[level-membership-for-neurology-category]
Chapter 83 Tumors of the Brain and Spine
Introduction
Primary central nervous system tumors comprise, in total, the second most common form of childhood cancer, exceeded only by the leukemias [CBTRUS, 2008; Jemal et al., 2008; Louis et al., 2007a; Preston-Martin, 1996; Ries et al., 1997]. Although relatively rare compared to other pediatric neurological conditions, these neoplasms cause significant neurologic morbidity and mortality. Unlike the situation in adults, where metastatic brain tumors outnumber primary central nervous system neoplasms by a ratio as high as 10 to 1, in the pediatric years, metastatic lesions are much less common and primary brain and spine tumors predominate [Bleyer, 1999; CBTRUS, 2008; Jemal et al., 2008; Louis et al., 2007b; Ries et al., 1997; Smith et al., 1998].
Tumors of the brain and spine constitute approximately one-quarter of all childhood cancers. In the United States, depending on what age is utilized to distinguish between adult and pediatric patients, between 3000 and 4000 children each year are diagnosed. Concepts and management of pediatric brain tumors are constantly evolving [Bleyer, 1999; Jemal et al., 2008; Louis et al., 2007a; Smith et al., 1998]. The classification of these neoplasms, on which treatment is based, is also changing, primarily due to the incorporation of molecular diagnostics.
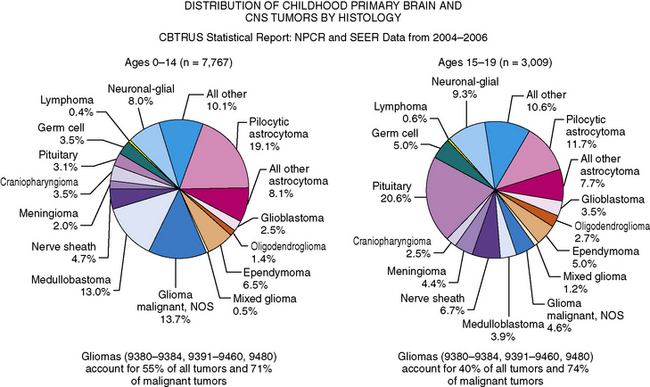
As in adults, the majority of primary central nervous system tumors in children are glial in origin; however, a significant proportion of tumors are embryonal (Figure 83-1) [CBTRUS, 2008; Jemal et al., 2008]. Embryonal, and mixed neuronal and glial tumors in infants are notoriously difficult to diagnose. The routine utilization of immunohistochemical analysis and, more recently, molecular analysis of tumors have altered concepts of diagnosis and classification, but their incorporation has lagged behind those in other childhood cancers, such as leukemia and neuroblastoma, and has led to the continued subjectivity of diagnosis.
Advances and refinements in therapy, to date, have resulted in a modest improvement in overall survival rate of patients. It has been estimated that there has been a 15–20 percent improvement in survival in patients diagnosed between 1975 and 1979, compared to those diagnosed and treated between 1996 and 2003 [CBTRUS, 2008; Jemal et al., 2008]. The complexities involved in the treatment of childhood brain and spine tumors have resulted in the development of regional centers focusing on the comprehensive management of children with these neoplasms. It is widely accepted that, for optimum management, a multidisciplinary approach is required, involving many specialties, including neurosurgery, neurology, oncology, neuropathology, neuroradiology, radiation oncology, ophthalmology, and rehabilitative specialists. In addition, given the relative infrequency of specific subtypes of childhood brain tumors, for progress to be made expeditiously, prospective multicentered, cooperative group-based trials are optimal. National and, in selected cases, international collaborations have become the cornerstone of modern management of childhood brain tumors.
Incidence
The incidence of childhood nervous system tumors is best estimated from population-based registries. Institution-based registries are potentially flawed by referral patterns; the tendency for larger programs to receive referrals of selective patients, skewing patient number; and the increasing mobility of patients, allowing individuals to seek second opinions and thus being included in statistics at multiple centers. The reported incidence rates of brain tumors for patients between 0 and 19 years ranges from 3.3 to 4.5 cases per 100,000 person years [CBTRUS, 2008]. This reported incidence is higher than the 2.7 cases per 100,000 person years reported in the late 1970s and early 1980s. It is believed that this higher incidence is partially due to the availability of magnetic resonance imaging (MRI), identifying more brain tumors earlier, and possibly the use of stereotactic biopsies to confirm the presence of primary central nervous system tumors [Bleyer, 1999; Smith et al., 1998]. Better reporting, the inclusion of lower-grade tumors in registries, and the wider utilization of epilepsy surgery confirming the diagnosis of low-grade neoplasms may also contribute to this increased incidence, rather than it representing a true increase in the number of brain and spine tumors. However, some studies have suggested that there is indeed a true increase in incidence. Among the major histological groupings, rates are highest for tumors of neuroepithelial tissue, with pilocytic astrocytomas and medulloblastomas being the most common individual subtypes of tumors.
Primary central nervous system tumors are more common in the first decade of life (Figure 83-2) [CBTRUS, 2008]. The reported incidence of brain tumors is higher in whites (4.7 per 100,000 person years) than in blacks (3.0 per 100,000 person years). The overall incidence for all brain tumors is highest among those between 0 and 4 years of age (5.2 per 100,000 person years), and lowest among those between 10 and 14 years of age (4.1 per 100,000 person years) [CBTRUS, 2008]. However, incidence does vary depending on tumor type, with ependymomas and medulloblastomas decreasing with age, and pilocytic astrocytomas peaking at 5–9 years of age. Germ cell tumors increase with age. In the 0–19 years age group, brain tumors are more common in boys, especially tumors of neuroepithelial origin and pineal germ cell tumors.
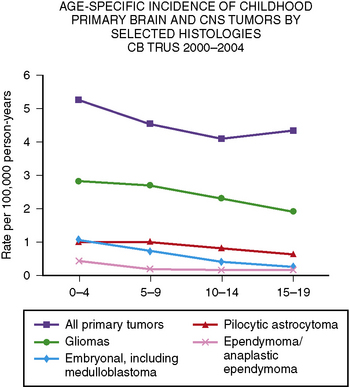
Overall, gliomas of multiple different subtypes, including pilocytic astrocytomas, astrocytomas not otherwise specified, and gliomas, are the most common type of childhood brain tumors, accounting for over 50 percent of all tumors in children between 0 and 14 years of age, and nearly 75 percent of all malignant tumors [CBTRUS, 2008]. Embryonal tumors, including medulloblastoma, comprise 15–20 percent of all tumors in children between 0 and 14 years, occurring in approximately 5 percent of those between 15 and 19 (see Figure 83-2) [CBTRUS, 2008].
Etiology
The causation of the majority of all childhood brain tumors is unknown [Bleyer, 1999; Bunin, 2000]. Despite this, there are genetic conditions and, to a lesser extent, environmental exposures that predispose to the development of tumors [Bleyer, 1999; Bunin, 2000]. Genetic syndromes related to increased development of specific tumor types have provided molecular genetic insights into childhood brain tumors that have been partially translatable to the study of apparently nongenetically inherited tumors. The known syndromes that are associated with the development of childhood brain tumors usually have an autosomal-dominant pattern of inheritance. At times, for unclear reasons, brain tumors will arise in multiple family members [Draper et al., 1977; Fitzgerald, 2000; Kuijten et al., 1993; Malmer et al., 2007; Moschovi et al., 1998].
Neurofibromatosis
Probably the most common genetic syndrome associated with development of primary central nervous system tumors is neurofibromatosis type 1, which is an autosomal-dominant disorder due to mutation of the NF1 gene [Burger et al., 1987; Cawthon et al., 1990; Listernick et al., 1999; Wallace et al., 1990]. The NF1 gene on the long arm of chromosome 17 encodes the neurofibromin protein, which, among other actions, is a tumor suppressor gene [Burger et al., 1987; Listernick et al., 1999; Pollack and Mulvihill, 1997]. Its absence allows ras activation and subsequent downstream intracellular signaling through the RAS/MAPK (MAP kinase) pathway. The gene is large, spanning 350 kb, and a mutation in the initially unaffected (wild-type) NF1 gene is thought to result in the development of a host of tumors. In the central nervous system, the most common are low-grade, most often pilocytic astrocytomas, which early in life have a marked predilection for the optic nerves and other parts of the visual pathway. Later in childhood, gliomas may develop in other central nervous system sites, especially midline areas, including the brainstem and corpus callosum.
Separation of low-grade tumors from areas of brain dysplasia, also common in neurofibromatosis type 1, can be difficult [Listernick et al., 1997]. These areas of abnormal signal on T2-weighted MRI are at least partially developmental, and many disappear as the child enters into puberty. Other central nervous system tumors, such as ependymomas and possibly medulloblastomas, are more common in patients with neurofibromatosis type 1. Neurofibromatosis type 1 occurs in as many as 1 in 3500 patients at risk, and brain tumors may occur in as high as 20 percent of patients, although, for unclear reasons, many affected patients do not require specific treatment.
Children with neurofibromatosis type 1 are also prone to development of neurofibromas, of which the most problematic in prepubertal patients are plexiform neurofibromas [Listernick et al., 1999; Packer and Rosser, 2002; Packer et al., 2002a]. Plexiform neurofibromas are congenital and may cause significant cosmetic disfigurement, organ dysfunction, and, dependent on location, neurologic compromise, including spinal cord compression. A percentage of plexiform neurofibromas, possibly as high as 10 percent, will undergo malignant transformation, although in the vast majority of patients this occurs after puberty, especially in the early adult years. Since patients with neurofibromatosis type 1 have an increased predilection for the development of other malignancies, including myelogenous leukemia and sarcomas, treatment of patients with neurofibromatosis type 1-associated brain tumors can be quite challenging. Chemotherapeutic alkylating drugs and radiation therapy may increase the risk of developing secondary tumors.
Neurofibromatosis type 2, which is also an autosomal-dominant disorder affecting approximately 1 in 25,000 to 1 in 400,000, is due to a gene abnormality on the short arm of chromosome 22 [Pollack and Mulvihill, 1997; Rouleau et al., 1993; Trofatter et al., 1993]. Patients with this condition are predisposed to developing schwannomas of the cranial nerves, spinal nerves, and peripheral nervous system. Bilateral acoustic schwannomas are essentially pathognomonic of the illness and are usually present by early adulthood. Gliomas and ependymomas, especially spinal ependymomas, also occur with increased frequency.
Tuberous Sclerosis and Other Autosomal-Dominant Conditions
Other autosomal-dominant neurogenetic diseases are related to specific tumor types. Tuberous sclerosis is associated with the development of subependymal giant cell astrocytomas, which tend to occur in the subependymal ventricular zone and most commonly cause obstructive hydrocephalus early in illness [Kandt et al., 1992; van Slegtenhorst et al., 1997]. Cerebellar gangliocytomas are associated with Cowden’s syndrome, caused by a germline mutation of the PTEN gene [Liaw et al., 1997]. The occurrence of dysplastic gangliocytoma of the cerebellum is also known as Lhermitte–Duclos disease. Caused by a mutation of the VHL gene, von Hippel–Landau syndrome is associated with the development of hemangioblastomas in the cerebellum, spinal cord, and retina [Duan et al., 1995; Latif et al., 1993].
