2 Using Cerebral Vaso-Reactivity in the Selection of Candidates for EC-IC Bypass Surgery
Introduction
Since the failure of the EC-IC (extracranial-intracranial) bypass study in showing decreased risk of stroke after bypass,1 the utility of this procedure, particularly in chronic occlusive vascular disease (OVD), has come into serious question. Despite that failure in an unselected group of patients, anecdotal and clinical experience has lead clinicians who treat patients with occlusive vascular disease to conclude that there must be a subgroup of patients who are at a much higher risk of stroke than the general population who would still benefit from this procedure. This has lead to a great deal of research into the underlying pathophysiology in OVD and subsequently the development of a number of physiology-based techniques that are able to predict patients at increased risk of stroke. Once a technique has been established to identify a high-risk group, it then remains to be proven to decrease subsequent stroke risk when used as a selection tool for bypass. In order to understand the rationale for using cerebral vaso-reactivity (CVR) studies as a rational tool for the selection of candidates for flow augmentation surgery, a review of the pathophysiology is first necessary.
Physiology of occlusive vascular disease
Central to the physiology in carotid occlusion is the concept of cerebral autoregulation and its interrelationship with chemoregulation. Cerebral autoregulation is an intrinsic property of the cerebral vasculature that allows for maintaining consistency of CBF (cerebral blood flow) over a wide range of cerebral perfusion pressures (CPPs). This property is not unique to the cerebral vasculature, and involves very tight molecular regulation, allowing very precise, second-to-second adaptation to changing stresses. It is important to emphasize that this is an intrinsic property of the vessels, and is separate from other chemical or neural mediators of CBF adaptation. The molecular mechanisms are signaled by stretch of the vasculature, which initiates a phospholipase C mediated cascade, resulting in inhibition of calcium-activated potassium channels in smooth muscle, leading to smooth muscle contraction. The opposing forces are likely related to the cellular metabolic activity within neural and glial cells leading to activation of the same potassium channels and vasodilatation.2
An alternate method of testing the state of autoregulation involves testing vascular reactivity. This approach involves performing a baseline study to assess CBF, followed by a vasodilatory challenge and repeat of the study. The challenge can be as simple as breath holding to increase pCO2 or inhalation of CO2 gas—both relying on the principle that increasing brain CO2 acidifies the tissue and leads to vasodilatation (called chemoregulation). Alternatively, a cerebral vasodilatory agent such as acetazolamide that blocks carbonic anhydrase can be administered to induce tissue acidification and vasodilatation. These methods do not alter the underlying state of intrinsic autoregulation or metabolism, but they are able to determine to what degree the vessels can respond to such a challenge. In normal brain vasculature, a vasodilatory agent should lead to an increase in CBF by a certain percentage over the baseline level (typically 15% to 40%). If the vasodilatory agent does not cause an increase in CBF, one can conclude that the vessels have reached a maximal level of autoregulatory vasodilatation. In some cases, one may even see in one or more vascular territories a paradoxical drop in CBF in response to a vasodilatory challenge. This has been referred to as a “steal” phenomenon, where the portion of the vasculature that has a normal blood supply dilates normally in response to the challenge while the vascular territory that is already maximally dilated, and most often dependent upon leptomeningeal collaterals, experiences a loss of collateral supply.3 This approach thereby identifies a vascular territory that has experienced a failure of autoregulation with a reduction of CBF and a state of maximal vasodilation, and potentially the tissue most at risk for infarction due to a progression of hemodynamic compromise.
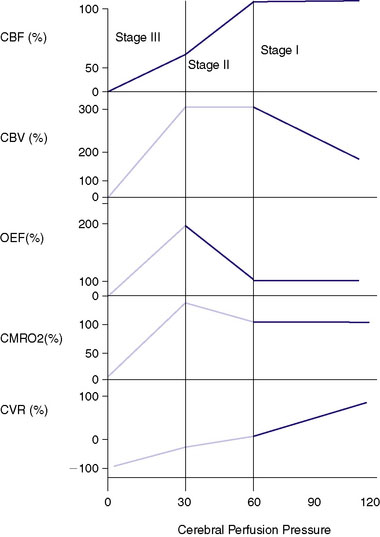
The cerebral vasculature has other adaptive mechanisms for compensating for decreasing perfusion pressure. Once the vessels are maximally dilated and the CPP continues to fall, the normally excessive levels of tissue nutrients (i.e., glucose and oxygen) carried within the blood supply begin to fall. A common measurement is the oxygen extraction fraction (OEF), which normally is less than 50% and rises with a progressive fall of CPP. With a fall of CBF below about 20cc/100 g/min, the oxygen reserves become exhausted and metabolism becomes compromised. The increase in OEF can be measured using positron emission tomography (PET), although there remains controversy regarding the most accurate method.4,5 This physiology underlies the now well accepted stages of hemodynamic insufficiency described by Powers et al.6,7 that with the progressive fall of CPP, CBF is maintained by vasodilatation with the rise of CBV. This is referred to as Stage I insufficiency. Stage II insufficiency involves the failure of autoregulation with a fall of CBF and a state of maximal vasodilatation and an increasing OEF. With a continued fall of CPP the brain enters Stage III, with progressive cellular death. This metabolic decline can be directly measured by the CMRO2 (cerebral metabolic rate of oxygen use) as a single PET study. A vasodilatory challenge study done in Stage III will show a loss of positive reactivity or a “steal” as described previously. Because OEF is a ratio of CBF and CMRO2, with progressive cellular injury the CMRO2 will fall and the OEF can potentially return to “normal” with an equally reduced CBF and CMRO2. This stage has been referred to as a progression of Stage III with “matched hypoperfusion”8-12,70 (Figure 2–1).
