Type 1 (Insulin-Dependent) Diabetes Mellitus
Etiology, Pathogenesis, Prediction, and Prevention
Type 1 (insulin-dependent) diabetes mellitus (T1DM) is associated with several immune abnormalities. Classically, when age at onset is young, classification of the disease is straightforward. Classification may be changing in the current obesity epidemic among the young, making differentiation between T1DM and type 2 diabetes mellitus (T2DM) more difficult.1,2 Although it has been known for decades that diabetes mellitus can occur in various degrees of severity, it was not until approximately 40 years ago that evidence was presented that indicated different modes of inheritance for what were then classified as “maturity-onset” and “juvenile-onset” types of diabetes.3 It is now evident that T1DM may occur at any age. The fact that T1DM in adults may fulfill clinical criteria of T2DM demonstrates the limitation of a disease classification based on clinical symptoms rather than on the etiology and pathogenesis. Despite the fact that autoantibodies are the main markers that predict and differentiate T1DM from other forms of diabetes, these markers are not yet included among the diagnostic criteria of T1DM.4
The diagnostic criteria used to differentiate between T1DM (insulin-dependent) and T2DM (non-insulin-dependent) are important for understanding the cause and pathogenesis of these two disease entities. The diagnostic criteria are primarily recommendations for the classification, the aim of which is better understanding of the cause of diabetes and optimization of care of diabetic patients. By using molecular genetics, it has been possible to clarify fully several monogenetic forms of diabetes. The etiology of these diabetes phenotypes has been explained by mutations in the genes for insulin, insulin receptor, glucokinase (maturity-onset diabetes in the young; MODY-2), HNF-4α (MODY-1), or HNF-1α (MODY-3).5 The clarification of the etiology of these diabetes phenotypes is a major advancement that has increased the diagnostic precision of diabetes syndromes.4,5 The understanding of the more complex and multifactorial T1DM syndrome also has undergone significant advances. This chapter is an attempt to present an evidence-based medicine review of the etiology, pathogenesis, and natural history of T1DM.
History
Early histologic studies of pancreatic tissue of diabetic patients who died shortly after clinical onset revealed that the pancreatic islets were altered by fibrosis, hyalinosis, atrophy, and infiltration of inflammatory cells.6 The inflammatory lesion of the islets of Langerhans was later described as insulitis,7 and quantitative studies of the pancreatic islets showed a specific loss of insulin-producing cells in association with clinical onset of T1DM.8,9 The rediscovery of insulitis9 was of major significance, especially because it was later observed that autoimmune thyroid disease often developed in diabetic patients treated with insulin or, conversely, that patients with diseases of autoimmune character (e.g., Grave’s disease, Hashimoto’s thyroiditis, pernicious anemia, and Addison’s disease) had an increased prevalence of T1DM.10 It was therefore suggested that the pathogenesis of T1DM involved autoimmune reactions directed toward the endocrine pancreas. This notion was supported by leukocyte migration inhibition to pancreatic islet antigens.11 Numerous studies have confirmed the presence of insulitis12,13; however, in spite of the assumption that T1DM is a T-cell mediated disease, reproducible and standardized tests of blood T-cell reactivity against islet autoantigens are yet to be established.14 Long-sought-for islet cell antibodies (ICA) were described in 1974,15,16 islet surface antibodies (ICSA) in 1978,17 and complement-dependent antibody-mediated islet cell cytotoxicity in 1980.18 The first antigen recognized by islet antibodies, an islet protein of 64,000 relative molecular mass (Mr), or 64K, was described in 1982.19,20 Later, the 64K protein was found to have glutamic acid decarboxylase (GAD) activity,20 but molecular cloning showed that the human islet GAD was a novel isoform, GAD65.21 Autoantibodies to insulin were demonstrated in 1983,22 to insulinoma-associated antigen 2 (IA-2), a receptor-type protein tyrosine phosphatase, in 1994,23 and to the ZnT8 transporter in 2007.24 In contrast to T-cell analyses, GAD65 and IA-2 autoantibodies may be determined in standardized assays.25 Taken together, ample evidence exists that islet cell autoimmunity is of major importance in the etiopathogenesis of T1DM.
Definition
T1DM results from selective destruction of the β cells in the pancreatic islets, leading to absolute lack of insulin and hyperglycemia.4 The β cells are principally destroyed by an aggressive autoimmune process, which is mediated but not limited to infiltration of CD4+ and CD8+ T cells, as well as macrophages, resulting in insulitis. An improved understanding of these processes is obtained in animals with spontaneous T1DM, most prominently the NOD mouse and the BB rat. These animals allow detailed genetic and experimental observations prior to onset of diabetes and facilitate preclinical trials to develop approaches of prevention and intervention, as recently reviewed.26
Symptoms and Signs
T1DM is often thought to be a disorder of acute onset, and the clinical onset may be dramatic. Nevertheless, T1DM may be discovered accidentally27 or associated with serious, life-threatening diabetic ketoacidosis (DKA). Over the years, however, numerous reports have noted that signs of subclinical diabetes precede the clinical onset. In addition, in adult diabetes patients classified with T2DM, a change sometimes occurs from an insulin-independent to insulin-dependent state.
T1DM is said to have four major clinical phases: preclinical diabetes, overt diabetes, partial remission phase (honeymoon), and chronic phase of lifelong dependency on injected insulin.28 It is now accepted that autoantibodies against GAD65, insulin, IA-2, or ZnT8, alone or in combination, may be present up to several years before the clinical onset of the disease.24,29,30 The possibility that islet autoimmunity might be present long before symptoms of hyperglycemia occur makes it difficult to define a causative factor. The clinical onset is not likely to occur until a major loss (80% to 90%) of the islet β cells has occurred. Currently it is not possible to determine the human β-cell mass or volume. β-Cell function tests are affected by both β-cell mass and insulin sensitivity.31 It is therefore not possible to relate clinical onset to β-cell mass, especially since some reports suggest that around 40% to 50% of β cells may be viable, owing to β-cell regeneration at the time of overt hyperglycemia.27
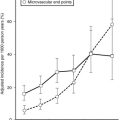
The occurrence of DKA is more commonly seen with new-onset disease, especially in children younger than 4 years of age (Fig. 14-1), but it is not specific to T1DM and may also occur in T2DM.32 DKA occurs invariably in different populations, ranging from 15% to 67% of patients in Europe and North America, whereas in the developing world, around 80% of children with T1DM present initially with DKA.33 Typically the child is acidotic with acetone fruity odor, air hunger and Kussmaul respiration, abdominal pain, nausea and vomiting, and polyuria and polydipsia (see Fig. 14-1). The child has hyperglycemia, glucosuria, ketonemia, and ketonuria. Without timely management, severe fluid and electrolyte depletion soon develop, with signs of hypoperfusion and deterioration in level of consciousness due to cerebral edema that may lead to coma and death (see Fig. 14-1).
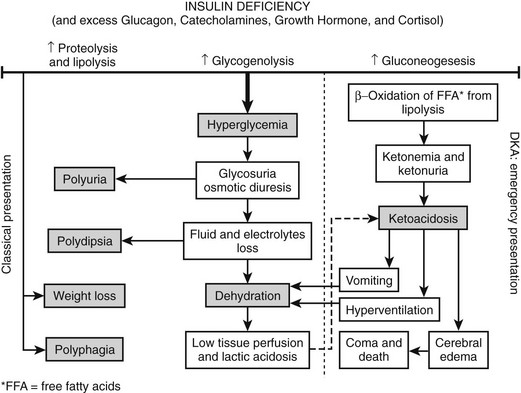
FIGURE 14-1 Pathophysiologic mechanisms and clinical presentations in type 1 diabetes mellitus. The clinical presentation in T1DM can be classical or emergency diabetic ketoacidosis (DKA), depending on the extent of insulin deficiency, effects of counteracting hormones, and other conditions. DKA can be viewed as an exaggerated compensatory physiologic mechanism.
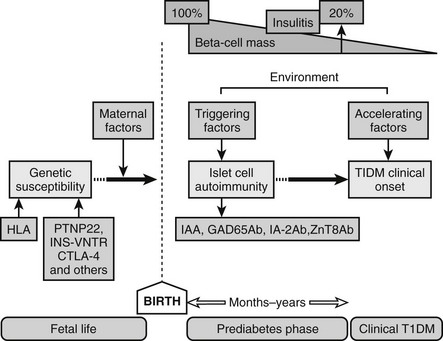
Because T1DM is detected in the majority of patients after a relatively short period of symptoms, such as increased thirst, polyuria, and unexplained weight loss, the natural history of the disease has been poorly defined until recently. With the current ability to define individuals at high risk for developing T1DM based on high-risk HLA and autoantibody positivity, our understanding of the prediabetic period is improving. Current prospective studies of children with increased risk for T1DM (TEDDY, DIPP; DAISY, DiPiS, BABYDiab, PANDA)34,35 have revealed that autoantibodies to GAD65, IA-2, or insulin and decreased ability to release insulin in response to glucose36 may develop several years before the clinical diagnosis. The sequence of events preceding the diagnosis of overt T1DM would include the following: (1) genetic predisposition; (2) overt immunologic abnormalities with normal glucose levels; (3) development of β-cell dysfunction; (4) development of overt hyperglycemia with detectable C peptide; and (5) the final stage of insulin dependency, with disappearance of C peptide (Fig. 14-2). The fact that T1DM develops in persons of all ages must be taken into account when studying the natural history in children and adults. It is of significance that T1DM is often associated with other autoimmune diseases such as autoimmune thyroiditis (15% to 30%), Addison’s disease (0.5%), and celiac disease (4% to 9%). Additionally, the risk of autoimmune diseases is increased among relatives to T1DM patients.37
Diagnostic Criteria
The basis for distinction between T1DM and T2DM is islet autoimmunity and the patient’s dependence on insulin. Most patients have a history of polyuria, polydipsia, and unexplained weight loss (see Fig. 14-1). The American Diabetes Association (ADA)4 and the International Society for Pediatric and Adolescent Diabetes (ISPAD)28 have recommended diagnostic criteria for T1DM (Table 14-1).
Table 14-1
Criteria for the Diagnosis of Diabetes
| 1. | Fasting plasma glucose (FPG) ≥126 mg/dL (7.0 mmol/L). Fasting is defined as no caloric intake for at least 8 hr.* |
| OR | |
| 2. | Symptoms of hyperglycemia and a casual plasma glucose ≥ 200 mg/dL (11.1 mmol/L). Casual is defined as any time of day without regard to time since last meal. The classic symptoms of hyperglycemia include polyuria, polydipsia, and unexplained weight loss. |
| OR | |
| 3. | 2-hr plasma glucose ≥200 mg/dL (11.1 mmol/L) during an oral glucose tolerance test (OGTT). The test should be performed as described by the World Health Organization, using a glucose load containing the equivalent of 75 g anhydrous glucose dissolved in water.* |
*In the absence of unequivocal hyperglycemia, these criteria should be confirmed by repeat testing on a different day.
From American Diabetes Association: Standards of medical care in diabetes–2008. Diabetes Care 31(Suppl. 1):S12–S54, 2008.
Evidence has been presented that adult-onset T2DM may progress to insulin dependence at a rate of 1% to 2% per year.3 This rate is increased in GAD65 autoantibody–positive patients classified with T2DM,38 and this form of T1DM has variably been termed type 1.5 diabetes, latent autoimmune diabetes in adults (LADA), or slowly progressive T1DM.39
Glucose tolerance tests, both oral and intravenous, are being used to evaluate diabetic states. The oral glucose tolerance test (OGTT) is a diagnostic criterion (see Table 14-1). Although glucose tolerance tests are more important in disorders of impaired glucose tolerance and T2DM, they may also find an increased use in the prediction and diagnosis of T1DM.40
Classification
T1DM can be clinically classified into two main types: immune-mediated, or type 1A, and the uncommon idiopathic, or type 1B (~10% of patients). Unlike immune-mediated T1ADM, T1BDM is not associated with islet-cell antibodies or HLA, although both forms can involve ketosis acidosis.4,5
Epidemiology
The prevalence of T1DM is low compared with that of T2DM. Among individuals aged 30 years or younger, the prevalence of T1DM does not usually exceed about 0.3%, compared with prevalence rates for T2DM of 4.2% worldwide and nearly 25% in certain high-risk populations.33 While current attention is directed toward the trends of the “epidemic” of T2DM, recent data suggest that there is a “parallel rise” in the incidence rates of both T1DM and T2DM.41 Both geographic and ethnic variations are seen in the prevalence rates. The International Diabetes Federation (IDF) estimates that in 2006 there were 440,000 cases of T1DM among children younger than 15 years of age, and that Southeast Asia contributes to a quarter of the prevalent cases, while Europe contributes with one fifth.33 It has been pointed out that the results of prevalence studies should be viewed with caution. Different age groups were studied,4,33 and although geographical variation may be related to variations in genetic predisposition, environmental risk factors, or both, it may also be due to some research methodological problems.33 In addition, it is unclear whether recently observed increases in incidence rates worldwide reflect a change in the age at onset of diabetes or a true increase in prevalence of T1DM.42
Incidence
Globally, the current rise in incidence rates by 3% (2% to 5%) is expected to increase 40% higher in 2010 compared to figures from 199843,44 (Fig. 14-3). Currently it is estimated that more than 70,000 new patients are diagnosed each year worldwide.33 There are more suggestions that the increasing incidence rates and changing trends are seen in populations with high incidence and populations with previously lower incidence rates.27 Over the last 3 decades, T1DM has shown temporal trends in most of the regions of the world except Central America and the West Indies, while European data showed that the main recent rise in incidence rates is in Central-Eastern countries.43 Other studies such as EURODIAB44 and DiaMond45 have attempted to better define the incidence rates in various populations by using prospective, population-based, geographically defined registries. Overall, the average annual incidence rate varies widely, from 0.1 per 100,000 children in parts of Asia and South America43,46 to 64.2 per 100,000 in Finland.47 Worldwide epidemiologic investigations of T1DM as a noncommunicable disease indicate a 4% to 6% annual increase in incidence rate in Scandinavia,33,44 and similar data are being collected in other countries.48 Pooled data from all sites in the EURODIAB study demonstrated an annual rate of increase in incidence of 3.4%, with more rapid rates of increase occurring in certain regions.43,44 About 1% of all children born in Scandinavian countries will manifest T1DM during their lifetimes. The causes of this rapid rise are not understood, and further studies are needed to define to what extent the rise may be due to environmental factors, genetic admixture, or both.
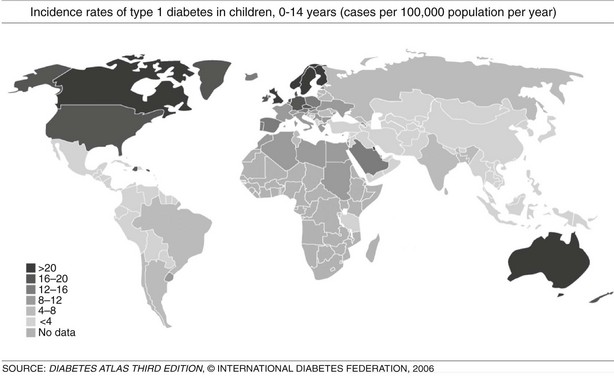
FIGURE 14-3 Incidence rates of type 1 diabetes mellitus (T1DM), 0 to 14 years: global trends in childhood T1DM. The incidence of T1DM varies widely, depending on genetic background and geographic location. (From Diabetes Atlas, 3rd ed. Brussels: International Diabetes Federation, 2006, with permission.)