Li–Fraumeni Syndrome
Li–Fraumeni syndrome is caused by a germline mutation in the multifunctional TP53 gene [Malkin et al., 1990; Srivastava et al., 1990]. The protein encoded by the gene has a role in cell-cycle control and in DNA integrity and repair. Gliomas, both high-grade and low-grade, are most common in patients with Li–Fraumeni syndrome, and such tumors may be multifocal. Other central nervous system tumors that occur in increased incidence in this syndrome include medulloblastomas, supratentorial neuroectodermal tumors, and choroid plexus tumors.
Gorlin’s Syndrome
Gorlin’s syndrome, also known as the nevoid basal cell carcinoma syndrome, accounts for probably less than 1 percent of all patients with medulloblastoma, but the relationship between this condition and the development of medulloblastoma has led to a better understanding of the molecular pathogenesis of a larger subset of patients with medulloblastoma [Amlashi et al., 2003; Stavrou et al., 2001; Taylor et al., 2000]. It has been estimated that 3–5 percent of patients with Gorlin’s syndrome will develop medulloblastoma. Gorlin’s syndrome is caused by an inherited germline mutation of the PATCHED 1 (PTCH1) gene on chromosome 9, which encodes the sonic hedgehog (SHH) receptor. PTCH1 normally suppresses sonic hedgehog signaling [Taylor et al., 2000]. The syndrome may be diagnosed early in life by characteristic dermatological and skeletal features, including odontogenic keratocysts of the jaw, bifid or fused ribs, macrocephaly, and calcification of the falx. The basal cell carcinomas tend to occur later in life. Mutations of the PTCH1 gene can be demonstrated in 20–40 percent of medulloblastomas, especially in the desmoplastic variant in infants. Since patients with Gorlin’s syndrome are predisposed to the development of basal cell carcinomas later in life, the use of craniospinal radiotherapy, which is the backbone of treatment for most patients with medulloblastoma, is problematic, as basal cell carcinomas may occur in large numbers in the radiation fields. The diagnosis of Gorlin’s syndrome is often made later in life, when the basal cell tumors become evident.
Turcot’s Syndrome
Turcot’s syndrome is another autosomal-dominant disorder due to mutations in the adenomatous polyposis coli (APC) gene (type 2), or secondary to mutations in DNA mismatch repair genes HPS2 and MLH1 (type 1) [Attard et al., 2007; Foulkes, 1995; Hamilton et al., 1995; Ng et al., 2005]. Patients with mutation of a mismatch repair gene are prone to developing colorectal carcinomas and gliomas. Patients with type 2 disease are at increased risk for developing medulloblastomas. Mutations in the APC gene result in abnormalities of the Wnt molecular pathway. This pathway has been noted to be abnormal in up to 15 percent of patients over 3 years of age with medulloblastoma, especially older patients, and may constitute a different, more favorable form of the disease.
Environmental Exposures, Including Radiation Therapy
Of the environment exposures that predispose to the development of brain tumors, the best documented is ionizing radiation [Bakhshi et al., 2003; Kimball Dalton et al., 1998; Rimm et al., 1987; Ron et al., 1988]. This association was demonstrated in children treated with radiation for tinea capitis during the 1940s and 1950s, who were found to have an increased incidence, later in life, of meningiomas, gliomas, and nerve sheath tumors [Ron et al., 1988]. Secondary brain tumors have been reported after cranial irradiation for leukemia, head and neck tumors, and primary brain tumors [Kimball Dalton et al., 1998; Kleinerman, 2009; Neglia et al., 1991; Rimm et al., 1987; Thierry-Chef et al., 2008]. The most common form of malignant brain tumor occurring after radiation is malignant gliomas, which tend to appear 5–15 years after radiation and are highly resistant to therapy. Meningiomas are the most common radiation-induced tumors, increase in frequency with time after radiotherapy, and do not begin to peak until ten years after exposure [Neglia et al., 1991; Rimm et al., 1987]. Other tumor types that have been reported include lower-grade gliomas, medulloblastomas, and ependymomas.
The effects of a multitude of environmental exposures, including maternal diet, on the occurrence of childhood brain tumors have not been conclusively shown [Bunin, 2000; Preston-Martin, 1996]. Factors that have been associated with a higher incidence of medulloblastoma in some, but not all, studies include maternal farm residence during pregnancy and pesticide exposure [Connelly and Malkin, 2007; Norman et al., 1996; Preston-Martin, 1996; Robison et al., 1995]. In some studies, mothers’ multivitamin use and food consumption during pregnancy have been found to be potentially protective against of the development of tumors, especially medulloblastoma. Initial studies suggested an association between significant exposure to electromagnetic waves or the use of cell phones in the development of gliomas, but other studies have not been able to confirm this relationship [Ali Kahn et al., 2003; Connelly and Malkin, 2007; Hardell et al., 2002]. Similarly, initial studies suggested that patients exposed to simian virus 40 (SV40), through immunizations, may have a higher incidence of developing medulloblastoma [Hayashi et al., 2001; Kim et al., 2002]. Subsequent studies could not confirm this association. Immunosuppression, either due to an underlying disorder of the immune system or after treatment with chronic immunosuppressive agents in situations such as organ transplantation, has been associated with an increased incidence of central nervous system lymphoma [Kuijten and Bunin, 1993; Penn, 1998].
Pathology and Classification
The present classification of primary central nervous system tumors dates back to the 1920s and, as devised by Bailey and Cushing, was based on a cell of origin concept and the theory that tumor cells developed from cells arrested in various stages of development [Bailey, 1925; Louis et al., 2007b]. For each developmental stage of a cell, a corresponding tumor could be identified. In the Bailey and Cushing classification, tumors were also classified on the basis of the morphology of the tumor [Bailey, 1925]. This classification was modified by histologic grading of predominantly glial tumors on a 1–4 scale, with 1 being the most benign and 4 being the most malignant. Over time, other systems have been devised, essentially to score tumors to connote their relative aggressivity and to link the nomenclature to prognosis.
At present, the most widely accepted classification schema is that of the World Health Organization (WHO) [Louis et al., 2007a]. This system, since its first publication in 1979, has included a grading scheme that basically attempts to be a malignancy scale. Generically, grade 1 tumors are those with low proliferative potential and the possibility of cure following surgical resection alone. Grade 2 lesions are believed to be more infiltrative in nature and may have a higher likelihood of recurring. Dependent on the tumor type, grade 2 tumors are also those considered to more likely to progress to higher grades, especially in the case of gliomas. Grade 3 is generally reserved for tumors with histological evidence of malignancy, which are less likely to be cured by surgery alone. Grade 4 tumors are those that are considered the most cytologically malignant and mitotically active. There have been attempts to modify the WHO classification for use in pediatrics. Although the WHO classification is applicable to pediatrics, the congenital nature of many tumors, often with elements that seem to show mixed lineage, such as neuronal and glial elements, can make classification difficult. In addition, in the pediatric modifications, which have not been fully accepted, tumor location is also a component of classification.
Probably the most controversial aspect of the classification of pediatric brain tumors has been the nomenclature utilized for central nervous system embryonal tumors [Rorke, 1983]. The most common embryonal tumor is the medulloblastoma, a term that was coined over 80 years ago and still exists, although it is now accepted that there is no such tumor cell as a medulloblast. The primary controversy centered on whether all histologically similar, primitive neuroectodermal tumors should be classified as one tumor type and then subclassified on the basis of other histological features, such as histological differentiation along one or more cellular lineages, or whether the tumor should be diagnosed based on both histological features and tumor location. The introduction of immunohistochemical techniques demonstrated the variable cell types that occur in embryonal tumors, although the majority of cells are small hyperchromatic round or oval cells (small blue cells) [Gould et al., 1990; Molenaar et al., 1989]. The availability of molecular genetic analysis helped settle this controversy, as it was shown that primitive embryonal tumors occurring in the cerebellum were molecularly distinct from those arising in the pineal region and from those arising in the cortex [Ellison, 2002; Haberler et al., 2006; Pomeroy and Sturla, 2003; Rorke et al., 1995].
The WHO classification does not separate tumor types on the basis of tumor location [Louis et al., 2007a]. However, by convention, childhood central nervous system gliomas are often discussed by location, and cerebellar astrocytomas, intrinsic brainstem gliomas, and visual pathway gliomas, especially optic nerve and/or chiasmatic tumors, are considered relatively distinct entities. There are few biologic data to support such separations, and there is probably little, if any, prognostic difference between a diffuse intrinsic brainstem glioma and a similar tumor involving the thalamus, or, for that matter, a juvenile pilocytic astrocytoma of the cerebrum versus juvenile pilocytic astrocytoma of the cerebellum.
Medulloblastoma and Supratentorial PNET
Isochromosome 17q, often in combination with loss of 17p, is the most common cytogenetic alteration in medulloblastoma, occurring in up to 50 percent of tumors [Biegel, 1999]. A clear association with clinical outcome has not been confirmed; however, recent evidence suggests that isochromosome 17q is an unfavorable prognostic indicator [Pan et al., 2005]. Supratentorial primitive neuroectodermal tumors (sPNET) display a unique set of genomic alterations that rarely include isochromosome 17q, indicating that sPNET are molecularly distinct from medulloblastoma, despite sharing a similar histological appearance [Bayani et al., 2000; Pfister et al., 2007].
High expression of the neurotrophin-3 receptor, TRKC, was the first molecular alteration identified as an independent predictor of favorable outcome in medulloblastoma [Grotzer et al., 2000; Segal et al., 1994]. Increased expression of ErbB2, a member of the epidermal growth factor receptor (EGFR) family, and MYCC oncogene independently correlates with a poor outcome [Gilbertson et al., 1995; Grotzer et al., 2001]. High-level amplifications of MYC family members are also seen in 5–15 percent of medulloblastomas, and are associated with the aggressive anaplastic medulloblastoma variant and unfavorable outcome [Herms et al., 2000].
Aberrant signaling through the developmental pathways, Hedgehog (Hh), Notch, and Wnt, has been linked to medulloblastoma formation and growth (Figure 83-3). The relationship between Hh and medulloblastoma was deduced from the observation that medulloblastomas arise in patients with nevoid basal cell carcinoma syndrome (Gorlin’s syndrome), a condition caused by a somatic mutation of the PATCHED (PTCH) gene [Wicking and Bale, 1997]. PTCH is an important regulator of the Hh mitogenic signaling pathway. Gorlin’s syndrome accounts for 1–2 percent of all medulloblastomas, most commonly of the desmoplastic subtype [Amlashi et al., 2003]. Mutations in Hh pathway genes have been identified in approximately 25 percent of sporadically occurring medulloblastomas, predominantly of the desmoplastic subtype [Dong et al., 2000; Pomeroy et al., 2002; Raffel et al., 1997].