Techniques used for testing CVR
Xenon/CT
Xenon has been thought to be an ideal technique for CBF determination due to its ability to provide quantitative data safely. 133Xenon may utilize individual gamma counters or an array that integrates information. Regional or tomographic SPECT (single photon emission tomography) techniques are utilized to record the washout of Xenon from the brain tissue with end tidal values being utilized to obtain an assessment of arterial content. A vast amount of information about CBF has been gained with this approach that is, however, limited by the need for precautions with radioactive Xenon and the inability to resolve information below the surface of the brain. Xenon/CT offers the advantage of providing high-resolution CBF information with equal validity in the center as well as the surface of the brain. Stable Xenon, which is radiodense and similar to iodine, is also a noble gas, thus not requiring any concerns about environmental contamination. The technique involves the measurement of both the arterial concentration and tissue arrival of Xenon on CT, allowing for solving of the Kety-Schmidt equation by integral math for each of the ∼24,000 CT pixels. The validity of the technique has been confirmed by cross-correlation with destructive (microspheres,13 iodoantipyrine14), and in vivo (133Xenon,15 PET,16 thermal dilution flow probes17) CBF technologies. No permanent morbidity or death has been reported despite its wide use for over 25 years, and Xenon gas is generally considered safe.18
Xenon techniques including 133Xenon SPECT and Xenon/CT have been used for examining CBF in carotid occlusion even before the failure of the EC-IC bypass trial.19 It was clearly recognized, even at that time, that physiological methods must have some role in selecting these patients. One observational study noted that only a small minority of patients (two of 20) showed increased CBF after bypass compared to the test done before bypass.20 Initially, decreased baseline CBF alone was used to select potential candidates for bypass and of 25 patients with normal baseline CBF, no early strokes were seen with no bypass, while bypass was shown to normalize decreased CBF levels in seven of eight patients. At the same time, other clinicians, mainly in Scandinavia, began to explore the concept of testing the CVR by administration of acetazolamide, as described previously, in selecting patients using regional 133Xenon who might benefit from bypass.21
The failure of the EC-IC bypass trial, however, meant that the financial and practical ability to perform bypass surgery for OVD was relegated to a primarily research role. As the group in St. Louis began a natural history study to define the stroke risk associated with increased OEF,22 similar work was being done with regard to CVR. Rogg23 first demonstrated that patients with symptomatic carotid occlusion or stenosis can have very different patterns of response to acetazolamide challenge. Forty-five percent of patients showed normal baseline CBF that augmented with acetazolamide ipsilateral to the occlusion. Thirty-one percent started with low baseline flow, but still augmented appropriately with acetazolamide, while 24% showed either no response or decreased response to acetazolamide, regardless of baseline CBF. Later, the same Pittsburg group, in a prospective trial, showed that patients with symptomatic carotid stenosis or occlusion can be divided more specifically to better predict subsequent stroke. Patients with normal baseline CBF and normal CVR had a stroke risk of 4.4% (2/46) in the mean 24-month follow-up period compared to 36% (8/22) patients with baseline CBF < 45 cc/100 g/min in any territory on the affected side as well as CVR showing negative reactivity (steal) of >5%. This translated to a 12.6 times increased risk of stroke in the group with a severe compromise of CVR. Furthermore, the two strokes in the normal reactivity group were contralateral to the affected carotid side, and were both in patients with carotid stenosis rather than occlusion.24 This represented some of the first data clearly linking the importance of poor CVR to subsequent stroke. Further data from this group confirmed this association further, especially in carotid occlusion, where the stroke rate in the 19.6-month mean follow-up period was 10% (2/26) in patients with normal CVR compared to 26% (10/38) in the patients with steal.25
This data has been cross-validated in other institutions as well, where the stroke rate was found to be 0.5% to 2.4% annually with normal CVR (measured by 133Xenon SPECT) compared to a 21.8% annual stroke rate if CVR was impaired in patients with carotid or MCA (middle cerebral artery) occlusion.26 Another group from Japan found a 6% (3/47) stroke rate in patients with normal CVR compared to 35% (8/23) with poor CVR. Decreased CVR was also found to be the only factor that the group measured that was associated with an increased stroke rate.27 There are some limitations in all of these data, particularly with regard to sampling error. None of these are long-term prospective observational studies, and the inclusion varies in terms of methodology, specific definition of impaired CVR, stenosis versus occlusion, intracranial versus extracranial disease, and clinical status of the patient. That said, there seems to be very convincing evidence from several sources that in patients with OVD there is a dramatic, statistically significant increase in risk of stroke predicted by impaired CVR.