Geographical Distribution
T1DM can occur in any region of the world, and recently it was observed that the incidence is increasing in countries with both high and low incidence rates; in the latter group, the rise is steeper.43,48 However, it is suggested that the incidence of T1DM increases with increasing distance away from the equator (see Fig. 14-3). The annual incidence of T1DM is higher in northern Europe than in the Mediterranean area, with the exception of Sardinia. Furthermore, the incidence in Iceland is lower than that in Sweden or Finland.43,44 Surprisingly, the T1DM incidence rate in Estonia is about 25% of the rate in Finland, in spite of their close geographic proximity.49 The incidence rates within countries also show stable differences. The eastern and southern parts of Finland,50 as well as the central and southern parts of Sweden,51 have higher incidence rates than the northern parts of those countries.
The cause of geographic variation remains unknown, but it has been speculated to be related to genetic factors primarily associated with different HLA-DR or DQ genotypes of the major histocompatibility complex (MHC) on chromosome 6. HLA class II molecules from the DQ and DR loci that are necessary (but not sufficient) for disease vary greatly between countries and thereby affect disease incidence.49,52,53 In addition, environmental factors are important in understanding the pathogenesis of T1DM and may also help to explain differences in geographic distribution. This is supported by the observation that monozygotic twins show less than 20% to 30% concordance rates for T1DM.54
In the European region, incidence rates show correlation with the frequency of HLA susceptibility genes in the general population.52,55 In this region, there is also wide variation in incidence rates, ranging from 6 to 62 cases/100,000/year among children 0 to 14 years of age, with higher rates observed in Scandinavian countries, especially Finland and Sweden.44,47 In countries with populations of European origins, there is less variation: Canada reports 21/100,000/year, the United States 16/100,000/year, and New Zealand and Australia have incidence rates ranging from 16 to 20 cases/100,000/year.43 In the Middle East and North Africa, few reliable data are available. The incidence rates range from 1.0/100,000/year in Pakistan to 8 to 12 cases/100,000/year in Egypt, Sudan, and Libya; however, reports from Kuwait showed a rise in incidence rates from 15.4 in 1992 to 20.9 in 1997. There are few published data from the Southeast Asian region, and data from the African region are mostly lacking. In South and Central America, the incidence rates are low, except for Argentina, 6.8, and Uruguay, 8.3 cases/100,000/year. The Western Pacific countries have low incidence rates, especially China, which has one of the lowest incidence rates worldwide, ranging from 0.1 to 0.6 cases/100,000/year (see Fig. 14-3).43,48
It has been documented that migration of people from relatively low incidence countries into countries with a “diabetogenic” environment (i.e., with high incidence rates) increases the risk among the migrant groups. Migrants, within a short period of time, assume comparative risks of the native population, and several studies confirmed the importance of country of origin in the risk of T1DM.56,57 Improved epidemiology and better diagnostic criteria to distinguish different forms of diabetes are critical in obtaining reliable incidence rates for various countries and states.
Variation with Age
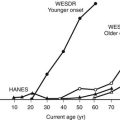
Until recently, T1DM was thought to occur almost exclusively in children and adolescents (Table 14-2). Epidemiologic studies with rigorous diagnostic criteria4 suggest, however, that the clinical onset of T1DM may occur at any age. The incidence rate varies with both age and gender (Fig. 14-4). The peak for both girls and boys, age 11 to 14 years, has been discernible in most studies and seems to be present irrespective of the country or area studied.44,46,48 This peak is associated with puberty and the maximal velocity of pubertal growth, and it may be associated with reduced insulin sensitivity due to hormonal factors related to growth. Previous studies have suggested that the annual rate of increase in incidence is higher in younger age groups, with rates of 6.4% in 0- to 4-year-olds, 3.1% in 5- to 9-year-olds, and 2.4% in 10- to 14-year-olds.44 In children, minor incidence peaks occur at 4 to 6 years58 and 7 to 8 years,59,60 which have been associated with entrance into preschool or school programs.61 Recent data suggest that around 50% to 60% of cases of T1DM in Western countries occur before the 15th birthday, and that more than 90% of childhood and adolescent diabetes is T1DM.55 Epidemiologic investigation has indicated that the incidence rate of T1DM in patients older than 20 years is lower than that seen in children, except for a possible peak at around 50 to 65 years.62 In addition to adults having what is considered a more classic T1DM clinical picture, some adult patients initially classified and treated as having T2DM may require insulin after 1 to 5 years of therapy with diet, exercise, or oral hypoglycemic agents.38,39 As noted earlier, this type of diabetes is referred to as type 1.5 diabetes, latent autoimmune diabetes in the adult (LADA), or slowly progressive insulin-dependent diabetes mellitus (SPIDDM).39,63 These patients are positive for islet autoantibodies and have lower body mass index and higher frequencies of high-risk HLA types compared with other “type 2” patients.63 It is speculated that these patients in fact have T1DM in addition to genetic factors that may inhibit rapid progression of β-cell killing.62
Table 14-2
Severity of Symptoms at Onset, A1C Goals, and Treatment for T1DM by Age Group

A1C, Hemoglobin A1c; PBG, plasma blood glucose; T1DM, type 1 diabetes mellitus.
Modified from American Diabetes Association: Standards of medical care in diabetes–2008. Diabetes Care 31(Suppl. 1):S12–S54, 2008.
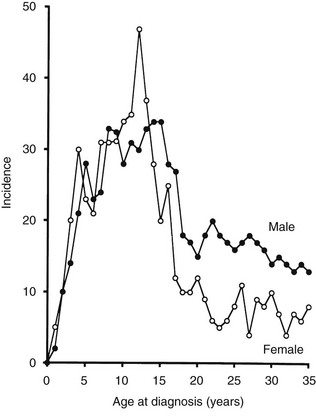
FIGURE 14-4 Age-adjusted incidence of insulin-dependent diabetes mellitus in relation to age at onset in male and female subjects. (Data from Nystrom L, Dahlquist G, Ostman J et al: Risk of developing insulin-dependent diabetes mellitus [IDDM] before 35 years of age: indications of climatological determinants for age at onset. Int J Epidemiol 21:352–358, 1992, with permission.)
Although rare, some forms of diabetes mellitus can occur in the newborn period, at birth, or during the first months of life. Neonatal diabetes (NDM) describes one kind of diabetes that occurs in the first month (up to 6 months) of life at a rate of 1 in 500,000 live births. It is characterized by insulin-sensitive hyperglycemia. About half of cases of NDM are transient (TNDM), but the remaining progress into a permanent (PNDM) type, with lifelong dependence on exogenous insulin. Clinically the infant is thirsty, with excessive urination and hyperglycemia and/or glycosuria, but the condition may progress into ketoacidosis. PNDM usually proceeds after the second year, and differentiation between the two forms can only be done through genetic testing. TNDM occurs at birth up to 3 to 6 months and is correlated to defects in specific genes: ZAC/HYMAI (ZAC pleomorphic adenoma gene–like 1, or PLAG1, and hydatidiform mole–associated and imprinted transcript, HYMAI), or KCNJ11, the moderately activating mutation of the Kir6.2 gene, or hepatocyte nuclear factor 1β (HNF1β) and SUR1 (sulfonylurea receptor 1). The PNDM, on the other hand, is related to several gene defects, such as pancreatic aplasia, activating mutations of KCNJ11 of the Kir6.2 gene that encodes for ATP potassium channel (KATP), the ABCC8 gene of sulfonylurea receptor 1 (SUR1), complete deficiency of glucokinase IPF1 (PDX1) gene, which encodes insulin promoter factor1, PTF1A, which encodes the pancreas transcription factor 1A, and also mutations in forehead box p3 (FOXP3) gene (also known as T-cell regulatory gene).64
Variation with Gender
It has been reported that the peak incidence in girls occurs earlier than in boys59 (see Fig. 14-4). If the clinical onset of T1DM is linked to pubertal growth, this difference in incidence rate can be explained by the fact that pubertal growth occurs earlier in girls. In children followed from birth because of T1DM genetic risk, the male-to-female ratio was 1.4 : 1.0 before 6 years of age at T1DM diagnosis. This ratio was increased (1.7 : 1.0) among children younger than 18 months of age.65 Prepubertal boys were found to be taller at the clinical onset of T1DM.60 In addition, newly diagnosed children of both genders showed advanced skeletal maturity.66 Even if boys tend to show an increased height compared with controls, their growth seems to cease about 35 weeks before the clinical onset of T1DM.60 It therefore is possible that processes affecting the pancreatic β-cell mass and the ability to produce insulin may have profound effects on body growth and function at a young age. Because these processes differ slightly between boys and girls, growth characteristics may offer a simple explanation for the differences seen in incidence rates between the genders.
A study that examined sex differences in T1DM incidence before the age of 15 showed a slight male preponderance in many but not all European countries, whereas a female preponderance was found in most African and Asian countries.67 Interestingly, the male excess was seen in all countries with the highest incidence rates (>20/105/yr), whereas a female excess was seen in all countries with low incidence rates (<4.5/l05/yr). Studies of older age groups have consistently shown a male preponderance for new cases of T1DM, with male/female ratios ranging from 1.3 to 2.5 : 1.68 One registry of T1DM among 15- to 34-year-olds (see Fig. 14-4) demonstrated that T1DM was 1.5 times more common among men than women.61 Further studies are necessary to document gender-dependent incidence rates and to explain their mechanisms.
Seasonal Variations
T1DM shows cyclic, sinusoidal appearance of seasonal variation in date of diagnosis, with a peak in winter months.48 This variation is proposed to be related to the timing of occurrences of certain precipitating factors such as viral infection and cold climate.48 It is of interest that islet cell autoantibodies appearing during the prediabetic state follow similar seasonal variations, being more prevalent in colder months and rare in summer and spring. Additionally, the triggering of β-cell autoimmunity appears to have variable trends over subsequent years and does not equally affect genetically susceptible siblings.69 These observations suggest that triggering of immunologic markers may be related to factors (most likely viruses) that prevail mostly in colder months with variable frequency of occurrence.
Etiology
The absence of an unambiguous mode of inheritance, the presence of a period of subclinical islet autoimmunity preceding clinical onset of disease, human leukocyte antigen (HLA) genes that control the immune response, and age and seasonal variation must be taken into account in attempts to explain the cause of T1DM. A defined etiologic factor, endogenous or exogenous, capable of causing T1DM remains to be identified (see Table 14-5). Because evidence exists of genetic heterogeneity in T1DM, it is possible that different causative factors are responsible. In experimental animals (see Table 14-5), both viral and chemical agents have been used to induce diabetes reproducibly, and certain strains of animals are at higher risk for developing diabetes due to genetic factors. In addition, only indirect evidence suggests that environmental factors that are clearly diabetogenic in animals are involved in initiating T1DM in humans. The following is a brief summary of possible genetic or environmental factors that are associated with the appearance of T1DM.
Inheritance
The mode of inheritance is complex, and around 80% to 85% of cases of T1DM occur sporadically without familial aggregation.52 Among HLA-identical siblings of T1DM-affected patients, about 20% eventually manifest the disease. The overall lifetime risk for first-degree relatives has been estimated at about 8% for siblings—15 times higher than the general population—and 5% for children of parents with T1DM.70 The risk of T1DM among offspring ranges from 2% to 4% if the mother was affected, compared to 6% to 9% if the father was affected, and the risk tolls to 30% when both parents are affected.52,70
Genetic Factors
T1DM is both genetically associated with and linked to certain HLA genetic factors of the major histocompatibility complex (MHC).71,72 By using DNA sequence information in the genetic analysis, it is found that more than 95% of all patients in whom T1DM onset occurred before age 30 years are positive for the chromosome 6 HLA haplotypes DRB1*04-DQAI*0301-BI*0302, DRBI*03-DQA1*0501-BI*0201, or both. Although some 40% to 50% of the background population carry these HLA factors, they represent necessary but insufficient prerequisites for the development of T1DM. It has been estimated that HLA contributes about 60% of T1DM risk among first-degree relatives to T1DM patients. Hence it is not surprising that less than 10% of genetically susceptible subjects develop the disease.68 Other genetic factors have indeed been identified,73,74 but none has shown a level of importance comparable to that of HLA.
The HLA haplotypes DRB1*04-DQAI*0301-BI*0302 and DRBI*03-DQA1*0501-BI*0201 are the two major-risk haplotypes71,75 (Table 14-3). The most important alleles are DQB1*0302 and DQB1*0201, along with DRB1*03. DRBI*04 is a large family of related molecules, and DRB*0401 confers an independent risk, whereas DRB1*0403 is negatively associated with T1DM and may protect or decelerate an ongoing disease process.75 DRB1*03 seems to be more important than DQB1*0201, as only DRBI*03-DQA1*0501-BI*0201, not DRBI*07-DQA1*0501-BI*0201, confers T1DM risk. DQB1*0401 and DQB1*0404 are susceptibility alleles on the DRB1*04-DQAI*0301-BI*0302 haplotype. DQB1*0604 and DQB1*0501 are susceptibility alleles, together with either DRBI*03-DQA1*0501-BI*0201 or DRB1*04-DQAI*0301-BI*0302. The genetic linkage and association between T1DM and HLA is remarkable in that certain HLA haplotypes are protective. Most prominently, DQA1*0102-B1*0602 and DQA1*0102-B1*0603 are protective before age 15 years (see Table 14-3). The detailed mechanisms by which HLA confers either risk or resistance is not fully understood.76 The function of these molecules is to display peptide antigens to be recognized by T-cell receptors (TCR). The disease association may therefore be related either to an inability to induce immunologic tolerance to certain autoantigens or to antigen presentation of an endogenous autoantigen. In the first case, the subject may be exposed to infectious agents that mimic autoantigens. Reactivity to the infectious agent sets off an immune response that cross-reacts with self. In the second case, the immune reaction may result in a direct attack on the individual’s own cells.
Table 14-3
Class II HLA Haplotype Risk Genes of Type 1 Diabetes in Caucasians
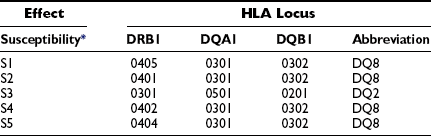

The commonest high-risk haplotype is: HLA DR4-DQ8 (DQB1*0302-A1*0301).
The commonest moderate-risk haplotype is: HLA DR3-DQ2 (DQB1*0201-A1*0501).
The commonest high-risk genotype is: heterozygous DR3-DQ2/DR4-DQ8.
*The five most susceptible (S).
†The four most protective (P).
Modified from Erlich H, Valdes AM, Noble J, et al: HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk. Analysis of the Type 1 Diabetes Genetics Consortium families. Diabetes 57:1084–1092, 2008.
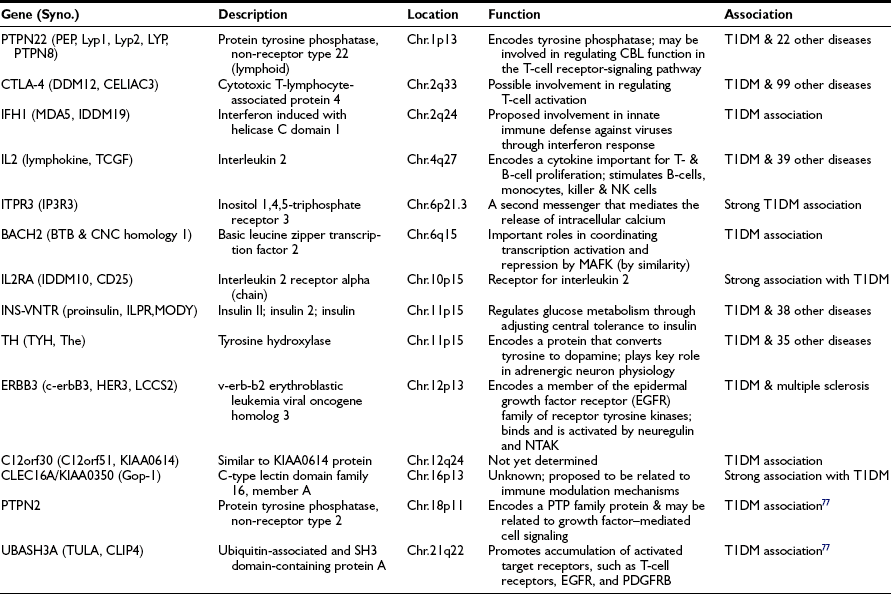
The genetics of T1DM is studied extensively because it represents a paradigm for genetically complex diseases. Genome screens and studies on candidate genes have provided evidence for genetic linkage between polymorphic DNA markers and more than 10 putative T1DM susceptibility genes (refer to T1DBase at http://www.t1dbase.org/page/Welcome/display). Currently, it has been shown that PTPN22 (chr.1p13), CTLA-4 (chr.2q33), IFH1 (chr.2q24), IL2 (chr.4q27), ITPR3 (chr.6p21), IL2RA (chr.10p15), INS-VNTR (chr.11p15), TH (chr.11p15), ERBB3 (chr.12p13), C12orf30 (chr.12q24), CLEC16A/KIAA0350 (chr.16p13), PTPN2 (chr.18p11) genes and two recently identified novel loci, BACH2 (chr.6q15) and UBASH3A (chr.21q22)77 are all associated with increased T1DM risk (Table 14-4). References to these individual genetic factors are found at the T1DBase website.
