Appetite Regulation and Thermogenesis
The classification of body weight as a regulated physiologic parameter is relatively novel. While obesity, including morbid obesity, has been recognized for thousands of years, the possibility that body weight was determined by a complex interaction between internal regulatory systems and the environment first received scientific attention in the mid-20th century. Studies of brain-lesioned animals indicated that disruption of the ventromedial hypothalamus produced a syndrome of obesity and hyperphagia,1–3 and ablation of the lateral hypothalamus resulted in aphagia, adipsia, and dramatic weight loss,1,4 suggesting that these areas were critical to the maintenance of energy balance. Findings also emerged demonstrating that obesity could be caused by administration of goldthioglucose,5 which damaged the ventromedial nucleus of the hypothalamus (VMH).6 Subsequent studies using monosodium glutamate–induced obesity indicated that the arcuate nucleus also played a role in maintaining energy balance.7 A series of parabiosis experiments performed between genetically obese mice, ob/ob and db/db, suggested that circulating factors might play a role in determining adiposity.8,9 In humans, the potential role of the hypothalamus in obesity came from the observation that patients with craniopharyngioma frequently became obese following resection of the tumor.10
Despite significant efforts along a number of lines of investigation which resulted in increased understanding of obesity, a full appreciation of obesity as an endocrine syndrome did not arrive until the discovery of leptin gene in 199411 and its receptor.12 Mutations in either gene served to explain two mouse obesity syndromes: the ob/ob mouse, which lacks the hormone leptin, and the db/db mouse, which lacks the long form of the leptin receptor. Recognition of the fact that mutations in these molecules led to similar phenotypes in humans rapidly followed and confirmed their importance.13,14 These discoveries led to a significant paradigm shift in the understanding of obesity and the nature of the fat cell. Existence of a hormone-specific obesity syndrome made it clear that body weight was subject to physiologic regulation. Furthermore, identification of the adipocyte as the source of the hormone changed the perception of the fat cell from that of a passive depot of energy stores to a regulator important to overall energy homeostasis.15,16 This overall shift redefined understanding of the processes involved in regulating overall energy balance in mammalian organisms, including humans. As a consequence, a sophisticated understanding of the interconnection of multiple organ systems in the brain and periphery and their interaction with the environment is currently evolving.
Components of Feeding
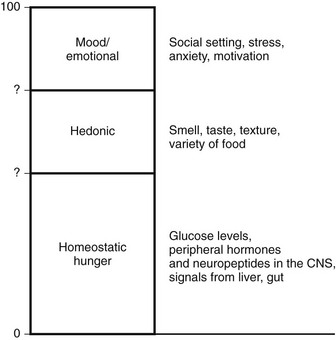
Intake of calories occurs for multiple reasons (Fig. 1-1). Perhaps the most important is the net caloric deficit that begins after the digestion of a meal. As calories are used, the increasing deficit that accrues eventually leads to hunger, food seeking, and food ingestion. This component of feeding may be called “homeostatic hunger” because it reflects a true metabolic deficit, and consumption is aimed at maintaining energy stores. Since this activity is critical to survival, it appears to be linked to reward pathways so that animals will “work” for food.17 For example, feeding involves flavor and texture and thus engages gustatory pathways that involve taste and smell. The rewards associated with ingestion of palatable flavors lead to eating past the point of metabolic repletion.18 Food variety also appears to engage reward pathways; increased variety prevents malnutrition. However, availability of an increased variety of energy-dense foods is associated with obesity.19 Motivation and reward pathways have been extensively explored with regard to drug addiction. While addiction to drugs of abuse has no homeostatic value, reinforcement of food rewards may occur through similar pathways. For example, agents that alter opioid and dopaminergic signals also act to modulate motivation for palatable food.20 Furthermore, eating is also linked to stress, which may predispose to hyperphagia in an environment where palatable food is readily available.19
Components of Energy Expenditure
Traditional models categorize energy expenditure (EE) into basal (obligate) and adaptive (or facultative) thermogenesis (AT). Obligate EE includes all pathways involved in the maintenance of basic metabolic and physiologic processes and is also referred to as resting metabolic rate (RMR) or more recently resting energy expenditure (REE). RMR includes both sleeping metabolic rate (SMR) and the increase in metabolic rate that is seen with arousal. AT includes cold and diet-induced thermogenesis. The cellular mechanisms that regulate obligatory and adaptive thermogenesis are often similar. Finally, physical activity represents a third category which has at least two components, nonexercise activity thermogenesis (NEAT) and sports-like exercise.21 These distinct categories of EE are in fact only approximate, and regulatory mechanisms are overlapping (Fig. 1-2). For example, thyroid hormone (TH) is required for up to 30% of basal EE, and adaptive increases in TH are required for normal cold-induced thermogenesis.22,23 Furthermore, physical activity (PA) can have long-lasting effects on REE, and PA may be enacted by stimuli that are traditionally considered stimulants of facultative thermogenesis, such as caloric excess.24 Approximate contributions for the components of energy expenditure are REE, 70%; PA, 20%; and facultative, 10%, with PA representing the most variable component.25
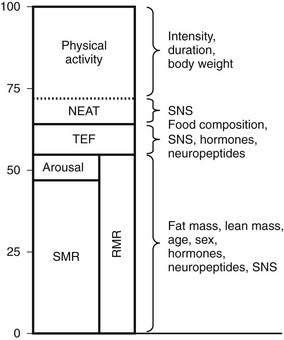
FIGURE 1-2 Approximate contribution of components of energy expenditure. Thermic effect of food (TEF) and resting metabolic rate (RMR) can be measured using a ventilatory hood. RMR and nonexercise activity thermogenesis (NEAT) can be measured in a respiratory chamber. Total energy expenditure can be measured using doubly labeled water. SMR, Sleeping metabolic rate.
Integration of Energy Balance
Inputs from a number of neuropeptides and neurotransmitters in the brain as well as peripheral signals integrate information to mediate energy balance.26 The interactions and pathways engaged by these signals are already complex, and understanding of the pathways is still evolving. Furthermore, it is not clear that all possible important signals have been identified. One interesting aspect of the signals involved is that many regulate both energy intake and energy expenditure in a coordinated fashion. Although a correlation of obesity with decreased sympathetic activity has been long recognized,27,28 the potential pathways regulating energy expenditure were thought to be separate from those regulating feeding and satiety. However, it is now recognized that peptides regulating appetite also play a role in regulating energy expenditure in an inverse manner. Thus, peptides that stimulate feeding decrease energy expenditure, promoting energy storage, while those that inhibit feeding increase energy expenditure (Table 1-1).
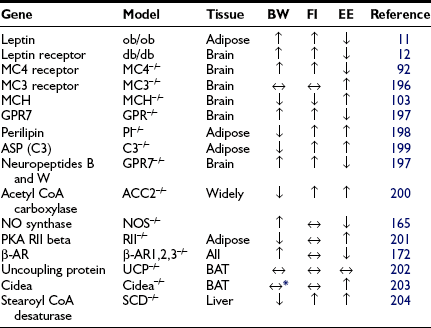
In the brain, both neurotransmitters and neuropeptides have complex functions.29 The role of neurotransmitters in regulating feeding behavior was recognized before the role of neuropeptides was appreciated. However, mechanisms of these neurotransmitters have been more difficult to define; they may act through multiple receptors, and effects may vary depending on the anatomic area targeted. Monoamine neurotransmitters may be stimulatory or inhibitory. Glutamate and γ-aminobutyric acid (GABA), which are the most abundant neurotransmitters in the hypothalamus, act to increase feeding, and some of the neurons expressing orexigenic neuropeptides appear to be GABAergic.30 One view of the interaction of transmitters and peptides is that peptides act as essential modulators of GABA and glutamate action.31 Serotonin, which acts through multiple receptors, is inhibitory,32 and some of these receptors are being considered as specific pharmacologic targets for the treatment of obesity.33 The roles of epinephrine, norepinephrine, and dopamine are more complex, and these transmitters may act to either stimulate or inhibit feeding.34,35 Although the mechanism of action is poorly understood, these pathways are the targets of the limited pharmacologic therapies that are currently available for the treatment of obesity. Biogenic amines currently in use include phentermine and sibutramine. Phentermine, an analog of amphetamine, acts to increase catecholamine release in the paraventricular nucleus of the hypothalamus. Mazindol has a similar mechanism of action. Sibutramine, which acts through its active metabolites, prevents reuptake of 5-HT but does not cause release. Sibutramine also inhibits noradrenaline reuptake.36
Recently, the importance of interaction of peptidergic systems with neurotransmitters has been recognized as playing a role. For example, leptin37,38 and MCH39,40 appear to regulate dopaminergic tone. In the arcuate, GABAergic AgRP/NPY neurons play a critical role in regulating pro-opiomelanocortin (POMC) neurons. Although ablation of AgRP or NPY individually or both peptides in combination does not result in an identifiable phenotype,41 ablation of the neurons in adult mice leads to rapid starvation.42 Consistent with the role of GABA regulating POMC neurons, mice with impaired GABA release are lean and resistant to diet-induced obesity.43
In addition to leptin, other signals from the periphery add to the complex pathways that are involved in the regulation of body weight. This includes multiple signals from the gut44,45 (Fig. 1-3). Cholecystokinin (CCK) is synthesized in the duodenum and jejunum and was recognized as a peptide capable of inhibiting appetite as early as 1973.46 CCK acts in the hindbrain to reduce meal size and duration.47 Peptide YY (PYY) is secreted by the distal portion of the gastrointestinal tract, and in addition to inhibiting gastric emptying,48 it also crosses the blood-brain barrier to act on arcuate nuclei and inhibit feeding.44 However, the role of PYY remains controversial, because when injected into the lateral ventricles of animals, PYY acts to increase food intake, and not all investigators have been able to reproduce the satiety effects.49 Glucagon-like peptide 1 (GLP-1) and oxyntomodulin are products of the preproglucagon gene and are synthesized in the gut and brain. Both act to inhibit food intake, through different mechanisms.50 Thus far, the only gut peptide known to stimulate appetite is ghrelin, which is secreted by the stomach and also acts on neurons in the arcuate nucleus.51
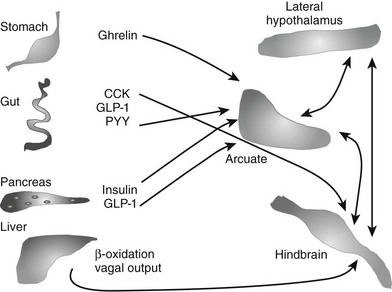
FIGURE 1-3 Schema of selected gut-to-brain signals that may play a role in mediating energy balance. A number of peptides from the gut play a role in mediating appetite and energy expenditure. Only one, ghrelin from the stomach, is orexigenic. All segments of the gut may also contribute to signals from vagal afferents. Finally, information of oxidation of fatty acids in the liver may also be transmitted by the vagus and play a role in mediating appetite. Within the brain, signaling acquires additional complexity; networks between the hypothalamus and hindbrain and cortex are all involved in regulating both intake and output.