Despite the fact that there is no randomized, controlled data to support the use of CVR, there has been nonrandomized data accruing over many years. As mentioned, the use of CVR in assessing patients with OVD began in the mid-1980s concurrent with the publication of the bypass trial. Vorstrup21 showed some of the first physiological data supporting the use of CVR in bypass selection. Adequate reactivity was defined as at least 13% flow augmentation from normal control data. Eighteen patients were bypassed for clinical reasons, nine of whom showed decreased CVR on acetazolamide testing, two of whom showed negative reactivity. Postoperatively, all of these patients showed improved CVR, but the two with negative reactivity were the only two whose baseline CBF increased. None of the remaining nine patients who were bypassed with normal CVR showed increased CBF postop, likely suggesting the lack of hemodynamic stress. The authors concluded that a randomized controlled trial was needed to assess this technique.21 Groups in Japan have also been using the technique in evaluating patients for bypass. In one study, 15 patients were studied before and after bypass. Three showed decreased CVR (again defined by normal controls), which improved after bypass. Furthermore, six patients had decreased baseline CBF and CVR pre-operatively. In three of these, the baseline CBF improved, and in four, the CVR improved postop.28 These early studies showed that in patients with impaired CVR, bypass can improve these physiologic parameters.
The next important step is to link whether bypass can decrease stroke risk in selected patients, and there have been some preliminary studies addressing this question. Kuroda29 first used CVR as a selection criterion for bypass in patients with OVD. Out of 32 patients, six who showed normal CBF and CVR were not bypassed, and there were no ischemic events in the mean 24-month follow-up. Of the nine patients with normal CBF but impaired CVR, eight were bypassed, and they showed no ischemic event and return of normal CVR on follow-up imaging. The one patient with poor CVR and without bypass suffered a CVA. Of the 11 patients with decreased baseline CBF and poor CVR, nine had bypass—of these, one suffered a perioperative CVA, one died of pneumonia, and CVR normalized in the rest with no further ischemic events. Of the two not bypassed, one suffered a CVA, and one had progressive deterioration followed by a basal ganglia hemorrhage. The final group consisted of patients with decreased baseline CBF, but normal CVR. Five of six were bypassed and none had subsequent CVA. The authors suggested that this may not, in fact, have represented a high-risk group, and that metabolic data may have shown decreased tissue metabolic demand.29 This study, although somewhat complicated and clearly limited due to the nonrandomized nature, seems to offer supportive evidence that normal CVR is protective, decreased CVR has a high rate of stroke when untreated (all three patients in this series), and bypass seems to offer significant protection from CVA with some risk of significant perioperative complications (two of 22 in this series).
A larger series looking specifically at this at-risk group based on decreased CVR followed again from the Pittsburg group.30 Forty-two patients with hemodynamic insufficiency were examined (baseline CBF < 45 cc/100 g/min and >5% negative reactivity). Thirty were treated medically and there were nine (30%) CVAs within 12 months (mean of 5 months). Twelve were bypassed only after developing an ischemic stroke and showed normalization of CVR in all, and there was no CVA in the 18-month follow-up with one asymptomatic hemorrhage. This was further strong evidence for both the dangerous natural history of these patients and the ability of bypass to not only reverse the hemodynamic findings, but decrease CVA risk with a very low risk of morbidity.30
Spect
Qualitative SPECT-based techniques have an instructive history in emphasizing the importance of quantitative measurements. With qualitative techniques, an index must be assigned based on the asymmetry, usually between the two hemispheres. These asymmetry indices have shown poor correlation with CBF patterns.31 The other practical limitation is that since these techniques assess the accumulation of the isotope in tissue over time, rather than the direct wash-in (as with Xenon/CT), an immediate acetazolamide or other challenge study cannot be done. Typically, to test response to acetazolamide, the SPECT study is repeated 3 days after the first, allowing for many potentially confounding variables. Given this, it does not seem surprising that Yokota was unable to determine a difference in stroke rates in patients with normal and “impaired” CVR by [123I] IMP(N-isopropyl-p-[123I] iodoamphetamine) SPECT.32 Another study using the tracer 99mTc-HMPAO also showed the ability to detect the increasing asymmetry and reported that one patient predicted to be at risk did subsequently have a stroke.33 A very interesting confirmation of the limitations of the qualitative tracer rather than simply the SPECT technique was reported by Ogasawara,34 who showed that in the same group of patients, CVR as measured by 133Xenon SPECT was strongly predictive of 5-year stroke risk, whereas CVR measured by [123I] IMP SPECT was not predictive. This study strongly refutes the conclusion drawn by Yokota that “reduced vasodilatory capacity does not play a major role in stroke recurrence.”32 For this reason, we strongly would discourage any extrapolation of data from quantitative techniques to qualitative techniques in the future. Qualitative CBF techniques cannot predict patients with OVD at risk for subsequent CVA and should not be used as criteria for bypass.
