[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 15
Type 2 Diabetes Mellitus
Etiology, Pathogenesis, and Natural History
Genetic versus Acquired Factors
Natural History of Type 2 Diabetes
Pathophysiology of Type 2 Diabetes Mellitus
Inflammatory Pathway Activation and Insulin Resistance
Other Contributors to the Insulin-Resistant Phenotype
Insulin-Mediated versus Non–Insulin-Mediated Glucose Uptake
Pathophysiology of Fasting versus Postprandial Hyperglycemia
Syndromes of Severe Insulin Resistance
Type 2 diabetes mellitus is the most common form of diabetes and is currently a major worldwide cause of morbidity and mortality. This is likely to worsen, given the rapidly increasing prevalence of this condition; therefore, an understanding of its etiology and pathogenesis is of considerable importance. By definition, patients with type 2 diabetes have neither autoimmune β cell destruction, as is found in type 1 diabetes, nor one of the other specific causes of diabetes described in Chapter 13. Type 2 diabetes is not a single disease process but instead represents a heterogeneous constellation of disease syndromes, all leading to the final common pathway of hyperglycemia. Many factors, alone or in combination, can cause hyperglycemia; thus, the complexity of the pathogenesis of type 2 diabetes reflects the heterogeneous genetic, pathologic, environmental, and metabolic abnormalities that can exist in different patients.
Normal glucose homeostasis relies on a balance between insulin secretion and tissue sensitivity to insulin. With respect to regulation of glucose metabolism, the tissue effects of insulin on skeletal muscle, liver, and adipose tissue are most important. Three major metabolic abnormalities coexist in type 2 diabetes,1–4 each contributing to the hyperglycemic state. These abnormalities are summarized in Fig. 15-1. To begin at the hepatic level, the role of the liver in the pathogenesis of type 2 diabetes is overproduction of glucose. Increased basal hepatic glucose production is characteristic of essentially all type 2 diabetic patients with fasting hyperglycemia.5–7 Skeletal muscle is depicted as the prototypic peripheral insulin target tissue, because 70% to 80% of all glucose is taken up by skeletal muscle in the in vivo insulin-stimulated state. Target tissues are insulin resistant in type 2 diabetes mellitus, and such resistance has been well described in many studies across a large variety of population groups.2–5,8–12 Finally, abnormal islet cell function plays a central role in the development of hyperglycemia; decreased β cell function and increased glucagon secretion are standard concomitants of the diabetic state.1,13,14 Taken together, abnormalities in these organ systems account for the syndrome of type 2 diabetes mellitus. In subsequent sections of this chapter, each of these abnormalities will be considered in further detail.
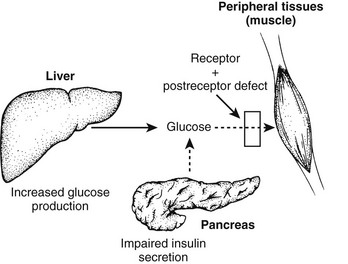
FIGURE 15-1 Summary of the metabolic abnormalities in type 2 diabetes mellitus that contribute to hyperglycemia. Increased hepatic glucose production, impaired insulin secretion, and insulin resistance caused by receptor and postreceptor defects all combine to generate the hyperglycemic state.
Genetic versus Acquired Factors
Abundant evidence supports the view that a strong genetic component contributes to type 2 diabetes. Although many patients have a positive family history for this disease, perhaps the strongest evidence comes from twin studies. In one study, 53 twin pairs were examined, in whom one twin was ascertained to have type 2 diabetes. On assessment of the other twin, type 2 diabetes had developed in 91% (48/53) of the co-twins.15 Although the five discordant twins were not overtly diabetic, they had mild glucose intolerance and abnormal insulin responses during oral glucose tolerance tests, suggesting that they too might ultimately progress to overt disease.
Further evidence for a genetic basis comes from striking differences in the prevalence of type 2 diabetes in various ethnic groups that are not explained by environmental factors. The prevalence of type 2 diabetes in the United States is 2% to 4% for Caucasians, but it is 4% to 6% for African Americans16 and 10% to 15% for Mexican Americans,17 and these numbers are increasing as the prevalence of obesity is rising at epidemic rates. More than 40% of the Pima Indians in Arizona have type 2 diabetes mellitus; this is the group with the highest incidence of type 2 diabetes in the world.18 Among the Pima Indians, 80% of 35- to 44-year-old offspring of two parents with type 2 diabetes mellitus before age 45 years have diabetes, and a positive family history of type 2 diabetes is a substantial risk factor for disease development. Clearly, this genetic predisposition interacts with adverse environmental influences, such as obesity and sedentary lifestyle, which are largely responsible for the sharp uptick in type 2 diabetes mellitus incidence in recent years.
Acquired Factors
Lifestyle: Diet, Exercise, and Obesity
Acquired factors play a major role in the development of type 2 diabetes in genetically predisposed individuals; this is clearly demonstrated by assessment of the impact of lifestyle changes on prevalence of diabetes in various ethnic populations. The prevalence of diabetes increases as ethnic groups migrate from lesser developed to more urbanized areas or simply change from an agrarian to a more sedentary, urban lifestyle. The former has been illustrated by surveys in Japanese subjects. In rural Japan, the prevalence of type 2 diabetes was approximately 4%,19 whereas among Japanese who have immigrated to the United States, the prevalence rises to more than 21%.20 This is also the case in the Pima Indians, who in Arizona have adopted a largely “Westernized” lifestyle, although those living in northwestern Mexico have remained agrarian. The Indians in Arizona have a prevalence of diabetes of 54% and 37% for men and women, respectively, whereas the Mexican Indians have a prevalence of 6% and 11%, respectively.21
It is likely that nutrition and lifestyle are the environmental factors that explain the difference in diabetes prevalence in genetically similar populations. Adoption of an urbanized, Westernized lifestyle is associated with change to a diet that has a higher content of total calories, fats, and refined carbohydrates. For example, the mean daily intake of fat among Japanese men living in Japan was reported to be 16.7 g; by contrast, in Japanese American men, the mean intake was 32.4 g.22 In addition, the level of physical activity is lower among ethnic groups living in the United States compared with the same ethnic groups living in their country of origin.23 These lifestyle changes obviously predispose to the development of obesity, and overwhelming evidence suggests that obesity is a major factor in the development of diabetes. The role of obesity in the pathogenesis of diabetes will be discussed in detail later in this chapter. Recent evidence supporting the role of lifestyle factors in the development of diabetes has come from the Diabetes Prevention Program.24 In this study, intensive lifestyle modification, consisting of dietary change and increased exercise, led to a 58% reduction in the progression of impaired glucose tolerance to diabetes over a 2.8 year period.
The extent of the contribution of these lifestyle factors, independent of their association with obesity, to the development of diabetes remains unclear. For example, it is not known whether specific dietary components, such as a diet that is rich in saturated fat or highly refined carbohydrates, play an independent role in the pathogenesis of type 2 diabetes. With regard to exercise, however, evidence from experimental and epidemiologic studies indicates that subjects with lower physical activity levels are more prone to develop diabetes, independent of obesity.25
Low Birth Weight
Low birth weight as a risk factor for the development of insulin resistance and diabetes mellitus later in life has been described in many populations over the past 2 decades.26–28 The mechanisms responsible for this association are unknown but may be related to epigenetic fetal adaptation to nutritional stimuli or excess fetal glucocorticoid exposure.29
Aging
Aging is associated with a decrease in glucose tolerance, which appears to be due to a decline in both insulin sensitivity and insulin secretion.30 However, age-related factors such as reduced physical activity and increased fat accumulation are at least in part responsible for this phenomenon. Obviously, type 2 diabetes incidence increases with age, but whether the aging process per se is contributory remains unclear.
Natural History of Type 2 Diabetes
The pathophysiologic findings depicted in Fig. 15-1 represent a single point in time after overt type 2 diabetes has developed. However, such an analysis does not reveal the progressive evolution of this disease. Fig. 15-2 presents a schematic description of the natural history or progression to type 2 diabetes. Evidence indicates that in most populations, those who evolve to type 2 diabetes begin with insulin resistance. Insulin resistance can be a primary inherited feature, but acquired factors such as obesity, sedentary lifestyle, and aging (particularly obesity) also can be causal or can exacerbate underlying genetic mechanisms of decreased insulin sensitivity. In an attempt to overcome insulin resistance, the β cell increases insulin secretion, resulting in hyperinsulinemia, which is able to maintain relatively normal glucose tolerance. In a subpopulation of subjects, however, this hyperinsulinemic response is insufficient to fully compensate for the prevailing insulin resistance, and impaired glucose tolerance (IGT) develops. Although a percentage of subjects with IGT may revert to normal glucose tolerance, IGT should be considered an intermediate stage in the development of type 2 diabetes, with many subjects eventually progressing to frank expression of the disease.
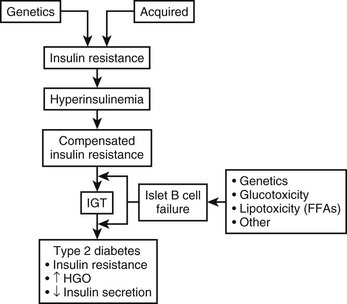
The proportion of IGT subjects who progress to type 2 diabetes depends on the particular ethnic groups studied and the methods of assessment used. For example, in the National Institutes of Health (NIH)-sponsored multicenter Diabetes Prevention Program, ≈10% of patients with IGT developed type 2 diabetes mellitus per year.31 During the transition from IGT to frank type 2 diabetes, at least three pathophysiologic changes can be observed. First is a marked fall in β cell function and insulin secretion. Whether this decrease is due to preprogrammed genetic abnormalities in β cell function, to acquired defects (such as glucotoxicity or lipotoxicity), or to both remains to be elucidated. Nevertheless, a marked decrease in β cell function accompanies this transition, and most believe that this decreased β cell function is the major contributor to the progression to type 2 diabetes mellitus. A second metabolic change is seen at the level of the liver. Subjects with IGT have normal basal rates of hepatic glucose output (HGO), whereas patients with fasting hyperglycemia have increased HGO. Thus, the capacity of the liver to overproduce glucose is an important contributory factor (albeit secondary) to the pathogenesis of type 2 diabetes. Finally, many but not all studies have indicated that patients with type 2 diabetes are more insulin resistant than are those with IGT. Most likely, this increase in insulin resistance is secondary to glucotoxicity or to other acquired factors.
Evidence implicating insulin resistance as a primary defect comes principally from studies examining subjects who are at increased risk for developing diabetes. One such group consists of individuals whose parents have type 2 diabetes. Using intravenous glucose tolerance tests (GTTs), Warram and coworkers evaluated 155 nondiabetic offspring whose parents both had type 2 diabetes.32 During a follow-up period averaging 13 years, type 2 diabetes developed in 16% of the total group. However, when offspring were categorized on the basis of insulin sensitivity at initial testing, the cumulative incidence of diabetes was 60% among subjects with preexisting insulin resistance and less than 5% in insulin-sensitive offspring. Thus, insulin resistance and hyperinsulinemia (rather than hypoinsulinemia) characterized the prediabetic state, and this occurred irrespective of obesity and antedated by many years the subsequent development of impaired insulin secretion and overt type 2 diabetes.32
Studies in ethnic populations with a high prevalence of type 2 diabetes also have provided data supporting a primary role for insulin resistance in diabetes development. Thus, among Pima Indians, hyperinsulinemia and an associated decrease in insulin-mediated glucose disposal are early abnormalities that predict the subsequent development of both IGT and type 2 diabetes.33,34 Pima Indians with IGT who progress to type 2 diabetes have lower insulin levels 2 hours after a glucose load than do those who continue to have IGT or who return to normal glucose tolerance. Similar results come from studies of Micronesians in Nauru, a population with a prevalence of type 2 diabetes of approximately 30%.35 Again, in this population, IGT and type 2 diabetes were most likely to develop in those with hyperinsulinemia at baseline, but progression from IGT to type 2 diabetes could be predicted by lower baseline insulin responsiveness to a glucose challenge.
Further evidence comes from studies in first-degree relatives of type 2 diabetic patients.36–38 In these studies, peripheral insulin resistance and hyperinsulinemia were found in normoglycemic relatives of diabetic patients. First-degree relatives with IGT displayed insulin resistance but also exhibited defects in insulin secretion that were not apparent in those with normal glucose tolerance.
Pathophysiology of Type 2 Diabetes Mellitus
Abnormal Pancreatic β Cell Function
Obesity is a major cause of insulin resistance, and as was described earlier, the β cell compensates for decreased insulin sensitivity by increasing insulin secretion. In normal glucose-tolerant subjects, the increase in insulin secretion that occurs with insulin resistance is described by a hyperbolic relationship39 (Fig. 15-3). Although this quantitative increase in insulin secretion in response to decreased insulin sensitivity should be viewed as an appropriate response, subtle qualitative changes in insulin secretion also are noted in the insulin-resistant state. Insulin normally is secreted in rapid regular pulses with a 5 to 15 minute frequency superimposed on slower ultradian oscillations every 80 to 150 minutes.40 These normal rapid, regular pulses are replaced by disordered pulses in obese subjects with insulin resistance and also in the insulin-resistant but glucose-tolerant offspring of subjects with type 2 diabetes.41 Indeed, insulin secretory pulse frequency has been shown to correlate inversely with peripheral insulin sensitivity.42,43
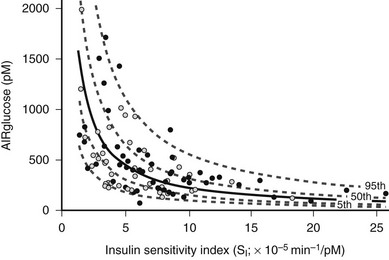
FIGURE 15-3 Relationship between insulin sensitivity and β cell function quantified as the first-phase insulin response (AIRglucose) in 93 (55 males and 38 females) apparently healthy, nondiabetic subjects younger than 45 years. The cohort demonstrates a broad range of insulin sensitivity and β cell function. The solid curve depicts the best fit relationship (50th percentile); the dashed curves represent the 5th, 25th, 75th, and 95th percentiles. The relationship is best described by a hyperbolic function, so that any change in insulin sensitivity is balanced by a reciprocal and proportionate change in β cell function. (Data from Kahn SE, Prigeon RL, McCulloch DK, et al: Quantification of the relationship between insulin sensitivity and β cell function in human subjects: evidence for a hyperbolic function, Diabetes 42:1663–1672, 1993.)
Insulin Secretion in Subjects With Impaired Glucose Tolerance
In subjects with IGT, both quantitative and qualitative defects in insulin secretion are usually present, although this can be variable.43,44 Part of this variability may be explained by the heterogeneity of this condition, as some subjects with IGT will revert to normal glucose tolerance, a proportion will progress to frank type 2 diabetes, and others will continue to have IGT for many years.45
With regard to the hyperbolic relationship between β cell function and insulin sensitivity, subjects with IGT secrete less insulin than is appropriate for their degree of insulin resistance.46 Furthermore, using the graded glucose infusion method, Polonsky has shown that subjects with IGT secrete less insulin at any given glucose level than do normoglycemic subjects matched for a similar degree of insulin resistance and obesity (Fig. 15-4).47 Insulin-resistant subjects with IGT who have progressive impairment in insulin secretion are most likely to develop full-blown type 2 diabetes.48
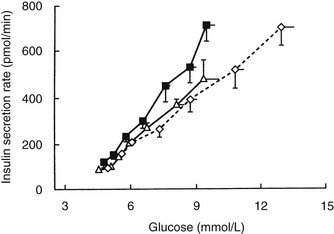
FIGURE 15-4 Relationship between average plasma glucose concentrations and insulin secretion rates during graded glucose infusion studies in a group of lean nondiabetic control subjects (open triangles), nondiabetic obese subjects (closed squares), and matched obese subjects with impaired glucose tolerance (open diamonds). The lowest glucose levels and insulin secretion rates were measured under basal conditions, and subsequent levels were obtained during glucose infusion rates of 1, 2, 3, 4, 6, and 8 mg/kg/min. Values are means ± standard error of the mean (SEM). (Data from Polonsky KS: The β-cell in diabetes: from molecular genetics to clinical research, Diabetes 44:705–717, 1995.)
Insulin secretion in response to a sustained intravenous glucose stimulus is normally biphasic: A rapid rise in insulin levels within 1 to 3 minutes (first phase) is followed by a return to baseline within 6 to 10 minutes with a subsequent gradual increase (second phase). Subjects with IGT have a reduction in both first- and second-phase responses49 to glucose, and a further qualitative defect in insulin secretion in IGT is the replacement of rapid regular secretory oscillations with disorganized pulses.41
Insulin Secretion in Subjects With Type 2 Diabetes
The abnormalities of insulin secretion described in subjects with IGT are also present in type 2 diabetes, although to a more marked degree. β cell function progressively deteriorates during the natural history of type 2 diabetes. This decline in β cell function is evident not only during the conversion from compensated insulin resistance to IGT and subsequently to overt type 2 diabetes, but also during the progressive course of established type 2 diabetes after its initial onset.50
Basal insulin levels usually are normal or increased in type 2 diabetes. Indeed, obese subjects with type 2 diabetes can have basal insulin levels severalfold higher than normal, but this does not mean that basal β cell secretory function is normal, because the prevailing plasma glucose level also must be taken into account.51–53 Hyperglycemia is the major stimulus for insulin secretion, and when normal individuals are made hyperglycemic by infusion of glucose, circulating insulin levels are much higher than those found in type 2 diabetes.51,52 Thus, patients with type 2 diabetes maintain normal or increased basal insulin levels only in the face of the enhanced stimulus of fasting hyperglycemia, which indicates an underlying impairment in the sensitivity of the β cell to glucose. Stimulated insulin levels in type 2 diabetes can be low, normal, or high depending on factors such as the severity of diabetes, the degree of obesity, and the preceding level of diabetic control.13,14,44
In Response to Intravenous Glucose: In type 2 diabetes, defects in the insulin secretory response to intravenous glucose are observed consistently. Once fasting plasma glucose levels exceed 126 mg/dL, the first-phase insulin response to intravenous glucose characteristically is completely absent. This relationship is shown in Fig. 15-5.1,54,55
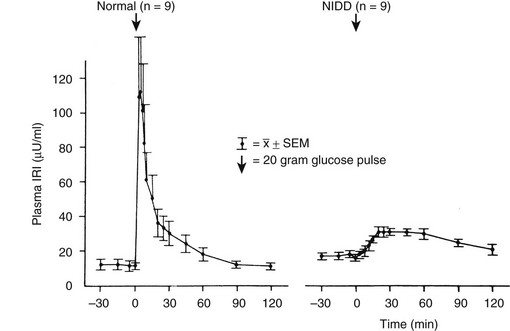
FIGURE 15-5 First-phase insulin release in response to the intravenous administration of glucose in normal and type 2 diabetic (non–insulin-dependent diabetes [NIDD]) subjects. Mean fasting plasma glucose concentrations were 83 ± 3 mg/dL in normal subjects and 160 ± 10 mg/dL in type 2 diabetic subjects. (Data from the American Diabetes Association, Inc., from Ward WWK, Beard JC, Halter JB, et al: Pathophysiology of insulin secretion in non-insulin-dependent diabetes mellitus, Diabetes Care 7:491–502, 1984.)
It is interesting to note that acute or first-phase insulin secretion in response to nonglucose stimuli such as arginine1 or isoproterenol56 is relatively preserved; this finding indicates a functionally selective β cell defect in response to glucose stimuli in type 2 diabetes. Because the acute insulin response to intravenously administered arginine or isoproterenol increases as the glucose concentration is raised, Porte and colleagues have attempted to quantitate this effect of glucose by plotting the increase in acute insulin response to arginine or isoproterenol pulses as a function of increasing plasma glucose level.1,14,51 The slope of this relationship is termed the glucose potentiation slope, and by this analysis, glucose potentiation of β cell function is also reduced in type 2 diabetes.1,14
Although absent first-phase insulin secretion may be a marker for β cell dysfunction, it is unlikely to be an important cause of glucose intolerance or hyperglycemia. Thus, mildly hyperglycemic and severely hyperglycemic type 2 diabetic patients are equally deficient in first-phase insulin secretion, implying that this deficiency does not play a role in further deterioration in glucose tolerance from mild to severe fasting hyperglycemia. Furthermore, in selected patients with normal glucose tolerance in whom type 1 diabetes eventually develops, the acute insulin response to intravenous glucose is absent during the normal stage and therefore does not cause hyperglycemia.57 Finally, α-adrenergic blockers can substantially restore the acute insulin response to intravenous glucose58 without major improvement in fasting glycemia or glucose tolerance.
Second-phase insulin secretion, which is assessed with the hyperglycemic clamp or the graded intravenous glucose infusion technique (see Fig. 15-4 for details), is markedly reduced in individuals with type 2 diabetes compared with normal subjects and those with IGT. In general, the more severe the diabetes, the lower is the second-phase insulin response.1,54
In Response to Oral Glucose and Mixed Meals: The insulin response in type 2 diabetes to oral ingestion of glucose or mixed meals is far more variable than the response to intravenous glucose. After oral glucose, insulin levels are usually subnormal in type 2 diabetes,14,36 although this might not always be the case in patients with mild hyperglycemia.44 This heterogeneity is depicted in Fig. 15-6, which summarizes the results of oral GTTs in a wide spectrum of normal and type 2 diabetic subjects.59 As can be seen, hyperinsulinemia frequently exists in mild states of glucose intolerance. In individuals with mild diabetes, insulin levels generally are in the “normal range,” although inappropriately or relatively low for the degree of hyperglycemia and insulin sensitivity. With more severe diabetes, absolute stimulated insulin levels are uniformly low.
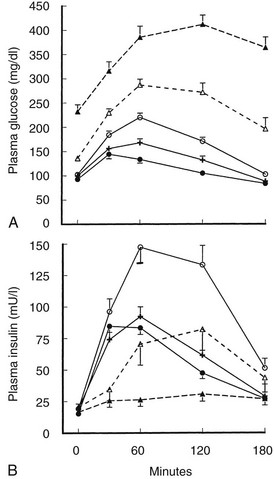
FIGURE 15-6 A, Mean (±standard error of the mean [SEM) plasma glucose response to oral glucose in the five subject groups. Closed circle, normal; cross, borderline tolerance; open circle, impaired glucose tolerance; open triangle and broken line, fasting hyperglycemia (110 to 150 mg/dL); closed triangle and broken line, fasting hyperglycemia (>150 mg/dL). B, Mean (±SEM) plasma insulin response to oral glucose in the five subject groups. Symbols are the same as in A. (Data from Reaven GM, Olefsky JM: Relationship between heterogeneity of insulin responses and insulin resistance in normal subjects, Diabetologia 13:201–206, 1977.)