Other factors derived from the gut that may play a role in appetite include fatty acid amides. For example, the cannabinoid receptor agonist, anandamide, is an ethanolamide synthesized in the gut and possibly regulated by nutritional status, since local concentrations increase with fasting.52 Anandamide may act centrally through cannabinoid receptors, which are known to promote appetite and increase energy homeostasis.53 In contrast oleoylethanolamide, which is also synthesized in the gut, acts to inhibit food intake and stimulate lipolysis through the activation of the peroxisome proliferator–activated receptor alpha (PPAR-α).54 Thus fatty acid–derived molecules may serve as an additional pathway of gut-mediated regulation of appetite.
Peripheral signals regarding the state of the gastrointestinal tract may also be integrated by the vagus nerve,55 which sends afferents to multiple brain areas, including the dorsal motor nucleus and the nucleus of the solitary tract. These areas appear to be involved in responses to neuropeptides, including ghrelin56 and α-MSH. The vagus may also play a role in conveying information on fatty acid oxidation in the liver. Fatty acid oxidation appears to play a role in mediating appetite; inhibition of this process is associated with increased appetite.57,58
Insulin may also play a role in inhibiting appetite, and it is known that neurons in the arcuate express insulin receptors59 and respond to insulin. Female mice lacking insulin receptor expression in the brain eat more than normal animals, and both genders develop mild obesity when placed on a high-fat diet.60 Insulin and glucose may play a role in meal initiation and meal termination.61
Thus, body weight is regulated by a complex interaction of signals involving both the gut and the brain. Additional complexity derives from the fact that these signals act through specific receptors. The receptors have anatomic-specific expression. Furthermore, some involve relatively large receptor families, as seen with the melanocortin receptors (see The Melanocortin System: α-MSH, Agouti-Related Peptide, and Central Melanocortin Receptors). For some of these pathways, the finding of spontaneous mutations associated with obesity in human populations has provided proof that body weight is regulated similarly in humans as in rodents (Table 1-2).
Table 1-2
Sample of Characterized Mutations Leading to Obesity in Humans
| Mutation | Reference |
| Leptin | 62, 13 |
| Leptin receptor | 14 |
| Melanocortin 4 receptor | 94, 95, 205 |
| Melanocortin 3 receptor | 206 |
| Prohormone convertase 1 | 207 |
| PPAR-γ | 208 |
| POMC | 96 |
POMC, Pro-opiomelanocortin; PPAR, peroxisome proliferator–activated receptor.
Specific Hormones and Neuropeptides
Leptin, identified through positional cloning of the ob gene,11 is a 167-amino-acid peptide hormone secreted by adipocytes. It signals through a membrane receptor that has six splice variants and belongs to the class I cytokine receptor family.12 Leptin signaling is required, although not sufficient alone, for normal energy balance. Animals, including mice and humans, lacking leptin62 or the leptin receptor14 have a syndrome of severe hyperphagia and obesity. In the case of leptin mutations, leptin administration leads to a marked resolution of the syndrome in both ob/ob mice63,64 and in rare human patients with leptin mutations.65,66 However, in the vast majority of obese mammals, leptin levels are elevated, correlating well with available fat stores,67,68 and administration of peripheral leptin has little effect on appetite. These findings revised the perception of leptin. While the complete absence of leptin has major consequences on appetite, the incremental increases in leptin that are seen with increased adiposity have little effect on the continued ingestion of calories or the storage of calories as fat. In contrast, repletion of leptin with fasting leads to attenuation of many of the neuroendocrine changes seen with fasting.69 In human females, leptin replacement leads to some of the abnormalities seen in hypothalamic amenorrhea secondary to strenuous exercise or low body weight.70 Thus the critical physiologic role of leptin appears to be to signal caloric deficiency and thus mediate the appropriate metabolic changes rather than to signal caloric excess.
Leptin targets specific neurons in the brain, specifically in the hypothalamus, although leptin receptors are also seen in other areas, including the ventral tegmental area (VTA). The best-characterized neurons are the NPY/AgRP and POMC neurons in the arcuate.30,71 NPY and AgRP are both orexigenic (appetite-inducing) peptides synthesized by the same population of neurons. POMC is expressed in a different population of arcuate neurons that process the preprohormone to a number of peptides, including α-MSH, which acts to suppress appetite. To date, the leptin-to-arcuate pathway represents the best-characterized pathway involved in the regulation of body weight, especially insofar as mutations disrupting this pathway have also been shown to be important in human obesity as well as rodent obesity (Fig. 1-4).
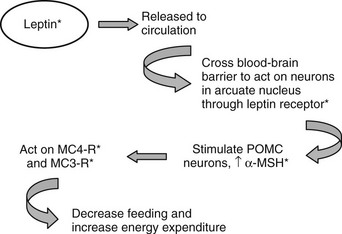
FIGURE 1-4 The leptin pathway. Leptin, a hormone secreted by adipocytes, crosses the blood-brain barrier to act on neurons in the arcuate nucleus. One set of target neurons are those synthesizing prepro-opiomelanocortin (POMC). Leptin acts to stimulate these neurons to synthesize POMC and release one of the POMC gene products, α-melanocyte-stimulating hormone (α-MSH). This peptide mainly acts through the melanocortin 4 receptor (MC4R) to decrease feeding, and through this receptor and the melanocortin 3 receptor (MC3R) to increase energy expenditure. Mutations in this pathway lead to disruption of the appropriate signals and to obesity. Known mutations leading to obesity in humans are marked with asterisks. (See text and Table 1-2 for details.)
Adipocytes synthesize many biologically active proteins with potential endocrine function (reviewed in Ref. 58 and Chapter 11). These include cytokines, immune-related proteins, complement and complement-related proteins, enzymes involved in steroid metabolism, and proteins of the rennin-angiotensin system. Furthermore, receptors for traditional endocrine hormones, nuclear hormones, cytokines, and catecholamines are all expressed by adipose tissue. These peptides are likely to form causal links between obesity, insulin resistance, and cardiovascular disease. Two recently discovered peptides, adiponectin and resistin, may play a role in modulating insulin resistance. Adiponectin inversely correlates with insulin resistance, declines with obesity, and increases with weight loss.72 In contrast, resistin impairs glucose tolerance and insulin sensitivity, and secretion increases with increasing adiposity.73 They may also be involved in determining predisposition to obesity and responses to a high-fat diet. Recently, excess adiposity has been associated with finding increased expression of multiple inflammatory markers in fat tissue. Thus, expression of interleukin 1 (IL-1), IL-5, plasminogen activator inhibitor 1 (PAI-1), tumor necrosis factor (TNF), and suppressor of cytokine signaling 3 (SOCS3) are all increased in obesity.74 These factors play a role in the decrease in insulin sensitivity associated with obesity; however, it is unclear (and seems unlikely) that any of these factors have direct effects on either appetite or energy expenditure.
The Hypothalamus
The potential role of neuropeptides in feeding behavior was first suggested by studies indicating that NPY was synthesized by arcuate75 neurons and elicited a robust feeding response when injected intracerebroventricularly (ICV).76,77 Chronic infusion of NPY leads to obesity in rats.78,79 Furthermore, expression of NPY increases with fasting, indicating that neurons making NPY respond to peripheral signals, signaling the state of energy balance.80,81 Interestingly, ablation of the NPY gene was not associated with altered body weight or feeding,82 although when mice lacking NPY were bred to mice lacking leptin, substantial attenuation of the ob/ob obesity syndrome was noted.83 However, animals without NPY show an abnormal response to refeeding after short-term fasting84 and also show an attenuated feeding response to hypoglycemia.85
The Melanocortin System: α-MSH, Agouti-Related Peptide, and Central Melanocortin Receptors
Humans and rodents require an intact melanocortin system in order to maintain normal body weight. The effect of α-MSH to decrease appetite was described in the late 1980s.86 However, the key role of melanocortins in the physiology of energy balance was not appreciated until the molecular mechanism of obesity of the yellow Ay mouse was identified. In this model, obesity is secondary to a mutation in the gene encoding a protein, agouti, which mediates coat color and leads to ectopic expression of the protein in all tissues, including the central nervous system.87 Subsequently it was discovered that agouti protein acted on melanocortin receptors to block melanocyte-stimulating hormone (MSH) action.88,89 These findings led to speculation that another protein normally expressed in the brain might have an action similar to that of agouti and to the discovery of agouti-related peptide, AgRP,90 which is expressed in the hypothalamus and interacts with the central melanocortin receptors, MC3R and MC4R.91 Overexpression of AgRP recapitulated an obesity syndrome similar to that seen in the Ay mouse, as did disruption of the MC4R.92 Mice with targeted disruption of the MC3R demonstrated a small increase in body fat and feeding efficiency, suggesting that at least in rodents, MC4 plays the dominant role in energy homeostasis.