Perfusion CT
With the ready availability of perfusion CT techniques at most institutions with modern CT scanners, a push has begun to find prognostic utility in these techniques, although definitive evidence is currently lacking. There are some very promising potential uses for this technique; however, there are also several limitations. First, the technique measures only intravascular contrast rather than tissue contrast as with Xenon. CT perfusion measures the arrival time of the intravascular contrast with about 50 scans at the same level within 1 minute as it is rapidly infused intravenously. There is no arterial input function, and so several assumptions are made in terms of calculating the resultant values. The CBF is calculated based on the MTT (mean transit time) or TTP (time to peak) and CBV values. In addition, there are serious risks of iodinated contrast as well as of radiation exposure for multiple studies that should not be overlooked.35,36 That said, one recent study demonstrated that after acetazolamide challenge in 15 patients with OVD, decreases in CBF and increases in MTT were noted.37 Another very small study also showed that there was increased MTT seen in three patients with OVD compared to one control with acetazolamide challenge.38 These preliminary data should suggest that perfusion CT may offer some insight into physiology in OVD, but at this point there are insufficient data to suggest that it is able to predict stroke or determine selection for bypass.
Perfusion MRI
Perfusion MRI is based on similar techniques as perfusion CT, and offers the same potential insights and limitations. Schreiber demonstrated the potential ability to detect a group of patients with impaired hemodynamics, showing that in five of eight patients with carotid occlusion, impaired CBF reactivity to acetazolamide was observed.39 Newer MR techniques may allow for some quantification of these values as well. In order to assess the validity of these measurements, perfusion MRI values were cross-correlated with Xenon/CT values. Since the MTT and the Tmax (maximum arrival time) are the only variables that are directly measured, these were thought to likely be the most accurate. It was found that the Tmax was slightly more accurate than MTT. The authors found that a Tmax of >4 sec showed 68% sensitivity, 80% specificity, and 77% accuracy for detecting a CBF of <20 sec by Xenon/CT.40 Although the authors demonstrated that Tmax and MTT were strongly associated with Xenon/CT values, it is unclear whether these moderately sensitive and specific values will translate into the ability to predict stroke or select patients for bypass.
TCD
TCD (transcranial Doppler) is a simple, noninvasive test that can be performed in a variety of situations, and for practical reasons, is a very attractive modality for assessing cerebral hemodynamics. The technique relies on measurement of the flow velocity in large proximal vessels such as the MCA. Although flow velocity alone has been shown to be poorly correlated with quantitative CBF,41,42 performing a vasodilatory challenge to assess CVR likely is closely correlated with CVR as tested with a CBF study.41,43 This is likely explained by the fact that large vessel morphology may be variable, and several factors including diameter and length will affect velocity besides CBF. When these are controlled for by testing the same vessel with a vasodilatory challenge, the velocity should then be reflective of the change in flow in the distal vasculature. It is clear that there is correlation of TCD CVR and Xenon-based CVR,41,43,44 although this is clearly not absolute. The correlation coefficient ranges from 0.4541 in asymptomatic patients (22.5% correlation) to 0.5941 and 0.6343 in symptomatic patients (35% to 40% correlation). For the average of the MCA territory by Xenon/CT, CVR measured by TCD of the MCA showed only 33% sensitivity, with 90.6% specificity.44
TCD-based CVR measurement is also limited in terms of the fact that there can be a great deal of interobserver variability. The reliability of the test is dependent on the TCD operator’s experience and consistency. In addition, a number of different techniques have been developed to measure CVR. Though an acetazolamide challenge is frequently used, just as with other techniques described above, attempts have been made to use even less invasive techniques such as breath-holding index (measured as a rise in end-tidal CO2 after breath holding), hyper- and hypo-ventilation, and breathing various concentrations of CO2 gas.45–47 All of these techniques rely on tissue acidification and cerebral chemoregulation, whether by varying the pCO2 or administering acetazolamide as described previously, but the variability in measurement techniques makes cross-comparison more difficult. It is clear that these various methods are closely correlated,45–47 although acetazolamide likely offers a slight advantage in distinguishing the degree of impairment.46,47
Despite the somewhat limited sensitivity and variety of vasodilatory stimuli, TCD has been shown to have utility in assessment of hemodynamics in patients with carotid occlusive vascular disease. Overall, worsened CVR compared to normal controls has been documented in patients with higher-grade carotid stenosis and occlusion.48,49 In addition, it is clear that worsened CVR is related to some surrogate measures of hemodynamic insufficiency including symptomatic patients, signs of ipsilateral low-flow infarction on CT,50 and neuropsychiatric testing.51 CVR testing of patients with high-grade stenosis has been used to determine if there is increased stroke risk in some patients with hemodynamic factors, and in fact, poor CVR is clearly associated with increased stroke risk.52–54 Although these studies seem to show that there is a flow-related component in some patients with carotid stenosis in addition to the well-known thromboembolic risk, it does not significantly change treatment strategies in these patients. It has been clearly shown that carotid endarterectomy (CEA) decreases stroke risk in patients with high-grade stenosis.55,56 It is interesting, however, that in patients with impaired CVR who undergo CEA, that these values normalize postoperatively.54 Furthermore, in patients with carotid occlusion and contralateral stenosis with poor CVR, the values often normalize on the occluded side after contralateral CEA.57