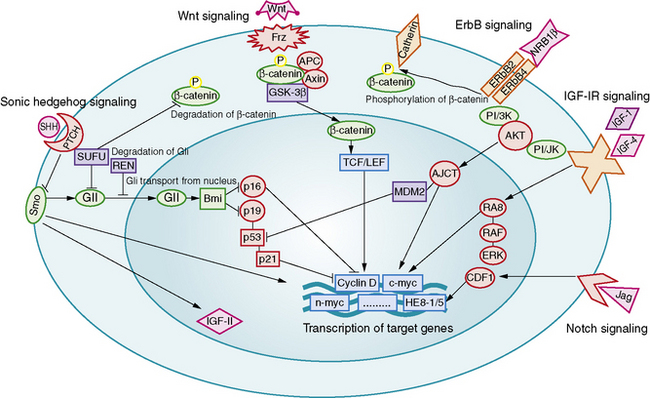
Increased copy numbers of Notch and mutations in the Wnt signaling pathway have each been detected in up to 15 percent of medulloblastomas. Expression of the Notch pathway target gene Hes1 is associated with shorter survival time, while nuclear immunoreactivity for beta-catenin, a marker of Wnt pathway activation, is associated with a highly favorable outcome [Ellison et al., 2005; Fan et al., 2004]. A more recent integrative genomic study identified five distinct molecular subtypes of medulloblastoma, including a Wnt signaling group associated with classic histology tumors in older children, and an Hh signaling group associated with desmoplastic tumors in younger children [Kool et al., 2008]. Both tumor groups shared increased expression of Notch and platelet-derived growth factor (PDGF) pathway genes.
Astrocytoma
Perhaps the most significant discovery to date for this group of tumors is the identification of BRAF oncogene-activating mutations in association with juvenile pilocytic astrocytoma (JPA) [Jones et al., 2008]. BRAF mutations appear to be rare in other types of pediatric low-grade glial tumors, and are associated primarily with JPA of the cerebellum and optic pathway [Jacob et al., 2009]. Low-grade gliomas, usually JPA of the optic nerves and chiasm, also occur in about 15 percent of children with neurofibromatosis type 1; however, BRAF mutations have not been identified in neurofibromatosis type 1-associated JPA [Jacob et al., 2009].
Evidence shows that childhood and adult malignant gliomas are molecularly distinct. Overexpression of EGFR is seen in 60–80 percent of high-grade gliomas in both children and adults; however, amplification of EGFR is found in 40 percent of adult tumors, but only rarely seen in childhood gliomas [Bredel et al., 1999]. TP53 is mutated in 12 percent of malignant gliomas in children aged less than 3 years, compared to 40 percent of the tumors in children older than 3 years [Pollack et al., 2001]. The 5-year survival is 17 percent in patients with high TP53 expression, and 44 percent in those with tumors that have low TP53 expression [Pollack et al., 2002]. PTEN mutations occur in only 8 percent of high-grade lesions in children, compared to 30 percent of adult high-grade tumors; however, expression of activated PDGF receptors and PTEN deficiency are associated with increasing malignant histology and decreased survival, respectively, among children with high-grade gliomas [Cheng et al., 1999; Raffel et al., 1999; Thorarinsdottir et al., 2008].
Other Childhood Brain Tumors
One of the most important molecular genetic alterations in childhood brain tumors is the discovery of hSNF5/INI1 gene mutations in association with atypical teratoid/rhabdoid tumors (AT/RT) [Biegel et al., 2002a]. AT/RT has sometimes been confused with medulloblastoma and PNET, but now screening for hSNF5/INI1 mutations can confirm the molecular diagnosis of AT/RT, which has important prognostic and therapeutic implications.
In ependymoma, chromosome 17p deletions have been found in 50–80 percent of tumors, but TP53 mutations have not been found to date [von Haken et al., 1996]. An integrative genomic study of ependymoma also revealed that supratentorial, posterior fossa, and spinal tumors have distinguishable molecular signatures, and that genes of supratentorial and spinal tumors are similarly expressed in mouse radial glial cells in the corresponding region of the CNS [Taylor et al., 2005].
Neuroradiology
The ability to stage tumor extent – especially to detect the presence of subarachnoid metastatic dissemination – is crucial. Many pediatric brain tumors have a propensity to disseminate throughout the subarachnoid spaces, often at the time of presentation. High-quality screening MRI studies of the entire neural axis are needed; clear outcome differences have been demonstrated with regard to pediatric brain tumors, depending on the presence or absence of dissemination [Packer et al., 2006]. Finally, repeated follow-up at appropriate time intervals is the best method for detecting early tumor recurrence; detection of smaller, presymptomatic recurrent tumor leads to improved outcomes [Saunders et al., 2003; Shaw et al., 1997].
Diffusion Imaging
Diffusion imaging (DI) investigates molecular translational movements (i.e., Brownian motion) of water molecules. Brownian motion produces signal loss proportional to the degree of molecular translation/diffusion. Within the cerebral milieu, cellular membranes and other macromolecular structures restrict water diffusion, which results in increased signal on DI. (Less diffusion means less signal loss secondary to spin dephasing.) Thus, signal on diffusion images can reflect cellular density and microarchitecture. The apparent diffusion coefficient (ADC) is a measure of the ability of tissue to restrict water diffusion. ADC adversely correlates with the grade of tumor and the cellular proliferation Ki-67 labeling index [Higano et al., 2006]. Increase in ADC, reflecting decrease in cellularity, has been observed as tumors respond to treatment.
In the intracellular and extracellular spaces of the brain, macromolecular proteins, intracellular organelles, cell walls, and myelin sheaths restrict or slow diffusion in certain directions. The most significant determinant of the preferred diffusion direction is the myelin sheath within cerebral white matter [Miller et al., 2003]. DI can therefore assess the microscopic structural integrity of white-matter tracts as the movement of water occurs preferentially along the long axis of the myelin sheath surrounding the axon. Anisotropy is an aggregate measure of the magnitude of displacement across all three dimensions; using information gleaned from the size, shape, orientation, and distribution (or spatial pattern) of diffusion ellipsoids within an imaged volume, one can characterize features of water diffusion within a voxel – the diffusion tensor. A voxel by voxel analysis of anisotropy can be performed, resulting in diffusion tensor imaging (DTI). Connectivity maps (i.e., tractography) can also be generated, which demonstrate the long association (axonal) pathways that convey cortical connections as tracklike structures.
DTI tractography is used to plan the surgical approach to a focal tumor. Important association pathways (e.g., pyramidal, optic tracts) can be localized in relation to a tumor; this helps the surgeon to avoid injury to these white-matter tracts. DTI also shows promise as a tool to evaluate for the presence and extent of tumoral invasion; whereas focal (well-marginated) tumors should displace but not disrupt axons outside of their borders, the microscopic tumor extension of infiltrative tumors is expected to disrupt local axonal structures [Helton et al., 2006; Talos et al., 2007].
Magnetic Resonance Spectroscopy
Spectroscopy can be performed with single-voxel or multivoxel techniques. Tissue composition within a voxel must be homogeneous, otherwise partial volume averaging of tissue of various compositions (e.g., normal, tumor, and necrotic tissue) occurs and the spectrum has little value. Single-voxel studies have improved spectral resolution over multivoxel techniques, though minimal voxel size for good signal to noise is about 4 cm3; further, in single-voxel imaging, the prescription (i.e., the determination of the voxel location) of the voxel is totally operator-dependent – and therefore not very reproducible. Spectroscopic analysis of a large volume of cerebral tissue is feasible with multivoxel techniques (i.e., two- or three-dimensional chemical shift imaging [CSI]), in which several subcentimeter voxels can be examined simultaneously. CSI is very helpful in the evaluation of small or heterogeneous tumors; in the evaluation of the extent infiltrative tumors where the tumor/normal brain boundary may be difficult to determine with conventional techniques; and to distinguish recurrent tumor from radiation necrosis. MR spectroscopy may be of help to monitor tumor response versus progression [Tzika et al., 2004]; prognosticate overall survival [Warren et al., 2000]; and improve preoperative identification of tumor type [Panigrahy et al., 2006].
Perfusion Imaging
Perfusion imaging can be performed with techniques based on dynamic susceptibility contrast (DSC) or on vascular permeability. DSC techniques assess cerebral tissue perfusion following a dynamic injection of gadolinium. During the first-pass transit of contrast through cerebral capillaries, which lasts anywhere from 5 to 15 seconds, gadolinium is (normally) restricted to the intravascular space. This intravascular restriction gives rise to signal inhomogeneity within image voxels (i.e., a susceptibility effect), which is reflected as signal loss on susceptibility-sensitive T2 (T2*) imaging techniques. A curve showing concentration of gadolinium over time can be generated. This enables a time versus signal intensity curve to be drawn, from which a variety of parameters can be inferred. These include mean transit time (MTT); time-to-peak; area under the curve, a measure of relative cerebral blood volume (rCBV); and relative cerebral blood flow (rCBF). rCBV corresponds to the volume of blood within brain tissue and can reflect the neovascularization associated with tumor growth (tumor angiogenesis). Perfusion imaging is therefore useful for the study of neovascularization and angiogenesis inhibition; for evaluation of tumor grade [Schmainda et al., 2004]; for targeting a lesion for biopsy; and to differentiate hypervascular tumors from tissue necrosis. DSC perfusion imaging may also be able to identify low-grade tumors that progress rapidly, and low-grade gliomas that have a propensity for malignant transformation [Law et al., 2006].
DSC perfusion studies do not consistently generate usable data. First, a larger-bore intravenous catheter must be placed for safe delivery of the injection of 3–4 cc/sec of gadolinium necessary to generate a well-defined first-pass curve; that is not always easy, especially in children with poor intravenous access. (Recent commercial introduction of ports and central lines that are safe to use with power injectors may alleviate this issue.) Second, susceptibility artifacts are not negligible, as DSC perfusion utilizes an echo planar technique (echo planar images are prone to image distortion by susceptibility artifacts); studies performed on children with dental braces or in the presence of significant amounts of intracranial/intratumoral hemorrhage are often useless. Finally, gadolinium contrast can leak through tumor capillaries; such leakage can result in an underestimation of rCBV, as the computations performed to process the susceptibility T2* signal assume that the contrast remains confined to the capillary [Cha et al., 2002].