Proinsulin Secretion
Another factor related to hyperinsulinemia in type 2 diabetes is circulating proinsulin levels. Proinsulin is secreted by β cells concomitantly with insulin and cross-reacts with insulin in most insulin immunoassays, thus contributing to the total measured immunoreactive insulin level. In normal subjects, proinsulin represents only a small portion (3% to 7%) of the insulin-like material secreted by β cells. However, using proinsulin-specific immunoassays, several groups have shown that in hyperinsulinemic states and in many cases of type 2 diabetes, an increased proportion of proinsulin is released and contributes to the measured insulin in standard immunoassays, so true insulin levels are overestimated.14,53,60 When corrected for this factor, basal insulin levels may be normal or moderately elevated in type 2 diabetes.
Mechanisms of β Cell Dysfunction
The overall mass of β cells changes in obesity and type 2 diabetes.61,62 β cell mass reflects the balance between new islet formation (neogenesis) and β cell loss due to apoptosis. Longitudinal studies in animal models suggest that β cell mass increases appropriately in response to a decrease in insulin sensitivity,62 and cross-sectional data from humans have long revealed an expanded β cell mass in obesity.63 In contrast, β cell mass is reduced in type 2 diabetes.61 On the basis of murine models and cross-sectional autopsy data from humans, diminished β cell mass is thought to be due to accelerated β cell apoptosis and the failure of islet neogenesis and β cell replication to compensate for this loss.62
It has been theorized that lipid accumulation in the β cell is implicated in the apoptotic process and the development of impaired insulin secretion in type 2 diabetes. This is referred to as lipotoxicity and may involve excess fatty acids entering β cells, thus triggering the apoptotic cellular response.64
A body of evidence also suggests a role for islet amyloid polypeptide (IAPP) in the loss of β cells. IAPP is synthesized in the β cell and is co-secreted with insulin.65 IAPP aggregates to form fibrils of amyloid, and islet amyloid is found at autopsy in up to 90% of subjects with type 2 diabetes.66 In vitro, IAPP is toxic, causing β cell apoptosis,67 and it may contribute to the reduced β cell mass that is associated with type 2 diabetes.
Insulin secretory abnormalities found in type 2 diabetes are often improved after a period of good blood glucose control, irrespective of the treatment used (diet, insulin, or oral hypoglycemic agents).68–70 This partial reversibility is consistent with the idea that, to some extent, the abnormalities may be secondary to hyperglycemia or some other factor associated with uncontrolled diabetes. Support for the “glucotoxicity” theory comes from a variety of in vivo and in vitro studies showing that chronic exposure of islets to hyperglycemia can result in a number of different defects in glucose-induced insulin secretion.71 It is important to note that when isolated human islets are incubated under euglycemic and hyperglycemic conditions, islets that are exposed to hyperglycemia demonstrate a marked defect in their ability to secrete insulin in response to subsequent glucose stimuli.72 Although the precise mechanism is unknown, it seems likely that glucotoxicity coupled with lipotoxicity plays some role in the impaired β cell function of type 2 diabetes.
An interesting finding from studies of mouse genetics is that insulin receptor signaling in β cells is important for normal function. Thus, mice in which the insulin receptor gene has been specifically deleted from β cells show a complete loss of first-phase insulin secretion in response to glucose but not arginine, reminiscent of the β cell defect in type 2 diabetes.73 Second-phase glucose-induced insulin secretion is also blunted in these mice, and they show an age-dependent progressive impairment in glucose tolerance. Glucose-stimulated insulin secretion involves transport of glucose into cells by a specific glucose transporter, termed GLUT2. Following uptake, glucose is phosphorylated by glucokinase, and subsequent intracellular metabolism of glucose-6-phosphate leads to stimulation of insulin secretion. In mouse studies, genetic deletion of GLUT2 leads to loss of glucose but not arginine-stimulated insulin secretion. It is interesting to note that feeding mice high-fat diets to induce obesity also leads to a decrease in β cell GLUT2 expression, suggesting another mechanism of interaction between acquired environmental factors and β cell dysfunction.
The decrease in β cell mass in type 2 diabetes is in the range of 30% to 50%. However, because sufficient insulin secretory reserve normally exists to sustain an 80% to 90% loss of β cells without the development of hyperglycemia, it follows that decreased functional capacity of the remaining β cells must exist in type 2 diabetes. Indeed, it has been shown that the maximal insulin secretory capacity may be reduced by as much as 80% in type 2 diabetic subjects.52 It is possible that the decrease in β cell mass in type 2 diabetes is somehow causally related to the decreased function of the remaining β cells. Thus, partially pancreatectomized rats and streptozotocin-treated rats display similar insulin secretory defects,71 which suggests that decreased glucose-stimulated insulin secretion with relative preservation of responsiveness to nonglucose stimuli may be a general type of abnormality that may occur in response to a variety of β cell insults.
Peripheral Insulin Resistance
Causes of Peripheral Insulin Resistance
Circulating Factors That Influence Insulin Action: Hormonal antagonists include all known counterregulatory hormones such as cortisol, growth hormone, glucagon, and catecholamines. In well-known syndromes (e.g., Cushing’s disease, acromegaly), elevated levels of these hormones can induce an insulin-resistant diabetic state. However, in the usual case of obesity or type 2 diabetes, excessive levels of these counterregulatory hormones are not an important contributory factor to insulin resistance.
Several years ago, Randle and coworkers hypothesized that the elevated circulating levels of free fatty acids (FFAs) found in obesity and type 2 diabetes impair peripheral glucose utilization.74 Substantial evidence indicates that FFAs do indeed contribute to insulin resistance, although the mechanisms differ from those originally proposed by Randle. FFAs also play an important role in the regulation of HGO and contribute to hepatic insulin insensitivity in obesity and type 2 diabetes. These mechanisms will be discussed in greater detail later in this chapter.
Impaired Access of Insulin to Target Cells: Because insulin must travel from the circulation to target tissues to elicit biological effects, any defect in this transfer could lead to functional insulin resistance. Compared with secretion into the circulation, the passage of insulin from the plasma compartment to tissue sites of action is markedly delayed, and in vivo effects of insulin in stimulating glucose disposal are well correlated with the appearance of insulin in the interstitial fluid.75 Lymph and interstitial insulin levels are ≈40% lower than those in plasma,75,76 which indicates that peripheral tissues are more sensitive to insulin than was previously recognized. Furthermore, the possibility arises that either the rate or the amount of insulin being transferred from the plasma to the interstitial compartment could be abnormal in type 2 diabetes or obesity, thereby contributing to the insulin-resistant state.77,78 Recent studies indicate that transport of insulin across the capillary in vivo occurs by diffusion76 and is not receptor mediated, as was previously suggested.79 Transport by diffusion fits with the finding that transcapillary passage is comparable in normal subjects, insulin-resistant nondiabetic subjects,80 and those with type 2 diabetes.81 Further evidence that the delayed activation of muscle glucose uptake in obesity and type 2 diabetes is not due to impaired transcapillary transport of insulin comes from a study by Nolan and coworkers.78 This study showed that the kinetic defect in the ability of insulin to stimulate leg glucose uptake was not accompanied by any delay in the activation of leg muscle insulin receptors by insulin, thus implying that the kinetic defect is distal to the insulin receptor.
Another physical factor that may relate to insulin resistance is muscle capillary density, which correlates with in vivo insulin sensitivity.82 Laakso and coworkers have shown that insulin, at least at pharmacologic levels, increases leg blood flow.83 Because tissue glucose uptake is a product of blood flow and the arteriovenous glucose difference, increased leg blood flow could contribute to overall glucose disposal. Similar studies performed in obese subjects and in subjects with type 2 diabetes revealed a decrease in the insulin-induced increase in leg blood flow, which may explain part of the decrease in total leg glucose uptake.83 However, others found no effect of insulin on blood flow,84 and Utriainen and colleagues used 15O [H2O] and positron emission tomography to confirm enhancement of leg muscle blood flow by pharmacologic insulin levels, but no difference in the response between type 2 diabetic and normal control subjects.85
Cellular Defects in Insulin Action: Available evidence points to a target tissue defect as the major cause of insulin resistance in type 2 diabetes. Before potential causes are considered, it is useful to review some general concepts concerning normal insulin action (Fig. 15-7). Insulin first binds to its cell surface receptor, a heterotetrameric glycoprotein composed of two α subunits (135 kDa) and two β subunits (95 kDa) linked by disulfide bonds.86–89 The α subunits are entirely extracellular and are responsible for insulin binding. The β subunits are transmembrane proteins containing a small extracellular domain and a larger cytoplasmic domain that includes insulin-regulated tyrosine kinase activity. Binding of insulin to the receptor rapidly induces tyrosine autophosphorylation of the β subunit involving three tyrosine residues in the kinase domain, in addition to tyrosine residues adjacent to the transmembrane domain and in the C terminus of the β subunit. Once the receptor has been autophosphorylated, its intrinsic tyrosine kinase catalytic activity is markedly enhanced, and it now can phosphorylate tyrosine residues on endogenous protein substrates. Activation of the insulin receptor tyrosine kinase is essential for transduction of the insulin signal and for internalization of the receptor. Patients with naturally occurring mutations in the tyrosine kinase domain of the insulin receptor have syndromes of severe insulin resistance.
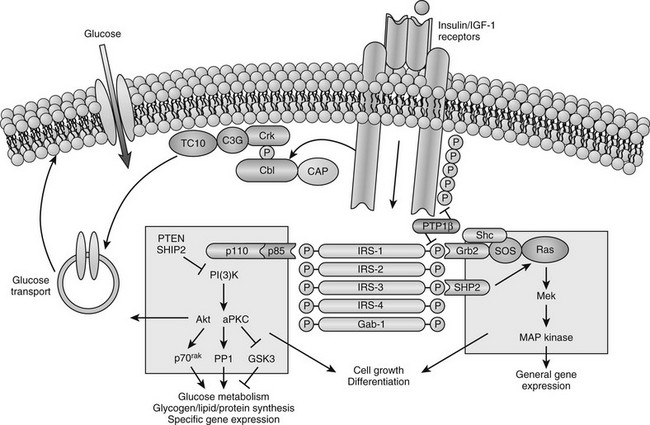
FIGURE 15-7 Model of cellular insulin action. (From Saltiel AR, Kahn CR: Insulin signalling and the regulation of glucose and lipid metabolism, Nature 414:799–806, 2001.)
In recent years, major advances have been made in our understanding of how the insulin signal is propagated downstream from the activated insulin receptor to various insulin-regulated enzymes, transporters, and insulin-responsive genes to mediate its metabolic and growth effects (see Fig. 15-7). This field is rapidly evolving and complex and is discussed only briefly here because it is covered comprehensively in Chapter 8. A large number of intermediate signaling molecules have been identified, and after activation of the insulin receptor kinase, more than one signaling pathway may be used (see Fig. 15-7). For example, some of the components in the pathways leading to mitogenic effects of insulin are distinct from those leading to activation of glucose transport. Even a single action of insulin such as stimulation of glucose transport can involve more than one signaling pathway. Several cytosolic protein substrates of the insulin receptors are phosphorylated on tyrosine residues within seconds of insulin binding to its receptor. The first of these substrates to be identified was insulin receptor substrate 1 (IRS-1).89,90 IRS-1 belongs to a growing family of proteins that includes IRS-2, IRS-3, IRS-4, and a protein termed shc, which are immediate substrates of the insulin receptor kinase involved in insulin signaling. These proteins have no enzymatic activity but act as docking proteins. Tyrosine phosphorylation of these substrates enhances their association with proteins that contain src homology-2 (SH2) domains. These SH2 domains contain ≈100 amino acids and can bind to specific short motifs that encompass a phosphotyrosine. The binding of specific SH2 domain–containing proteins to tyrosine-phosphorylated IRS proteins or shc generates multicomponent signaling complexes, which, in turn, modulate the activities of phosphoinositide-3-kinase (PI3K), several serine kinases, and phosphatases that act on key insulin-regulated enzymes and transcription factors.
One of the most important effects of insulin with respect to type 2 diabetes is stimulation of glucose uptake into skeletal muscle, adipocytes, and heart muscle. Under most physiologic circumstances, glucose transport in these tissues is rate limiting for overall glucose disposal.91–93 Tissue glucose uptake is mediated by a family of at least five facilitative glucose transporters, each derived from a separate gene. These transporters show a high degree of homology, but each has tissue-specific distribution.94,95 One of them, GLUT4, or the insulin-sensitive glucose transporter, is uniquely expressed in skeletal muscle, adipose tissue, and cardiac muscle. In the unstimulated state, most of the GLUT4 proteins are located in an intracellular vesicular pool. Upon insulin stimulation, recruitment or translocation of these glucose transporter-rich vesicles to the cell surface causes insertion of GLUT4 proteins into the plasma membrane, where they begin to transport glucose into the cell.96–100
Characteristics of Insulin Resistance in Subjects With Impaired Glucose Tolerance or Type 2 Diabetes
The frequency of insulin resistance increases as the degree of carbohydrate intolerance worsens.101 Thus, many, but not all, subjects with IGT are insulin resistant, whereas essentially every type 2 diabetic patient with fasting hyperglycemia displays this abnormality. Numerous studies indicate that insulin resistance is more marked in type 2 diabetes than in the prediabetic IGT state.3,5,36 However, other reports show only a modest increase in the degree of insulin resistance going from IGT to type 2 diabetes. Because most type 2 diabetic patients are overweight, obesity-induced insulin resistance is clearly a major contributing factor in these patients. However, obesity is not the only cause, in that the insulin resistance in obese type 2 diabetic patients exceeds that caused by obesity alone, and nonobese type 2 diabetic patients are also insulin resistant.3,5,101
All methods of assessing insulin resistance in vivo rely on measurement of the ability of a fixed dose or concentration of insulin to promote glucose disposal. Thus, a blunted decline in plasma glucose concentration after administration of intravenous insulin has been demonstrated in type 2 diabetes.102,103 Another approach has been to infuse insulin and glucose at fixed rates while endogenous insulin secretion is inhibited by a combination of epinephrine and propranolol or by somatostatin.102,104 With this method, the resulting steady-state plasma glucose level reflects the action of concomitantly infused insulin; the higher the steady-state plasma glucose, the greater is the degree of insulin resistance. Bergman and colleagues’ minimal model is yet another method of assessing in vivo insulin resistance.102 This method entails computer modeling of plasma glucose and insulin levels after an intravenous glucose bolus to generate an index of insulin sensitivity. With all these methods, type 2 diabetic subjects exhibit a significant decrease in insulin sensitivity compared with controls.3,102,105,106
More detailed studies of in vivo insulin resistance have been carried out with the euglycemic glucose clamp method.102 With this approach, insulin is infused at a constant rate, resulting in a given steady-state plasma insulin level, while plasma glucose is kept constant at a predetermined level by a feedback-controlled variable infusion of glucose. The insulin normally lowers the plasma glucose level by suppressing HGO and by stimulating tissue glucose uptake. During the insulin infusion, the amount of glucose that has to be infused to keep plasma glucose levels constant increases gradually until a steady state is reached. Under these steady-state conditions, the glucose disposal rate provides an excellent quantitative assessment of the biological effect of a particular steady-state insulin level. If a radioactive or stable isotope of glucose is also infused during the study, HGO during the clamp can be quantified. In type 2 diabetes, glucose disposal rates are 30% to 60% lower than those in normal subjects at any given insulin infusion rate. If several studies at different insulin levels are performed in a given subject, dose response curves for insulin-stimulated glucose disposal and suppression of HGO can be constructed. Patients with type 2 diabetes (obese and nonobese) exhibit both a rightward shift in their dose response curve (diminished sensitivity) and a marked decrease in their maximal rate of glucose disposal (decreased responsiveness) (Fig. 15-8). These changes tend to be more pronounced in obese diabetic patients, particularly at maximal glucose disposal rates (see Fig. 15-8). The insulin resistance of obese type 2 diabetic patients is significantly greater than that of nondiabetic obese subjects. Subjects with IGT tend to have a rightward shift in their dose response curves with normal maximal glucose disposal rates (see Fig. 15-8).
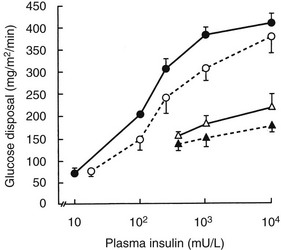
FIGURE 15-8 Mean insulin dose response curves for control subjects (closed circles), subjects with impaired glucose tolerance (open circles), and nonobese (open triangles) and obese (closed triangles) type 2 diabetic subjects. The group with impaired glucose tolerance has a rightward shift in the dose response curve without a change in maximal response (i.e., decreased sensitivity). Lean and obese patients with type 2 diabetes have both a rightward shift and a reduction in the response to a maximally stimulating concentration of insulin (i.e., decreased sensitivity and decreased insulin responsiveness). (Data from Kolterman OG, Gray RS, Griffin J, et al: Receptor and post-receptor defects contribute to the insulin resistance in non-insulin dependent diabetes mellitus, J Clin Invest 68:957–969, 1981.)
Because skeletal muscle is responsible for the great majority of in vivo insulin-stimulated glucose uptake, this tissue must be the major site for resistance to insulin-stimulated glucose disposal. This conclusion is evidenced by the demonstration of insulin resistance in type 2 diabetic patients during forearm perfusion studies.107,108 Leg catheterization studies have shown that skeletal muscle accounts for ≈80% of whole-body insulin-mediated glucose uptake, and that leg skeletal muscle is markedly resistant to the ability of insulin to stimulate glucose uptake in type 2 diabetes.109–111 Thus, although other insulin target tissues display decreased insulin sensitivity, they do not account for a significant proportion of overall glucose uptake, and one can conclude that all measures of in vivo insulin action on glucose disposal largely assess the resistance of skeletal muscle to take up glucose under the influence of insulin.
Pathophysiologic Abnormalities in Insulin Target Tissues
Mechanisms of Skeletal Muscle Insulin Resistance: As the first step in insulin action, it is apparent that a decrease in cellular insulin receptors could lead to insulin resistance. However, this potential relationship is not as clear as it would seem because a maximal insulin effect is achieved at insulin concentrations that occupy a fraction of the surface receptors, giving rise to the concept of “spare” receptors. A maximal response of glucose transport in adipocytes and muscle is achieved with only 10% to 20% of the receptors occupied.112,113 Once the critical number of receptors needed to generate a maximal response is activated, additional increases in the prevailing insulin concentration lead to increases in receptor occupancy with no further increase in biological response, because a step (or steps) distal to the receptor is now rate limiting. The functional significance is that a decreased number of insulin receptors leads to a rightward shift in the insulin biological function dose response curve, with decreased responses at all submaximal insulin concentrations but a normal maximal response. A reduction in the maximal insulin response generally denotes the presence of a postbinding abnormality. In this context, the term postbinding defect includes abnormalities of insulin receptor function that affect its transmembrane signaling function, such as its kinase activity. A postreceptor defect refers to any abnormality in a step distal to the insulin receptor.
Early studies showed decreased insulin binding to circulating monocytes from obese and IGT subjects and from both obese and nonobese type 2 diabetic patients.114,115 This decreased binding was due to a decrease in insulin receptor number with no change in affinity. Similar results were subsequently obtained, when isolated adipocytes, hepatocytes, and skeletal muscle from obese subjects and patients with type 2 diabetes were used.116,117 The decrease in cellular insulin receptors in obesity and type 2 diabetes may well be secondary to hyperinsulinemia, inasmuch as elevated circulating insulin levels can downregulate receptor number.
A reduction in insulin receptor tyrosine kinase activity in type 2 diabetes generally has been found in patients with normal kinase activity in IGT.78,91,117 The receptor autophosphorylation/kinase defect appears to be generalized to all insulin target tissues and relatively specific for the hyperglycemic insulin-resistant state that is seen in type 2 diabetes.
Emerging evidence points to a role for IRS proteins in the development of insulin resistance. A defect in insulin-stimulated IRS-1 tyrosine phosphorylation is found in skeletal muscle from type 2 diabetic patients, although overall IRS-1 expression is unchanged.118 Serine/thr phosphorylation of IRS proteins is closely associated with reduction of signaling through IRS. Two potential mechanisms may underlie this phenomenon. First, serine phosphorylation may block the interaction of IRS-1 with its target proteins.119 Second, proteasomal-mediated degradation of IRS-1 may be increased.120 Several intermediary lipid metabolites and cytokines have been shown to activate a variety of serine/threonine kinases that induce serine phosphorylation of IRS-1. Serine kinases implicated include c-Jun NH2-terminal kinase (JNK),121 IκB kinase (IKK),122 PKC theta, S6K, and MTOR. It is interesting to note that this places IRS-1 at the intersection of a variety of intracellular pathways, including inflammation, endoplasmic reticulum (ER) stress, and nutrient sensing, all of which can activate serine/threonine kinases that may phosphorylate IRS-1.
IRS-2 is also important for insulin signaling and glucose homeostasis. In mice with disruption of the IRS-2 gene, profound defects in both insulin action (predominantly in the liver) and β cell function develop, progressing to diabetes.123
PI3K plays a key role in mediating the effects of insulin on glucose metabolism.124 Insulin-stimulated PI3K activity in skeletal muscle is reduced in both obese nondiabetic subjects125 and patients with type 2 diabetes.118 This reduction in PI3K activity correlates with the decrease in whole-body glucose disposal.118 An interesting twist on this relates to the regulatory subunits of PI3K. PI3K consists of a catalytic, 110 kDa subunit, as well as a family of regulatory subunits (p50, p55, and p85). If expression of the regulatory and catalytic subunits is unbalanced, with excess levels of the regulatory subunits, then monomeric p50/55/85 can bind through their SH2 domains to tyrosine phosphorylated substrates, such as IRS-1. When this happens, they can compete out binding of the dimeric p110.p85 PI3K complex, thus inhibiting PI3K signaling. Activated PI3K stimulates pyruvate dehydrogenase kinase (PDK1) in the plasma membrane, which then leads to activation of AKT and PKC λ/ζ. Both of these latter enzymes are upstream of GLUT4 translocation and are important regulators of glucose transport stimulation. Decreased insulin-induced AKT and PKC λ/ζ activation are widely described in insulin-resistant skeletal muscle from a variety of states.
Insulin-stimulated glucose transport in isolated muscle fibers and adipocytes from type 2 diabetic patients is markedly reduced at all insulin concentrations.10,126 What is the mechanism of this decrease? In adipocytes from type 2 diabetic subjects, decreased GLUT4 levels have been reported. In contrast, skeletal muscle GLUT4 mRNA and protein levels are normal in type 2 diabetes.127,128 Because the muscle of type 2 diabetic patients is not deficient in GLUT4 protein, it appears that the defect in insulin-stimulated glucose transport reflects a decrease in the ability of insulin to signal translocation of GLUT4 to the cell surface.
