Insulin Secretion
Regulated insulin secretion from pancreatic beta cells is critical to health. Both insufficient insulin secretion (resulting in diabetes mellitus) and excess insulin secretion (leading to hypoglycemia) are life threatening. The complexity of regulated insulin secretion in health becomes apparent with the difficulty of reproducing it in patients with insulin deficiency. Appropriate regulated insulin secretion depends on several components. First, development and maintenance of an appropriate number of functional insulin-secreting β cells, often collectively referred to as the β cell mass, are required.1 Second, β cells need to sense the key regulators of insulin secretion, most importantly, the prevailing blood glucose concentration.2 Third, proinsulin synthesis and processing (see Chapter 6) must proceed at a rate adequate to provide sufficient insulin for secretion, the insulin being targeted to insulin vesicles that are available for secretion (secretion competent).3 Because most insulin secretory granules are not secretion competent (presumably because of aging or other factors),4–6 the focus for regulation of insulin secretion is the pool of insulin secretory vesicles that are primed, docked, and available for secretion.5 Finally, minute-by-minute changes in insulin release from these primed and docked vesicles must be tightly linked to the regulating signals that affect the β cell. Predominant among these is the circulating glucose concentration.7 In addition, other circulating fuels (free fatty acids, amino acids)8–11; other circulating hormones such as glucagon-like peptide-1 (GLP-1),12–14 glucose-dependent insulinotropic polypeptide (GIP),15 and epinephrine16; innervation by adrenergic and cholinergic fibers17–19; and paracrine effects such as islet amyloid polypeptide (IAPP) and insulin itself20–23 are important regulators of insulin secretion.
Our understanding of these complex processes that underlie successful regulated insulin secretion is hampered by the complexity of the anatomy of the endocrine organ that subserves regulated insulin secretion (Fig. 7-1). The islet of Langerhans was named after Paul Langerhans (1847-1888) (Fig. 7-2), who when he was a medical student first described the appearance of these islets scattered in the pancreas.24 Langerhans died at the age of 41 of tuberculosis, most likely contracted by performing autopsies. The link between the pancreas and the regulation of glucose homeostasis was finally established in 1889 by Joseph von Mering (1849-1908) and Oscar Minkowski (1858-1931), who in order to explore the involvement of the pancreas in fat absorption performed pancreas resections in dogs,25 resulting in polyuria and polydipsia due to glycosuria and, eventually, recognition of the role of the pancreas in glucose regulation.
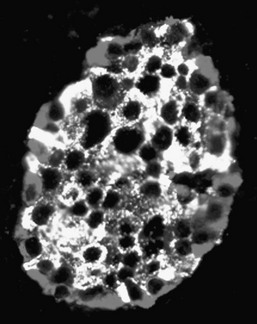
FIGURE 7-1 A human islet of Langerhans stained by immunofluoresence for insulin (green) and glucagon (blue).


FIGURE 7-2 A, Paul Langerhans (shown here in a rare family photograph) died young from tuberculosis. B, The face page of the thesis of Paul Langerhans defended on February 18, 1869. (From Schadewaldt H: Geschichte des diabetes mellitus. Berlin, Springer-Verlag, 1975, pp 52–53.)
Islet Structure in Health
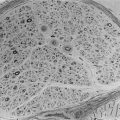
In humans, approximately one million islets of Langerhans are scattered in the pancreas.26–29 Islets vary greatly in size, with larger islets providing most of the insulin-secreting β cells and typically containing ≈2000 β cells.26,27,30 Each islet has its own complex anatomy, with the core consisting mainly of β cells that are tightly interconnected by gap junctions31–33 (Fig. 7-3), surrounded by a mantle of other endocrine cells, including glucagon-secreting α cells, somatostatin-secreting δ cells, and pancreatic polypeptide–secreting (PP) cells.29,34–36 In humans, ≈35% of islet cells are nonendocrine cells. The nature of the extracellular matrix proteins is also important in the function and development of the islet.37,38 Regional heterogeneity is seen in the pattern of endocrine cells in islets, for example, with glucagon-secreting α cells more abundant in the body and the tail of the pancreas, in contrast to the more frequent PP-secreting cells present in the head of the pancreas.39,40 Islets are richly vascularized, receiving ≈10% of pancreatic blood flow, despite accounting for only ≈1% of pancreatic mass.27,41–43 Islets are also richly innervated by nerve fibers tracking with vessels44–46 (Fig. 7-4). The arteriolar input to the islet initially supplies the β cell–rich islet core before it is further distributed to the α cell and/or the PP-enriched mantle.42 The consequence of this is that non–β cell endocrine cells in the islet are exposed to very high paracrine insulin concentrations that may be important in normal function.47–49 The development of the endocrine pancreas is addressed in detail elsewhere (see Chapter 5).50 It is clear that β cell mass is regulated in adult rodents, increasing in response to hyperglycemia, obesity, and pregnancy.51–54 β Cell mass is also greater in obese versus lean humans, but the increment is much less marked (≈0.5-fold vs. ≈10-fold) than in obese rodents.55 Although an adaptive increase in β cell mass in mice is largely accomplished by an increase in β cell replication, β cell replication in vivo is very rare in humans.51,54,55 Because β cell replication appears to be rare in adult humans in contrast to mice, the question arises as to whether there is an alternative source of new β cells in adult humans. It has been proposed that new islets may be formed during adult life from ductal precursors recapitulating the pattern observed during development.50,56,57 Although islet buds are frequently seen on exocrine ducts in adult life in humans and rodents,55,57 it is difficult to prove that these are newly forming islets rather than products of arrested development. Interest now is focused on the possibility that stem cells might provide an ongoing source of β cells in health and may be harnessed therapeutically in diabetes. Data are conflicting regarding whether marrow-derived stem cells are potential precursors for β cells in rodents.58,59 As yet, no data are available on humans.
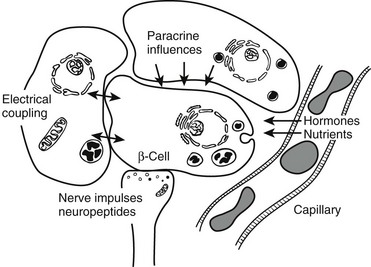
FIGURE 7-3 Schematic diagram of β cell with vascular, neural, and paracrine influences. (Adapted with permission from Hellerstrom C: The life story of the pancreatic β cell. Diabetologia 26:395, 1984.)
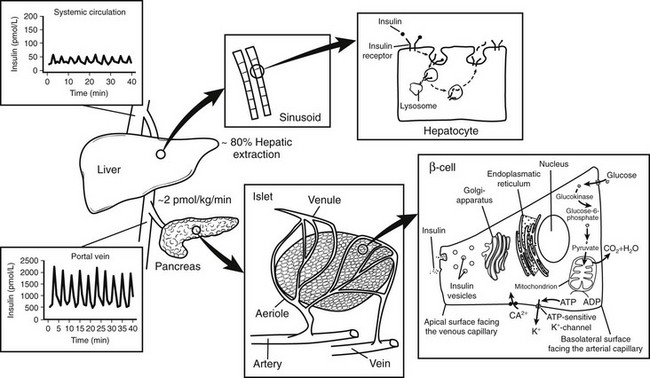
FIGURE 7-4 Relationship between insulin secretion from the islet, insulin clearance, and insulin action at the hepatocytes. Insulin is secreted at a rate of approximately 2 pmol/L/min from the islets of Langerhans into the portal vein. Approximately 80% of the total amount of insulin secreted is extracted from the liver sinusoids. Thus, oscillations in insulin secretion in the portal vein (≈2000 pmol/L) largely exceed those measured in the peripheral circulation (≈50 pmol/L). Following insulin binding, the insulin receptor-ligand complex is internalized into the cytosol of the hepatocyte. While insulin mainly undergoes enzymatic degradation in the lysosomes, the insulin receptor is reinserted into the plasma membrane within approximately 5 minutes.
Islet Function in Health
Given the critical importance of avoiding hypoglycemia, it is not surprising that the circulating glucose concentration is so predominant in the regulating of insulin secretion. Indeed the glucose dose response curve for insulin secretion by isolated human islets is remarkably similar to that of humans in vivo.60–62 So that beta cells can “sense” the prevailing blood glucose, islets need to be well vascularized, and the cytosol of β cells should be readily accessible to glucose. This is accomplished by rich islet vascularization with fenestrated vessels and abundant glucose-2-transporter proteins on the β cell surface.27,41–43,63,64 The latter allow rapid equilibrium of glucose between extracellular and intracellular concentrations. Given this rapid access of circulating glucose to the β cell cytosol, β cells “sense” the circulating glucose concentration by the rate-limiting step in glucose metabolism—phosphorylation of glucose to glucose-6-phosphate.65,66 This is accomplished in β cells by the expression of a glucokinase isoform with a Km of ≈150 mg/dL (7 mM) in the middle of the physiologic glucose concentration range.67 Thus the rate of provision of glucose-6-phosphate into the glycolytic pathway and the subsequent provision of pyruvate for the tricarboxylic acid cycle are closely linked to the plasma glucose concentration.66 The resultant mitochondrial pyruvate oxidation generates adenosine triphosphate (ATP), which in turn activates ATP-sensitive potassium channels (closing these channels), leading to cell depolarization and an influx of ionized calcium.68,69 The ionized calcium is believed to interact with primed docked insulin secretory vesicles that then discharge their contents either wholly (by exocytosis) or in part (by “kiss and run”) into the extracellular space.70,71 This rich vascular supply and the fenestrated vessels ensure rapid delivery of secreted insulin into the pancreatic venous efflux and then to the hepatic portal vein.