The profound effects caused by disruption of the melanocortin pathway stimulated a search of MC4R mutations in humans, especially in children with early-onset obesity and a strong family history of obesity. Several such mutations were readily identified,93,94 and currently it is estimated that 5% of persons with severe familial early-onset obesity have MC4 mutations.95 In humans, obesity has also been associated with mutations in the POMC gene, which encodes multiple transcripts. Disruption of this gene leads to deficiency in both adrenocorticotropic hormone (ACTH) and MSH, and patients present with adrenal insufficiency and obesity. Since MSH expression outside of the central nervous system mediates hair color, patients with POMC mutations will frequently also have red hair.96
Melanin-Concentrating Hormone
Melanin-concentrating hormone is a 19-amino-acid peptide synthesized in magnocellular neurons of the lateral hypothalamus; it plays a key role in maintaining energy balance in animals.97,98 The peptide structure and anatomic distribution is highly conserved, and the sequence is identical in rodents, sheep, and humans. When injected ICV in rats, MCH induces an acute increase in feeding behavior. Chronic infusions in mice lead to a syndrome of mild obesity associated with decreased energy expenditure.99 Deletion of both the MCH and the MCH receptor genes are associated with leanness.100–102 In the case of the receptor knockouts, leanness is secondary to increased expenditure, because animals without the receptor eat as much or more than wild-type animals. Deletion of MCH from mice lacking leptin leads to a marked attenuation of the obesity phenotype, which is secondary to changes in energy expenditure rather than feeding.103 Pharmacologic blockade of the MCH receptor leads to leanness and reduces meal size.104 Chronic infusions of MCH agonists also lead to obesity similar to that seen with MCH infusion.105 The importance of the MCH system has not been validated in humans, since a phenotype of MCH deficiency would present with a lean phenotype. However, the homology of MCH in all strains of mammals examined strongly suggests that MCH will play a role in humans.
The Gut
Ghrelin, produced in the stomach, was identified as the endogenous ligand for the receptor responsible for growth hormone secretion.106 Subsequently it was found to produce adiposity in rodents, an effect that is independent of its ability to stimulate growth hormone secretion.107 Although infusions of ghrelin induce hunger and increased feeding,108 endogenous levels are low in obese individuals and increase with weight loss.109 This rise is not seen after gastric bypass surgery, which may help to explain the success of this procedure in mediating weight loss in obese humans.110,111 Ghrelin levels are extremely high in the Prader-Willi syndrome of genetic obesity.112 Ghrelin is transported into the brain, where it acts to stimulate NPY/AgRP neurons in the arcuate nucleus and is thus part of a circuit mediating energy homeostasis involving the stomach and the hypothalamus.113
Peptide YY
Peptide YY is synthesized and secreted throughout the intestine, although concentrations are higher in the distal portion, particularly in the colon and rectum, and the 3-36 form crosses the blood-brain barrier. Food intake stimulates PYY release, and higher serum concentrations are seen after fatty meals. As release occurs prior to nutrients reaching the distal parts of the gastrointestinal tract, neural reflexes may act to stimulate release, possibly through the vagus. PYY 1-36 has structural similarity to NPY and binds with high affinity to all five NPY receptors; the 3-36 form binds preferentially to the Y2 receptor. PYY acts on both the intestine and the brain. In the intestine, it increases fluid absorption and delays gastric emptying. In the brain, the 3-36 form has substantial effects on appetite. When given intravenously to human volunteers, it reduces caloric intake and increases the sensation of satiety.114 Similar effects have been reported in rats115; however, this effect is controversial because other investigators have been unable to reproduce the satiating effect.49 ICV injection of PYY clearly increases feeding, presumably through targeting a different receptor subset.116
PYY levels are low in patients with morbid obesity. One report suggests that levels rise after weight loss secondary to gastric bypass surgery.117 This suggests a potential role of PYY in the treatment of obesity.
Glucagon-Like Peptide-1 and Oxyntomodulin
GLP-1 and oxyntomodulin, along with GLP-2, are products of the preproglucagon gene and result from posttranslational processing by prohormone convertases. The preproglucagon gene is expressed in the central nervous system, in intestinal L cells, and in the pancreas. Both GLP-1 and oxyntomodulin act as satiety signals through the GLP-1 receptor.50,118,119 Release from the small intestine is seen after food ingestion; however, the peptides are rapidly cleaved by dipeptidyl peptidase IV and thus have a short half-life. GLP-2 does not affect satiety.120 GLP-1 has effects on insulin secretion and beta cell mass, while GLP-2 affects the growth of intestinal epithelial cells.121 GLP-1 has therapeutic potential in the treatment of obesity and type 2 diabetes, although this could be limited because of the short half-life. One approach aimed at circumventing this problem is the use of peptidase inhibitors and long-acting analogs such as exenatide.122
The Role of Energy Expenditure in Body Weight Regulation
Measuring ingested food is straightforward, at least in experimental animals. However, measuring energy expenditure is complicated because one must account for all components as well as the relative contribution of each component. The most accurate method for determination of EE is by direct calorimetry, which can be measured either by water immersion or closed-chamber heat convection, but both of these methods are cumbersome and are rarely used. Ingestion of doubly labeled water allows interpolation of EE from the amount of 2H218O ingested, and the amount of 2H and 18O released as water and carbon dioxide has been shown to be accurate to within 5% of indirect calorimetry.123 However, these techniques require use of both expensive non-radioactive isotopes and instrumentation. Since more than 90% of oxygen consumption arises from mitochondrial metabolism,124 O2 consumption can be used as an index of energy expenditure. For rodent studies, indirect calorimeters consist of closed multichambered modules with oxygen and CO2 sensors that measure gas exchange. Most are also equipped with laser beam break systems that allow simultaneous monitoring of spontaneous locomotor activity. Models with continuous access to food and water are available, and these can be used to monitor animals over intervals as long as days or weeks. Software is supplied to calculate VO2, VCO2, and RER, as well as total heat production based on VO2. In humans, respirometers such as the Deltatrac or Cosmed are most commonly used. A significant limitation of these monitors is that observation requires wearing space helmet–like head gear which results in a limited observation period. Some clinical research facilities have closed room-sized chambers which measure gas exchange using methodology similar to that used by rodent monitors.
Comparing Energy Expenditure Between Individual Subjects
Using indirect calorimetry to compare EE among organisms that differ in body weight has inherent inaccuracies. Differences in body weight are usually associated with differences in tissue distribution; for example, animals that are obese will have similar lean body mass but have increased adipose tissue. Energy expenditure of different tissues varies over a broad range,125 and it is not possible to calculate the contribution of each organ. While fat has a relatively low EE, in obese animals, it may represent a relatively high proportion of total mass. Many investigators normalize VO2 for body weight or, preferably, lean body mass that more closely reflects total EE.126 However, there is no clear-cut agreement about how EE is best expressed. To control for this confounder, some perform pair feeding experiments. When two groups of animals are fed the same amount of food, and one group loses more weight, the group experiencing the greater weight loss either had greater EE or lower energy absorption in the intestine. If digestive loses are accounted for, one can conclude that the group that lost more weight had higher EE. An example of pair feeding (PF) is shown in Fig. 1-5.127 The PF paradigm also has flaws, possibly owing to a relative state of semistarvation that is perceived by PF animals. For example, compared to leptin-treated rats that have decreased food intake, PF rats are relatively hypothyroid due to decreased hepatic conversion of T4 to T3.128
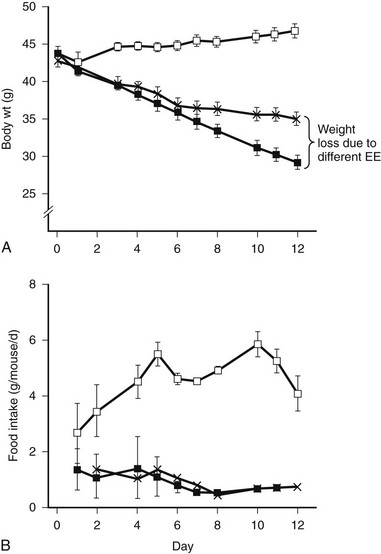
FIGURE 1-5 Pair feeding in ob/ob mice results in less weight loss, implying increased energy expenditure (EE) from leptin treatment. Leptin-deficient ob/ob mice are hyperphagic (control open squares) and respond to leptin treatment by reducing food intake and increasing EE. The increased body weight loss in the ob/ob leptin-treated group (closed squares) compared to the pair-fed group is due to increased EE. (From Levin N, Nelson C, Gurney A et al: Decreased food intake does not completely account for adiposity reduction after ob protein infusion, Proc Natl Acad Sci U S A 93:1726–1730, 1996.)
How Does Energy Expenditure Contribute to Obesity?
Small discrepancies between energy intake and EE over a long time can result in net positive energy balance and obesity. For example, assuming constant energy expenditure, the current epidemic of obesity could theoretically be stabilized by lowering food intake by 100 kcal per day according to one estimate.129 By analogy, if food intake is constant, a small increase of activity leading to an increased expenditure of 100 calories would have the same effect. Increasing energy expenditure is known to be an important contributor to resistance to diet-induced obesity in rodents and appears to be strain specific, even in mice eating the same diet and total calories. When an obesity-susceptible strain is placed on a high-fat diet, there is a very small increase in oxygen consumption. In contrast, the obesity-resistance strain shows a substantial increase in oxygen consumption both secondary to increased RMR and increased spontaneous locomotor activity.130
The role of EE in the pathogenesis of obesity remains unresolved. A number of studies support the hypothesis that reduced EE plays a pathogenic role in the development of obesity. Early studies, based on the comparison of self-reported food diaries to weight gain, suggested that obese patients have significantly lower EE compared to lean patients. However, these differences could not be confirmed in studies using the doubly labeled water technique,123,131 and differences were attributed to underreporting of food consumption. Nonetheless, there is compelling evidence that lower EE and predisposition to obesity is genetically determined. Obesity itself has a strong genetic component,132 and resting metabolic rate also appears to be an inherited trait, and is independent of fat-free mass, age, and sex as a predictor of EE.133 Overfeeding studies in monozygotic twins show a high degree of similarity in weight gain between but not among twins and also argue strongly that genetic factors play a major role in controlling EE.134 Also, the environment in which identical twins are raised has little influence over eventual body mass index (BMI).135 Finally, direct and indirect measurements of EE and respiratory quotient (RQ) have shown small but measurable differences between obese and lean patients, particularly in certain ethnic groups such as Pima Indians.136,137 Longitudinal studies have confirmed that differences in EE are associated with tendency to develop obesity over a period of years.138
In contrast, other studies failed to find significant differences in EE between obese and lean human patients. For example, EE increases linearly with increasing BMI, so increased EE at higher body weight would function to resist further body weight change.25 Similarly, children aged 5 to 10 with varying known susceptibility to obesity have similar increases in RMR.139 Furthermore, no differential activity between lean and obese individuals in systems regulating body weight has been reported; this includes sympathetic nervous system (SNS) activity,140 catecholamine turnover,141 lipolysis,142 the thermic effect of food,25 and thyroid hormones.143 In summary, some studies have reported data to support the hypothesis that relatively low EE contributes to the development of obesity. These findings, in addition to clear defects in EE that are seen in obese rodent models, suggest that defects in EE may be attractive targets for antiobesity treatments.
