vomiting, and myalgias [Leech et al., 1981]. Coma, paralysis, seizures, and other more severe neurologic complications, although infrequent in adults, were common in infants affected during the 1941 epidemic [Medovy, 1943]. Infants can acquire the virus transplacentally and become symptomatic during the neonatal period [Shinefield and Townsend, 1953]. The diagnosis can be established by detecting western equine encephalitis virus-specific IgM in serum or CSF, or by using PCR [Calisher et al., 1986; Lambert et al., 2003]. Despite few reports of neurodiagnostic studies, EEG slowing and normal or nonspecific neuroimaging findings can be anticipated. Most children with western equine encephalitis recover completely, although developmental delay, an increased risk for seizures, and behavioral disturbance are potential sequelae [Earnest et al., 1971].
Venezuelan Equine Encephalitis Virus
Venezuelan equine encephalitis virus causes influenzalike disease, encephalitis, and intrauterine infection among inhabitants of Latin America [CDC, 1993a, 1995a]. During major epidemics, equine and human cases have been observed as far north as the southern United States. A major outbreak of Venezuelan equine encephalitis occurred in 1995, affecting 75,000–100,000 persons in Colombia and Venezuela, but this epidemic did not spread to North America [Rivas et al., 1997]. Encephalitis affects approximately 10 percent of infected persons and tends to be mild, with headache, somnolence, and occasionally convulsions. Intrauterine infection, a rare complication of maternal Venezuelan equine encephalitis virus infection, can cause fetal death or severe encephaloclastic CNS lesions, including hydranencephaly [Wenger, 1977]. The diagnosis can be established serologically or by virus isolation. Therapy is supportive; Venezuelan equine encephalitis cannot currently be prevented by vaccination [Paessler and Weaver, 2009].
Rubella Virus
Clinical features
Rubella virus causes neurologic dysfunction during acute childhood infection and in the offspring of women who are infected with rubella virus during the first trimester. These complications are rare in the United States and other regions with compulsory vaccination against rubella virus (Figure 81-10). Before vaccine licensure in 1969, rubella epidemics occurred at 6- to 9-year intervals, and during the last major pandemic in the mid-1960s, 20,000 cases of congenital rubella syndrome were reported. Between 10 and 20 percent of adults living in regions with compulsory immunization remain susceptible to rubella, however, and outbreaks of rubella and congenital rubella syndrome [Panagiotopoulos and Georgakopoulou, 2004] indicate that the rubella virus continues to pose a real threat in many regions of the world [Spika et al., 2003]. Rarely, maternal reinfection with rubella virus causes fetal infection [Robinson et al., 1994].
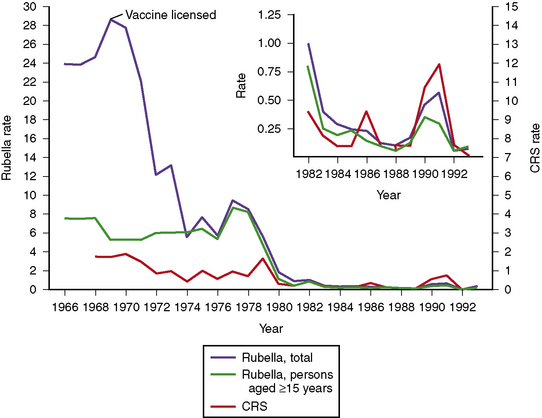
Rubella virus produces fever, coryza, lymphadenopathy of the posterior auricular and occipital nodes, and a maculopapular rash involving the face and trunk. Rare neurologic complications after acquired rubella virus infection consist of headache, seizures, or Guillain–Barré syndrome. The clinical features of intrauterine rubella virus infection, recognized first by Gregg in 1941, include cataracts, congenital heart disease, sensorineural hearing loss, microcephaly, intrauterine growth retardation, hepatosplenomegaly, petechial rash, osteopathy, jaundice, and pneumonitis [Bale, 2009; Dudgeon, 1975]. Sequelae relate directly to the gestational timing of maternal infections, with severe defects common before 12 weeks, when the risk of congenital rubella syndrome approaches 90 percent [Ueda et al., 1979]. By contrast, maternal infections after 24 weeks of gestation tend to be benign.
Diagnosis
The diagnosis of rubella virus infection can be established serologically by isolating the rubella virus from urine or nasopharyngeal secretions, or by detecting rubella virus RNA in CSF or other fluids by using reverse transcription PCR [Bosma et al., 1995]. Direct hyperbilirubinemia, elevated serum transaminases, and thrombocytopenia are potential laboratory abnormalities after intrauterine infection. Imaging of infants with congenital rubella syndrome reveals cerebral atrophy, intracranial calcifications, periventricular cysts, or leukomalacia [Yamashita et al., 1991].
Treatment and outcome
At present, therapy for rubella virus infections, including intrauterine infection, consists of supportive care. Survivors of intrauterine rubella virus infection have high rates of sequelae affecting vision, hearing, cardiac function, and intellect [Forrst et al., 2002; Givens et al., 1993]. Potential delayed sequelae of congenital rubella syndrome include endocrinopathies (diabetes mellitus, thyroid dysfunction, and growth hormone deficiency), progressive hearing loss, glaucoma, vascular abnormalities, and postrubella panencephalitis [Sever et al., 1985; Townsend et al., 1975]. Congenital rubella syndrome can be prevented by compulsory immunization programs.
Flaviviruses
The flaviviruses, another large family of arthropod-borne viruses, include Japanese encephalitis virus; St. Louis encephalitis virus; West Nile virus; and several agents associated with tick-borne encephalitis in Europe, Asia, and North America [Sejvar, 2006]. Japanese encephalitis, the most common arthropod-borne encephalitis worldwide, causes several thousand encephalitis cases annually in endemic areas of eastern Asia, although vaccine programs have reduced the incidence of Japanese encephalitis in several regions [CDC, 1993b].Tick-borne encephalitis cases are infrequent in the United States and Canada, but relatively common in Europe and Asia [Sejvar, 2006].The CDC received reports of nearly 5000 cases of St. Louis encephalitis between 1964 and 2004 [CDC, 2009g], but nearly half of these were observed during the massive epidemic of 1975, involving 29 states and the District of Columbia [Creech, 1977]. Beginning in 1999, West Nile virus spread throughout the United States, causing systemic and neurologic illness among adults and children [Hayes and O’Leary, 2004], and by late 2009, nearly 30,000 cases of West Nile virus infections had been reported to the CDC during the first 10 years of U.S. West Nile virus activity [CDC, 2009d]. Of these, 25–50 percent have been neuroinvasive.
Japanese Encephalitis Virus
Clinical features
Japanese encephalitis, a mosquito-borne virus endemic throughout much of eastern Asia, causes as many as 50,000 cases of encephalitis each year, usually in children younger than 15 years of age [CDC, 1993b, 2009g]. Disease activity peaks from May to October, although some countries, such as Indonesia, Malaysia, and India, experience cases year-round. The virus is maintained in a bird–vertebrate cycle involving Culex mosquitoes, domestic pigs, and wading birds [CDC, 1993b, 2009g]. Children with Japanese encephalitis have an abrupt, fulminant illness with fever, convulsions, and coma [Kumar et al., 1990]. Headache, vomiting, increased muscle tone, breathing irregularities, and occasionally focal neurologic deficits are additional clinical manifestations [Kumar et al., 1990]. The prodromal phase is less than 24 hours long in most children with Japanese encephalitis, but occasionally exceeds 7 days.
Diagnosis
The diagnosis of Japanese encephalitis can be confirmed by isolating the virus from CSF or brain tissue, detecting serologic responses to the virus, or detecting Japanese encephalitis virus RNA in CSF using reverse transcription PCR [Meiyu et al., 1997]. MRI can reveal bilateral lesions of the thalami or basal ganglia [Kalita and Misra, 2000; Kumar et al., 1997].
Treatment and outcome
Mortality rates for Japanese encephalitis range from 8 to 40 percent; 40–60 percent of the survivors have neurologic sequelae consisting of mental retardation, seizure disorders, motor deficits, or subtle behavioral and intellectual abnormalities [Kumar et al., 1993; Ooi et al., 2008]. An inactivated Japanese encephalitis virus vaccine prevents infection. The vaccine has an 80 percent efficacy after two doses [CDC, 1993b]; adverse reactions occur at a rate of approximately 15 per 100,000 doses [Berg et al., 1997; Takahashi et al., 2000]. Widespread immunization programs in Japan and other Asian nations have reduced the annual incidence of Japanese encephalitis by 90 percent or more [Arai et al., 2008].
St. Louis Encephalitis Virus
Clinical features
St. Louis encephalitis virus, a mosquito-borne virus endemic to the midwestern United States, occasionally causes epidemics of aseptic meningitis, encephalitis, and influenza-like illness, often involving older adults in urban or suburban areas. The 1975 epidemic, the largest to date, affected more than 2000 persons [Creech, 1977]. The attack rates and disease severity of St. Louis encephalitis virus infections, similar to those of West Nile virus, increase with advancing age [Meehan et al., 2000]. The virus is maintained in a cycle involving Culex mosquitoes and pigeons, sparrows, doves, and other birds [Johnson, 1998].
Clinical syndromes associated with St. Louis encephalitis virus infection include nonspecific febrile illnesses, aseptic meningitis, and encephalitis, although most infections are asymptomatic [Creech, 1977]. After an incubation period of 21 days, children with St. Louis encephalitis may have a biphasic illness, with low-grade fever, diarrhea, vomiting, and malaise preceding the neurologic signs and symptoms [Kaplan et al., 1978]. In patients with encephalitis, representing approximately two-thirds of symptomatic infections, headache, vomiting, fever, neck stiffness, and lethargy or agitation begin abruptly. Tremor can be present; focal neurologic findings are infrequent, at least among reported adult cases. Fever may reach 41°C and can persist 7 or more days. Clinical improvement usually begins within 7–10 days of disease onset.
Diagnosis, Treatment, And Outcome
The CSF reveals a pleocytosis in most patients with St. Louis encephalitis. Neuroimaging studies are normal or nonspecifically abnormal, and EEGs may indicate diffuse slowing. The diagnosis can be established by detecting serologic responses in serum or CSF [Monath et al., 1984] or by nucleic acid amplification methods [CDC, 2009g]. Treatment consists of supportive care. Mortality, ranging from 8 to 20 percent [Creech, 1977; Meehan et al., 2000], is highest among persons older than age 50 years. Potential sequelae include seizures, parkinsonism, and intellectual deficits.
West Nile Virus
Clinical Features
First identified in Uganda in 1937, West Nile virus later appeared in Egypt and Israel in the 1950s [Marberg et al., 1956]. Beginning in the mid-1990s, human outbreaks became more frequent and severe, causing meningitis, encephalitis, and fatalities among persons living in Israel, Algeria, and Romania [Hubalek and Halouzka, 1999]. In 1999, cases of West Nile virus infection occurred in the United States, marking the first appearance of the virus in the Western hemisphere. An encephalitis outbreak in the New York City metropolitan area, linked to a West Nile virus strain closely related to a strain identified in Israel in 1998, caused seven deaths among adults [Lanciotti et al., 1999; Nash et al., 2001].
In 2000, West Nile virus activity was detected in 12 states surrounding New York State, but by the end of 2001, 27 states and the District of Columbia reported the presence of West Nile virus [Petersen et al., 2002]. West Nile virus spread rapidly among North American birds and mosquitoes, and led to a massive human outbreak in 2003 that affected nearly 10,000 inhabitants of states from New York to California (Figure 81-11). Only two western states (Oregon and Washington) did not report West Nile virus activity in that year [CDC, 2004a, c]. The epidemic continued, and by late 2009, nearly 30,000 cases of human disease due to West Nile virus had been reported to the CDC [CDC, 2009d].
This was the peak year of West Nile virus activity in the United States in the last decade.
(From the Centers for Disease Control and Prevention. Available at: http://www.cdc.gov/ncidod/dvbid/westnile/mapsactivity/surv&control03maps.htm. Accessed November 15, 2009.)
West Nile virus is maintained in a bird–mosquito cycle, involving rural and urban wild birds and Culex and Aedes mosquitoes; humans represent dead-end hosts in most instances, although human-to-human transmission by organ transplantation and blood transfusion can occur [CDC, 2005a]. Asymptomatic human infections with West Nile virus outnumber symptomatic cases by approximately 5 to 1 [CDC, 2004c]. Neurologic manifestations, including meningitis, encephalitis, myelitis, poliolike illness, and others, occur in approximately 1 of every 150 infected persons [CDC, 2009d]. Adults constitute the majority of cases. After an incubation period of 2–14 days, systemic symptoms of fever, malaise, headache, nausea, and vomiting occur in symptomatic infections. Of symptomatic patients, 50 percent have a maculopapular rash and lymphadenopathy. To date, most patients with the neurologic complications of West Nile virus infections have been older than age 25 years.
Approximately 60 percent of persons with West Nile virus-induced neurologic disease have encephalitis [Sejvar et al., 2003]. Encephalitis can begin with the usual systemic prodrome, or more abruptly, with high fever and coma [Campbell et al., 2002]. Typical abnormalities in West Nile virus encephalitis include altered mental status, coma, diffuse muscle weakness (proximal greater than distal), respiratory paralysis, and hyporeflexia. Tremor, myoclonus, and parkinsonian features are additional components of the acute illness [Sejvar et al., 2003]. Less common neurologic manifestations of West Nile virus infection include aseptic meningitis, myelitis, optic neuritis, and Guillain–Barré syndrome [Campbell et al., 2002; Sejvar et al., 2003]. Involvement of the anterior horn cells can produce a poliomyelitis-like disorder with asymmetric paralysis of the upper or lower extremities [Li et al., 2003; Sejvar, 2004]. This disorder produces clinical, histopathologic, and electrophysiologic abnormalities comparable to those of classic poliomyelitis [Sejvar, 2004]. Although patients with West Nile virus-induced meningitis and encephalitis gradually recover, patients with limb paralysis usually have permanent deficits [Li et al., 2003; Sejvar et al., 2003].
A single case report indicates that West Nile virus can be transmitted to the fetus, causing intrauterine infection of the CNS [CDC, 2002]. In late 2002, an infant was born to a woman who had experienced meningoencephalitis during her pregnancy. At birth, the infant was found to have bilateral chorioretinitis and cystic encephalomalacia. A subsequent epidemiological study that evaluated the outcomes of West Nile virus infections during the pregnancies of 71 women identified no additional cases of congenital West Nile virus infections [O’Leary et al., 2006], indicating that congenital West Nile virus infection is an uncommon event.
Diagnosis, treatment, and outcome
Laboratory abnormalities include mild anemia and peripheral leukocytosis or leukopenia. The CSF of West Nile virus encephalitis resembles that of most arboviral infections, consisting of a mild, lymphocytic pleocytosis (30–100 cells/mm3), elevated protein content (80–100 mg/dL), and normal glucose content. Cranial CT is normal, whereas MRI can reveal leptomeningeal enhancement, periventricular inflammation, or abnormalities of the spinal cord [Campbell et al., 2002]. The diagnosis of West Nile virus infection can be confirmed by serologic studies that detect neutralizing antibodies to the virus in acute or convalescent serum samples or West Nile virus-specific IgM in serum or CSF. West Nile virus-specific IgM persists in serum for at least 16 months in more than half of infected persons [Campbell et al., 2002]. Infected patients experience brief periods of viremia, but infectious virus is rarely isolated from serum or CSF. Reverse transcription PCR can be used to detect West Nile virus in clinical samples [Lanciotti and Kersat, 2001]. A CDC protocol guides the evaluation of infants with suspected intrauterine West Nile virus infection [CDC, 2004f].