The value of CVR measured by TCD has been established in patients with carotid occlusion as well. In one group using inhaled CO2 gas, only four of 48 (8%) patients with normal CVR suffered ipsilateral ischemic events compared to 12 of 33 (32%) with impaired reactivity in the mean 38-month follow-up.58 Another group showed that breath holding index of <0.69 was the only factor of their measured variables associated with subsequent stroke.59 This group later showed that this impaired CVR was also associated with a lack of angiographic collaterals, with a 32.7% stroke rate with no major collateral vessels. All of these patients had impaired CVR. If patients have one or two collateral vessels, the CVR pattern was variable, and impaired CVR was associated with subsequent stroke risk, pointing out the utility of physiologic assessment in these patients. Furthermore, none of their patients with normal CVR had ischemic events in the mean 24-month follow-up.60 The concentration of inhaled CO2 may affect the sensitivity of measurement as well, and 8% CO2 seemed to show the best predictive value, with a Kaplan-Meyer log rank statistic of 7.81 (p = 0.0052) and annual ischemic event rate of 8.53%.61
PET
The power of PET data is, that in addition to quantitative CBF data, it also provides metabolic data. Unfortunately, access to the oxygen isotope needed for the cerebral studies requires an on-site cyclotron, and is limited at many institutions. That said, the technique offers powerful, quantitative insight into cerebral hemodynamics. Quantitative CBF can be obtained using PET by injection of 15O [H2O]. The tissue arrival time is calculated by the PET detectors, and the arterial concentration (input function) is calculated by measuring the arterial concentration of the radioactive tracer either continuously or by frequent sampling.62 Although this is slightly more labor intensive than measuring the end tidal Xenon with Xenon/CT, a repeat study can be performed to determine the response to an intervention. By administering acetazolamide as described previously, the CVR can be determined tomographically.63
The majority of data using PET in predicting patients at hemodynamic risk in carotid occlusion has focused on the ability to detect Stage II hemodynamic insufficiency by measuring an increased OEF. This is the basis of a study funded by the National Institutes of Health (Carotid Occlusion Surgery Study [COSS]) to use increased OEF as the entry criteria in symptomatic patients with carotid occlusion followed by randomization to bypass surgery or medical management.64,65 It is clear that increased OEF has been shown to be predictive of subsequent stroke risk.5,6,22,66 In 2001, Derdeyn5 proposed using qualitative OEF data rather than quantitative data and that became the model for the COSS trial as of 2003.64
It has come into question as to why there is discordance between the patients detected to be at risk by CVR-based techniques and OEF. Yamauchi using quantitative OEF found a 50% PPV of increased OEF in detecting patients with impaired CVR as measured by PET.67 Likewise, Nemoto found 37.5% of patients with poor CVR by Xenon/CT but normal OEF,8 and Kuroda found 57%.11 These data then beg the question as to whether CVR techniques detect some patients without hemodynamic impairment or whether OEF is missing some patients with hemodynamic impairment. To address this question, surrogate markers of severe impairment have been used. Nemoto showed that in the patients with impaired CVR but normal OEF, there remained a highly significant association with a subcortical, low-flow related pattern of infarction on MRI.8 Likewise, Kuroda found that OEF did not correlate with subcortical white matter changes,11 suggesting that many patients with normal OEF may still have signs of hemodynamic insufficiency.
These data lend support to an addition to the concept of stages of hemodynamic failure, in that there is likely a stage after Stage II, where CBF continues to fall, reactivity continues to worsen or even become negative (steal), and OEF falls along with baseline cellular metabolism8 (see Figures 2–1 and 2–2). This clarifies the seeming discordant physiology conceptually, and explains why patients who seem to have some of the most impaired hemodynamics, as measured by CVR and confirmed with presence of white matter changes, may in fact show “normal” OEF.
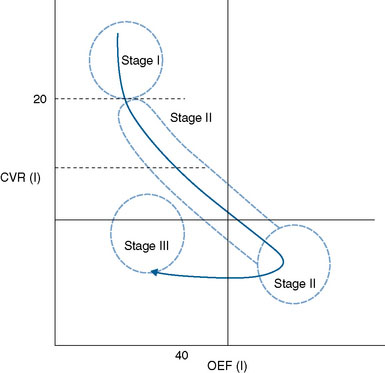
In order to measure these physiologic discrepancies, the change in OEF after acetazolamide challenge can be determined. The OEF should decrease if there is an increase in CBF (normal reactivity) with acetazolamide. The OEF reactivity (OEFR) has been found to correlate with CVR much better than OEF alone,10,68,69 likely identifying patients with severe hemodynamic compromise despite normal baseline OEF values. In these preliminary studies, two patients with the poorest CVR values and severe white matter changes were found to have normal baseline OEF, but were identified as compromised using OEFR.68 Furthermore, based on these data it has been proposed that demonstration of positive OEFR definitively indicates a state of hemodynamic impairment, where as a negative OEFR demonstrates a state of hemodynamic reserve.69 This may offer an alternative to the difficulty with assigning threshold values for OEF.7
Despite the evidence that patients with poor CVR and normal OEF may still have hemodynamic insufficiency, it is unclear if this is predictive of future stroke. This question was recently addressed by Hokari in a small preliminary study that showed a stroke rate in mean 45.6-month follow-up of three of nine patients with poor CVR and increased OEF compared to 0/11 with poor CVR and normal OEF.69 One may surmise based on these data that when this group of patients enters the state of matched hypometabolism, where OEF begins to fall along with metabolism, that the decreased metabolic demand may be protective in terms of hemodynamic stroke. That is to say that as the tissue enters a state of severe hemodynamic insufficiency, decreased CBF is needed to maintain tissue viability. Clearly, further studies are needed to assess stroke risk and therapeutic interventions in this group.