Permeability-based perfusion techniques take advantage of leakage of gadolinium through tumor vessel walls. In healthy cerebral capillaries, the blood–brain barrier prevents gadolinium leakage into the interstitium. Tumor capillaries have disrupted or absent blood–brain barriers, which allows contrast to leak outside the vessel. The rate of leakage is proportional to the permeability of the capillary wall. Permeability imaging, also known as relaxivity imaging, is a T1-weighted technique in which a curve of dynamic signal intensity (i.e., enhancement) versus time is generated. The slope of enhancement reflects the degree of permeability of capillaries. The intensity of initial enhancement reflects tissue vascularity; fractional blood volume (fBV) can be extrapolated [Roberts et al., 2000]. fBV is a fairly good estimate of the rCBV data obtained with susceptibility perfusion [Haroon et al., 2007].
Positron Emission Tomography
Positron emission tomography (PET), with use of (18)F-fluorodeoxyglucose (FDG), allows for the evaluation of regional cerebral glucose metabolism. PET is useful in the determination of tumor metabolism/grade and of tumor extent, and to distinguish persistent or recurrent tumor from radiation necrosis [Pirotte et al., 2007]. Metabolic changes can be measured with a spatial resolution approaching 2–3 mm in the newer scanners. Images can be co-acquired with a CT scan (CT-PET), or can be co-registered with MR images using fusion software. Metabolic activity of tumor sites can be determined with quantitative, as well as qualitative (comparing FDG uptake in a tumor to uptake of normal gray or white matter), assessment. Higher-grade tumors tend to be more glucose-avid than lower-grade tumors, except for juvenile pilocytic astrocytomas, which show very high glucose uptake. For lesions that do not show abnormal glucose uptake, of (11)C-methionine PET scanning is helpful to differentiate neoplastic from non-neoplastic types. Thallium-201 single-photon emission CT (SPECT) now has a decreasing role in evaluation of pediatric brain tumors, given its more limited anatomic resolution.
Staging and Stratification
Pre- and postoperative staging is the cornerstone of therapy for most embryonal childhood central nervous system tumors and, to a lesser extent, glial neoplasms [Gajjar et al., 2004; Packer et al., 1999a]. The utility of staging for both determination of prognosis and decisions concerning most appropriate therapy has been best shown for medulloblastoma, as staging has been an accepted component of management since the mid-1980s. The first widely accepted staging system was the “Chang” classification schema, which has been extensively modified over time. All these schemas share the property of being based on degree of surgical resection or, more specifically, the amount of tumor left after surgery and the extent of tumor spread outside the primary tumor site. The latter is determined by a combination of results from MRI of the entire brain and spine, and cerebrospinal fluid cytological examination, preferably from the lumbar space, if deemed medically safe [Deutsch and Reigel, 1980; Gajjar et al., 2004; Packer, 1999; Packer et al., 1999a]. Embryonal tumors and glial neoplasms rarely spread to non-neural sites; thus, evaluation for disease outside the primary nervous system, at the time of diagnosis, is usually not indicated. The classical TNM (tumor, nodes, metastasis) staging system used for non-central nervous system tumors is replaced by a TM system for brain and spine tumors. Although the need for lumbar cerebrospinal fluid cytological examination has been debated, studies have shown that it provides complementary information to imaging results [Packer et al., 1999a; Zeltzer et al., 1999].
A complicating factor has been the fact that the performance and interpretation of imaging results have been variable in national and international studies [Packer et al., 2006]. In the most recent medulloblastoma studies, approximately 10 percent of studies were inadequately performed, primarily due to movement artifact or incomplete imaging (primarily of the spine), and another 5–10 percent of studies are considered, upon central review, to be incorrectly interpreted. Tumors that are nonenhancing at the primary site may result in nonenhancing dissemination along the neuroaxis, which is difficult to assess and leads to under-assessment of the extent of disease. In addition, normal superficial spinal cord vascularity can be misinterpreted as leptomeningeal enhancement.
Histology has been incorporated into risk stratification for many tumor types. For embryonal tumors, such as medulloblastoma, histological features, including desmoplasia/nodularity in infants and anaplasia in infants and older children, are now increasingly accepted as important components of staging and risk-stratification classifications, although the independent significance of such features has been related variably to outcome [Eberhart et al., 2004; Giangaspero et al., 1999, 2006; Grotzer et al., 2007]. For glial tumors or ependymomas, histological features that have been translated into grades are an accepted component of classification and are used both for risk stratification and for determinations of treatment.
It has become clear for the embryonal tumors, especially medulloblastoma, that molecular genetic parameters provide extremely important information [Ellison et al., 2005; Fernandez-Teijeiro et al., 2004; Gajjar et al., 2004; Grotzer et al., 2007; Pfister et al., 2009; Ray et al., 2004]. In retrospective studies, a host of chromosomal and molecular genetic findings had been related to outcome. In some cases, these findings show a strong correlation with other parameters, such as the tendency to disseminate or features of anaplasia, but in other studies they are of independent prognostic value.
Clinical Presentation
Nonlocalizing Signs and Symptoms
Nonlocalizing signs and symptoms predominate early in the course of illness in almost all types of childhood nervous system tumors. This is due to a variety of factors, including the predilection of childhood brain tumors to arise in the posterior fossa and cause obstruction of cerebrospinal fluid early in the course of illness, resulting in the nonspecific symptoms of headaches, nausea, vomiting, and lethargy. Separation of headaches caused by neoplasms, as opposed to the more common headaches children develop, can be extremely difficult [Glaser et al., 1971; Honig and Charney, 1982]. The majority of children with brain tumors, especially those with posterior fossa tumors and large supratentorial tumors, will have headaches by the time of diagnosis. However, headaches are notoriously nonspecific early in the course of illness. A change from a nonspecific headache pattern to morning headaches should raise concern that a mass lesion is present. Headaches that wake a child from sleep also suggest the possibility of increased intracranial pressure. Although this is true in general terms, recognizing such changes, in practicality, can be quite difficult for the child or family, and migraine headaches can wake a child from sleep. Headaches are especially difficult to characterize in very young children.
Localizing Symptoms
Supratentorial Tumors
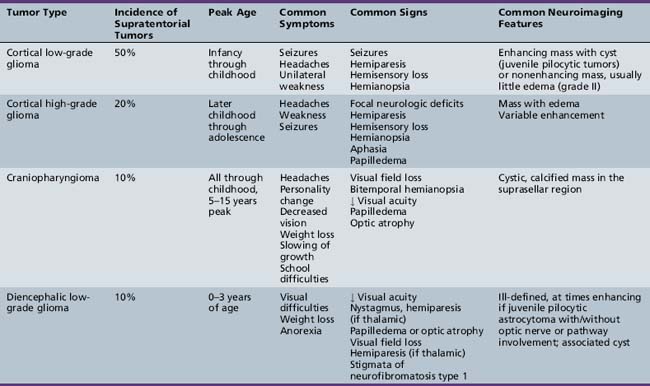
The diagnosis of primary central nervous system tumors is usually easier when there are associated localizing signs and symptoms (Table 83-1). Supratentorial tumors present with symptoms and signs similar to those seen in older patients and adults. Headaches are the most common initial complaint and are often nonlocalizing early in the course of illness [Hirsch et al., 1989]. Seizures are the second most common symptom encountered and approximately one-quarter of children with supratentorial tumors will have seizures as their initial symptom. This is especially true with tumors of the frontal or temporal lesion. Slower-growing cortical tumors, especially gliomas, are more likely to result in convulsions early in the course of illness. Although patients with more aggressive cortical gliomas may have seizures, focal neurologic deficits, such as hemiparesis or, in a patient amenable to careful examination, hemisensory loss or visual field loss, are more common.
Posterior Fossa Tumors
Five tumor types are most likely to occur in the posterior fossa, as noted in Table 83-2. Since cerebellar astrocytomas are more laterally placed and are slower-growing tumors, their symptoms tend to be more indolent than those of medulloblastomas, which fill the fourth ventricle early in the course of illness and result in obstructive hydrocephalus. However, midline cerebellar astrocytomas may mimic medulloblastomas [Packer et al., 1999a]. Brainstem gliomas are an insidious lesion and are likely to cause cranial nerve neuropathies associated with motor, cerebellar, and sensory deficits. Ependymomas are the great mimickers of other tumor types in the posterior fossa and may present as slow-growing, medulloblastoma-like tumors or, if the tumor involves the cerebellopontine angle, can mimic brainstem gliomas and result in multiple cranial neuropathies, especially sixth, seventh, and eighth nerve palsies.
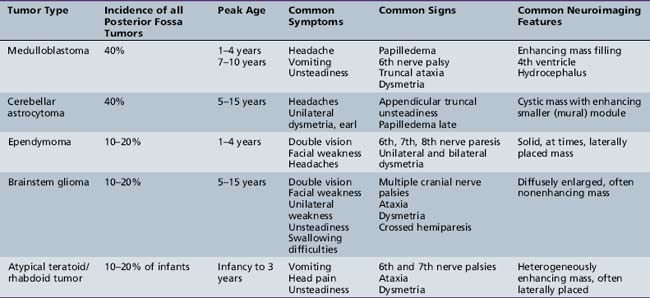
Midline Tumors
The infiltrating nature of midline tumors may also make diagnosis difficult [Albright et al., 1984; Glaser et al., 1971]. Many of these neoplasms are low-grade and classically result in insidious, nonspecific, and often nonlocalizing symptoms early in the course of illness. In the school-age child, infiltrating tumors and occasionally midline compressive masses, such as craniopharyngiomas, may also result in the nonspecific finding of declining academic performance, associated with fatigue and personality change.
General Aspects of Treatment
Surgery
Surgery remains the initial step in the treatment for the overwhelming majority of childhood central nervous system tumors, and the degree of resection, even for malignant neoplasms that require other forms of adjuvant therapy, is often an important factor in determining outcome [Albright et al., 1995; Campbell et al., 1996; Chen et al., 2007; Horn et al., 1999; Packer et al., 1999a; Roujeau et al., 2007]. As regards diagnosis, attainment of a representative portion of the neoplasm for histological and increasingly biological analysis is crucial. Exceptions to the need for tissue for diagnosis include neuroradiographic characteristic diffuse intrinsic brainstem tumors; chiasmatic/hypothalamic infiltrating gliomas in children with neurofibromatosis type 1 and possibly for those without; and germ cell tumors, predominantly mixed germ cell tumors, which secrete diagnostic proteins into cerebrospinal fluid and blood.