Regulation of Energy Expenditure
Physical Activity
Physical activity (PA) is the most variable component of daily EE, ranging from nearly zero kcal/day in sedentary adults to thousands of kcal/day in endurance athletes. PA has effects on EE both acutely, with large increases in maximal oxygen consumption, and chronically, via improved respiratory capacity. Thus, PA represents an ideal mechanism to resist obesity in the setting of increased food intake. Only 10% of the variability in human body weight is estimated to be due to differences in PA, however.144 Studies in Pima Indian children aged 5 to 10 have shown a negative relationship between PA and eventual obesity, but PA was not predictive.139,145 Other studies have shown decreased PA but no change in overall EE between lean and obese adolescents.146
Coordinated PA is a complex behavior that is regulated by numerous mechanisms at multiple sites in the CNS.24 Recently, the demonstration that PA is regulated via specific neuropeptides in specific sites in the CNS, such as the leptin and melanocortin pathways, suggest that PA is a component of the “adipostatic” system.147 For example, mice lacking MC4R not only consume more calories but fail to enact PA on running wheels in response to caloric excess.147 The effects of AgRP administration include not just increased food intake but also a decrease in spontaneous locomotor activity.116 Mice lacking both MCH and MCH receptor show increases in baseline physical activity.130,148 These effects are likely to be mediated by effects of the neuropeptide on striatal pathways. It is known that MCH ablation is associated with altered dopaminergic tone.40 Physical activity clearly increases EE and can alter RMR in the postexercise period as well. In humans, sustained weight loss is most successful with a combination of decreased food intake and PA.149 Further investigation into the regulation of PA as a specific mechanism to control body fat stores will clearly be of great importance in the field of obesity.
Nonexercise activity thermogenesis (NEAT), as an independent component of physical activity, may be an important mediator of increased EE with increasing body weight. Careful overfeeding studies in lean humans showed that the majority of increased EE in response to caloric excess occurs not via increases in thermic effect of food, RMR, or coordinated PA, but rather most likely in NEAT.150 While formally a subclass of PA, NEAT includes all tasks of daily living, including posture, fidgeting, and even chewing gum.151 NEAT can be accurately measured by sensors in humans and rodents.152 At least a portion of increased EE in hyperthyroidism is attributable to increase in NEAT.153
Regulation of Thermogenesis
Brown Adipose Tissue and Mitochondria
At the tissue level, brown adipose tissue (BAT) plays a critical role in thermogenesis and is essential for the response of animals to cold exposure through induction of non-shivering thermogenesis.154 BAT is densely packed with mitochondria, and cells express high levels of uncoupling protein 1 (UCP1). Generation of heat occurs as UCP1 uncouples respiration from adenosine triphosphate (ATP) formation and results in heat production. BAT also appears to play a role in energy balance, since complete ablation of BAT results in obesity.155 In mice housed at thermoneutrality, ablation of UCP1 is also associated with an obese phenotype in mice which is exacerbated by feeding of a high-fat diet.156
Although regulation of EE through BAT is well established in mice, the possibility that an analogous process exists in humans has been debated, and many have claimed that brown fat does not exist in man. Recent studies using positron emission tomography (PET) have now confirmed that BAT exists in humans. In 2007, a retrospective analysis of published PET studies provided evidence for BAT.157 A recent study evaluating 3640 consecutive fluorine-18-labeled (18F)-fluorodeoxyglucose (FDG) PET, as well as PET-CT performed for clinical diagnosis coupled with analysis of clinical surgical specimens for UCP1, confirmed the presence of BAT in a small percentage of both men and women.158 The regulation of these depots by cold exposure has also been reported.159 Finally, another group utilized PET scanning and prospective biopsy sampling and also identified BAT depots in man.160
Brown fat function is regulated through the SNS (Fig. 1-6) by norepinephrine, both directly, through action on β3 receptors which increase cAMP, and indirectly, through induced lipolysis which ultimately increases peroxisome proliferator–activated receptor γ coactivator 1α (PGC1α) and thus stimulates UCP1,154 leading to uncoupling of mitochondrial oxidation (see later). BAT is also regulated by thyroid hormone, but the molecular mechanism for this is poorly understood. A number of transcriptional factors regulate BAT cell development.161 For example, PGC1α is required for cold-induced thermogenesis, a process which is impaired in animals lacking this factor.162 The protein PDRM16 plays a critical role in BAT differentiation and maintenance of the BAT phenotype.163
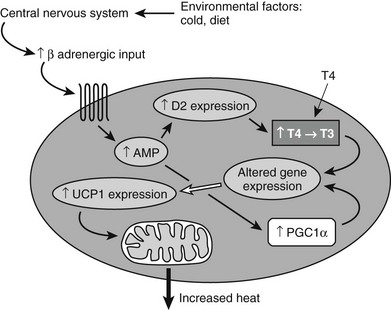
FIGURE 1-6 Signals from the environment such as diet or cold exposure alter function in the central nervous system, which then changes sympathetic outflow. Increased sympathetic activity acts through a Gs-coupled β-adrenergic receptor in brown fat to alter gene expression (including induction of deiodinase 2 [D2]) and to increase expression of UCP1. PGC1α is also activated. Induction of D2 leads to increased conversion of T4 to T3, which mediates additional changes in gene expression. Increased UCP1 and increased activity of PGC1α all act to increase heat production. PGC1α, Peroxisome proliferator–activated receptor γ coactivator 1α; UCP1, uncoupling protein 1.
At the organelle level, mitochondria generate most of the energy produced by a eukaryotic cell through a complex process that couples oxidative phosphorylation of carbon fuels to the production of ATP and a proton pump which creates an electrochemical gradient. As electrons flow through mitochondrial complexes, protons are pumped out. Influx of protons back through the gradient is coupled to the phosphorylation of adenosine diphosphate (ADP) to ATP. Using mitochondrial inhibitors, it has been shown that oxidative phosphorylation in mitochondria accounts for 90% of energy expenditure.124 In some organisms, oxidative phosphorylation can be uncoupled from ATP generation to generate heat. In mammals, this process is important for adaptation to cold and occurs primarily in BAT, which expresses a unique inducible uncoupling protein, UCP1, that lowers the mitochondrial membrane potential.164 Uncoupling thus leads to altered EE. The degree of uncoupling can be theoretically controlled at a number of levels, including through regulation of UCP levels, UCP activity, or via wholesale changes in mitochondrial protein levels, biogenesis, and electron transport.16 Defective mitochondrial biogenesis, for example, has recently been shown to affect EE and body weight regulation.165
Absolute mitochondrial number in a number of tissues, including BAT and muscle, can also affect REE. Mitochondrial number is regulated through multiple factors that affect biogenesis.166 Physiologic regulators of biogenesis include exercise training, an effect that is probably signaled through combined effects of calcium signaling, adenosine monophosphate–activated protein kinase (AMPK), and nitric oxide. Chronic exercise leads to an elevation of PGC1α. Calorie restriction also leads to biogenesis, presumably through SIRT1 activation.167
Regulatory Inputs Acting on Energy Expenditure
Some of the pathways involved in regulating EE have been defined,168 and as noted earlier, most factors that regulate feeding also regulate energy expenditure in an inverse manner, that is, increased feeding is coupled to decreased energy expenditure (Fig 1-7). Interestingly, food consumption itself is associated with changes in EE. Overfeeding is associated with physiologic adaptations, including increased SNS activity, decreased parasympathetic nervous system activity, and the inferred form of PA known as nonexercise activity thermogenesis, or NEAT (Fig. 1-8).
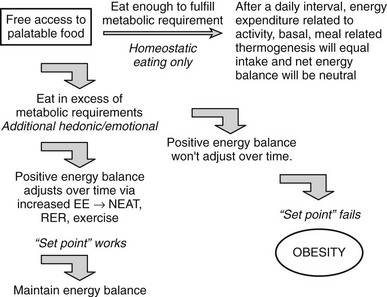
FIGURE 1-7 Physiologic changes that accompany weight gain and loss in mammals. EE, Energy expenditure; PA, physical activity; PNS, parasympathetic nervous system; SNS, sympathetic nervous system; T3, triiodothyronine. (Adapted from Rosenbaum M, Keibel RL, Hirsch J: Obesity, N Engl J Med 337:396–406, 1997.)
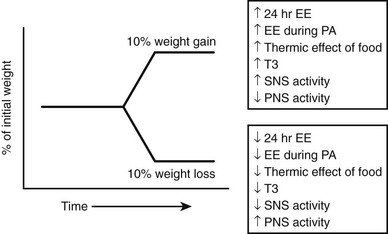
FIGURE 1-8 In an environment where there is free access to palatable food, some individuals will eat largely in response to homeostatic hunger; that is, they will largely consume calories necessary to sustain metabolic and physical activity. At the beginning of each day, they will be in neutral energy balance with respect to the previous day. These individuals are represented in the 30% to 40% of Western populations who maintain normal body weight (or if overweight, are able to maintain a stable weight over a long period of time). Some individuals will eat excess calories beyond what is necessary to sustain metabolic requirements. This extra eating will occur in response to the setting the food is served in or to the hedonic aspects of the food. Some of these individuals will utilize a limited number of extra calories by adjustments in resting metabolic rate (RMR) or small changes in the thermic effect of food or nonexercise activity thermogenesis (NEAT). Some individuals may also adapt consciously by increasing energy expenditure (EE) through physical activity, thereby “assisting” their set-point. These individuals will also be represented in a group that maintains stable body weight over a long period of time. In contrast, some individuals will overeat. Overeating may involve small caloric increments that, for unclear reasons, the individual cannot make adequate adjustments for through increased RMR or NEAT. Some overeating may involve a large number of calories, the utilization of which would require increased physical activity or diet. Over time, these individuals will gain weight. Rate of weight gain may be slow (2 to 3 pounds each year over decades), but weight gain can be substantial, resulting in obesity.