When islets are stimulated by an abrupt increase in glucose concentration in vitro (perifusion) or in vivo (intravenous glucose tolerance test), the resultant insulin secretion is biphasic60,72–74 (Fig. 7-5). An immediate first phase of insulin secretion occurs over ≈3 minutes and is followed by a more prolonged second phase of insulin secretion. This observation led to the concept proposed by Grodsky of distinct subcellular pools of insulin.5,75,76 More recently, these hypothetical pools have developed a likely anatomic basis. First-phase insulin secretion appears to reflect the immediate discharge of primed and docked insulin secretory vesicles, while second-phase insulin secretion most likely requires priming and mobilization of insulin vesicles before the time of their discharge.77 The exact molecular processes involved in the priming and mobilization of insulin vesicles remain unknown but may include provision of ATP following mitochondrial oxidation of pyruvate. An ATP-independent pathway for glucose-mediated insulin secretion also has been proposed, given the observation that when the KATP channel is defective as the result of mutations in the sulphonylurea receptor, some degree of glucose-mediated insulin secretion prevails.78
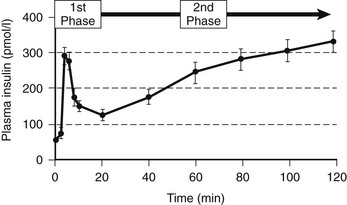
FIGURE 7-5 The relationship between the timing of the introduction of an intravenous glucose infusion (t = 0 min) and plasma insulin concentrations. A rapid first phase of insulin secretion is followed by second-phase secretion. (From Pratley RE, Weyer C: The role of impaired early insulin secretion in the pathogenesis of type II diabetes mellitus. Diabetologia 44:931, 2001.)
It has been argued that there is no physiologic counterpart of first-phase insulin secretion in vivo, given the intravenous glucose challenge used to elicit it. To the contrary, almost all insulin secretion in vivo is likely released from the same pool, as ≈90% of insulin secretion is derived from discrete insulin secretory bursts that occur at ≈4 minute intervals.79,80 Thus regulation of ≈90% of insulin secretion can be accomplished through changes in size (secretory burst mass) or frequency of these discrete insulin pulses. The pacemaker for this high-frequency pulsatile insulin secretion is unknown, although it is present in individual islets, in that isolated independent islets secrete insulin in pulses every ≈4 minutes.60,73,81,82 Whatever the basis for the pacemaker, it is remarkably robust and does not appear to change under almost any conditions. Under almost all conditions studied, regulated changes in insulin secretion are accomplished exclusively through changes in the insulin secretory burst mass. For example, enhanced insulin secretion as a result of glucose ingestion or glucose infusion, infusion or ingestion of sulphonylurea drugs, and GLP-1 infusion is accomplished by an increase in the insulin secretory burst mass60,83–86 (Fig. 7-6). Suppression of insulin secretion by somatostatin and insulin-like growth factor-1 (IGF-1) is accomplished by a reduction in insulin burst mass.84,87 The one circumstance in which pulse frequency has been shown to change is induction of general anesthesia. Induction of general anesthesia profoundly suppresses insulin secretion, but although this is accomplished by inhibition of insulin burst mass, insulin pulse frequency increases under these circumstances.88
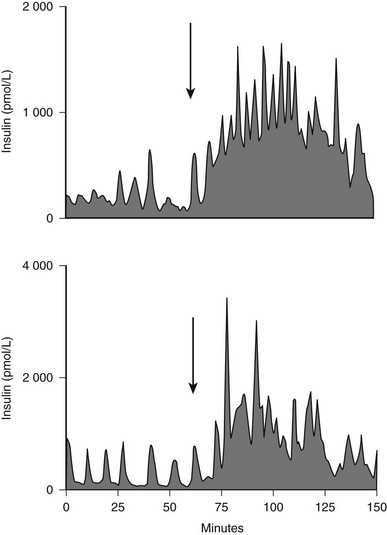
FIGURE 7-6 The portal vein insulin concentration before (0-60) and after meal ingestion (arrow) in two representative dogs. The portal vein insulin concentration excursions vary from approximately 300 to 1000 pmol/L before meal ingestion to 2000 to 4000 pmol/L after meal ingestion. Similar concentration profiles have been seen in the human portal vein. (With permission from Porksen N, Munn S, Steers J, Veldhuis JD, Butler PC: Effects of glucose ingestion versus infusion on pulsatile insulin secretion: The incretin effect is achieved by amplification of insulin secretory burst mass. Diabetes 45:1317–1323, 1996.)
Because most insulin secretion in vivo arises from these discrete insulin secretory bursts, the approximately one million islets scattered in the exocrine pancreas must be coordinated to discharge their insulin secretory bursts synchronously. This coordination is accomplished at least in part by the intrinsic neural network in the pancreas, analogous to the intrinsic neural network in the gut, which allows coordinated peristalsis; it probably also occurs through entrainment by the oscillating glucose concentration, which presumably arises as a consequence of insulin pulses.89–93 As a consequence of this coordination, the insulin concentration wavefront that affects the liver each ≈4 minutes measures ≈2000 pmol/L in the fasting state and as much as 5000 pmol/L after meal ingestion.94 The amplitude of this concentration wavefront is greatly attenuated (≈50 pmol/L) by the time the insulin is released into the systemic circulation, presumably as a result of both dilution of portal vein insulin in the systemic circulation and selective extraction of insulin pulses in the liver (see Figs. 7-4 and 7-6). Although no studies have knowingly reproduced these dramatic portal vein insulin concentration dynamics in vivo, early-phase insulin secretion after glucose ingestion, which likely approximates these kinetics in the portal vein, may be important in suppressing hepatic glucose production, an important adaptive response to ingested glucose to minimize the postprandial increase in glucose concentrations (see Chapter 10). It is unknown to what extent exposure of the liver to these dramatic oscillations in insulin concentration is important for insulin sensitivity. Infusions of much smaller insulin pulses versus a continuous insulin infusion have been shown to enhance insulin action.93,95,96 Insulin is also secreted in an ultradian rhythm with a frequency of ≈20 minutes.97
Under conditions of daily living, numerous factors involved in regulating insulin secretion are integrated to provide a rate of insulin secretion of ≈2 pmol/kg/min in the fasting state, increasing to ≈10 pmol/kg/min after meal ingestion.98–101 The wide range in these rates is based on the insulin sensitivity of the individual (see Chapter 15). Thus with aging and obesity, and in response to exercise, adaptive changes in the rate of insulin secretion occur.79,101–107 In health, insulin secretion adaptively changes according to insulin requirements.62,104,108 The most prevalent need for increased insulin secretion involves the insulin resistance consequent upon obesity.79,101 In response to obesity, the daily insulin requirement increases by as much as 10-fold. However, in humans, the β cell mass in obese versus lean individuals is increased only ≈0.5-fold.55 This implies that the most important adaptive change needed to meet chronically increased insulin secretion requirements in insulin-resistant humans is an increase in insulin secretion per β cell, rather than simply an increase in the number of β cells.
Insulin Clearance
Insulin is secreted in the portal vein, where it is delivered directly to the liver. As a consequence of rapid blood flow in the portal vein (≈0.8 L/min) and fenestrations in the hepatic sinusoids, hepatocytes are directly exposed to the high-amplitude oscillations that arise from insulin secretory bursts within seconds of secretion. After insulin binding to the receptors at the hepatocyte membrane, the insulin receptor-ligand complex is rapidly internalized to form an intracytoplasmic vesicle.109–114 Although insulin undergoes enzymatic degradation in the endosomes,115,116 the insulin receptor is reinserted into the plasma membrane within ≈5 minutes to become available for the next insulin burst.113,117 In health in humans, ≈80% of endogenous insulin secretion is cleared by the liver with the first pass through the liver.118–121 Following oral glucose ingestion, insulin extraction is diminished by ≈50%.122–124 The major factor that determines the rate of insulin clearance appears to be the amount of insulin presented to the liver.125,126 In fact, a close relationship between insulin secretion and hepatic insulin uptake has been described,119,127 consistent with the idea of a finite number of insulin receptors present on hepatocytes. The extent of hepatic extraction also appears to depend on the amplitude of insulin oscillations presented to the liver.84,128 Since the mean interval of insulin pulses presented to the liver almost coincides with the time period calculated for insulin receptor recycling (≈5 minutes),113 pulsatile delivery of insulin may prevent the liver from desensitization. This, together with the fact that selective extraction of insulin pulses by the liver is evident, implies that varying the pattern of insulin delivery to the liver may provide the β cell an opportunity to regulate end-organ actions of insulin.