For some tumor types, surgery is curative and complete resection obviates the need for alternative therapies [Schneider et al., 1992]. This is primarily true for low-grade gliomas, especially pilocytic astrocytomas of the cerebellum or cerebrum, gangliogliomas, other mixed neuronal-glial tumors, and possibly supratentorial low-grade ependymomas [Poussaint and Rodriguez, 2006]. For other, more aggressive tumor types, surgery may be a crucial component of multimodality treatment, as outcome is statistically better for patients with nondisseminated medulloblastomas, posterior fossa ependymomas, and cortical malignant gliomas if total or near-total resection can be obtained [Albright et al., 1989; Packer et al., 1992; Wisoff et al., 1998]. For those tumors that located in eloquent areas of brain or are disseminated, the preoperative assessment of whether “total” resection or an attempt at total resection provides a survival benefit is usually the prime determinant of whether aggressive tumor removal is tried or less aggressive approaches, including open or stereotactic needle biopsy, will suffice. Stereotactic biopsy techniques, be they CT- or MRI-guided, are highly accurate in targeting deep-seated lesions and are associated with morbidities of less than 5 percent and mortalities of less than 1 percent [Broggi et al., 1983]. The advent of biologic therapies, often requiring fresh frozen tissue for molecular analysis, has resulted in the need, in certain circumstances, for additional tissue that cannot be obtained by needle biopsy alone.
Intraoperative Techniques
For those tumors in which extent of resection is a major determinant of outcome, improvements in neurosurgical technology have made “total resections” more likely and possibly safer. Progressive improvements in the operative microscope, which facilitates the visualization of the interface between the neoplasm and normal brain, have allowed for more extensive resections. Frame-based and frameless stereotactic guidance systems are now routinely used for improved preoperative targeting of the tumor and to decrease injury to the surrounding normal brain [Barnett et al., 1993; Kelly, 1990]. Similarly, ultrasonic guidance has allowed for better intraoperative localization. A recent addition to the neurosurgical armamentarium has been the availability of intraoperative MRI, which allows real-time evaluation for extent of surgical resection [Martin et al., 2000]. Intraoperative MRI is performed both with low-field units, which require interruption of the operation for set-up and visualization, and with high-field units, which utilize MRI-compatible surgical equipment and allow the operation to be carried out essentially within the MRI unit. These techniques are most useful for circumscribed benign tumors; however, for infiltrating neoplasms the extent of resection of the neoplasm continues to be difficult to determine.
In functionally critical areas of the brain, intraoperative monitoring of the visual, auditory, and somatosensory pathways can be used to improve the safety of tumor resection [Schneider et al., 1993]. Similarly, for intrinsic and extrinsic brainstem lesions, intraoperative monitoring of cranial nerve function is an important adjunct [Berger et al., 1990]. Maintenance of function intraoperatively does not assure normal function postoperatively, as edema and apparent delayed damage may occur. Awake craniotomy can be used in older children, but it is usually of limited utility in the majority of pediatric brain tumor cases.
Perioperative Considerations
It has been suggested by many that improvements in preoperative and postoperative care have resulted in lower surgical morbidity and mortality. However, what constitutes acceptable morbidity is extremely difficult to define, and obviously, one of the goals of surgery is to have no mortality and limited morbidity. Preoperatively, in children with increased intracranial pressure, every attempt is made to try to decrease pressure and stabilize the patient. In patients with hydrocephalus, this often includes the placement of a temporary external ventricular drain [Dias and Albright, 1989]. In most centers, in patients with posterior fossa tumors and hydrocephalus, a temporary drain is utilized and, within 24–72 hours, the tumor is removed. Removal may restore adequate cerebrospinal fluid flow to avoid and the need for permanent ventriculoperitoneal diversion (ventriculoperitoneal shunting). The minority of patients with posterior fossa lesions, usually fewer than 40 percent, require permanent external drainage. Third ventriculostomy has been proven to be a useful adjunct in reducing the need for permanent ventriculoperitoneal drainage, but the effect of this technique on long-term outcome has not yet been shown [Hopf et al., 1999; Jones et al., 1990].
In children with cortical tumors, prophylactic anticonvulsants are often utilized, even if there is no history of seizures. The need for such treatment and the duration of treatment, postoperatively, are unclear [Foy et al., 1992]. Phenytoin is often used preoperatively due to its lack of significant sedation in most patients, its ability to be given and loaded intravenously, and the ease of switching from intravenous to oral dosing. In patients on steroids, due to drug metabolism interactions, maintenance of therapeutic levels of phenytoin may be difficult, especially when patients are switched to oral dosing. In children who may require postoperative chemotherapy, levetiracetam is an effective alternative, since it also can be used orally or intravenously, and has the added advantage that it does not undergo hepatic metabolism and, thus, does not interact with the metabolism of most antineoplastic chemotherapeutic agents.
Postoperative Morbidity
Surgery in the suprasellar area is frequently associated with increased postoperative visual impairments, which may include decreases in visual acuity and increased deficits in visual fields [Sanford, 1994]. Hypothalamic abnormalities and significant hormonal deficits secondary to disruption of the hypothalamic–pituitary axis also frequently occur. In large tumors that extend to the subfrontal area, removal of the tumor may cause additional damage and both early and late emotional/personality changes. Hypothalamic injury may also result in abnormalities in satiety control and excessive weight gain, primarily in children with craniopharyngiomas.
Surgery for posterior fossa tumors may result in direct brainstem damage, with resultant cranial nerve deficits and damage to the cerebellum or cerebellar peduncles, with increased ataxia and dysmetria. Such direct brainstem damage may be difficult to separate from the increasingly recognized posterior fossa mutism syndrome, which is a constellation of the delayed onset of mutism (characteristically, a few hours to a day after surgery), hypotonia, cerebellar deficits, supranuclear palsies, emotional lability, and severe irritability [Doxey et al., 1999; Packer et al., 1992; Pollack, 1997]. The exact pathogenesis of the posterior fossa mutism syndrome is unknown. Seen primarily, but not exclusively, after medulloblastoma resection, it is thought to be secondary to unilateral or bilateral cerebellar dentate nuclei damage and/or disruption of critical pathways between the cerebellum and the cortex, especially the premotor and supplementary motor cortices. This syndrome results in permanent sequelae in approximately 50 percent of initially symptomatic patients, including persistent abnormalities in tone, coordination, speech, and cognitive abilities [Robertson et al., 2006; Wells et al., 2008]. The presence of lower motor neuron cranial nerve deficits or MRI features of immediate brainstem injury or cerebellar damage makes direct brainstem injury more likely. Postoperative brainstem edema has been reported to be more common in patients with medulloblastoma who develop posterior fossa mutism syndrome compared to patients who do not.
Radiation Therapy
Radiation therapy is an integral component of treatment for many forms of childhood brain tumors. For children with benign tumors, its use is usually limited to those tumors that are not amenable to gross total resections. For children with more aggressive or malignant tumors, radiation therapy has been the backbone of most curative and palliative therapies, even in those patients whose tumors are totally resected. For medulloblastoma, other embryonal tumors, and germ cell tumors, delivery of radiotherapy to the entire neuroaxis (craniospinal radiotherapy), supplemented with boost radiotherapy to the primary tumor site, is routinely employed to treat neuroaxis sanctuary disease and prevent exoprimary disease relapse, even in patients without evidence of tumor dissemination [Packer et al., 1999b]. Craniospinal radiotherapy has been associated with improved survival, but also significant long-term endocrinologic, growth, and cognitive sequelae. In very young children, especially infants, radiotherapy may result in even more severe sequelae and attempts have been made to delay, if not obviate, the need for radiotherapy by the use of chemotherapy. Chemotherapy is often utilized with radiotherapy in older patients, not only to increase survival, but also to decrease the dose or volume of radiation therapy required for disease control or cure.
Changing Techniques/Delivery
As in other forms of medicine, radiation therapy techniques continue to evolve. Focal and craniospinal radiation treatment planning is increasingly dependent on sophisticated three-dimensional computerized analysis [Ciernik et al., 2003; Hug et al., 2002; Kirsch and Tarbell, 2004; Thornton et al., 1991]. “Conformal” radiation therapy is the standard of care and such three-dimensional treatment planning involves multiple individual fields delivered in a variety of different orientations. Intensity modulated radiation therapy (IMRT) is a form of conformal radiation that, depending on the situation, may provide some advantages over other conformal planning techniques [Leibel et al., 1994]. Stereotactic radiosurgery is a further refinement in the delivery of radiation therapy and can be multifractionated, but it is frequently given in a single fraction, termed “radiosurgery.” Radiosurgery systems that deliver hypofractionated radiotherapy, such as the gamma knife, cyber knife, and modified linear accelerated units (LINAC), are designed to deliver a high radiation dose to a small intracranial target in few fractions (often one fraction) by focusing multiple small radiation beams on the target [Boethius et al., 2002; Chiou et al., 2001; Hadjipanayis et al., 2002]. Radiosurgery is usually utilized to treat smaller tumors, normally 3 cm or smaller, but overlapping fields can be used in somewhat larger tumors. Its use in pediatrics has been relatively limited, but it has been employed with good short-term control for small intrinsic tumors, such as low-grade (e.g., pilocytic) gliomas, and other benign tumors, such as craniopharyngiomas. Radiosurgery results in a greater likelihood of acute and subacute reactions, and has less utility in regions of brain where critical areas could be injured by acute necrosis, or in infiltrating tumors.
Radiation therapy is conventionally delivered by photon particles. Other types of particle beam irradiation have been used in selected circumstances. Electron particles, which have a lesser depth of penetrance, have been utilized for delivery of the spinal portion of craniospinal radiation [Pai Panandiker et al., 2007]. Such beams have the disadvantage of not being able to penetrate as deeply as photon irradiation, but have the advantage of having less radiation scatter. Proton beam irradiation is more widely used. It can be modulated to release its energy at a specific depth, unlike photon beams that deliver dose as they enter and exit the target [Hug et al., 2002; MacDonald et al., 2008; Merchant et al., 2008; Plowman et al., 1999]. Dosimetry studies demonstrate that the surrounding areas receive less irradiation after proton treatment compared to photons. For patients undergoing craniospinal radiotherapy, this decrease in “scatter” can spare radiation to tissues surrounding neuroaxis, such as the thyroid gland, colon, and heart. There are also advantages in some posterior fossa tumors, with sparing of dose to the cochlea, especially in treatment of children with posterior fossa ependymomas. Proton beam units were initially almost prohibitively expensive and few units were available to treat children. With new technologies, there will be greater availability in the near future.
In addition to hypofractionated delivery techniques, such as the gamma knife, there has been interest and experience in using a smaller dose of radiotherapy multiple times during the day. Such “hyperfractionated” radiotherapy is based on the concepts that tumor cells are more likely to enter growth phases when they are radiosensitive than non-tumor cells, and that normal cells are better able to repair radiation-induced damage than tumor cells [Freeman, 1996; Mandell et al., 1999; Packer et al., 1993; Thames et al., 1983]. Radiation delivered more frequently allows the normal cells, but not the tumor cells, to undergo cellular repair and, thus, higher doses of radiotherapy can be “safely” utilized, with less toxicity and more efficacy. Early experience with this approach, using dose fractions ranging from 100–120 cGy, twice daily, to total doses as high as 7800 cGy in pediatrics, were disappointing in children with brainstem gliomas [Packer et al., 1993]. There was no improvement in survival, but an apparently higher incidence of acute sequelae, due to brainstem necrosis. Studies using hyperfractionated radiotherapy, especially in delivery of craniospinal radiotherapy, are on-going, with completed studies showing no clear-cut increased acute toxicity and, possibly, improved disease control.