Indeed, clear evidence suggests that this is the case. For example, Kelley and coworkers used quantitative confocal laser scanning microscopy to examine insulin-stimulated recruitment of GLUT4 to the sarcolemma in muscle biopsies from patients with type 2 diabetes.98 In the basal state, sarcolemmal GLUT4 labeling was similar in diabetic and normal subjects, but in response to insulin, the increase in GLUT4 in type 2 diabetic subjects was only 25% of that in control subjects. A quantitatively similar defect in GLUT4 translocation was found in obese nondiabetic subjects. In both type 2 diabetic subjects and obese nondiabetic subjects, the defect in GLUT4 translocation was associated with marked impairment in insulin-stimulated muscle glucose transport as determined by positron emission tomography. Others using biochemical muscle subfractionation techniques have found a defect in GLUT4 translocation in patients with type 2 diabetes.99
Trafficking of GLUT4 involves a complex system analogous to synaptic vesicle movement, and an expanding list of proteins involved in the regulation of GLUT4 trafficking are being identified.100 Clearly, impaired GLUT4 translocation could be due to a defect in one or more of these GLUT4 vesicle-trafficking proteins, which is an area requiring intensive investigation.
Oxidative and Nonoxidative Glucose Metabolism in Skeletal Muscle: By performing indirect calorimetry during glucose clamp studies, one can determine the intracellular fate of glucose by measuring the percentage of glucose that is oxidized versus that which undergoes nonoxidative glucose metabolism (consisting of storage as glycogen plus glycolysis). The insulin concentrations necessary for half-maximal stimulation of glucose oxidation (≈50 mU/L in normal subjects) are lower than those required for stimulation of glucose uptake and storage as glycogen (≈100 mU/L).129 Thus, at low physiologic insulin levels, oxidative glucose disposal is quantitatively more important, but at higher insulin levels, nonoxidative glucose metabolism predominates.12 Defects in both oxidative and nonoxidative glucose metabolism exist in type 2 diabetes, although the decrease in nonoxidative metabolism is greater.9,36,111,130,131 Shulman and coworkers, using nuclear magnetic resonance (NMR) spectroscopy of the gastrocnemius muscle during an infusion of 13C-enriched glucose, showed that during a hyperinsulinemic hyperglycemic clamp study, nonoxidative glucose disposal is highly correlated with rates of skeletal muscle glycogen deposition.9 Moreover, a 50% reduction in the rate of muscle glycogen synthesis in type 2 diabetic patients was found, and defects in nonoxidative glucose metabolism and muscle glycogen synthesis correlated well with the decrease in whole-body glucose uptake.9
The reduced muscle glycogen synthesis rate in type 2 diabetes could be the result of a decrease in glucose transport, impaired glucose phosphorylation, or an abnormality in the glycogen synthetic pathway, and decreases in GLUT4 translocation, hexokinase II, and glycogen synthase have been reported. To identify the primary site of the intracellular block in glycogen synthesis, Rothman and coworkers used 31P-NMR during glucose clamp studies to measure glucose-6-phosphate concentrations in gastrocnemius muscle.132 They reasoned that a primary block in glycogen synthesis (e.g., resulting from decreased glycogen synthase) would lead to increased glucose-6-phosphate levels, whereas if the decreased flux of glucose to glycogen reflected impaired glucose transport and/or phosphorylation, then glucose-6-phosphate levels would be low. They found a lower steady-state glucose-6-phosphate concentration in type 2 diabetes, indicating that the reduced rate of glycogen synthesis was secondary to impaired glucose transport, hexokinase activity, or both. In additional muscle NMR studies, these investigators detected a very low intracellular free glucose concentration during hyperinsulinemic hyperglycemic clamp studies in both normal and type 2 diabetic patients.81 This finding strongly suggested that glucose transport is the rate-controlling step in insulin-stimulated muscle glycogen synthesis, indicating that decreased insulin-mediated glucose transport is the major defect in the muscle insulin resistance of type 2 diabetes.
Skeletal Muscle Lipid Metabolism: Increased free fatty acid flux is a consistent finding in type 2 diabetic patients, as well as in obese insulin-resistant nondiabetic subjects. This usually is accompanied by elevated circulating FFA levels, particularly in the postprandial state, and increased FFA uptake into skeletal muscle, as well as liver, can be a cause of decreased insulin sensitivity. After cellular uptake, FFAs are converted to long-chain fatty acyl CoAs (LCFA-CoAs), which can be transported into the mitochondria by carnitine palmitoyltransferases to undergo β oxidation (Fig. 15-9). If not transported into the mitochondria, LCFA-CoAs can be reesterified with glycerol-3-phosphate (G-3-P) to form phosphatidic acid (PA), which is converted to diacylglyercol (DAG) and then to triglycerides. When levels of fatty acyl CoAs (particularly palmitoyl CoA) are high, the ability of cells to oxidize or store this lipid intermediate may be limiting, which leads to increased conversion of fatty acyl CoA to ceramide. With increased uptake of FFA into the cell, intracellular levels of LCFA-CoAs and intermediates such as DAG, ceramide, and triglyceride are increased.133,134 Evidence exists to indicate that these fatty acid intermediates and metabolites can impair insulin signaling by activating inhibitory serine/threonine kinases such as JNK1, PKCθ, IKKβ, and MTOR/p70S6K. In turn, these inhibitory serine kinases phosphorylate IRS-1, impairing the ability of IRS-1 to propagate downstream insulin signaling. In addition, accumulation of ceramide has an additional effect of impairing AKT activation, thus further inhibiting insulin signaling.
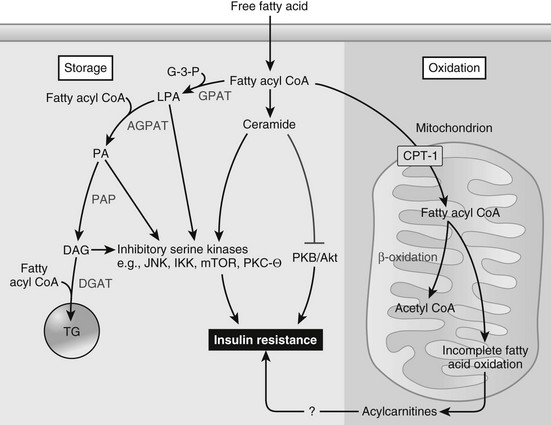
FIGURE 15-9 Fatty acid metabolism and insulin action in skeletal muscle or liver. Obesity results in an increased flux of free fatty acids into the circulation and uptake by the myocyte or hepatocyte. Activated fatty acids (i.e., fatty acyl-CoAs) are “metabolized” primarily via one of two pathways, oxidation or storage. When fatty acid flux exceeds the ability of these pathways to dispose of fatty acyl-CoAs, intermediaries of fatty acid metabolism (e.g., diacylglycerol (DAG), phosphatidic acid (PA), lysophosphatidic acid (LPA), ceramide) accumulate. In turn, these fatty acid intermediates can activate a number of different serine kinases that can negatively regulate insulin action. Ceramide can also impair insulin action through interactions with PKB/Akt. An inability to completely oxidize fatty acids through β-oxidation, which leads to an accumulation of acylcarnitines, has also been hypothesized to cause insulin resistance, although the precise mechanisms leading to insulin resistance are, to date, unknown. AGPAT, acyl glycerol-3-phosphate acyltransferase; PAP, phosphatidic acid hydrolyase; PA, phosphatidic acid. (From Schenk S, Saberi M, Olefsky JM: Insulin sensitivity: modulation by nutrients and inflammation, J Clin Invest 118:2992–3002, 2008.)
Intramyocellular triglyceride content, measured by muscle biopsy or NMR spectroscopy, is increased in obesity and type 2 diabetes135 and is a strong predictor of insulin resistance in both animals and humans.136,137 It is likely that increased intramyocellular triglyceride content does not, by itself, impair insulin signaling, but acts as a marker of increased intracellular LCFA-CoAs, DAGs, and other lipid intermediates. A strongly negative correlation has been demonstrated between whole-body insulin sensitivity, as determined by the glucose clamp, and the content of LCFA-CoAs and DAGs, as measured in muscle biopsy samples.138
Kinetic Defects in Insulin Action: Although most quantitative assessments of in vivo insulin resistance report impaired insulin action based on steady-state measurements, kinetic defects in insulin action in obesity have been demonstrated. Thus, the rate of activation of the effect of insulin in stimulating glucose disposal is decreased, and the rate of deactivation of the effect of insulin is increased.77 Given that under physiologic postprandial conditions, insulin is secreted in a phasic rather than a steady-state manner, it is likely that kinetic defects in insulin action are of functional importance, and that steady-state measurements of insulin action underestimate the functional defect in insulin sensitivity. This has been demonstrated by measuring glucose disposal during phasic administration of insulin during a glucose clamp, designed to mimic the pattern of insulin secretion during oral glucose tolerance tests. Total insulin-stimulated glucose disposal during the “phasic” clamp was reduced by 64% in obese subjects compared with lean controls.139 This is greater than the 20% to 50% decrease in steady-state insulin-mediated glucose disposal that was observed in glucose clamp studies in these same subjects, confirming the functional importance of kinetic abnormalities in insulin action.77,140
Liver
Hepatic Glucose Output: After an overnight fast, normal basal glucose production rates are 1.8 to 2.2 mg/kg/min, with about 90% of the glucose that is released into the circulation coming from the liver. After glucose ingestion, HGO must be suppressed promptly to limit the rise in plasma glucose levels; as intestinal glucose delivery wanes, HGO rates must be restored to meet the obligatory glucose needs of tissues such as the brain. These changes in HGO are mediated largely by changes in insulin and counterregulatory hormones (predominantly glucagon). The rate of basal HGO is increased in both obese and nonobese type 2 diabetic patients,6,7,11 but not in subjects with IGT (Fig. 15-10A). The fasting plasma glucose level and HGO are closely correlated in type 2 diabetic patients (Fig. 15-10B), which indicates that the rate of basal glucose production by the liver directly modulates the level of fasting hyperglycemia in type 2 diabetes. Gluconeogenesis is the predominant source of HGO following an overnight fast,141 and most studies suggest that gluconeogenesis is increased in type 2 diabetes.142,143 Thus, enhanced gluconeogenesis is the proximate cause of increased HGO in type 2 diabetes, and although the mechanism is unclear, it is probably a multifactorial defect. Glucagon levels are elevated in type 2 diabetes, and the effect of glucagon in stimulating the synthesis and release of glucose by the liver is well known. Hyperglycemia normally exerts a suppressive effect on α cell glucagon secretion, and the presence of hyperglucagonemia in the face of hyperglycemia implies that pancreatic α cells in type 2 diabetes are resistant to the inhibitory effects of glucose. Other factors are possible, but regardless of the mechanisms, increased α cell function in type 2 diabetes is a consistent abnormality, and suppression of plasma glucagon levels by infusion of somatostatin lowers plasma glucose levels in both normal and type 2 diabetic subjects.144
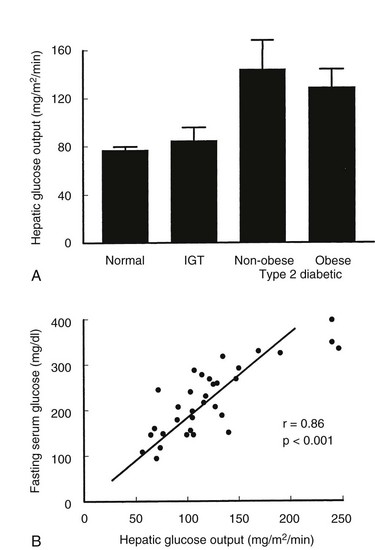
FIGURE 15-10 A, Rates of hepatic glucose production in the basal state (7:00 to 9:00 am after an overnight fast) in normal subjects, subjects with impaired glucose tolerance (IGT), and obese or nonobese subjects with type 2 diabetes. Hepatic glucose output is normal in subjects with IGT but is markedly increased in type 2 diabetes. B, Relationship between the individual hepatic glucose production rate and fasting plasma glucose level in type 2 diabetic subjects. (Data from Kolterman OG, Gray RS, Griffin J, et al: Receptor and post-receptor defects contribute to the insulin resistance in non-insulin dependent diabetes mellitus, J Clin Invest 68:957–969, 1981.)
Hepatic glucose production can be suppressed completely by high physiologic or supraphysiologic insulin levels in type 2 diabetes, but the sensitivity of HGO to lower concentrations of insulin is reduced.12 This reduced insulin sensitivity also contributes to the overall increase in glucose production in these patients. The ability of insulin to suppress HGO may in part occur indirectly77,145 through suppression of adipose tissue lipolysis and plasma FFA levels. Thus, impaired insulin-induced suppression of HGO in type 2 diabetes may in part be secondary to impaired inhibition of plasma FFA levels by insulin.13
Hepatic Glucose Uptake: Although earlier data held that most orally ingested glucose was extracted by the liver and was largely converted to glycogen,146 more recent studies indicate that skeletal muscle is quantitatively the most important tissue for disposal of an oral glucose load, with 50% to 60% of total glucose disposal being accounted for by skeletal muscle.147 Only 20% to 35% of oral glucose is taken up by the liver. In contrast to direct stimulation of muscle glucose uptake by insulin and incorporation into glycogen, insulin plays a permissive role in promoting hepatic glucose uptake (HGU). Thus, insulin does not cause net HGU or stimulation of liver glycogen deposition without an increased portal venous glucose concentration. The main determinant of glucose transport into and out of the liver is the glucose concentration gradient between sinusoids and hepatocytes.148 After glucose ingestion, uptake of glucose by the liver (newly absorbed and recirculating) is impaired in type 2 diabetes.149 This may be due to decreased hepatic glucokinase activity, and this defect in HGU accounts for ≈25% of postprandial hyperglycemia in type 2 diabetes.150
Mechanisms of Hepatic Insulin Resistance: Various defects in the cellular actions of insulin have been described in livers of type 2 diabetic patients, or in relevant animal models, and, in most ways, these defects are similar to what is seen in skeletal muscle and other tissues. These include a decrease in insulin receptor tyrosine kinase activity, decreased IRS-1/PI3K signaling, and impaired activation of AKT. An important pathophysiologic element unique to the liver involves the transcriptional control of gluconeogenesis. Gluconeogenesis is finely controlled by a complex transcriptional network that allows the liver to adjust rapidly to changes in glucose need. Glucagon stimulates adenylcyclase activity, leading to increased intracellular cyclic adenosine monophosphate (AMP) levels, with activation of PKA and subsequent phosphorylation and activation of the nuclear transcription factor CREB. CREB is a master positive regulator of gluconeogenic gene expression and the effects of CREB are facilitated by two nuclear co-activators, TORC2 and CBP. In addition, PGC1α, FOX01, and HNF4a positively activate the gluconeogenic program. It is important to note that TORC2, CBP, and FOX01 are highly insulin regulated. Insulin-induced phosphorylation of these co-factors, through activation of AKT/PKCλ, causes nuclear exclusion of these proteins with inhibition of gluconeogenesis. This provides the mechanism for the ability of insulin to downmodulate gluconeogenesis. Once the liver is insulin resistant, phosphorylation of these co-factors is impaired and high levels of nuclear TORC2, CBP, and FOX01 exist, leading to increased gluconeogenesis. From a therapeutic point of view, controlled inhibition of hepatic gluconeogenesis is a highly valuable approach to the treatment of type 2 diabetes.
Adipose Tissue
Adipokines: As was noted earlier, adipose tissue represents the body’s largest endocrine organ. Indeed, adipose tissue is responsible for secretion of a large number of polypeptide factors, some of which are exclusively elaborated by adipocytes and are termed adipokines. Some of the major adipokines include leptin, adiponectin, and resistin.
Adiponectin is secreted by adipocytes and circulates as a large multimeric protein complex consisting of up to 18 monomers.151 Two receptors for adiponectin have been identified (Adipo-R1 and Adipo-R2) with somewhat different tissue expression patterns. Adipo-R1 is more highly expressed in liver, whereas Adipo-R2 is predominant in muscle. Through these receptors, adiponectin stimulates AMP kinase activity, leading to a variety of cellular effects that all serve to improve tissue insulin sensitivity. Thus, adiponectin is a bonafide insulin-sensitizing hormone and may play an important role in pathophysiologic states. For example, adiponectin levels are usually decreased in obesity and other insulin-resistant states, and circulating high-molecular-weight adiponectin levels are inversely correlated with the magnitude of insulin resistance. Furthermore, administration of adiponectin to animals leads to improvement in insulin sensitivity. Treatment of animals and insulin-resistant humans with insulin-sensitizing thiazolidinediones leads to an increase in adiponectin secretion from adipocytes, consistent with a role for increased adiponectin in the insulin-sensitizing effects of this class of therapeutics.
Resistin is another adipokine, and although it is much less well studied than adiponectin, previous reports suggest that resistin leads to a decrease in insulin sensitivity, and elevated levels of resistin may track with insulin-resistant states.152 Leptin is perhaps the best known of the adipokines, and it exerts its effects largely by reducing food intake and increasing thermogenesis.153 Obviously, in so far as leptin helps to control body weight, it has a profound effect on obesity-induced insulin resistance. Independent of its effects on obesity, some evidence indicates that leptin may directly improve insulin sensitivity, although this is less certain and requires further investigation. This is an interesting and evolving field, and a number of other adipokines have been identified that have effects on diverse physiologic systems.154–160
Impact of Regional Fat Distribution and Adipocyte Size: Adipose tissue is responsible for only a small fraction of overall whole-body glucose disposal. Although glucose uptake per kilogram of tissue is reduced in insulin-resistant states, overall glucose disposal in adipose tissue probably is not significantly altered, given the expanded fat mass in most insulin-resistant subjects.161
Intraperitoneal (visceral) adipose tissue may be particularly deleterious to glucose homeostasis. Because of its anatomic location, visceral fat drains directly to the liver via the portal vein, thereby exposing the liver to high concentrations of FFA from this depot. Furthermore, visceral adipocytes appear to be more responsive to catecholamine-stimulated lipolysis and less responsive to suppression of lipolysis by insulin.162,163 It has long been recognized that excess fat in the upper part of the body (central or abdominal), termed android obesity, is associated with increased risk for type 2 diabetes, dyslipidemia, and increased mortality compared with lower-body (gluteofemoral), or gynoid, obesity.164–166 Although the relationship between visceral fat and cardiovascular risk has been established, the association of insulin sensitivity with visceral versus subcutaneous truncal adipose tissue remains controversial. Visceral fat area, as determined by computed tomography scan, is correlated with decreased insulin action, as measured by the glucose clamp. On the other hand, with the use of similar techniques, the total volume of subcutaneous truncal adipose tissue was reported to be a better predictor of insulin resistance than was visceral fat.167 Because subcutaneous truncal adipose tissue contributes a greater quantity of overall FFAs to the systemic circulation than does visceral fat, it might have a more important influence on peripheral insulin action.
Subjects with deficiency of adipose tissue amount (lipoatrophy) or distribution (lipodystrophy) are also insulin resistant, with excess triglyceride deposition in skeletal muscle and the liver.168 In a transgenic animal model with absence of white adipose tissue, insulin resistance is associated with lipid deposition in skeletal muscle and liver169—a phenotype that can be reversed by surgical implantation of normal adipose tissue.170 These findings suggest that adipose tissue plays a pivotal role in buffering of fatty acid flux, with insufficient buffering leading to “ectopic triglyceride” storage in muscle and the liver, resulting in deleterious metabolic effects.171 The more common scenario, however, is of excess adipose tissue in obesity; in this situation, the antilipolytic effects of insulin are impaired. This could result in increased FFA flux into muscle and liver, contributing to the increased intramyocellular and hepatic triglyceride content and insulin resistance observed in this condition.172
Another aspect of lipid metabolism that influences insulin action is that of adipocyte size. Larger adipocytes are more resistant to insulin-stimulated glucose uptake and to insulin suppression of lipolysis,173,174 and larger subcutaneous adipocytes may predict the development of type 2 diabetes, independent of insulin resistance.175 Smaller fat cells may be more efficient at fatty acid uptake and better able to buffer lipid flux. Indeed, it has been hypothesized that failure of adipogenic precursor cells to differentiate into adipocytes results in glucose intolerance,176 which may be due to inefficient handling of lipid flux by the remaining large adipocytes.
Inflammatory Pathway Activation and Insulin Resistance
In recent years, the concept has emerged that chronic, low-grade activation of proinflammatory pathways within insulin target tissues is an important cause of obesity-related insulin resistance.177 Thus, for many years, it has been known that elevated levels of proinflammatory cytokines, such as tumor necrosis factor (TNF)α and interleukin (IL)-6, as well as C-reactive protein (CRP), exist in patients with type 2 diabetes mellitus and insulin resistance. Activation of intracellular inflammatory pathways within myocytes, hepatocytes, or adipocytes involves increased expression and/or activity of several serine kinases such as JNK1, IKKβ, and PKCθ, all of which can inhibit insulin action. Consistent with this, neutralization of TNFα improves insulin sensitivity in obese rodents, and genetic knockout or chemical inhibition of JNK1178 or IKKβ179 improves insulin sensitivity in various mouse models. Perhaps more important, salicylates are well-known anti-inflammatory compounds that inhibit the IKKβ/NFκB pathway, and treatment of insulin-resistant rodents and type 2 diabetic patients with these agents leads to improved insulin action with a corresponding reduction in hyperglycemia.180
The expanding adipose tissue mass in obesity and type 2 diabetes is a central focus for this chronic tissue inflammatory response. Activation of adipose tissue inflammatory pathways leads to a variety of adverse metabolic changes, including (1) increased lipolysis with elevated circulating FFA levels, (2) release of cytokines, which can impair insulin action, and (3) changes in the mix of adipokines (e.g., decreased adiponectin and increased resistin levels); all of these factors can impair insulin sensitivity.177
Chronic inflammation in the liver can be a cause of hepatic insulin resistance. Thus, in obese and insulin-resistant states, inflammatory pathways are activated in liver tissue, with increased JNK1, IKKβ, and PKCθ activity, and these serine kinases blunt insulin signaling in hepatocytes. Attenuation of insulin action results in increased levels of gluconeogenesis and glycogenolysis and elevated hepatic glucose production rates. In obese subjects, this situation often is accompanied by steatosis or steatohepatitis.181
It is likely that this chronic inflammatory process contributes to skeletal muscle insulin resistance through changes in circulating concentrations of adipokines, or cytokines released from adipose tissue and/or liver. In addition, adipose tissue is interspersed within skeletal muscle, and these intermuscular adipose depots are increased in obese patients.137 This intermuscular adipose tissue contains macrophages and other immune cells, and the numbers of these cells are increased in obesity.182 It is possible that cytokines released by these immune cells could have local paracrine effects that contribute to skeletal muscle insulin resistance.