Therapy for the neurologic complications of West Nile virus infection currently consists of supportive care, with close observation for respiratory decompensation. Ribavirin possesses activity against the West Nile virus in vitro, but there are no controlled trials on which to base management decisions. There were 264 deaths among the 9862 cases reported to the CDC in 2003, corresponding to a mortality rate of approximately 2.7 percent [CDC, 2009d]. Deaths have been more common among the elderly. Approximately one-third of survivors have permanent sequelae affecting cognition, behavior, or motor function.
Tick-Borne Flaviviruses
Several closely related, tick-borne flaviviruses of Europe, Asia, and North America can produce encephalitis or aseptic meningitis in children and adults living in these regions [CDC, 2009h; Kaiser, 2008; Lesnicar et al., 2003; Mansfield et al., 2009]. Tick-borne encephalitis represents a major public health concern among unvaccinated persons in Eastern Europe and Asia. Patients with European tick-borne encephalitis often have a biphasic illness, experiencing headache, malaise, myalgia, and low-grade fever during the initial viremic phase. Approximately 20–30 percent of infected individuals have a second, neurologic phase with headache, high fever, somnolence, coma, convulsions, cranial nerve palsies, or paralysis that mimics Guillain–Barré syndrome [CDC, 2009h]. Such patients have relatively high rates of neurologic sequelae [CDC, 2009h]. Cases of tick-borne encephalitis have also been described after consumption of unpasteurized, virus-contaminated milk, or in travelers to endemic regions [CDC, 2009h]. Vaccination can prevent infection among children inhabiting or visiting endemic regions, although vaccine side effects are common [CDC, 2009h; Kunze et al., 2004].
In North America, the tick-borne Powassan virus, maintained in a vertebrate–mosquito cycle in eastern Canada and the northeastern United States, has been associated with cases of encephalitis, sometimes in children [CDC, 2009h; Embil et al., 1983; Romero and Newland, 2003; Smith et al., 1974]. Clinical features consist of seizures, somnolence, or coma; occasionally, children can have focal features, necessitating acyclovir therapy [Embil et al., 1983]. CSF examination usually reveals a mixed pleocytosis; EEGs may be slow or epileptiform. The diagnosis is established serologically. Children generally recover completely with supportive care, although sequelae and death have been described.
Bunyaviruses
La Crosse Virus
Clinical features
La Crosse virus, a California serogroup virus maintained in the wild through a mosquito–vertebrate animal cycle, causes encephalitis, known as La Crosse encephalitis, among children living in the midwestern and eastern United States. Before the emergence of West Nile virus in the United States, La Crosse encephalitis represented the most common arthropod-borne encephalitis in America (see Figure 81-3), accounting for approximately 70–100 reported cases among children per year [CDC, 2009i; McJunkin et al., 2001; Rust et al., 1999]. Nonspecific clinical features, consisting of fever, vomiting, headache, abdominal pain, and malaise, begin 3–7 days after exposure to the virus. Neurologic features appear 2–4 days later and consist of seizures (60 percent), lethargy or coma (50 percent), and focal deficits (20 percent) [McJunkin et al., 2001; Rust et al., 1999]. Neurologic symptoms usually resolve during the subsequent week.
Diagnosis
Laboratory findings include a peripheral leukocytosis (often with a neutrophil predominance) and mixed CSF pleocytosis ranging from 10 to 500 leukocytes/mL in approximately 90 percent of children. EEGs can be diffusely or focally slow, whereas CT and MRI are usually normal. The diagnosis of California encephalitis can be established serologically using IgM capture assays of serum or CSF [McJunkin et al., 2001]. The diagnosis also has been established by brain biopsy [McJunkin et al., 1997], but invasive diagnostic procedures are rarely necessary. Reverse transcription PCR can detect the RNA of California group viruses [Kuno et al., 1996].
Treatment and outcome
Because most cases of La Crosse encephalitis are mild, and current antiviral agents, with the possible exception of ribavirin [McJunkin et al., 1997], lack anti-California encephalitis virus activity, therapy consists of supportive care. Focal EEG or clinical manifestations of La Crosse encephalitis mimic herpes simplex virus and necessitate acyclovir therapy while awaiting the results of microbiologic studies. Most children with California encephalitis recover completely, although residual behavioral changes, seizure tendencies, or EEG abnormalities have been reported in 10–15 percent of children with proven California encephalitis [Balkhy and Schreiber, 2000; Grabow et al., 1969; McJunkin et al., 2001; Rust et al., 1999].
Reoviruses
Colorado Tick Fever Virus
Colorado tick fever, a disorder endemic in the western United States, causes an influenzalike illness that begins approximately 5 days after exposure to Colorado tick fever-infected Rocky Mountain wood ticks, Dermacentor andersoni [Goodpasture et al., 1978]. The disorder usually appears in May through July in young adults who engage in outdoor recreational activities and often report tick exposure. Typical signs and symptoms include fever, headache, malaise, myalgia, abdominal pain, and vomiting. CNS manifestations, consisting of meningitis or mild encephalitis, affect less than 5 percent of children with Colorado tick fever [Goodpasture et al., 1978; Spruance and Bailey, 1973]. Such patients may have peripheral leukocytosis, a mixed CSF pleocytosis, and diffusely slow EEGs [Spruance and Bailey, 1973]. The diagnosis can be established by virus isolation during the prolonged viremic phase [Goodpasture et al., 1978] or serologic studies. Reverse transcription PCR holds promise as a rapid, specific diagnostic method [Johnson et al., 1997]. Patients require supportive care and usually recover without sequelae.
Rotavirus
Rotavirus commonly infects infants or young children during the winter months and causes fever, diarrhea, vomiting, and dehydration. Approximately 1–4 percent of children with rotavirus gastroenteritis experience seizures, either febrile or afebrile, or altered sensorium [Lloyd et al., 2007; Motoyama et al., 2009; Yoshida et al., 1995]. CSF findings in children with rotavirus-associated CNS complications include a normal glucose content, normal protein content, and normal or elevated CSF white blood cell count [Yoshida et al., 1995]. The EEG may reveal diffuse, nonspecific slowing, whereas neuroimaging studies are usually normal. Rotavirus gastroenteritis can be diagnosed by detecting rotavirus antigen in stool samples, and CNS involvement can be established by detecting rotavirus RNA in CSF by using reverse transcription PCR [Yoshida et al., 1995]. Children with rotavirus infections have benign, self-limited illnesses, although fluid status must be monitored carefully.
Paramyxoviruses
The paramyxovirus family, containing the mumps and measles viruses, has contributed historically to considerable mortality and neurologic morbidity [CDC, 1997d; Knipe et al., 2007; Hughes, 1977; Johnson, 1998]. Consequently, vaccines were developed for these viruses and introduced in the late 1960s. In the United States, currently, 150 or fewer cases of measles are reported annually to the CDC, a greater than 99 percent reduction compared with the number in the prevaccine era [CDC, 2004d]. Subacute sclerosing panencephalitis, a post-measles condition with an incidence of approximately 1 case per million cases of measles [Modlin et al., 1979], has virtually disappeared in many regions as a direct consequence of measles vaccination.
Although measles cases have declined in the United States and other countries with compulsory immunization programs, measles still caused nearly 200,000 deaths in 2007, the most recent year with available data [Henao-Restrepo et al., 2003; WHO, 2009a]. Measles caused 5 percent of all deaths worldwide in children younger than 5 years in 2000. In 2009, the World Health Organization and other organizations targeted measles for global eradication by 2010, a goal that proved elusive. Members of the closely related orthomyxovirus family, the influenza viruses, have been associated with Reye’s syndrome, a postinfectious disorder, and meningoencephalitis, albeit rarely.
Measles Virus
Clinical features
Despite the availability of the measles vaccine, a safe and effective live-virus preparation, measles remains a major worldwide public health concern [CDC, 1997d; Henao-Restrepo et al., 2003; WHO, 2009a]. Measles virus spreads via the respiratory route, with contagion highest during the prodromal catarrhal stage that precedes the rash. Before vaccine availability, measles typically caused disease in infants and young children during the winter and spring. Symptoms of measles infection include fever, cough, coryza, and conjunctivitis, beginning approximately 12 days after exposure in susceptible persons. The characteristic maculopapular exanthem appears on the face and spreads centrifugally to involve the trunk and extremities.
Neurologic complications of measles conform to three distinct disorders:
Measles encephalomyelitis, an immune-mediated disorder with an incidence of approximately 1 case per 1000 cases of measles, begins within 7 days after the onset of the measles rash [Johnson et al., 1984], and children younger than 10 years constitute most cases. Clinical manifestations include headache, irritability, somnolence, coma, and seizures, although some children have ataxia, choreoathetosis, or focal deficits [Johnson et al., 1984]. Subacute measles encephalopathy, first reported in the 1970s [Murphy and Yunis, 1976], begins 1–7 months after measles infection or immunization in an immunocompromised host, often a child with acute leukemia [Mustafa et al., 1993]. Altered consciousness and generalized or focal seizures, including epilepsia partialis continua, are typical clinical signs; 15–40 percent of patients have hemiparesis, hemiplegia, ataxia, aphasia, or visual symptoms [Mustafa et al., 1993].
Subacute sclerosing panencephalitis, a rare disorder in areas with measles immunization programs, begins 5–10 years after measles infection and usually affects young children at an average age of 7 years at disease onset [Anlar et al., 2001; Campbell et al., 2005; Garg, 2008; Honarmand et al., 2004; Modlin et al., 1979]. The disorder displays relatively stereotyped clinical stages, beginning with insidious declines in behavior and cognition that may mimic a psychiatric disorder. Myoclonus, a prominent component of the next phase of subacute sclerosing panencephalitis, involves the extremities, trunk, or head, and generalized or focal seizures may begin concurrently [Risk and Haddad, 1979]. As the disorder progresses, speech and intellectual function deteriorate; myoclonus intensifies; and other neurologic abnormalities, such as choreoathetosis, bradykinesia, or rigidity, appear. The typical course ends in debility, coma, and death. Approximately half of patients have chorioretinitis and visual impairment; a small proportion of subacute sclerosing panencephalitis cases begin in adulthood [Singer et al., 1997], or have atypical features with rapid deterioration and death [Silva et al., 1981].
Diagnosis
The diagnosis of measles encephalomyelitis can be established by detecting measles virus IgG titer elevations in acute and convalescent serum, or measles-specific IgM in serum or CSF. Measles virus also can be detected in clinical samples by using reverse transcription PCR [Shimizu et al., 1993]. Brain MRI may reveal white-matter lesions compatible with acute demyelination. In subacute measles encephalopathy, the CSF, although usually normal, may reveal a lymphocytic pleocytosis. EEGs typically reveal focal or generalized slowing or epileptiform discharges. Cranial CT is usually normal, but brain MRI can detect nonspecific changes involving the cortex, white matter, or deep nuclei (Figure 81-12) [Chen et al., 1994]. Paramyxovirus particles or measles virus RNA can be detected in brain tissue.
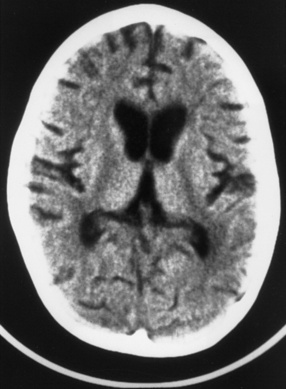
Fig. 81-12 T1-weighted magnetic resonance image from an immunocompromised child with measles encephalitis.
The pathogenesis of subacute sclerosing panencephalitis, although still obscure, reflects defective replication of the measles virus in neural tissues [Garg, 2008]. Patients with subacute sclerosing panencephalitis have elevated CSF immunoglobulin levels, oligoclonal bands, and increased IgG synthesis rates, indicating massive production of measles-specific IgG. High CSF titers of measles-specific IgG establish the diagnosis of subacute sclerosing panencephalitis, and measles virus RNA can be detected in CSF or plasma by reverse transcription PCR [Nakayama et al., 1995]. EEGs show bilaterally synchronous spike-wave or slow-wave bursts that assume a suppression-burst pattern over time. MRI commonly exhibits T2 prolongation of subcortical and periventricular white matter and eventually cortical atrophy (Figure 81-13) [Lum et al., 1986; Anlar et al., 1996].
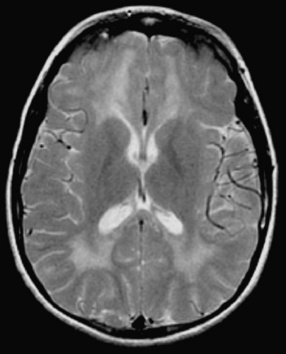
Treatment and outcome
Treatment of measles encephalomyelitis consists of supportive care. Outcome varies, ranging from complete recovery to survival with impaired intellectual function, motor deficits, or death. Although one report suggests transient improvement after ribavirin therapy [Mustafa et al., 1993], most children with subacute measles encephalopathy die or have severe neurologic sequelae. Subacute sclerosing panencephalitis progresses to death in 1–3 years in most cases, although some patients have extended plateau phases, either spontaneously or coincident with inosine pranobex (inosiplex; isoprinosine) therapy [Garg, 2008; Risk and Haddad, 1979; Yalaz et al., 1992]; intraventricular or intravenous ribavirin has also been used with uncertain benefits [Campbell et al., 2005]. A multinational trial indicated that approximately 35 percent of patients with subacute sclerosing panencephalitis stabilize during inosine pranobex therapy, but this study found no added benefit from interferon-alpha [Gascon, 2003].
Mumps Virus
Clinical features
Before vaccine licensure, mumps virus infection accounted for most cases of aseptic meningitis and mild encephalitis in the United States and many other regions. More than 150,000 cases of mumps were reported in the United States in 1968, with aseptic meningitis in 10 percent and encephalitis in 0.2 percent, whereas fewer than 2000 cases of mumps were reported in 1993, a 99 percent decrease [CDC, 1995b]. By the early 1980s, mumps meningoencephalitis had disappeared in many regions of the world [Koskiniemi et al., 1991]. However, large numbers of people remain susceptible to mumps virus infection, even in regions with compulsory immunization programs. In 2006, more than 6500 persons, predominantly between the ages of 18 and 24 years, and living in eight midwestern states of the USA, contracted mumps [Dayan et al., 2008], an outbreak attributed to vaccine failure.
Clinical features appear 2 weeks after acquisition and consist of anorexia, malaise, and low-grade fever; parotitis is a cardinal clinical feature. One-third or more of mumps virus infections are asymptomatic. Aseptic meningitis or encephalitis, heralded by headache, vomiting, and high fever, is associated with stiff neck, somnolence, and, occasionally, seizures, coma, or focal deficits [Koskiniemi et al., 1983]. Additional neurologic complications of mumps include acute cerebellar ataxia and hydrocephalus secondary to mumps virus-induced ependymitis [Bray, 1972].