Islet Structure and Function in Type 1 Diabetes
In type 1 diabetes, a marked deficiency in β cell mass is seen.56,129–133 Before diabetes develops, a prolonged period occurs during which the autoimmune disease is thought to be active.134–136 During this latent period, a progressive decline in first-phase insulin secretion has been documented, as have impaired insulin pulses.135,137–140 By the time of clinical presentation, ≈90% of β cells have been lost; however, a substantial capacity for endogenous insulin secretion remains, although a relatively rapid further loss of this occurs over the next 2 years.56,131,133,141,142 The mechanisms underlying this further loss of insulin secretion likely include both further loss of β cell mass and decreased insulin secretion per β cell, each in large part a consequence of the hyperglycemia.143,144 It is important here to distinguish human islets from rodent islets, about which of necessity much research is carried out. Human β cells exposed to glucose concentrations typically present in patients with even relatively well-controlled diabetes (≈150 mg/dL; 8 mM) have an increased frequency of apoptosis, induced in part by endogenous expression of interleukin-1β (rat islets do not have an increased rate of β cell apoptosis until glucose concentrations increase to ≈360 mg/dL or 20 mM).143 In addition, insulin secretion by human islets is impaired within 96 hours after exposure to these levels of glucose concentration.145 Chronic exposure of islets to this relatively modest level of glucose appears to preferentially deplete the primed and docked insulin secretory vesicles in that both glucose-induced first-phase insulin secretion and glucose-induced insulin secretory burst mass are greatly attenuated.145 Evidence also suggests that an increased workload of β cells may accelerate the autoimmune-mediated destruction. Taken together, these factors have been called glucose toxicity.144,146 The reversibility of these factors at least in the short term is illustrated by the partial recovery of insulin secretion and glycemic control in patients treated who are brought toward normal blood glucose concentrations with exogenous insulin therapy.147–150
Not only is some residual insulin secretion detected in patients with recent-onset type 1 diabetes, but with increasingly sensitive assays, insulin secretion may be detected many years after onset of type 1 diabetes. Moreover, detectable β cells are commonly present in the pancreas of patients with even longstanding type 1 diabetes.56 The question arises whether this residual insulin secretion results from a small pool of β cells that are relatively protected from β cell destruction, or from newly formed β cells. This distinction is important because the latter would imply that a novel approach to restoration of β cell mass in patients with type 1 diabetes would be to suppress ongoing β cell destruction. Preservation or restoration of even an inadequate amount of insulin secretion to allow insulin independence still would have great potential clinical benefit in that microvascular complications are decreased in patients with residual β cell function.151
Islet Structure and Function in Type 2 Diabetes
Most but not all studies indicate that there is an ≈60% decrease in β cell mass in humans with type 2 diabetes (Fig. 7-7).55,152,153 This decrease in β cell mass appears to be due to an increased frequency of β cell apoptosis; therefore type 2 diabetes can be considered to have much in common with type 1 diabetes.54,55,154,155 The most important distinction appears to be the absence of an autoimmune-mediated cause for the accelerated β cell apoptosis and the more modest degree of β cell deficiency. The importance of an ≈60% deficit in β cell mass might be questioned because a similar defect does not lead to diabetes in rodents.156 Although juvenile rodents have a remarkable capacity for β cell regeneration after a partial pancreatectomy,51,54 surgical reduction of β cell mass in adult humans does not prompt β cell regeneration.157 A deficit in β cell mass comparable with that seen in humans with type 2 diabetes does lead to diabetes in large animal species that are potentially more representative of humans, including the pig, the dog, and nonhuman primates.128,158–160 Indeed a comparable β cell deficit leads to loss of first-phase insulin secretion, a deficit in insulin pulse mass, and decreased hepatic insulin clearance in the pig,128,161 thereby reproducing the pattern of abnormal insulin secretion and insulin clearance seen in type 2 diabetes.162 The increased frequency of β cell apoptosis in type 2 diabetes has been ascribed to glucose toxicity and to increased concentrations of free fatty acids, free radicles, and oligomers of islet amyloid polypeptide (IAPP, also known as amylin).143,163,164 The key question as to whether this increase precedes the development of hyperglycemia or is a consequence of it remains unknown.
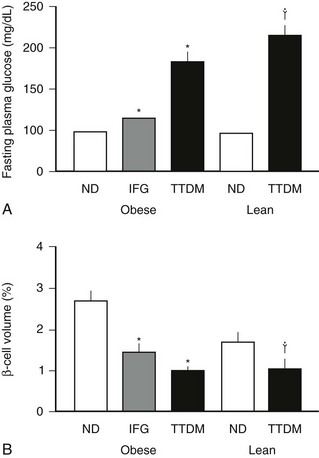
FIGURE 7-7 The mean blood glucose (A) and relative pancreatic β cell volume (B) in obese versus lean human subjects with type 2 diabetes (TTDM), those with impaired fasting glucose (IFG), and nondiabetic patients (ND). The relative β cell volume is increased in obese versus lean ND by approximately 50%. The relative β cell volume is decreased by approximately 65% in obese TTDM versus controls. (From Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC: β-Cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes 52:105, 2003.)
The functional defects in insulin secretion seen in type 2 diabetes have been reviewed elsewhere.74 In brief, when glucose concentrations are matched, there is a major defect in insulin secretion in both basal and glucose-stimulated (hyperglycemic clamp or oral glucose load) insulin secretion165–169 (Figs. 7-8 through 7-10). Defects in insulin secretion in response to different stimuli, including glucose, arginine, and GIP, can be detected in individuals at high risk for developing type 2 diabetes, such as first-degree relatives or women with a history of gestational diabetes.166,170–176 A decrease in pancreatic insulin stores is noted in patients with type 2 diabetes, but given the small proportion of insulin vesicles that undergo secretion, it is clear that there must be a specific defect in the availability of primed, docked insulin secretory vesicles for glucose stimulation to occur.77 This is supported by marked defects in early insulin secretion after meal ingestion, first-phase insulin secretion after glucose ingestion, and defective glucose-mediated insulin secretory burst mass in type 2 diabetes (see Figs. 7-10 and 7-11).74,177–179 The increased ratio of circulating proinsulin to insulin characteristic of type 2 diabetes has been ascribed to both defective proinsulin processing and increased insulin demand leading to secretion of immature insulin vesicles.180–182 The defects in first-phase insulin secretion, insulin pulse mass, and proinsulin/insulin processing all can be reversed in patients with type 2 diabetes by overnight inhibition of insulin secretion (see Fig. 7-11).183 The pattern of insulin secretion defects present in type 2 diabetes can be recapitulated in pigs through induction of a deficit in β cell mass comparable with that seen in type 2 diabetes.128,161 It is of interest to compare the normal adaptive responses of β cell mass and insulin secretion to insulin resistance in obese nondiabetic humans versus the deficits in these parameters in obese humans with type 2 diabetes (Fig. 7-12). In nondiabetic humans, a modest adaptation in β cell mass (≈50% increased) and a much greater increase (≈300%) in insulin secretion are seen, so that in the setting of an adequate β cell mass and normal blood glucose concentrations, β cells show considerable capacity for sustained increased secretion. In contrast, in type 2 diabetes, a rather comparable deficit in β cell mass and insulin secretion (≈60%) is noted under conditions of daily living (as in Fig. 7-12), although the deficit in insulin secretion can be considered much greater at matched glucose concentrations (as in Fig. 7-8).
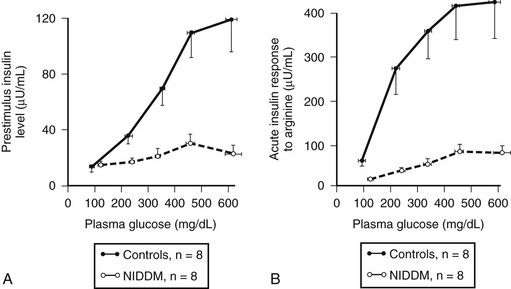
FIGURE 7-8 The plasma insulin concentration in patients with type 2 diabetes (NIDDM) and nondiabetic controls in relation to a graded glucose infusion (A) and following an arginine bolus at graded glucose concentrations (B), revealing marked impairment of insulin secretion to both glucose and arginine when glucose values are matched. (With permission from Ward WK, Bolgiano DC, McKnight B, Halter JB, Porte D Jr: Diminished β cell secretory capacity in patients with noninsulin-dependent diabetes mellitus. J Clin Invest 74:1318, 1984.)
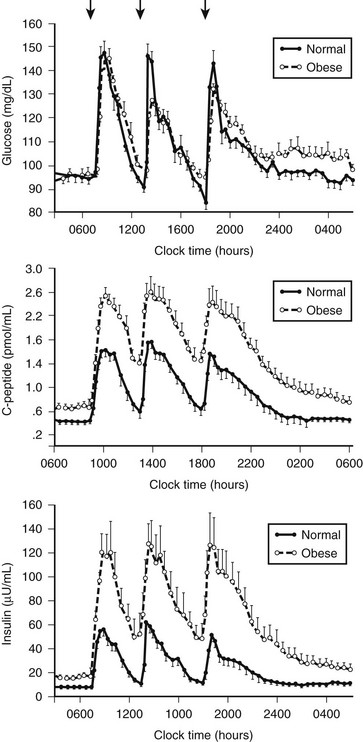
FIGURE 7-9 Twenty-four-hour pattern of insulin secretion in normal-weight and obese nondiabetic humans. In most obese humans, insulin plasma glucose concentrations are maintained at a comparable concentration by increased insulin secretion to compensate for insulin resistance. (From Polonsky KS, Given BD, Van Cauter E: Twenty-four-hour profiles and pulsatile patterns of insulin secretion in normal and obese subjects. J Clin Invest 81:433, 1988.)