Although low-grade, chronic tissue inflammation can cause insulin resistance in obese and type 2 diabetic subjects, how does this inflammatory process begin? One possibility is that nutrient overload of adipocytes and liver initiates stress pathways, which activate some of the serine kinases (JNK1, IKKβ) that inhibit insulin action.183,184 Another important possibility relates to tissue macrophages. In obesity, the content of tissue macrophages is increased in adipose tissue and liver (Kupffer cells). These immune cells are highly proinflammatory and release a variety of cytokines, which act in a paracrine fashion on neighboring insulin target cells to cause insulin resistance.185,186 In this scenario, the chronic obese and type 2 diabetic state leads to chronic accumulation of activated tissue macrophages, which, in turn, release inhibitory cytokines that act locally in a paracrine fashion, thus contributing to the overall insulin-resistant state.
Other Contributors to the Insulin-Resistant Phenotype
Insulin-Mediated Versus Non–Insulin-Mediated Glucose Uptake
Under basal conditions, a near steady state of glucose flux is approximated, and the rate of glucose appearance (HGO) equals the overall rate of glucose disposal. To understand the significance of increased basal HGO in type 2 diabetes, it is important to distinguish between insulin-dependent and insulin-independent processes of glucose disposal. By definition, insulin-mediated glucose uptake (IMGU) occurs in insulin target tissues under the influence of insulin. Non–insulin-mediated glucose uptake (NIMGU) consists of all glucose uptake that is not under the influence of insulin, and this has two components. NIMGU occurs in tissues (primarily the central nervous system) that are not targets for insulin action; it also involves insulin target cells and comprises the basal rate (non–insulin mediated) of glucose disposal by these tissues. Total glucose disposal (Rd) equals the sum of NIMGU and IMGU. NIMGU can be assessed in vivo by measuring Rd under conditions of severe insulinopenia induced by an infusion of somatostatin.187 Thus, after measurement of basal Rd (at basal or fasting insulin and glucose levels), somatostatin is administered to inhibit insulin secretion to negligible levels. Rd gradually falls to a new steady state that equals NIMGU because insulin action is absent under these conditions. With this approach, the proportion of basal Rd that is NIMGU is approximately two thirds in normal individuals at euglycemia and in type 2 diabetic subjects studied at their basal level of hyperglycemia.187 This means that at all levels of basal glycemia (normal and diabetes), most of the glucose is disposed of by NIMGU mechanisms, and the elevated rates of basal HGO that prevail in type 2 diabetes are associated with increased rates of NIMGU.
Pathophysiology of Fasting Versus Postprandial Hyperglycemia
Once type 2 diabetes develops, peripheral insulin resistance, impaired insulin secretion, and increased HGO all contribute to fasting hyperglycemia, but increased HGO predominates. This conclusion derives from the known physiology of glucose homeostasis in the basal state. Thus, in the postabsorptive fasting state, insulin levels are low, and approximately 70% of basal glucose uptake (Rd) is non–insulin mediated in both normal and hyperglycemic type 2 diabetic subjects.187 Because skeletal muscle accounts for only 15% to 20% of basal glucose Rd, it follows that an impairment in insulin-mediated glucose uptake by muscle will have little effect on overall basal glucose Rd or on the fasting plasma glucose level. Major increases in fasting glucose levels do not occur unless the rate of glucose entry into the systemic circulation (Ra) increases, and in the basal state, glucose Ra essentially equals HGO. In type 2 diabetes, increases in glucose Ra readily lead to increases in fasting glucose levels because, in the setting of peripheral insulin resistance and impaired insulin secretion, the ability of IMGU to rise and accommodate an increase in Ra is severely curtailed. To illustrate, if basal Ra = Rd = 2 mg/kg/min at euglycemia and basal NIMGU = 1.4 mg/kg/min (70%) with a basal IMGU of 0.6 mg/kg/min (30%), a modest increase in HGO to 2.6 mg/kg/min would require doubling of IMGU (to 1.2 mg/kg/min) to maintain Rd = Ra (HGO) at euglycemia. Because type 2 diabetic subjects are insensitive to insulin, a much larger increase in insulin secretion would be necessary to produce euglycemia than in normal individuals. Because insulin secretion is impaired in type 2 diabetes, the ability of a type 2 diabetic subject to increase IMGU in response to a rise in Ra is greatly restricted. Instead, to raise Rd to the level of the new Ra and bring the system back into balance, the fasting glucose level must rise until Rd increases by mass action and equals Ra. This line of reasoning is strongly supported by the available data, which demonstrate close direct relationships between fasting plasma glucose levels and basal HGO in large groups of type 2 diabetic subjects under a variety of conditions.7 Thus, decreased insulin secretion and action provide the setting that allows glucose Ra to regulate fasting plasma glucose, leading to the principle that fasting hyperglycemia is largely due to increased HGO.
Gastrointestinal Incretins and Intestinal Bypass Surgery
An incretin is a gastrointestinal hormone that increases insulin release from the β cell in response to a meal or an oral glucose load. The “incretin effect” was first suggested following the observation that an oral glucose load stimulates insulin secretion to a significantly higher degree than does intravenous glucose. In subjects with type 2 diabetes mellitus, this incretin effect is greatly diminished, suggesting that this may play a significant role in the pathogenesis of type 2 diabetes mellitus.188
Although many peptides produced in the stomach and/or small intestine have a role in the regulation of food intake, the two dominant gut-derived incretins are GIP and GLP-1. GIP is secreted from enteroendocrine cells (“K cells”), which have highest density in the duodenum but are found throughout the small intestinal mucosa.189 Secretion of GIP is stimulated by carbohydrates and by lipids, and a 10- to 20-fold elevation in plasma concentration is seen following a meal. The GIP receptor is a Gαs-coupled G protein coupled receptor (GPCR), and activation leads to enhanced insulin secretion. GLP-1 is derived from the polypeptide product of the glucagon gene through a process of differential proteolytic cleavage within intestinal “L cells.”190 GLP-1 secretion is stimulated by the presence of nutrients in the gut lumen (predominantly distal small intestine and colon), and its secretion is highly correlated with insulin release. Similar to GIP, it interacts with a Gαs-coupled GPCR on β cells and is a potent insulin secretagogue.
In subjects with type 2 diabetes mellitus, GIP levels appear to be unchanged, whereas a pronounced defect in GLP-1 secretion is noted.191 It is interesting to note that type 2 diabetic subjects have a pronounced resistance to exogenous GIP infusion but have a normal response to GLP-1 infusion.192,193 Novel incretin-based treatment agents have focused on reducing the metabolism of endogenous GLP-1 and GIP through inhibition of dipeptidyl peptidase 4 (DPP-4), or through agonism of the GLP-1 receptor.194
Intestinal Bypass Surgery
Gastric bypass surgery is used increasingly for the treatment of morbid obesity. Over the years, clinical observations have demonstrated that morbidly obese type 2 diabetic patients undergoing bypass surgery exhibit rapid (within days) resolution of the diabetic state even before significant weight loss occurs. This rapid resolution of hyperglycemia has been reported within days after several types of upper intestinal bypass surgery, including Roux-en-Y gastric bypass, duodenal-jejunal bypass, and ileal transposition. This rapid insulin-sensitizing effect is not seen with adjustable gastric banding or with caloric restriction alone, both of which typically have a more delayed onset of effect. Although some differences in technique exist, bypass surgeries have in common the anatomic displacement of the duodenum and the proximal jejunum. Based on this, two competing hypotheses have emerged to describe the mechanisms underlying glucose normalization in response to these surgeries.195 The “foregut hypothesis” states that intestinal bypass surgery removes an as yet unidentified diabetogenic factor that is present in the bypassed duodenal/jejunal segment. The “hindgut hypothesis” states that increased/rapid delivery of food to the distal jejunum and ileum (hindgut) directly stimulates secretion of an antidiabetic factor, possibly GLP-1.
Although limited, recent experimental data in rats seem to support the foregut hypothesis in this model. In the Goto-Kakazaki rat model of lean type 2 diabetes, duodenal-jejunal bypass surgery led to improved insulin sensitivity, independent of GLP-1 levels. Further, reoperation to reestablish duodenal continuity with the stomach led to return of diabetes, even in animals in which a second bypass conduit was created to simultaneously induce rapid nutrient passage to the distal small intestine.195 Conversely, in obese human subjects undergoing Roux-en-Y gastric bypass, GLP-1 levels were increased within 2 days postoperatively, and this elevation was correlated with reduced appetite.196
Syndromes of Severe Insulin Resistance
Some patients demonstrate a severe degree of resistance to endogenous or exogenous insulin. These patients may require extremely high treatment doses of insulin to normalize blood glucose levels and represent a clinically distinct group designated as having “severe insulin resistance.”197 Definitive laboratory values to designate the presence of severe insulin resistance have not been agreed upon, although fasting insulin levels above 50 to 70 µU/mL or peak (post–oral glucose tolerance test [OGTT]) insulin levels above 350 µU/mL may be acceptable diagnostic thresholds.198 In addition to insulin resistance, certain clinical features are often present with many of these syndromes, including acanthosis nigricans and, in women, symptoms of ovarian hyperandrogenism.
Lipoatrophic Diabetes
Loss of subcutaneous fat (lipoatrophy)199 can be associated with severe insulin resistance. The mechanism underlying insulin resistance in these conditions is multifactorial, related to ectopic deposition of triglycerides in peripheral tissues, increased circulating lipid metabolites such as free fatty acids, and decreased levels of adipokines (adipose-derived hormones) such as adiponectin and leptin. Although all lipoatrophy syndromes are associated with insulin resistance to a degree, the severity of metabolic disturbance generally is correlated with the extent of adipose tissue loss. In this regard, severe insulin resistance refractory to insulin therapy may be seen in syndromes of generalized lipoatrophy but are unusual with partial lipoatrophy.
The most common cause of lipoatrophy is that associated with antiretroviral therapy in patients with human immunodeficiency virus (HIV) infection. However, many other disease states may present with partial or generalized lipoatrophy, and these may in turn be acquired conditions or may have a genetic basis. The topics of lipoatrophy and lipodystrophy are covered comprehensively in Chapter 12.
Defects in Insulin Receptor Signaling
Patients with type B resistance have extreme levels of insulin resistance associated with the presence of anti-insulin receptor antibodies.200 First described in 1975, patient-derived serum and serum immunoglobulins may block insulin binding in a variety of tissues and subjects.201 As with other autoimmune disorders, this condition is seen more commonly in women, and among ethnic groups is most common in blacks. Mean age of onset is 40, but reported cases have ranged from ages 12 to 78.
The main clinical features include hyperglycemia, acanthosis nigricans, and clinical signs of autoimmunity. These may include leukopenia; increases in antinuclear antibody, erythrocyte sedimentation rate, and serum immunoglobulin (Ig)G; proteinuria; alopecia; arthritis; vitiligo; Raynaud’s phenomenon; and enlarged salivary glands. Insulin resistance may be extreme, with patients requiring >1000 U/day. However, some patients may present with postprandial hyperglycemia coupled with fasting hypoglycemia, or isolated fasting hypoglycemia.202 In cases with hypoglycemia, insulin levels may be elevated as the result of an antibody-induced reduction in insulin clearance, and this may be confused with insulinoma.203 Hypoglycemia may be due in part to antibody-mediated activation of insulin receptor (IR) signaling.
The clinical course can be variable, ranging from persistent hyperglycemia not amenable to insulin treatment to spontaneous remission months to years after disease onset. Patients have rarely progressed from severe insulin resistance to severe hypoglycemia over several weeks to months. Glucocorticoids and/or plasmapheresis may be useful therapy in severe cases.204
The dominant site for antibody binding is a limited region in the receptor α subunit between amino acid residues 450 and 601.205 Although initial studies demonstrated that insulin receptor antibodies blocked insulin binding to its receptor in a wide variety of tissues from several species, the biological activity of insulin receptor antibodies is complex. Virtually all antibodies isolated from hyperglycemic or hypoglycemic patients acutely stimulate insulin receptor activity, such as glucose uptake and metabolism.206 Over time, cells become refractory to these effects, and this may underlie the clinical heterogeneity of this syndrome.207
Type A Insulin Resistance
This clinical definition is applied to individuals with severe, apparently inherited insulin resistance, in the absence of growth defects or lipodystrophy. The most common features are the peripubertal onset of insulin resistance and acanthosis nigricans and, in women, symptoms of ovarian hyperandrogenism. Body habitus is varied but can be accompanied by prominent musculature, which may result from hyperandrogenism or from insulin-induced anabolism via the insulin-like growth factor (IGF)-1 receptor. Many of these patients harbor heterozygous mutations in the insulin receptor gene or have homozygous mutations that cause relatively mild defects in receptor function.208,209 Mutations may be associated with impaired binding of insulin to cells and tissues from affected patients,210,211 or with normal insulin binding but impaired receptor tyrosine kinase activity.212 The syndrome may display autosomal dominant or recessive inheritance. Other patients with clinical type A insulin resistance syndrome do not have mutations in the insulin receptor gene and may have as yet unidentified mutations in downstream intracellular signaling molecules.
Other Congenital Syndromes of Insulin Receptor Signaling
1. Leprechaunism (Donohue’s syndrome): First described in 1954 in two siblings with intrauterine and postnatal growth retardation, sparse subcutaneous fat, acanthosis nigricans, and early death.213 Patients have characteristic facies with large ears and micrognathia. Fasting hyperglycemia and marked hyperinsulinemia are always present, and survival past the first year is uncommon. The precise molecular defect in leprechaunism is not known; however, insulin binding to cells is markedly reduced,214,215 and inactivating mutations in the insulin receptor locus have been reported.216
2. Rabson-Mendenhall syndrome: A clinical syndrome that includes severe insulin resistance and acanthosis. Patients present with characteristic features of short stature, protruberant abdomen, abnormal dentition and nails, and pineal hyperplasia. Insulin binding is reduced, which may be associated with reduced receptor synthesis.217 This in turn is due to impaired cleavage of the insulin proreceptor, which is associated with a point mutation that affects the proteolytic cleavage site.218 The degree of insulin resistance is intermediate between that seen with leprechaunism and type A insulin resistance.
3. Pseudoacromegaly: Severe insulin resistance associated with pathologic tissue growth similar to that seen in acromegaly, but with normal IGF-1 and growth hormone (GH) levels. The initial report from Flier et al. in 1993219 described a 19-year-old patient with acromegaloid features, a history of accelerated linear growth, and a normal GH/IGF-1 axis. The patient was found to have severe fasting hyperinsulinemia and lacked a hypoglycemic response to an IV insulin bolus. Cultured skin fibroblasts had normal insulin-stimulated mitogenic responses but displayed a marked reduction in insulin-stimulated glucose uptake. No functional insulin receptor or GLUT4 mutations were identified. Additional studies suggest that the defect resides in the activation of phosphoinositide-3-kinase.220 The acromegaloid features seen in this syndrome are hypothesized to result from very high insulin levels coupled with the normal insulin anabolic and mitogenic signaling pathways. The genetic defect underlying this syndrome remains unknown. In a recent report, pericentric inversion of chromosome 11 segregated with acromegaloid features in one family, although candidate genes affected by this inversion remain to be determined.221
Defects Related to the Insulin Molecule
The seminal studies of Berson and Yalow222 demonstrated the presence of anti-insulin antibodies, which caused insulin resistance in a series of insulin-treated patients. As a complication of insulin therapy, insulin resistance due to anti-insulin antibodies is associated with a very high antibody titer223 and was prevalent with the widespread intermittent use of beef- and pork-derived insulin of limited purity, containing proinsulin, C-peptide, and other peptide contaminants. Anti-insulin antibodies may develop in persons treated with human insulin as well but are associated with clinically significant insulin resistance to therapy only in rare instances.224 Immune insulin resistance tends to be self-limited; 50% of cases last less than 6 months, and 75% less than 1 year. Switching insulin preparations is considered the first line of treatment. In refractory cases, prednisone (60 to 80 mg/day for 2 to 3 weeks) has resulted in lower insulin requirements and on occasion has been associated with dramatic responses, including hypoglycemia.225
Rare patients have been described who demonstrate antibodies in the absence of previous insulin therapy, a condition called the insulin autoimmune syndrome. This condition is associated with extremely high serum levels of endogenous insulin but normal serum levels of glucose and is usually seen in conjunction with other autoimmune diseases. The most frequent presentation is postprandial hypoglycemia.226
Increased Insulin Degradation
Some patients demonstrate severe resistance to insulin administered subcutaneously but normal responses to insulin delivered intravenously or intraperitoneally. To account for this finding, it has been proposed that these patients have increased insulin-degrading enzyme (insulinase) activity in subcutaneous tissues. A previous attempt to directly demonstrate increased skin insulinase activity, or to directly measure the presence of insulin degradation products in skin from these patients, was not successful.227
Mutant Insulins
Several families have been described in which one or more members have missense mutations in the insulin gene causing amino acid substitutions within the proinsulin molecule, leading to biologically defective insulin.228–235 All patients have marked fasting hyperinsulinemia or hyperproinsulinemia, but with normal glucose levels or with mild hyperglycemia. These patients respond normally to exogenous insulin and therefore are “insulin resistant” only to the endogenously produced hormone.
Molecular Genetics of Type 2 Diabetes Mellitus
Candidate Gene Approach
Most candidate gene studies have focused on genes that encode proteins in the pathways of pancreatic β cell insulin secretion, or insulin signal transduction. Many studies in this regard have been undertaken, and lack of reproducibility has limited their interpretation. Earlier studies in this area had problems with poor matching of cases and controls, testing of a limited number of markers per gene, and small study population size with consequently little power to detect genuine effects. As a result, few associations have been replicated in additional populations.236 Several studies examined the insulin receptor, glycogen synthase, and GLUT4 as potential candidate genes in type 2 diabetes. Except in rare individuals, the primary sequences of these genes are normal, eliminating them as diabetes gene loci. In some populations, IRS-1 variants may be two to three times more common in patients with type 2 diabetes than in normal subjects, and in some studies, an association has been found between polymorphisms affecting the region of IRS-1 important for PI3K binding and a reduction in insulin sensitivity,237 although not all studies have confirmed this association.238
Genome-Wide Association Studies
Because the candidate gene approach was not productive in identifying type 2 diabetes mellitus genes, it was largely supplanted by genome-wide association (GWA) studies involving linkage analysis.239 In a GWA study, DNA samples are obtained from two groups of participants: subjects with disease, and matched subjects without disease. The genomes then are scanned for markers of genetic variation such as SNPs. If specific genomic markers are more frequent in subjects with the disease phenotype, they are termed disease-associated. These markers then can be used to direct researchers to specific chromosomal loci, and positional cloning techniques are used to identify the specific gene. However, initial attempts at performing GWA studies to identify major susceptibility loci in type 2 diabetes were limited by the prohibitive technical (e.g., there are ≈10 million common SNPs) and financial hurdles involved in studying the large populations needed to attain sufficient power to detect the small effects of individual genes.
Recent advances have greatly increased the power of GWA studies to detect disease susceptibility alleles/haplotypes. Dense genotyping chips, containing sets of hundreds of thousands of SNPs that provide coverage of much of the human genome, are now available, so that for the first time, GWA studies of thousands of cases and controls are feasible. Most important has been the development of the International HapMap resource.240–242 The HapMap is based on the idea that the human genome can be organized into haplotype blocks, that is, sizable regions of DNA over which little evidence for historical recombination has been found (all SNPs found within a haplotype are inherited together as a group owing to their proximity to each other, rather than inherited randomly, as occurs with recombination). Within each haplotype block, a “tag SNP” has been identified that uniquely identifies that haplotype. The number of tag SNPs that contain most of the information about the patterns of genetic variation is estimated to be 300,000 to 600,000—far fewer than the 10 million common SNPs. Thus, these haplotype frameworks provide substantial statistical power in studies of common genetic variation, and the construction of a haplotype map of the human genome has greatly facilitated comprehensive genetic association studies of human disease.
The potential power of these molecular genetic tools to discover novel mechanisms underlying the development of type 2 diabetes has been demonstrated in several recent studies. The Wellcome Trust Case Control Consortium (WTCCC) recently completed a GWA scan in 1924 cases of type 2 diabetes mellitus and 2938 population controls from the United Kingdom, using the Affymetrix GeneChip Human Mapping 500 k Array Set (Affymetrix Research Services Lab, Santa Clare, CA).243 The strongest GWA signals were observed for SNPs in TCF7L2 (transcription factor 7-like 2), which acts within the WNT signaling pathway and may affect diabetes risk through pancreatic β cell dysfunction.244 Other association signals were reported within genes for KCNJ11 (encodes for the inwardly rectifying Kir6.2 component of the pancreatic β cell adenosine triphosphate [ATP]-dependent potassium channel), P12A PPARγ2, FTO (fat mass and obesity associated), CDKAL1 (CDK regulatory subunit associated protein 1-like 1), HHEX (homeobox, hematopoietically expressed), IDE (insulin-degrading enzyme), IGF2BP2 (insulin-like growth factor 2 binding protein 2), and CDKN2A/CDKN2B (cyclin-dependent kinase 2A/2B). Integration of data from similar studies performed by the Diabetes Genetics Initiative,245 the Finland–United States Investigation of NIDDM Genetics,246 and WTCCC/UKT2D247 is shown in Table 15-1.
Maturity-Onset Diabetes of the Young (MODY)
MODY refers to disorders due to monogenic defects in β cell function, with little or no defect in insulin action. They usually are transmitted as an autosomal dominant trait and are characterized by the onset of overt diabetes, usually before 25 years of age. The MODY phenotype is associated with abnormalities in at least six genetic loci on different chromosomes. The gene for MODY-1 was initially localized to the long arm of chromosome 20, close to the adenosine deaminase locus, by Bell and colleagues, on the basis of extensive family studies of the RW pedigree.248 The gene was subsequently identified as the transcription factor hepatocyte nuclear factor 4a (HNF-4a).249 MODY-2 is due to mutations in the glucokinase gene that catalyzes the formation of glucose-6-phosphate from glucose in islet β cells and hepatocytes. More than 40 different mutations have been described.250,251 The missense mutations that co-segregate with diabetes in families with MODY alter the affinity of the enzyme for glucose or the maximal activity for glucose phosphorylation. Because insulin secretion is intimately linked to the metabolism of glucose-6-phosphate, glucokinase is thought to play a key role as the “glucose sensor” of the β cell. Impaired hepatic glucose phosphorylation also may contribute to hyperglycemia in patients with MODY-2. It is interesting to note that most subjects with glucokinase mutations have very mild hyperglycemia, a normal first-phase insulin response to intravenous glucose, and a nonprogressive course. MODY-3 is the most common subtype. It is associated with mutations on chromosome 12 in a gene encoding HNF-1a, a liver transcription factor that also is expressed in β cells.252 In contrast to patients with MODY-2, hyperglycemia in patients with MODY-3 tends to be progressive. Because they frequently present in adolescence with symptomatic hyperglycemia, type 1 diabetes mellitus might be diagnosed. MODY-4 is due to mutations in yet another transcription factor gene, insulin promoter factor 1 (IPF-1), which in its homozygous form leads to total pancreatic agenesis.253 Additional mutations have more recently been described that involve HNF-1b254 and NeuroD1/BETA2.255 In “garden variety” type 2 diabetes, mutations in the glucokinase and hepatic nuclear transcription factor genes are rare.256 Point mutations of IPF-1, however, have been associated with increased risk for type 2 diabetes.257
Concluding Overview
With regard to the sequential development of type 2 diabetes, population-based and prospective studies indicate that insulin resistance is the initial defect, although in some populations, insulin secretory abnormalities may be primary.13,258 In a number of different ethnic groups, insulin resistance has been found to exist in the prediabetic state in the absence of any impairment in insulin secretion. Thus, insulin resistance and hyperinsulinemia characterize most individuals in whom type 2 diabetes is destined to develop. However, except in extreme cases, insulin resistance alone is not sufficient to cause the full-blown type 2 diabetic phenotype. Decreased β cell function eventually must supervene to cause the full-blown syndrome. This setting leads to the scheme depicted in Fig. 15-2, in which insulin resistance (genetic and/or acquired) leads to hyperinsulinemia and compensated glucose metabolism. Eventually, in some of these individuals, perhaps those with a coexisting genetically determined β cell defect, the ability of β cells to compensate by sustaining hyperinsulinemia declines, and insulin secretory defects appear, along with decreased β cell mass. At this stage, glucose metabolism decompensates, and the hyperglycemic diabetic state emerges. This scheme (see Fig. 15-2) describes most patients, but because type 2 diabetes is a heterogeneous disease, this scenario may not apply to all.