Diagnosis, treatment, and outcome
CSF pleocytosis is common in patients with mumps virus meningitis or encephalitis. CSF protein content is usually normal, and the glucose content can be slightly reduced [Koskiniemi et al., 1983]. The diagnosis can be confirmed by detecting mumps virus-specific IgM or IgG. EEGs in children with encephalitis may reveal generalized or focal slowing. Children require supportive care; most recover completely.
Rhabdoviruses
Rabies Virus
Clinical features
Dreaded since antiquity, rabies poses a threat to individuals in many regions of the world, especially in the developing countries of Africa and Asia, where the incidence of 1 in 100,000 to 1 million inhabitants corresponds to approximately 50,000 deaths annually [Fishbein and Robinson, 1993; Plotkin, 2000; WHO, 2009b]. In these regions, unvaccinated domestic dogs transmit most rabies virus infections to humans, and children constitute most human rabies cases. By contrast, human and animal rabies has been eradicated in a few regions as a result of quarantine and vaccination of domestic animals [CDC, 1997a; Johnson, 1998]; for example, rabies does not exist in Hawaii.
In the continental United States, rabies remains endemic in certain wild animals, including raccoons, skunks, foxes, and bats. In the northeastern states, the number of infected raccoons has increased steadily since the 1970s [CDC, 1997a, 2009j; Fishbein and Robinson, 1993; Rupprecht et al., 1995]; in 2008, there were 6841 cases of animal rabies reported to the CDC [CDC, 2009j]. This epizootic constitutes a potential health hazard and an economic burden for postexposure prophylaxis; the cost of human rabies detection, control, and prevention in the United States now exceeds $300 million annually. The CDC receives reports of 1–3 indigenous cases of human rabies annually in the United States. Many cases have no apparent history of animal bites and have been linked by molecular analysis to the rabies virus strain carried by the insectivorous silver-haired bat [CDC, 2009j].
After a variable incubation period, averaging 20–60 days, human rabies begins with chills, fever, headache, sore throat, malaise, nausea, or abdominal pain [Johnson, 1998; Plotkin, 2000; WHO, 2009b]. Pain or itching corresponding to the site of inoculation is common. Neurologic symptoms and signs begin thereafter; symptoms and signs are cerebral in 80 percent and Guillain–Barré syndrome-like in the remainder. Patients with cerebral symptoms have marked behavioral changes, with agitation, hypersalivation, delirium, and opisthotonic posturing [WHO, 2009b]. Hydrophobia, present in 20–50 percent of patients, produces spasms of the laryngeal muscles, diaphragm, and accessory respiratory muscles, which can lead to respiratory arrest and death. Seizures ensue in many cases. Eventually, patients with rabies encephalitis lapse into a fatal coma. Rabies-induced Guillain–Barré syndrome-like disease produces bulbar dysfunction, facial diplegia, and, often, respiratory arrest [Chopra et al., 1980]. Cardiac dysrhythmias and irreversible coma are additional causes of death in paralytic cases.
Diagnosis
Because of its rarity in the United States and Canada, human rabies is often unsuspected [Noah et al., 1998; CDC, 2009j], especially during the early, nonspecific phase. Consequently, rabies usually is identified at postmortem examination. Premortem diagnosis can be established by immunofluorescence examination of skin from the nape of the neck, detection of rabies virus antibody in CSF or serum (assuming the patient was not vaccinated previously), or isolation of rabies virus from saliva or CSF [Plotkin, 2000; Smith, 1996]. Reverse transcription PCR can be used to detect rabies virus and determine the molecular profile and animal origin of the rabies virus strain. CSF may be normal or reveal nonspecific changes, with a mixed or lymphocytic pleocytosis and mild protein elevation [Chopra et al., 1980; Plotkin, 2000]. CT is normal; MRI can demonstrate signal abnormalities of the basal ganglia or thalami [Awasthi et al., 2001].
Treatment and outcome
Nearly all persons with symptomatic rabies die, despite supportive care, and only two survivors with “good” neurologic status have been reported in the medical literature over the past four decades [Wilde et al., 2008]. In a highly publicized and therapeutically important case, an unvaccinated teenager treated aggressively with ribavirin, amantadine, and drug-induced coma survived, with moderate neurological sequelae consisting of choreoathetosis, dysarthria, and a gait disorder [Willoughby et al., 2005]. Children with suspected or possible rabies exposures, including anyone with exposure to live or dead bats, should receive postexposure prophylaxis in accordance with CDC and local public health guidelines [CDC, 2009k], taking into account the animal species and the nature of the exposure. All postexposure treatment should begin with immediate and thorough cleansing of the wound with soap and water [Plotkin, 2000]. Bites are more likely to transmit the virus than scratches or other encounters [Fishbein and Robinson, 1993].
Because of the strong epidemiologic relationship between bat rabies strains and human rabies, prophylaxis should be administered after contact with bats, unless testing of the bat proves negative for rabies [CDC, 2009k]. Human-to-human transmission of rabies virus has been reported after corneal and organ transplantation [CDC, 2004e; Houff et al., 1979], but transmission by body fluids has not been described. None the less, persons with suspected rabies should be maintained in strict isolation to minimize contact with rabies virus-contaminated fluids and to reduce the number of health-care workers who require postexposure prophylaxis. An average of 54 persons (family members and health-care workers) per patient have required postexposure prophylaxis after contact with U.S. human rabies cases [Noah et al., 1998].
Arenavirisues
Lymphocytic choriomeningitis virus, a rodent-borne arenavirus, infects humans in temperate regions throughout Europe and North America, and outbreaks of human lymphocytic choriomeningitis virus disease have been linked to hamsters and wild or laboratory mice [Peters et al., 1996]. Although many infections occur without symptoms, lymphocytic choriomeningitis virus can induce a nonspecific, influenza-like disorder, with fever, headache, myalgia, nausea, vomiting, backache, and cough. Occasional patients, including children, have prominent CNS symptoms, with aseptic meningitis or mild meningoencephalitis [Kang and McGavern, 2008]. CSF findings include a mixed pleocytosis, sometimes including eosinophils, normal or elevated protein content, and a normal or depressed glucose content. CT can show hydrocephalus secondary to the meningeal and ependymal inflammation.
Lymphocytic choriomeningitis virus causes an intrauterine infection, with clinical and radiographic features that resemble intrauterine cytomegalovirus disease or intrauterine toxoplasmosis [Barton and Mets, 2001; Bonthius et al., 2007b; Schulte et al., 2006; Wright et al., 1997]. Infants with this disorder frequently display chorioretinitis, microcephaly, or macrocephaly [Barton and Mets, 2001; Bonthius et al., 2007b; Wright et al., 1997], and imaging studies reveal hydrocephalus, cortical dysplasia, cerebral atrophy, or intracranial calcifications (Figure 81-14 and Figure 81-15). The diagnosis of congenital lymphocytic choriomeningitis virus infection can be established by detecting lymphocytic choriomeningitis virus-specific antibodies in CSF or serum, and potentially by detecting lymphocytic choriomeningitis virus RNA in CSF with reverse transcriptase PCR [Read et al., 1997]. Patients with acquired infections recover completely, whereas congenitally infected infants have high rates of neurodevelopmental sequelae [Barton and Mets, 2001; Bonthius et al., 2007b; Wright et al.,1997]. Current antiviral drugs do not possess clinically useful antilymphocytic choriomeningitis virus activity.
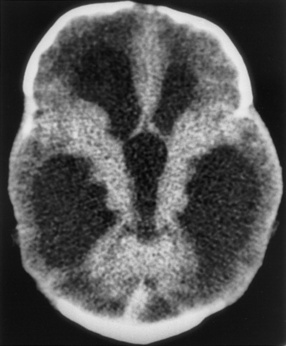
The scan reveals massive ventriculomegaly compatible with aqueductal obstruction.
(From Wright R et al. Congenital lymphocytic choriomeningitis virus syndrome: A disease that mimics congenital toxoplasmosis or cytomegalovirus infection. Pediatrics 1997;100:1.)
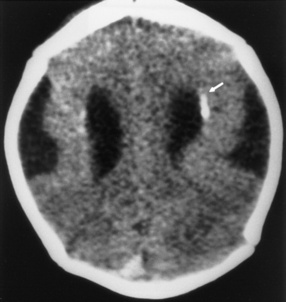
Retroviruses
Human immunodeficiency virus type 1 (HIV-1) and human T-cell lymphotropic virus type 1 (HTLV-1), human retroviruses linked in the 1980s to HIV/AIDS and progressive myelopathy, respectively, contribute to considerable neurologic morbidity among humans worldwide [Varmus, 1988]. Maintained in human reservoirs and transmitted via virus-contaminated human fluids or blood products, these retroviruses chronically infect the host and produce neurologic disease coincident with primary infection or subsequently during virus reactivation. Although the understanding of the virology, immunobiology, neuropathogenesis, and therapy of retroviral infections has advanced considerably since the discovery of these viruses, cures or vaccines to prevent these infections have not yet been identified.
Human Immunodeficiency Virus
Clinical features
Cases of AIDS in children first appeared in the early 1980s before the causative agent, HIV-1, was identified [Falloon et al., 1989]. Early cases of childhood AIDS were linked to blood transfusions, hemophilia requiring factor replacement, or having a parent with AIDS or an AIDS-related condition. Subsequently, most HIV-1 infections in infants and children were linked to mothers harboring HIV-1. The offspring of untreated, HIV-1-infected women have a 15–30 percent risk of acquiring the virus, whereas current combination antiretroviral treatment and management strategies in HIV-1-infected mothers and their infants can reduce the risk of perinatal transmission to 2 percent or less [American Academy of Pediatrics, 2009; CDC, 1994b; Connor et al., 1994; Havens and Waters, 2004].
By the late 1980s, AIDS cases had been identified in more than 100 countries or territories, and the total number of AIDS cases worldwide rose nearly exponentially from approximately 12,000 cases in 1984 to more than 100,000 cases by the end of that decade. Although the rates of HIV-1 infection have plateaued or declined since in the United States and other regions, HIV-1 infection remains a major threat to the world’s population [De Cock et al., 2000; UNAIDS, 2002; WHO, 2008]. Currently, more than 90 percent of new HIV-1 infections occur among persons living in Asia and Africa (Figure 81-16), and the number of people living with HIV/AIDS globally remains enormous – approximately 33 million in 2007 [WHO, 2008]. The World Health Organization data indicate that approximately 7500 persons (approximately 1000 of these being children) acquire HIV-1 infection each day, corresponding to approximately 3 million persons with new HIV infections in 2007 [WHO, 2008]. More than 20 million infants, children, and adults have died since the first cases of AIDS were identified in 1981.

Infants infected with HIV-1 vertically can become symptomatic after the third month of life, manifesting hepatomegaly, lymphadenopathy, failure to thrive, interstitial pneumonitis, opportunistic infections (especially with Pneumocystis jiroveci [formerly, Pneumocystis carinii] or cytomegalovirus), or neurologic disease [Falloon et al., 1989]. The severity and spectrum of the clinical manifestations are modified substantially, however, by antiparasitic, antiviral, and antiretroviral therapies. In some children, HIV-1 infection can remain dormant, and 10 or more years may elapse before the symptoms of HIV-1 infection appear. Before current combination antiretroviral therapies, a child infected vertically with HIV-1 had a 50 percent probability of severe HIV/AIDS-related disease by age 5 years [Barnhart et al., 1996].
Neurologic complications were noted among adults early in the AIDS epidemic, and by the mid-1980s, similar disorders were reported in children [Belman et al., 1985; Epstein et al., 1985]. These early reports emphasized the dramatic, progressive decline in motor and intellectual skills in HIV-1-infected children, sometimes coinciding with the appearance of opportunistic infections with cytomegalovirus, P. jiroveci, Toxoplasma gondii, or other microorganisms. Early studies suggested that HIV-1-associated progressive encephalopathy developed in approximately 50 percent of children with advanced HIV-1 disease, whereas improved antiretroviral therapies have modified substantially the natural history of encephalopathy in HIV-1-infected children [Belman, 2002; Patel et al., 2009]. In utero exposure to HIV-1 without evidence of perinatal infection does not adversely affect early neurologic development [Belman et al., 1996].
Before the availability of effective antiretroviral therapies, infants and children with HIV-1 encephalopathy exhibited progressive motor dysfunction, cognitive abnormalities, developmental delay, and acquired microcephaly [Belman et al., 1985, 1986; Epstein et al., 1985]. Typical neurologic findings, attributable to the direct CNS effects of HIV-1, consist of apathy, dementia, ataxia, hyperreflexia, weakness, seizures, or myoclonus. Current antiretroviral therapies prevent or delay the onset of certain HIV-1-related neurologic complications [Gavin and Yogev, 1999; Belman, 2002; Patel et al., 2009]. HIV-1 invades the CNS early after intrauterine or postnatal infection and displays tropism for microglia/macrophages and astrocytes; microglia and macrophages are the principal reservoirs of HIV-1 infection within the CNS [Epstein and Gendelman, 1993; Epstein and Sharer, 1994]. Certain CNS cell populations, including neurons and oligodendrocytes, contain few or no HIV-1 nucleic acids, indicating that HIV-1 causes dysfunction and apoptosis of such cells through perturbations of proinflammatory cytokines, excitotoxins, and CNS neurotransmitters [Epstein and Sharer, 1994; Mitchell, 2006]. CNS disease, either due directly to HIV-1 or caused indirectly through opportunistic infections, reflects the degree of immunodeficiency [Belman, 2002] and the viral load of infected children.
Additional disorders linked directly to HIV-1 infection of the CNS or peripheral nervous system are aseptic meningitis, meningoencephalitis, myopathy, and Guillain–Barré syndrome-like conditions [Srugo et al., 1992]. HIV/AIDS can lead to numerous secondary CNS complications, including stroke [Park et al., 1990], primary CNS lymphoma, and CNS infections with T. gondii, cytomegalovirus [Griffiths, 2006], varicella zoster virus, Mycobacterium tuberculosis, fungi, and JC virus, the cause of progressive multifocal leukoencephalopathy [Wrzolek et al., 1995]. The last-mentioned condition occurs in approximately 4 percent of adults with AIDS and some children, and causes weakness, visual loss, cognitive decline, gait abnormalities, and sensory disturbances [Berger et al., 1992]. PCR can be used to detect JC virus in clinical samples [McGuire et al., 1995].