Consumption of a meal also leads to increased thermogenesis, a phenomenon known as dietary-induced thermogenesis (DIT). This may be caused in part by the energy required to digest the meal. Some have proposed that DIT evolved in mammals to allow wasting of energy after consumption of large quantities of low-quality diets necessary to obtain sufficient essential amino acids.169,170 The thermic response of BAT to diet has long been appreciated.171 Recently, DIT in rodents was shown to be a critically important antiobesity mechanism, especially in response to caloric excess,172 but the role of DIT in humans remains unclear.
Interestingly in rodents, consumption of a very high-fat ketogenic diet also stimulates energy expenditure, and mice eating equal calories of such a diet versus a high-fat/high-sucrose diet show evidence of increased uncoupling in BAT, fail to gain excess weight, and are more insulin sensitive than control animals fed chow.19
Effects of the Sympathetic Nervous System on EE and Obesity
Many lines of evidence support a role for the SNS in the regulation of body weight via control of EE. Lower baseline SNS activity can be associated with propensity for future weight gain.173,174 The SNS is controlled by the CNS, and postganglionic neurons release either norepinephrine (sympathetic) or acetylcholine (parasympathetic) from their terminals (reviewed in Ref. 162). A popular model for regulation of EE in response to caloric excess is as follows: brain → SNS → βARs → thermogenesis. Indeed, low SNS activity is seen in most obese rodent models, and activation of this pathway by βAR agonists is effective in reducing obesity.175,176 Numerous attempts to perturb SNS function (surgical, chemical, immunologic, genetic) have not affected body weight, however, and thus the importance of SNS-mediated induced thermogenesis is difficult to interpret.177–180 In contrast, in mice, elimination of all adrenergic signaling from BAT through the ablation of all three βARs results in a “β-less” mouse with a phenotype of obesity that appears to be due purely to a decrease in EE.172 Furthermore, while β-less mice are mildly obese on a regular diet, they become massively obese when challenged with a high-fat diet. The normal response (increased VO2) to a high-fat diet seen in wild-type mice is completely absent in β-less mice, demonstrating that βARs are required for obesity resistance and increased thermogenesis in response to a high-fat diet.
Thyroid Hormone
Thyroid hormone (thyroxine [T4] and triiodothyronine [T3]) plays a critical role in regulating obligatory REE and AT. The role of TH on body weight homeostasis under normal circumstances is unclear, but hyperthyroidism is commonly associated with weight loss, even in the context of increased food intake. Indeed, based on this finding, thyroid extracts were used with some success in the treatment of obesity beginning a century ago; early studies correlated TH treatment with basal metabolic rate and weight loss.181,182 The actions of TH on metabolism are multiple and quite complex.
In humans, energy expenditure correlates with TH levels, and substantial changes of up to 50% increases in hyperthyroidism and 50% decreases in hypothyroidism have been reported.183 Approximately 30% of basal thermogenesis is TH mediated.23 In fact, hypothyroidism was formerly diagnosed by whole-body oxygen consumption prior to the availability of biochemical tests of for pituitary thyroid-stimulating hormone (TSH) and serum TH. In mammals, the main function of TH is to maintain temperature homeostasis and not adipose stores.23 Numerous diverse pathways, including anabolic and catabolic metabolism of lipids, carbohydrates, and proteins, are stimulated in response to TH (reviewed by Silva23 and Bianco et al.183). Also, the increase in ATP turnover and heat are probably derived from the baseline increase in flux of many cellular pathways, such as the maintenance of ion gradients, ion cycling, and uncoupling, the sum of which is to increase EE.184 Noting that the level of T4 sufficiency, as measured by TSH, correlates well with REE.185 Thyroid hormone plays a critical role in adaptive thermogenesis response to environmental cold, fever, and overfeeding. In response to cold signals, the SNS stimulates BAT, and a rapid increase in T3 levels are seen,183 mediated by the induction of type 2 deiodinase (D2), which converts T4 into its more active congener, T3,186 under SNS control.187 These events ultimately result in increased UCP1. D2 is essential for cold-induced thermogenesis,22,188 a response which is impaired in hypothyroid animals.189
Although TH is critical to the regulation of REE and adaptive thermogenesis, its effects on body weight in the absence of pathology such as hyperthyroidism or hypothyroidism are difficult to determine. TH levels are affected by nutritional status and fall rapidly with fasting but are also under the control of leptin, which can rescue this fall.69 Thyroid function is normal in obesity, although positive correlation of TSH and BMI have been reported.190 Loss of function of D2 via gene disruption is critical to thermogenesis, but it has no demonstrable effect on body weight in mice191; hypothyroid rats are capable of increasing thermogenesis in response to overfeeding a high-fat feeding.192
Nonetheless, because of the significant effects of TH in pharmacologic doses on REE, there is considerable interest in TH mimetics as a possible treatment for obesity. Such analogs would need to exert effects on energy expenditure and thermogenesis without adverse effects on other organ systems such as heart and bone.193 Such agents have been proven effective in studies with rodents and monkeys.194,195 An overview of some of the inputs into regulation of BAT and mitochondrial function is shown in Fig. 1-6.
References
1. Anand, B, Brobeck, J. Localization of a “feeding center” in the hypothalamus of the rat. Proc Soc Exper Biol Med. 1951;77:323–324.
2. Hetherington, A, Ranson, S. Hypothalamic lesions and adiposity in the rat. Anat Rec. 1940;78:149–172.
3. Mayer, J, Barrnett, R. Obesity following unilateral hypothalamic lesions in rats. Science. 1955;121:599–600.
4. Morrison, S, Mayer, J. Adipsia and aphagia in rats after lateral subthalamic lesions. Am J Physiol. 1957;219:1397–1402.
5. Owen, JJ, Parson, W, Crispell, K. Dietary dilution studies in gold thioglucose induced obesity in the mouse. Metabolism. 1953;2:363–366.
6. Debons, AF, Krimsky, I, From, A, et al. Site of action of gold thioglucose in the hypothalamic satiety center. Am J Physiol. 1970;219:1397–1402.
7. Nemeroff, CB, Lipton, MA, Kizer, JS. Models of neuroendocrine regulation: use of monosodium glutamate as an investigational tool. Dev Neurosci. 1978;1:102–109.
8. Coleman, DL. Effects of parabiosis of obese with diabetes and normal mice. Diabetologia. 1973;9:294–298.
9. Coleman, DL, Hummel, KP. Effects of parabiosis of normal with genetically diabetic mice. Am J Physiol. 1969;217:1298–1304.
10. Sklar, CA. Craniopharyngioma: endocrine sequelae of treatment. Pediatr Neurosurg. 1994;21(Suppl 1):120–123.
11. Zhang, Y, Proenca, R, Maffei, M, et al. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432.
12. Tartaglia, LA, Dembski, M, Weng, X, et al. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83:1263–1271.
13. Montague, CT, Farooqi, IS, Whitehead, JP, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387:903–908.
14. Clement, K, Vaisse, C, Lahlou, N, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392:398–401.
15. Flier, JS. The adipocyte: storage depot or node on the energy information superhighway? Cell. 1995;80:15–18.
16. Spiegelman, BM, Flier, JS. Obesity and the regulation of energy balance. Cell. 2001;104:531–543.
17. Saper, CB, Chou, TC, Elmquist, JK. The need to feed: homeostatic and hedonic control of eating. Neuron. 2002;36:199–211.
18. Yeomans, MR, Blundell, JE, Leshem, M. Palatability: response to nutritional need or need-free stimulation of appetite? Br J Nutr. 2004;92(Suppl 1):S3–14.
19. Kennedy, E. Dietary diversity, diet quality, and body weight regulation. Nutr Rev. 2004;62:S78–S81.
20. Zhang, M, Balmadrid, C, Kelley, AE. Nucleus accumbens opioid, GABAergic, and dopaminergic modulation of palatable food motivation: contrasting effects revealed by a progressive ratio study in the rat. Behav Neurosci. 2003;117:202–211.
21. Levine, JA. Nonexercise activity thermogenesis (NEAT): environment and biology. Am J Physiol Endocrinol Metab. 2004;286:E675–E685.
22. de Jesus, LA, Carvalho, SD, Ribeiro, MO, et al. The type 2 iodothyronine deiodinase is essential for adaptive thermogenesis in brown adipose tissue. J Clin Invest. 2001;108:1379–1385.
23. Silva, JE. The thermogenic effect of thyroid hormone and its clinical implications. Ann Intern Med. 2003;139:205–213.
24. Kotz, CM, Teske, JA, Billington, CJ. Neuroregulation of nonexercise activity thermogenesis and obesity resistance. Am J Physiol Regul Integr Comp Physiol. 2008;294:R699–R710.
25. Ravussin, E, Swinburn, BA. Pathophysiology of obesity. Lancet. 1992;340:404–408.
26. Gao, Q, Horvath, TL. Neurobiology of feeding and energy expenditure. Annu Rev Neurosci. 2007;30:367–398.
27. Bray, GA. 1989 McCollum Award lecture. Genetic and hypothalamic mechanisms for obesity–finding the needle in the haystack. Am J Clin Nutr. 1989;50:891–902.
28. Bray, GA. Reciprocal relation of food intake and sympathetic activity: experimental observations and clinical implications. Int J Obes Relat Metab Disord. 2000;24(Suppl 2):S8–17.
29. Kalra, SP, Dube, MG, Pu, S, et al. Interacting appetite-regulating pathways in the hypothalamic regulation of body weight. Endocr Rev. 1999;20:68–100.
30. Cowley, MA, Smart, JL, Rubinstein, M, et al. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484.
31. van den Pol, AN. Weighing the role of hypothalamic feeding neurotransmitters. Neuron. 2003;40:1059–1061.
32. Blundell, JE. Serotonin and appetite. Neuropharmacology. 1984;23:1537–1551.
33. Garfield, AS, Heisler, LK. Pharmacological targeting of the serotonergic system for the treatment of obesity. J Physiol. 2009;587:49–60.
34. Leibowitz, SF. Brain monoamines and peptides: role in the control of eating behavior. Fed Proc. 1986;45:1396–1403.
35. Nelson, DL, Gehlert, DR. Central nervous system biogenic amine targets for control of appetite and energy expenditure. Endocrine. 2006;29:49–60.