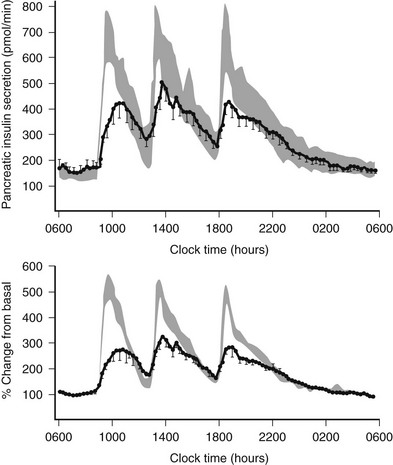
FIGURE 7-10 Twenty-four-hour insulin secretion profiles in type 2 diabetes (solid line) versus normal range (mean ± 1 SEM) for matched subjects. Measured insulin secretion rates are comparable in those with type 2 diabetes mellitus and in controls under fasting conditions, but this is, of course, defective in the setting of hyperglycemia. Following meal ingestion, a marked defect in TTDM is seen despite the marked hyperglycemia. (With permission from Polonsky KS, Given BD, Hirsch LJ, et al: Abnormal patterns of insulin secretion in non-insulin-dependent diabetes mellitus. N Engl J Med 318:1231,1988.)
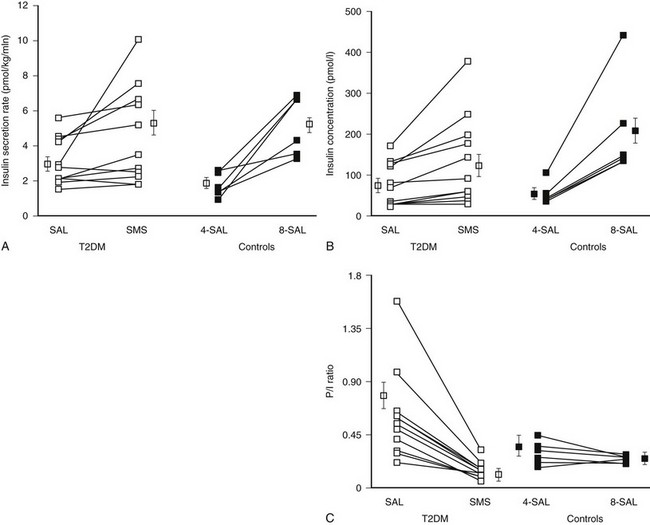
FIGURE 7-11 Insulin secretion (A), insulin concentration (B), and proinsulin/insulin ratio (C) in patients with type 2 diabetes (T2DM) and controls after prior overnight saline (SAL) or somatostatin (SMS) infusion at glucose 8 mmol/L versus nondiabetic controls at glucose 4 or 8 mmol/L. Prior overnight inhibition of insulin secretion by somatostatin restored glucose-induced insulin secretion and the proinsulin-to-insulin ratio to normal in type 2 diabetes. (With permission from Laedtke T, Kjems L, Porksen N, Schmitz O, Veldhuis J, Kao PC, et al: Overnight inhibition of insulin secretion restores pulsatility and proinsulin/insulin ratio in type 2 diabetes. Am J Physiol 279:E523 and E526, 2000.)
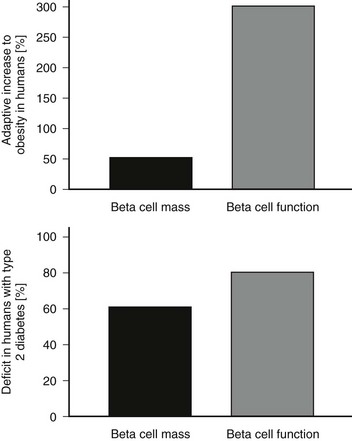
FIGURE 7-12 Comparison of the appropriate adaptive response of the islet to insulin resistance (top panel, lean vs. obese nondiabetic humans) and the failed adaptive response in obese humans with type 2 diabetes (bottom panel). The percent adaptive increase in β cell mass and insulin secretion are shown in the top panel and percent deficits are shown in the bottom panel.
Taken together, these data imply that once the β cell mass has been diminished to a critical threshold, the availability of primed docked insulin secretory vesicles that can be discharged by exocytosis and/or a kiss and run-in response to an increment in glucose are deficient. This concept is supported further by the observation that defective glucose-induced first-phase insulin and pulsatile insulin secretion can be developed similarly in human islets exposed to a glucose concentration of 150 mg/dL (8 mM) for 96 hours, but that this defect is prevented by the concurrent addition of a potassium channel opener to the islets that prevents high rates of insulin secretion during exposure to high glucose.145 However, the concept becomes more complex when one considers the almost immediate restoration of first-phase insulin secretion, pulsatile secretion, and glycemic regulation accomplished in patients with type 2 diabetes infused with the incretin hormone GLP-1.86,184–188 Also, although first-phase insulin secretion in response to intravenous arginine is still defective in patients with type 2 diabetes compared with controls, when blood glucose is considered, the magnitude of the defect is much less than that seen in response to glucose.168 This discordance might be due in part to the secondary defects associated with glucokinase function secondary to hyperglycemia. Also, increased expression of uncoupling protein-2 targeted to the mitochondrial membrane in response to chronic hyperglycemia may attenuate the glucose-induced signal for secretion due to attenuation of the mitochondrial proton gradient as accomplished by pyruvate oxidation.189
Islet Turnover and Regeneration
Although β cells have long been held to be in an irreversibly postmitotic state in postnatal humans, evidence for the presence of islet renewal is seen until adulthood.190 Thus, although the frequency of β cell replication declines with increasing age,191 occasional replicating β cells can be detected even in humans of advanced age.192 Furthermore, the presence of β cells in the pancreas of patients with longstanding type 1 diabetes despite continued autoimmune destruction supports the postulate of ongoing β cell turnover in such individuals.193 Whether such new β cells are formed primarily through the mechanism of replication of existing β cells, or whether they arise from replication-independent sources, such as through differentiation from pancreatic progenitor cells, is controversial. However, although β cell replication can be detected in human pancreatic tissue by the use of specific markers (e.g., expression of Ki67), as yet no methods have been developed to detect β cell formation from putative pancreatic stem cells in humans.
The continued turnover of β cells in adult humans directly leads to the question as to why β cell mass ultimately fails to regenerate in humans with type 1 and type 2 diabetes; two points seem important in this regard. First, although β cell replication is present to some extent in the human pancreas, the frequency (and presumably rate) of replication is considerably lower in adult humans than in young rodents.55,154 Second, because replicating β cells are vulnerable to apoptosis,164,194 any attempted β cell regeneration is likely to fail unless the respective cause of cell death (i.e., autoimmune infiltration or the detrimental effects of hIAPP, free fatty acids, or hyperglycemia) has been prevented successfully. In light of this, future therapeutic approaches that aim to restore β cell mass in diabetes not only should focus on enhancing new β cell formation, but at the same time should seek to inhibit β cell apoptosis. Finally, given the slow rate of β cell turnover seen in adult humans, any such intervention will likely require long treatment durations, probably over several years, for a significant gain in β cell mass to be achieved.
References
1. Weir, GC, Bonner-Weir, S, Leahy, JL. Islet mass and function in diabetes and transplantation. Diabetes. 1990;39(4):401–405.
2. Meglasson, MD, Burch, PT, Berner, DK, et al. Chromatographic resolution and kinetic characterization of glucokinase from islets of Langerhans. Proc Natl Acad Sci U S A. 1983;80(1):85–89.
3. Rubenstein, AH, Clark, JL, Melani, F, et al. Secretion of proinsulin and C-peptide by pancreatic beta-cells and its circulation in blood. Nature. 1969;224:697–699.
4. Hellman, B, Sehlin, J, Taljedal, IB. Calcium and secretion: distinction between two pools of glucose-sensitive calcium in pancreatic islets. Science. 1976;194(4272):1421–1423.
5. Gold, G, Gishizky, ML, Grodsky, GM. Evidence that glucose “marks” beta cells resulting in preferential release of newly synthesized insulin. Science. 1982;218(4567):56–58.
6. Daniel, S, Noda, M, Straub, SG, et al. Identification of the docked granule pool responsible for the first phase of glucose-stimulated insulin secretion. Diabetes. 1999;48(9):1686–1690.
7. Gabbay, KH, Korff, J, Schneeberger, EE. Vesicular binesis: glucose effect on insulin secretory vesicles. Science. 1975;187(4172):177–179.
8. Crespin, SR, Greenough, WB, 3rd., Steinberg, D. Stimulation of insulin secretion by infusion of free fatty acids. J Clin Invest. 1969;48(10):1934–1943.
9. Raptis, S, Dollinger, HC, Schroder, KE, et al. Differences in insulin, growth hormone and pancreatic enzyme secretion after intravenous and intraduodenal administration of mixed amino acids in man. N Engl J Med. 1973;288(23):1199–1202.
10. Boden, G, Chen, X, Iqbal, N. Acute lowering of plasma fatty acids lowers basal insulin secretion in diabetic and nondiabetic subjects. Diabetes. 1998;47(10):1609–1612.
11. Dobbins, RL, Chester, MW, Stevenson, BE, et al. A fatty acid-dependent step is critically important for both glucose- and non-glucose-stimulated insulin secretion. J Clin Invest. 1998;101(11):2370–2376.