Diagnosis
Passive transfer of maternal antibody complicates detection of HIV-1 infection during the first 18 months of life, and 30 percent or more of exposed infants serorevert [American Academy of Pediatrics, 2009; Read, 2007]. Consequently, detection of HIV-1 nucleic acids (DNA or RNA) by the PCR is necessary to diagnose HIV-1 infection in young infants [American Academy of Pediatrics, 2009; Belman, 2002; Bremer et al., 1996; Read, 2007]. HIV-1 infection in exposed infants (i.e., infants with positive antibody tests shortly after birth or infants born to HIV-1-seropositive mothers) can be confirmed by serial serum PCR assays, with the first test in the immediate newborn period, a second test during the first or second month of life, and a third test at 3–6 months of age. If two samples are positive for HIV-1, the infant is considered infected; two successive negative tests indicate that infection is unlikely [American Academy of Pediatrics, 2009; Read, 2007]. In children and adolescents, serologic studies using enzyme-linked immunosorbent assay and Western blotting can identify HIV-1 infection. Reverse transcriptase PCR monitoring of virus loads guides the treatment of HIV-1-infected infants, children, and adolescents [American Academy of Pediatrics, 2009].
The CSF is usually normal in HIV-1-associated progressive encephalopathy, whereas the EEG may exhibit diffuse slowing. Neuroimaging studies reveal cortical atrophy in vertically or horizontally acquired infections (Figure 81-17). Calcifications of the basal ganglia or frontal white matter – best detected by CT – and nonspecific abnormalities of white or gray matter – most evident by MRI – occur in vertically acquired HIV infections [DeCarli et al., 1993; Belman et al., 1986]. CT, MRI, and studies of CSF or blood can identify secondary infectious and neoplastic complications in HIV-1-infected children. Combination antiretroviral therapy has improved the prognosis for many of the opportunistic infections in persons with HIV/AIDS [American Academy of Pediatrics, 2009; Belman, 2002; Clifford et al., 1999].
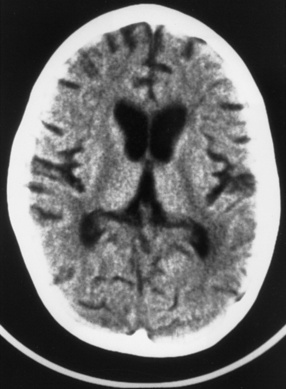
Treatment and outcome
Combination antiretroviral therapy and refined treatments for the infectious or neoplastic complications of AIDS have enhanced the survival of HIV-1-infected persons greatly [American Academy of Pediatrics, 2009; Belman, 2002; Melvin, 1997; van Rossum et al., 2002; Patel et al., 2009]. The mean survival time for treated children with HIV/AIDS exceeds 10 years, which is substantially greater than that of HIV-1 infected children who were treated in the early 1990s; the risk of death has thus declined considerably [Palumbo, 2000; Patel et al., 2009; Pizzo et al., 1988]. Although children comprise a very small proportion of AIDS deaths in the United States, achieving similar outcomes remains challenging in resource-poor countries. Worldwide, a child dies with HIV/AIDS approximately every minute, and of the more than 2 million children living with HIV/AIDS worldwide, only 10 percent were receiving antiretroviral therapy in 2007 [WHO, 2008].
Several infectious disorders, including cytomegalovirus retinitis, cerebral toxoplasmosis, P. jiroveci, herpes zoster, and CNS or systemic fungal infections, respond favorably to current antiviral, antiparasitic, and antifungal therapies [American Academy of Pediarics, 2009]. Intravenous immunoglobulin also benefits selected children with hypogammaglobulinemia or recurrent, serious bacterial infections [Spector et al., 1994]. Certain complications, such as progressive multifocal leukoencephalopathy or primary CNS lymphoma, respond poorly to the available therapies; the prolonged survival of HIV-1-infected patients appears to increase their lifetime risk of developing progressive multifocal leukoencephalopathy or CNS lymphoma.
Current strategies uses combinations of at least three different antiretroviral drugs from the classes of nucleoside/nucleotide reverse transcriptase inhibitors, protease inhibitors, and non-nucleoside reverse transcriptase inhibitors. The goal of the therapeutic strategies is to reduce the HIV-1 viral load to undetectable levels and to restore immune function. Combination therapy produces immunologic and neurocognitive improvement, even in advanced HIV-1 infection [American Academy of Pediatrics, 2009; Department of Health and Human Services, 2008; Essajee et al., 1999; Patel et al., 2009]. Assessments at the start of antiretroviral therapy should include testing for CD4 T-cell count, plasma HIV-1 RNA, complete blood cell count, serum chemistry profile (especially tests of renal and liver function), and opportunistic infections, including T. gondii, hepatitis viruses, and tuberculosis. Frequent measurements of CD4 T-cell counts and HIV-1 RNA load by quantitative reverse transcriptase PCR, as well as routine laboratory studies (complete cell count, urinalysis, and serum chemistry profiles), have essential roles in monitoring HIV-1-infected children while they are undergoing antiretroviral therapy [Department of Health and Human Services, 2008].
The development of drug resistance remains a serious potential threat for HIV-1-infected patients and necessitates testing upon entry into treatment and when response to treatment is suboptimal [Boden et al., 1999; Department of Health and Human Services, 2008; Hirsch et al., 2008]. As many as 15 percent of HIV-1-infected adults display HIV-1 drug resistance prior to antiretroviral therapy [Department of Health and Human Services, 2008]. Therapy-associated immune reconstitution in patients with AIDS can lead to immune restoration disease, a condition characterized by paradoxical exacerbation of secondary infections during the initial 6 months of treatment [French et al., 2004; Stoll and Schmidt, 2003]. Immune restoration disease may occur in 30 percent of AIDS patients with low CD4+ cell counts who begin combination antiretroviral therapy. Children who undergo combination antiretroviral therapy can experience lipodystrophy, insulin resistance, osteopenia, and growth failure [Leonard and McComsey, 2003]. Secondary infectious complications of AIDS, such as cytomegalovirus retinitis, T. gondii encephalitis, and cryptococcal or tuberculous meningitis, require therapy with appropriate antimicrobial agents. Treatment of primary CNS lymphoma in persons with AIDS remains problematic [Belman, 2002].
Because treatment of HIV/AIDS continues to evolve, individuals and centers experienced in HIV/AIDS treatment should be consulted regarding current therapeutic approaches [Kitahata et al., 1996]. For updated, downloadable information regarding the available antiretroviral therapies and current evaluation and treatment guidelines for infants, children, and adolescents, see www.aidsinfo.nih.gov (accessed November 15, 2009). Despite the remarkable success of combined antiretroviral therapy, current drugs do not eradicate HIV-1 from infected persons [Lambotte et al., 2003]. Persons who reach end-stage AIDS, despite therapy, frequently experience AIDS-defining conditions, such as dementia/encephalopathy, progressive multifocal leukoencephalopathy, lymphoma, or invasive cytomegalovirus infections, during the 12 months before death [Welch and Morse, 2002]. Vaccines to prevent HIV/AIDS, although the focus of considerable research, are not yet available.
Human T-Cell Lymphotropic Virus Type I
HTLV-1, originally detected in adults with T-cell leukemia, causes a progressive myelopathy known as HTLV-1-associated myelopathy/tropical spastic paraparesis, an endemic disorder in Japan, certain Caribbean Islands, the Seychelles, Africa, India, and certain regions of Latin America [Levin and Jacobson, 1997; Osame et al., 1987; Román et al., 1987]. Transmission of HTLV-1 parallels that of HIV-1, involving vertical (mother-to-child) and horizontal (person-to-person or transfusion of blood products) routes. Infection during childhood occurs commonly in endemic regions. Although most reported patients have been adults, a few have had onset of symptoms during late childhood or early adolescence [Bittencourt et al., 2006; de Oliveira et al., 2004; Janssen et al., 1991; LaGrenade et al., 1995]. Clinical manifestations of HTLV-1-associated myelopathy/tropical spastic paraparesis are those of a chronic, progressive myelopathy, with spastic paraparesis, lower-extremity hyperreflexia, sensory dysfunction, and bladder dysfunction. Infective dermatitis, an eczematoid rash usually involving the scalp, is a common feature in pediatric cases [Bittencourt et al., 2006]. Although some patients respond temporarily to corticosteroid therapy, no curative antiviral therapy is currently available. Disease in HTLV-1-infected children and adolescents progresses more rapidly than in adults [Bittencourt et al., 2006].
Emerging Viral Infections
Nipah Virus
In the fall of 1998, an outbreak of severe encephalitis appeared in Malaysia [Chua, 2003; Chua et al., 2000; Johnson, 2003]. Electron microscopic studies of infected tissues identified a paramyxovirus, named Nipah virus after Kampung Sungei Nipah, the Malaysian village in which the virus was first detected. During the first outbreak, 265 cases of encephalitis occurred in humans ranging in age from 10 to nearly 70 years. The clinical features included fever, headache, vomiting, cough, cerebellar signs, pupillary abnormalities, seizures, autonomic dysfunction, and coma [Bellini et al., 2005; Goh et al., 2000]. Among the 265 human cases, there were 105 deaths, corresponding to a mortality rate of nearly 40 percent; ribavirin therapy was associated with reduced mortality [Chong et al., 2001].
Case-control studies indicated that Nipah encephalitis in humans resulted from contact with pigs or with pig excrement [Parashar et al., 2000], and after slaughter and disposal of pigs, the initial outbreak ended [Chua et al., 2000]. Fruit bats (genus Pteropus) serve as a natural reservoir of the virus. Virus persistence in endemic areas has led to additional outbreaks in India and Bangladesh, and during recent outbreaks, person-to-person spread, as well as food-borne transmission, has been documented [Chadha et al., 2006; Gurley et al., 2007; Hsu et al., 2004]. The disease remains highly fatal; neurocognitive deficits are common among survivors [Sejvar et al., 2007].
Dengue Virus
Dengue virus, an arthropod-borne flavivirus with four serotypes, exists endemically in tropical and subtropical regions worldwide, and at least 2.5 billion of the world’s inhabitants are at risk of infection [CDC, 2009i]. The CDC estimates that more than 100 million persons are infected with dengue virus annually [CDC, 2009i]. Spread by the bites of Aedes aegypti or Aedes albopictus mosquitoes, dengue virus commonly causes dengue fever (breakbone fever), a self-limited disorder associated with high fever, headache, myalgia, bone pain, rash, and easy bruising [CDC, 2009i]. The more severe form of infection, dengue hemorrhagic fever, is associated with a second phase, usually as the fever abates, with severe abdominal pain, dyspnea, and persistent vomiting’ in severe cases, death occurs due to shock or congestive heart failure. Petechiae, bruising, and frank hemorrhage, due to dengue virus-induced thrombocytopenia, can accompany this stage.
Dengue virus infections have been associated recently with encephalitis in endemic regions, although such cases appear to be rare, given the ubiquitous nature of dengue virus infections [Wasay et al., 2008]. Individuals with this condition experience fever, seizures, weakness, and alterations in consciousness, and have neurodiagnostic findings including CSF pleocytosis, slowing on EEG, and cerebral edema or focal areas of T2 hyperintensity on MRI [Wasay et al., 2008]. Myositis also occurs [Misra et al., 2006]. Cases of dengue virus encephalitis or encephalopathy have been described in both children and adults [Kumar et al., 2008; Misra et al., 2006; Wasay et al., 2008]. The diagnosis can be established by isolating the virus from clinical specimens (serum or autopsy tissues), detecting serologic responses using IgM antibody capture enzyme-linked immunosorbent assay (MAC-ELISA), or by detecting viral nucleic acids in serum or CSF using reverse transcription PCR [CDC, 2009i]. Virus can be isolated from serum during the first 5 days of fever; after this, MAC-ELISA becomes a highly sensitive and specific means to detect dengue virus infections [CDC, 2009l]. At present, treatment consists of fluid replacement, supportive care, and avoidance of medications, such as aspirin, that might potentiate the hemorrhagic complications.
Parechoviruses
Human parechoviruses, members of the picornavirus family and previously considered echoviruses, were linked to neurological disease of infants and young children early in the current millennium [Boivin et al., 2005; Ito et al., 2004]. In the initial case report, a 1-year-old Japanese girl experienced transient paralysis during a self-limited illness associated with fever and diarrhea [Ito et al., 2004]. Subsequently, parechovirus infections were described in Canadian neonates with high fever, rash, and respiratory distress; the diagnosis of parechovirus was established by growth of the viruses in cell culture and by analysis of the isolates by reverse transcription PCR [Boivin et al., 2005]. Most recently, parechovirus infections have been linked to neonatal encephalitis and injury to cerebral white matter [Levorson et al., 2009; Verboon-Maciolek et al., 2008a, b]. The cases to date indicate that parechovirus infections should be considered in infants who have sepsislike illnesses, especially when these are accompanied by rash, irritability, and seizures. Although CSF should be obtained to exclude other conditions, including neonatal herpes simplex virus infections, CSF pleocytosis appears to be an uncommon feature of this infection [Verboon-Maciolek et al., 2008b]. Parechovirus infection can be confirmed by reverse transcription PCR analysis of blood or CSF, or by culturing the virus from CSF, stool, or nasopharyngeal secretions. Given the propensity of parechoviruses to damage the white matter, infants with confirmed infections should undergo serial examination by MRI.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Abd-Allah S.A., Jansen P.W., Ashwal S., et al. Intravenous immunoglobulin as therapy for pediatric Guillain-Barré syndrome. J Child Neurol. 1997;12:376-380.
Abzug M.J., Cloud G., Bradley J., et al. Double blind placebo-controlled trial of pleconaril in infants with enterovirus meningitis. Pediatr Infect Dis J. 2003;22:335-341.
Adams L.A., Deboer B., Jeffrey G., et al. Ganciclovir and the treatment of Epstein-Barr virus hepatitis. J Gastroenterol Hepatol. 2006;21:1758-1760.
Alexander J.P., Baden L., Pallansch M.A., et al. Enterovirus 71 infections and neurologic disease – United States, 1977–1991. J Infect Dis. 1994;169:905-908.
Alkalay A.L., Pomerance J.J., Rimoin D.L. Fetal varicella syndrome. J Pediatr. 1987;111:320-323.
American Academy of Pediatrics. 2009 Red Book: Report of the Committee on Infectious Diseases, ed 28. Elk Grove Village, IL: American Academy of Pediatrics; 2009.
Andreoni M., El-Sawaf G., Rezza G., et al. High seroprevalence of antibodies to human herpesvirus-8 in Egyptian children: Evidence of nonsexual transmission. J Natl Cancer Inst. 1999;91:465-469.
Anlar B., Köse G., Gürer Y., et al. Changing epidemiologic features of subacute sclerosing panencephalitis. Infection. 2001;29:192-195.
Anlar B., Saatçi I., Köse G., et al. MRI findings in subacute sclerosing panencephalitis. Neurology. 1996;47:1278-1283.
Arai S., Matsunaga Y., Takasaki T., et al. Vaccine Preventable Diseases Surveillance Program of Japan. Japanese encephalitis: surveillance and elimination effort in Japan from 1982 to 2004. Jpn J Infect Dis. 2008;61:333-338.
Askalan R., Laughlin S., Mayank S., et al. Chickenpox and stroke in childhood: A study of frequency and causation. Stroke. 2001;32:1257-1262.
Atkinson C., Walter S., Sharland M., et al. Use of stored dried blood spots for retrospective diagnosis of congenital CMV. J Med Virol. 2009;81:1394-1398.
Awasthi M., Parmar H., Patankar T., et al. Imaging findings in rabies encephalitis. Am J Neuroradiol. 2001;22:677-680.