Once type 2 diabetes has been established, the abnormal metabolic and hormonal milieu leads to secondary changes with worsening hyperglycemia. Thus, insulin resistance tends to be more marked in type 2 diabetes than in the prediabetic IGT state,3,5,36 and insulin secretion continues to decline throughout the course of established type 2 diabetes.50 Therefore, many patients, even if they initially are well controlled by diet or oral hypoglycemic agents, eventually require insulin for control of their diabetes. Glucotoxicity may well be the causal link here. Thus, strong evidence indicates that chronic hyperglycemia per se can lead to secondary worsening of both insulin resistance and insulin secretion, with the potential for a vicious cycle in which hyperglycemia begets more hyperglycemia. Support for this concept comes from numerous observations showing that control of hyperglycemia by insulin therapy, weight loss, or oral agents leads to improvements in both insulin secretion and insulin action. Obesity and aging, as well as other factors, are acquired conditions that can cause or contribute to insulin resistance and β cell dysfunction.
References
1. Ward, W, Beard, J, Porte, D. Clinical aspects of islet β-cell function in non-insulin-dependent diabetes mellitus. Diabetes Metab Rev. 1986;2:297–313.
2. DeFronzo, R. The triumvirate: β-cell, muscle, liver: a collusion responsible for NIDDM. Diabetes. 1988;37:667–687.
3. Seely, B, Olefsky, J. Cellular and genetic mechanisms for insulin resistance in common disorders of obesity and diabetes. In: Moller D, ed. Insulin Resistance and Its Clinical Disorders. New York: John Wiley and Sons, 1993.
4. Reaven, G. Role of insulin resistance in human disease. Diabetes. 1988;7:1595–1607.
5. Kolterman, O, Gray, R, Griffin, J, et al. Receptor and post-receptor defects contribute to the insulin resistance in non-insulin dependent diabetes mellitus. J Clin Invest. 1981;68:957–969.
6. Dineen, S, Gerich, J, Rizza, R. Carbohydrate metabolism in non-insulin-dependent diabetes mellitus. N Engl J Med. 1992;327:707–713.
7. Ferrannini, E, Groop, L. Hepatic glucose production in insulin resistant states. Diabetes Metab Rev. 1989;5:711–725.
8. Bogardus, C, Lillioja, S, Howard, B, et al. Relationships between insulin secretion, insulin action, and fasting plasma glucose concentration in non-diabetic and non-insulin-dependent diabetic subjects. J Clin Invest. 1984;74:1238–1246.
9. Shulman, G, Rothman, D, Jue, T, et al. Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. N Engl J Med. 1990;322:223–228.
10. Ciaraldi, T, Kolterman, O, Scarlett, J, et al. Role of glucose transport in the postreceptor defect of non-insulin-dependent diabetes mellitus. Diabetes. 1982;31:1016–1022.
11. Firth, R, Bell, P, Rizza, R. Insulin action in non-insulin-dependent diabetes mellitus: the relationship between hepatic and extrahepatic insulin resistance and obesity. Metabolism. 1987;36:1091–1095.
12. Groop, L, Bonadonna, R, DelPrato, S, et al. Glucose and free fatty acid metabolism in non-insulin-dependent diabetes mellitus: evidence for multiple sites of insulin resistance. J Clin Invest. 1989;84:205–213.
13. Efendic, S, Luft, R, Wajngot, A. Aspects of the pathogenesis of type 2 diabetes. Endocr Rev. 1984;5:395–410.
14. Porte, D. Banting lecture 1990: β-cells in type II diabetes mellitus. Diabetes. 1991;40:166–190.
15. Barnett, A, Eff, C, Leslie, R, et al. Diabetes in identical twins: a study of 200 pairs. Diabetologia. 1981;20:87–93.
16. Rich, S. Mapping genes in diabetes: genetic epidemiological perspective. Diabetes. 1990;39:1315–1319.
17. Haffner, S, Sern, M, Mitchell, B, et al. Incidences of type II diabetes in Mexican Americans predicted by fasting insulin and glucose levels, obesity, and body fat distribution. Diabetes. 1990;39:283–288.
18. Knowler, W, Bennett, P, Pettitt, D, et al. Diabetes incidence in Pima Indians: contributions of obesity and parental diabetes. Am J Epidemiol. 1981;113:144–156.
19. Toyota, T, Kudo, M, Goto, Y, et al. Prevalence of diabetes mellitus in rural and urban population of Japan. In: Baba S, Goto Y, Fukui I, eds. Diabetes Mellitus in Asia: Ecological Aspects of Epidemiology, Complications and Treatment. Amsterdam: Excerpta Medica; 1976:35–40.
20. Fujimoto, W, Leonetti, D, Kinyoun, J, et al. Prevalence of complications among second-generation Japanese-American men with diabetes, impaired glucose tolerance, or normal glucose tolerance. Diabetes. 1987;36:730–739.
21. Ravussin, E, Valencia, E, Esparza, J, et al. Effects of a traditional lifestyle on obesity in Pima Indians. Diabetes Care. 1994;17:1067–1074.
22. Lands, W, Hamazaki, T, Yamazaki, K, et al. Changing dietary patterns. Am J Clin Nutr. 1990;51:991–993.
23. Lee, MM, Wu-Williams, A, Whittemore, AS, et al. Comparison of dietary habits, physical activity and body size among Chinese in North America and China. Int J Epidemiol. 1994;23:984–990.
24. Diabetes Prevention Program Research Group. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403.
25. Helmrich, S, Ragland, D, Leung, R, et al. Physical activity and reduced occurrence of non-insulin dependent diabetes mellitus. N Engl J Med. 1991;325:147–152.
26. Hales, C, Barker, D, Clark, P, et al. Fetal and infant growth and impaired glucose tolerance at age 64. BMJ. 1991;303:1019–1022.
27. Carlsson, S, Persson, P, Alvarsson, M, et al. Low birth weight, family history of diabetes, and glucose intolerance in Swedish middle-aged men. Diabetes Care. 1999;22:1043–1047.
28. Xiao, X, Zhang, Z-X, Cohen, HJ, et al. Evidence of a relationship between infant birth weight and later diabetes and impaired glucose regulation in a Chinese population. Diabetes Care. 2008;31:483–487.
29. Barker, D. The developmental origins of insulin resistance. Horm Res. 2005;64(suppl 3):2–7.
30. Chen, M, Bergman, R, Pacini, G, et al. Pathogenesis of age-related glucose intolerance in man: insulin resistance and decreased β-cell function. J Clin Endocrinol Metab. 1985;60:13–20.
31. Orchard, TJ, Temprosa, M, Goldberg, R, et al. The effect of metformin and intensive lifestyle intervention on the metabolic syndrome: the Diabetes Prevention Program randomized trial. Ann Intern Med. 2005;142:611–619.
32. Warram, JH, Martin, BC, Krolewski, AS, et al. Slow glucose removal rate and hyperinsulinemia precede the development of type II diabetes in the offspring of diabetic parents. Ann Intern Med. 1990;13:909–915.
33. Lillioja, S, Mott, D, Howard, B, et al. Impaired glucose tolerance as a disorder of insulin action: longitudinal and cross-sectional studies in Pima Indians. N Engl J Med. 1988;318:1217–1225.
34. Saad, M, Knowler, W, Pettitt, D, et al. The natural history of impaired glucose tolerance in the Pima Indians. N Engl J Med. 1988;319:1500–1505.
35. Serjeantson, S, Zimmet, P. Genetics of non-insulin dependent diabetes mellitus in 1990. Baillieres Clin Endocrinol Metab. 1991;5:477–493.
36. Eriksson, J, Franssila-Kallunki, A, Ekstrand, A, et al. Early metabolic defects in persons at increased risk for non-insulin-dependent diabetes mellitus. N Engl J Med. 1989;321:337–343.
37. Vaag, A, Henriksen, J, Beck-Nielsen, H. Decreased insulin activation of glycogen synthase in skeletal muscles in young nonobese Caucasian first-degree relatives of patients with non-insulin-dependent diabetes mellitus. J Clin Invest. 1992;89:782–788.
38. Perseghin, G, Ghosh, S, Gerow, K, et al. Metabolic defects in lean nondiabetic offspring of NIDDM parents: a cross-sectional study. Diabetes. 1997;46:1010–1016.
39. Kahn, S, Prigeon, R, McCulloch, D, et al. Quantification of the relationship between insulin sensitivity and β-cell function in human subjects: evidence for a hyperbolic function. Diabetes. 1993;46:1663–1672.
40. Lang, D, Matthews, D, Peto, J, et al. Cyclic oscillations of basal plasma glucose and insulin concentrations in human beings. N Engl J Med. 1979;301:1023–1027.
41. O’Rahilly, S, Turner, R, Matthews, D. Impaired pulsatile secretion of insulin in relatives of patients with non-insulin-dependent diabetes. N Engl J Med. 1988;318:1225–1230.
42. Hunter, S, Atkinson, A, Ennis, C, et al. Association between insulin secretory pulse frequency and peripheral insulin action in NIDDM and normal subjects. Diabetes. 1996;45:683–686.
43. Fajans, S, Cloutier, M, Crowther, R. Clinical and etiologic heterogeneity of idiopathic diabetes mellitus. Diabetes. 1978;27:1112–1125.
44. Reaven, G, Bernstein, R, Davis, B, et al. Nonketotic diabetes mellitus: insulin deficiency or insulin resistance? Am J Med. 1976;60:80–88.
45. Alberti, K. The clinical implications of impaired glucose tolerance. Diabet Med. 1996;13:927–937.
46. Cavaghan, M, Ehrmann, D, Byrne, M, et al. Treatment with the oral antidiabetic agent troglitazone improves beta cell responses to glucose in subjects with impaired glucose tolerance. J Clin Invest. 1997;100:530–537.
47. Polonsky, K. The beta-cell in diabetes: from molecular genetics to clinical research. Diabetes. 1995;44:705–717.
48. Cook, J, Page, R, Levy, J, et al. Hyperglycaemic progression in subjects with impaired glucose tolerance: association with decline in beta cell function. Diabet Med. 1993;10:321–326.
49. van Haeften, TW, Pimenta, W, Mitrakou, A, et al. Relative contributions of [beta]-cell function and tissue insulin sensitivity to fasting and postglucose-load glycemia. Metabolism. 2000;49:1318–1325.
50. U.K. Prospective Diabetes Study Group. U.K. prospective diabetes study 16: Overview of 6 years’ therapy of type II diabetes: a progressive disease. Diabetes. 1995;44:1249–1258.
51. Halter, J, Graf, R, Porte, DJ. Potentiation of insulin secretory responses by plasma glucose levels in man: evidence that hyperglycemia in diabetes compensates for impaired glucose potentiation. J Clin Endocrinol Metab. 1979;48:946–954.
52. Ward, W, Bolgiano, D, McKnight, B, et al. Diminished β cell secretory capacity in patients with noninsulin-dependent diabetes mellitus. J Clin Invest. 1984;74:1318–1328.
53. Reaven, G, Chen, Y, Hollenbeck, C, et al. Plasma insulin, C-peptide, and proinsulin concentrations in obese and nonobese individuals with varying degrees of glucose tolerance. J Clin Endocrinol Metab. 1993;76:44–48.
54. Brunzell, J, Robertson, R, Lerner, R, et al. Relationships between fasting plasma glucose levels and insulin secretion during intravenous glucose tolerance tests. J Clin Endocrinol Metab. 1976;42:222–229.
55. Ward, W, Beard, J, Halter, J, et al. Pathophysiology of insulin secretion in non-insulin-dependent diabetes mellitus. Diabetes Care. 1984;7:491–502.
56. Robertson, R, Porte, D. The glucose receptor: a defective mechanism in diabetes mellitus distinct from the beta-adrenergic receptor. J Clin Invest. 1976;52:870–876.
57. Srikanta, S, Ganda, O, Eisenbarth, G, et al. Islet-cell antibodies and beta-cell function in monozygotic triplets and twins initially discordant for type I diabetes mellitus. N Engl J Med. 1983;308:322–325.
58. Robertson, R, Halter, J, Porte, DJ. A role for alpha-adrenergic receptors in abnormal insulin secretion in diabetes mellitus. J Clin Invest. 1976;57:791–795.
59. Reaven, G, Olefsky, J. Relationship between heterogeneity of insulin responses and insulin resistance in normal subjects. Diabetologia. 1977;13:201–206.
60. Temple, R, Clark, P, Nagi, D, et al. Radioimmunoassay may overestimate insulin in non-insulin-dependent diabetics. Clin Endocrinol (Oxf). 1990;32:689–693.
61. Clark, A, Wells, C, Buley, I, et al. Islet amyloid, increased A-cells, reduced β-cells and exocrine fibrosis: quantitative changes in the pancreas in type 2 diabetes. Diabetes Res. 1988;9:151–160.
62. Butler, AE, Janson, J, Bonner-Weir, S, et al. β-Cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110.
63. Ogilvie, R. The islets of Langerhans in 19 cases of obesity. J Pathol. 1933;37:473–481.
64. Shimabukuro, M, Zhou, Y-T, Levi, M, et al. Fatty acid induced β cell apoptosis: a link between obesity and diabetes. Proc Natl Acad Sci U S A. 1998;95:2498–2502.
65. Kahn, S, D’Alessio, D, Schwartz, M, et al. Evidence of co-secretion of islet amyloid polypeptide and insulin by beta-cells. Diabetes. 1990;39:634–638.
66. Kahn, S, Andrikopoulos, S, Verchere, C. Islet amyloid: a long-recognized but underappreciated pathological feature of type 2 diabetes. Diabetes. 1999;48:241–253.
67. Lorenzo, A, Razzaboni, B, Weir, G, et al. Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature. 1994;368:756–760.
68. Kolterman, O, Gray, R, Shapiro, G, et al. The acute and chronic effects of sulfonylurea therapy in type II diabetics. Diabetes. 1984;33:346–354.
69. Garvey, W, Olefsky, J, Griffin, J, et al. The effect of insulin treatment on insulin secretion and insulin action in type II diabetes mellitus. Diabetes. 1985;34:222–234.
70. Henry, R, Wallace, P, Olefsky, J. The effects of weight loss on the mechanisms of hyperglycemia in obese noninsulin-dependent diabetes mellitus. Diabetes. 1986;35:990–998.
71. Leahy, J, Bonner-Weir, S, Weir, G. β-Cell dysfunction induced by chronic hyperglycemia. Diabetes Care. 1992;15:442–455.
72. Eizirik, D, Korbutt, G, Hellerstrom, C. Prolonged exposure of human pancreatic islets to high glucose concentrations in vitro impairs the β-cell function. J Clin Invest. 1992;90:1263–1268.
73. Kulkarni, R, Bruning, J, Winnay, J, et al. Tissue-specific knockout of the insulin receptor in pancreatic β cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell. 1999;96:329–339.
74. Randle, P, Hales, C, Garland, P, et al. The glucose fatty-acid cycle: its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1:785–789.
75. Yang, Y, Hope, J, Ader, M, et al. Insulin transport across capillaries is rate limiting for insulin action in dogs. J Clin Invest. 1989;84:1620–1628.
76. Steil, G, Ader, M, Moore, D, et al. Transendothelial insulin transport is not saturable in vivo: no evidence for a receptor-mediated process. J Clin Invest. 1996;97:1497–1503.
77. Prager, R, Wallace, P, Olefsky, J. In vivo kinetics of insulin action on peripheral glucose disposal and hepatic glucose output in normal and obese subjects. J Clin Invest. 1986;78:472–481.
78. Nolan, J, Ludvik, B, Baloga, J, et al. Mechanisms of the kinetic defect in insulin action in obesity and NIDDM. Diabetes. 1997;46:994–1000.
79. King, G, Johnson, S. Receptor-mediated transport of insulin across endothelial cells. Science. 1985;227:1583–1586.
80. Castillo, C, Bogardus, C, Bergman, R, et al. Interstitial insulin concentrations determine glucose uptake rates but not insulin resistance in lean and obese men. J Clin Invest. 1994;93:10–16.
81. Cline, GW, Petersen, KF, Krssak, M, et al. Impaired glucose transport as a cause of decreased insulin-stimulated muscle glycogen synthesis in type 2 diabetes. N Engl J Med. 1999;341:240–246.
82. Lillioja, S, Young, A, Culter, C, et al. Skeletal muscle capillary density and fiber type are possible determinants of in vivo insulin resistance in man. J Clin Invest. 1987;80:415–424.
83. Laakso, M, Edelman, S, Brechtel, G, et al. Impaired insulin-mediated skeletal muscle blood flow in patients with NIDDM. Diabetes. 1992;41:1076–1083.
84. Natali, A, Buzzugoli, G, Taddei, S, et al. Effects of insulin on hemodynamics and metabolism in human forearm. Diabetes. 1990;39:490–500.
85. Utriainen, T, Nuutila, P, Takala, T, et al. Intact insulin stimulation of skeletal muscle blood flow, its heterogeneity and redistribution, but not of glucose uptake in non-insulin-dependent diabetes mellitus. J Clin Invest. 1997;100:777–785.
86. Kahn, C, White, M. The insulin receptor and the molecular mechanisms of insulin action. J Clin Invest. 1988;82:1151–1156.
87. Olefsky, J. The insulin receptor: a multi-functional protein. Diabetes. 1990;39:1009–1016.
88. Wilden, PA, Siddle, K, Haring, E, et al. The role of insulin receptor kinase domain autophosphorylation in receptor-mediated activities: analysis with insulin and anti-receptor antibodies. J Biol Chem. 1992;267:13719–13727.
89. White, M. The insulin signalling system and the IRS proteins. Diabetologia. 1997;40(suppl):2–17.
90. Virkamaki, A, Ueki, K, Kahn, C. Protein-protein interaction in insulin signaling and the molecular mechanisms of insulin resistance. J Clin Invest. 1999;103:931–943.
91. Fink, R, Wallace, P, Brechtel, G, et al. Evidence that glucose transport is rate-limiting for in vivo glucose uptake. Metabolism. 1992;41:897–902.
92. Furler, S, Jenkins, A, Storlien, L, et al. In vivo location of the rate-limiting step of hexose uptake in muscle and brain tissue of rats. Am J Physiol. 1991;261:E337–E347.
93. Ren, JM, Marshall, BA, Gulve, EA, et al. Evidence from transgenic mice that glucose transport is rate-limiting for glycogen deposition and glycolysis in skeletal muscle. J Biol Chem. 1993;268:16113–16115.
94. Pessin, J, Bell, G. Mammalian facilitative glucose transporter family: structure and molecular regulation. Annu Rev Physiol. 1992;84:911–930.
95. Shepherd, P, Kahn, B. Glucose transporters and insulin action. N Engl J Med. 1999;341:248–257.
96. Karnieli, E, Zarnowski, MJ, Hissin, PJ, et al. Insulin-stimulated translocation of glucose transport systems in the isolated rat adipose cell: time course, reversal, insulin concentration dependency, and relationship to glucose transport activity. J Biol Chem. 1981;256:4772–4777.
97. Kono, T, Suzuki, K, Dansey, LE, et al. Energy-dependent and protein synthesis-independent recycling of the insulin-sensitive glucose transport mechanism in fat cells. J Biol Chem. 1981;256:6400–6407.
98. Kelley, D, Mintun, M, Watkins, S, et al. The effect of non-insulin-dependent diabetes mellitus and obesity on glucose transport and phosphorylation in skeletal muscle. J Clin Invest. 1996;97:2705–2713.
99. Garvey, W, Maianu, L, Zhu, J-H, et al. Evidence for defects in the trafficking and translocation of Glut 4 glucose transporters in skeletal muscle as a cause of human insulin resistance. J Clin Invest. 1998;101:2377–2386.
100. Pessin, JE, Thurmond, DC, Elmendorf, JS, et al. Molecular basis of insulin-stimulated GLUT4 vesicle trafficking. LOCATION! LOCATION! LOCATION!. J Biol Chem. 1999;274:2593–2596.
101. Olefsky, J, Ciaraldi, T. The insulin receptor: basic characteristics and its role in insulin resistant state. In: Brownlee M, ed. Diabetes Mellitus. New York: Garland Press; 1980:73–115.
102. Bergman, R, Finegood, D. Assessment of insulin sensitivity in vivo. Endocr Rev. 1985;1:45–86.
103. Himsworth, H, Kerr, R. Insulin-sensitive and insulin-insensitive types of diabetes mellitus. Clin Sci. 1939;4:119–152.
104. Jones, CNO, Pei, D, Staris, P, et al. Alterations in the glucose-stimulated insulin secretory dose-response curve and in insulin clearance in nondiabetic insulin-resistant individuals. J Clin Endocrinol Metab. 1997;82:1834–1838.
105. Ginsberg, H, Kimmerling, G, Olefsky, J, et al. Demonstration of insulin resistance in maturity onset diabetic patients with fasting hyperglycemia. J Clin Invest. 1975;55:454–460.
106. Welch, S, Gebhart, S, Bergman, R, et al. Minimal model analysis of intravenous glucose tolerance test-derived insulin sensitivity in diabetic subjects. J Clin Endocrinol Metab. 1990;71:1508–1518.
107. Bonadonna, R, Del Prato, S, Saccomani, M, et al. Transmembrane glucose transport in skeletal muscle of patients with non-insulin-dependent diabetes. J Clin Invest. 1993;92:486–494.