1 The EC/IC Bypass Study Group. Failure of extracranial-intracranial arterial bypass to reduce the risk of ischemic stroke. Results of an international randomized trial. N Engl J Med. 1985;313:1191-1200.
2 Harder D.R., Roman R.J., Gebremedhin D., et al. A common pathway for regulation of nutritive blood flow to the brain: arterial muscle membrane potential and cytochrome P450 metabolites. Acta Physiol Scand. 1998;164:527-532.
3 Yonas H., Kromer H., Jungreis C. Compromised vascular reserves does predict subgroups with carotid occlusion and an increased stroke risk. J Stroke Cereb Dis. 1997;6:458.
4 Lin R., Uchino K., Zaidi S., et al. Quantitative versus qualitative OEF identification of hemodynamic compromise in stroke patients with large artery occlusion. J Cereb Blood Flow Metab. 2009;29:S593.
5 Derdeyn C.P., Videen T.O., Grubb R.L.Jr, et al. Comparison of PET oxygen extraction fraction methods for the prediction of stroke risk. J Nucl Med. 2001;42:1195-1197.
6 Grubb R.L.Jr, Derdeyn C.P., Fritsch S.M., et al. Importance of hemodynamic factors in the prognosis of symptomatic carotid occlusion. JAMA. 1998;280:1055-1060.
7 Derdeyn C.P., Grubb R.L.Jr, Powers W.J. Re: Stages and thresholds of hemodynamic failure. Stroke. 2003;34:589.
8 Nemoto E.M., Yonas H., Kuwabara H., et al. Identification of hemodynamic compromise by cerebrovascular reserve and oxygen extraction fraction in occlusive vascular disease. J Cereb Blood Flow Metab. 2004;24:1081-1089.
9 Nemoto E.M., Yonas H., Kuwabara H., et al. Hemodynamic compromise in occlusive vascular disease by PET OEF with acetazolamide. Stroke. 2004;35:269-270.
10 Nemoto E.M., Yonas H., Kuwabara H., et al. Differentiating hemodynamic compromise by the OEF response to acetazolamide in occlusive vascular disease. Oxygen Trans Tissue. 2005;26(566):135-141.
11 Kuroda S., Shiga T., Houkin K., et al. Cerebral oxygen metabolism and neuronal integrity in patients with impaired vasoreactivity attributable to occlusive carotid artery disease. Stroke. 2006;37:393-398.
12 Pindzola R.R., Sashin D., Nemoto E.M., et al. Identifying regions of compromised hemodynamics in symptomatic carotid occlusion by cerebrovascular reactivity and oxygen extraction fraction. Neurol Res. 2006;28:149-154.
13 Gur D., Yonas H., Jackson D.L., et al. Simultaneous measurements of cerebral blood flow by the Xenon/CT method and the microsphere method: a comparison. Invest Radiol. 1985;20:672-677.
14 Wolfson S.K.Jr, Clark J., Greenberg J.H., et al. Xenon-enhanced computed tomography compared with [14C]iodoantipyrine for normal and low cerebral blood flow states in baboons. Stroke. 1990;21:751-757.
15 Matsuda M., Lee H., Kuribayashi K., et al. Comparative study of regional cerebral blood flow values measured by Xe CT and Xe SPECT. Acta Neurol Scand Suppl. 1996;166:13-16.
16 Nariai T. Comparison of measurement between Xe/CT CBF and PET in cerebrovascular disease and brain tumor. Acta Neurol Scand Suppl. 1996;166:10-12.
17 Valadka A.B., Hlatky R., Furuya Y., et al. Brain tissue PO2: correlation with cerebral blood flow. Acta Neurochir Suppl. 2002;81:299-301.
18 Latchaw R.E., Yonas H., Pentheny S.L., et al. Adverse reactions to Xenon-enhanced CT cerebral blood flow determination. Radiology. 1987;163:251-254.
19 Yonas H., Gur D., Good B.C., et al. Stable Xenon CT blood flow mapping for evaluation of patients with extracranial-intracranial bypass surgery. J Neurosurg. 1985;62:324-333.
20 Vorstrup S., Lassen N.A., Henriksen L., et al. CBF before and after extracranial-intracranial bypass surgery in patients with ischemic cerebrovascular disease studied with 133Xe-inhalation tomography. Stroke. 1985;16:616-626.
21 Vorstrup S., Brun B., Lassen N.A. Evaluation of the cerebral vasodilatory capacity by the acetazolamide test before EC-IC bypass surgery in patients with occlusion of the internal carotid artery. Stroke. 1986;17:1291-1298.
22 Powers W.J., Tempel L.W., Grubb R.L.Jr. Influence of cerebral hemodynamics on stroke risk: one-year follow-up of 30 medically treated patients. Ann Neurol. 1989;25:325-330.
23 Rogg J., Rutigliano M., Yonas H., et al. The acetazolamide challenge: imaging techniques designed to evaluate cerebral blood flow reserve. Am J Roentgenol. 1989;153:605-612.
24 Yonas H., Smith H.A., Durham S.R., et al. Increased stroke risk predicted by compromised cerebral blood flow reactivity. J Neurosurg. 1993;79:483-489.