Bale J.F. Fetal infections and brain development. Clin Perinatol. 2009;36:639-653.
Bale J.F. Human herpesviruses and neurological disorders of childhood. Semin Pediatr Neurol. 1999;4:278-287.
Bale J.F., Miner L.J. Herpes simplex virus infections of the newborn. Curr Treat Options Neurol. 2005;7:151-156.
Balkhy H.H., Schreiber J.R. Severe La Crosse encephalitis with significant neurologic sequelae. Pediatr Infect Dis J. 2000;19:77-80.
Barnhart H.X., Caldwell M.B., Thomas P., et al. Natural history of human immunodeficiency virus disease in perinatally infected children: An analysis from the Pediatric Spectrum of Disease Project. Pediatrics. 1996;97:710-716.
Barone S.R., Kaplan M.H., Krilov L.R. Human herpesvirus-6 infection in children with first febrile seizures. J Pediatr. 1995;127:95-97.
Barthez-Carpentier M.A., Rozenberg F., Dussaix E., et al. Relapse of herpes simplex encephalitis. J Child Neurol. 1995;10:363-368.
Barton L.L., Mets M.B. Congenital lymphocytic choriomeningitis virus infection: Decade of rediscovery. Clin Infect Dis. 2001;33:370-374.
Baskin H.J., Hedlund G. Neuroimaging of herpesvirus infections in children. Pediatr Radiol. 2007;37:949-963.
Bellini W.J., Harcourt B.H., Bowden N., et al. Nipah virus: an emergence paramyxovirus causing severe encephalitis in humans. J Neurovirol. 2005;11:481-487.
Belman A.L. HIV infection and AIDS. Neurol Clin. 2002;20:983-1011.
Belman A.L., Lantos G., Horoupian D., et al. AIDS: Calcifications of the basal ganglia in infants and children. Neurology. 1986;36:1192-1199.
Belman A.L., Muenz L.R., Marcus J.C., et al. Neurologic status of human immunodeficiency virus 1-infected infants and their controls: A prospective study from birth to 2 years. Pediatrics. 1996;98:1109-1118.
Belman A.L., Ultmann M.H., Horoupian D., et al. Neurological complications in infants and children with acquired immune deficiency syndrome. Ann Neurol. 1985;18:560-566.
Berger J., Albrecht J., Belman A.L., et al. Progressive multifocal leukoencephalopathy in children with HIV infection. AIDS. 1992;6:837-841.
Bergman I., Painter M.J., Wald E.R., et al. Outcome in children with enteroviral meningitis during the first year of life. J Pediatr. 1987;110:705-709.
Bergström T., Vahlne A., Alestig K., et al. Primary and recurrent herpes simplex virus type 2 induced meningitis. J Infect Dis. 1990;162:322-330.
Berg S.W., Mitchell B.S., Hanson R.K., et al. Systemic reactions in U.S. Marine Corps personnel who received Japanese encephalitis vaccine. Clin Infect Dis. 1997;24:265-266.
Bittencourt A.L., Primo J., Oliveira M.F. Manifestations of the human T-cell lymphotropic virus type I infection in childhood and adolescence. J Pediatr (Rio J). 2006;82:411-420.
Blanton J.D., Robertson K., Palmer D., et al. Rabies surveillance in the US during 2008. J Am Vet Med Assoc. 2009;235:676-689.
Boden D., Hurley A., Zhang L., et al. HIV-1 drug resistance in newly infected individuals. JAMA. 1999;282:1135-1141.
Boivin G., Abed Y., Boucher F.D. Human parechovirus 3 and neonatal infections. Emerg Infect Dis. 2005;11:103-107.
Bonthius D.J., Nichols B., Harb H., et al. Lymphocytic choriomeningitis virus infection of the developing brain: role of host age. Ann Neurol. 2007;62:356-374.
Bonthius D.J., Wright R., Tseng B., et al. Congenital lymphocytic choriomeningitis virus infection: spectrum of disease. Ann Neurol. 2007;62:347-355.
Boppana S.B., Fowler K.B., Vaid Y., et al. Neuroradiographic finding in the newborn period and long-term outcome in children with symptomatic congenital cytomegalovirus infection. Pediatrics. 1997;99:409-414.
Bosma T.J., Corbett K.M., Eckstein M.B., et al. Use of PCR for prenatal and postnatal diagnosis of congenital rubella. J Clin Microbiol. 1995;33:2881-2887.
Boyle G.J., Michaels M.G., Webber S.T., et al. Posttransplantation lymphoproliferative disorders in pediatric thoracic organ recipients. J Pediatr. 1997;131:309-313.
Bray P.F. Mumps – a cause of hydrocephalus? Pediatrics. 1972;49:446-449.
Bremer J.W., Lew J.F., Cooper E., et al. Diagnosis of infection with human immunodeficiency virus type 1 by a DNA polymerase chain reaction assay among infants enrolled in the Women and Infant’s Transmission Study. J Pediatr. 1996;129:198-207.
Brunnell P. Varicella in pregnancy, the fetus, and the newborn: Problems in management. J Infect Dis. 1992;166:S42.
Burgess R.C., Michaels L., Bale J.F., et al. Polymerase chain reaction amplification of herpes simplex viral DNA from the geniculate ganglion of a patient with Bell’s palsy. Ann Otol Rhinol Laryngol. 1994;103:775-779.
Calisher C.H. Medically important arboviruses of the United States. Clin Microbiol Rev. 1994;7:89-116.
Calisher C.H., Berardi V.P., Muth D.J., et al. Specificity of immunoglobulin M and G antibody response in humans infected with eastern and western equine encephalitis viruses: Applications to rapid serodiagnosis. J Clin Microbiol. 1986;23:369-371.
Campbell C., Levin S., Humphreys P., et al. Subacute sclerosing panencephalitis: results of the Canadian Paediatric Surveillance Program and review of the literature. BMC Pediatr. 2005;5:47.
Campbell G.L., Marfin A.A., Lanciotti R.S., et al. West Nile virus. Lancet Infect Dis. 2002;20:519-529.
Centers for Disease Control and Prevention. Western equine encephalitis in the United States and Canada, 1987. Morb Mortal Wkly Rep. 1987;36:655-659.
Centers for Disease Control and Prevention: Arboviral Diseases ? United States, 1992. Morb Mortal WKly Rep. 1993;42:467-468.
Centers for Disease Control and Prevention. Inactivated Japanese encephalitis virus vaccine. Morb Mortal Wkly Rep. 1993;42(RR-1):1-15.
Centers for Disease Control and Prevention. Certification of poliomyelitis eradication – the Americas, 1994. Morb Mortal Wkly Rep. 1994;43:720-722.
Centers for Disease Control and Prevention. Recommendations of the U.S. Public Health Service task force on the use of zidovudine to reduce perinatal transmission of human immunodeficiency virus. Morb Mortal Wkly Rep. 1994;43(RR-11):1.
Centers for Disease Control and Prevention. Venezuelan encephalitis – Colombia, 1995. Morb Mortal Wkly Rep. 1995;44:721-724.
Centers for Disease Control and Prevention. Mumps surveillance – United States, 1988-1993. Morb Mortal Wkly Rep. 1995;44(SS-3):1-14.
Centers for Disease Control and Prevention. Update: Raccoon rabies epizootic – United States, 1996. Morb Mortal Wkly Rep. 1997;45:1117-1120.
Centers for Disease Control Prevention. Paralytic poliomyelitis – United States, 1980–1994. Morb Mortal Wkly Rep. 1997;46:79-83.
Centers for Disease Control and Prevention. Measles eradication: Recommendations from a meeting cosponsored by the World Health Organization, the Pan American Health Organization, and CDC. Morb Mortal Wkly Rep. 1997;46(RR-11):1-20.
Centers for Disease Control and Prevention. Nonpolio enterovirus surveillance – United States, 1997–1999. Morb Mortal Wkly Rep. 2000;49:913-916.
Centers for Disease Control and Prevention. Intrauterine West Nile Virus infection – New York, 2002. Morb Mortal Wkly Rep. 2002;51:1135-1136.
Centers for Disease Control and Prevention. www.cdc.gov/ncidod/dvbid/westnile/surv&controlCaseCount03_detailed.htm, 2004.
Centers for Disease Control and Prevention. Progress toward poliomyelitis eradication – Nigeria, January 2003-March 2004. Morb Mortal Wkly Rep. 2004;53:343-346.
Centers for Disease Control and Prevention. www.cdc.gov/ncidod/dvbid/westnile/background.html, 2004.
Centers for Disease Control and Prevention. Epidemiology of measles – United States, 2001–2003. Morb Mortal Wkly Rep. 2004;53:713-716.
Centers for Disease Control and Prevention. Update: Investigation of rabies infections in organ donor and transplant recipients – Alabama, Arkansas, Oklahoma, and Texas, 2004. Morb Mortal Wkly Rep. 2004;53:615-616.
Centers for Disease Control and Prevention. Interim guidelines for the evaluation of infants born to mothers infected with West Nile virus during pregnancy. Morb Mortal Wkly Rep. 2004;53:154-157.
Centers for Disease Control and Prevention. West Nile virus infections in organ transplant recipients. Morb Mortal Wkly Rep. 2005;54:1-3.
Centers for Disease Control and Prevention. Elimination of rubella and congenital rubella syndrome – United States, 1969–2004. Morb Mortal Wkly Rep. 2005;54:279-282.
Centers for Disease Control and Prevention (CDC). Enterovirus surveillance – United States. 2002–2004. Morb Mortal Wkly Rep. 2006;55:153-156.
Centers for Disease Control and Prevention (CDC). Progress toward interruption of wild poliovirus transmission – worldwide, 2008. Morb Mortal Wkly Rep. 2009;18(58):308-312.
Centers for Disease Control and Prevention (CDC). Update on vaccine-derived poliomyelitis – worldwide, January 2008-June 2009. Morb Mortal Wkly Rep. 2009;18(58):1002-1006.
Centers for Disease Control and Prevention. http://www.cdc.gov/EasternEquineEncephalitis/Epi.html, 2009. Accessed November 14, 2009
Centers for Disease Control and Prevention. http://www.cdc.gov/ncidod/dvbid/westnile/surv&control.htm, 2009. Accessed November 8, 2009
Centers for Disease Control and Prevention. http://www.cdc.gov/ncidod/dvbid/arbor/weefact.htm, 2009. Accessed November 14, 2009
Centers for Disease Control and Prevention. http://www.cdc.gov/ncidod/dvbid/sle/Sle_FactSheet.html, 2009. Accessed November 14, 2009
Centers for Disease Control and Prevention. http://www.cdc.gov/ncidod/dvrd/Spb/mnpages/dispages/TBE.htm, 2009. Accessed November 14, 2009
Centers for Disease Control and Prevention. http://www.cdc.gov/ncidod/dvbid/arbor/lacfact.htm, 2009. Accessed November 14, 2009
Centers for Disease Control and Prevention. http://www.cdc.gov/dengue/clinicalLab/laboratory.html, 2009. Accessed November 22, 2009
Centers for Disease Control and Preventionl. http://www.cdc.gov/rabies/epidemiology.html, 2009. Accessed November 14, 2009
Centers for Disease Control and Prevention. http://www.cdc.gov/rabies/exposure/postexposure.html, 2009. Accessed November 14, 2009
Chadha M.S., Comer J.A., Lowe L., et al. Nipah virus-associated encephalitis outbreak. Siliguiri, India. Emerg Infect Dis. 2006;12:235-240.
Chang Y., Cesarman E., Pessin M.S., et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma patients. Science. 1994;266:1865-1869.
Chayavichitsilp P., Buckwalter J.V., Krakowski A.C., et al. Herpes simplex. Pediatr Rev. 2009;30:119-129.
Chen C.Y., Chang Y.C., Huang C.C., et al. Acute flaccid paralysis in infants and young children with enterovirus 71 infections: MR imaging findings and clinical correlates. Am J Neuroradiol. 2001;22:200-205.
Chen R.E., Ramsay D.A., deVeber L.L., et al. Immunosuppressive measles encephalitis. Pediatr Neurol. 1994;10:325-327.
Chong H.T., Kamarulzaman A., Tan C.T., et al. Treatment of acute Nipah encephalitis with ribavirin. Ann Neurol. 2001;49:810-813.
Chopra J.S., Banerjee A.K., Murthy J.M.K., et al. Paralytic rabies: A clinic – pathological study. Brain. 1980;103:789-802.
Chua K.B. Nipah virus outbreak in Malaysia. J Clin Virol. 2003;26:265-275.
Chua K.B., Bellini W.J., Rota P.A., et al. Nipah virus: A recently emergent deadly paramyxovirus. Science. 2000;288:1432-1435.
Chuang Y.Y., Chiu C.H., Wong K.S., et al. Severe adenovirus infection in children. J Microbiol Immunol Infect. 2003;36:37-40.
Clifford D.B., Buller R.S., Mohammed S., et al. Use of polymerase chain reaction to demonstrate cytomegalovirus DNA in CSF of patients with human immunodeficiency virus infection. Neurology. 1993;43:75-79.
Clifford D.B., Yiannoutsos C., Glicksman M., et al. HAART improves prognosis in HIV-associated progressive multifocal leukoencephalopathy. Neurology. 1999;52:623-625.
Connelly K.P., DeWitt L.D. Neurology complications of infectious mononucleosis. Pediatr Neurol. 1994;10:181-184.
Connolly A.M., Dodson W., Prensky A.L., et al. Course and outcome of acute cerebellar ataxia. Ann Neurol. 1994;35:673-679.
Connor E.M., Sperling R.S., Gelber R., et al. Reduction of maternal-infant transmission of human immunodeficiency virus type 1 with zidovudine treatment. N Engl J Med. 1994;331:1173-1180.
Conrady C.D., Drevets D.A., Carr D.J. Herpes simplex virus type 1 (HSV-1) infection of the central nervous system: Is an immune response a good thing? J Neuroimmunol. 2009. Oct 8
Corey L., Spear P.G. Infections with herpes simplex viruses. N Engl J Med. 1986;314:686-691.
Corey L., Wald A. Maternal and neonatal herpes simplex virus infections. N Engl J Med. 2009;361:1376-1385.
Crawford J.R., Santi M.R., Thorarinsdottir H.K., et al. Detection of human herpesvirus-6 variants in pediatric brain tumors: association of viral antigen in low grade gliomas. J Clin Virol. 2009;46:37-42.
Creech W.B. St. Louis encephalitis in the United States, 1975. J Infect Dis. 1977;135:1014-1016.
Darin N., Bergstrom T., Fast A., et al. Clinical, serological, and PCR evidence of cytomegalovirus infection in the central nervous system of childhood. Neuropediatrics. 1994;25:316-322.
Dayan H.D., Quinlisk M.P., Parker A.A., et al. Recent resurgence of mumps in the United States. N Engl J Med. 2008;358:1580-1589.
DeCarli C., Civitello L.A., Brouwers P., et al. The prevalence of computed tomographic abnormalities of the cerebrum in 100 consecutive children symptomatic with human immune deficiency virus. Ann Neurol. 1993;34:198-205.