Radiation Therapy-Associated Sequelae
The determination of the optimal, often maximal, safe dose and volume of radiotherapy that can be used is dependent on the tumor type and the radiosensitivity of structures in the central nervous system, tempered by the age of the child [Halperin et al., 1994]. Younger children are more sensitive to the detrimental effects of radiation on the brain. In general, larger daily dose fractions cause more acute and possibly long-term neurologic toxicity; standard fractionation is 150–180 cGy daily [Emami et al., 1991; Kao et al., 1994; Mulhern et al., 1989; Schultheiss et al., 1995]. Preoperative conditions, such as hydrocephalus and pre-existing brain injury, may affect the tolerance of the developing brain to radiotherapy. Even in children of similar ages with similar diseases, there can be significant differences in how radiotherapy can be tolerated, suggesting that there are molecular genetic differences that affect host vulnerability; these genetic predispositions are, however, largely unknown [Kao et al., 1994].
The detrimental effects of radiation therapy have been relatively crudely divided into three major types: acute reactions occurring during treatment; subacute or early delayed reactions occurring a few weeks to months after irradiation; and late reactions occurring several months to years after treatment (Table 83-3) [Halperin et al., 1994]. Acute reactions are considered to be secondary to acute cellular injury and are often transient, with associated cytokine release, inflammatory changes, and edema. An increased incidence of acute reactions has been associated with higher dose fractions and possibly with total dose. Subacute reactions include the so-called “somnolence syndrome,” which is manifest by irritability and fatigue, often associated with headaches, occurring a few weeks after the completion of extensive focal or whole-brain irradiation therapy. Transient radiation myelitis is a spinal cord subacute radiation therapy reaction, often manifested by Lhermitte’s sign, which consists of electric shocks down the spine; it also occurs a few weeks after completion of spinal radiation. The etiology of these subacute effects is unknown and it has been postulated that they are due to transient oligodendroglioma damage. Subacute reactions tend to be transient, lasting for 7–10 days, and have not been clearly associated with long-term sequelae. Delayed reactions are more clearly related to total dose and volume of radiotherapy, and are more likely to result in permanent damage. Delayed reactions include a spectrum of radiation injuries ranging from focal radiation necrosis to more common cognitive defects [Bowers et al., 2006; Robison et al., 2005]. The etiology of such late changes again is multifactorial, and is thought to result, in great part, from vascular injury of large- or small-caliber vessels.
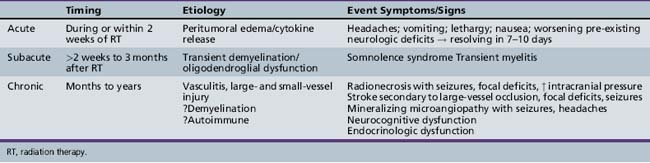
The tolerance of the different areas of the brain to radiotherapy varies [Leibel, 1987]. In general, the brainstem is considered relatively resistant to standard doses of radiotherapy (5400–6000 cGy). The spinal cord is considered somewhat more radiosensitive, and often radiotherapy doses are reduced to 4500–5000 cGy in attempts to decrease the likelihood of postradiation myelopathy. The cerebral cortex can usually, in older patients, tolerate focal doses of radiotherapy in the 5500–6000 cGy range, but such doses can cause delayed brain injury. Whole-brain doses of 2400–3600 cGy or higher usually result in age-dependent neurocognitive damage in children, as will be discussed later. Areas outside the primary brain parenchyma are also important determinants of the dose and volume of radiotherapy. Cranial nerves are relatively resistant to radiation-induced damage [Leibel, 1987]. Although there are concerns about irradiating the optic nerves, in studies, doses less than 6000 cGy were relatively well tolerated, especially when lower-dose fractions of radiotherapy were employed [Jiang et al., 1994; Parsons et al., 1994]. Auditory toxicity secondary to damage to the eighth nerve and/or auditory apparatus may also occur; the cochlea is especially sensitive to radiation damage, which is potentiated by the use of ototoxic drugs, particularly cisplatin [Grau and Overgaard, 1996; Merchant et al., 2004a; Miettinen et al., 1997]. The retina may also be in the radiation field and is sensitive to vascular damage [Miettinen et al., 1997]. Vision may deteriorate years after radiation and there does seem to be a dose–response relationship, with doses less than 4500 cGy to the retina causing less injury [Merchant et al., 2002a]. Cataracts may also increase visual morbidity [Deeg et al., 1984; Packer et al., 2003].
Irradiation of the hypothalamic–pituitary axis often is unavoidable because of location of the tumor, and can result in permanent endocrinologic damage [Constine et al., 1993; Gurney et al., 2003; Merchant et al., 2002b; Moshang et al., 1996; Packer et al., 2001, 2003; Shalet et al., 1987]. Growth hormone secretion is usually the most sensitive to the effects of radiotherapy, followed, in order of degree of sensitivity, by thyroid stimulating hormone, follicle stimulating hormone-luteinizing hormone (FSH-LH), and adrenocorticotropic hormone (ACTH). There does seem to be a dose relationship to such damage, with significant impairment of growth hormone secretion occurring more commonly after 2400 cGy, especially in doses greater than 2900 cGy, to the hypothalamus.
The severity of neurocognitive sequelae secondary to whole-brain radiotherapy, usually given as part of craniospinal radiotherapy and most frequently employed in tumors such as medulloblastoma and germinoma, is dependent on dose, age of the child, poorly understood host vulnerabilities, and probably pre-existing brain injury [Kao et al., 1994; Mulhern et al., 1989]. Craniospinal radiation therapy, especially in children less than 7 years of age, and particularly in those less than 5 years of age, results in a demonstrable drop in overall intelligence, as well as a host of selective learning, attentional, and executive disabilities [Johnson et al., 1994; Packer et al., 1989; Palmer et al., 2001; Silber et al., 1992]. In older children, depending on dose, falls in overall intelligence may not occur, but selective learning and executive disabilities are common. For very young children, especially infants, craniospinal irradiation has been associated with a strong likelihood of frank mental retardation. For this reason, infants are rarely treated with craniospinal irradiation therapy, unless there are no other options for disease control. There is no clear-cut age at which radiation therapy becomes safe or, for that matter, unsafe, and there is no consensus concerning the age at which radiotherapy should be employed, even in a tumor such as medulloblastoma, in which radiotherapy has been shown to be an extremely effective means of tumor control, but also a frequent cause of significant sequelae.
Another long-term sequela of radiotherapy that has been discussed earlier is development of secondary neoplasms. A host of different tumors may arise. with high-grade gliomas being most frequent 5–15 years after radiotherapy. and meningiomas increasing in incidence after 10 years [Neglia et al., 2006]. Children who are younger at diagnosis are more likely to develop tumors [Neglia et al., 2006].
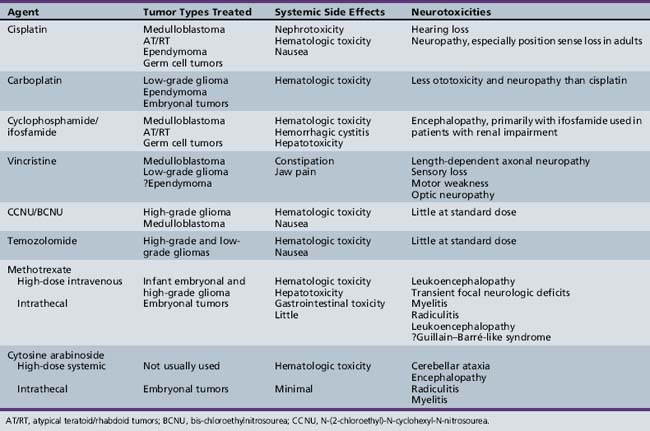
Chemotherapy
Within the last decade, chemotherapy has gained an expanded role in the treatment of childhood brain tumors (Table 83-4). The most common chemotherapeutic agents utilized for pediatric central nervous system tumors are in the class of DNA alkylators, such as cisplatin, carboplatin, lomustine, cyclophosphamide, and ifosfamide, which typically serve as the backbone of the treatment of medulloblastoma, low-grade glioma, ependymoma, and germ cell tumors; and temozolomide, which is widely used for the treatment of high-grade glioma. Plant alkaloids, such as the mitotic inhibitors, vincristine and vinblastine, or the topoisomerase inhibitor, etoposide, are also commonly employed within these regimens. Yet, the overall success of chemotherapy for pediatric central nervous system tumors has remained somewhat limited. Two major reasons for this limitation are impediments to drug delivery across the blood–brain barrier and resistance to chemotherapy. Newer therapeutic strategies have thus been designed to overcome these barriers or to utilize drugs that specifically target tumor biology. Only issues relating to these aspects of delivery of chemotherapy will be discussed here, since the indications for the use of specific chemotherapy and the current outcomes are discussed separately for each tumor type.
High-Dose Systemic Chemotherapy
The goal of high-dose chemotherapy (HDCT) is effectively to increase the delivery of cytotoxic agents to the tumor by overcoming the limited permeability of the blood–brain barrier. Several trials have been conducted in children with primary and refractory malignant brain tumors, utilizing single or sequential cycles of myeloablative or myelosuppressive HDCT, with either autologous bone marrow transplant (ABMT) or peripheral blood stem cell (PBSC) support [Dunkel et al., 1998; Jakacki et al., 1998; Kalifa et al., 1992; Mason et al., 1998]. Classic lipid-soluble alkylating agents (e.g.. thiotepa), which have nonoverlapping hematologic toxicities and show little cross-resistance, have been predominantly investigated. The most impressive responses have been noted in medulloblastoma and supratentorial PNET; however, in some studies, toxicity has been excessive and the overall efficacy of this strategy remains unproven [Butturini et al., 2009; Choi et al., 2008; Fangusaro et al., 2008; Gururangan et al., 2008; Rosenfeld et al., 2009; Shih et al., 2008].
Regional Chemotherapy
Intrathecal or intraventricular administration of cytotoxic agents is an alternative method to increase tumor exposure to chemotherapy, as well as to control leptomeningeal tumor dissemination. To date, these methods have been limited by the lack of available active agents that can be given by this route of administration. The availability of mafosfamide, a pre-activated derivative of cyclophosphamide, and a soluble form of busulfan has led to several phase 1 clinical trials in children with central nervous system tumors [Blaney et al., 2005; Gururangan et al., 2006]. Convection-enhanced intracavitary and intratumoral delivery of drug therapy and biologically active nanoparticles is the most recent regional strategy to be investigated [Allard et al., 2009].