36. Clapham, JC, Arch, JR, Tadayyon, M. Anti-obesity drugs: A critical review of current therapies and future opportunities. Pharmacol Ther. 2001;89:81–121.
37. Fulton, S, Pissios, P, Manchon, RP, et al. Leptin regulation of the mesoaccumbens dopamine pathway. Neuron. 2006;51:811–822.
38. Hommel, JD, Trinko, R, Sears, RM, et al. Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron. 2006;51:801–810.
39. Georgescu, D, Sears, RM, Hommel, JD, et al. The hypothalamic neuropeptide melanin-concentrating hormone acts in the nucleus accumbens to modulate feeding behavior and forced-swim performance. J Neurosci. 2005;25:2933–2940.
40. Pissios, P, Frank, L, Kennedy, AR, et al. Dysregulation of the mesolimbic dopamine system and reward in MCH−/− mice. Biol Psychiatry. 2008;64:184–191.
41. Qian, S, Chen, H, Weingarth, D, et al. Neither agouti-related protein nor neuropeptide Y is critically required for the regulation of energy homeostasis in mice. Mol Cell Biol. 2002;22:5027–5035.
42. Luquet, S, Perez, FA, Hnasko, TS, et al. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science. 2005;310:683–685.
43. Tong, Q, Ye, CP, Jones, JE, et al. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci. 2008;11:998–1000.
44. Halatchev, IG, Ellacott, KL, Fan, W, et al. Peptide YY3-36 inhibits food intake in mice through a melanocortin-4 receptor-independent mechanism. Endocrinology. 2004;145:2585–2590.
45. Woods, SC. Gastrointestinal satiety signals I. An overview of gastrointestinal signals that influence food intake. Am J Physiol Gastrointest Liver Physiol. 2004;286:G7–13.
46. Gibbs, J, Young, RC, Smith, GP. Cholecystokinin elicits satiety in rats with open gastric fistulas. Nature. 1973;245:323–325.
47. Moran, TH, Kinzig, KP. Gastrointestinal satiety signals II. Cholecystokinin. Am J Physiol Gastrointest Liver Physiol. 2004;286:G183–G188.
48. Moran, TH, Smedh, U, Kinzig, KP, et al. Peptide YY(3-36) inhibits gastric emptying and produces acute reductions in food intake in rhesus monkeys. Am J Physiol Regul Integr Comp Physiol. 2005;288:R384–R388.
49. Tschop, M, Castaneda, TR, Joost, HG, et al. Physiology: does gut hormone PYY3–36 decrease food intake in rodents? Nature. 2004;430:1. [p following 165; discussion 2 p following 165].
50. Baggio, LL, Huang, Q, Brown, TJ, et al. Oxyntomodulin and glucagon-like peptide-1 differentially regulate murine food intake and energy expenditure. Gastroenterology. 2004;127:546–558.
51. Inui, A, Asakawa, A, Bowers, CY, et al. Ghrelin, appetite, and gastric motility: the emerging role of the stomach as an endocrine organ. FASEB J. 2004;18:439–456.
52. Gomez, R, Navarro, M, Ferrer, B, et al. A peripheral mechanism for CB1 cannabinoid receptor-dependent modulation of feeding. J Neurosci. 2002;22:9612–9617.
53. Kunos, G, Osei-Hyiaman, D, Liu, J, et al. Endocannabinoids and the control of energy homeostasis. J Biol Chem. 2008;283:33021–33025.
54. Proulx, K, Cota, D, Castaneda, TR, et al. Mechanisms of oleoylethanolamide-induced changes in feeding behavior and motor activity. Am J Physiol Regul Integr Comp Physiol. 2005;289:R729–R737.
55. Thorens, B, Larsen, PJ. Gut-derived signaling molecules and vagal afferents in the control of glucose and energy homeostasis. Curr Opin Clin Nutr Metab Care. 2004;7:471–478.
56. Williams, DL, Grill, HJ, Cummings, DE, et al. Vagotomy dissociates short- and long-term controls of circulating ghrelin. Endocrinology. 2003;144:5184–5187.
57. Friedman, MI, Harris, RB, Ji, H, et al. Fatty acid oxidation affects food intake by altering hepatic energy status. Am J Physiol. 1999;276:R1046–R1053.
58. Horn, CC, Ji, H, Friedman, MI. Etomoxir, a fatty acid oxidation inhibitor, increases food intake and reduces hepatic energy status in rats. Physiol Behav. 2004;81:157–162.
59. Marks, JL, Porte, D, Jr., Stahl, WL, et al. Localization of insulin receptor mRNA in rat brain by in situ hybridization. Endocrinology. 1990;127:3234–3236.
60. Bruning, JC, Gautam, D, Burks, DJ, et al. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000;289:2122–2125.
61. Woods, SC, Schwartz, MW, Baskin, DG, et al. Food intake and the regulation of body weight. Annu Rev Psychol. 2000;51:255–277.
62. Farooqi, S, Rau, H, Whitehead, J, et al. Ob gene mutations and human obesity. Proc Nutr Soc. 1998;57:471–475.
63. Halaas, JL, Gajiwala, KS, Maffei, M, et al. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546.
64. Stephens, TW, Basinski, M, Bristow, PK, et al. The role of neuropeptide Y in the antiobesity action of the obese gene product. Nature. 1995;377:530–532.
65. Farooqi, IS, Jebb, SA, Langmack, G, et al. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med. 1999;341:879–884.
66. Farooqi, IS, Matarese, G, Lord, GM, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. 2002;110:1093–1103.
67. Considine, RV, Sinha, MK, Heiman, ML, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med. 1996;334:292–295.
68. Frederich, RC, Hamann, A, Anderson, S, et al. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med. 1995;1:1311–1314.
69. Ahima, RS, Prabakaran, D, Mantzoros, C, et al. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382:250–252.
70. Welt, CK, Chan, JL, Bullen, J, et al. Recombinant human leptin in women with hypothalamic amenorrhea. N Engl J Med. 2004;351:987–997.
71. Elias, CF, Aschkenasi, C, Lee, C, et al. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron. 1999;23:775–786.
72. Nawrocki, AR, Scherer, PE. The delicate balance between fat and muscle: adipokines in metabolic disease and musculoskeletal inflammation. Curr Opin Pharmacol. 2004;4:281–289.
73. Steppan, CM, Lazar, MA. The current biology of resistin. J Intern Med. 2004;255:439–447.
74. Shoelson, SE, Goldfine, AB. Getting away from glucose: fanning the flames of obesity-induced inflammation. Nat Med. 2009;15:373–374.
75. Chronwall, BM. Anatomy and physiology of the neuroendocrine arcuate nucleus. Peptides. 1985;6(Suppl 2):1–11.
76. Stanley, BG, Leibowitz, SF. Neuropeptide Y injected in the paraventricular hypothalamus: a powerful stimulant of feeding behavior. Proc Natl Acad Sci U S A. 1985;82:3940–3943.
77. Allen, LG, Kalra, PS, Crowley, WR, et al. Comparison of the effects of neuropeptide Y and adrenergic transmitters on LH release and food intake in male rats. Life Sci. 1985;37:617–623.
78. Beck, B, Stricker-Krongrad, A, Nicolas, JP, et al. Chronic and continuous intracerebroventricular infusion of neuropeptide Y in Long-Evans rats mimics the feeding behaviour of obese Zucker rats. Int J Obes Relat Metab Disord. 1992;16:295–302.
79. Zarjevski, N, Cusin, I, Vettor, R, et al. Chronic intracerebroventricular neuropeptide-Y administration to normal rats mimics hormonal and metabolic changes of obesity. Endocrinology. 1993;133:1753–1758.
80. Billington, CJ, Briggs, JE, Harker, S, et al. Neuropeptide Y in hypothalamic paraventricular nucleus: a center coordinating energy metabolism. Am J Physiol. 1994;266:R1765–R1770.
81. Wilding, JP, Gilbey, SG, Bailey, CJ, et al. Increased neuropeptide-Y messenger ribonucleic acid (mRNA) and decreased neurotensin mRNA in the hypothalamus of the obese (ob/ob) mouse. Endocrinology. 1993;132:1939–1944.
82. Erickson, JC, Clegg, KE, Palmiter, RD. Sensitivity to leptin and susceptibility to seizures of mice lacking neuropeptide Y. Nature. 1996;381:415–421.
83. Erickson, JC, Hollopeter, G, Palmiter, RD. Attenuation of the obesity syndrome of ob/ob mice by the loss of neuropeptide Y. Science. 1996;274:1704–1707.
84. Segal-Lieberman, G, Trombly, DJ, Juthani, V, et al. NPY ablation in C57BL/6 mice leads to mild obesity and to an impaired refeeding response to fasting. Am J Physiol Endocrinol Metab. 2003;284:E1131–E1139.
85. Sindelar, DK, Ste Marie, L, Miura, GI, et al. Neuropeptide Y is required for hyperphagic feeding in response to neuroglucopenia. Endocrinology. 2004;145:3363–3368.
86. Tsujii, S, Bray, GA. Acetylation alters the feeding response to MSH and beta-endorphin. Brain Res Bull. 1989;23:165–169.
87. Miller, MW, Duhl, DM, Vrieling, H, et al. Cloning of the mouse agouti gene predicts a secreted protein ubiquitously expressed in mice carrying the lethal yellow mutation. Genes Dev. 1993;7:454–467.
88. Michaud, EJ, Bultman, SJ, Klebig, ML, et al. A molecular model for the genetic and phenotypic characteristics of the mouse lethal yellow (Ay) mutation. Proc Natl Acad Sci U S A. 1994;91:2562–2566.
89. Yen, TT, Gill, AM, Frigeri, LG, et al. Obesity, diabetes, and neoplasia in yellow A(vy)/− mice: ectopic expression of the agouti gene. FASEB J. 1994;8:479–488.
90. Shutter, JR, Graham, M, Kinsey, AC, et al. Hypothalamic expression of ART, a novel gene related to agouti, is up-regulated in obese and diabetic mutant mice. Genes Dev. 1997;11:593–602.
91. Tota, MR, Smith, TS, Mao, C, et al. Molecular interaction of Agouti protein and Agouti-related protein with human melanocortin receptors. Biochemistry. 1999;38:897–904.