De Cock K.M., Fowler M.G., Mercier E., et al. Prevention of mother-to-child HIV transmission in resource-poor countries: Translating research into policy and practice. JAMA. 2000;283:1175-1182.
Demmler G.J. Summary of a workshop on surveillance for congenital cytomegalovirus disease. Rev Infect Dis. 1991;13:315-329.
Demmler G.J., Buffone G.J., Schimbor C.M., et al. Detection of cytomegalovirus in urine from newborns by using polymerase chain reaction DNA amplification. J Infect Dis. 1988;158:1177-1184.
de Oliveira MdeF, Bittencourt A.L., Brites C., et al. HTLV-I associated myelopathy/tropical spastic paraparesis in a 7-year-old boy associated with infective dermatitis. J Neurol Sci. 2004;222:35-38.
Department of Health and Human Services. Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. November 3, 2008:1-139. http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. Accessed November 15, 2009
Deresiewicz R.L., Thaler S.J., Hsu L., et al. Clinical and neuroradiographic manifestations of eastern equine encephalitis. N Engl J Med. 1997;336:1867-1874.
Desmond R.A., Accortt N.A., Talley L., et al. Enteroviral meningitis: natural history and outcome of pleconaril therapy. Antimicrob Agents Chemother. 2006;50:2409-2414.
de Vries L.S., Gunardi H., Barth P.G., et al. The spectrum of cranial ultrasound and magnetic resonance imaging abnormalities in congenital cytomegalovirus infection. Neuropediatrics. 2004;35:113-119.
Dewhurst S. Human herpesvirus type 6 and human herpesvirus type 7 infections of the central nervous system. Herpes. 2004;11(Suppl 2):105A-111A.
Dhillon G.P., Raina V.K. Epidemiology of Japanese encephalitis in context with Indian scenario. J Indian Med Assoc. 2008;106:660-663.
Doja A., Bitnun A., Jones E.L., et al. Pediatric Epstein-Barr Virus-Associated Encephalitis: 10-Year Review. J Child Neurol. 2006;21:385-391.
Domachowske J.B., Cunningham C.K., Cummings D.L., et al. Acute manifestations and neurologic sequelae of Epstein-Barr virus encephalitis in children. Pediatr Infect Dis J. 1996;15:871-875.
Domingues R.B., Fink M.C., Tsanaclis S.M., et al. Diagnosis of herpes simplex encephalitis by magnetic resonance imaging and polymerase chain reaction assay of cerebrospinal fluid. J Neurol Sci. 1998;157:148-153.
Dudgeon J.A. Congenital rubella. J Pediatr. 1975;87:1078-1086.
Dutta J.K. Human rabies in India: Epidemiologic features, management and current methods of prevention. Trop Doct. 1999;29:196-201.
Earnest M.P., Goolishian H.A., Calverley J.R., et al. Neurologic, intellectual and psychologic sequelae following western encephalitis. Neurology. 1971;21:969-974.
Ellstein–Shturman R., Borkowsky W., Fish I., et al. Myelitis associated with genetial herpes in a child. J Pediatr. 1976;88:523.
Embil J.A., Camfield P., Artsob H., et al. Powassan virus encephalitis resembling herpes simplex encephalitis. Arch Intern Med. 1983;143:341-343.
Epstein L.G., Gendelman H.E. Human immunodeficiency virus type 1 infection of the central nervous system: Pathogenetic mechanisms. Ann Neurol. 1993;33:429-436.
Epstein L.G., Sharer L.R. Neurological manifestations of perinatally acquired HIV-1 infection. Semin Pediatr Neurol. 1994;1:50-57.
Epstein L.G., Sharer L.R., Joshi V.V., et al. Progressive encephalopathy in children with acquired immune deficiency syndrome. Ann Neurol. 1985;17:488-496.
Essajee S.M., Kim M., Gonzalez C., et al. Immunologic and virologic responses to HAART in severely immunocompromised HIV-1-infected children. AIDS. 1999;13:2523-2532.
Falloon J., Eddy J., Wiener L., et al. Human immunodeficiency virus infection in children. J Pediatr. 1989;114:1-30.
Feigin R.D., Shackelford P.G. Value of repeat lumbar puncture in the differential diagnosis of meningitis. N Engl J Med. 1973;289:571-574.
Fishbein D.B., Robinson L.E. Rabies. N Engl J Med. 1993;329:1632-1638.
Forrst J.M., Turnbull F.M., Sholler G.F., et al. Gregg’s congenital rubella patients 60 years later. Med J Aust. 2002;177:664-667.
Fowler K.B., McCollister F.P., Dahle A.J., et al. Progressive and fluctuating sensorineural hearing loss in children with asymptomatic congenital cytomegalovirus infection. J Pediatr. 1997;130:624-630.
Fowler K.B., Stagno S., Pass R.F. Maternal immunity and prevention of congenital cytomegalovirus infection. JAMA. 2003;289:1008-1011.
Fowlkes A.L., Honarmand S., Glaser C., et al. Enterovirus-associated encephalitis in the California encephalitis project, 1998–2005. J Infect Dis. 2008;198:1685-1691.
French M.A., Price P., Stone S.F. Immune restoration disease after antiretroviral therapy. AIDS. 2004;18:1615-1627.
Frey T. Optic neuritis in children infections mononucleosis as an etiology. Doc Ophthalmol. 1973;34:183-188.
Garg R.K. Subacute sclerosing panencephalitis. J Neurol. 2008;255:1861-1871.
Gascon G.G., International Consortium on Subacute Sclerosing Panencephalitis. Randomized treatment study of inosiplex versus combined inosiplex and intraventricular interferon-alpha in subacute sclerosing panencephalitis (SSPE): International multicenter study. Child Neurol. 2003;18:819-827.
Gavin P., Yogev R. Central nervous system abnormalities in pediatric human immunodeficiency virus infection. Pediatr Neurosurg. 1999;31:115-123.
Gewurz B.E., Marty F.M., Baden L.R., et al. Human herpesvirus 6 encephalitis. Curr Infect Dis Rep. 2008;10:292-299.
Gilden D. Varicella zoster virus and central nervous system syndromes. Herpes. 2004;11(Suppl 2):89a-94a.
Givens K.T., Lee D.A., Jones T., et al. Congenital rubella syndrome: Ophthalmic manifestations and associated systemic disorders. Br J Ophthalmol. 1993;77:358-363.
Goh K.J., Tan C.T., Chew N.K., et al. Clinical features of Nipah virus encephalitis among pig farmers in Malaysia. N Engl J Med. 2000;342:1229-1235.
Goodpasture H.C., Poland J.D., Francy D.B., et al. Colorado tick fever: Clinical, epidemiologic, and laboratory aspects of 228 cases in Colorado in 1973–1974. Ann Intern Med. 1978;88:303-310.
Grabow J.D., Matthews C.G., Chun R.W., et al. The electroencephalogram and clinical sequelae of California arbovirus encephalitis. Neurology. 1969;19:394-404.
Griffiths P.D. CMV as a cofactor enhancing progression of AIDS. J Clin Virol. 2006;35:489-492.
Grose C. Congenital infections caused by varicella zoster virus and herpes simplex virus. Semin Pediatr Neurol. 1994;1:43.
Grose C., Bonthius D., Afifi A.K. Chickenpox and the geniculate ganglion: Facial nerve palsy, Ramsay Hunt syndrome and acyclovir treatment. Pediatr Infect Dis J. 2002;21:615-617.
Guess H.A., Broughton D.D., Melton L.J., et al. Epidemiology of herpes zoster in children and adolescents. Pediatrics. 1985;76:512-517.
Gurley E.S., Montgomery J.M., Hossain M.J., et al. Person to person transmission of Nipah virus in a Bangladeshi community. Emerg Infect Dis. 2007;13:1031-1037.
Gutman L.T., Wilfert C.M., Eppes S. Herpes simplex virus encephalitis in children: Analysis of cerebrospinal fluid and progressive neurodevelopmental deterioration. J Infect Dis. 1986;154:415-421.
Hall C.B., Long C.E., Schnabel K.C., et al. Human herpesvirus-6 infection in children. N Engl J Med. 1994;331:432-450.
Hamilton M.S., Jackson M.A., Abel D. Clinical utility of polymerase chain reaction testing for enteroviral meningitis. Pediatr Infect Dis J. 1999;18:533-537.
Hanshaw J.B., Dudgeon. Viral Diseases of the Fetus and Newborn. Philadelphia: WB Saunders Co; 1978.
Hausler M., Schaade L., Kemeny S., et al. Encephalitis related to primary varicella zoster virus infection in immunocompetent children. J Neurol Sci. 2002;195:111-116.
Havens P.L., Waters D. Management of the infant born to a mother with HIV infection. Pediatr Clin North Am. 2004;51:909-937.
Hayes E.B., O’Leary D.R. West Nile virus: A pediatric perspective. Pediatrics. 2004;113:1375-1381.
Henao-Restrepo A.M., Strebel P., Hoekstra E.J. Experience in global measles control, 1990–2001. J Infect Dis. 2003;187(Suppl 1):S-15.
Hilt D.C., Buchholz D., Krumholz A., et al. Herpes zoster ophthalmicus and delayed contralateral hemiparesis caused by cerebral angiitis: Diagnosis and management approaches. Ann Neurol. 1983;14:543-553.
Hilty M.D., Haynes R.E., Azimi P.H., et al. California encephalitis in children. Am J Dis Child. 1972;124:530-533.
Hirsch M.S., Günthard H.F., Schapiro J.M., et al. Antiretroviral drug resistance testing in adult HIV-1 infection: 2008 recommendations of an International AIDS Society – USA panel. Clin Infect Dis. 2008;47:266-285.
Holland N.R., Power C., Mathews V., et al. Cytomegalovirus encephalitis in acquired immunodeficiency syndrome (AIDS). Neurology. 1994;44:507-514.
Honarmand S., Glaser C.A., Chow E., et al. Subacute sclerosing panencephalitis in the differential diagnosis of encephalitis. Neurology. 2004;63:1489-1493.
Horwitz C.A., Henle W., Henle G., et al. Clinical and laboratory evaluation of cytomegalovirus-induced mononucleosis in previously healthy individuals. Medicine. 1986;66:124-134.
Houff S.A., Burton R.C., Wilson R.W., et al. Human-to-human transmission of rabies virus by corneal transplant. N Engl J Med. 1979;300:603.
Hsieh W.B., Chiu N.C., Hu K.C., et al. Outcome of herpes simplex encephalitis in children. J Microbiol Immunol Infect. 2007;40:34-38.
Hsu V.P., Hossain M.J., Parashar U.D., et al. Nipah virus reemergence. Bangladesh. Emerg Infect Dis. 2004;10:2082-2087.
Huang C.C., Liu C.C., Chang Y.C., et al. Neurologic complications in children of enterovirus 71 infection. N Engl J Med. 1999;341:936.
Hubalek Z., Halouzka J. West Nile fever – a reemerging mosquito-borne viral disease in Europe. Emerg Infect Dis. 1999;5:643-650.
Hughes S.S. The virus: A history of the concept. London: Heinemann Educational Books; 1977.
Hutto C., Arvin A., Jacobs R., et al. Intrauterine herpes simplex virus infections. J Pediatr. 1987;110:97-101.
Huttunen P., Lappalainen M., Salo E., et al. Differential diagnosis of acute central nervous system infections in children using modern microbiological methods. Acta Paediatr. 2009;98:1300-1306.
Iannetti P., Nigro G., Spalice A., et al. Cytomegalovirus infection and schizencephaly: Case reports. Ann Neurol. 1998;43:123-127.
Ito M., Yamashita T., Tsuzuki H., et al. Isolation and identification of a novel human parechovirus. J Gen Virol. 2004;85:391-398.
Jacobson M.A. Treatment in cytomegalovirus retinitis in patients with the acquired immunodeficiency syndrome. N Engl J Med. 1997;337:105-114.
James S.H., Kimberlin D.W., Whitley R.J. Antiviral therapy for herpesvirus central nervous system infections: neonatal herpes simplex virus infection, herpes simplex encephalitis, and congenital cytomegalovirus infection. Antiviral Res. 2009;83:207-213.
Jansen P.W., Perkin R.M., Ashwal S. Guillain-Barré syndrome in childhood: Natural course and efficacy of plasmapheresis. Pediatr Neurol. 1992;9:16-20.
Janssen R.S., Kaplan J.E., Khabbaz R.F., et al. HTLV-I associated myelopathy/tropical spastic paraparesis in the United States. Neurology. 1991;41:1355-1357.
Johnson A.J., Karabatsos N., Lanciotti R.S. Detection of Colorado tick fever virus by using reverse transcriptase PCR and application of the technique in laboratory diagnosis. J Clin Microbiol. 1997;35:1203-1208.
Johnson R.T. Acute encephalitis. Clin Infect Dis. 1996;23:219-224.
Johnson R.T. Emerging infections of the nervous system. J Neurovirol. 2003;9:140-147.
Johnson R.T. Viral infections of the nervous system, ed 2. New York: Lippincott-Raven; 1998.
Johnson R.T., Griffin D.E., Hirsch R.L., et al. Measles encephalomyelitis – clinical and immunologic studies. N Engl J Med. 1984;310:137-141.
Kaiser R. Tick-borne encephalitis. Infect Dis Clin North Am. 2008;22:561-575.
Kalita J., Misra U.K. Comparison of CT scan and MRI findings in the diagnosis of Japanese encephalitis. J Neurol Sci. 2000;174:3-8.
Kang S.S., McGavern D.B. Lymphocytic choriomeningitis infection of the central nervous system. Front Biosci. 2008;13:4529-4543.
Kaplan A.M., Longhurst W.L., Randall D.L. St. Louis encephalitis in children. West J Med. 1978;128:279-281.
Kido S., Ozaki T., Asada H., et al. Detection of varicella zoster virus (VZV) DNA in clinical samples from patients with VZV by the polymerase chain reaction. J Clin Microbiol. 1991;29:76-79.
Kimberlin D.W. Herpes simplex virus, meningitis, and encephalitis in neonates. Herpes. 2004;11(Suppl 2):65A.
Kimberlin D.W., Lakeman F.D., Arvin A.M., et al. Application of the polymerase chain reaction to the diagnosis and management of neonatal herpes simplex virus disease. J Infect Dis. 1996;174:1162-1167.
Kimberlin D.W., Lin C.-Y., Jacobs R.F., et al. Natural history of neonatal herpes simplex virus infections in the acyclovir era. Pediatrics. 2001;108:223-227.
Kimberlin D.W., Lin C.Y., Jacobs R.F., et al. Safety and efficacy of high-dose intravenous acyclovir in the management of neonatal herpes simplex virus infections. Pediatrics. 2001;108:230-238.
Kimberlin D.W., Lin C.Y., Sanchez P.J., et al. Effect of ganciclovir therapy on hearing in symptomatic congenital cytomegalovirus disease involving the central nervous system: A randomized, controlled trial. J Pediatr. 2003;143:16-25.
Kitahata M.M., Koepsell T.D., Deyo R.A., et al. Physicians’ experience with the acquired immunodeficiency syndrome as a factor in patients’ survival. N Engl J Med. 1996;334:701-706.