108. Capaldo, B, Napoli, R, Di Marino, L, et al. Quantitation of forearm glucose and free fatty acid (FFA) disposal in normal subjects and type II diabetic patients: evidence against an essential role for FFA in the pathogenesis of insulin resistance. J Clin Endocrinol Metab. 1988;67:893–898.
109. DeFronzo, R, Jacot, E, Jequier, E, et al. The effect of insulin on the disposal of intravenous glucose: results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes. 1981;30:1000–1007.
110. Edelman, S, Laakso, M, Wallace, P, et al. Kinetics of insulin mediated and non-insulin mediated glucose uptake in man. Diabetes. 1990;39:955–964.
111. Kelley, D, Mokan, M, Mandarino, L. Intracellular defects in glucose metabolism in obese patients with NIDDM. Diabetes. 1992;41:698–706.
112. Kono, T, Barham, F. The relationship between the insulin-binding capacity of fat cells and the cellular response to insulin: studies with intact and trypsin-treated fat cells. J Biol Chem. 1971;246:6210–6216.
113. Kolterman, O, Scarlett, J, Olefsky, J. Insulin resistance in non-insulin-dependent, type II diabetes mellitus. J Clin Endocrinol Metab. 1982;11:363–388.
114. Olefsky, J, Reaven, G. Decreased insulin binding to lymphocytes from diabetic patients. J Clin Invest. 1974;54:1323–1328.
115. Olefsky, J, Reaven, G. Insulin binding in diabetes: relationships with plasma insulin levels and insulin sensitivity. Diabetes. 1977;26:680–688.
116. Freidenberg, G, Reichart, D, Olefsky, J, et al. Reversibility of defective adipocyte insulin receptor kinase activity in non-insulin dependent diabetes mellitus: effect of weight loss. J Clin Invest. 1990;82:1398–1406.
117. Maegawa, H, Shigeta, Y, Egawa, K, et al. Impaired autophosphorylation of insulin receptors from abdominal skeletal muscles in nonobese subjects with NIDDM. Diabetes. 1991;40:815–819.
118. Bjornholm, M, Kawano, Y, Lehtihet, M, et al. Insulin receptor substrate-1 phosphorylation and phosphatidylinositol 3-kinase activity in skeletal muscle from NIDDM subjects after in vivo insulin stimulation. Diabetes. 1997;46:524–527.
119. Paz, K, Hemi, R, LeRoith, D, et al. A molecular basis for insulin resistance: elevated serine/threonine phosphorylation of IRS-1 and IRS-2 inhibits their binding to the juxtamembrane region of the insulin receptor and impairs their ability to undergo insulin-induced tyrosine phosphorylation. J Biol Chem. 1997;272:29911–29918.
120. Pederson, TM, Kramer, DL, Rondinone, CM. Serine/threonine phosphorylation of IRS-1 triggers its degradation: possible regulation by tyrosine phosphorylation. Diabetes. 2001;50:24–31.
121. Aguirre, V, Uchida, T, Yenush, L, et al. The c-Jun NH2-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser307. J Biol Chem. 2000;275:9047–9054.
122. Gao, Z, Hwang, D, Bataille, F, et al. Serine phosphorylation of insulin receptor substrate 1 by inhibitor kappa B kinase complex. J Biol Chem. 2002;277:48115–48121.
123. Withers, DJ, Gutierrez, JS, Towery, H, et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998;391:900–904.
124. Shepherd, P, Withers, D, Siddle, K. Phosphoinositide 3-kinase: the key switch mechanism in insulin signaling. Biochem J. 1998;333:471–490.
125. Goodyear, L, Giorgino, F, Sherman, L, et al. Insulin receptor phosphorylation, insulin receptor substrate-1 phosphorylation, and phosphatidylinositol 3-kinase activity are decreased in intact skeletal muscle strips from obese subjects. J Clin Invest. 1995;95:2195–2204.
126. Zierath, J, He, L, Guma, A, et al. Insulin action on glucose transport and plasma membrane Glut 4 content in skeletal muscle from patients with NIDDM. Diabetologia. 1996;39:1180–1189.
127. Eriksson, J, Koranyi, L, Bourey, R, et al. Insulin resistance in type 2 (non-insulin-dependent) diabetic patients and their relatives is not associated with a defect in the expression of the insulin-responsive glucose transporter (GLUT-4) gene in human skeletal muscle. Diabetologia. 1992;35:143–147.
128. Garvey, W, Maianu, L, Hancock, J, et al. Gene expression of GLUT4 in skeletal muscle from insulin-resistant patients with obesity, IGT, GDM, and NIDDM. Diabetes. 1992;41:465–475.
129. Yki-Jarvinen, H, Bogardus, C, Howard, B. Hyperglycemia stimulates carbohydrate oxidation in humans. Am J Physiol. 1987;253:E376–E382.
130. Golay, A, DeFronzo, R, Ferrannini, E, et al. Oxidative and non-oxidative glucose metabolism in non-obese type 2 (non-insulin dependent) diabetic patients. Diabetologia. 1988;31:585–591.
131. Del Prato, S, Bonadonna, R, Bonora, E, et al. Characterization of cellular defects of insulin action in type 2 (non-insulin-dependent) diabetes mellitus. J Clin Invest. 1993;91:484–494.
132. Rothman, D, Shulman, R, Shulman, G. 31P nuclear magnetic resonance measurements of muscle glucose-6-phosphate. J Clin Invest. 1992;89:1069–1075.
133. Oakes, N, Cooney, G, Camilleri, S, et al. Mechanisms of liver and muscle insulin resistance induced by chronic high-fat feeding. Diabetes. 1997;46:1768–1774.
134. Schmitz-Peiffer, C, Browne, C, Oakes, N, et al. Alterations in the expression and cellular localization of protein kinase C isozymes epsilon and theta are associated with insulin resistance in skeletal muscle of the high-fat-fed rat. Diabetes. 1997;46:169–178.
135. Anderwald, C, Bernroider, E, Krssak, M, et al. Effects of insulin treatment in type 2 diabetic patients on intracellular lipid content in liver and skeletal muscle. Diabetes. 2002;51:3025–3032.
136. Kraegen, E, Clark, P, Jenkins, A, et al. Development of muscle insulin resistance after liver insulin resistance in high-fat-fed rats. Diabetes. 1991;40:1397–1403.
137. Pan, D, Lillioja, S, Kriketos, A, et al. Skeletal muscle triglyceride levels are inversely related to insulin action. Diabetes. 1997;46:983–988.
138. Ellis, BA, Poynten, A, Lowy, AJ, et al. Long-chain acyl-CoA esters as indicators of lipid metabolism and insulin sensitivity in rat and human muscle. Am J Physiol Endocrinol Metab. 2000;279:E554–E560.
139. Prager, R, Wallace, P, Olefsky, J. Hyperinsulinemia does not compensate for peripheral insulin resistance in obesity. Diabetes. 1987;36:327–334.
140. Kolterman, O, Insel, L, Saekow, M, et al. Mechanisms of insulin resistance in human obesity: evidence for receptor and post-receptor defects. J Clin Invest. 1980;65:1272–1284.
141. Rothman, D, Magnusson, I, Katz, L, et al. Quantitation of hepatic glycogenolysis and gluconeogenesis in fasting humans with 13C NMR. Science. 1991;254:573–576.
142. Consoli, A, Nurjhan, N, Reilly, J, et al. Mechanism of increased gluconeogenesis in non-insulin-dependent diabetes mellitus: role of alterations in systemic, hepatic, and muscle lactate and alanine metabolism. J Clin Invest. 1990;86:2038–2045.
143. Magnusson, I, Rothman, D, Katz, L, et al. Increased rate of gluconeogenesis in type II diabetes mellitus: a 13C nuclear magnetic resonance study. J Clin Invest. 1992;90:1323–1327.
144. Baron, A, Schmeiser, L, Shragg, G, et al. The role of hyperglucagonemia in the maintenance of increased rates of hepatic glucose output in type II diabetics. Diabetes. 1987;36:274–283.
145. Rebrin, K, Steil, G, Getty, L, et al. Free fatty acid as a link in the regulation of hepatic glucose output by peripheral insulin. Diabetes. 1995;44:1038–1045.
146. Felig, P, Wahren, J, Hendler, R. Influence of oral glucose ingestion on splanchnic glucose and gluconeogenic substrate metabolism in man. Diabetes. 1975;24:468–475.
147. Ferrannini, E, Bjorkman, O, Reichard, G. The disposal of an oral glucose load in healthy subjects: a quantitative study. Diabetes. 1985;34:580–588.
148. Niewoehner, C, Nuttall, F. Relationship of hepatic glucose uptake to intrahepatic glucose concentration in fasted rats after glucose load. Diabetes. 1988;37:1559–1566.
149. Ludvik, B, Nolan, J, Roberts, A, et al. Evidence for decreased splanchnic glucose uptake after oral glucose administration in non-insulin-dependent diabetes mellitus. J Clin Invest. 1997;100:2354–2361.
150. Basu, A, Basu, R, Shah, P, et al. Type 2 diabetes impairs splanchnic uptake of glucose but does not alter intestinal glucose absorption during enteral glucose feeding: additional evidence for a defect in hepatic glucokinase activity. Diabetes. 2001;50:1351–1362.
151. Scherer, PE, Williams, S, Fogliano, M, et al. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem. 1995;270:26746–26749.
152. Steppan, C, Bailey, S, Bhat, S, et al. The hormone resistin links obesity to diabetes. Nature. 2001;409:307–312.
153. Zhang, Y, Proenca, R, Maffei, M, et al. Positional cloning of the mouse obese gene and its human orthologue. Nature. 1994;372:425–432.
154. Boucher, J, Masri, B, Daviaud, D, et al. Apelin, a newly identified adipokine up-regulated by insulin and obesity. Endocrinology. 2005;146:1764–1771.
155. Yang, Q, Graham, TE, Mody, N, et al. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature. 2005;436:356–362.
156. Berndt, J, Kloting, N, Kralisch, S, et al. Plasma visfatin concentrations and fat depot-specific mRNA expression in humans. Diabetes. 2005;54:2911–2916.
157. Hida, K, Wada, J, Eguchi, J, et al. Visceral adipose tissue-derived serine protease inhibitor: a unique insulin-sensitizing adipocytokine in obesity. Proc Nat Acad Sci U S A. 2005;102:10610–10615.
158. Yang, R-Z, Lee, M-J, Hu, H, et al. Identification of omentin as a novel depot-specific adipokine in human adipose tissue: possible role in modulating insulin action. Am J Physiol Endocrinol Metab. 2006;290:E1253–E1261.
159. Goralski, KB, McCarthy, TC, Hanniman, EA, et al. Chemerin, a novel adipokine that regulates adipogenesis and adipocyte metabolism. J Biol Chem. 2007;282:28175–28188.
160. Yan, Q-W, Yang, Q, Mody, N, et al. The adipokine lipocalin 2 is regulated by obesity and promotes insulin resistance. Diabetes. 2007;56:2533–2540.
161. Virtanen, KA, Lonnroth, P, Parkkola, R, et al. Glucose uptake and perfusion in subcutaneous and visceral adipose tissue during insulin stimulation in nonobese and obese humans. J Clin Endocrinol Metab. 2002;87:3902–3910.
162. Bolinder, J, Krager, L, Ostman, J, et al. Differences at the receptor and post-receptor levels between human omental and subcutaneous adipose tissue in the action of insulin on lipolysis. Diabetes. 1983;32:117–123.
163. Rebuffe-Scrive, M, Andersson, B, Olbe, L, et al. Metabolism of adipose tissue in intraabdominal depots of nonobese men and women. Metabolism. 1989;38:453–458.
164. Vague, J. La differénciation sexuelle, facteur determinant des formes de l’obésité. Presse Méd. 1947;55:339–340.
165. Lapidus, L, Bengtsson, C, Larsson, B, et al. Distribution of adipose tissue and risk of cardiovascular disease and death: 12 year follow-up of participants in the study of women in Gothenburg, Sweden. Br Med J. 1984;289:1257–1261.
166. Ohlson, L, Larsson, B, Svärdsudd, K, et al. The influence of body fat distribution on the incidence of diabetes mellitus: 13.5 years of follow-up of the participants in the study of men born in 1913. Diabetes. 1985;34:1055–1058.
167. Goodpaster, B, Thaete, F, Simoneau, J-A, et al. Subcutaneous abdominal fat and thigh muscle composition predict insulin sensitivity independently of visceral fat. Diabetes. 1997;46:1579–1585.
168. Robbins, D, Horton, E, Tulp, O, et al. Familial partial lipodystrophy: complications of obesity in the non-obese? Metabolism. 1982;31:445–452.
169. Kim, JK, Gavrilova, O, Chen, Y, et al. Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. J Biol Chem. 2000;275:8456–8460.
170. Gavrilova, O, Marcus-Samuels, B, Graham, D, et al. Surgical implantation of adipose tissue reverses diabetes in lipoatrophic mice. J Clin Invest. 2000;105:271–278.
171. Frayn, K. Adipose tissue as a buffer for daily lipid flux. Diabetologia. 2002;45:1201–1210.
172. Groop, L, Saloranta, C, Shank, M, et al. The role of free fatty acid metabolism in the pathogenesis of insulin resistance in obesity and non-insulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1991;72:96–107.
173. Czech, M. Cellular basis of insulin insensitivity in large rat adipocytes. J Clin Invest. 1976;57:1523–1532.
174. Olefsky, J. Insensitivity of large rat adipocytes to the antilipolytic effects of insulin. J Lipid Res. 1977;18:459–464.
175. Weyer, C, Foley, J, Bogardus, C, et al. Enlarged subcutaneous abdominal adipocyte size, but not obesity itself, predicts Type II diabetes independent of insulin resistance. Diabetologia. 2000;43:1498–1506.
176. Danforth, EJ. Failure of adipocyte differentiation causes Type II diabetes mellitus? Nat Genet. 2000;26:13.
177. Schenk, S, Saberi, M, Olefsky, J. Insulin sensitivity: modulation by nutrients and inflammation. J Clin Invest. 2008;118:2992–3002.
178. Hirosumi, J, Tuncman, G, Chang, L, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336.
179. Arkan, M, Hevener, A, Greten, F, et al. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–198.
180. Yuan, M, Konstantopoulos, N, Lee, J, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science. 2001;293:1673–1677.
181. Maher, J, Leon, P, Ryan, J. Beyond insulin resistance: innate immunity in nonalcoholic steatohepatitis. Hepatology. 2008;48:670–678.
182. Weisberg, S, McCann, D, Desai, M, et al. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808.
183. Hosogai, N, Fukuhara, A, Oshima, K, et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes. 2007;56:901–911.
184. Wang, B, Wood, I, Trayhurn, P. Dysregulation of the expression and secretion of inflammation-related adipokines by hypoxia in human adipocytes. Pflügers Arch. 2007;455:479–492.
185. Lumeng, CN, DeYoung, SM, Bodzin, JL, et al. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes. 2007;56:16–23.
186. Nguyen, MTA, Favelyukis, S, Nguyen, A-K, et al. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem. 2007;282:35279–35292.
187. Baron, A, Kolterman, O, Bell, J, et al. Rates of non-insulin mediated glucose uptake are elevated in type II diabetic subjects. J Clin Invest. 1985;76:1782–1788.
188. Nauck, M, Stockmann, F, Ebert, R, et al. Reduced incretin effect in type 2 (non-insulin dependent) diabetes. Diabetologia. 1986;29:46–52.
189. Mortensen, K, Christensen, LL, Holst, JJ, et al. GLP-1 and GIP are colocalized in a subset of endocrine cells in the small intestine. Regul Pept. 2003;114:189–196.
190. Holst, J. Glucagon-like peptide-1: from extract to agent. The Claude Bernard Lecture 2005. Diabetologia. 2006;49:253–260.
191. Nauck, MA, Baller, B, Meier, JJ. Gastric inhibitory polypeptide and glucagon-like peptide-1 in the pathogenesis of type 2 diabetes. Diabetes. 2004;53(suppl 3):S190–S196.
192. Nauck, M, Heimesaat, M, Orskov, C, et al. Preserved incretin activity of glucagon-like peptide 1 (7–36 amide) but not of synthetic human gastric inhibitory polypeptide in patients with type 2 diabetes mellitus. J Clin Invest. 1993;91:301–307.
193. Elahi, D, McAloon-Dyke, M, Fukagawa, N, et al. The insulinotropic actions of glucose-dependent insulinotropic peptide (GIP) and glucagon-like peptide-1 (7–37) in normal and diabetic subjects. Regul Pept. 1994;61:63–74.
194. Chia, CW, Egan, JM. Special features: incretin-based therapies in type 2 diabetes mellitus. J Clin Endocrinol Metab. 2008;93:3703–3716.
195. Rubino, F, Forgione, A, Cummings, D, et al. The mechanism of diabetes control after gastrointestinal bypass surgery reveals a role of the proximal small intestine in the pathophysiology of type 2 diabetes. Ann Surg. 2006;244:741–749.
196. le Roux, C, Welbourn, R, Werling, M, et al. Gut hormones as mediators of appetite and weight loss after Roux-en-Y gastric bypass. Ann Surg. 2007;246:780–785.
197. Tritos, NA, Mantzoros, CS. Syndromes of severe insulin resistance. J Clin Endocrinol Metab. 1998;83:3025–3030.
198. Vandal-Puig, A, Moller, D. Insulin resistance: classification, prevalence, clinical manifestations, and diagnosis. In: Azziz R, Nestler JE, Dewailly D, eds. Androgen Excess Disorders in Women. Philadelphia: Lippincott Raven; 1997:227–236.
199. Garg, A. Acquired and inherited lipodystrophies. N Engl J Med. 2004;350:1220–1234.
200. Arioglu, E, Andewelt, A, Diablo, C, et al. Clinical course of the syndrome of autoantibodies to the insulin receptor (type B insulin resistance): a 28-year perspective. Medicine (Baltimore). 2002;81:87–100.
201. Flier, J, Kahn, CR, Roth, J, et al. Antibodies that impair insulin receptor binding in an unusual diabetic syndrome with severe insulin resistance. Science. 1975;190:63–65.
202. Taylor, S, Grunberger, G, Marcus-Samuels, B, et al. Hypoglycemia associated with antibodies to the insulin receptor. N Engl J Med. 1982;307:1422–1426.
203. Taylor, S, Barbetti, F, Accili, D, et al. Syndromes of autoimmunity and hypoglycemia: autoantibodies directed against insulin and its receptor. Endocrinol Metab Clin North Am. 1989;18:123–143.
204. Page, K, Dejardin, S, Kahn, C, et al. A patient with type B insulin resistance syndrome, responsive to immune therapy. Nat Clin Pract Endocrinol Metab. 2007;3:835–840.
205. Zhang, B, Roth, R. A region of the insulin receptor important for ligand binding (residues 450–601) is recognized by patients’ autoimmune antibodies and inhibitory monoclonal antibodies. Proc Natl Acad Sci U S A. 1991;88:9858–9862.
206. Kahn, C, Baird, K, Flier, J, et al. Effects of autoantibodies to the insulin receptor on isolated adipocytes. J Clin Invest. 1977;60:1094–1106.
207. Karlsson, F, Van Obberghen, E, Grunfeld, C, et al. Desensitization of the insulin receptor at an early postreceptor step by prolonged exposure to anti-receptor antibody. Proc Natl Acad Sci U S A. 1979;76:809–813.
208. Taylor, SI, Arioglu, E. Genetically defined forms of diabetes in children. J Clin Endocrinol Metab. 1999;84:4390–4396.
209. O’Rahilly, S, Moller, D. Mutant insulin receptors in syndromes of insulin resistance. Clin Endocrinol (Oxf). 1992;36:121–132.
210. Bar, R, Muggeo, M, Kahn, C, et al. Characterization of insulin receptors in patients with the syndrome of insulin resistance and acanthosis nigricans. Diabetologia. 1980;18:209–216.
211. Podskalny, J, Kahn, C. Cell culture studies on patients with extreme insulin resistance. I. Receptor defects on cultured fibroblasts. J Clin Endocrinol Metab. 1982;54:261–268.
212. Grigorescu, F, Flier, JS, Kahn, CR. Defect in insulin receptor phosphorylation in erythrocytes and fibroblasts associated with severe insulin resistance. J Biol Chem. 1984;259:15003–15006.
213. Donohue, W, Uchida, I. Leprechaunism: a euphemism for a rare familial disorder. J Pediatr. 1954;45:505–519.
214. Sethu-Kumar Reddy, S, Lauris, V, Kahn, C. Insulin receptor function in fibroblasts from patients with Leprechaunism: differential alterations in binding, autophosphorylation, kinase activity, and receptor-mediated internalization. J Clin Invest. 1988;82:1359–1365.
215. Taylor, S, Roth, J, Blizzard, R, et al. Qualitative abnormalities in insulin binding in a patient with extreme insulin resistance. Proc Natl Acad Sci U S A. 1981;178:7157–7161.
216. Kadowaki, T, Bevins, C, Cama, A, et al. Two mutant alleles of the insulin receptor gene in a patient with extreme insulin resistance. Science. 1988;240:787–790.
217. Moncada, V, Hedo, JA, Serrano-Rios, M, et al. Insulin receptor biosynthesis in culture lymphocytes from an insulin-resistant patient (Rabson-Mendenhall syndrome): evidence for a defect before insertion of receptor into plasma membrane. Diabetes. 1986;35:802–807.
218. Yoshimasa, Y, Seino, S, Whittaker, J, et al. Insulin-resistant diabetes due to a point mutation that prevents insulin proreceptor processing. Science. 1988;240:784–787.
219. Flier, JS, Moller, DE, Moses, AC, et al. Insulin-mediated pseudoacromegaly: clinical and biochemical characterization of a syndrome of selective insulin resistance. J Clin Endocrinol Metab. 1993;76:1533–1541.
220. Dib, K, Whitehead, J, Humphreys, P, et al. Impaired activation of phosphoinositide 3-kinase by insulin in fibroblasts from patients with severe insulin resistance and pseudoacromegaly. J Clin Invest. 1998;101:1111–1120.
221. Stratakis, CA, Turner, ML, Lafferty, A, et al. A syndrome of overgrowth and acromegaloidism with normal growth hormone secretion is associated with chromosome 11 pericentric inversion. J Med Genet. 2001;38:338–343.
222. Berson S, Yalow R, Ellenberg M, Rifkin H, eds. Diabetes mellitus: theory and practice. McGraw-Hill: New York, 1980:388–423.
223. Francis, A, Hanning, I, Alberti, K. The influence of insulin antibody levels on the plasma profiles and action of subcutaneously injected human and bovine short acting insulins. Diabetologia. 1985;28:330–334.
224. Schernthaner, G. Immunogenicity and allergenic potential of animal and human insulins. Diabetes Care. 1993;16(suppl 3):155–165.
225. Fineberg, SE, Kawabata, TT, Finco-Kent, D, et al. Immunological responses to exogenous insulin. Endocr Rev. 2007;28:625–652.