25 Webster M.W., Makaroun M.S., Steed D.L., et al. Compromised cerebral blood flow reactivity is a predictor of stroke in patients with symptomatic carotid artery occlusive disease. J Vasc Surg. 1995;21:338-344. discussion 44–5
26 Kuroda S., Houkin K., Kamiyama H., et al. Long-term prognosis of medically treated patients with internal carotid or middle cerebral artery occlusion: can acetazolamide test predict it? Stroke. 2001;32:2110-2116.
27 Ogasawara K., Ogawa A., Yoshimoto T. Cerebrovascular reactivity to acetazolamide and outcome in patients with symptomatic internal carotid or middle cerebral artery occlusion: a Xenon-133 single-photon emission computed tomography study. Stroke. 2002;33:1857-1862.
28 Yamashita T., Kashiwagi S., Nakano S., et al. The effect of EC-IC bypass surgery on resting cerebral blood flow and cerebrovascular reserve capacity studied with stable XE-CT and acetazolamide test. Neuroradiology. 1991;33:217-222.
29 Kuroda S., Kamiyama H., Abe H., et al. Acetazolamide test in detecting reduced cerebral perfusion reserve and predicting long-term prognosis in patients with internal carotid artery occlusion. Neurosurgery. 1993;32:912-918. discussion 8–9
30 Przybylski G.J., Yonas H., Smith H.A. Reduced stroke risk in patients with compromised cerebral blood flow reactivity treated with superficial temporal artery to distal middle cerebral artery bypass surgery. J Stroke Cerebrovasc Dis. 1998;7:302-309.
31 Witt J.P., Yonas H., Jungreis C. Cerebral blood flow response pattern during balloon test occlusion of the internal carotid artery. Am J Neuroradiol. 1994;15:847-856.
32 Yokota C., Hasegawa Y., Minematsu K., et al. Effect of acetazolamide reactivity on long-term outcome in patients with major cerebral artery occlusive diseases. Stroke. 1998;29:640-644.
33 Knop J., Thie A., Fuchs C., et al. 99mTc-HMPAO-SPECT with acetazolamide challenge to detect hemodynamic compromise in occlusive cerebrovascular disease. Stroke. 1992;23:1733-1742.
34 Ogasawara K., Ogawa A., Terasaki K., et al. Use of cerebrovascular reactivity in patients with symptomatic major cerebral artery occlusion to predict 5-year outcome: comparison of Xenon-133 and iodine-123-IMP single-photon emission computed tomography. J Cereb Blood Flow Metab. 2002;22:1142-1148.
35 Krol A.L., Dzialowski I., Roy J., et al. Incidence of radiocontrast nephropathy in patients undergoing acute stroke computed tomography angiography. Stroke. 2007;38:2364-2366.
36 Wang C.L., Cohan R.H., Ellis J.H., et al. Frequency, outcome, and appropriateness of treatment of nonionic iodinated contrast media reactions. AJR Am J Roentgenol. 2008;191:409-415.
37 Chen A., Shyr M.H., Chen T.Y., et al. perfusion imaging with acetazolamide challenge for evaluation of patients with unilateral cerebrovascular steno-occlusive disease. AJNR Am J Neuroradiol. 2006;27:1876-1881.
38 Smith L.M., Elkins J.S., Dillon W.P., et al. Perfusion-CT assessment of the cerebrovascular reserve: a revisit to the acetazolamide challenges. J Neuroradiol. 2008;35:157-164.
39 Schreiber W.G., Guckel F., Stritzke P., et al. Cerebral blood flow and cerebrovascular reserve capacity: estimation by dynamic magnetic resonance imaging. J Cereb Blood Flow Metab. 1998;18:1143-1156.
40 Olivot J.M., Mlynash M., Zaharchuk G., et al. Perfusion MRI (Tmax and MTT) correlation with Xenon CT cerebral blood flow in stroke patients. Neurology. 2009;72:1140-1145.
41 Piepgras A., Schmiedek P., Leinsinger G., et al. A simple test to assess cerebrovascular reserve capacity using transcranial Doppler sonography and acetazolamide. Stroke. 1990;21:1306-1311.
42 Sekhar L.N., Wechsler L.R., Yonas H., et al. Value of transcranial Doppler examination in the diagnosis of cerebral vasospasm after subarachnoid hemorrhage. Neurosurgery. 1988;22:813-821.
43 Dahl A., Lindegaard K.F., Russell D., et al. A comparison of transcranial Doppler and cerebral blood flow studies to assess cerebral vasoreactivity. Stroke. 1992;23:15-19.
44 Pindzola R.R., Balzer J.R., Nemoto E.M., et al. Cerebrovascular reserve in patients with carotid occlusive disease assessed by stable Xenon-enhanced CT cerebral blood flow and transcranial Doppler. Stroke. 2001;32:1811-1817.
45 Markus H.S., Harrison M.J.G. Estimation of cerebrovascular reactivity using transcranial Doppler, including the use of breath-holding as the vasodilatory stimulus. Stroke. 1992;23:668-673.
46 Muller M., Voges M., Piepgras U., et al. Assessment of cerebral vasomotor reactivity by transcranial Doppler ultrasound and breath-holding: a comparison with acetazolamide as vasodilatory stimulus. Stroke. 1995;26:96-100.