Inhibition of Resistance to Chemotherapy
Strategies to overcome tumor resistance to drug therapy have been employed with some success. A key example of this has been with the alkylator, temozolomide, which has shown activity against malignant gliomas in adults [Friedman et al., 2000]. A major resistance mechanism against this drug is DNA alkylation repair via the enzyme MGMT. The drug O6-benzylguanine depletes tumor MGMT levels, thus rendering the tumor more sensitive to temozolomide [Friedman et al., 2002]. These findings led to the phase I investigation of the systemic combination of these two agents in children with CNS tumors [Broniscer et al., 2007].
Biologic Therapy
Inhibitors of Receptor Tyrosine Kinases and Cellular Signal Transduction
Given the molecular evidence for the high expression of EGFR family members (ErbB1–4) in pediatric malignant glioma, ependymoma, and medulloblastoma, and the correlation of ErbB2 expression with poor outcome, the initial phase I trials of kinase inhibitors in childhood central nervous system tumors centered on targeting EGFR family members with the agents gefitinib, lapatinib, and erlotinib [Broniscer et al., 2009; Jakacki et al., 2008]. Trials have also been conducted to investigate the use of lonafarnib and tipifarnib, farnesyl transferase inhibitors that block Ras oncogene activation and Ras-mediated signaling [Fouladi et al., 2007; Haas-Kogan et al., 2008; Kieran et al., 2007]. Other clinical research studies have focused on targeting endothelial signaling to prevent angiogenesis and tumor growth, such as imatinib, which inhibits PDGF rector kinase function; cilengitide, which targets endothelial integrins; and bevacizumab and other similar agents that target the vascular endothelial growth factor receptor signaling axis [Kieran et al., 2009; Pollack et al., 2007].
The most recent studies have focused on targeting the aberrant developmental pathways believed to be critical for the growth and survival of specific subsets of medulloblastomas. Phase I trials utilizing drugs aimed at blocking Hh and Notch pathway signaling are currently under way, and the preliminary results of the Hh antagonists have been encouraging [Yauch et al., 2009].
Immunotherapy
Immunotherapy is a potentially attractive way to treat childhood brain tumors, as therapy, theoretically, can be selective and designed to attack only the tumor cell [Barker and Billingham, 1977; Pollack et al., 2000]. The central nervous system is a relatively privileged site immunologically, due to its lack of lymphatic drainage and its blood–brain barrier, but there is evidence that, to some degree, peripheral T-cell activity may be carried into the central nervous system. A variety of different approaches have been utilized in the immunotherapy of central nervous system tumors – primarily in adults, but also, to some degree, in pediatrics [Heimberger and Sampson, 2009; Liau et al., 1999; Okada et al., 2003; Plautz et al., 1998, 2000; Pollack et al., 2000; Yamanaka et al., 2003]. These include administration of cytokines such as interleukin and interferon, expansion of tumor-specific T cells, the use of monoclonal antibodies, and, most recently, cancer vaccines.
Interferon-alpha, beta, and gamma have been used in pediatric trials, with unclear efficacy [Mahaley et al., 1988, 1989; Nagai and Arai, 1984; Yates et al., 1985; Yung et al., 1987]. Studies continue to utilize interferon with other modalities of treatment, especially radiation therapy, in attempts to improve outcome. Adoptive immunotherapy techniques, such as the intratumoral administration of interleukin-2 to boost antitumor cellular immune response and the delivery of lymphokine-activated killer cells, have been used almost exclusively in adults, again with limited efficacy to date.
Monoclonal antibodies have been, and continue to be, evaluated in both adults and children with central nervous system tumors [Colapinto et al., 1990; Lashford et al., 1988; Neuwelt et al., 1987]. The majority of studies have utilized radionuclide-conjugated antibodies, delivered either systemically or intrathecally. There is significant concern that such radionuclide techniques will be limited by the blood–brain barrier, and the majority of trials are now utilizing direct delivery to the cerebrospinal fluid via either intrathecal or intraventricular means. Intrathecal delivery is limited by abnormalities in cerebrospinal fluid flow and essentially cannot be used in children with ventriculoperitoneal shunts, unless they have on/off valves, which must be closed for hours to allow better drug distribution. Many patients cannot tolerate this.
Tumor vaccines are being studied actively, in both children and adults [Liau et al., 1999, 2005; Okada et al., 2003; Yamanaka et al., 2003]. Initially, these vaccines required fresh tissue to be obtained from the patient, and then a vaccine to be produced for that individual patient. Some recent vaccine studies have utilized vaccines produced to target antigens thought to be expressed commonly on tumor cells, but not on normal cells. These vaccines rely on the tumor specificity of these antigens, with the understanding that some patients who receive the vaccine will not have tumors that express such antigens. Vaccine trials include those that use cytokine-transfected tumor cells, adoptive transfer of tumor-activated T cells, and antigen-pulsed dendritic cells. In patients with bulk disease, it is likely that these vaccines, at best, will be an adjuvant to other forms of therapy.
Gene Therapy
Introduced with great enthusiasm over 20 years ago, gene transfer therapy has yet to show efficacy in the majority of adults or children with central nervous system tumors. It is a process in which genetic material is transferred into cells and carries with it the information either to stop or to slow the ability of these cells to multiply [Culver et al., 1992; Lowenstein et al., 1999; Ram et al., 1997]. Gene therapy may also utilize genetic information that, after the tumor cells take up the gene material, makes them sensitive to other forms of treatment. Almost all of the gene transfer therapies have utilized a virus-mediated delivery system, and it was believed that the environment of the central nervous system, which has few dividing nontumorous cells, might allow the specific targeting of the viral vectors to enter mitotically active cells of the malignancy. It is unclear whether the gene transferred into the cell will be able, through the release of cellular toxins, to kill surrounding tumor cells (the bystander effect), and also whether the delivery, dependent on the type of virus utilized, will be selective and not harm the surrounding normal cells. To date, gene therapy techniques have required the direct installation or injection of the viral particles or the gene product into tumor tissue. The capability of the virus to transfect an adequate number of cells to result in tumor control is a major limitation, and has led most gene therapy approaches to focus on the rim of well-resected tumors. Due to concerns over the development of viral meningitis, patients with tumors that are open to, or close to, the ventricular surface have been ineligible for studies. Additional immunomodulatory or antiangiogenic benefits of gene therapy are unproven.
The gene therapy studies that have been completed in pediatrics essentially have been limited to the use of the herpes simplex virus thymidine kinase type 1 gene, which, when transfected into a tumor cell, can act as a suicide gene if exposed to systemically administered ganciclovir [Packer et al., 2000]. The gene product produced causes phosphorylation of the drug, resulting in cell death. Using a replicated deficient retrovirus, this approach has been shown to be feasible, but may result in transient adverse symptomatology, including seizures, headaches, and cerebral edema. In the one published pediatric study to date, the symptoms resolved spontaneously or after a short course of systemic corticosteroids. Due to lack of availability of this gene transfer therapy product, a phase II study was never completed [Packer et al., 2000].
One of the main unknowns of gene therapy studies is whether it is mandatory for all the tumor cells to take up the gene product, and, if there are associated bystander effects, whether they are due to release of cytokines or immunomodulatory effects that can make therapy more effective [Culver et al., 1992; Lowenstein et al., 1999; Ram et al., 1997]. In studies to date, the efficiency of tumor cell transfection has been extremely variable, ranging from 10 to 90 percent. Another major unknown associated with the viral vector delivery is what vector will be safe for the child. Viruses such as the adeno-associated virus may result in a better transfection rate than replicative-defective viruses, but it is unclear what the long-term effects of this virus will be on the brain. Viruses that are replicative-competent may result in encephalitis, meningoencephalitis, and systemic illness, especially in patients who are immunosuppressed due to prior use of radiation, chemotherapy, or corticosteroids. Replication-deficient viruses may be safer, but may result in a lower transfection rate. With increasing safety guidelines, gene therapy studies are once again under way in pediatric patients with brain tumors.
Management of Specific Tumors
Embryonal Tumors
According to the World Health Organization classification of central nervous system tumors, embryonal tumors constitute a collection of biologically heterogeneous lesions that are malignant and share the tendency to disseminate within the nervous system, predominantly the cerebrospinal fluid pathways, early in the course of illness [Louis et al., 2007a]. Such lesions may arise anywhere in the central nervous system, but the most common site is the posterior fossa, presumably because the most common such tumor is the medulloblastoma. As noted in Box 83-1, the WHO recognizes five types of embryonal tumors. A histologically similar tumor, the pineoblastoma, arises in the pineal region. According to the WHO classification, pineoblastoma is considered a pineal tumor rather than an embryonal tumor. Pineoblastoma is therefore discussed in the section on pineal tumors.







Medulloblastoma
Medulloblastoma, which by definition arises in the posterior fossa, is by far the most common embryonal tumor, and is also the single most common form of malignant brain tumor of childhood [Louis et al., 2007b; Packer et al., 1999a]. Medulloblastomas are highly cellular tumors with deeply basophilic nuclei of variable size and shape, little cytoplasm, and abundant mitoses, and may show histologic heterogeneity (Figure 83-4) [Packer et al., 1999a; Rorke, 1999]. A variety of different subtypes of medulloblastoma have been noted, and the importance of further subdivision is primarily to connote a difference in prognosis between histological subtypes [Gould et al., 1990; Molenaar et al., 1989]. Two medulloblastoma variants that connote a different prognosis are the nodular/desmoplastic variant, which carries an excellent prognosis in infants, and the large cell/anaplastic variant, which connotes a poorer prognosis [Eberhart et al., 2004; Giangaspero et al., 1992, 1999]. Of all the childhood malignant tumors, the biology of medulloblastoma has been best delineated. With current means of treatment, the majority of children with medulloblastoma can be expected to be alive 5 years after diagnosis, and many of these children are cured [Packer et al., 1994, 2006]. Increasing emphasis recently has been placed on the quality of life of survivors and on the potential means to reduce treatment-related sequelae without decreasing the likelihood of survival.
As a whole, medulloblastomas constitute 20 percent of all primary central nervous system tumors and have a bimodal incidence peak, being most common in children 3–4 years of age, and then again becoming more frequent between the ages of 7 and 10 [CBTRUS, 2008; McNeil et al., 2002]. Between 15 and 20 percent of all medulloblastomas are diagnosed in the first 2 years of life. For unclear reasons, they are more likely to occur in Caucasians at a ratio 1.7 to 1, and there is a male predominance.