Environmental Factors
T1DM is characterized by a multifactorial web of environmental factors (Table 14-5). The interaction between genetic and environmental factors and the aggressive islet autoimmune phenomena is complex. The fact that the disease has a relatively long latent period, in which autoimmunity is triggered long before the onset of the clinical syndrome (see Fig. 14-2), indicates a role of environmental factors as initiators or promoters in relation to genetic predisposition.68 Several environmental factors are incriminated in the etiology of T1DM, but linking is indirect, and a definite proof of confirmed association for each factor is yet to be achieved, since the majority of investigations have been carried out at the time of clinical diagnosis and not in relation to the time of islet autoimmunity.
Table 14-5
Viruses and Other Environmental Factors Implicated In or Able to Induce Type 1 (Insulin-Dependent) Diabetes

Viral Infection
There are numerous reports of individual cases in which T1DM onset followed an acute viral infection, borne out by subsequent animal studies in which viruses have been shown to have diabetogenic activity.78 The first case linking T1DM to an acute viral infection was reported in the late 19th century; the onset of T1DM appeared to be precipitated by a mumps infection in a child.79 Many similar reports have followed since,78,80 and taken together, these reports suggest a relation between the clinical onset of T1DM and several viruses, including rubella, mumps, coxsackievirus B, rotavirus, cytomegalovirus, and Epstein-Barr virus, have been implicated.78 The true relation between these viral diseases and the clinical onset of T1DM remains conjectural.
Viruses are thought to increase the risk of T1DM through different proposed mechanisms. Whereas some viruses induce rapid and direct effect, causing β-cell destruction and subsequent insulin deficiency, other viruses induce slower, longstanding effect through activation of autoreactive T cells. Viruses may also inhibit insulin production by inducing interferons and HLA antigen expression, or they may mimic autoantigens of β cells.78,80
T1DM develops in individuals who are positive for HLA-DR3-DQ2, DR4-DQ8, or both (discussed earlier). The DQ6 haplotype containing DQB1*0602 confers resistance among children. Because these specificities are present in about half the population, a virus inducing either islet autoimmunity, clinical onset of T1DM, or both, may not be spread effectively enough to cause disease. In addition, variations in the annual incidence rate are often taken as evidence of an involvement of virus. An annual variation was found in a population of 15- to 34-year-olds, with lower numbers of new patients being identified during the summer months.48,69 In the group of children younger than 6 or 7 years who have T1DM, however, this annual variation is not always present.51,81
Earlier studies suggested that the congenital rubella syndrome was strongly associated with T1DM; approximately 10% to 20% of children develop autoimmune T1DM, and around 50% of them develop autoimmune antibodies.80 However, most countries with high incidence, such as Finland and Sweden, had successfully implemented vaccination programs against rubella virus. Vaccination practices have prevented rubella epidemics, but they have not affected the incidence rate of T1DM.
Maternal enteroviral infection during pregnancy also appears to be a risk factor for childhood T1DM.82 It is therefore possible that gestational infections by many types of viruses affect the maturation of the immune system, causing certain children to be more predisposed to autoimmunity and thereby increasing the risk for T1DM. Prior exposure to measles, mumps, and rubella, but not vaccination, decreased prevalence of pancreatic and thyroid autoantibodies.83 Maternal viral infections or reduced exposure to natural infections may be associated with an increase in T1DM.
Enteroviruses, in particular the coxsackie B serotype have been extensively studied at the time of clinical onset84 but rarely in relation to the appearance of islet autoimmunity.80 Coxsackie B4 virus was isolated from the pancreas of a child who died at presentation with T1DM, propagated in the in-vitro cultures of endocrine pancreatic cells, and then shown to have diabetic activity in certain mouse strains.85 Coxsackievirus infection in the mouse seems to be associated with virus replication in the β cells, followed by the formation of GAD65 antibodies.86,87 Two important hypotheses follow from these experiments. One hypothesis is that the coxsackievirus induces β-cell neoantigens, which initiate an (auto)immune reaction. This hypothesis can be tested by analyzing the appearance of such neoantigens. The neoantigen may initiate a devastating reaction if its structure mimics a self-protein. In line with this hypothesis, a sequence in GAD65 is identical to that in a coxsackievirus antigen.88,89 Another hypothesis is that Coxsackie B virus replication in β cells results in β-cell necrosis and the formation of antibodies against β-cell constituents or “hidden antigens” not normally surveyed by the immune system.90 An autoimmune reaction is initiated that may escalate with time.
Other viruses incriminated in T1DM etiology include rotaviruses, which are common causes of childhood gastroenteritis. These viruses showed peptide sequences similar to T-cell epitopes in IA-2 and GAD antibodies, suggesting their role in T1DM.82 Cytomegalovirus is a DNA virus that is thought to induce autoimmunity through a molecular mimicry mechanism. Mumps virus was correlated to autoimmunity and even to overt T1DM. Human endogenous retroviruses (HERV) were also reported to correlate with T1DM.80
In experimental animals (see Table 14-4), several viruses are known to induce T1DM, either through direct effects on the β cells, causing rapid destruction (e.g., encephalomyocarditis virus EMC-D) or through the disruption of the normal immune regulatory mechanisms (e.g., Kilham rat virus [KRV], which causes autoimmune T1DM in diabetes-resistant BioBreeding [DR-BB] rats).78 Other viruses (e.g., reovirus) were found to affect primarily the immune system of the host to induce a polyclonal autoantibody response. The production of autoantibodies appeared to be closely related to the pathogenesis of disease in mice.86,87 Captured bank voles may develop T1DM with islet autoantibodies and signs of parechovirus infection, raising the question whether there are zoonotic virus–inducing islet autoimmunity, T1DM, or both.91
The virus-induced disease in mice depends on strain, because some mouse strains are resistant to the pathogenesis of a virus that induces T1DM in another strain. Similar strain dependency to the β-cytotoxic agent streptozotocin, followed by the inoculation of a diabetogenic virus, rendered otherwise virus-resistant mice diabetic.92 This observation may be significant to humans because it is possible that repeated injuries to the pancreatic β cells over several years of life may eventually induce T1DM. An additional role of HLA has been suggested, because it cannot be excluded that repeated injuries are particularly detrimental if the T1DM-associated HLA alleles are linked with a poor regenerative capacity of the pancreatic β cells.
Hygiene Hypothesis
The hygiene hypothesis proposes that as living environment is improved, children become less exposed to infectious agents, which leads to inadequate maturation of their immune systems. This hypothesis suggests that early exposure to pathogens may enhance the immune responses of those children, thereby suppressing autoimmune reactions involved in T1DM pathogenesis. The latter suggestion is based on increasing incidences of diseases like asthma or other atopic disorders, in addition to the fact that T1DM is more prevalent in developed societies. The hypothesis also suggests that younger children are more prone to infections, since they did not acquire antibodies against viruses (e.g., enterovirus antibodies from their mothers), resulting in an increased risk for T1DM or other autoimmune diseases.80 Indeed, other studies reported higher maternal enterovirus antibodies in countries with lower incidence of T1DM compared to countries with high incidence.82
Dietary Factors
The dietary hypothesis is based on earlier observations that breastfed children have a lesser risk of T1DM. It was also noted that early exposure of infants younger than 6 months to cow’s milk proteins may double the T1DM risk, particularly in HLA high-risk children. Other studies described an association between islet autoimmunity and shorter duration of breastfeeding, as well as early exposure of infants to bovine protein.93 The Diabetes Prediction and Prevention (DIPP) study of Finland reported a fivefold higher risk of multiple islet autoantibodies among children with HLA DQB1*0302 who received cow’s milk formula before the age of 4 months. Similar associations between islet autoimmunity or T1DM and intake of glutens, foods rich in protein, carbohydrates, and nitrosamine compounds were reported.94 Additionally, early exposure to bovine insulin in cow’s milk may lead to formation of autoantibodies against human insulin through cross-reaction.95 These findings are controversial. The Diabetes Auto-Immunity Study in the Young (DAISY) found no evidence correlating cow’s milk, enteroviral infection, or vaccinations with increased T1DM risk.96 Similarly, the EURODIAB Substudy-2 Group suggested that rapid growth, rather than cow’s milk or early introduction of solid food, would explain the increased incidence rate of T1DM.97 The islet autoimmune processes in the spontaneously diabetic NOD mouse and BB rat are easily inhibited by dietary manipulation.98 Studies in inbred and pathogen-free laboratory rodents underscore the possible importance of the gut immune system in T1DM.
Maternal Factors
Several maternal factors were reported to increase the risk of T1DM in children. Higher maternal age (and to a lesser extent, paternal age) at the time of delivery increased the risk of T1DM in the offspring. Exposure of the mother, as mentioned earlier, to enteroviral infection, colder season of delivery, ABO blood group incompatibility, maternal islet autoantibodies, or eclampsia all increased the risk of T1DM.99 An analysis of nondiabetic mothers with islet autoantibodies at delivery indicated that an association between HLA and islet autoimmunity may depend on environmental exposure during pregnancy.30,82 Follow-up of mothers and children will determine the risk of T1DM. However, mothers who developed T1DM after 8 years of age confer lower risk on their offspring compared to fathers with T1DM. On the other hand, birth order seems to have positive association with the risk of T1DM, since the first child carries the highest risk, and this risk declines by 15% with each new child.99 In the NOD mouse, it was reported that obliteration of the maternal transmission of antibodies inhibited the T-cell-mediated destruction of islet β cells in NOD mice.100 These and other investigations, in humans as well as in laboratory animals, underscore the importance of clarifying whether transmission of maternal autoantibodies affects the incidence of T1DM in susceptible offspring.
Birth Weight and Growth Rate
Several studies suggest a positive association between T1DM HLA risk and an increase in perinatal and postnatal growth. Children with higher weight-for-height or BMI during infancy or childhood have greater T1DM risk.101 It has also been suggested that high energy intake (particularly disaccharides such as sucrose) may be important to hyperinsulinemia resulting in rapid growth and T1DM susceptibility.102 The “accelerator hypothesis” suggests that factors like rapid growth, weight gain, and high energy intake would stress β cells, leading to acceleration (not triggering) of cell-mediated autoimmune destruction28 and that this may explain the increasing incidence of T1DM among young age groups.
Psychological Stress
Case-controlled studies reported that negative life events occurring during the first 2 years of life, difficult adaptation, or child behavioral deviances may increase the risk of T1DM.102 It has also been reported that psychological stress, measured as psychosocial strain during pregnancy, seems to be involved in the induction or progression of β-cell-related autoimmunity in cord blood and during the first year of life.103 Additionally, high parenting stress, experiences of serious life events, foreign origin of the mother, and low socioeconomic status were all associated with β-cell-related autoimmunity in young children.104 It was hypothesized that parent-infant relationship may be a “psychobiological regulator” and that early disruption of this relationship may cause stress and affect the hypothalamic-pituitary-adrenal axis. Such changes may affect the immature autonomic nervous system of the child and subsequently impair the regulation of autoimmunity.105
Toxic Substances
Alloxan and streptozotocin are widely used to induce β-cell destruction and diabetes in experimental animals.106 Some species including man are more resistant to these drugs than others. Streptozotocin is commonly used in treating certain gastroenteric tumors, including glucagonomas; however, T1DM rarely develops in these patients. A number of compounds structurally related to streptozotocin and alloxan have been implicated as possible environmental agents contributing to T1DM. The nitrosamine moiety of the streptozotocin molecule may be diabetogenic when present on molecules other than D-glucose.107 Pyriminil (Vacor), an effective rodenticide, is highly diabetogenic in humans.106 Individuals in whom diabetes developed after ingestion of Vacor had islet cell surface antibodies, indicating that β-cell-related autoimmunity may develop after β-cell destruction. The TEDDY study (The Environmental Determinants of Diabetes in the Young) is an international effort to substantiate the possibility that environmental chemicals are causative factors in the development of T1DM.108
Pathology
Studies of the pancreas in newly diagnosed T1DM patients have indicated that the gland is diminished in size compared with that in matched controls.8,109 An atrophic pancreas with little or negligible residual insulin is typical of a patient with longstanding T1DM. The atrophy affects primarily the tail of the pancreas. In this part, the composition of the endocrine pancreas is dominated by β and α cells, whereas in the pancreatic head, β and pancreatic polypeptide (PP) cells predominate. In addition to insulin deficiency, the pancreatic atrophy appears to be reflected in lower serum levels of pancreatic trypsin and isoamylase.110,111 It is speculated that the pancreas atrophy is a result of an intrapancreatic insulin deficiency affecting the growth of the pancreas.
The insulin deficiency is the result of a specific loss of the β cells, which affects the total mass of the endocrine pancreas in T1DM (Table 14-6). The islets are small and appear pseudoatrophic.112,113 In contrast to the normal pancreas, in which the β cells predominate, the endocrine cells in the diabetic pancreas are primarily α, δ, and PP cells. Lobular variation has been described, and areas with β-cell-containing islets without pancreatic atrophy also have been detected, suggesting that parts of the pancreas may be spared112 in individuals with newly diagnosed T1DM. Sometimes β cells are seen in close proximity to duct cells, suggesting neoformation of cells. The understanding of the developing endocrine pancreas has improved, and a number of transcription factors controlling the process have been identified.114 However, one of the characteristics of the pancreas in many children with newly diagnosed T1DM is the presence of inflammatory cells adjacent to islets or cords of newly formed β cells.112
Table 14-6
Morphometric Analysis of the Endocrine Pancreas in Control Individuals and Patients with Type 1 Diabetes Mellitus
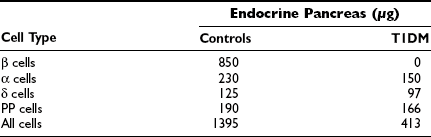
Adapted from Rachier J, Goebbles RM, Henquin JC: Cellular composition of the human diabetic pancreas. Diabetologia 24:336–371, 1983, with permission.