12. Holst, JJ, Orskov, C, Nielsen, OV, et al. Truncated glucagon-like peptide I, an insulin-releasing hormone from the distal gut. FEBS Lett. 1987;211(2):169–174.
13. Kreymann, B, Williams, G, Ghatei, MA, et al. Glucagon-like peptide-1 7–36: a physiological incretin in man. Lancet. 1987;2(8571):1300–1304.
14. Nauck, MA, Bartels, E, Orskov, C, et al. Additive insulinotropic effects of exogenous synthetic human gastric inhibitory polypeptide and glucagon-like peptide-1-(7–36) amide infused at near-physiological insulinotropic hormone and glucose concentrations. J Clin Endocrinol Metab. 1993;76(4):912–917.
15. Pederson, RA, Schubert, HE, Brown, JC. The insulinotropic action of gastric inhibitory polypeptide. Can J Physiol Pharmacol. 1975;53(2):217–223.
16. Miller, RE. Pancreatic neuroendocrinology: peripheral neural mechanisms in the regulation of the Islets of Langerhans. Endocr Rev. 1981;2(4):471–494.
17. Giugliano, D, Cerciello, T, Giannetti, G, et al. Impaired insulin secretion in human diabetes mellitus. I. The effect of alpha-adrenergic inhibition. Pharmacol Res Commun. 1982;14(3):217–225.
18. Havel, PJ, Taborsky, GJ, Jr. The contribution of the autonomic nervous system to changes of glucagon and insulin secretion during hypoglycemic stress. Endocr Rev. 1989;10(3):332–350.
19. Ortiz-Alonso, FJ, Herman, WH, Gertz, BJ, et al. Effect of an oral alpha 2-adrenergic blocker (MK-912) on pancreatic islet function in non-insulin-dependent diabetes mellitus. Metabolism. 1991;40(11):1160–1167.
20. Beischer, W, Schmid, M, Kerner, W, et al. Does insulin play a role in the regulation of its own secretion? Horm Metab Res. 1978;10(2):168–169.
21. Grasso, S, Messina, A, Saporito, N, et al. Serum-insulin response to glucose and amino acids in the premature infant. Lancet. 1968;2(7571):755–756.
22. Degano, P, Silvestre, RA, Salas, M, et al. Amylin inhibits glucose-induced insulin secretion in a dose-dependent manner. Study in the perfused rat pancreas. Regul Pept. 1993;43(1–2):91–96.
23. Garvey, WT, Revers, RR, Kolterman, OG, et al. Modulation of insulin secretion by insulin and glucose in type II diabetes mellitus. J Clin Endocrinol Metab. 1985;60(3):559–568.
24. Langerhans, P, Beitrag zur mikroskopischen Anatomie der Bauchspeicheldruse. Berlin, 1869.
25. Mering, Jv, Minkowski, O. Diabetes mellitus nach Pankreasexstirpation. Zschr klin Med. 1889;14:404–423.
26. Stefan, Y, Orci, L, Malaisse-Lagae, F, et al. Quantitation of endocrine cell content in the pancreas of nondiabetic and diabetic humans. Diabetes. 1982;31(8 Pt 1):694–700.
27. Williams, JA, Goldfine, ID. The insulin-pancreatic acinar axis. Diabetes. 1985;34(10):980–986.
28. Rahier, J, Goebbels, RM, Henquin, JC. Cellular composition of the human diabetic pancreas. Diabetologia. 1983;24(5):366–371.
29. Bonner-Weir, S. Anatomy of the islet of Langerhans. In: Samols E, ed. The Endocrine Pancreas. New York: Raven Press, Ltd.; 1991:15–27.
30. Deng, S, Vatamaniuk, M, Huang, X, et al. Structural and functional abnormalities in the islets isolated from type 2 diabetic subjects. Diabetes. 2004;53(3):624–632.
31. Orci, L, Malaisse-Lagae, F, Amherdt, M, et al. Cell contacts in human islets of Langerhans. J Clin Endocrinol Metab. 1975;41(5):841–844.
32. Meissner, HP. Electrophysiological evidence for coupling between beta cells of pancreatic islets. Nature. 1976;262(5568):502–504.
33. In’t Veld, PA, Pipeleers, DG, Gepts, W. Glucose alters configuration of gap junctions between pancreatic islet cells. Am J Physiol. 1986;251(2 Pt 1):C191–C196.
34. Orci, L, Unger, RH. Functional subdivision of islets of Langerhans and possible role of D cells. Lancet. 1975;2(7947):1243–1244.
35. Orci, L. Macro- and micro-domains in the endocrine pancreas. Diabetes. 1982;31(6 Pt 1):538–565.
36. Stagner, JI, Samols, E. The vascular order of islet cellular perfusion in the human pancreas. Diabetes. 1992;41(1):93–97.
37. Montesano, R, Mouron, P, Amherdt, M, et al. Collagen matrix promotes reorganization of pancreatic endocrine cell monolayers into islet-like organoids. J Cell Biol. 1983;97(3):935–939.
38. Hayek, A, Beattie, GM, Cirulli, V, et al. Growth factor/matrix-induced proliferation of human adult beta-cells. Diabetes. 1995;44(12):1458–1460.
39. Wittingen, J, Frey, CF. Islet concentration in the head, body, tail and uncinate process of the pancreas. Ann Surg. 1974;179(4):412–414.
40. Clark, A, Holman, RR, Matthews, DR, et al. Non-uniform distribution of islet amyloid in the pancreas of ‘maturity-onset’ diabetic patients. Diabetologia. 1984;27(5):527–528.
41. Wharton, GK. The blood supply of the pancreas, with special reference to that of the islands of Langerhans. Anat Rec. 1932;53:55–81.
42. Lifson, N, Kramlinger, KG, Mayrand, RR, et al. Blood flow to the rabbit pancreas with special reference to the islets of Langerhans. Gastroenterology. 1980;79(3):466–473.
43. Bonner-Weir, S, Orci, L. New perspectives on the microvasculature of the islets of Langerhans in the rat. Diabetes. 1982;31(10):883–889.
44. Legg, PG. The fine structure and innervation of the beta and delta cells in the islet of Langerhans of the cat. Z Zellforsch Mikrosk Anat. 1967;80(3):307–321.
45. Bloom, SR. Blood glucose control by direct islet innervation. Horm Metab Res Suppl. 1976;6:85–90.
46. Creutzfeldt, W. The incretin concept today. Diabetologia. 1979;16(2):75–85.
47. Asplin, CM, Paquette, TL, Palmer, JP. In vivo inhibition of glucagon secretion by paracrine beta cell activity in man. J Clin Invest. 1981;68(1):314–318.
48. Maruyama, H, Hisatomi, A, Orci, L, et al. Insulin within islets is a physiologic glucagon release inhibitor. J Clin Invest. 1984;74(6):2296–2299.
49. Hope, KM, Tran, PO, Zhou, H, et al. Regulation of alpha-cell function by the beta-cell in isolated human and rat islets deprived of glucose: the “switch-off” hypothesis. Diabetes. 2004;53(6):1488–1495.
50. Edlund, H. Pancreatic organogenesis–developmental mechanisms and implications for therapy. Nat Rev Genet. 2002;3(7):524–532.
51. Bonner-Weir, S, Deery, D, Leahy, JL, et al. Compensatory growth of pancreatic beta-cells in adult rats after short-term glucose infusion. Diabetes. 1989;38(1):49–53.
52. Swenne, I, Eriksson, U. Diabetes in pregnancy: islet cell proliferation in the fetal rat pancreas. Diabetologia. 1982;23(6):525–528.
53. Marynissen, G, Aerts, L, Van Assche, FA. The endocrine pancreas during pregnancy and lactation in the rat. J Dev Physiol. 1983;5(6):373–381.
54. Butler, AE, Janson, J, Soeller, WC, et al. Increased beta-cell apoptosis prevents adaptive increase in beta-cell mass in mouse model of type 2 diabetes: evidence for role of islet amyloid formation rather than direct action of amyloid. Diabetes. 2003;52(9):2304–2314.
55. Butler, AE, Janson, J, Bonner-Weir, S, et al. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52(1):102–110.
56. Gepts, W. Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes. 1965;14(10):619–633.
57. Bonner-Weir, S, Baxter, LA, Schuppin, GT, et al. A second pathway for regeneration of adult exocrine and endocrine pancreas. A possible recapitulation of embryonic development. Diabetes. 1993;42(12):1715–1720.
58. Ianus, A, Holz, GG, Theise, ND, et al. In vivo derivation of glucose-competent pancreatic endocrine cells from bone marrow without evidence of cell fusion. J Clin Invest. 2003;111(6):843–850.
59. Steptoe, RJ, Ritchie, JM, Harrison, LC. Transfer of hematopoietic stem cells encoding autoantigen prevents autoimmune diabetes. J Clin Invest. 2003;111(9):1357–1363.
60. Ritzel, RA, Veldhuis, JD, Butler, PC. Glucose stimulates pulsatile insulin secretion from human pancreatic islets by increasing secretory burst mass: dose-response relationships. J Clin Endocrinol Metab. 2003;88(2):742–747.
61. Perley, MJ, Kipnis, DM. Plasma insulin responses to oral and intravenous glucose: studies in normal and diabetic subjects. J Clin Invest. 1967;46(12):1954–1962.