92. Huszar, D, Lynch, CA, Fairchild-Huntress, V, et al. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141.
93. Yeo, GS, Farooqi, IS, Aminian, S, et al. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet. 1998;20:111–112.
94. Vaisse, C, Clement, K, Guy-Grand, B, et al. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat Genet. 1998;20:113–114.
95. Vaisse, C, Clement, K, Durand, E, et al. Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J Clin Invest. 2000;106:253–262.
96. Krude, H, Biebermann, H, Luck, W, et al. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet. 1998;19:155–157.
97. Qu, D, Ludwig, DS, Gammeltoft, S, et al. A role for melanin-concentrating hormone in the central regulation of feeding behaviour. Nature. 1996;380:243–247.
98. Whitlock, BK, Daniel, JA, McMahon, CD, et al. Intracerebroventricular melanin-concentrating hormone stimulates food intake in sheep. Domest Anim Endocrinol. 2005;28:224–232.
99. Ito, M, Gomori, A, Ishihara, A, et al. Characterization of MCH-mediated obesity in mice. Am J Physiol Endocrinol Metab. 2003;284:E940–E945.
100. Shimada, M, Tritos, NA, Lowell, BB, et al. Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature. 1998;396:670–674.
101. Marsh, DJ, Weingarth, DT, Novi, DE, et al. Melanin-concentrating hormone 1 receptor-deficient mice are lean, hyperactive, and hyperphagic and have altered metabolism. Proc Natl Acad Sci U S A. 2002;99:3240–3245.
102. Chen, Y, Hu, C, Hsu, CK, et al. Targeted disruption of the melanin-concentrating hormone receptor-1 results in hyperphagia and resistance to diet-induced obesity. Endocrinology. 2002;143:2469–2477.
103. Segal-Lieberman, G, Bradley, RL, Kokkotou, E, et al. Melanin-concentrating hormone is a critical mediator of the leptin-deficient phenotype. Proc Natl Acad Sci U S A. 2003;100:10085–10090.
104. Astrand, A, Bohlooly, YM, Larsdotter, S, et al. Mice lacking melanin-concentrating hormone receptor 1 demonstrate increased heart rate associated with altered autonomic activity. Am J Physiol Regul Integr Comp Physiol. 2004;287:R749–R758.
105. Shearman, LP, Camacho, RE, Sloan Stribling, D, et al. Chronic MCH-1 receptor modulation alters appetite, body weight and adiposity in rats. Eur J Pharmacol. 2003;475:37–47.
106. Kojima, M, Hosoda, H, Date, Y, et al. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402:656–660.
107. Tschop, M, Smiley, DL, Heiman, ML. Ghrelin induces adiposity in rodents. Nature. 2000;407:908–913.
108. Wren, AM, Seal, LJ, Cohen, MA, et al. Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab. 2001;86:5992.
109. Cummings, DE, Shannon, MH. Roles for ghrelin in the regulation of appetite and body weight. Arch Surg. 2003;138:389–396.
110. Tritos, NA, Mun, E, Bertkau, A, et al. Serum ghrelin levels in response to glucose load in obese subjects post-gastric bypass surgery. Obes Res. 2003;11:919–924.
111. Cummings, DE, Weigle, DS, Frayo, RS, et al. Plasma ghrelin levels after diet-induced weight loss or gastric bypass surgery. N Engl J Med. 2002;346:1623–1630.
112. Haqq, AM, Stadler, DD, Rosenfeld, RG, et al. Circulating ghrelin levels are suppressed by meals and octreotide therapy in children with Prader-Willi syndrome. J Clin Endocrinol Metab. 2003;88:3573–3576.
113. Cowley, MA, Smith, RG, Diano, S, et al. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron. 2003;37:649–661.
114. Batterham, RL, Le Roux, CW, Cohen, MA, et al. Pancreatic polypeptide reduces appetite and food intake in humans. J Clin Endocrinol Metab. 2003;88:3989–3992.
115. Batterham, RL, Cowley, MA, Small, CJ, et al. Gut hormone PYY(3-36) physiologically inhibits food intake. Nature. 2002;418:650–654.
116. Tang-Christensen, M, Vrang, N, Ortmann, S, et al. Central administration of ghrelin and agouti-related protein (83–132) increases food intake and decreases spontaneous locomotor activity in rats. Endocrinology. 2004;145:4645–4652.
117. Alvarez Bartolome, M, Borque, M, Martinez-Sarmiento, J, et al. Peptide YY secretion in morbidly obese patients before and after vertical banded gastroplasty. Obes Surg. 2002;12:324–327.
118. Flint, A, Raben, A, Astrup, A, et al. Glucagon-like peptide 1 promotes satiety and suppresses energy intake in humans. J Clin Invest. 1998;101:515–520.
119. Gutzwiller, JP, Goke, B, Drewe, J, et al. Glucagon-like peptide-1: a potent regulator of food intake in humans. Gut. 1999;44:81–86.
120. Schmidt, PT, Naslund, E, Gryback, P, et al. Peripheral administration of GLP-2 to humans has no effect on gastric emptying or satiety. Regul Pept. 2003;116:21–25.
121. Brubaker, PL, Drucker, DJ. Minireview: Glucagon-like peptides regulate cell proliferation and apoptosis in the pancreas, gut, and central nervous system. Endocrinology. 2004;145:2653–2659.
122. Winchester, P, Kendall, K, Peters, H, et al. The effect of therapeutic horseback riding on gross motor function and gait speed in children who are developmentally delayed. Phys Occup Ther Pediatr. 2002;22:37–50.
123. Ravussin, E, Harper, IT, Rising, R, et al. Energy expenditure by doubly labeled water: validation in lean and obese subjects. Am J Physiol. 1991;261:E402–E409.
124. Rolfe, DF, Brown, GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev. 1997;77:731–758.
125. Wang, Z, O’Connor, TP, Heshka, S, et al. The reconstruction of Kleiber’s law at the organ-tissue level. J Nutr. 2001;131:2967–2970.
126. Ravussin, E, Lillioja, S, Anderson, TE, et al. Determinants of 24-hour energy expenditure in man. Methods and results using a respiratory chamber. J Clin Invest. 1986;78:1568–1578.
127. Levin, N, Nelson, C, Gurney, A, et al. Decreased food intake does not completely account for adiposity reduction after ob protein infusion. Proc Natl Acad Sci U S A. 1996;93:1726–1730.
128. Cusin, I, Rouru, J, Visser, T, et al. Involvement of thyroid hormones in the effect of intracerebroventricular leptin infusion on uncoupling protein-3 expression in rat muscle. Diabetes. 2000;49:1101–1105.
129. Hill, JO, Wyatt, HR, Reed, GW, Peters, JC. Obesity and the environment: where do we go from here? Science. 2003;299:853–855.
130. Kokkotou, E, Jeon, JY, Wang, X, et al. Mice with MCH ablation resist diet-induced obesity through strain-specific mechanisms. Am J Physiol Regul Integr Comp Physiol. 2005;289:R117–R124.
131. Livingstone, MB. Assessment of food intakes: are we measuring what people eat? Br J Biomed Sci. 1995;52:58–67.
132. Andersson, LB. Genes and obesity. Ann Med. 1996;28:5–7.
133. Bogardus, C, Lillioja, S, Ravussin, E, et al. Familial dependence of the resting metabolic rate. N Engl J Med. 1986;315:96–100.
134. Bouchard, C, Tremblay, A, Despres, JP, et al. The response to long-term overfeeding in identical twins. N Engl J Med. 1990;322:1477–1482.
135. Stunkard, AJ, Harris, JR, Pedersen, NL, et al. The body-mass index of twins who have been reared apart. N Engl J Med. 1990;322:1483–1487.
136. Zurlo, F, Lillioja, S, Esposito-Del Puente, A, et al. Low ratio of fat to carbohydrate oxidation as predictor of weight gain: study of 24-h RQ. Am J Physiol. 1990;259:E650–E657.
137. Tataranni, PA, Harper, IT, Snitker, S, et al. Body weight gain in free-living Pima Indians: effect of energy intake vs expenditure. Int J Obes Relat Metab Disord. 2003;27:1578–1583.
138. Ravussin, E, Lillioja, S, Knowler, WC, et al. Reduced rate of energy expenditure as a risk factor for body-weight gain. N Engl J Med. 1988;318:467–472.
139. Salbe, AD, Weyer, C, Harper, I, et al. Assessing risk factors for obesity between childhood and adolescence: II. Energy metabolism and physical activity. Pediatrics. 2002;110:307–314.
140. Scherrer, U, Randin, D, Tappy, L, et al. Body fat and sympathetic nerve activity in healthy subjects. Circulation. 1994;89:2634–2640.
141. Rumantir, MS, Vaz, M, Jennings, GL, et al. Neural mechanisms in human obesity-related hypertension. J Hypertens. 1999;17:1125–1133.
142. Jansson, PA, Larsson, A, Smith, U, et al. Glycerol production in subcutaneous adipose tissue in lean and obese humans. J Clin Invest. 1992;89:1610–1617.
143. Kokkoris, P, Pi-Sunyer, FX. Obesity and endocrine disease. Endocrinol Metab Clin North Am. 2003;32:895–914.
144. Ravussin, E, Bogardus, C. Energy balance and weight regulation: genetics versus environment. Br J Nutr. 2000;83(Suppl 1):S17–S20.
145. Salbe, AD, Weyer, C, Harper, I, et al. Relation between physical activity and obesity. Am J Clin Nutr. 2003;78:193–194.
146. Ekelund, U, Aman, J, Yngve, A, et al. Physical activity but not energy expenditure is reduced in obese adolescents: a case-control study. Am J Clin Nutr. 2002;76:935–941.
147. Butler, AA, Marks, DL, Fan, W, et al. Melanocortin-4 receptor is required for acute homeostatic responses to increased dietary fat. Nat Neurosci. 2001;4:605–611.
148. Zhou, D, Shen, Z, Strack, AM, et al. Enhanced running wheel activity of both Mch1r- and Pmch-deficient mice. Regul Pept. 2005;124:53–63.