The presence of inflammatory cells in the pancreas of diabetes patients with short-duration disease was initially demonstrated at the turn of the 20th century. The term insulitis was first introduced in 1940,7 but the phenomenon was not established until later quantitative investigations reported insulitis in 16 of 23 individuals with T1DM who died within 6 months of diagnosis.8 It is believed that insulitis is not detected in a greater percentage of new-onset T1DM patients because the total mass of β cells is already markedly reduced at the time of clinical diagnosis.13 Immunocytochemical investigation in rare specimens of pancreas from patients who died shortly after the clinical onset of T1DM indicates that all cell types considered part of the immune system populate the islets to form the insulitis115; T lymphocytes, B lymphocytes, macrophages, and occasional natural killer (NK) cells may be seen. In agreement with earlier studies, examination of pancreatic islets in laparoscopically obtained biopsy specimens from newly diagnosed T1DM patients116,117 demonstrated insulitis in about 60% of patients, with a T cell-predominant infiltration and increased expression of HLA class I.118 Notably, the presence of insulitis was strongly correlated with the presence of other autoimmune features, specifically positivity for GAD65 or IA-2 autoantibodies.118 In contrast, recent examination of specimens from 1507 pancreatic donors aged 25 to 60 years revealed 62 (4%) islet autoantibody–positive subjects. Insulitis was found in only 2 of the 62 (3%) subjects; the 2 subjects were positive for multiple islet autoantibodies and susceptible HLA. None of the 62 matched controls. These results suggest that positivity for islet autoantibodies does not immediately mean that insulitis is present. Islet autoantibodies may precede insulitis, which therefore may require the concurrent presence of multiple islet autoantibodies.119
Individuals at risk for development of T1DM have been found to have hyperproinsulinemia, a condition thought to reflect maximally stimulated or perhaps exhausted β cells.120,121 When compared with matched controls, HLA-identical siblings of patients with T1DM have been found to have evidence of both insulin resistance and impaired β-cell function.122 To estimate the residual mass of insulin-producing cells in humans, it is important to take into account such factors as the maximal rate of insulin release, extraction of insulin in the liver and peripheral tissues, and resistance to insulin action.123 It has been shown that intensive therapy for T1DM helps sustain endogenous insulin secretion, which in turn is associated with better metabolic control and lower risk for hypoglycemia and chronic complications.124,125
The role of immune cells in the process leading to the disappearance of β cells is well established in the NOD mouse and BB rat.26 It is possible to transfer diabetes adoptively with clonal T lymphocytes in both species. Similarly, transfer of T1DM between HLA-identical siblings has been achieved by bone marrow transplantation.126
Pathogenesis
The pathogenesis of T1DM is strongly associated with several immune abnormalities. These immune abnormalities, in particular autoantibodies directed against specific islet cell autoantigens such as insulin, GAD65, IA-2 and ZnT8, are dynamic markers of an ongoing disease process. These pathogenic markers are useful to predict and classify T1DM. The rate of β-cell destruction appears to be influenced by HLA. The HLA DQA1*0301-B1*0302/DQA1*0501–BI*0201 genotype may accelerate β-cell destruction.127 The DQB1*0602 or 0603 alleles are dominantly protective, although the negative association is attenuated with increased age at onset.128 In DQB1*0602/0603-positive patients, the second haplotype is most often DQA1*0201-B1*0302,129 suggesting that HLA may influence the tempo of the disease process. Because the HLA molecules are important determinants in regulating the immune response in humans, the susceptibility to T1DM is speculated to be conferred by the functional importance of these molecules in the immune response. The role of the class II molecules in the immune response is to convey cell-cell interactions between T-helper (CD4+) lymphocytes and antigen-presenting cells (APC) or B lymphocytes. The CD4+ T cell provides help to cytotoxic T(CD8+) lymphocytes as well as to B lymphocytes. The importance of the immune system to T1DM pathogenesis is underscored by recent genome-wide association studies, which suggest that variants of genetic factors important to the immune response increase the risk of T1DM (see Table 14-4).
Immunologic Abnormalities
Cellular Immunopathophysiology
It is possible that the APC activity is altered in patients or in individuals susceptible to T1DM. Blood monocytes in T1DM patients may be defective in prostaglandin synthetase,130 and dendritic cells from patients with T1DM have a maturational and functional defect.131 APC from T1DM patients secrete markedly higher levels of proinflammatory cytokines in response to a nonantigenic stimulus.132 Also, healthy HLA DR3-positive subjects have an abnormally prolonged Fc receptor–mediated mononuclear phagocyte system.133
T lymphocytes are also important to the pathogenesis of T1DM. The earliest demonstration involved tests for leukocyte migration inhibition or blast formation, suggesting that T1DM patients may be sensitized to pancreatic antigens.10,11,134 It was also noted that a delayed hypersensitivity reaction developed in these patients to subcutaneously injected pancreatic homogenate.11,134 Several disorders of autoimmune character have been found to have an imbalance in the peripheral blood between T-helper (CD4+), cytotoxic (CD8+), and regulatory (CD4+CD25+) T cells.135 Recent clinical trials with anti-T-cell drugs, such as cyclosporine,136 or humanized monoclonal antibodies against CD3,137 have been shown to preserve residual β-cell function in newly diagnosed T1DM patients. Clinical trials with alum-formulated GAD65 showed preservation of C-peptide and an increase in T regulatory cells.138,139 However, assays specific to T cells recognizing insulin, GAD65, or IA-2 have proven difficult to establish and standardize.14,140 Immunodominant epitopes of GAD65141,142 and IA-2143 have been identified and should be useful in improving cellular assays in T1DM.144–146 The use of HLA tetramers makes it possible to detect147 and clone autoantigen-specific human T cells.148 Other approaches to detect T-cell reactivity against islet cell antigens, such as ELISPOT, which measures cytokine secretion from individual T cells, are being explored.149 Several reports indicate an increased frequency of in-vitro proliferating T cells in patients with T1DM, compared with levels in control subjects, in response to autoantigens such as GAD89,150,151 and IA-2.143
Humoral Immunopathophysiology
Islet cell antibodies (ICA) detected by indirect immunofluorescence on frozen sections of human blood group O pancreas was the first indication of autoantibodies in T1DM.16 Islet cell surface antibodies (ICSA; see Table 14-7) in newly diagnosed T1DM patients were demonstrated in dispersed-cell preparations of rat or mouse pancreatic islets17,152 Antibodies in T1DM sera preferentially bind to β cells if the disease was diagnosed before age 30 years.153 The observation that antibodies are capable of binding to living β cells is important because it allows testing of the possibility that surface-bound antibodies either mediate immune effector mechanisms or directly affect the function of the β cells. The former phenomena included complement-mediated cytotoxicity or antibody-dependent cellular cytotoxicity.154 Either mechanism could contribute to killing pancreatic β cells, provided that the in-vitro phenomenon also is occurring in vivo. However, not knowing the autoantigen was a shortcoming in these types of autoantibody studies. Several assay systems are now in use to determine the presence of antibodies reactive with pancreatic islet cell autoantigens (Table 14-7). Autoantibodies to GAD65, IA-2, insulin, and the zinc transporter ZnT8 have replaced ICA.24,25 One or more of these autoantibodies may be present in more than 95% of newly diagnosed T1DM patients, and they present variably during the prodrome of islet autoimmunity long before the clinical onset. Two or more of these autoantibodies have a high predictive value of T1DM.27 Therefore, it is critical that reliable tests be available to detect these autoantibodies.
Glutamic Acid Decarboxylase Antibodies
Glutamic acid decarboxylase is an enzyme present mainly in pancreatic β cells and neuron cells. This enzyme produces γ-aminobutyric acid (GABA), which is stored in small neurotransmitter vesicles. Two isoforms are known: GAD65 and GAD67. Immunoprecipitation of human islet proteins has revealed the presence of autoantibodies against a 64K protein19,155 identified as GAD but found to represent an isoenzyme, GAD65, coded for by a gene on human chromosome 10.21 The previously known GAD67 on chromosome 2 shares 65% of the amino acids, but this isoform is not expressed in human β cells.156,157
The frequency of GAD65 antibodies (GAD65Ab) in children with new-onset T1DM is 70% to 80% (Fig. 14-5). Patients with GAD67 antibodies are almost invariably GAD65-antibody positive. The GAD65 autoantibody (GAD65Ab) frequency in new-onset patients is little affected by the age at onset; however, in children younger than 10 years, more girls than boys have GAD65Ab. GAD65Ab are evanescent but less so than ICA (Fig. 14-6). GAD65Ab tend to be the most prevalent and persist for longer duration after the diagnosis of T1DM when compared to other autoantigens. Almost 50% of patients with disease duration of 10 years may still be GAD65Ab positive (see Fig. 14-6).158,159 GAD65Ab have high sensitivity but lesser specificity, owing to variations in the titer, which is usually low at time of diagnosis.
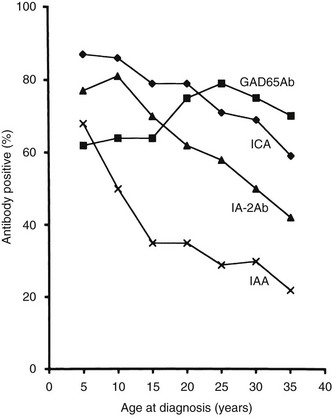
FIGURE 14-5 Frequency of autoantibodies against glutamic acid decarboxylase (GAD)65 (GAD65Ab), insulinoma-associated antigen 2 (IA-2Ab), and insulin autoantibodies (IAA) at the time of clinical diagnosis of type 1 diabetes mellitus in relation to age at onset.
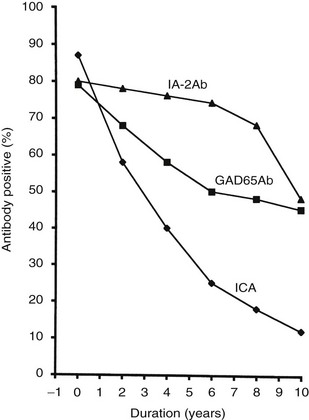
FIGURE 14-6 Frequency of autoantibodies against glutamic acid decarboxylase (GAD)65 (GAD65Ab) and insulinoma-associated antigen 2 (IA-2Ab), as well as islet cell antibodies (ICA), in relation to the duration of type 1 diabetes mellitus.
Any assessment of an individual’s risk of developing T1DM depends on the ability to determine accurately the presence of antibodies in a prospective analysis. This is particularly important because GAD65Ab may appear temporarily in healthy individuals.160 Persistent GAD65Ab are more often detected in DRBI*03-DQA1*0501-B1*0201-positive than in DRB1*04-DQ A1*0301-B1*0302-positive subjects.161 GAD65Ab may be detected in about 1% of the general population, but the frequency increases to about 8% among first-degree relatives of patients with T1DM.162,163 The positive predictive value of GAD65Ab for T1DM is about 50% or higher among first-degree relatives. It may be as high as 20% in schoolchildren with high-risk HLA.164 GAD65Ab also predict stiff-man syndrome, although the GAD65Ab epitopes are different from those in T1DM.165,166 T1DM is best predicted when the GAD65Ab is combined with insulin autoantibodies, IA-2Ab, or ZnT8Ab.27,167,168 In studies of GAD65 epitopes with human monoclonal GAD65 antibodies, it was found that anti-idiotypic GAD65Ab were reduced in T1DM patients. It was suggested that the major marker for T1DM is rather the absence of specific anti-idiotypic antibodies than the presence of GAD65Ab.169
In order to provide unified comparative assays, standardization of GAD65Ab (and IA-2A) has been done in the DASP (Diabetes Antibody Standardization Program).25 Both the radiobinding assay (RBA) and enzyme-linked immunosorbent assay (ELISA) performance have improved and are able to discriminate controls from patients (see Table 14-7). These results imply the value of GAD65Ab (and IA-2Ab) both for classification of patients with diabetes and for screening high-risk populations.
Studies in the BB rat and the NOD mouse have indicated that GAD65Ab may be detected in these animals.170 However, an international workshop on lessons from animal models for human T1DM questioned the nature of GAD65Ab (and IA-2A) in the NOD mouse.170 ELISA- and RBA-detected GAD65Ab in some mice seemed mostly to represent nonspecific binding. The role of GAD65 in spontaneous or transgenic diabetic mice remains an enigma, as GAD65 is not expressed in mouse beta cells.
Insulin Autoantibodies
Insulin is produced after the C-peptide has been cleaved from proinsulin. The molecular weight of insulin is 5.8 kD, and the molecule is composed of the A and B chain. It was demonstrated in a radioligand-binding assay that approximately 50% of patients with newly diagnosed but untreated T1DM had insulin autoantibodies (IAA).22 Among children at T1DM genetic risk and younger than 5 years of age, IAA were reported to be the first islet autoantibody to appear but fluctuated more than ICA, GAD65Ab, or IA-2Ab.171 Proinsulin is therefore viewed as a primary specific autoantigen, especially since it is unique to the β cells. IAA react with both insulin and proinsulin, indicating that they possess shared epitopes as targets of IAA.
IAA were positively associated with DR4, perhaps because they are in linkage disequilibrium with DQA1*0301-DQB1*0302.161,172 A first analysis of putative IAA epitopes indicates that the amino acids B1 to B3 and A8 to A13 are important.173 IAA and insulin antibodies that appear after insulin therapy have similar binding characteristics.174
It has been proposed that IAA predict T1DM better in children (see Fig. 14-5) than in adults.175,176 Current prospective, population-based studies of children followed from birth are therefore required to establish the possible association between HLA, including the DR4 subtypes, environmental exposures, and the appearance of IAA.
IAA assays have been standardized, and only the fluid-phase radioimmunoassay (but not ELISA) was found to have high diagnostic sensitivity and specificity for T1DM.177,178 The fluid-phase radioimmunoassay for IAA is still lacking an international standard, and the IAA test is therefore yet to be standardized.177 However, insulin antibodies developed after initiation of insulin therapy have higher specificity for insulin and may therefore be detected by ELISA.179
Insulin appears to be an important autoantigen in the NOD mouse but not in the BB rat. IAA measured by sensitive RBA were found to be markers of autoimmunity in NOD mice.170 Furthermore, it was found that CD4 T-cells in the NOD mouse specifically target the β chain of insulin, and administration of the whole insulin molecule or its β chain in the prediabetes phase may prevent or delay disease onset in susceptible mice.179 It has also been proposed that proinsulin acts as the primary autoantigen that triggers a pathologic autoimmune reaction against the β cells. This reaction is propagated into other antigens such as IGRP (islet-specific glucose-6-phosphatase catalytic subunit–related protein) through epitope spreading.180
Islet Antigen-2 Antibodies (Insulinoma-Associated Antigen-2 Antibodies)
Trypsin treatment of the 64K immunoprecipitate revealed 40K and 37K fragments of islet antigen181,182 yielding two isoforms of the tyrosine phosphatase–like protein islet antigen-2 antibodies (IA-2Ab), IA-2 (ICA512)23 and IA-2β (phogrin),183–185 respectively. IA-2 and IA-2β are both associated with the secretion granule membrane.182,186,187 These isoforms share high structural similarities but lack enzymatic activity, and their functions in neuroendocrine tissues such as the hypothalamus and pancreatic islets are not fully understood. IA-2Ab are detected in 60% to 70% of patients with new-onset T1DM.188 These antibodies are less frequent with increasing age at onset (see Fig. 14-5).189 The IA-2Ab would therefore better predict young age at onset of T1DM. Longitudinal studies of first-degree relatives suggest that IA-2Ab tend to appear closer to the clinical onset of T1DM compared with GAD65Ab, which tend to appear earlier in the prodrome.188 The IA-2Ab are evanescent (see Fig. 14-6), but when present in patients who are young at disease onset, as many as 50% may still be IA-2Ab positive after having had diabetes for 10 years (see Fig. 14-6).