62. O’Rahilly, SO, Hosker, JP, Rudenski, AS, et al. The glucose stimulus-response curve of the beta-cell in physically trained humans, assessed by hyperglycemic clamps. Metabolism. 1988;37(10):919–923.
63. Kvietys, PR, Perry, MA, Granger, DN. Permeability of pancreatic capillaries to small molecules. Am J Physiol. 1983;245(4):G519–G524.
64. Guillam, MT, Hummler, E, Schaerer, E, et al. Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nat Genet. 1997;17(3):327–330.
65. Morita, H, Yano, Y, Niswender, KD, et al. Coexpression of glucose transporters and glucokinase in Xenopus oocytes indicates that both glucose transport and phosphorylation determine glucose utilization. J Clin Invest. 1994;94(4):1373–1382.
66. Matschinsky, FM. Banting Lecture 1995. A lesson in metabolic regulation inspired by the glucokinase glucose sensor paradigm. Diabetes. 1996;45(2):223–241.
67. Roche, E, Assimacopoulos-Jeannet, F, Witters, LA, et al. Induction by glucose of genes coding for glycolytic enzymes in a pancreatic beta-cell line (INS-1). J Biol Chem. 1997;272(5):3091–3098.
68. MacDonald, PE, Wheeler, MB. Voltage-dependent K(+) channels in pancreatic beta cells: role, regulation and potential as therapeutic targets. Diabetologia. 2003;46(8):1046–1062.
69. Henquin, JC, Ravier, MA, Nenquin, M, et al. Hierarchy of the beta-cell signals controlling insulin secretion. Eur J Clin Invest. 2003;33(9):742–750.
70. Tsuboi, T, Rutter, GA. Insulin secretion by ‘kiss-and-run’ exocytosis in clonal pancreatic islet beta-cells. Biochem Soc Trans. 2003;31(Pt 4):833–836.
71. Tsuboi, T, McMahon, HT, Rutter, GA. Mechanisms of dense core vesicle recapture following “kiss and run” (“cavicapture”) exocytosis in insulin-secreting cells. J Biol Chem. 2004;279:45115–45124.
72. DeFronzo, RA, Tobin, JD, Andres, R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol. 1979;237(3):E214–E223.
73. Song, SH, Kjems, L, Ritzel, R, et al. Pulsatile insulin secretion by human pancreatic islets. J Clin Endocrinol Metab. 2002;87(1):213–221.
74. Pratley, RE, Weyer, C. The role of impaired early insulin secretion in the pathogenesis of Type II diabetes mellitus. Diabetologia. 2001;44(8):929–945.
75. Grodsky, G, Landahl, H, Curry, D, et al. A two-compartmental model for insulin secretion. Adv Metab Disord. 1970;1(Suppl 1):45–50.
76. O’Connor, MD, Landahl, H, Grodsky, GM. Comparison of storage- and signal-limited models of pancreatic insulin secretion. Am J Physiol. 1980;238(5):R378–R389.
77. Rorsman, P, Renstrom, E. Insulin granule dynamics in pancreatic beta cells. Diabetologia. 2003;46(8):1029–1045.
78. Shiota, C, Larsson, O, Shelton, KD, et al. Sulfonylurea receptor type 1 knock-out mice have intact feeding-stimulated insulin secretion despite marked impairment in their response to glucose. J Biol Chem. 2002;277(40):37176–37183.
79. Polonsky, KS, Given, BD, Van Cauter, E. Twenty-four-hour profiles and pulsatile patterns of insulin secretion in normal and obese subjects. J Clin Invest. 1988;81(2):442–448.
80. Porksen, N, Nyholm, B, Veldhuis, JD, et al. In humans at least 75% of insulin secretion arises from punctuated insulin secretory bursts. Am J Physiol. 1997;273(5 Pt 1):E908–E914.
81. Cunningham, BA, Deeney, JT, Bliss, CR, et al. Glucose-induced oscillatory insulin secretion in perifused rat pancreatic islets and clonal beta-cells (HIT). Am J Physiol. 1996;271(4 Pt 1):E702–E710.
82. Porksen, N, Hollingdal, M, Juhl, C, et al. Pulsatile insulin secretion: detection, regulation, and role in diabetes. Diabetes. 2002;51(Suppl 1)):S245–S254.
83. Porksen, N, Munn, S, Steers, J, et al. Effects of glucose ingestion versus infusion on pulsatile insulin secretion. The incretin effect is achieved by amplification of insulin secretory burst mass. Diabetes. 1996;45(10):1317–1323.
84. Porksen, N, Munn, SR, Steers, JL, et al. Effects of somatostatin on pulsatile insulin secretion: elective inhibition of insulin burst mass. Am J Physiol. 1996;270(6 Pt 1):E1043–E1049.
85. Porksen, N, Grofte, B, Nyholm, B, et al. Glucagon-like peptide 1 increases mass but not frequency or orderliness of pulsatile insulin secretion: Effects of somatostatin on pulsatile insulin secretion: elective inhibition of insulin burst mass. Diabetes. 1998;47(1):45–49.
86. Ritzel, R, Schulte, M, Porksen, N, et al. Glucagon-like peptide 1 increases secretory burst mass of pulsatile insulin secretion in patients with type 2 diabetes and impaired glucose tolerance. Diabetes. 2001;50(4):776–784.
87. Porksen, N, Hussain, MA, Bianda, TL, et al. IGF-I inhibits burst mass of pulsatile insulin secretion at supraphysiological and low IGF-I infusion rates. Am J Physiol. 1997;272(3 Pt 1):E352–E358.
88. Vore, SJ, Aycock, ED, Veldhuis, JD, et al. Anesthesia rapidly suppresses insulin pulse mass but enhances the orderliness of insulin secretory process. Am J Physiol Endocrinol Metab. 2001;281(1):E93–E99.
89. Matthews, DR, Lang, DA, Burnett, MA, et al. Control of pulsatile insulin secretion in man. Diabetologia. 1983;24(4):231–237.
90. Stagner, JI, Samols, E. Role of intrapancreatic ganglia in regulation of periodic insular secretions. Am J Physiol. 1985;248(5 Pt 1):E522–E530.
91. Porksen, N, Munn, S, Ferguson, D, et al. Coordinate pulsatile insulin secretion by chronic intraportally transplanted islets in the isolated perfused rat liver. J Clin Invest. 1994;94(1):219–227.
92. Sha, L, Westerlund, J, Szurszewski, JH, et al. Amplitude modulation of pulsatile insulin secretion by intrapancreatic ganglion neurons. Diabetes. 2001;50(1):51–55.
93. Porksen, N. The in vivo regulation of pulsatile insulin secretion. Diabetologia. 2002;45(1):3–20.
94. Song, SH, McIntyre, SS, Shah, H, et al. Direct measurement of pulsatile insulin secretion from the portal vein in human subjects. J Clin Endocrinol Metab. 2000;85(12):4491–4499.
95. Matthews, DR, Naylor, BA, Jones, RG, et al. Pulsatile insulin has greater hypoglycemic effect than continuous delivery. Diabetes. 1983;32(7):617–621.
96. Paolisso, G, Sgambato, S, Torella, R, et al. Pulsatile insulin delivery is more efficient than continuous infusion in modulating islet cell function in normal subjects and patients with type 1 diabetes. J Clin Endocrinol Metab. 1988;66(6):1220–1226.
97. Simon, C, Brandenberger, G. Ultradian oscillations of insulin secretion in humans. Diabetes. 2002;51(Suppl 1):S258–S261.
98. Eaton, RP, Allen, RC, Schade, DS, et al. Prehepatic insulin production in man: kinetic analysis using peripheral connecting peptide behavior. J Clin Endocrinol Metab. 1980;51(3):520–528.
99. Polonsky, KS, Pugh, W, Jaspan, JB, et al. C-peptide and insulin secretion. Relationship between peripheral concentrations of C-peptide and insulin and their secretion rates in the dog. J Clin Invest. 1984;74(5):1821–1829.
100. Polonsky, KS, Rubenstein, AH. C-peptide as a measure of the secretion and hepatic extraction of insulin. Pitfalls and limitations. Diabetes. 1984;33(5):486–494.
101. Polonsky, KS, Given, BD, Hirsch, L, et al. Quantitative study of insulin secretion and clearance in normal and obese subjects. J Clin Invest. 1988;81(2):435–441.
102. Walton, C, Godsland, IF, Proudler, AJ, et al. Effect of body mass index and fat distribution on insulin sensitivity, secretion, and clearance in nonobese healthy men. J Clin Endocrinol Metab. 1992;75(1):170–175.
103. King, DS, Dalsky, GP, Clutter, WE, et al. Effects of exercise and lack of exercise on insulin sensitivity and responsiveness. J Appl Physiol. 1988;64(5):1942–1946.
104. Dela, F, Mikines, KJ, Tronier, B, et al. Diminished arginine-stimulated insulin secretion in trained men. J Appl Physiol. 1990;69(1):261–267.
105. Haffner, SM, Stern, MP, Hazuda, HP, et al. Hyperinsulinemia in a population at high risk for non-insulin-dependent diabetes mellitus. N Engl J Med. 1986;315(4):220–224.
106. Numata, K, Tanaka, K, Saito, M, et al. Very low calorie diet-induced weight loss reverses exaggerated insulin secretion in response to glucose, arginine and glucagon in obesity. Int J Obes Relat Metab Disord. 1993;17(2):103–108.