149. Jakicic, JM. Exercise in the treatment of obesity. Endocrinol Metab Clin North Am. 2003;32:967–980.
150. Levine, JA, Eberhardt, NL, Jensen, MD. Role of nonexercise activity thermogenesis in resistance to fat gain in humans. Science. 1999;283:212–214.
151. Levine, J, Baukol, P, Pavlidis, I. The energy expended in chewing gum. N Engl J Med. 1999;341:2100.
152. Levine, J, Melanson, EL, Westerterp, KR, et al. Measurement of the components of nonexercise activity thermogenesis. Am J Physiol Endocrinol Metab. 2001;281:E670–E675.
153. Levine, JA, Nygren, J, Short, KR, et al. Effect of hyperthyroidism on spontaneous physical activity and energy expenditure in rats. J Appl Physiol. 2003;94:165–170.
154. Cannon, B, Nedergaard, J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84:277–359.
155. Lowell, BB, V, SS, Hamann, A, et al. Development of obesity in transgenic mice after genetic ablation of brown adipose tissue. Nature. 1993;366:740–742.
156. Feldmann, HM, Golozoubova, V, Cannon, B, et al. UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 2009;9:203–209.
157. Nedergaard, J, Bengtsson, T, Cannon, B. Unexpected evidence for active brown adipose tissue in adult humans. Am J Physiol Endocrinol Metab. 2007;293:E444–E452.
158. Cypess, AM, Lehman, S, Williams, G, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1509–1517.
159. van Marken Lichtenbelt, WD, Vanhommerig, JW, Smulders, NM, et al. Cold-activated brown adipose tissue in healthy men. N Engl J Med. 2009;360:1500–1508.
160. Virtanen, KA, Lidell, ME, Orava, J, et al. Functional brown adipose tissue in healthy adults. N Engl J Med. 2009;360:1518–1525.
161. Seale, P, Kajimura, S, Spiegelman, BM. Transcriptional control of brown adipocyte development and physiological function–of mice and men. Genes Dev. 2009;23:788–797.
162. Lin, J, Wu, PH, Tarr, PT, et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–135.
163. Seale, P, Bjork, B, Yang, W, et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454:961–967.
164. Nicholls, DG. A history of UCP1. Biochem Soc Trans. 2001;29:751–755.
165. Nisoli, E, Clementi, E, Paolucci, C, et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896–899.
166. Hock, MB, Kralli, A. Transcriptional control of mitochondrial biogenesis and function. Annu Rev Physiol. 2009;71:177–203.
167. Lagouge, M, Argmann, C, Gerhart-Hines, Z, et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109–1122.
168. Rosenbaum, M, Keibel, RL, Hirsch, J. Obesity. N Engl J Med. 1997;337:396–406.
169. Rothwell, NJ, Stock, MJ. A role for brown adipose tissue in diet-induced thermogenesis. Nature. 1979;281:31–35.
170. Stock, MJ. Gluttony and thermogenesis revisited. Int J Obes Relat Metab Disord. 1999;23:1105–1117.
171. Glick, Z, Teague, RJ, Bray, GA. Brown adipose tissue: thermic response increased by a single low protein, high carbohydrate meal. Science. 1981;213:1125–1127.
172. Bachman, ES, Dhillon, H, Zhang, CY, et al. betaAR signaling required for diet-induced thermogenesis and obesity resistance. Science. 2002;297:843–845.
173. Tataranni, PA, Young, JB, Bogardus, C, et al. A low sympathoadrenal activity is associated with body weight gain and development of central adiposity in Pima Indian men. Obes Res. 1997;5:341–347.
174. Snitker, S, Macdonald, I, Ravussin, E, et al. The sympathetic nervous system and obesity: role in aetiology and treatment. Obes Rev. 2000;1:5–15.
175. Arch, JR, Ainsworth, AT, Cawthorne, MA, et al. Atypical beta-adrenoceptor on brown adipocytes as target for anti-obesity drugs. Nature. 1984;309:163–165.
176. Himms-Hagen, J, Cui, J, Danforth, E, Jr., et al. Effect of CL-316,243, a thermogenic beta 3-agonist, on energy balance and brown and white adipose tissues in rats. Am J Physiol. 1994;266:R1371–R1382.
177. Levin, BE, Triscari, J, Marquet, E, et al. Dietary obesity and neonatal sympathectomy. I. Effects on body composition and brown adipose. Am J Physiol. 1984;247:R979–R987.
178. Rohrer, DK, Chruscinski, A, Schauble, EH, et al. Cardiovascular and metabolic alterations in mice lacking both beta1- and beta2-adrenergic receptors. J Biol Chem. 1999;274:16701–16708.
179. Thomas, SA, Palmiter, RD. Thermoregulatory and metabolic phenotypes of mice lacking noradrenaline and adrenaline. Nature. 1997;387:94–97.
180. Susulic, VS, Frederich, RC, Lawitts, J, et al. Targeted disruption of the beta 3-adrenergic receptor gene. J Biol Chem. 1995;270:29483–29492.
181. Lisser, H. The frequency of endogenous endocrine obesity and its treatment by glandular therapy. Cal West Med. 1924;22:509–514.
182. Mason, E. The Treatment of Obesity, Can. J Med. 1924;14:1052–1056.
183. Bianco, AC, Maia, AL, da Silva, WS, et al. Adaptive activation of thyroid hormone and energy expenditure. Biosci Rep. 2005;25:191–208.
184. Lebon, V, Dufour, S, Petersen, KF, et al. Effect of triiodothyronine on mitochondrial energy coupling in human skeletal muscle. J Clin Invest. 2001;108:733–737.
185. al-Adsani, H, Hoffer, LJ, Silva, JE. Resting energy expenditure is sensitive to small dose changes in patients on chronic thyroid hormone replacement. J Clin Endocrinol Metab. 1997;82:1118–1125.
186. Leonard, JL, Mellen, SA, Larsen, PR. Thyroxine 5′-deiodinase activity in brown adipose tissue. Endocrinology. 1983;112:1153–1155.
187. Silva, JE, Larsen, PR. Adrenergic activation of triiodothyronine production in brown adipose tissue. Nature. 1983;305:712–713.
188. Ribeiro, MO, Carvalho, SD, Schultz, JJ, et al. Thyroid hormone–sympathetic interaction and adaptive thermogenesis are thyroid hormone receptor isoform specific. J Clin Invest. 2001;108:97–105.
189. Bianco, AC, Silva, JE. Optimal response of key enzymes and uncoupling protein to cold in BAT depends on local T3 generation. Am J Physiol. 1987;253:E255–E263.
190. Reinehr, T. Obesity and thyroid function. Mol Cell Endocrinol. 2009. [[in press]].
191. Schneider, MJ, Fiering, SN, Pallud, SE, et al. Targeted disruption of the type 2 selenodeiodinase gene (DIO2) results in a phenotype of pituitary resistance to T4. Mol Endocrinol. 2001;15:2137–2148.
192. Curcio, C, Lopes, AM, Ribeiro, MO, et al. Development of compensatory thermogenesis in response to overfeeding in hypothyroid rats. Endocrinology. 1999;140:3438–3443.
193. Baxter, JD, Webb, P. Thyroid hormone mimetics: potential applications in atherosclerosis, obesity and type 2 diabetes. Nat Rev Drug Discov. 2009;8:308–320.
194. Villicev, CM, Freitas, FR, Aoki, MS, et al. Thyroid hormone receptor beta-specific agonist GC-1 increases energy expenditure and prevents fat-mass accumulation in rats. J Endocrinol. 2007;193:21–29.
195. Grover, GJ, Mellstrom, K, Ye, L, et al. Selective thyroid hormone receptor-beta activation: a strategy for reduction of weight, cholesterol, and lipoprotein (a) with reduced cardiovascular liability. Proc Natl Acad Sci U S A. 2003;100:10067–10072.
196. Butler, AA, Kesterson, RA, Khong, K, et al. A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology. 2000;141:3518–3521.
197. Ishii, M, Fei, H, Friedman, JM. Targeted disruption of GPR7, the endogenous receptor for neuropeptides B and W, leads to metabolic defects and adultonset obesity. Proc Natl Acad Sci U S A. 2003;100:10540–10545.
198. Martinez-Botas, J, Anderson, JB, Tessier, D, et al. Absence of perilipin results in leanness and reverses obesity in Lepr(db/db) mice. Nat Genet. 2000;26:474–479.
199. Xia, Z, Stanhope, KL, Digitale, E, et al. Acylation-stimulating protein (ASP)/complement C3adesArg deficiency results in increased energy expenditure in mice. J Biol Chem. 2004;279:4051–4057.
200. Abu-Elheiga, L, Matzuk, MM, Abo-Hashema, KA, et al. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science. 2001;291:2613–2616.
201. Cummings, DE, Brandon, EP, Planas, JV, et al. Genetically lean mice result from targeted disruption of the RII beta subunit of protein kinase A. Nature. 1996;382:622–626.
202. Energack, S, Jacobsson, A, Simpson, EM, et al. Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obsess. Nature. 1997;387:90–94.
203. Zhou, Z, Yon Toh, S, Chen, Z, et al. Cidea-deficient mice have lean phenotype and are resistant to obesity. Nat Genet. 2003;35:49–56.
204. Cohen, P, Miyazaki, M, Socci, ND, et al. Role for stearoyl-CoA desaturase-1 in leptin-mediated weight loss. Science. 2002;297:240–243.
205. Hinney, A, Schmidt, A, Nottebom, K, et al. Several mutations in the melanocortin-4 receptor gene including a nonsense and a frameshift mutation associated with dominantly inherited obesity in humans [In Process Citation]. J Clin Endocrinol Metab. 1999;84:1483–1486.
206. Lee, YS, Poh, LK, Loke, KY. A novel melanocortin 3 receptor gene (MC3R) mutation associated with severe obesity. J Clin Endocrinol Metab. 2002;87:1423–1426.
207. Jackson, RS, Creemer, JW, Ohagi, S, et al. Obesity and impaired prohomone processing associated with mutations in the human prohormone convertase 1 gene [see comments]. Nat Genet. 1997;16:303–306.
208. Ristom, M, Muller-Wieland, D, Pfeiffer, A, et al. Obesity associated with a mutation in a genetic regulator of adipocyte differentiation. N Engl J Med. 1998;399:953–959.