IA-2Ab were associated primarily with DRB1*0401 and not with DQA1*0301-DQBI*0302,161 and the risk for this antibody is increased in males.189 Surprisingly, the presence of IA-2Ab in T1DM was negatively associated with DQA1*0501-DQBI*0201.128 The mechanisms by which the IA-2 proteins become recognized as autoantigens are therefore not yet understood. The frequency of IA-2Ab in the general population is less than 1%.39 In young children with genetic susceptibility, progression to clinical T1DM onset was associated with broad epitope response and juxtamembrane reactivity to the IA-2 antigen, while the occurrence of IgE-IA-2 antibodies provided relative protection.190
The predictive value of IA-2A for T1DM is best estimated in combination with GAD65Ab and IAA.27,168 The fact that sera from T1DM patients could precipitate GAD65, IA-2, and IA-2β simultaneously using RBA made it possible to develop standardized assays for these autoantibodies with high levels of sensitivity and specificity.25 ELISA tests for IA-2Ab are available with similar diagnostic sensitivity and specificity to radioligand assays, and combination ELISA tests using biotin-labeled preparations of both IA-2Ab and GAD65Ab have been developed. IA-2Ab performance with ELISA was improved in DASP to achieve levels of equivalent to in-house results of RIA (see Table 14-7).25
At onset of diabetes in NOD mice, IA-2Ab had higher signals (36% to 47%) in RBA than control mice, and these results were concordant with an IA-2Ab ELISA (50%). However, although both GAD65Ab and IA-2Ab had strong concordance in individual mice, they seemed to represent nonspecific binding.170 IA-2Ab have not been reported in the BB rat or in other spontaneously diabetic laboratory rodents; however, these antibodies were detected in bank voles developing diabetes in captivity.91 The BB rat developed lymphopenia and diabetes because of a mutation in the GIMAP 5 gene,191 and it was therefore of interest that high titers of IA-2Ab were associated with a genetic polymorphism in the human GIMAP5 gene.192
ZnT8 Transporter (SLC30A8)
Insulin used to be thought of as the only β-cell-specific autoantigen, but the β-cell-specific ZnT8 transporter (zinc transporter isoform-8) was found not only to be the target of islet autoimmunity but also a genetic marker for T2DM.24,193 The ZnT8 transporter is important to the formation of insulin crystals in that it facilitates Zn2+ transportation into the β-cell insulin granules. This antigen is encoded by the SLC30A8 gene, which is one of nine human genes for multispanning transmembrane proteins.194 Epitope mapping of these autoantibodies showed that ZnT8Ab primarily react with the C-terminal end of the protein. Polymorphic variants of ZnT8 at amino acid position 325 in the cytosolic tail were found to be either arginine (ZnT8-R) or tryptophan (ZnT8-W),193,194 both representing markers of ZnT8Ab susceptibility with potential diagnostic, therapeutic, and prognostic uses.24
ZnT8Ab were detected in 26% of subjects who were reported negative for GAD65Ab, IA-2Ab, and IAA. On the other hand, ZnT8Ab were found to be positive in 60% to 80% of newly diagnosed T1DM patients but in only 2% of controls and less than 3% of T2DM patients.168,193
ZnT8Ab were also found during the islet autoimmune prodrome, often after 2 years of age and independently of other autoantibodies but mainly later than GAD65Ab and IAA.24,193 The high specificity and independent appearance of ZnT8Ab highlight their usefulness for prediction, especially in older children and adolescents, since ZnT8Ab prevalence and titer levels increase with age.
Following similar principles of GAD65Ab and IA-2Ab standardization, fluid-phase radioassays of ZnT8Ab (C-terminal) were standardized and validated in the DASP workshop (see Table 14-7).24,168
In summary, currently available methods for detecting autoantigen-specific autoantibodies (GAD65Ab, IA-2Ab, IAA, and ZnT8Ab) are of value in predicting T1DM.27,168 Standardized assays for these autoantibodies25,168 are important when selecting participants in prevention and intervention trials.195 How the uptake, processing, and presentation of these autoantigens by APC that initiate the formation of islet cell autoantigen–reactive T and B lymphocytes are accomplished are important questions that remain to be answered.
Candidate (Minor) Autoantigens
Several substances or molecules attributed to the autoimmunity of T1DM may represent candidate or minor autoantigens. Autoantigens reported to be associated with T1DM include imogen 38,196 vesicle-associated membrane protein-2 (VAMP2),197 neuropeptide-Y (NPY),197 glima-38,198 heat shock protein 60 (HSP60),199 ICA69,200 carboxypeptidase H (CPH),201 DNA topoisomerase II,202 CD38 (ADP ribosyl cyclase/cyclic ADP-ribose hydrolase),203,204 Glut-2 (Type-2 glucose transporter),205 ICA12/SOX13,206,207 and others (Table 14-8). The role of these candidate autoantigens in islet autoimmunity, T1DM, or both, remains to be determined.
Table 14-8
Candidate (Minor) Autoantigens in Type 1 Diabetes Mellitus
| Candidate Autoantigens | Notes | Reference |
| Imogen-38 | Mitochondrial 38-kD protein found in β cells more frequently than in α cells. No antibodies are yet detected against imogen-38, but it may be a target for bystander autoimmune attack in diabetes rather than a primary autoantigen. | Arden196 |
| Vesicle-associated membrane protein-2 (VAMP2) | A protein associated with secretary vesicles of pancreatic β cells. Anti-VAMP-2 antibodies detected in sera of 21% of T1DM patients and 4% of controls. | Hirai197 |
| Neuropeptide Y (NPY) | In the pancreas, it inhibits insulin secretion stimulated by glucose secretion. NPY was detected in 9% of sera from T1DM patients and 2% of controls. | Hirai197 |
| Glima 38 | Antibodies against this amphiphilic glycated β-cell membrane protein are found in 20% of new-onset T1DM, 14% of pre-diabetic individuals, and 0% of healthy controls. | Aanstoot198 |
| Heat shock protein 60 (HSP60) | Antibodies against this protein are found in 15% of T1DM patients and 20% of rheumatoid arthritis patients. | Ozawa199 |
| Islet cell autoantibody 69 ICA69 | Antibodies to this novel peptide are found in 5%-30% of new-onset T1DM but can also be found in 6% of normal controls and 20% of rheumatoid arthritis patients. | Martin200 |
| Carboxypeptidase H (CPH) | A glycoprotein functions in processing proinsulin to insulin within islet granules. CPHAb were detected in 20% of ICA+ relatives and 0% of controls. | Castano201 |
| DNA topoisomerase II | TopIIAb can be found in 50% of T1DM patients. | Chang202 |
| CD38 (ADP ribosyl cyclase/cyclic ADP-ribose hydrolase) | CD38Ab were found in 13% of T1DM patients, 10% of T2DM, and 1.3% of controls among Caucasian populations. It was also reported in 14% of T2DM Japanese patients. | Pupilli203 Ikehata204 |
| Glut-2 (type-2 glucose transporter) | A glucose transporter that was found in 32%-80% of newly diagnosed T1DM patients but only 6.6% of controls. | Inman205 |
| ICA12/SOX13 | Autoantibodies to ICA12/SOX13 were demonstrated in 10%–30% of T1DM and 6%–9% of T2DM patients but in only 2%-4% of controls from different populations. | Kasimiotis206 Törn207 |
Prediction
Highly effective, precise, and reproducible screening assays for HLA genotypes, as well as for GAD65Ab, lA-2Ab, ZnT8Ab, and possibly IAA, are in use to detect individuals who are carriers of islet cell autoimmunity with an increased risk for T1DM (Table 14-9). Early detection of T1DM serves several purposes. Among them is the distinct possibility that ketoacidosis associated with a dramatic and traumatic onset of T1DM, in some cases leading to death, would be forestalled. In addition, immunotherapeutic modalities for disease prevention of β-cell destruction are under active investigation, with the hope that T1DM may someday be a preventable disease.
Table 14-9
Autoantibodies and Immune Complexes Found With Increased Frequency Among Type 1 Diabetes Patients and Their First-Degree Relatives
| Target | Autoantigen |
| Organ-Specific | |
| Islets | GAD65, IA-2, insulin, ZnT8 |
| Thyroid | Thyroid peroxidase (TPO), thyroglobulin (Tg) |
| Parathyroid | NACHT leucine-rich-repeat protein 5 (NALP5) |
| Stomach | H+/K+ ATPase, intrinsic factor |
| Adrenals | 21-hydroxylase |
| Pituitary | Enolase is a candidate autoantigen |
| Non-Organ-Specific | |
| Peripheral lymphocytes | Lymphocytotoxic |
| Nucleic acids | Single-stranded RNA, double-stranded RNA |
| Cell constituents | Tubulin, insulin receptor |
| Plasma proteins | Albumin |
| Immune complexes | Solid-phase C1q-binding, Raji cell binding |
ATP, Adenosine triphosphate; GAD, glutamic acid decarboxylase.
It is well established that the development of islet autoimmunity and T1DM may be triggered during early infancy or even intrauterine life among genetically susceptible subjects.29,30 Transplacentally-transferred autoantibodies may be detected in cord blood of newborn infants of diabetic mothers, but they mainly disappear by 6 months. On the other hand, nondiabetic mothers with detectable antibodies confer even higher disease risk to their offspring.208
The fact that clinical onset of T1DM is preceded by a long “prediabetes” phase marked by islet autoimmunity markers makes T1DM a candidate for prediction and prevention. The predictive value of these autoantibodies varies with age, and combining more than one autoantibody is increasing the positive predictive value. The improved ability to measure autoantibodies against GAD65, IA-2, and insulin made it possible to predict T1DM by up to 98% accuracy when ZnT8Ab were added to the arsenal of islet autoantibody tests.168 A high predictive value of the screening tests is critical to designing interventional studies. This strategy is most useful in high-risk groups such as first-degree relatives, where high diagnostic specificity is required. The levels of autoantibodies reflect the degree of aggressiveness of the autoimmune reaction and are related to persistence of positivity and subsequent progression into clinical disease.209
HLA testing alone is not a primary indicator for the prediction of T1DM, but it has a significant role when used with autoimmune markers to increase sensitivity of prediction among subgroups with moderate risk. However, adding HLA-DQ did not affect the diagnostic sensitivity of islet autoantibodies.210 Several other genetic loci have also been reported to be associated with T1DM genetic risk, but their contribution was less than HLA (see Table 14-4).
The prediction of β-cell function is assessed through metabolic tests using the first-phase insulin response (FPIR) as a marker among susceptible individuals tested following an intravenous glucose tolerance test (IVGTT). Low levels of FPIR below 50 mU/L confer a risk of 85% within 5 years and predict a 92% risk in the presence of one autoantibody, especially ICA or IAA.211
Prevention
The ability to prevent or reverse autoimmune diabetes in laboratory animals such as the NOD mouse and BB rat using different therapeutic approaches has incited several prevention/intervention studies in humans.26 However, the possibilities of success in humans are significantly different from these animals. Studies in the NOD mouse and the BB rat showed that treatment with insulin or nicotinamide reduced spontaneous diabetes. These studies initiated large clinical trials, such as the Diabetes Prevention Trial-1, the Diabetes Prediction and Prevention Project, and the European Nicotinamide Diabetes Intervention Trial, all without effects (see following discussion). Preclinical studies in rodents may therefore not always be suitable to guide human trials. On the other hand, the spontaneous T1DM rodents may be useful to dissect mechanisms of preventive studies, previously shown to be successful in humans.
Whether to choose a preventive or intervention strategy is a matter of debate, which is related to several factors. First, it has been suggested that intervention trials should be reserved for therapies with greater risk that need to be used on a small number of newly diagnosed patients within limited follow-up periods. Second, prevention trials may be implemented on safer therapies that can be used in the preclinical phase with longer periods of follow-up.34 Prevention trials are often grouped into antigen-specific and non-antigen-specific therapies.35
Primary prevention trials would target high-risk groups (such as infants with high HLA risk or positive family history) prior to the initiation of the autoimmune insult. Environmental factors such as nutritional factors are of interest especially from birth up to 6 years of age; the period when autoimmunity is mostly triggered and highest incidence rise has been observed.44 The Trial to Reduce IDDM in the Genetically At Risk (TRIGR) is a randomized, placebo-controlled, double-blind trial which is testing the effects of cow’s milk in genetically-at-risk infants with positive family history. Infants were randomized to receive cow’s milk or hydrolyzed casein formula following 6 to 8 months of breastfeeding. TRIGR is implemented in 17 countries and expected to provide results in 2009.212 The Nutritional Intervention to Prevent Diabetes (NIP-Diabetes) is recruiting pregnant women and high-risk infants younger than 5 months of age. NIP is using oral docosahexaenoic acid (DHA), a fatty acid thought to prevent islet autoimmunity, given to the mothers as a supplement. Infants will be followed up till 9 years of age. Results from this study will decide future larger trials.35,212 The gluten-free diet trial, PREVFIN, tested the possible effect of eliminating gluten from the diet on the risk of T1DM. It ended in 2002 with no observed effects on islet autoantibodies or disease progression.35 Meta-analysis of results from several earlier observational studies suggested that a vitamin D supplement given to children significantly reduces T1DM risk. There also was some evidence that this association was affected by the dose and timing of supplementation.213 A randomized, open-label trial initiated in 2003 tested the use of 2000 IU/day of vitamin D3 (cholecalciferol), compared to the currently used supplement dose of 400 IU/day, as a possible preventive factor. This pilot study (Manitoba, Canada) recruited infants up to 4 weeks of age who had high HLA genetic risk; results from phase 1 are awaited.35
Secondary prevention strategies recruit children with high HLA genetic risk with positive autoantibodies and children and young adults with multiple autoantibodies. The aim is to retard and suppress β-cell destruction by aggressive immunity. Antigen-based secondary prevention trials using insulin as a specific antigen have been carried out. The Diabetes Prevention Trial-1 (DPT-1) was a large efficacy randomized trial that had two arms: parenteral insulin (high-risk subject, no placebo, completed in 2002) and oral insulin (moderate-risk subject, placebo-controlled, completed in 2005 and repeat study is ongoing).35,212 Both arms showed no effect on the progression of T1DM, although results from subanalysis of the oral arm gave strong evidence on reducing the incidence of T1DM in relatives with insulin autoimmunity (IAA ≥ 80 U).35 Nicotinamide (vitamin B3) was used orally as non-antigen-specific agent in the European Nicotinamide Diabetes Intervention Trial (ENDIT). The results of ENDIT showed no effect on T1DM outcome.35,212 The Diabetes Prediction and Prevention Project (DIPP) was a randomized, placebo-controlled trial implemented in Finland; it recruited high-genetic-risk children to test the efficacy of intranasal insulin on the prevention of T1DM. Lately released results showed that intranasal insulin did not delay or prevent T1DM.214 Other trials that used cyclosporine, vitamin E, or intensive insulin treatment in combination with nicotinamide were also without effects. Other prevention trials tested some drugs such as ketotifen, a histamine antagonist, with no effect observed.35,211,212
Intervention
Intervention or tertiary prevention trials target newly diagnosed T1DM patients, aiming at preserving the residual β-cell mass and enhancing their functionality and regeneration. Like prevention trials, intervention trials can also be either antigen-specific or non-antigen specific. Immunologic vaccination with specific pancreatic antigens has been adopted by a group of trials. The NBI-6024 is a currently ongoing, randomized, placebo-controlled, double-blind trial testing the effects of the altered peptide ligand (APL) insulin B:9-23 vaccine, which is a significant islet autoantigen. This genetically engineered vaccine is injected subcutaneously to adolescents and adults with new-onset T1DM.35
The DiaPep277 was also a randomized, placebo-controlled, double-blind trial that used an immunomodulatory peptide heat shock protein 60 (HSP60) given subcutaneously to subjects with new-onset T1DM. Results from DiaPep277 suggested prevention of β-cell destruction and preservation of C-peptide levels within 12 to 18 months in adults, but a similar effect was not found in T1DM children.34,35
GAD56 formulated with alum has been used in randomized, placebo-controlled immunomodulation trials in LADA215 and 10- to 18-year-old T1DM patients.216 Subcutaneous injection of 20 µg GAD-alum resulted in preservation of residual insulin secretion.215,216 Other trials are using proinsulin-peptide vaccine or proinsulin-based DNA vaccine (BHT-3021) among high-risk children.34,35
The non-antigen-specific approach has been used in numerous interventional studies. Randomized, placebo-controlled trials with oral cyclosporin-A showed reduced loss of residual C-peptide and insulin dosages; however, the effects were transient, and side effects outweighed the benefits.35,212 Although azathioprine trials showed no effects when used alone, its combination with prednisone showed partial, temporary remission of new-onset T1DM.35 Antithymocyte globulin (aTG), used in organ transplantation, showed reduction in insulin requirements when combined with prednisone, but severe, transient thrombocytopenia emerged as a major side effect.35 CD3 monoclonal antibodies including hOKT3gl (Ala-Ala) and ChAglyCD3 (TRX4), which suppress T-cell activity by blocking their proliferation and differentiation, showed preserved C-peptide levels and lowered insulin requirements for 18 to 24 months after therapy.35,35 Trials with CD3 monoclonal antibodies have continued. The Anti-CD20, rituximab, is a monoclonal antibody which reduces blood β cells by blocking the CD20 receptors. Rituximab has shown effects in other autoimmune diseases such as rheumatoid arthritis, with possible benefits in T1DM.34,212 Diazoxide, an inhibitor of insulin secretion, was used to allow residual β cells to rest and protect them from further immune-mediated destruction, but no effect was reported. Similarly, a pilot study with subcutaneous BCG vaccine showed no effects.35
Newly designed trials using autologous umbilical cord blood cells or gene-engineered dendritic cells have been lately launched as tertiary-level interventional trials.212 Mesenchymal stem cells (MSC) have immunomodulatory effects and may be used to prevent and cure T1DM.217 MSC may contribute through direct effects on regulatory T cells and autoreactive T cells and through correction of B cells and natural killer cells, or through indirect effects by regulating the function of dendritic cells or enhancing peripheral immunologic tolerance.217 Tertiary intervention includes both whole-organ pancreatic transplantation and isolated islet transplantation. Both approaches have proved successful in terms of restoring glycemic levels and insulin independence, but isolated islet transplantation had lesser glycemic control, transient effects, and increased morbidity but higher costs.218
References
1. Pinhas-Hamiel, O, Dolan, LM, Zeitler, PS. Diabetic ketoacidosis among obese African-American adolescents with NIDDM. Diabetes Care. 1997;20:484–486.