107. Chang, AM, Halter, JB. Aging and insulin secretion. Am J Physiol Endocrinol Metab. 2003;284(1):E7–E12.
108. Stumvoll, M, Tataranni, PA, Stefan, N, et al. Glucose allostasis. Diabetes. 2003;52(4):903–909.
109. Bergeron, JJ, Cruz, J, Khan, MN, et al. Uptake of insulin and other ligands into receptor-rich endocytic components of target cells: the endosomal apparatus. Annu Rev Physiol. 1985;47:383–403.
110. Carpentier, JL, Gazzano, H, Van Obberghen, E, et al. Internalization and recycling of 125I-photoreactive insulin-receptor complexes in hepatocytes in primary culture. Mol Cell Endocrinol. 1986;47(3):243–255.
111. Halperin, ML, Cheema-Dhadli, S, Haynes, FJ, et al. A theoretical analysis of the turnover of the hepatic insulin receptor in the rat. Clin Invest Med. 1986;9(2):141–143.
112. Levy, JR, Olefsky, JM. The trafficking and processing of insulin and insulin receptors in cultured rat hepatocytes. Endocrinology. 1987;121(6):2075–2086.
113. Goodner, CJ, Sweet, IR, Harrison, HC, Jr. Rapid reduction and return of surface insulin receptors after exposure to brief pulses of insulin in perifused rat hepatocytes. Diabetes. 1988;37(10):1316–1323.
114. Duckworth, WC. Insulin degradation: mechanisms, products, and significance. Endocr Rev. 1988;9(3):319–345.
115. Yonezawa, K, Yokono, K, Yaso, S, et al. Degradation of insulin by insulin-degrading enzyme and biological characteristics of its fragments. Endocrinology. 1986;118(5):1989–1996.
116. Hamel, FG, Mahoney, MJ, Duckworth, WC. Degradation of intraendosomal insulin by insulin-degrading enzyme without acidification. Diabetes. 1991;40(4):436–443.
117. Knutson, VP. Cellular trafficking and processing of the insulin receptor. Faseb J. 1991;5(8):2130–2138.
118. Waldhausl, W, Bratusch-Marrain, P, Gasic, S, et al. Insulin production rate following glucose ingestion estimated by splanchnic C-peptide output in normal man. Diabetologia. 1979;17(4):221–227.
119. Eaton, RP, Allen, RC, Schade, DS. Hepatic removal of insulin in normal man: dose response to endogenous insulin secretion. J Clin Endocrinol Metab. 1983;56(6):1294–1300.
120. Ferrannini, E, Wahren, J, Faber, OK, et al. Splanchnic and renal metabolism of insulin in human subjects: a dose-response study. Am J Physiol. 1983;244(6):E517–E527.
121. Shah, P, Vella, A, Basu, A, et al. Effects of free fatty acids and glycerol on splanchnic glucose metabolism and insulin extraction in nondiabetic humans. Diabetes. 2002;51(2):301–310.
122. Shapiro, ET, Tillil, H, Miller, MA, et al. Insulin secretion and clearance. Comparison after oral and intravenous glucose. Diabetes. 1987;36(12):1365–1371.
123. Shuster, LT, Go, VLW, Rizza, RA, et al. Incretin effect due to increased secretion and decreased clearance of insulin in normal humans. Diabetes. 1988;37:200–203.
124. Nauck, MA, Homberger, E, Siegel, EG, et al. Incretin effects of increasing glucose loads in man calculated from venous insulin and C-peptide responses. J Clin Endocrinol Metab. 1986;63:492–498.
125. Harding, PE, Bloom, G, Field, JB. Effect of infusion of insulin into portal vein on hepatic extraction of insulin in anesthetized dogs. Am J Physiol. 1975;228(5):1580–1588.
126. Polonsky, K, Jaspan, J, Emmanouel, D, et al. Differences in the hepatic and renal extraction of insulin and glucagon in the dog: evidence for saturability of insulin metabolism. Acta Endocrinol (Copenh). 1983;102(3):420–427.
127. Tillil, H, Shapiro, ET, Rubenstein, AH, et al. Reduction of insulin clearance during hyperglycemic clamp. Dose-response study in normal humans. Diabetes. 1988;37(10):1351–1357.
128. Kjems, LL, Kirby, BM, Welsh, EM, et al. Decrease in beta-cell mass leads to impaired pulsatile insulin secretion, reduced postprandial hepatic insulin clearance, and relative hyperglucagonemia in the minipig. Diabetes. 2001;50(9):2001–2012.
129. Warren, S, Root, HF. The pathology of diabetes, with special reference to pancreatic regeneration. Am J Pathol. 1925;1:415–430.
130. Kloppel, G, Drenck, CR, Oberholzer, M, et al. Morphometric evidence for a striking B-cell reduction at the clinical onset of type 1 diabetes. Virchows Arch A Pathol Anat Histopathol. 1984;403(4):441–452.
131. Junker, K, Egeberg, J, Kromann, H, et al. An autopsy study of the islets of Langerhans in acute-onset juvenile diabetes mellitus. Acta Pathol Microbiol Scand [A]. 1977;85(5):699–706.
132. Lohr, M, Kloppel, G. Residual insulin positivity and pancreatic atrophy in relation to duration of chronic type 1 (insulin-dependent) diabetes mellitus and microangiopathy. Diabetologia. 1987;30(10):757–762.
133. Pipeleers, D, Ling, Z. Pancreatic beta cells in insulin-dependent diabetes. Diabetes Metab Rev. 1992;8(3):209–227.
134. Gorsuch, AN, Spencer, KM, Lister, J, et al. Evidence for a long prediabetic period in type I (insulin-dependent) diabetes mellitus. Lancet. 1981;2(8260–8261):1363–1365.
135. Srikanta, S, Ganda, OP, Jackson, RA, et al. Type I diabetes mellitus in monozygotic twins: chronic progressive beta cell dysfunction. Ann Intern Med. 1983;99(3):320–326.
136. Atkinson, MA, Eisenbarth, GS. Type 1 diabetes: new perspectives on disease pathogenesis and treatment. Lancet. 2001;358(9277):221–229.
137. Srikanta, S, Ganda, OP, Gleason, RE, et al. Pre-type I diabetes. Linear loss of beta cell response to intravenous glucose. Diabetes. 1984;33(8):717–720.
138. Soeldner, JS, Tuttleman, M, Srikanta, S, et al. Insulin-dependent diabetes mellitus and autoimmunity: islet-cell autoantibodies, insulin autoantibodies, and beta-cell failure. N Engl J Med. 1985;313(14):893–894.
139. Lo, SS, Hawa, M, Beer, SF, et al. Altered islet beta-cell function before the onset of type 1 (insulin-dependent) diabetes mellitus. Diabetologia. 1992;35(3):277–282.
140. Chaillous, L, Rohmer, V, Maugendre, D, et al. Differential beta-cell response to glucose, glucagon, and arginine during progression to type I (insulin-dependent) diabetes mellitus. Metabolism. 1996;45(3):306–314.
141. Clarson, C, Daneman, D, Drash, AL, et al. Residual beta-cell function in children with IDDM: reproducibility of testing and factors influencing insulin secretory reserve. Diabetes Care. 1987;10(1):33–38.
142. Hanafusa, T, Miyazaki, A, Miyagawa, J, et al. Examination of islets in the pancreas biopsy specimens from newly diagnosed type 1 (insulin-dependent) diabetic patients. Diabetologia. 1990;33(2):105–111.
143. Maedler, K, Sergeev, P, Ris, F, et al. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest. 2002;110(6):851–860.
144. Robertson, RP. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J Biol Chem. 2004;279:42351–42354.
145. Ritzel, RA, Hansen, JB, Veldhuis, JD, et al. Induction of beta-cell rest by a Kir6.2/SUR1-selective K(ATP)-channel opener preserves beta-cell insulin stores and insulin secretion in human islets cultured at high (11 mM) glucose. J Clin Endocrinol Metab. 2004;89(2):795–805.
146. Robertson, RP, Harmon, J, Tran, PO, et al. Glucose toxicity in beta-cells: type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes. 2003;52(3):581–587.
147. Madsbad, S, Krarup, T, Reguer, L, et al. Effect of strict blood glucose control on residual B-cell function in insulin-dependent diabetics. Diabetologia. 1981;20(5):530–534.
148. Madsbad, S. Prevalence of residual B cell function and its metabolic consequences in Type 1 (insulin-dependent) diabetes. Diabetologia. 1983;24(3):141–147.
149. The DCCT Research Group. Effects of age, duration and treatment of insulin-dependent diabetes mellitus on residual beta-cell function: observations during eligibility testing for the Diabetes Control and Complications Trial (DCCT). J Clin Endocrinol Metab. 1987;65(1):30–36.
150. The Diabetes Control and Complications Trial Research Group. Effect of intensive therapy on residual beta-cell function in patients with type 1 diabetes in the diabetes control and complications trial. A randomized, controlled trial. Ann Intern Med. 1998;128(7):517–523.
151. Sjoberg, S, Gunnarsson, R, Gjotterberg, M, et al. Residual insulin production, glycaemic control and prevalence of microvascular lesions and polyneuropathy in long-term type 1 (insulin-dependent) diabetes mellitus. Diabetologia. 1987;30(4):208–213.