Knipe D.M., Howley P.M., Griffin D.E., et al. Fields Virology. Philadelphia, PA: Lippincott, Williams & Wilkins; 2007.
Koch L.H., Fisher R.G., Chen C., et al. Congenital herpes simplex virus infection: two unique cutaneous presentations associated with probable intrauterine transmission. J Am Acad Dermatol. 2009;60:312-315.
Koskiniemi M., Donner M., Pettay O. Clinical appearance and outcome in mumps encephalitis in children. Acta Paediatr Scand. 1983;72:603-609.
Koskiniemi M., Happonen J.M., Järvenpää A.J., et al. Neonatal herpes simplex virus infection: A report of 43 patients. Pediatr Infect Dis J. 1989;8:830-835.
Koskiniemi M., Rantalaiho T., Piiparinen H., et al. Infections of the central nervous system of suspected viral origin: A collaborative study from Finland. J Neurovirol. 2001;7:400-408.
Koskiniemi M., Rautonen J., Lehtokoski-Lehtiniemi E., et al. Epidemiology of encephalitis in children: A 20 year survey. Ann Neurol. 1991;29:492-497.
Kumar R., Mathur A., Kumar A., et al. Clinical features and prognostic indicators of Japanese encephalitis in children in Lucknow (India). Indian J Med Res. 1990;91:321-327.
Kumar R., Mathur A., Singh K.B., et al. Clinical sequelae of Japanese encephalitis in children. Indian J Med Res. 1993;97:9-13.
Kumar R., Tripathi S., Tambe J.J., et al. Dengue encephalopathy in children in Northern India: clinical features and comparison with non-Dengue. J Neurol Sci. 2008;269:41-48.
Kumar S., Misra U.K., Kalita J., et al. MRI in Japanese encephalitis. Neuroradiology. 1997;39:180-184.
Kuno G., Mitchell C.J., Chang G.J., et al. Detecting bunyaviruses of the Bunyamwera and California serogroups by a PCR technique. J Clin Microbiol. 1996;34:1184-1188.
Kunze U., Asokliene L., Bektimirov T., et al. Tick-borne encephalitis in childhood – consensus 2004. Wien Med Wochenschr. 2004;154:242-245.
LaGrenade L., Morgan C., Carberry C., et al. Tropical spastic paraparesis occurring HTLV-I associated infective dermatitis: Report of two cases. West Indian Med J. 1995;44:34-35.
Lakeman F.D., Whitley R.J. Diagnosis of herpes simplex encephalitis: Application of polymerase chain reaction to cerebrospinal fluid from brain biopsied patients and correlation with disease. J Infect Dis. 1995;171:857-863.
Lambert A.J., Martin D.A., Lanciotti R.S. Detection of North American eastern and western equine encephalitis viruses by nucleic acid amplification assays. J Clin Microbiol. 2003;41:379-385.
Lambotte O., Deiva K., Tardieu M. HIV-1 persistence, viral reservoir, and the central nervous system in the HAART era. Brain Pathol. 2003;13:95-103.
Lanciotti R.S., Kersat A.J. Nucleic acid sequence-based amplifications assays for rapid detection of West Nile and St. Louis encephalitis viruses. J Clin Microbiol. 2001;39:4506-4513.
Lanciotti R.S., Roehrig J.T., Deubel V., et al. Origin of the West Nile virus responsible for an outbreak of encephalitis in the northeastern United States. Science. 1999;286:2333-2337.
Landgren M., Kyllerman M., Bergström T., et al. Diagnosis of Epstein Barr virus-induced central nervous system infections by DNA amplification from cerebrospinal fluid. Ann Neurol. 1994;35:631-635.
Leach C.T., Sumaya C.V., Brown N.A. Human herpesvirus-6: Clinical implications of a recently discovered, ubiquitous agent. J Pediatr. 1992;121:173-181.
Leech R.W., Harris J.C., Johnson R.H. 1975 encephalitis epidemic in North Dakota and Western Minnesota. Minn Med. 1981;64:545-548.
Leis A.A., Butler I.J. Infantile herpes zoster ophthalmicus and acute hemiparesis following intrauterine chickenpox. Neurology. 1987;37:1537-1538.
Leonard E.G., McComsey G.A. Metabolic complications of antiretroviral therapy in children. Pediatr Infect Dis J. 2003;22:77-84.
Leonard J.R., Moran C.J., Cross D.T., et al. MR imaging of herpes simplex type 1 encephalitis in infants and young children: A separate pattern of findings. Am J Roentgenol. 2000;174:1651-1655.
Lesnicar G., Poljak M., Seme K., et al. Pediatric tick-borne encephalitis in 371 cases from an endemic region in Slovenia, 1959 to 2000. Pediatr Infect Dis J. 2003;22:612-617.
Levin M.C., Jacobson S. HTLV-I associated myelopathy/tropical spastic paraparesis (HAM/TSP): A chronic progressive neurologic disease associated with immunologically mediated damage to the central nervous system. J Neurovirol. 1997;3:126-140.
Levorson R., Jantausch B., Weiderman B.L., et al. Human parechovirus-3 infection: emerging pathogen in neonatal sepsis. Pediatr Infect Dis J. 2009;28:545-547.
Li J., Loeb J.A., Shy M.E., et al. Asymmetric flaccid paralysis: A neuromuscular presentation of West Nile virus infection. Ann Neurol. 2003;53:703-710.
Liaw S.-B., Shen E.-Y. Alice in Wonderland syndrome as presenting symptom of EBV infection. Pediatr Neurol. 1991;7:464-466.
Lloyd M., Lloyd J., Gesteland P., et al. Association between rotavirus and seizures in children hospitalized in Utah, 2002–2006. Ann Neurol. 2007;62:S142.
Lum G.B., Williams J.P., Dyken P.R., et al. Magnetic resonance and CT imaging correlated with clinical status in SSPE. Pediatr Neurol. 1986;2:75-79.
Malm G. Neonatal herpes simplex virus infection. Semin Fetal Neonatal Med. 2009;14:204-208.
Malm G., Engman M.L. Congenital cytomegalovirus infections. Semin Fetal Neonatal Med. 2007;12:154-159.
Mansfield K.L., Johnson N., Phipps L.P., et al. Tick-borne encephalitis virus – a review of an emerging zoonosis. J Gen Virol. 2009;90:1781-1794.
Marberg K., Goldblum N., Sterk V.V., et al. The natural history of the West Nile fever: 1. Clinical observations during an epidemic in Israel. Am J Hyg. 1956;64:259-269.
Martin J.N., Ganem D.E., Osmond D.H., et al. Sexual transmission and the natural history of human herpesvirus 8 infection. N Engl J Med. 1998;338:948-954.
Maschke M., Kastrup O., Forsting M., et al. Update on neuroimaging of infectious central nervous system disease. Curr Opin Neurol. 2004;17:475-480.
McCutchan J.A. Clinical impact of cytomegalovirus infections of the nervous system in patients with AIDS. Clin Infect Dis. 1995;21:S196-S201.
McGuire D., Barhite S., Hollander H., et al. JC virus DNA in cerebrospinal fluid in human immunodeficiency virus-infected patients: Predictive value for progressive multifocal leukoencephalopathy. Ann Neurol. 1995;37:395-399.
McJunkin J.E., de los Reyes E.C., Irazuzta J.E., et al. La Crosse encephalitis in children. N Engl J Med. 2001;344:801-807.
McJunkin J.E., Khan R., de-los Reyes E.C., et al. Treatment of severe La Crosse encephalitis with intravenous ribavirin following diagnosis by brain biopsy. Pediatrics. 1997;99:261-267.
Mease P.J., Ochs H.D., Wedgwood R.J. Successful treatment of meningoencephalitis and myositis-fasciitis with intravenous immune globulin therapy in a patient with X-linked agammaglobulinemia. N Engl J Med. 1981;304:1278.
Medovy H. Western equine encephalomyelitis in infants. J Pediatr. 1943;22:308.
Meehan P.J., Wells D.L., Paul W., et al. Epidemiological features of and public health response to a St. Louis encephalitis epidemic in Florida, 1990–1. Epidemiol Infect. 2000;125:181-188.
Meiyu F., Huosheng C., Cuihua C., et al. Detection of flaviviruses by reverse transcriptase-polymerase chain reaction with the universal primer set. Microbiol Immunol. 1997;41:209-213.
Melvin A.J., Mohan K.M., Arcuino L.A., et al. Clinical, virologic and immunologic responses of children with advanced human immunodeficiency virus type 1 disease treated with protease inhibitors. Pediatr Infect Dis J. 1997;16:968-974.
Mikati M.A., Feraru E., Krishnamoorthy K., et al. Neonatal herpes simplex meningoencephalitis. Neurology. 1990;40:1433-1437.
Miller E.T., Marsh R.H., Harris N.S. Rabies exposure – implications for wilderness travelers. Wilderness Environ Med. 2009;20:290-296.
Miller R.F., Lucas S.B., Hall-Craggs M.A., et al. Comparison of magnetic resonance imaging with neuropathological findings in the diagnosis of HIV and CMV associated CNS disease in AIDS. J Neurol Neurosurg Psychiatry. 1997;62:346-351.
Miravet E., Danchaivijitr N., Basu H., et al. Clinical and radiological features of childhood cerebral infarction following varicella zoster virus infection. Dev Med Child Neurol. 2007;49:417-422.
Misra U.K., Kalita J., Syam U.K., et al. Neurological manifestations of dengue virus infection. J Neurol Sci. 2006;244:117-122.
Mitchell C.D. HIV-1 encephalopathy among perinatally infected children: Neuropathogenesis and response to highly active antiretroviral therapy. Ment Retard Dev Disabil Res Rev. 2006;12(3):216-222.
Mitchell P.S., Espy M.J., Smith T.F., et al. Laboratory diagnosis of central nervous system infections with herpes simplex virus by PCR performed in cerebrospinal fluid specimens. J Clin Microbiol. 1997;35:2873-2877.
Mizrahi E.M., Tharp B.R. A characteristic EEG pattern in neonatal herpes simplex encephalitis. Neurology. 1982;32:1215-1220.
Modlin J.F., Halsey N.A., Eddins D.L., et al. Epidemiology of subacute sclerosing panencephalitis. J Pediatr. 1979;94:231-236.
Monath T.P., Nystrom R.R., Bailey R.E., et al. Immunoglobulin M antibody capture enzyme-linked immunosorbent assay for diagnosis of St. Louis encephalitis. J Clin Microbiol. 1984;20:784-790.
Motoyama M., Ichiyama T., Matsushige T., et al. Clinical characteristics of benign convulsions with rotavirus gastroenteritis. J Child Neurol. 2009;24:557-561.
Murph J.R., Bale J.F. The natural history of acquired cytomegalovirus infection among children in group day care. Am J Dis Child. 1988;142:843-846.
Murphy J.V., Yunis E.J. Encephalopathy following measles infection in children with chronic illness. J Pediatr. 1976;88:937-942.
Mustafa M.M., Weitman S.D., Winick N.J., et al. Subacute measles encephalitis in the young immunocompromised host: Report of two cases diagnosed by polymerase chain reaction and treated with ribavirin and review of the literature. Clin Infect Dis. 1993;16:654-660.
Nakayama T., Mori T., Yamaguchi S., et al. Detection of measles virus genome directly from clinical samples by reverse-transcriptase-polymerase chain reaction and genetic variability. Virus Res. 1995;35:1-16.
Nash D., Mostashari F., Fine A., et al. The outbreak of West Nile virus infection in the New York City area in 1999. N Engl J Med. 2001;344:1807-1814.
Negrini B., Kelleher K.J., Wald E.R. Cerebrospinal fluid findings in aseptic versus bacterial meningitis. Pediatrics. 2000;105:316-319.
Nicolosi A., Hauser W.A., Beghi E., et al. Epidemiology of central nervous system infections in Olmstead County, Minnesota, 1950–1981. J Infect Dis. 1986;154:399-408.
Noah D.L., Drenzek C.L., Smith J.S., et al. Epidemiology of human rabies in the United States, 1980 to 1996. Ann Intern Med. 1998;128:922-930.
Noyola D.E., Demmler G.J., Nelson C.T., et al. Early predictors of neurodevelopmental outcome in symptomatic congenital cytomegalovirus infection. J Pediatr. 2001;138:325-331.
O’Leary D.R., Kuhn S., Kniss K.L., et al. Birth outcomes following West Nile Virus infection of pregnant women in the United States: 2003–2004. Pediatrics. 2006;11:e537-e545.
Ooi M.H., Lewthwaite P., Lai B.F., et al. The epidemiology, clinical features, and long-term prognosis of Japanese encephalitis in central Sarawak, Malaysia, 1997–2005. Clin Infect Dis. 2008;47:458-468.
Osame M., Matsumoto M., Usuku K., et al. Chronic progressive myelopathy associated with elevated antibodies to human T-lymphotropic virus type I and adult T-cell leukemia-like cells. Ann Neurol. 1987;21:117-122.
Paessler S., Weaver S.C. Vaccines for Venezuelan equine encephalitis. Vaccine. 2009;27(Suppl 4):D80-D85.
Palumbo P.E. Antiretroviral therapy of HIV infection in children. Pediatr Clin North Am. 2000;47:155-169.
Panagiotopoulos T., Georgakopoulou T. Epidemiology of rubella and congenital rubella syndrome in Greece, 1994–2003. Eur Surveill. 2004;1:17-19.
Parashar U.D., Sunn L.M., Ong F., et al. Case-control study of risk factors for human infection with a new zoonotic paramyxovirus, Nipah virus during 1998–1999 outbreak of severe encephalitis in Malaysia. J Infect Dis. 2000;181:1755-1759.
Park Y.D., Belman A.L., Kim T.S., et al. Stroke in pediatric acquired immunodeficiency syndrome. Ann Neurol. 1990;28:303-311.
Pass R.F., Stagno S., Myers G.J., et al. Outcome of symptomatic congenital cytomegalovirus infection: Results of long-term longitudinal follow-up. Pediatrics. 1980;66:758-762.
Pass R.F., Zhang C., Evans A., et al. Vaccine prevention of maternal cytomegalovirus infection. N Engl J Med. 2009;360:1191-1199.
Patel K., Ming X., Williams P.L., et al. Impact of HAART and CNS-penetrating antiretroviral regimens on HIV encephalopathy in perinatally infected children and adolescents. AIDS. 2009;23:1893-1901.
Peters C.J., Buchmeir M., Rollin P.E., et al. Arenaviruses. In: Fields B.N., Knipe D.M., Howley P.M., et al, editors. Fields virology. Philadelphia: Lippincott-Raven, 1996.
Petersen L.R., Roehrig J.T., Hughes J.M. West Nile virus encephalitis. N Engl J Med. 2002;347:1225-1226.
Pizzo P.A., Eddy J., Falloon J., et al. Effect of continuous intravenous infusion of zidovudine (AZT) in children with symptomatic HIV infection. N Engl J Med. 1988;319:889-896.
Plotkin S.A. Rabies. Clin Infect Dis. 2000;30:4.