2. Libman, IM, Becker, DJ. Coexistence of type 1 and type 2 diabetes mellitus: “Double” Diabetes? Pediatr Diabetes. 2003;4:110–113.
3. Köbberling, J, Tattersall, B. The Genetics of Diabetes Mellitus. London: Academic Press; 1982.
4. American Diabetes Association. Standards of Medical Care in Diabetes—2008. Diabetes Care. 2008;31(Suppl. 1):S12–S54.
5. American Diabetes Association. Diagnosis and Classification of Diabetes Mellitus. Diabetes Care. 2007;30(Suppl. 1):S42–S47.
6. Weichselbaum, A. Über die veränderungen des pankreas bei diabetes mellitus. Sitzungsber Akad Wiss Wien Math Naturw Klasse. 1910;119:73–281.
7. Von Meyenburg, H. Über “insulitis” bei diabetes. Schweitz Med Wochenschr. 1940;21:554–561.
8. Gepts, W. Pathologic anatomy of the pancreas in juvenile diabetes mellitus. Diabetes. 1965;14:619–633.
9. Gepts, W, De Mey, J. Islet cell survival determined by morphology. Diabetes. 1978;27:251–261.
10. Nerup, J, Binder, C. Thyroid, gastric and adrenal autoimmunity in diabetes mellitus. Acta Endocrinol. 1973;72:279–286.
11. Nerup, J, Andersen, OO, Bendixen, G. Antipancreatic cellular hypersensitivity in diabetes mellitus: Experimental induction of an anti-pancreatic, cellular hypersensitivity and associated morphological β-cell changes in the rat. Acta Allergol. 1973;28:231–249.
12. Pipeleers, D, Ling, Z. Pancreatic beta cells in insulin-dependent diabetes. Diabetes Metab Rev. 1992;8:209–227.
13. Lernmark, Å, Klöppel, G, Stenger, D, et al. Heterogeneity of islet pathology in two infants with recent onset diabetes mellitus. Virchows Arch. 1995;425:631–640.
14. Tree, T IM, Roep, BO, Peakman, M. A mini meta-analysis of studies on CD4+CD25+ T cells in human type 1 diabetes. Report of the Immunology of Diabetes Society T Cell Workshop. Ann NY Acad Sci. 2006;1079:9–18.
15. MacCuish, AC, Barnes, EW, Irvine, WJ, et al. Antibodies to pancreatic islet cells in insulin-dependent diabetics with coexistent autoimmune disease. Lancet. 1974;2:1529–1531.
16. Bottazzo, GF, Florin-Christensen, A, Doniach, D. Islet cell antibodies in diabetes mellitus with autoimmune polyendocrine deficiencies. Lancet. 1974;2:1279–1283.
17. Lernmark, Å, Freedman, ZR, Hofmann, C, et al. Islet-cell-surface antibodies in juvenile diabetes mellitus. N Engl J Med. 1978;299:375–380.
18. Dobersen, MJ, Scharff, JE, Ginsberg-Fellner, F, et al. Cytotoxic autoantibodies to beta-cells in the serum of patients with insulin-dependent diabetes mellitus. N Engl J Med. 1980;303:1493–1498.
19. Baekkeskov, S, Nielsen, JH, Marner, B, et al. Autoantibodies in newly diagnosed diabetic children immunoprecipitate human pancreatic islet cell proteins. Nature. 1982;298:167–169.
20. Baekkeskov, S, Aanstoot, HJ, Christgau, S, et al. Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature. 1990;347:151–156.
21. Karlsen, AE, Hagopian, WA, Grubin, CE, et al. Cloning and primary structure of a human islet isoform of glutamic acid decarboxylase from chromosome 10. Proc Natl Acad Sci U S A. 1991;88:8337–8341.
22. Palmer, JP, Asplin, CM, Clemons, P, et al. Insulin antibodies in insulin-dependent diabetics before insulin treatment. Science. 1983;222:1337–1339.
23. Lan, MS, Lu, J, Goto, Y, et al. Molecular cloning and identification of a receptor-type protein tyrosine phosphatase, IA-2, from human insulinoma. DNA Cell Biol. 1994;13:505–514.
24. Wenzlau, JM, Juhl, K, Yu, L, et al. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc Natl Acad Sci USA. 2007;104(43):17040–17045.
25. Törn, C, Mueller, PW, Schlosser, M, et al. Diabetes Antibody Standardization Program: evaluation assays for autoantibodies to glutamic acid decarboxylase and islet antigen-2. Diabetologia. 2008;51:846–852.
26. Yang, Y, Santamaria, P. Lessons on autoimmune diabetes from animal models. Clinical Science. 2006;110:627–639.
27. Haller, MJ, Atkinson, MA, Schatz, D. Type 1 diabetes Mellitus: Etiology, Presentation, and Management. Pediatr Clin N Am. 2005;52:1553–1578.
28. Couper, JJ, Donaghue, KC. ISPAD Clinical Practice Consensus Guidelines 2006–2007. Phases of diabetes. Pediatric Diabetes. 2007;8:44–47.
29. Gorsuch, AN, Spencer, KM, Lister, J, et al. Evidence for a long prediabetic period in type 1 (insulin-dependent) diabetes mellitus. Lancet. 1981;2:1363–1365.
30. Lynch, KF, Lernmark, B, Merlo, J, et al. Cord blood islet autoantibodies and seasonal association with the type 1 diabetes high-risk genotype. Journal of Perinatology. 2008;28:211–217.
31. Klinke, DJ. Extent of beta cell destruction is important but insufficient to predict the onset of type 1 diabetes mellitus. PLoS ONE. 2008;3:e1374.
32. Wang, ZH, Kihl-Selstam, E, Eriksson, JW. Ketoacidosis occurs in both type 1 and type 2 diabetes—a population-based study from Northern Sweden. Diabetic Medicine. 2008;25:867–870.
33. International Diabetes Federation (IDF). World Atlas of Diabetes. www.eatlas.idf.org, 2006.
34. Haller, MJ, Gottliebb, PA, Schatz, DA. Type 1 diabetes intervention trials 2007: where are we and where are we going? Curr Opin Endocrinol Diabetes Obes. 2007;14:283–287.
35. Staeva-Vieira, T, Peakman, M, Herrath, MV. Translational mini-review series on type 1 diabetes: immune-based therapeutic approach for type 1 diabetes. Clinical and Experimental Immunology. 2007;148:17–31.
36. Keskinen, P, Korhonen, S, Kupila, A, et al. First-phase insulin response in young healthy children at genetic and immunological risk for Type 1 diabetes. Diabetologia. 2002;45:1639–1648.
37. Barker, JM. Type 1 diabetes-associated autoimmunity: natural history, genetic associations, and screening. J Clin Endocrinol Metab. 2006;91:1210–1217.
38. Hagopian, WA, Karlsen, AE, Gottsater, A, et al. Quantitative assay using recombinant human islet glutamic acid decarboxylase (GAD65) shows that 64K autoantibody positivity at onset predicts diabetes type. J Clin Invest. 1993;91:368–374.
39. Palmer, JP, Hirsch, III. What’s in a name: latent autoimmune diabetes of adults, type 1.5, adult-onset, and type 1 diabetes. Diabetes Care. 2003;26:536–538.
40. Sosenko, JM, Palmer, JP, Greenbaum, CJ, et al. Increasing the accuracy of oral glucose tolerance testing and extending its application to individuals with normal glucose tolerance for the prediction of type 1 diabetes. The Diabetes Prevention Trial-Type 1. Diabetes Care. 2007;30:38–42.
41. Molbak, AG, Christau, B, Marner, B, et al. Incidence of insulin-dependent diabetes mellitus in age groups over 30 years in Denmark. Diabet Med. 1994;11:650–655.
42. Pundziute-Lycka, A, Dahlquist, G, Nystrom, L, et al. The incidence of type 1 diabetes has not increased but shifted to a younger age at diagnosis in the 0–34 years group in Sweden 1983–1998. Diabetologia. 2002;45:783–791.
43. International Diabetes Federation (IDF). Diabetes in children: epidemiology. Pediatric Diabetes. 2007;8(Suppl. 8):10–18.
44. EURODIAB ACE Study Group. Variation and trends in incidence of childhood diabetes in Europe. Lancet. 2000;355:873–876.
45. DIAMOND Project Group. Incidence and trends of childhood Type 1 diabetes worldwide 1990–1999. Diabet Med. 2006;23(8):857–866.
46. Karvonen, M, Viik-Kajander, M, Moltchanova, E, et al. Incidence of childhood type 1 diabetes worldwide. Diabetes Mondiale (DiaMond) project group. Diabetes Care. 2000;23:1516–1526.
47. Harjutsalo, V, Sjöberg, L, Tuomilehto, J. Time trends in the incidence of type 1 diabetes in Finnish children: a cohort study. The Lancet. 2008;371:1777–1782.
48. Soltesz, G, Patterson, CC, Dahlquist, G. Worldwide childhood type 1 diabetes incidence—what can we learn from epidemiology? Pediatric Diabetes. 2007;8(Suppl. 6):6–14.
49. Tuomilehto, J. Epidemiology of childhood diabetes in the Baltic area. Nord Med. 1992;107:244–246.
50. Reunanen, A, Åkerblom, HK, Käär, ML. Prevalence and ten-year (1970–1979) incidence of insulin-dependent diabetes mellitus in children and adolescents in Finland. Acta Paediatr Scand. 1982;71:893–899.
51. Dahlquist, G, Blom, L, Tuvemo, T, et al. The Swedish Childhood Diabetes Study: results from a nine year case register and one year case-referent study indicating that type 1 (insulin-dependent) diabetes mellitus is associated with both type 2 (noninsulin-dependent) diabetes mellitus and autoimmune disorders. Diabetologia. 1989;32:2–6.
52. Todd, JA, Farrall, M. Panning for gold: genome-wide scanning for linkage in type 1 diabetes. Hum Mol Genet. 1996;5:1443–1448.
53. Awata, T, Kuzuya, T, Matsuda, A, et al. Genetic analysis of HLA class II alleles and susceptibility to type 1 (insulin-dependent) diabetes mellitus in Japanese subjects. Diabetologia. 1992;35:419–424.
54. Kyvik, KO, Green, A, Beck-Nielsen, H. Concordance rates of insulin dependent diabetes mellitus: a population based study of young Danish twins. Br Med J. 1995;311:913–917.
55. Craig, ME, Hattersley, A, Donaghue, K. ISPAD Clinical Practice Consensus Guidelines 2006–2007. Definition, epidemiology and classification. Pediatric Diabetes. 2006;7:343–351.
56. Serrano-Rìos, M, Goday, A, Larrad, TM. Migrant populations and the incidence of type 1 diabetes mellitus: an overview of the literature with a focus on the Spanish-heritage countries in Latin America. Diabetes Metab Res Rev. 1999;15:113–132.
57. Hjern, A, Söderström, U. Parental country of birth is a major determinant of childhood type 1 diabetes in Sweden. Pediatric Diabetes. 2008;9:35–39.
58. Feltbower, RG, McKinney, PA, Parslow, RC, et al. Type 1 diabetes in Yorkshire, UK: time trends in 0–14 and 15–29-year-olds, age at onset and age-period-cohort modelling. Diabetes Med. 2003;20:437–441.
59. Dahlquist, G, Blom, L, Lönnberg, G. The Swedish Childhood Diabetes Study: a multivariate analysis of risk determinants for diabetes in different age groups. Diabetologia. 1991;34:757–762.
60. Blom, L, Persson, LA, Dahlquist, G. Growth velocity and development of insulin-dependent diabetes mellitus. Diabetologia. 1992;35:528–533.
61. Nyström, L, Dahlquist, G, Ostman, J, et al. Risk of developing insulin-dependent diabetes mellitus (IDDM) before 35 years of age: indications of climatological determinants for age at onset. Int J Epidemiol. 1992;21:352–358.
62. Lorenzen, T, Pociot, E, Hougaard, P, et al. Long-term risk of IDDM in first-degree relatives of patients with IDDM. Diabetologia. 1994;37:321–327.
63. Tuomi, T, Carlsson, A, Li, H, et al. Clinical and genetic characteristics of type 2 diabetes with and without GAD antibodies. Diabetes. 1999;48:150–157.
64. Aguilar-Bryan, L, Bryan, J. Neonatal Diabetes Mellitus. Endocrine Reviews. 2008;29:265–291.
65. Larsson, HE, Hansson, G, Carlsson, A, et al. for the DiPiS Study Group: Children developing type 1 diabetes before 6 years of age have increased linear growth independent of HLA genotypes. Diabetologia. 2008;51:1623–1630.
66. Edelsten, AD, Hughes, IA, Oakes, S. Height and skeletal maturity in children with newly diagnosed juvenile-onset diabetes. Arch Dis Child. 1981;56:40–44.
67. Karvonen, M, Pitkaniemi, M, Pitkaniemi, J, et al. Sex difference in the incidence of insulin-dependent diabetes mellitus: an analysis of the recent epidemiological data: World Health Organization DIAMOND Project Group. Diabetes Metab Rev. 1997;13:275–291.
68. Gale, EA, Gillespie, KM. Diabetes and gender. Diabetologia. 2001;44:3–15.
69. Knip, M, Veijola, R, Virtanen, SM, et al. Environmental triggers and determinants of type 1 diabetes. Diabetes. 2005;54(Suppl. 2):S125–S136.
70. Åkesson, K, Nyström, L, Färnkvist, L, et al. and the DISS study group: Increased risk of diabetes among relatives of female insulin-treated patients diagnosed at 15–34 years of age. Diabetic Medicine. 2005;22:1551–1557.
71. Schranz, D, Lernmark, Å. Immunology in diabetes: an update. Diabetes Metab Rev. 1998;14:3–29.
72. Nepom, GT. Immunogenetics and IDDM. Diabetes Rev. 1993;1:93–103.
73. Davies, JL, Kawaguchi, Y, Bennett, ST, et al. A genome-wide search for human type 1 diabetes susceptibility genes. Nature. 1994;371:130–136.