47 Ringelstein E.B., Vaneyck S., Mertens I. Evaluation of cerebral vasomotor reactivity by various vasodilating stimuli: comparison of CO2 to acetazolamide. J Cereb Flow Metab. 1992;12:162-168.
48 Chimowitz M.I., Furlan A.J., Jones S.C., et al. Transcranial Doppler assessment of cerebral perfusion reserve in patients with carotid occlusive disease and no evidence of cerebral infarction. Neurology. 1993;43:353-357.
49 Orosz L., Fulesdi B., Hoksbergen A., et al. Assessment of cerebrovascular reserve capacity in asymptomatic and symptomatic hemodynamically significant carotid stenoses and occlusions. Surg Neurol. 2002;57:333-339.
50 Ringelstein E.B., Sievers C., Ecker S., et al. Noninvasive assessment of CO2-induced cerebral vasomotor response in normal individuals and patients with internal carotid artery occlusions. Stroke. 1988;19:963-969.
51 Silvestrini M., Paolino I., Vernieri F., et al. Cerebral hemodynamics and cognitive performance in patients with asymptomatic carotid stenosis. Neurology. 2009;72:1062-1068.
52 Silvestrini M., Vernieri F., Pasqualetti P., et al. Impaired cerebral vasoreactivity and risk of stroke in patients with asymptomatic carotid artery stenosis. JAMA. 2000;283:2122-2127.
53 Gur A.Y., Bova I., Bornstein N.M. Is impaired cerebral vasomotor reactivity a predictive factor of stroke in asymptomatic patients? Stroke. 1996;27:2188-2190.
54 Widder B., Paulat K., Hackspacher J., et al. Transcranial Doppler CO2 test for the detection of hemodynamically critical carotid artery stenoses and occlusions. Eur Arch Psychiatry Neurol Sci. 1986;236:162-168.
55 Barnett H.J., Taylor D.W., Eliasziw M., et al. Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis. North American Symptomatic Carotid Endarterectomy Trial Collaborators. N Engl J Med. 1998;339:1415-1425.
56 Randomised trial of endarterectomy for recently symptomatic carotid stenosis: final results of the MRC European Carotid Surgery Trial (ECST). Lancet. 1998;351:1379-1387.
57 Vernieri F., Pasqualetti P., Diomedi M., et al. Cerebral hemodynamics in patients with carotid artery occlusion and contralateral moderate or severe internal carotid artery stenosis. J Neurosurg. 2001;94:559-564.
58 Kleiser B., Widder B. Course of carotid artery occlusions with impaired cerebrovascular reactivity. Stroke. 1992;23:171-174.
59 Vernieri F., Pasqualetti P., Passarelli F., et al. Outcome of carotid artery occlusion is predicted by cerebrovascular reactivity. Stroke. 1999;30:593-598.
60 Vernieri F., Pasqualetti P., Matteis M., et al. Effect of collateral blood flow and cerebral vasomotor reactivity on the outcome of carotid artery occlusion. Stroke. 2001;32:1552-1558.
61 Markus H., Cullinane M. Severely impaired cerebrovascular reactivity predicts stroke and TIA risk in patients with carotid artery stenosis and occlusion. Brain. 2001;124:457-467.
62 Huang S.C., Carson R.E., Hoffman E.J., et al. Quantitative measurement of local cerebral blood-flow in humans by positron computed-tomography and O-15-water. J Cereb Blood Flow Metab. 1983;3:141-153.
63 Kuwabara Y., Ichiya Y., Sasaki M., et al. Time dependency of the acetazolamide effect on cerebral hemodynamics in patients with chronic occlusive cerebral arteries. Early steal phenomenon demonstrated by [15O]H2O positron emission tomography. Stroke. 1995;26:1825-1829.
64 Grubb R.L.Jr, Powers W.J., Derdeyn C.P., et al. The carotid occlusion surgery study. Neurosurg Focus. 2003;14:e9.
65 Adams H.P.Jr, Powers W.J., Grubb R.L.Jr, et al. Preview of a new trial of extracranial-to-intracranial arterial anastomosis: the carotid occlusion surgery study. Neurosurg Clin N Am. 2001;12:613-624. ix–x
66 Derdeyn C.P., Grubb R.L.Jr, Powers W.J. Cerebral hemodynamic impairment: methods of measurement and association with stroke risk. Neurology. 1999;53:251-259.
67 Yamauchi H., Okazawa H., Kishibe Y., et al. Oxygen extraction fraction and acetazolamide reactivity in symptomatic carotid artery disease. J Neurol Neurosurg Psychiatry. 2004;75:33-37.
68 Nemoto E.M., Yonas H., Pindzola R.R., et al. PET OEF reactivity for hemodynamic compromise in occlusive vascular disease. J Neuroimaging. 2007;17:54-60.
69 Jumaa M., Hammer M., Uchino K., et al. Hemodynamic compromise identified by oxygen extraction fraction response (OEFR) to acetazolamide in stroke patients with large artery occlusion. J Cereb Blood Flow Metab. 2009;29:S592.
70 Nemoto E.M., Yonas H., Chang Y. Stages and thresholds of hemodynamic failure. Stroke. 2003;34:2-3.