Pollak L., Klein C., Schiffer J., et al. Electroencephalographic abnormalities in aseptic meningitis and noninfectious headache. A comparative study. Headache. 2001;41:79-83.
Przelomski M.M., O’Rourke E., Grady G.F., et al. Eastern equine encephalitis in Massachusetts. Neurology. 1988;38:736-739.
Rathore M.H., Mercurio K., Halstead D. Herpes simplex type 1 meningitis. Pediatr Infect Dis J. 1996;15:824-828.
Read J.S., Committee on Pediatric AIDS. American Academy of Pediatrics. Diagnosis of HIV-1 infection in children younger than 18 months in the United States. Pediatrics. 2007;120:e1547-e1562.
Read S.J., Jeffery K.J., Bangham C.R. Aseptic meningitis and encephalitis: The role of PCR in the diagnostic laboratory. J Clin Microbiol. 1997;35:691-696.
Risk W.S., Haddad F.S. The variable natural history of subacute sclerosing panencephalitis. Arch Neurol. 1979;36:610-614.
Rivas F., Diaz L.A., Cardenas V.M., et al. Epidemic Venezuelan equine encephalitis in La Guajira, Colombia, 1995. J Infect Dis. 1997;175:828-832.
Rivera L.B., Boppana S.B., Fowler K.B., et al. Predictors of hearing loss in children with symptomatic congenital cytomegalovirus infection. Pediatrics. 2002;110:762-767.
Robinson J., Lemay M., Vaudry W.L. Congenital rubella after anticipated maternal immunity: Two cases and a review of the literature. Pediatr Infect Dis J. 1994;13:812-815.
Román G.C., Schoenberg B.S., Madden D.L., et al. Human T-lymphotrophic virus type I antibodies in the serum of patients with tropical spastic paraparesis in the Seychelles. Arch Neurol. 1987;44:605-607.
Romero J.R., Newland J.G. Viral meningitis and encephalitis: traditional and emerging agents. Semin Pediatr Infect Dis. 2003;14:72-82.
Rorabaugh M.L., Berlin L.E., Heldrich F., et al. Aseptic meningitis in infants younger than 2 years of age: Acute illness and neurologic complications. Pediatrics. 1993;92:206-211.
Rosenfeld J., Taylor C.L., Atlas S.W. Myelitis following chickenpox: A case report. Neurology. 1993;43:1834-1836.
Rupprecht C.E., Smith J.S., Fekadu M., et al. The ascension of wildlife rabies: A cause for public concern or intervention? Emerg Infect Dis. 1995;4:107-114.
Rust R. Multiple sclerosis, acute disseminated encephalomyelitis, and related conditions. Semin Pediatr Neurol. 2000;7:66-90.
Rust R., Thompson W.H., Mathews C.G., et al. La Crosse and other forms of California encephalitis. J Child Neurol. 1999;14:1-14.
Sauerbrei A.. Varicella zoster infections during pregnancy. Mushahwa I.K., editor. Congenital and Other Related Infectious Diseases of the Newborn. Perspectives in Medical Virology. vol 13. Amsterdam: Elsevier; 2007:51-73.
Sawyer M.H. Enterovirus infections: Diagnosis and treatment. Semin Pediatr Infect Dis. 2002;13:40-47.
Schlesinger Y., Buller R.S., Brunstrom J.E., et al. Expanded spectrum of herpes simplex encephalitis in childhood. J Pediatr. 1995;126:234-241.
Schlesinger Y., Sawyer M.H., Storch G.A. Enteroviral meningitis in infancy: Potential role for polymerase chain reaction in patient management. Pediatrics. 1994;94:157-162.
Schroth G., Gawehn J., Thron A., et al. Early diagnosis of herpes simplex encephalitis by MRI. Neurology. 1987;37:179-183.
Schulte D.J., Comer J.A., Erickson B.R., et al. Congenital lymphocytic choriomeningitis virus: an underdiagnosed cause of neonatal hydrocephalus. Pediatr Infect Dis J. 2006;25:560-562.
Sebire G., Meyer L., Chabrier S. Varicella as a risk factor for cerebral infarction in childhood: A case-control study. Ann Neurol. 1999;45:679-680.
Sejvar J.J. The evolving epidemiology of viral encephalitis. Curr Opin Neurol. 2006;19:350-357.
Sejvar J.J. West Nile and “poliomyelitis”. Neurology. 2004;63:206-207.
Sejvar J.J., Haddad M.B., Tierney B.C., et al. Neurologic manifestations and outcome of West Nile virus infection. JAMA. 2003;290:511-515.
Sejvar J.J., Hossain J., Saha S.K., et al. Long term neurological and functional outcome in Nipah virus infection. Ann Neurol. 2007;62:235-242.
Sever J.L., South M.A., Shaver K.A. Delayed manifestations of congenital rubella. Rev Infect Dis. 1985;7:S164-S169.
Shimizu H., McCarthy C.A., Smaron M.F., et al. Polymerase chain reaction for detection of measles virus in clinical samples. J Clin Microbiol. 1993;31:1034-1039.
Shinefield H.R., Townsend T.E. Transplacental transmission of western equine encephalomyelitis. J Pediatr. 1953;43:21-25.
Silva C.A., Paula-Barbosa M.M., Pereira S., et al. Two cases of rapidly progressive subacute sclerosing panencephalitis. Arch Neurol. 1981;38:109-113.
Singer C., Lang A.E., Suchowersky O. Adult-onset subacute sclerosing panencephalitis: case reports and review of the literature. Mov Disord. 1997;12:342-353.
Smith J.S. New aspects of rabies with emphasis on epidemiology, diagnosis and prevention of disease in the United States. Clin Microbiol Rev. 1996;9:166-176.
Smith R., Woodall J.P., Whitney E., et al. Powassan virus infection: a report of three human cases of encephalitis. Am J Dis Child. 1974;127:691-693.
Spector S.A., Gelber R.D., McGrath N., et al. A controlled trial of intravenous immune globulin for the prevention of serious bacterial infections in children receiving zidovudine for advanced human immunodeficiency virus infection. Pediatric AIDS Clinical Trials Group. N Engl J Med. 1994;331:1181-1187.
Spika J.S., Wassilak S., Pebody R. Measles and rubella in the world health organization European region: Diversity creates challenges. J Infect Dis. 2003;187(Suppl 1):S191-S195.
Spruance S.L., Bailey A. Colorado tick fever: a review of 115 laboratory-confirmed cases. Arch Intern Med. 1973;131:288-293.
Srugo I., Wittek A.E., Israele V., et al. Meningoencephalitis in a neonate congenitally infected with human immunodeficiency virus type 1. J Pediatr. 1992;120:93-95.
Stoll M., Schmidt R.E. Immune restoration inflammatory syndromes: The dark side of successful antiretroviral treatment. Curr Infect Dis Rep. 2003;5:266-276.
Studahl M., Ricksten A., Sandberg T., et al. Cytomegalovirus encephalitis in four immunocompetent patients. Lancet. 1992;340:1045-1046.
Suga S., Yoshikawa T., Asano Y., et al. Clinical and virological analyses of 21 infants with exanthem subitum (roseola infantum) and central nervous system complications. Ann Neurol. 1993;33:597-603.
Sumaya C.V. Serologic testing for Epstein-Barr virus – developments in interpretation. J Infect Dis. 1985;151:984-987.
Sumaya C.V., Ench Y. Epstein-Barr virus infectious mononucleosis in children: I. Clinical and general laboratory findings. Pediatrics. 1985;75:1003-1010.
Sumaya C.V., Ench Y. Epstein-Barr virus infectious mononucleosis in children: II. Heterophil antibody and viral specific responses. Pediatrics. 1985;75:1011-1019.
Sunaga Y., Hikima A., Ostuka T., et al. Acute cerebellar ataxia with abnormal MRI lesions after varicella infection. Pediatr Neurol. 1995;13:340-342.
Swisher C.N., Boyer K., McLeod R., et al. Congenital toxoplasmosis. Semin Pediatr Neurol. 1994;1:4-25.
Takahashi H., Pool V., Tsail T.F., et al. Adverse events after Japanese encephalitis vaccination: Review of post-marketing surveillance data from Japan and the United States. The VAERS Working Group. Vaccine. 2000;18:2963-2969.
Tenembaum S., Chitnis T., Ness J., et al. International Pediatric MS Study Group. Acute disseminated encephalomyelitis. Neurology. 2007;68(16 Suppl 2):S23-S36.
Tien R.D., Felsberg G.J., Osumi A.K. Herpesvirus infections of the CNS: MR findings. Am J Radiol. 1993;161:167-176.
Torigoe S., Koide W., Yamada M., et al. Human herpesvirus 7 infection associated with central nervous system manifestations. J Pediatr. 1996;129:301-305.
Townsend J.J., Baringer J.R., Wolinsky J.S., et al. Progressive rubella panencephalitis. N Engl J Med. 1975;292:990-993.
Ueda K., Nishida Y., Oshima K., et al. Congenital rubella syndrome: Correlation of gestational age at time of maternal rubella with type of defect. J Pediatr. 1979;94:763-765.
UNAIDS. A global overview of the epidemic. Barcelona Report, 2002;22.
van der Knaap M.S., Vermeulen G., Barkhof F., et al. Pattern of white matter abnormalities at MR imaging: use of polymerase chain reaction testing of Guthrie cards to link pattern with congenital cytomegalovirus infection. Radiology. 2004;230:529-536.
van Rossum A.M., Fraaij P.L., de Groot R. Efficacy of highly active antiretroviral therapy in HIV-1 infected children. Lancet Infect Dis. 2002;2:93-102.
Varmus H. Retroviruses. Science. 1988;240:1427-1435.
Verboon-Maciolek M.A., Groenendaal F., Hahn C.D., et al. Human parechovirus causes encephalitis with white matter injury in neonates. Ann Neurol. 2008;64:266-273.
Verboon-Maciolek M.A., Krediet T.G., Gerards L.J., et al. Severe Neonatal Parechovirus Infection and Similarity With Enterovirus Infection. Pediatr Infect Dis J. 2008;27:241-245.
Verma N.A., Zheng X.T., Harris M.U., et al. Outbreak of life-threatening coxsackievirus B1 myocarditis in neonates. Clin Infect Dis. 2009;49:759-763.
Volpi A. Epstein-Barr virus and human herpesvirus type 8 infections of the central nervous system. Herpes. 2004;11(Suppl 2):120A-127A.
Vossough A., Zimmerman R.A., Bilaniuk L.T., et al. Imaging findings of neonatal herpes simplex virus type 2 encephalitis. Neuroradiology. 2008;50:355-366.
Wasay M., Channa R., Jumani M., et al. Encephalitis and myelitis associated with dengue viral infection. clinical and neuroimaging features. 2008;110:635-640.
Welch K., Morse A. Adult Spectrum of Disease Project in New Orleans. The clinical profile of end-stage AIDS in the era of highly active antiretroviral therapy. AIDS Patient Care STDS. 2002;16:75-81.
Weller T.H. Varicella and herpes zoster. N Engl J Med. 1983;309:1362-1368.
Wenger F. Venezuelan equine encephalitis. Teratology. 1977;16:359-362.
Whitley R., Arvin A., Prober C., et al. A controlled trial comparing vidarabine with acyclovir in neonatal herpes simplex virus infection. N Engl J Med. 1991;324:444-449.
Whitley R.J., Arvin A., Prober C., et al. Predictors of mortality in neonates with herpes simplex virus infections. N Engl J Med. 1991;324:450-454.
Whitley R.J. Virus encephalitis. N Engl J Med. 1990;323:242-250.
Whitley R.J., Alford C.A., Hirsch M.S., et al. Vidarabine versus acyclovir therapy in herpes simplex encephalitis. N Engl J Med. 1986;314:144-149.
Whitley R.J., Cobbs C.G., Alford C.A., et al. Diseases that mimic herpes simplex encephalitis. JAMA. 1989;262:234-239.
Whitley R.J., Corey L., Arvin A., et al. Changing presentation of herpes simplex virus infection in neonates. J Infect Dis. 1988;158:109-116.
Whitley R.J., Gnann J.W. Viral encephalitis: Familiar infections and emerging pathogens. Lancet. 2002;359:507-513.
Whitley R.J., Lakeman F. Herpes simplex virus infections of the central nervous system: Therapeutic and diagnostic considerations. Clin Infect Dis. 1995;20:414-420.
Whitley R.J., Soong S.J., Linneman C., et al. Herpes simplex encephalitis. JAMA. 1982;247:317-320.
Wilde H., Hemachudha T., Jackson A.C. Viewpoint: management of human rabies. Trans R Soc Trop Med Hyg. 2008;102:979-982.
Wilfert C.M., Buckley R.H., Mohanakumar T., et al. Persistent and fatal central nervous system echovirus infections in patients with agammaglobulinemia. N Engl J Med. 1977;296:1485-1489.
Willoughby R.E.Jr, Tieves K.S., Hoffman G.M., et al. Survival after treatment of rabies with induction of coma. N Engl J Med. 2005;352:2508-2514.
Windebank A.J., Litchy W.J., Daube J.R., et al. Late effects of paralytic poliomyelitis in Olmstead County, Minnesota. Neurology. 1991;41:501-507.
World Health Organization. http://www.who.int/hiv/data/2008_global_summary_AIDS_ep.png, 2008.
World Health Organization. http://www.who.int/mediacentre/factsheets/fs286/en/index.html, 2009. Accessed November 14, 2009
World Health Organization. http://www.who.int/mediacentre/factsheets/fs099/en/, 2009. Accessed November 14, 2009
Wright R., Johnson D., Neumann M., et al. Congenital lymphocytic choriomeningitis virus syndrome: A disease that mimics congenital toxoplasmosis or cytomegalovirus infection. Pediatrics. 1997;100:E9.
Wrzolek M.A., Brudkowska J., Kozlowski P.B., et al. Opportunistic infections of the central nervous system in children with HIV infection: Report of 9 cases and review of the literature. Clin Neuropathol. 1995;14:187-196.
Yalaz K., Anlar B., Oktem F., et al. Intraventricular interferon and oral inosiplex in the treatment of subacute sclerosing panencephalitis. Neurology. 1992;42:488-491.
Yamashita Y., Matsuishi T., Murakami Y., et al. Neuroimaging findings (ultrasonography, CT, MRI) in 3 infants with congenital rubella syndrome. Pediatr Radiol. 1991;21:547-549.
Yoshida A., Kawamitu T., Tanaka R., et al. Rotavirus encephalitis: Detection of the virus genomic RNA in the cerebrospinal fluid of a child. Pediatr Infect Dis J. 1995;14:914-916.
Yoshikawa T., Ihira M., Suzuki K., et al. Invasion by human herpesvirus 6 and human herpesvirus 7 of the central nervous system in patients with neurological signs and symptoms. Arch Dis Child. 2000;83:170-171.
Zhang S.Y., Jouanguy E., Sancho-Shimizu V., et al. Human Toll-like receptor-dependent induction of interferons in protective immunity to viruses. Immunol Rev. 2007;220:225-236.
Zhang S.Y., Jouanguy E., Ugolini S., et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317(Sep 14):1522-1527.

{kind=link}
{kind=link}