74. Concannon, P, Gogolin-Ewens, KJ, Hinds, DA, et al. A second-generation screen of the human genome for susceptibility to insulin-dependent diabetes mellitus. Nat Genet. 1998;19:292–296.
75. Erlich, H, Valdes, AM, Noble, J, et al. HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk. Analysis of the Type 1 Diabetes Genetics Consortium families. Diabetes. 2008;57:1084–1092.
76. Thorsby, E. HLA-associated disease susceptibility: which genes are primarily involved? Immunologist. 1995;3(2):51–58.
77. Grant, S, Qu, H-Q, Bradfield, JP, et al. Follow up analysis of genome-wide association data identifies novel loci for type 1 diabetes. Diabetes. 2008.
78. Jun, HS, Yoon, JW. A new look at viruses in type 1 diabetes. Diabetes Metab Res Rev. 2002;19:8–31.
79. Harris, HF. A case of diabetes mellitus quickly following mumps on the pathological alterations of salivary glands, closely resembling those found in pancreas, in a case of diabetes mellitus. Boston Med Surg J CXL. 1898;465:591–601.
80. Van Der-Werf, N, Kroese, FGM, Rozing, J, et al. Viral infections as potential triggers of type 1 diabetes. Diabetes Metab Res Rev. 2007;23:169–183.
81. Mason, DR, Scott, RS, Darlow, BA. Epidemiology of insulin-dependent diabetes mellitus in Canterbury, New Zealand. Diabetes Res Clin Pract. 1987;3:21–29.
82. Viskari, H, Ludvigsson, J, Uibo, R, et al. Relationship between the incidence of type 1 diabetes and maternal enterovirus antibodies: time trends and geographical variation. Diabetologia. 2005;48:1280–1287.
83. Lindberg, B, Ahlfors, K, Carlsson, A, et al. Previous exposure to measles, mumps and rubella—but not vaccination during adolescence correlates to the prevalence of pancreatic and thyroid autoantibodies. Pediatrics. 1999;104:e12.
84. Haverkos, HW, Battula, N, Drotman, DP, et al. Enteroviruses and type 1 diabetes mellitus. Biomed Pharmacother. 2003;57:379–385.
85. Yoon, JW, Austin, M, Onodera, T, et al. Virus-induced diabetes mellitus: isolation of a virus from the pancreas of a child with diabetic ketoacidosis. N Engl J Med. 1979;300:1173–1179.
86. Gerling, I, Chatterjee, NK, Nejman, C. Coxsackie virus B4-induced development of antibodies to 64,000 Mr islet autoantigen and hyperglycemia in mice. Autoimmunity. 1991;6:49–56.
87. Hou, J, Sheikh, S, Martin, DL, et al. Coxsackievirus B4 alters pancreatic glutamate decarboxylase expression in mice soon after infection. J Autoimmun. 1993;6:529–542.
88. Kaufman, DL, Erlander, MG, Clare-Salzler, M, et al. Autoimmunity to two forms of glutamate decarboxylase in insulin-dependent diabetes mellitus. J Clin Invest. 1992;89:283–292.
89. Atkinson, MA, Bowman, MA, Campbell, L, et al. Cellular immunity to a determinant common to glutamate decarboxylase and coxsackievirus in insulin-dependent diabetes. J Clin Invest. 1994;94:2125–2129.
90. Horwitz, MS, Bradley, LM, Harbertson, J, et al. Diabetes induced by coxsackievirus: initiation by bystander damage and not molecular mimicry. Nat Med. 1998;4:781–785.
91. Niklasson, B, Heller, KE, Schønecker, B, et al. Development of type 1 diabetes in wild bank voles associated with islet autoantibodies and the novel Ljungan virus. Experimental Diab Res. 2003;4:35–44.
92. Toniolo, A, Onodera, T, Yoon, J-W, et al. Introduction of diabetes by cumulative environmental insults from viruses and chemicals. Nature. 1980;297:87–89.
93. Gerstein, HC. Cow’s milk exposure and type I diabetes mellitus: a critical overview of the clinical literature. Diabetes Care. 1994;17:13–19.
94. Vaarala, O. Environmental causes: dietary causes. Endocrinol Metab Clin N Am. 2004;33:17–26.
95. Vaarala, O, Paronen, J, Otonkoski, T, et al. Cow milk feeding induces antibodies to insulin in children—a link between cow milk and insulin-dependent diabetes? Scand J Immunol. 1998;47:131–135.
96. Norris, JM, Beaty, B, Klingensmith, G, et al. Lack of association between early exposure to cow’s milk protein and β-cell autoimmunity: diabetes autoimmunity study in the young (DAISY). JAMA. 1999;276:609–614.
97. The EURODIAB Substudy-2 Group. Rapid early growth is associated with increased risk of childhood type 1 diabetes in various European populations. Diabetes Care. 2002;25:1755–1760.
98. Vaarala, O. Is it dietary Insulin? Ann NY Acad Sci. 2006;1079:350–359.
99. Hawa, MI, Leslie, RGD. Early induction of type 1 diabetes. Clin Exp Immunol. 2001;126:181–183.
100. Greeley, SA, Katsumata, M, Yu, L, et al. Elimination of maternally transmitted autoantibodies prevents diabetes in nonobese diabetic mice. Nat Med. 2002;8:399–402.
101. Hyppönen, E, Kenward, MG, Virtanen, SM, et al. the Childhood Diabetes in Finland (DiMe) Study Group: Infant feeding, early weight gain, and risk of type 1 diabetes. Diabetes Care. 1999;22:1961–1965.
102. Thernlund, GM, Dahlquist, G, Hansson, K, et al. Psychological stress and the onset of IDDM in children. Diabetes Care. 1995;18:1323–1329.
103. Lernmark, B, Lynch, K, Lernmark, Å. Cord blood islet autoantibodies are related to stress in the mother during pregnancy. Ann N Y Acad Sci. 2006;1079:345–349.
104. Sepa, A, Wahlberg, J, Vaarala, O, et al. Psychological stress may induce diabetes-related autoimmunity in infancy. Diabetes Care. 2005;28:290–295.
105. Mead, VP. A new model for understanding the role of environmental factors in the origins of chronic illness: a case study of type 1 diabetes mellitus. Medical Hypotheses. 2004;63:1035–1046.
106. Szkudelski, T. The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol Res. 2001;50:536–546.
107. Helgason, T, Jonasson, MR. Evidence for a food additive as a cause of ketosis-prone diabetes. Lancet. 1981;2:716–720.
108. Hagopian, W, Lernmark, Å, Rewers, M, et al. TEDDY—the environmental determinants of diabetes in the young: an observational clinical trial. Ann NY Acad Sci. 2006;1079:320–326.
109. Klöppel, G, Drenck, CR, Oberholzer, M, et al. Morphometric evidence for a striking β-cell reduction at the clinical onset of type 1 diabetes. Virchows Arch A Pathol Anat Histopathol. 1984;403:441–452.
110. Dandona, P, Elias, E, Beckett, AG. Serum trypsin concentrations in diabetes mellitus. Br Med J. 1978;2:1125–1127.
111. Landin-Olsson, M, Borgstrom, A, Blom, L, et al. Immunoreactive trypsin(ogen) in the sera of children with recent-onset insulin-dependent diabetes and matched controls: The Swedish Childhood Diabetes Group. Pancreas. 1990;5:241–247.
112. Gepts, W, LaCompte, PM. The pancreatic islets in diabetes. Am J Med. 1981;70:105–115.
113. Rahier, J. The diabetic pancreas: a pathologist’s view. In: Lefebvre P, Pipeleers D, eds. The Pathology of the Endocrine Pancreas. Berlin: Springer-Verlag; 1988:17–40.
114. Edlund, H. Transcribing pancreas. Diabetes. 1998;47:1817–1823.
115. Bottazzo, GE, Dean, BM, McNally, JM, et al. In situ characterization of autoimmune phenomena and expression of HLA molecules in the pancreas in diabetic insulitis. N Engl J Med. 1985;313:353–360.
116. Hanafusa, T, Miyazaki, A, Miyagawa, J. Examination of islets in the pancreas biopsy specimens from newly diagnosed type 1 (insulin-dependent) diabetic patients. Diabetologia. 1990;33:105–111.
117. Itoh, N, Hanafusa, T, Miyazaki, A, et al. Mononuclear cell infiltration and its relation to the expression of major histocompatibility complex antigens and adhesion molecules in pancreas biopsy specimens from newly diagnosed insulin-dependent diabetes mellitus patients. J Clin Invest. 1993;92:2313–2322.
118. Imagawa, A, Hanafusa, T, Tamura, S, et al. Pancreatic biopsy as a procedure for detecting in situ autoimmune phenomena in type 1 diabetes: close correlation between serological markers and histological evidence of cellular autoimmunity. Diabetes. 2001;50:1269–1273.
119. In’t Veld, P, Lievens, D, De Grijse, J, et al. Screening for insulitis in adult autoantibody-positive organ donors. DIABETES. 2007;56:2400–2404.
120. Heaton, DA, Millward, BA, Gray, IP, et al. Increased proinsulin levels as an early indicator of β-cell dysfunction in non-diabetic twins of type 1 (insulin-dependent) diabetic patients. Diabetologia. 1988;31:182–184.
121. Roder, ME, Knip, M, Hartling, SG, et al. Disproportionately elevated proinsulin levels precede the onset of insulin-dependent diabetes mellitus in siblings with low first phase insulin responses: The Childhood Diabetes in Finland Study Group. J Clin Endocrinol Metab. 1994;79:1570–1575.
122. Johnston, C, Raghu, P, McCulloch, D, et al. β-Cell function and insulin sensitivity in nondiabetic HLA-identical siblings of insulin-dependent diabetics. Diabetes. 1987;36:829–837.
123. Kahn, SE, Prigeon, RL, McCulloch, DK, et al. Quantification of the relationship between insulin sensitivity and β-cell function in human subjects: evidence for a hyperbolic function. Diabetes. 1993;42:1663–1672.
124. Ludvigsson, J, Heding, LG, Larsson, Y, et al. C-peptide in juvenile diabetics beyond the post-initial remission period: relation to clinical manifestations at onset of diabetes, remission and diabetic control. Acta Pediatr Scand. 1977;66:177–184.
125. The Diabetes Control and Complications Trial Research Group. Effect of intensive therapy on residual beta-cell function in patients with type 1 diabetes in the diabetes control and complications trial: a randomized, controlled trial. Ann Intern Med. 1998;128:517–523.
126. Lampeter EE Homberg, M, Quabeck, K, et al. Transfer of insulin-dependent diabetes between HLA-identical siblings by bone marrow transplantation. Lancet. 1993;342:174.
127. Knip, M, Ilonen, J, Mustonen, A, et al. Evidence of an accelerated β-cell destruction in HLA-Dw3/Dw4 heterozygous children with type 1 (insulin-dependent) diabetes. Diabetologia. 1986;29:347–351.
128. Graham, J, Kockum, I, Sanjeevi, CB, et al. Negative association between type 1 diabetes and HLA DQB1*0602-DQAl*0102 is attenuated with age at onset: The Swedish Childhood Diabetes Study Group. Fur J Immunogenet. 1999;26:117–127.
129. Sanjeevi, CB, Landin-Olsson, M, Kockum, I, et al. Effects of the second HLA-DQ haplotype on the association with childhood insulin dependent diabetes mellitus. Tissue Antigens. 1994;45:148–152.
130. Litherland, SA, Xie, XT, Hutson, AD, et al. Aberrant prostaglandin synthase 2 expression defines an antigen-presenting cell defect for insulin-dependent diabetes mellitus. J Clin Invest. 1999;104:515–523.
131. Jansen, A, van Hagen, M, Drexhage, HA. Defective maturation and function of antigen-presenting cells in type 1 diabetes. Lancet. 1995;345:491–492.
132. Plesner, A, Greenbaum, CJ, Gaur, LK, et al. Macrophages from high-risk HLA-DQB1*0201/*0302 type 1 diabetes mellitus patients are hypersensitive to lipopolysaccharide stimulation. Scand J Immunol. 2002;56:522–529.
133. Lawley, TJ, Hall, RP, Fauci, AS, et al. Defective Fc-receptor functions associated with the HLA-B8/DRw3 haplotype. N Engl J Med. 1981;304:185–192.
134. MacCuish, AC, Jordan, J, Campbell, CJ, et al. Cell-mediated immunity to human pancreas in diabetes mellitus. Diabetes. 1974;23:693–697.
135. Lernmark, Å. Cell-mediated immunity in type 1 (insulin-dependent) diabetes: update 84. In: Andreani D, di Mario U, Federlin KF, et al, eds. Immunology in Diabetes. London: Kimpton Medical Publications; 1984:121–131.
136. Bougneres, PF, Carel, JC, Castano, L, et al. Factors associated with early remission of type 1 diabetes in children treated with cyclosporine. N Engl J Med. 1988;318:663–6670.
137. Harold, KC, Hagopian, W, Auger, JA, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002;346:1692–1698.
138. Agardh, CD, Cilio, CM, Lethagen, ÅL, et al. Clinical evidence for the safety of GAD65 immunomodulation in adult-onset autoimmune diabetes. Journal of Diabetes and Its Complications. 2005;19:238–246.
139. Ludvigsson, J, Faresjo, M, Hjorth, M, et al. GAD Treatment and insulin secretion in recent-onset type 1 diabetes. N Engl J Med. 2008 Oct 8.
140. Roep, BO. The role of T-cells in the pathogenesis of type 1 diabetes: From cause to cure. Diabetologia. 2003;46:305–321.
141. Wicker, LS, Chen, SL, Nepom, GT, et al. Naturally processed T cell epitopes from human glutamic acid decarboxylase identified using mice transgenic for the type 1 diabetes-associated human MHC class II allele, DRB1*040L. J Clin Invest. 1996;98:2597–2603.
142. Novak, EJ, Liu, AW, Gebe, JA, et al. Tetramer-guided epitope mapping: rapid identification and characterization of immunodominant CD4+ T cell epitopes from complex antigens. J Immunol. 2001;166:6665–6670.
143. Endl, J, Otto, H, Jung, G, et al. Identification of naturally processed T cell epitopes from glutamic acid decarboxylase presented in the context of HLA-DR alleles by T lymphocytes of recent onset IDDM patients. J Clin Invest. 1997;99:2405–2415.
144. Sonderstrup, G, McDevitt, H. Identification of autoantigen epitopes in MHC class II transgenic mice. Immunol Rev. 1998;164:129–138.
145. Geluk, A, van Meijgaarden, KE, Schloot, NC, et al. HLA-DR binding analysis of peptides from islet antigens in IDDM. Diabetes. 1998;47:1594–1601.
146. Congia, M, Patel, S, Cope, AP, et al. T cell epitopes of insulin defined in HLA-DR4 transgenic mice are derived from preproinsulin and proinsulin. Proc Natl Acad Sci U S A. 1998;95:3833–3838.
147. Reijonen, H, Novak, EJ, Kochik, S, et al. Detection of GAD65-specific T-cells by major histocompatibility complex class II tetramers in type 1 diabetic patients and at-risk subjects. Diabetes. 2002;51:1375–1382.
148. Papadopoulos, G, Petersen, J, Andersen, V. Spontaneous in-vitro immunoglobulin secretion at the diagnosis of insulin-dependent diabetes. Acta Endocrinol. 1984;105:521–527.
149. Schloot, NC, Meierhoff, G, Karlsson-Faresjo, M, et al. Comparison of cytokine Elispot assay formats for the detection of islet antigen autoreactive T cells: report of the third immunology of diabetes society T-cell workshop. J Autoimmun. 2003;21:365–376.