226. Cavaco, B, Uchigata, Y, Porto, T, et al. Hypoglycaemia due to insulin autoimmune syndrome: report of two cases with characterisation of HLA alleles and insulin autoantibodies. Eur J Endocrinol. 2001;145:311–316.
227. Schade, D, Duckworth, W. In search of the subcutaneous-insulin-resistance syndrome. N Engl J Med. 1986;315:147–153.
228. Steiner, D, Tager, H, Chang, S, et al. Lessons learned from molecular biology of insulin-gene mutations. Diabetes Care. 1990;13:600–609.
229. Tager, H, Given, B, Baldwin, D, et al. A structurally abnormal insulin causing human diabetes. Nature. 1979;281:122–125.
230. Haneda, M, Polonsky, K, Bergenstal, R, et al. Familial hyperinsulinemia due to a structurally abnormal insulin: definition of an emerging new clinical syndrome. N Engl J Med. 1984;310:1288–1294.
231. Nanjo, K, Miyano, M, Kondo, M, et al. Insulin Wakayama: familial mutant insulin syndrome in Japan. Diabetologia. 1987;30:87–92.
232. Gabbay, K, Bergenstal, R, Wolff, J, et al. Familial hyperproinsulinemia: partial characterization of circulating proinsulin-like material. Proc Natl Acad Sci U S A. 1979;76:2881–2885.
233. Robbins, D, Blix, P, Rubenstein, A, et al. A human proinsulin variant at arginine 65. Nature. 1981;291:679–681.
234. Robbins, D, Shoelson, S, Rubenstein, A, et al. Familial hyperproinsulinemia: two cohorts secreting indistinguishable type II intermediates of proinsulin conversion. J Clin Invest. 1984;73:714–719.
235. Shoelson, S, Haneda, M, Blix, P, et al. Three mutant insulins in man. Nature. 1983;302:540–543.
236. Barroso, I. Genetics of type 2 diabetes. Diabet Med. 2005;22:517–535.
237. Virkamaki, A, Ueki, K, Kahn, R. Protein-protein interaction in insulin signaling and the molecular mechanisms of insulin resistance. J Clin Invest. 1999;103:931–943.
238. Hager, J, Zouali, H, Velho, G, et al. Insulin receptor substrate (IRS-1) gene polymorphisms in French NIDDM families. Lancet. 1993;342:1430.
239. Permutt, M, Hattersley, A. Searching for type 2 diabetes genes in the post-genome era. Trends Endocrinol Metab. 2000;11:383–393.
240. Thorisson, GA, Smith, AV, Krishan, L, et al. The International HapMap Project Web site. Genome Res. 2005;15:1592–1593.
241. International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945.
242. Manolio, T, Brooks, L, Collins, F. A HapMap harvest of insights into the genetics of common disease. J Clin Invest. 2008;118:1590–1605.
243. The Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;47:661–678.
244. Saxena, R, Gianniny, L, Burtt, NP, et al. Common single nucleotide polymorphisms in TCF7L2 are reproducibly associated with type 2 diabetes and reduce the insulin response to glucose in nondiabetic individuals. Diabetes. 2006;55:2890–2895.
245. Diabetes Genetics Initiative. Genome-wide association identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336.
246. Scott, LJ, Mohlke, KL, Bonnycastle, LL, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345.
247. Zeggini, E, Weedon, MN, Lindgren, CM, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–1341.
248. Bell, GI, Xiang, KS, Newman, MV, et al. Gene for non-insulin-dependent diabetes mellitus (maturity-onset diabetes of the young subtype) is linked to DNA polymorphism on human chromosome 20q. Proc Nat Acad Sci U S A. 1991;88:1484–1488.
249. Yamagata, K, Furuta, J, Oda, N, et al. Mutations in the hepatocyte nuclear factor 4α gene in maturity-onset diabetes of the young (MODY 1). Nature. 1996;384:458–460.
250. Permutt, M, Chiu, K, Tanizawa, Y. Glucokinase and NIDDM: a candidate gene that paid off. Diabetes. 1992;41:1367–1372.
251. Miller, S, Anand, G, Karschnia, E, et al. Characterization of glucokinase mutations associated with maturity-onset diabetes of the young type 2 (MODY-2): different glucokinase defects lead to a common phenotype. Diabetes. 1999;48:1645–1651.
252. Yamagata, K, Oda, N, Kaisaki, P, et al. Mutations in the hepatocyte nuclear factor-1α gene in maturity onset diabetes of the young (MODY 3). Nature. 1996;384:455–458.
253. Stoffers, D, Ferrer, J, Clarke, W, et al. Early-onset type II diabetes mellitus (MODY 4) linked to IPF1. Nat Genet. 1997;117:138–139.
254. Horikawa, Y, Iwasaki, N, Hara, M, et al. Mutation in hepatocyte nuclear factor-1β gene (TCF2) associated with MODY. Nat Genet. 1997;17:384–385.
255. Malecki, M, Jhala, U, Antonellis, A, et al. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet. 1999;23:323–328.
256. Frayling, TM, McCarthy, MI, Walker, M, et al. No evidence for linkage at candidate type 2 diabetes susceptibility loci on chromosomes 12 and 20 in United Kingdom Caucasians. J Clin Endocrinol Metab. 2000;85:853–857.
257. Macfarlane, W, Frayling, T, Ellard, S, et al. Missense mutations in the insulin promoter factor-1 gene predispose to type 2 diabetes. J Clin Invest. 1999;104:R33–R39.
258. Pimenta, W, Korytkowski, M, Mitrakou, A, et al. Pancreatic beta-cell dysfunction as the primary genetic lesion in NIDDM: evidence from studies in normal glucose-tolerant individuals with a first-degree NIDDM relative. JAMA. 1995;273:1855–1861.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 15
Type 2 Diabetes Mellitus
Etiology, Pathogenesis, and Natural History
Genetic versus Acquired Factors
Natural History of Type 2 Diabetes
Pathophysiology of Type 2 Diabetes Mellitus
Inflammatory Pathway Activation and Insulin Resistance
Other Contributors to the Insulin-Resistant Phenotype
Insulin-Mediated versus Non–Insulin-Mediated Glucose Uptake
Pathophysiology of Fasting versus Postprandial Hyperglycemia
Syndromes of Severe Insulin Resistance
Type 2 diabetes mellitus is the most common form of diabetes and is currently a major worldwide cause of morbidity and mortality. This is likely to worsen, given the rapidly increasing prevalence of this condition; therefore, an understanding of its etiology and pathogenesis is of considerable importance. By definition, patients with type 2 diabetes have neither autoimmune β cell destruction, as is found in type 1 diabetes, nor one of the other specific causes of diabetes described in Chapter 13. Type 2 diabetes is not a single disease process but instead represents a heterogeneous constellation of disease syndromes, all leading to the final common pathway of hyperglycemia. Many factors, alone or in combination, can cause hyperglycemia; thus, the complexity of the pathogenesis of type 2 diabetes reflects the heterogeneous genetic, pathologic, environmental, and metabolic abnormalities that can exist in different patients.
Normal glucose homeostasis relies on a balance between insulin secretion and tissue sensitivity to insulin. With respect to regulation of glucose metabolism, the tissue effects of insulin on skeletal muscle, liver, and adipose tissue are most important. Three major metabolic abnormalities coexist in type 2 diabetes,1–4 each contributing to the hyperglycemic state. These abnormalities are summarized in Fig. 15-1. To begin at the hepatic level, the role of the liver in the pathogenesis of type 2 diabetes is overproduction of glucose. Increased basal hepatic glucose production is characteristic of essentially all type 2 diabetic patients with fasting hyperglycemia.5–7 Skeletal muscle is depicted as the prototypic peripheral insulin target tissue, because 70% to 80% of all glucose is taken up by skeletal muscle in the in vivo insulin-stimulated state. Target tissues are insulin resistant in type 2 diabetes mellitus, and such resistance has been well described in many studies across a large variety of population groups.2–5,8–12 Finally, abnormal islet cell function plays a central role in the development of hyperglycemia; decreased β cell function and increased glucagon secretion are standard concomitants of the diabetic state.1,13,14 Taken together, abnormalities in these organ systems account for the syndrome of type 2 diabetes mellitus. In subsequent sections of this chapter, each of these abnormalities will be considered in further detail.
FIGURE 15-1 Summary of the metabolic abnormalities in type 2 diabetes mellitus that contribute to hyperglycemia. Increased hepatic glucose production, impaired insulin secretion, and insulin resistance caused by receptor and postreceptor defects all combine to generate the hyperglycemic state.
Genetic versus Acquired Factors
Abundant evidence supports the view that a strong genetic component contributes to type 2 diabetes. Although many patients have a positive family history for this disease, perhaps the strongest evidence comes from twin studies. In one study, 53 twin pairs were examined, in whom one twin was ascertained to have type 2 diabetes. On assessment of the other twin, type 2 diabetes had developed in 91% (48/53) of the co-twins.15 Although the five discordant twins were not overtly diabetic, they had mild glucose intolerance and abnormal insulin responses during oral glucose tolerance tests, suggesting that they too might ultimately progress to overt disease.
Further evidence for a genetic basis comes from striking differences in the prevalence of type 2 diabetes in various ethnic groups that are not explained by environmental factors. The prevalence of type 2 diabetes in the United States is 2% to 4% for Caucasians, but it is 4% to 6% for African Americans16 and 10% to 15% for Mexican Americans,17 and these numbers are increasing as the prevalence of obesity is rising at epidemic rates. More than 40% of the Pima Indians in Arizona have type 2 diabetes mellitus; this is the group with the highest incidence of type 2 diabetes in the world.18 Among the Pima Indians, 80% of 35- to 44-year-old offspring of two parents with type 2 diabetes mellitus before age 45 years have diabetes, and a positive family history of type 2 diabetes is a substantial risk factor for disease development. Clearly, this genetic predisposition interacts with adverse environmental influences, such as obesity and sedentary lifestyle, which are largely responsible for the sharp uptick in type 2 diabetes mellitus incidence in recent years.
Acquired Factors
Lifestyle: Diet, Exercise, and Obesity
Acquired factors play a major role in the development of type 2 diabetes in genetically predisposed individuals; this is clearly demonstrated by assessment of the impact of lifestyle changes on prevalence of diabetes in various ethnic populations. The prevalence of diabetes increases as ethnic groups migrate from lesser developed to more urbanized areas or simply change from an agrarian to a more sedentary, urban lifestyle. The former has been illustrated by surveys in Japanese subjects. In rural Japan, the prevalence of type 2 diabetes was approximately 4%,19 whereas among Japanese who have immigrated to the United States, the prevalence rises to more than 21%.20 This is also the case in the Pima Indians, who in Arizona have adopted a largely “Westernized” lifestyle, although those living in northwestern Mexico have remained agrarian. The Indians in Arizona have a prevalence of diabetes of 54% and 37% for men and women, respectively, whereas the Mexican Indians have a prevalence of 6% and 11%, respectively.21
It is likely that nutrition and lifestyle are the environmental factors that explain the difference in diabetes prevalence in genetically similar populations. Adoption of an urbanized, Westernized lifestyle is associated with change to a diet that has a higher content of total calories, fats, and refined carbohydrates. For example, the mean daily intake of fat among Japanese men living in Japan was reported to be 16.7 g; by contrast, in Japanese American men, the mean intake was 32.4 g.22 In addition, the level of physical activity is lower among ethnic groups living in the United States compared with the same ethnic groups living in their country of origin.23 These lifestyle changes obviously predispose to the development of obesity, and overwhelming evidence suggests that obesity is a major factor in the development of diabetes. The role of obesity in the pathogenesis of diabetes will be discussed in detail later in this chapter. Recent evidence supporting the role of lifestyle factors in the development of diabetes has come from the Diabetes Prevention Program.24 In this study, intensive lifestyle modification, consisting of dietary change and increased exercise, led to a 58% reduction in the progression of impaired glucose tolerance to diabetes over a 2.8 year period.
The extent of the contribution of these lifestyle factors, independent of their association with obesity, to the development of diabetes remains unclear. For example, it is not known whether specific dietary components, such as a diet that is rich in saturated fat or highly refined carbohydrates, play an independent role in the pathogenesis of type 2 diabetes. With regard to exercise, however, evidence from experimental and epidemiologic studies indicates that subjects with lower physical activity levels are more prone to develop diabetes, independent of obesity.25
Low Birth Weight
Low birth weight as a risk factor for the development of insulin resistance and diabetes mellitus later in life has been described in many populations over the past 2 decades.26–28 The mechanisms responsible for this association are unknown but may be related to epigenetic fetal adaptation to nutritional stimuli or excess fetal glucocorticoid exposure.29
Aging
Aging is associated with a decrease in glucose tolerance, which appears to be due to a decline in both insulin sensitivity and insulin secretion.30 However, age-related factors such as reduced physical activity and increased fat accumulation are at least in part responsible for this phenomenon. Obviously, type 2 diabetes incidence increases with age, but whether the aging process per se is contributory remains unclear.
Natural History of Type 2 Diabetes
The pathophysiologic findings depicted in Fig. 15-1 represent a single point in time after overt type 2 diabetes has developed. However, such an analysis does not reveal the progressive evolution of this disease. Fig. 15-2 presents a schematic description of the natural history or progression to type 2 diabetes. Evidence indicates that in most populations, those who evolve to type 2 diabetes begin with insulin resistance. Insulin resistance can be a primary inherited feature, but acquired factors such as obesity, sedentary lifestyle, and aging (particularly obesity) also can be causal or can exacerbate underlying genetic mechanisms of decreased insulin sensitivity. In an attempt to overcome insulin resistance, the β cell increases insulin secretion, resulting in hyperinsulinemia, which is able to maintain relatively normal glucose tolerance. In a subpopulation of subjects, however, this hyperinsulinemic response is insufficient to fully compensate for the prevailing insulin resistance, and impaired glucose tolerance (IGT) develops. Although a percentage of subjects with IGT may revert to normal glucose tolerance, IGT should be considered an intermediate stage in the development of type 2 diabetes, with many subjects eventually progressing to frank expression of the disease.
The proportion of IGT subjects who progress to type 2 diabetes depends on the particular ethnic groups studied and the methods of assessment used. For example, in the National Institutes of Health (NIH)-sponsored multicenter Diabetes Prevention Program, ≈10% of patients with IGT developed type 2 diabetes mellitus per year.31 During the transition from IGT to frank type 2 diabetes, at least three pathophysiologic changes can be observed. First is a marked fall in β cell function and insulin secretion. Whether this decrease is due to preprogrammed genetic abnormalities in β cell function, to acquired defects (such as glucotoxicity or lipotoxicity), or to both remains to be elucidated. Nevertheless, a marked decrease in β cell function accompanies this transition, and most believe that this decreased β cell function is the major contributor to the progression to type 2 diabetes mellitus. A second metabolic change is seen at the level of the liver. Subjects with IGT have normal basal rates of hepatic glucose output (HGO), whereas patients with fasting hyperglycemia have increased HGO. Thus, the capacity of the liver to overproduce glucose is an important contributory factor (albeit secondary) to the pathogenesis of type 2 diabetes. Finally, many but not all studies have indicated that patients with type 2 diabetes are more insulin resistant than are those with IGT. Most likely, this increase in insulin resistance is secondary to glucotoxicity or to other acquired factors.
Evidence implicating insulin resistance as a primary defect comes principally from studies examining subjects who are at increased risk for developing diabetes. One such group consists of individuals whose parents have type 2 diabetes. Using intravenous glucose tolerance tests (GTTs), Warram and coworkers evaluated 155 nondiabetic offspring whose parents both had type 2 diabetes.32 During a follow-up period averaging 13 years, type 2 diabetes developed in 16% of the total group. However, when offspring were categorized on the basis of insulin sensitivity at initial testing, the cumulative incidence of diabetes was 60% among subjects with preexisting insulin resistance and less than 5% in insulin-sensitive offspring. Thus, insulin resistance and hyperinsulinemia (rather than hypoinsulinemia) characterized the prediabetic state, and this occurred irrespective of obesity and antedated by many years the subsequent development of impaired insulin secretion and overt type 2 diabetes.32
Studies in ethnic populations with a high prevalence of type 2 diabetes also have provided data supporting a primary role for insulin resistance in diabetes development. Thus, among Pima Indians, hyperinsulinemia and an associated decrease in insulin-mediated glucose disposal are early abnormalities that predict the subsequent development of both IGT and type 2 diabetes.33,34 Pima Indians with IGT who progress to type 2 diabetes have lower insulin levels 2 hours after a glucose load than do those who continue to have IGT or who return to normal glucose tolerance. Similar results come from studies of Micronesians in Nauru, a population with a prevalence of type 2 diabetes of approximately 30%.35 Again, in this population, IGT and type 2 diabetes were most likely to develop in those with hyperinsulinemia at baseline, but progression from IGT to type 2 diabetes could be predicted by lower baseline insulin responsiveness to a glucose challenge.
Further evidence comes from studies in first-degree relatives of type 2 diabetic patients.36–38 In these studies, peripheral insulin resistance and hyperinsulinemia were found in normoglycemic relatives of diabetic patients. First-degree relatives with IGT displayed insulin resistance but also exhibited defects in insulin secretion that were not apparent in those with normal glucose tolerance.
Pathophysiology of Type 2 Diabetes Mellitus
Abnormal Pancreatic β Cell Function
Obesity is a major cause of insulin resistance, and as was described earlier, the β cell compensates for decreased insulin sensitivity by increasing insulin secretion. In normal glucose-tolerant subjects, the increase in insulin secretion that occurs with insulin resistance is described by a hyperbolic relationship39 (Fig. 15-3). Although this quantitative increase in insulin secretion in response to decreased insulin sensitivity should be viewed as an appropriate response, subtle qualitative changes in insulin secretion also are noted in the insulin-resistant state. Insulin normally is secreted in rapid regular pulses with a 5 to 15 minute frequency superimposed on slower ultradian oscillations every 80 to 150 minutes.40 These normal rapid, regular pulses are replaced by disordered pulses in obese subjects with insulin resistance and also in the insulin-resistant but glucose-tolerant offspring of subjects with type 2 diabetes.41 Indeed, insulin secretory pulse frequency has been shown to correlate inversely with peripheral insulin sensitivity.42,43
FIGURE 15-3 Relationship between insulin sensitivity and β cell function quantified as the first-phase insulin response (AIRglucose) in 93 (55 males and 38 females) apparently healthy, nondiabetic subjects younger than 45 years. The cohort demonstrates a broad range of insulin sensitivity and β cell function. The solid curve depicts the best fit relationship (50th percentile); the dashed curves represent the 5th, 25th, 75th, and 95th percentiles. The relationship is best described by a hyperbolic function, so that any change in insulin sensitivity is balanced by a reciprocal and proportionate change in β cell function. (Data from Kahn SE, Prigeon RL, McCulloch DK, et al: Quantification of the relationship between insulin sensitivity and β cell function in human subjects: evidence for a hyperbolic function, Diabetes 42:1663–1672, 1993.)
Insulin Secretion in Subjects With Impaired Glucose Tolerance
In subjects with IGT, both quantitative and qualitative defects in insulin secretion are usually present, although this can be variable.43,44 Part of this variability may be explained by the heterogeneity of this condition, as some subjects with IGT will revert to normal glucose tolerance, a proportion will progress to frank type 2 diabetes, and others will continue to have IGT for many years.45
With regard to the hyperbolic relationship between β cell function and insulin sensitivity, subjects with IGT secrete less insulin than is appropriate for their degree of insulin resistance.46 Furthermore, using the graded glucose infusion method, Polonsky has shown that subjects with IGT secrete less insulin at any given glucose level than do normoglycemic subjects matched for a similar degree of insulin resistance and obesity (Fig. 15-4).47 Insulin-resistant subjects with IGT who have progressive impairment in insulin secretion are most likely to develop full-blown type 2 diabetes.48
FIGURE 15-4 Relationship between average plasma glucose concentrations and insulin secretion rates during graded glucose infusion studies in a group of lean nondiabetic control subjects (open triangles), nondiabetic obese subjects (closed squares), and matched obese subjects with impaired glucose tolerance (open diamonds). The lowest glucose levels and insulin secretion rates were measured under basal conditions, and subsequent levels were obtained during glucose infusion rates of 1, 2, 3, 4, 6, and 8 mg/kg/min. Values are means ± standard error of the mean (SEM). (Data from Polonsky KS: The β-cell in diabetes: from molecular genetics to clinical research, Diabetes 44:705–717, 1995.)
Insulin secretion in response to a sustained intravenous glucose stimulus is normally biphasic: A rapid rise in insulin levels within 1 to 3 minutes (first phase) is followed by a return to baseline within 6 to 10 minutes with a subsequent gradual increase (second phase). Subjects with IGT have a reduction in both first- and second-phase responses49 to glucose, and a further qualitative defect in insulin secretion in IGT is the replacement of rapid regular secretory oscillations with disorganized pulses.41
Insulin Secretion in Subjects With Type 2 Diabetes
The abnormalities of insulin secretion described in subjects with IGT are also present in type 2 diabetes, although to a more marked degree. β cell function progressively deteriorates during the natural history of type 2 diabetes. This decline in β cell function is evident not only during the conversion from compensated insulin resistance to IGT and subsequently to overt type 2 diabetes, but also during the progressive course of established type 2 diabetes after its initial onset.50
Basal insulin levels usually are normal or increased in type 2 diabetes. Indeed, obese subjects with type 2 diabetes can have basal insulin levels severalfold higher than normal, but this does not mean that basal β cell secretory function is normal, because the prevailing plasma glucose level also must be taken into account.51–53 Hyperglycemia is the major stimulus for insulin secretion, and when normal individuals are made hyperglycemic by infusion of glucose, circulating insulin levels are much higher than those found in type 2 diabetes.51,52 Thus, patients with type 2 diabetes maintain normal or increased basal insulin levels only in the face of the enhanced stimulus of fasting hyperglycemia, which indicates an underlying impairment in the sensitivity of the β cell to glucose. Stimulated insulin levels in type 2 diabetes can be low, normal, or high depending on factors such as the severity of diabetes, the degree of obesity, and the preceding level of diabetic control.13,14,44
In Response to Intravenous Glucose: In type 2 diabetes, defects in the insulin secretory response to intravenous glucose are observed consistently. Once fasting plasma glucose levels exceed 126 mg/dL, the first-phase insulin response to intravenous glucose characteristically is completely absent. This relationship is shown in Fig. 15-5.1,54,55
FIGURE 15-5 First-phase insulin release in response to the intravenous administration of glucose in normal and type 2 diabetic (non–insulin-dependent diabetes [NIDD]) subjects. Mean fasting plasma glucose concentrations were 83 ± 3 mg/dL in normal subjects and 160 ± 10 mg/dL in type 2 diabetic subjects. (Data from the American Diabetes Association, Inc., from Ward WWK, Beard JC, Halter JB, et al: Pathophysiology of insulin secretion in non-insulin-dependent diabetes mellitus, Diabetes Care 7:491–502, 1984.)