152. Clark, A, Wells, CA, Buley, ID, et al. Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: quantitative changes in the pancreas in type 2 diabetes. Diabetes Res. 1988;9(4):151–159.
153. Sakuraba, H, Mizukami, H, Yagihashi, N, et al. Reduced beta-cell mass and expression of oxidative stress-related DNA damage in the islet of Japanese Type II diabetic patients. Diabetologia. 2002;45(1):85–96.
154. Butler, AE, Jang, J, Gurlo, T, et al. Diabetes due to a progressive defect in beta-cell mass in rats transgenic for human islet amyloid polypeptide (HIP Rat): a new model for type 2 diabetes. Diabetes. 2004;53(6):1509–1516.
155. Donath, MY, Storling, J, Maedler, K, et al. Inflammatory mediators and islet beta-cell failure: a link between type 1 and type 2 diabetes. J Mol Med. 2003;81(8):455–470.
156. Bonner-Weir, S, Trent, DF, Weir, GC. Partial pancreatectomy in the rat and subsequent defect in glucose-induced insulin release. J Clin Invest. 1983;71(6):1544–1553.
157. Menge, BA, Tannapfel, A, Belyaev, O, et al. Partial pancreatectomy in adult humans does not provoke beta-cell regeneration. Diabetes. 2008;57(1):142–149.
158. Ward, WK, Wallum, BJ, Beard, JC, et al. Reduction of glycemic potentiation. Sensitive indicator of beta-cell loss in partially pancreatectomized dogs. Diabetes. 1988;37(6):723–729.
159. van der Burg, MP, Gooszen, HG, Guicherit, OR, et al. Contribution of partial pancreatectomy, systemic hormone delivery, and duct obliteration to glucose regulation in canine pancreas. Importance in pancreas transplantation. Diabetes. 1989;38(9):1082–1089.
160. Stagner, JI, Samols, E. Deterioration of islet beta-cell function after hemipancreatectomy in dogs. Diabetes. 1991;40(11):1472–1479.
161. Larsen, MO, Gotfredsen, CF, Wilken, M, et al. Loss of beta-cell mass leads to a reduction of pulse mass with normal periodicity, regularity and entrainment of pulsatile insulin secretion in Gottingen minipigs. Diabetologia. 2003;46(2):195–202.
162. Lang, DA, Matthews, DR, Burnett, M, et al. Brief, irregular oscillations of basal plasma insulin and glucose concentrations in diabetic man. Diabetes. 1981;30(5):435–439.
163. Robertson, RP, Harmon, J, Tran, PO, et al. Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes. 2004;53(Suppl 1):S119–S124.
164. Ritzel, RA, Butler, PC. Replication increases beta-cell vulnerability to human islet amyloid polypeptide-induced apoptosis. Diabetes. 2003;52(7):1701–1708.
165. Temple, RC, Carrington, CA, Luzio, SD, et al. Insulin deficiency in non-insulin-dependent diabetes. Lancet. 1989;1(8633):293–295.
166. Pimenta, W, Korytkowski, M, Mitrakou, A, et al. Pancreatic beta-cell dysfunction as the primary genetic lesion in NIDDM. Evidence from studies in normal glucose-tolerant individuals with a first-degree NIDDM relative. JAMA. 1995;273(23):1855–1861.
167. Kipnis, DM. Insulin secretion in diabetes mellitus. Ann Intern Med. 1968;69:891–901.
168. Ward, WK, Bolgiano, DC, McKnight, B, et al. Diminished B cell secretory capacity in patients with noninsulin-dependent diabetes mellitus. J Clin Invest. 1984;74(4):1318–1328.
169. Davies, MJ, Rayman, G, Grenfell, A, et al. Loss of the first phase insulin response to intravenous glucose in subjects with persistent impaired glucose tolerance. Diabet Med. 1994;11(5):432–436.
170. O’Rahilly, S, Turner, RC, Matthews, DR. Impaired pulsatile secretion of insulin in relatives of patients with non-insulin-dependent diabetes. N Engl J Med. 1988;318(19):1225–1230.
171. Eriksson, J, Franssila-Kallunki, A, Ekstrand, A, et al. Early metabolic defects in persons at increased risk for non-insulin-dependent diabetes mellitus. N Engl J Med. 1989;321(6):337–343.
172. Humphriss, DB, Stewart, MW, Berrish, TS, et al. Multiple metabolic abnormalities in normal glucose tolerant relatives of NIDDM families. Diabetologia. 1997;40(10):1185–1190.
173. Meier, JJ, Hücking, K, Holst, JJ, et al. Reduced insulinotropic effect of gastric inhibitory polypeptide in first-degree relatives of patients with type 2 diabetes. Diabetes. 2001;50:2497–2504.
174. Ward, WK, Johnston, CL, Beard, JC, et al. Insulin resistance and impaired insulin secretion in subjects with histories of gestational diabetes mellitus. Diabetes. 1985;34(9):861–869.
175. Ryan, EA, Imes, S, Liu, D, et al. Defects in insulin secretion and action in women with a history of gestational diabetes. Diabetes. 1995;44(5):506–512.
176. Gerich, JE. The genetic basis of type 2 diabetes mellitus: impaired insulin secretion versus impaired insulin sensitivity. Endocrine Rev. 1998;19:491–503.
177. Polonsky, KS, Given, BD, Hirsch, LJ, et al. Abnormal patterns of insulin secretion in non-insulin-dependent diabetes mellitus. N Engl J Med. 1988;318(19):1231–1239.
178. Nauck, M, Stöckmann, F, Ebert, R, et al. Reduced incretin effect in Type 2 (non-insulin-dependent) diabetes. Diabetologia. 1986;29:46–54.
179. Schmitz, O, Porksen, N, Nyholm, B, et al. Disorderly and nonstationary insulin secretion in relatives of patients with NIDDM. Am J Physiol. 1997;272(2 Pt 1):E218–E226.
180. Ward, WK, LaCava, EC, Paquette, TL, et al. Disproportionate elevation of immunoreactive proinsulin in type 2 (non-insulin-dependent) diabetes mellitus and in experimental insulin resistance. Diabetologia. 1987;30(9):698–702.
181. Porte, D, Jr., Kahn, SE. Hyperproinsulinemia and amyloid in NIDDM. Clues to etiology of islet beta-cell dysfunction? Diabetes. 1989;38(11):1333–1336.
182. Rhodes, CJ, Alarcon, C. What beta-cell defect could lead to hyperproinsulinemia in NIDDM? Some clues from recent advances made in understanding the proinsulin-processing mechanism. Diabetes. 1994;43(4):511–517.
183. Laedtke, T, Kjems, L, Porksen, N, et al. Overnight inhibition of insulin secretion restores pulsatility and proinsulin/insulin ratio in type 2 diabetes. Am J Physiol Endocrinol Metab. 2000;279(3):E520–E528.
184. Gutniak, MK, Holst, JJ, Ørskov, C, et al. Antidiabetogenic effect of glucagon-like peptide-1 (7–36)amide in normal subjects and patients with diabetes mellitus. N Engl J Med. 1992;326:1316–1322.
185. Nauck, MA, Kleine, N, Ørskov, C, et al. Normalization of fasting hyperglycaemia by exogenous glucagon-like peptide 1 (7–36 amide) in type 2 (non-insulin-dependent) diabetic patients. Diabetologia. 1993;36:741–744.
186. Rachman, J, Gribble, FM, Barrow, BA, et al. Normalization of insulin responses to glucose by overnight infusion of glucagon-like peptide 1 (7–36) amide in patients with NIDDM. Diabetes. 1996;45:1524–1530.
187. Kjems, LL, Holst, JJ, Vølund, A, et al. The influence of GLP-1 on glucose-stimulated insulin secretion: effects on beta-cell sensitivity in type 2 and nondiabetic subjects. Diabetes. 2003;52:380–386.
188. Meier, JJ, Gallwitz, B, Salmen, S, et al. Normalization of glucose concentrations and deceleration of gastric emptying after solid meals during intravenous glucagon-like peptide 1 in patients with type 2 diabetes. J Clin Endocrinol Metab. 2003;88:2719–2725.
189. Chan, CB, De Leo, D, Joseph, JW, et al. Increased uncoupling protein-2 levels in beta-cells are associated with impaired glucose-stimulated insulin secretion: mechanism of action. Diabetes. 2001;50(6):1302–1310.
190. Meier, JJ. Beta cell mass in diabetes: a realistic therapeutic target? Diabetologia. 2008;51(5):703–713.
191. Meier, JJ, Butler, AE, Saisho, Y, et al. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes. 2008;57(6):1584–1594.
192. Meier, JJ, Lin, JC, Butler, AE, et al. Direct evidence of attempted beta-cell regeneration in an 89-year=old patient with recent onset type 1 diabetes. Diabetologia. 2006;49:1838–1844.
193. Meier, JJ, Bhushan, A, Butler, AE, et al. Sustained beta cell apoptosis in patients with long-standing type 1 diabetes: indirect evidence for islet regeneration? Diabetologia. 2005;48(11):2221–2228.
194. Meier, JJ, Ritzel, RA, Maedler, K, et al. Increased vulnerability of newly forming beta cells to cytokine-induced cell death. Diabetologia. 2006;49(1):83–89.