150. Lohmann, T, Leslie, RDG, Hawa, M, et al. Immunodominant epitopes of glutamic acid decarboxylase 65 and 67 in insulin-dependent diabetes mellitus. Lancet. 1994;343:1607–1608.
151. Lohmann, T, Halder, T, Engler, J, et al. T cell reactivity to DR*0401- and DQ*0302-binding peptides of the putative autoantigen IA-2 in type 1 diabetes. Exp Clin Endocrinol Diabetes. 1999;107:166–171.
152. Van de Winkel, M, Smets, G, Gepts, W, et al. Islet cell surface antibodies from insulin-dependent diabetics bind specifically to pancreatic β cells. J Clin Invest. 1982;70:41–49.
153. Dobersen, MJ, Scharff, JE. Preferential lysis of pancreatic β cells by islet cell surface antibodies. Diabetes. 1982;31:449–462.
154. Gilliam, LK, Palmer, JP, Lernmark, Å. Autoantibodies and the disease process of type 1 diabetes mellitus. In: LeRoith D, Taylor SI, Olefsky JM, eds. Diabetes Mellitus: A Fundamental and Clinical Text. Philadelphia: Lippincott Williams & Wilkins; 2003:499–518.
155. Petersen, JB, Russel, S, Marshall, MO, et al. Differential expression of glutamic acid decarboxylase in rat and human islets. Diabetes. 1993;42:484–495.
156. Karlsen, AE, Hagopian, WA, Petersen, JS, et al. Recombinant glutamic acid decarboxylase representing a single isoform expressed in human islets detects IDDM-associated 64K autoantibodies. Diabetes. 1992;41:1355–1359.
157. Christie, MR, Vohra, G, Champagne, P, et al. Distinct antibody specificities to a 64K islet cell antigen in type 1 diabetes as revealed by trypsin treatment. J Exp Med. 1990;172:789–794.
158. Borg, H, Gottsater, A, Fernlund, P, et al. A 12-year prospective study of the relationship between islet antibodies and beta-cell function at and after the diagnosis in patients with adult-onset diabetes. Diabetes. 2002;51:1754–1762.
159. Sanjeevi, CB, Hagopian, WA, Landin-Olsson, M, et al. Association between autoantibody markers and subtypes of DR4 and DR4-DQ in Swedish children with insulin-dependent diabetes reveals closer association of tyrosine pyrophosphatase autoimmunity with DR4 than DQ8. Tissue Antigens. 1998;51:281–286.
160. Yu, L, Chase, HP, Falorni, A, et al. Sexual dimorphism in transmission to offspring of expression of islet autoantibodies. Diabetologia. 1995;38:1353–1357.
161. Rolandsson, O, Hägg, E, Hampe, C, et al. Levels of glutamate decarboxylase (GAD65) and tyrosine phosphatase-like protein (IA-2) autoantibodies in the general population are related to glucose intolerance and body mass index. Diabetologia. 1999;42:555–559.
162. Bonifacio, E, Genovese, S. Braghi S, et at: Islet autoantibody markers in IDDM: risk-assessment strategies yielding high sensitivity. Diabetologia. 1995;38:816–822.
163. Kim, J, Namchuck, M, Bugawan, T, et al. Higher autoantibody levels and recognition of a linear NH2-terminal epitope in the autoantigen GAD65, distinguish stiff-man syndrome from insulin-dependent diabetes mellitus. J Exp Med. 1994;180:595–606.
164. Eisenbarth, GS, Jackson, GS. Insulin autoimmunity: the rate-limiting factor of pre-type 1 diabetes. J Autoimmun. 1992;5:214–246.
165. Li, L, Hagopian, WA, Brashear, HR, et al. Identification of autoantibody epitopes of glutamic acid decarboxylase in stiff-man syndrome patients. J Immunol. 1994;152:930–934.
166. Leslie, RD, Atkinson, MA, Notkins, AL. Autoantigens IA-2 and GAD in type 1 (insulin-dependent) diabetes. Diabetologia. 1999;42:3–14.
167. Bingley, PJ, Bonifacio, E, Williams, AJK, et al. Prediction of IDDM in the general population: strategies based on combinations of autoantibody markers. Diabetes. 1997;46:1701–1710.
168. Eisenbarth, GS, Jeffrey, J. The natural history of type 1A diabetes. Arq Bras Endrocrinol Metab. 52(2), 2008.
169. Oak, S, Gilliam, LK, Landin-Olsson, M, et al. The lack of anti-idiotypic antibodies, not the presence of the corresponding autoantibodies to glutamate decarboxylase, defines type 1 diabetes. PNAS. 2008;105:5471–5476.
170. Bonifacio, E, Atkinson, M, Eisenbarth, G, et al. International workshop on lessons from animal models for human type 1 diabetes: identification of insulin but not glutamic acid decarboxylase or IA-2 as specific autoantigens of humoral autoimmunity in nonobese diabetic mice. Diabetes. 2001;50:2451–2458.
171. Kukko, M, Kimpimäki, T, Korhonen, S, et al. Dynamics of diabetes-associated autoantibodies in young children with human leukocyte antigen-conferred risk of type 1 diabetes recruited from the general population. J Clin Endocrinol Metab. 2005;90:2712–2717.
172. Castano, L, Ziegler, A, Ziegler, R, et al. Characterization of insulin autoantibodies in relatives of patients with insulin-dependent diabetes mellitus. Diabetes. 1993;42:1202–1209.
173. Brooks-Worrell, BM, Nielson, D, Palmer, JP. Insulin autoantibodies and insulin antibodies have similar binding characteristics. Proc Assoc Am Physicians. 1999;111:92–96.
174. Arslanian, SA, Becker, DJ, Rabin, B, et al. Correlates of insulin antibodies in newly diagnosed children with insulin-dependent diabetes before insulin therapy. Diabetes. 1985;34:926–930.
175. Ziegler, AG, Ziegler, R, Vardi, P, et al. Life-table analysis of progression to diabetes of anti-insulin autoantibody-positive relatives of individuals with type 1 diabetes. Diabetes. 1989;38:1320–1325.
176. Eisenbarth, GS, Gianani, R, Yu, L, et al. Dual-parameter model for prediction of type 1 diabetes mellitus. Proc Assoc Am Physicians. 1998;110:126–135.
177. Verge, CF, Stenger, D, Bonifacio, E, et al. Combined use of autoantibodies (IA-2 autoantibody, GAD autoantibody, insulin autoantibody, cytoplasmic islet cell antibodies) in type 1 diabetes: combinatorial islet autoantibody workshop. Diabetes. 1998;47:1857–1866.
178. Greenbaum, CJ, Palmer, JP, Kuglin, B. et at: Insulin autoantibodies measured by radioimmunoassay methodology are more related to insulin-dependent diabetes mellitus than those measured by enzyme-linked immunosorbent assay: results of the Fourth International Workshop on the Standardization of Insulin Autoantibody Measurement. J Clin Endocrinol Metab. 1992;74:1040–1044.
179. Gianani, R, Eisenbarth, GS. The stages of type1A diabetes. Immunol Rev. 2005;204:232–249.
180. Krishnamurthy, B, Dudek, NL, McKenzie, MD, et al. Responses against islet antigens in NOD mice are prevented by tolerance to proinsulin but not IGRP. J Clin Invest. 2006;116:3258–3265.
181. Christie, MR, Genovese, S, Cassidy, D, et al. Antibodies to islet 37K antigen, but not to glutamate decarboxylase, discriminate rapid progression to IDDM in endocrine autoimmunity. Diabetes. 1994;43:1254–1259.
182. Wasmeier, C, Hutton, JC. Molecular cloning of phogrin, a protein-tyrosine phosphatase homologue localized to insulin secretory granule membranes. J Biol Chem. 1996;271:18161–18170.
183. Cui, L, Yu, W-P, DeAizpurua, HJ, et al. Cloning and characterization of islet cell antigen-related protein-tyrosine phosphatase (PTP), a novel receptor-like PTP and autoantigen in insulin-dependent diabetes. J Biol Chem. 1996;271:24817–24823.
184. LaGasse, J, Jelinek, L, Sexson, S, et al. An islet-cell protein tyrosine phosphatase is a likely precursor to the 37K autoantigen in type 1 diabetes: human and macaque sequences, tissue distribution, unique and shared epitopes, and predictive autoantibodies. Mot Med. 1997;3:163–173.
185. Hagopian, WA, Sanjeevi, CB, Kockum, I, et al. Glutamate decarboxylase-, insulin- and islet cell-antibodies and HLA typing to detect diabetes in a general population-based study of Swedish children. J Clin Invest. 1995;95:1505–1511.
186. Landin-Olsson, M, Palmer, JP, Lernmark, Å, et al. Predictive value of islet cell and insulin autoantibodies for type 1 (insulin-dependent) diabetes mellitus in a population-based study of newly-diagnosed diabetic and matched control children. Diabetologia. 1992;35:1068–1073.
187. Kawasaki, E, Eisenbarth, G, Wasmeier, C, et al. Autoantibodies to protein tyrosine phosphatase-like protein in type 1 diabetes: overlapping specificities to phogrin and ICA512/IA-2. Diabetes. 1996;45:1344–1349.
188. Graham, J, Hagopian, WA, Kockum, I, et al. Genetic effects on age-dependent onset and islet cell autoantibody markers in type 1 diabetes. Diabetes. 2002;51:1346–1555.
189. Solimena, M, Dirkx, R, Jr., Hermel, JM, et al. ICA 512, an autoantigen of type 1 diabetes, is an intrinsic membrane protein of neurosecretory granules. EMBO J. 1996;15:2102–2114.
190. Hoppu, S, Härkönen, T, Ronkainen, MS, et al. IA-2 antibody isotypes and epitope specificity during the prediabetic process in children with HLA-conferred susceptibility to type 1 diabetes. Clinical and Experimental Immunology. 2006;144:59–66.
191. MacMurray, AJ, Moralejo, DH, Kwitek, AE, et al. Lymphopenia in the BB rat model of type 1 diabetes due to a mutation in a novel immune-associated nucleotide (IAN)-related gene. Genome Res. 2002;12:1029–1039.
192. Shin, J-H, Janer, M, McNeney, B, et al. IA-2 autoantibodies in incident type I diabetes patients are associated with a polyadenylation signal polymorphism in GIMAP5. Genes and Immunity. 2007;8:503–512.
193. Wenzlau, JM, Hutton, JC, Davidson, HW. New antigenic targets in type 1 diabetes. Current Opinion in Endocrinology. Diabetes & Obesity. 2008;15:315–320.
194. Wenzlau, JM, Liu, Y, Yu, L, et al. A common non-synonymous single nucleotide polymorphism in the SLC30A8 gene determines ZnT8 autoantibody specificity in type 1 diabetes. Diabetes. 2008;57:2693–2697.
195. Grubin, CE, Daniels, T, Toivola, B, et al. A novel radioligand binding assay to determine diagnostic accuracy of isoform-specific glutamic acid decarboxylase antibodies in childhood IDDM. Diabetologia. 1994;37:344–350.
196. Arden, SD, Roep, BO, Neophytou, PI, et al. Imogen 38: a novel 38-kD islet mitochondrial autoantigen recognized by T cells from a newly diagnosed type 1 diabetic patient. J Clin Invest. 1996;97:551–561.
197. Hirai, H, Miura, J, Hu, Y, et al. Selective screening of secretory vesicle-associated proteins for autoantigens in type 1 diabetes: VAMP2 and NPY are new minor autoantigens. Clinical Immunology. 2008;127:366–374.
198. Aanstoot, HJ, Kang, SM, Kim, J, et al. Identification and characterization of glima 38, a glycosylated islet cell membrane antigen, which together with GAD65 and IA2 marks the early phases of autoimmune response in type 1 diabetes. J Clin Invest. 1996;97:2772–2783.
199. Ozawa, Y, Kasuga, A, Nomaguchi, H, et al. Detection of autoantibodies to the pancreatic islet heat shock protein 60 in insulin-dependent diabetes mellitus. J Autoimmun. 1996;9:517–524.
200. Martin, S, Kardorf, J, Schulte, B, et al. Autoantibodies to the islet antigen ICA69 occur in IDDM and in rheumatoid arthritis. Diabetologia. 1995;38:351–355.
201. Castano, L, Russo, E, Zhou, L, et al. Identification and cloning of a granule autoantigen (carboxypeptidase-H) associated with type 1 diabetes. J Clin Endocrinol Metab. 1991;73:1197.
202. Chang, YH, Hwang, J, Shang, HF, et al. Characterization of human topoisomerase II as an autoantigen recognized by patients with IDDM. Diabetes. 1996;45:408.
203. Pupilli, C, Giannini, S, Marchetti, P, et al. Autoantibodies to CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase) in Caucasian patients with diabetes: effects on insulin release from human islets. Diabetes. 1999;48:2309.
204. Ikehata, F, Satoh, J, Nata, K, et al. Autoantibodies against CD38 (ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase) that impair glucose-induced insulin secretion in noninsulin-dependent diabetes patients. J Clin Invest. 1998;102:395.
205. Inman, LR, MaAllister, CT, Chen, L, et al. Autoantibodies to the GLUT-2 glucose transporter of beta cells in insulin-dependent diabetes mellitus of recent onset. Proc Natl Acad Sci USA. 1993;90:1281.
206. Kasimiotis, H, Fida, S, Rowley, MJ, et al. Antibodies to SOX13 (ICA12) are associated with type 1 diabetes. Autoimmunity. 2001;33:95.
207. Törn, C, Shtauvere-Brameus, A, Sanjeevi, CB, et al. Increased autoantibodies to SOX13 in Swedish patients with type 1 diabetes. Ann NY Acad Sci. 2002;958:218.
208. Hämäläinen, AM, Knip, M. Autoimmunity and familial risk of type 1 diabetes. Current Diabetes Reports. 2002;2:347–353.
209. Barker, JM, Barriga, KJ, Yu, L. Prediction of autoantibody positivity and progression to type 1 diabetes: diabetes autoimmunity study in the young (DAISY). J Clin Endocrinol Metab. 2004;89:3896–3902.
210. Casu, A, Trucco, M, Pietropaolo, M. A look to the future: prediction, prevention, and cure including islet transplantation and stem cell therapy. Pediatr Clin N Am. 2005;52:1779–1804.
211. Vendrame, F, Gottlieb, PA. Prediabetes: prediction and prevention trials. Pediatr Clin N Am. 2004;33:75–92.
212. Kishiyama, CM, Chase, HP, Barker, MB. Prevention strategies for type 1 diabetes. Rev Endocr Metab Disord. 2006;7:215–224.
213. Zipitis, CS, Akobeng, AK. Vitamin D supplementation in early childhood and risk of type 1 diabetes: a systematic review and meta-analysis. Arch Dis Child. 2008;93:512–517.
214. Näntö-Salonen, K, Kupila, A, Simell, S, et al. Nasal insulin to prevent type 1 diabetes in children with HLA genotypes and autoantibodies conferring increased risk of disease: a double-blind, randomised controlled trial. Lancet. 2008.
215. Bekris, LM, Jensen, RA, Lagerquist, E, et al. GAD65 autoantibody epitopes in adult patients with latent autoimmune diabetes following GAD65 vaccination. Diabetic Medicine. 2007;24:521–526.
216. Ludvigsson, J, Faresjö, M, Hjorth, M, et al. GAD treatment and insulin secretion in recent-onset type 1 diabetes. N Engl J Med. 2008;359:1909–1920.
217. Abdi, R, Fiorina, P, Adra, CN, et al. Immunomodulation by mesenchymal stem cells: a potential therapeutic strategy for type 1 diabetes. Diabetes. 2008;57:1759–1767.
218. Frank, A, Deng, S, Huang, X, et al. Transplantation for type I diabetes: comparison of vascularized whole-organ pancreas with isolated pancreatic islets. Ann Surg. 2004;240:631–643.