It is interesting to note that acute or first-phase insulin secretion in response to nonglucose stimuli such as arginine1 or isoproterenol56 is relatively preserved; this finding indicates a functionally selective β cell defect in response to glucose stimuli in type 2 diabetes. Because the acute insulin response to intravenously administered arginine or isoproterenol increases as the glucose concentration is raised, Porte and colleagues have attempted to quantitate this effect of glucose by plotting the increase in acute insulin response to arginine or isoproterenol pulses as a function of increasing plasma glucose level.1,14,51 The slope of this relationship is termed the glucose potentiation slope, and by this analysis, glucose potentiation of β cell function is also reduced in type 2 diabetes.1,14
Although absent first-phase insulin secretion may be a marker for β cell dysfunction, it is unlikely to be an important cause of glucose intolerance or hyperglycemia. Thus, mildly hyperglycemic and severely hyperglycemic type 2 diabetic patients are equally deficient in first-phase insulin secretion, implying that this deficiency does not play a role in further deterioration in glucose tolerance from mild to severe fasting hyperglycemia. Furthermore, in selected patients with normal glucose tolerance in whom type 1 diabetes eventually develops, the acute insulin response to intravenous glucose is absent during the normal stage and therefore does not cause hyperglycemia.57 Finally, α-adrenergic blockers can substantially restore the acute insulin response to intravenous glucose58 without major improvement in fasting glycemia or glucose tolerance.
Second-phase insulin secretion, which is assessed with the hyperglycemic clamp or the graded intravenous glucose infusion technique (see Fig. 15-4 for details), is markedly reduced in individuals with type 2 diabetes compared with normal subjects and those with IGT. In general, the more severe the diabetes, the lower is the second-phase insulin response.1,54
In Response to Oral Glucose and Mixed Meals: The insulin response in type 2 diabetes to oral ingestion of glucose or mixed meals is far more variable than the response to intravenous glucose. After oral glucose, insulin levels are usually subnormal in type 2 diabetes,14,36 although this might not always be the case in patients with mild hyperglycemia.44 This heterogeneity is depicted in Fig. 15-6, which summarizes the results of oral GTTs in a wide spectrum of normal and type 2 diabetic subjects.59 As can be seen, hyperinsulinemia frequently exists in mild states of glucose intolerance. In individuals with mild diabetes, insulin levels generally are in the “normal range,” although inappropriately or relatively low for the degree of hyperglycemia and insulin sensitivity. With more severe diabetes, absolute stimulated insulin levels are uniformly low.
FIGURE 15-6 A, Mean (±standard error of the mean [SEM) plasma glucose response to oral glucose in the five subject groups. Closed circle, normal; cross, borderline tolerance; open circle, impaired glucose tolerance; open triangle and broken line, fasting hyperglycemia (110 to 150 mg/dL); closed triangle and broken line, fasting hyperglycemia (>150 mg/dL). B, Mean (±SEM) plasma insulin response to oral glucose in the five subject groups. Symbols are the same as in A. (Data from Reaven GM, Olefsky JM: Relationship between heterogeneity of insulin responses and insulin resistance in normal subjects, Diabetologia 13:201–206, 1977.)
Proinsulin Secretion
Another factor related to hyperinsulinemia in type 2 diabetes is circulating proinsulin levels. Proinsulin is secreted by β cells concomitantly with insulin and cross-reacts with insulin in most insulin immunoassays, thus contributing to the total measured immunoreactive insulin level. In normal subjects, proinsulin represents only a small portion (3% to 7%) of the insulin-like material secreted by β cells. However, using proinsulin-specific immunoassays, several groups have shown that in hyperinsulinemic states and in many cases of type 2 diabetes, an increased proportion of proinsulin is released and contributes to the measured insulin in standard immunoassays, so true insulin levels are overestimated.14,53,60 When corrected for this factor, basal insulin levels may be normal or moderately elevated in type 2 diabetes.
Mechanisms of β Cell Dysfunction
The overall mass of β cells changes in obesity and type 2 diabetes.61,62 β cell mass reflects the balance between new islet formation (neogenesis) and β cell loss due to apoptosis. Longitudinal studies in animal models suggest that β cell mass increases appropriately in response to a decrease in insulin sensitivity,62 and cross-sectional data from humans have long revealed an expanded β cell mass in obesity.63 In contrast, β cell mass is reduced in type 2 diabetes.61 On the basis of murine models and cross-sectional autopsy data from humans, diminished β cell mass is thought to be due to accelerated β cell apoptosis and the failure of islet neogenesis and β cell replication to compensate for this loss.62
It has been theorized that lipid accumulation in the β cell is implicated in the apoptotic process and the development of impaired insulin secretion in type 2 diabetes. This is referred to as lipotoxicity and may involve excess fatty acids entering β cells, thus triggering the apoptotic cellular response.64
A body of evidence also suggests a role for islet amyloid polypeptide (IAPP) in the loss of β cells. IAPP is synthesized in the β cell and is co-secreted with insulin.65 IAPP aggregates to form fibrils of amyloid, and islet amyloid is found at autopsy in up to 90% of subjects with type 2 diabetes.66 In vitro, IAPP is toxic, causing β cell apoptosis,67 and it may contribute to the reduced β cell mass that is associated with type 2 diabetes.
Insulin secretory abnormalities found in type 2 diabetes are often improved after a period of good blood glucose control, irrespective of the treatment used (diet, insulin, or oral hypoglycemic agents).68–70 This partial reversibility is consistent with the idea that, to some extent, the abnormalities may be secondary to hyperglycemia or some other factor associated with uncontrolled diabetes. Support for the “glucotoxicity” theory comes from a variety of in vivo and in vitro studies showing that chronic exposure of islets to hyperglycemia can result in a number of different defects in glucose-induced insulin secretion.71 It is important to note that when isolated human islets are incubated under euglycemic and hyperglycemic conditions, islets that are exposed to hyperglycemia demonstrate a marked defect in their ability to secrete insulin in response to subsequent glucose stimuli.72 Although the precise mechanism is unknown, it seems likely that glucotoxicity coupled with lipotoxicity plays some role in the impaired β cell function of type 2 diabetes.
An interesting finding from studies of mouse genetics is that insulin receptor signaling in β cells is important for normal function. Thus, mice in which the insulin receptor gene has been specifically deleted from β cells show a complete loss of first-phase insulin secretion in response to glucose but not arginine, reminiscent of the β cell defect in type 2 diabetes.73 Second-phase glucose-induced insulin secretion is also blunted in these mice, and they show an age-dependent progressive impairment in glucose tolerance. Glucose-stimulated insulin secretion involves transport of glucose into cells by a specific glucose transporter, termed GLUT2. Following uptake, glucose is phosphorylated by glucokinase, and subsequent intracellular metabolism of glucose-6-phosphate leads to stimulation of insulin secretion. In mouse studies, genetic deletion of GLUT2 leads to loss of glucose but not arginine-stimulated insulin secretion. It is interesting to note that feeding mice high-fat diets to induce obesity also leads to a decrease in β cell GLUT2 expression, suggesting another mechanism of interaction between acquired environmental factors and β cell dysfunction.
The decrease in β cell mass in type 2 diabetes is in the range of 30% to 50%. However, because sufficient insulin secretory reserve normally exists to sustain an 80% to 90% loss of β cells without the development of hyperglycemia, it follows that decreased functional capacity of the remaining β cells must exist in type 2 diabetes. Indeed, it has been shown that the maximal insulin secretory capacity may be reduced by as much as 80% in type 2 diabetic subjects.52 It is possible that the decrease in β cell mass in type 2 diabetes is somehow causally related to the decreased function of the remaining β cells. Thus, partially pancreatectomized rats and streptozotocin-treated rats display similar insulin secretory defects,71 which suggests that decreased glucose-stimulated insulin secretion with relative preservation of responsiveness to nonglucose stimuli may be a general type of abnormality that may occur in response to a variety of β cell insults.
Peripheral Insulin Resistance
Causes of Peripheral Insulin Resistance
Circulating Factors That Influence Insulin Action: Hormonal antagonists include all known counterregulatory hormones such as cortisol, growth hormone, glucagon, and catecholamines. In well-known syndromes (e.g., Cushing’s disease, acromegaly), elevated levels of these hormones can induce an insulin-resistant diabetic state. However, in the usual case of obesity or type 2 diabetes, excessive levels of these counterregulatory hormones are not an important contributory factor to insulin resistance.
Several years ago, Randle and coworkers hypothesized that the elevated circulating levels of free fatty acids (FFAs) found in obesity and type 2 diabetes impair peripheral glucose utilization.74 Substantial evidence indicates that FFAs do indeed contribute to insulin resistance, although the mechanisms differ from those originally proposed by Randle. FFAs also play an important role in the regulation of HGO and contribute to hepatic insulin insensitivity in obesity and type 2 diabetes. These mechanisms will be discussed in greater detail later in this chapter.
Impaired Access of Insulin to Target Cells: Because insulin must travel from the circulation to target tissues to elicit biological effects, any defect in this transfer could lead to functional insulin resistance. Compared with secretion into the circulation, the passage of insulin from the plasma compartment to tissue sites of action is markedly delayed, and in vivo effects of insulin in stimulating glucose disposal are well correlated with the appearance of insulin in the interstitial fluid.75 Lymph and interstitial insulin levels are ≈40% lower than those in plasma,75,76 which indicates that peripheral tissues are more sensitive to insulin than was previously recognized. Furthermore, the possibility arises that either the rate or the amount of insulin being transferred from the plasma to the interstitial compartment could be abnormal in type 2 diabetes or obesity, thereby contributing to the insulin-resistant state.77,78 Recent studies indicate that transport of insulin across the capillary in vivo occurs by diffusion76 and is not receptor mediated, as was previously suggested.79 Transport by diffusion fits with the finding that transcapillary passage is comparable in normal subjects, insulin-resistant nondiabetic subjects,80 and those with type 2 diabetes.81 Further evidence that the delayed activation of muscle glucose uptake in obesity and type 2 diabetes is not due to impaired transcapillary transport of insulin comes from a study by Nolan and coworkers.78 This study showed that the kinetic defect in the ability of insulin to stimulate leg glucose uptake was not accompanied by any delay in the activation of leg muscle insulin receptors by insulin, thus implying that the kinetic defect is distal to the insulin receptor.
Another physical factor that may relate to insulin resistance is muscle capillary density, which correlates with in vivo insulin sensitivity.82 Laakso and coworkers have shown that insulin, at least at pharmacologic levels, increases leg blood flow.83 Because tissue glucose uptake is a product of blood flow and the arteriovenous glucose difference, increased leg blood flow could contribute to overall glucose disposal. Similar studies performed in obese subjects and in subjects with type 2 diabetes revealed a decrease in the insulin-induced increase in leg blood flow, which may explain part of the decrease in total leg glucose uptake.83 However, others found no effect of insulin on blood flow,84 and Utriainen and colleagues used 15O [H2O] and positron emission tomography to confirm enhancement of leg muscle blood flow by pharmacologic insulin levels, but no difference in the response between type 2 diabetic and normal control subjects.85
Cellular Defects in Insulin Action: Available evidence points to a target tissue defect as the major cause of insulin resistance in type 2 diabetes. Before potential causes are considered, it is useful to review some general concepts concerning normal insulin action (Fig. 15-7). Insulin first binds to its cell surface receptor, a heterotetrameric glycoprotein composed of two α subunits (135 kDa) and two β subunits (95 kDa) linked by disulfide bonds.86–89 The α subunits are entirely extracellular and are responsible for insulin binding. The β subunits are transmembrane proteins containing a small extracellular domain and a larger cytoplasmic domain that includes insulin-regulated tyrosine kinase activity. Binding of insulin to the receptor rapidly induces tyrosine autophosphorylation of the β subunit involving three tyrosine residues in the kinase domain, in addition to tyrosine residues adjacent to the transmembrane domain and in the C terminus of the β subunit. Once the receptor has been autophosphorylated, its intrinsic tyrosine kinase catalytic activity is markedly enhanced, and it now can phosphorylate tyrosine residues on endogenous protein substrates. Activation of the insulin receptor tyrosine kinase is essential for transduction of the insulin signal and for internalization of the receptor. Patients with naturally occurring mutations in the tyrosine kinase domain of the insulin receptor have syndromes of severe insulin resistance.
FIGURE 15-7 Model of cellular insulin action. (From Saltiel AR, Kahn CR: Insulin signalling and the regulation of glucose and lipid metabolism, Nature 414:799–806, 2001.)
In recent years, major advances have been made in our understanding of how the insulin signal is propagated downstream from the activated insulin receptor to various insulin-regulated enzymes, transporters, and insulin-responsive genes to mediate its metabolic and growth effects (see Fig. 15-7). This field is rapidly evolving and complex and is discussed only briefly here because it is covered comprehensively in Chapter 8. A large number of intermediate signaling molecules have been identified, and after activation of the insulin receptor kinase, more than one signaling pathway may be used (see Fig. 15-7). For example, some of the components in the pathways leading to mitogenic effects of insulin are distinct from those leading to activation of glucose transport. Even a single action of insulin such as stimulation of glucose transport can involve more than one signaling pathway. Several cytosolic protein substrates of the insulin receptors are phosphorylated on tyrosine residues within seconds of insulin binding to its receptor. The first of these substrates to be identified was insulin receptor substrate 1 (IRS-1).89,90 IRS-1 belongs to a growing family of proteins that includes IRS-2, IRS-3, IRS-4, and a protein termed shc, which are immediate substrates of the insulin receptor kinase involved in insulin signaling. These proteins have no enzymatic activity but act as docking proteins. Tyrosine phosphorylation of these substrates enhances their association with proteins that contain src homology-2 (SH2) domains. These SH2 domains contain ≈100 amino acids and can bind to specific short motifs that encompass a phosphotyrosine. The binding of specific SH2 domain–containing proteins to tyrosine-phosphorylated IRS proteins or shc generates multicomponent signaling complexes, which, in turn, modulate the activities of phosphoinositide-3-kinase (PI3K), several serine kinases, and phosphatases that act on key insulin-regulated enzymes and transcription factors.
One of the most important effects of insulin with respect to type 2 diabetes is stimulation of glucose uptake into skeletal muscle, adipocytes, and heart muscle. Under most physiologic circumstances, glucose transport in these tissues is rate limiting for overall glucose disposal.91–93 Tissue glucose uptake is mediated by a family of at least five facilitative glucose transporters, each derived from a separate gene. These transporters show a high degree of homology, but each has tissue-specific distribution.94,95 One of them, GLUT4, or the insulin-sensitive glucose transporter, is uniquely expressed in skeletal muscle, adipose tissue, and cardiac muscle. In the unstimulated state, most of the GLUT4 proteins are located in an intracellular vesicular pool. Upon insulin stimulation, recruitment or translocation of these glucose transporter-rich vesicles to the cell surface causes insertion of GLUT4 proteins into the plasma membrane, where they begin to transport glucose into the cell.96–100
Characteristics of Insulin Resistance in Subjects With Impaired Glucose Tolerance or Type 2 Diabetes
The frequency of insulin resistance increases as the degree of carbohydrate intolerance worsens.101 Thus, many, but not all, subjects with IGT are insulin resistant, whereas essentially every type 2 diabetic patient with fasting hyperglycemia displays this abnormality. Numerous studies indicate that insulin resistance is more marked in type 2 diabetes than in the prediabetic IGT state.3,5,36 However, other reports show only a modest increase in the degree of insulin resistance going from IGT to type 2 diabetes. Because most type 2 diabetic patients are overweight, obesity-induced insulin resistance is clearly a major contributing factor in these patients. However, obesity is not the only cause, in that the insulin resistance in obese type 2 diabetic patients exceeds that caused by obesity alone, and nonobese type 2 diabetic patients are also insulin resistant.3,5,101
All methods of assessing insulin resistance in vivo rely on measurement of the ability of a fixed dose or concentration of insulin to promote glucose disposal. Thus, a blunted decline in plasma glucose concentration after administration of intravenous insulin has been demonstrated in type 2 diabetes.102,103 Another approach has been to infuse insulin and glucose at fixed rates while endogenous insulin secretion is inhibited by a combination of epinephrine and propranolol or by somatostatin.102,104 With this method, the resulting steady-state plasma glucose level reflects the action of concomitantly infused insulin; the higher the steady-state plasma glucose, the greater is the degree of insulin resistance. Bergman and colleagues’ minimal model is yet another method of assessing in vivo insulin resistance.102 This method entails computer modeling of plasma glucose and insulin levels after an intravenous glucose bolus to generate an index of insulin sensitivity. With all these methods, type 2 diabetic subjects exhibit a significant decrease in insulin sensitivity compared with controls.3,102,105,106
More detailed studies of in vivo insulin resistance have been carried out with the euglycemic glucose clamp method.102 With this approach, insulin is infused at a constant rate, resulting in a given steady-state plasma insulin level, while plasma glucose is kept constant at a predetermined level by a feedback-controlled variable infusion of glucose. The insulin normally lowers the plasma glucose level by suppressing HGO and by stimulating tissue glucose uptake. During the insulin infusion, the amount of glucose that has to be infused to keep plasma glucose levels constant increases gradually until a steady state is reached. Under these steady-state conditions, the glucose disposal rate provides an excellent quantitative assessment of the biological effect of a particular steady-state insulin level. If a radioactive or stable isotope of glucose is also infused during the study, HGO during the clamp can be quantified. In type 2 diabetes, glucose disposal rates are 30% to 60% lower than those in normal subjects at any given insulin infusion rate. If several studies at different insulin levels are performed in a given subject, dose response curves for insulin-stimulated glucose disposal and suppression of HGO can be constructed. Patients with type 2 diabetes (obese and nonobese) exhibit both a rightward shift in their dose response curve (diminished sensitivity) and a marked decrease in their maximal rate of glucose disposal (decreased responsiveness) (Fig. 15-8). These changes tend to be more pronounced in obese diabetic patients, particularly at maximal glucose disposal rates (see Fig. 15-8). The insulin resistance of obese type 2 diabetic patients is significantly greater than that of nondiabetic obese subjects. Subjects with IGT tend to have a rightward shift in their dose response curves with normal maximal glucose disposal rates (see Fig. 15-8).
FIGURE 15-8 Mean insulin dose response curves for control subjects (closed circles), subjects with impaired glucose tolerance (open circles), and nonobese (open triangles) and obese (closed triangles) type 2 diabetic subjects. The group with impaired glucose tolerance has a rightward shift in the dose response curve without a change in maximal response (i.e., decreased sensitivity). Lean and obese patients with type 2 diabetes have both a rightward shift and a reduction in the response to a maximally stimulating concentration of insulin (i.e., decreased sensitivity and decreased insulin responsiveness). (Data from Kolterman OG, Gray RS, Griffin J, et al: Receptor and post-receptor defects contribute to the insulin resistance in non-insulin dependent diabetes mellitus, J Clin Invest 68:957–969, 1981.)
Because skeletal muscle is responsible for the great majority of in vivo insulin-stimulated glucose uptake, this tissue must be the major site for resistance to insulin-stimulated glucose disposal. This conclusion is evidenced by the demonstration of insulin resistance in type 2 diabetic patients during forearm perfusion studies.107,108 Leg catheterization studies have shown that skeletal muscle accounts for ≈80% of whole-body insulin-mediated glucose uptake, and that leg skeletal muscle is markedly resistant to the ability of insulin to stimulate glucose uptake in type 2 diabetes.109–111 Thus, although other insulin target tissues display decreased insulin sensitivity, they do not account for a significant proportion of overall glucose uptake, and one can conclude that all measures of in vivo insulin action on glucose disposal largely assess the resistance of skeletal muscle to take up glucose under the influence of insulin.
Pathophysiologic Abnormalities in Insulin Target Tissues
Mechanisms of Skeletal Muscle Insulin Resistance: As the first step in insulin action, it is apparent that a decrease in cellular insulin receptors could lead to insulin resistance. However, this potential relationship is not as clear as it would seem because a maximal insulin effect is achieved at insulin concentrations that occupy a fraction of the surface receptors, giving rise to the concept of “spare” receptors. A maximal response of glucose transport in adipocytes and muscle is achieved with only 10% to 20% of the receptors occupied.112,113 Once the critical number of receptors needed to generate a maximal response is activated, additional increases in the prevailing insulin concentration lead to increases in receptor occupancy with no further increase in biological response, because a step (or steps) distal to the receptor is now rate limiting. The functional significance is that a decreased number of insulin receptors leads to a rightward shift in the insulin biological function dose response curve, with decreased responses at all submaximal insulin concentrations but a normal maximal response. A reduction in the maximal insulin response generally denotes the presence of a postbinding abnormality. In this context, the term postbinding defect includes abnormalities of insulin receptor function that affect its transmembrane signaling function, such as its kinase activity. A postreceptor defect refers to any abnormality in a step distal to the insulin receptor.
Early studies showed decreased insulin binding to circulating monocytes from obese and IGT subjects and from both obese and nonobese type 2 diabetic patients.114,115 This decreased binding was due to a decrease in insulin receptor number with no change in affinity. Similar results were subsequently obtained, when isolated adipocytes, hepatocytes, and skeletal muscle from obese subjects and patients with type 2 diabetes were used.116,117 The decrease in cellular insulin receptors in obesity and type 2 diabetes may well be secondary to hyperinsulinemia, inasmuch as elevated circulating insulin levels can downregulate receptor number.
A reduction in insulin receptor tyrosine kinase activity in type 2 diabetes generally has been found in patients with normal kinase activity in IGT.78,91,117 The receptor autophosphorylation/kinase defect appears to be generalized to all insulin target tissues and relatively specific for the hyperglycemic insulin-resistant state that is seen in type 2 diabetes.
Emerging evidence points to a role for IRS proteins in the development of insulin resistance. A defect in insulin-stimulated IRS-1 tyrosine phosphorylation is found in skeletal muscle from type 2 diabetic patients, although overall IRS-1 expression is unchanged.118 Serine/thr phosphorylation of IRS proteins is closely associated with reduction of signaling through IRS. Two potential mechanisms may underlie this phenomenon. First, serine phosphorylation may block the interaction of IRS-1 with its target proteins.119 Second, proteasomal-mediated degradation of IRS-1 may be increased.120 Several intermediary lipid metabolites and cytokines have been shown to activate a variety of serine/threonine kinases that induce serine phosphorylation of IRS-1. Serine kinases implicated include c-Jun NH2-terminal kinase (JNK),121 IκB kinase (IKK),122 PKC theta, S6K, and MTOR. It is interesting to note that this places IRS-1 at the intersection of a variety of intracellular pathways, including inflammation, endoplasmic reticulum (ER) stress, and nutrient sensing, all of which can activate serine/threonine kinases that may phosphorylate IRS-1.
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