Although the etiology of medulloblastoma is unknown for the majority of patients, as reviewed previously in this chapter, several familial syndromes have been associated with an increased risk of developing medulloblastoma, including Gorlin’s and Turcot’s syndromes [Foulkes, 1995; Hamilton et al., 1995; Taylor et al., 2000]. Initially, investigations suggested that medulloblastomas arose from at least two types of progenitor cells in the cerebellum, one arising in the midline ventricular zone of the fourth ventricle, and others from the more laterally positioned, cerebellar external granular layer [de Bont et al., 2008; Read et al., 2006]. Those tumors emanating from the external granular layer are believed to arise from a more restricted neuronal differentiated precursor cell and are more likely to have mutations of the PATCHED-1 gene on chromosome 9, with resultant activation of the normally suppressed sonic hedgehog signaling pathway, as discussed in the biology section of this chapter. The development and growth of tumors are thought to arise from stem cells in the ventricular zone and seem dependent on a variety of growth factors and resultant activated intracellular pathways, including the RAS/MAPK cellular signaling pathway [MacDonald et al., 2001]. Biologic studies have clearly shown that most medulloblastomas are biologically distinct from other embryonal tumors.
Using chromosomal data, information obtained from immunohistochemical analysis, and both RNA- and DNA-based gene array studies, medulloblastomas have been biologically separated into multiple subgroups [de Bont et al., 2008; de Haas et al., 2008; Eberhart et al., 2003, 2004; Ellison et al., 2005; Grotzer et al., 2007; Khalili et al., 2003; Packer et al., 1999a; Pfister et al., 2009; Ray et al., 2004; Read et al., 2006; Takei et al., 2009; Thompson et al., 2006]. Overlap exists between biologic subgroups and it is unclear how many distinct subgroups of medulloblastoma will ultimately be identified. However, it is clear that medulloblastoma is not a biologically homogeneous disease. The importance of identification of the growth factors and the cellular pathways driving the propagation of medulloblastoma is not only for better stratification of the disease, but also potentially for treatment. There are presently multiple agents in testing that target growth factors and intracellular signaling pathways, and their addition to medulloblastoma therapeutic approaches may dramatically alter therapy and prognosis in the future.
Clinical presentation and diagnosis
Medulloblastoma most commonly presents with nonspecific findings of vomiting and headache, which occur in 80 percent of patients by the time of diagnosis [Packer et al., 1999a]. This is usually associated with obstruction of cerebrospinal fluid flow at either the third or fourth ventricular outlets, and hydrocephalus. Symptom duration is classically 1–3 months before diagnosis; patients with metastatic disease are more likely to be diagnosed earlier and have a poorer prognosis. Usually, by diagnosis, the headache has transformed into one more classically associated with increased intracranial pressure, occurring upon wakening, and accompanied by morning nausea and vomiting. Unsteadiness is noted in 50–80 percent of patients at diagnosis and is usually truncal, with significant gait abnormalities.
Medulloblastoma may present acutely with a severe alteration in consciousness and even coma [Fine, 2002; Packer et al., 1999a]. This is usually due to macroscopic hemorrhage into the tumor, and rapid tumor expansion with acute hydrocephalus and/or compression of the brainstem.
Medulloblastomas are usually radiographically distinguishable from other posterior fossa tumors, as they tend to be relatively well-defined masses arising in the medullary velum or roof of the fourth ventricle, often with some invasion of the middle cerebellar peduncle and compression or invasion of the brainstem [Akay et al., 2003; Jaiswal et al., 2004; MacDonald et al., 2003]. The tumor is hyperdense compared to normal cerebellum on CT, which distinguishes it from juvenile pilocytic astrocytoma. Calcifications may be present in up to 20 percent of cases, but are usually not conspicuous. On MRI, medulloblastomas tend to be homogenous with iso- to hypointense signal on T1-weighted images and hypointense signal on T2-weighted images (Figure 83-5). The majority of medulloblastomas enhance with contrast enhancement, although 5–10 percent do not enhance. In infancy, medulloblastoma with extensive nodularity may occur and display discrete contrasting-enhancing masses with almost a grape cluster appearance.
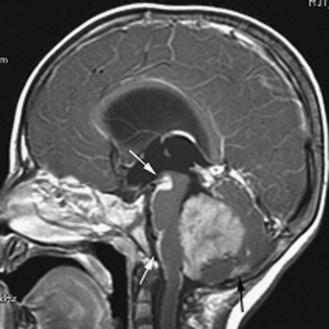
Additional neuroimaging techniques that help to distinguish medulloblastoma from other posterior tumors include DI and MR spectroscopy [Erdem et al., 2001]. The ADC values of medulloblastomas are usually lower than those of other posterior fossa tumors. Single-voxel MR spectroscopy demonstrates high levels of choline and a lower, or absent, NAA peak [Wang et al., 1995].
Since between 15 and 30 percent of medulloblastomas are disseminated to other regions of the nervous system prior to diagnosis, neuroradiographic evaluation of the entire neuroaxis is indicated, if possible, before surgery [Zeltzer et al., 1999]. Evaluation of dissemination can be difficult after surgery, due to changes caused by postoperative blood. Pitfalls in the evaluation of extent of disease include nonenhancing dissemination, especially in patients with initially nonenhancing primary site tumors, and spinal cord vascularity being interpreted as leptomeningeal disease.
Management and outcome
The treatment of children with medulloblastoma is multidisciplinary and, in most cases, multimodality, requiring surgery, radiation, and chemotherapy [MacDonald et al., 2003; Packer et al., 1999b]. Treatment is also dependent on the age of the child, predominantly due to the potential neurotoxic effects of radiotherapy, and disease stratification.
In non-disseminated patients, who comprise 70–80 percent of all patients with medulloblastoma, multiple studies have demonstrated that the extent of surgical resection is prognostic of outcome, with those patients undergoing more complete or near-total resections having better outcome than patients whose tumors were subtotally resected [Albright et al., 1996, 2000; Evans et al., 1990; Hughes et al., 1988; Tait et al., 1990]. This has been shown both in infants and in older children, but has not been clearly demonstrated in children with disseminated disease. With current means of adjuvant therapy, there is no clear-cut statistical difference in outcome between patients who undergo a total resection and those undergoing a near-total resection [Packer et al., 1994, 2006]. Patients who undergo a minimal resection or biopsy have an extremely poor prognosis.
Although the goal of surgery is to remove all the tumor, tumor resections (be they partial or complete) can be associated with significant immediate or somewhat delayed postoperative complications. Direct brainstem and cerebellar damage has been noted in 5–10 percent of patients. Increasingly, the posterior mutism syndrome has been recognized. Nearly 25 percent of patients in one recent North American trial had the posterior mutism syndrome, initially thought to be a rare entity, and one-half of all patients with this syndrome had sequelae 1 year later [Robertson et al., 2006; Wells et al., 2008]. Also, patients with this syndrome have poorer long-term neurocognitive outcomes.
Following surgery, patients with medulloblastoma have been prospectively staged, based on the amount of residual tumor after surgery; extent of dissemination assessed on both neuroimaging evaluation of the entire neuroaxis and cerebrospinal fluid cytology; examination; and, in some classification schemes, histology [Packer et al., 1999a]. Historically, children 3 years of age or older at diagnosis, with medulloblastomas, have been classified as having either average-risk or poor-risk disease (Table 83-5) [MacDonald et al., 2003; Packer et al., 1999a]. Patients with average-risk disease are those with non-disseminated, totally or near-totally resected tumors, with a non-anaplastic histology. All other patients are considered to have poor-risk disease.
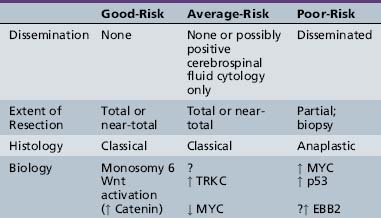
The staging systems used to date have not incorporated biologic data, primarily because these molecular parameters have not been available in real time [Fernandez-Teijeiro et al., 2004; Gajjar et al., 2004; MacDonald et al., 2003; Pfister et al., 2009; Ray et al., 2004]. The use of biologic parameters will no doubt dramatically change stratification and, subsequently, approaches to treatment. There is a growing consensus that medulloblastoma in older patients will likely be divided into at least three categories (see Table 83-5).
In younger children, separation into major risk groups is also accepted (Table 83-6) [Giangaspero et al., 2006; MacDonald et al., 2003; Packer et al., 1999a] Young children with extensively nodular/desmoplastic medulloblastoma, presumably whose tumors are driven by signaling of the sonic hedgehog pathway, have a better prognosis.
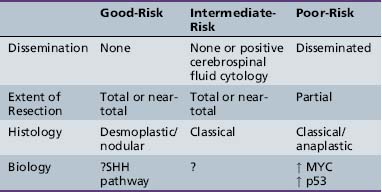
Standard treatment for children with average-risk disease, over 3 years of age, includes radiation and chemotherapy [Packer et al., 1994, 1999b, 2006]. Survival rates improved dramatically when craniospinal irradiation therapy became a standard component of medulloblastoma treatment, independent of whether there was evidence on staging studies of dissemination. Initial doses of craniospinal radiation were chosen arbitrarily; conventionally, following surgery, average-risk patients were treated with 3600 cGy of craniospinal radiation and 1800–1960 cGy of boost radiotherapy to the primary tumor site, which, in most patients, included the entire posterior fossa (local tumor dose 5400–5580 cGy). With such treatment, 5-year progression-free survival rates were between 55 and 65 percent, with some, but not all, 5-year survivors being cured of their disease [Hughes et al., 1988; Thomas et al., 2000]. Attempts to reduce the craniospinal dose of radiation therapy to 2340 cGy were considered unsuccessful because there was an increased early incidence of leptomeningeal disease failure; however, long-term follow-up of the cohort of patients treated on a randomized study comparing 3600 cGy to 2340 cGy did not show a statistical difference in 8-year survival in patients treated with lower-dose radiotherapy, as compared to those who received higher-dose treatment [Thomas et al., 2000].
The addition of chemotherapy has improved survival for children with average-risk medulloblastoma; treatment with craniospinal radiation therapy and chemotherapy, given during and after radiation therapy via agents including vincristine (given during radiation therapy) and CCNU, cisplatin, cyclophosphamide, and vincristine post radiotherapy, has demonstrated progression-free survival rates at 5 years in the 80–85 percent range, with few patients relapsing after 5 years [Packer et al., 1994, 1999b, 2006]. The sequencing of radiation and chemotherapy seems critical, as studies that have delayed radiation by the use of pre-radiation chemotherapy have reported poorer survival rates [Allen et al., 2009; Bailey et al., 1995; Kortmann et al., 2000] (Figure 83-6). In a recent prospective trial, the post-radiation combination of CCNU (N-(2-chloroethyl)-N-cyclohexyl-N-nitrosourea; Lomustine), cisplatin, and vincristine was found to be equivalent to a cyclophosphamide, cisplatin, and vincristine regimen [Packer et al., 2006]. Other regimens, including higher-dose regimens utilized over a shorter period of time with similar agents, have shown comparable survival figures [Gajjar et al., 2006].
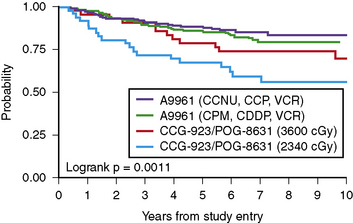
The addition of chemotherapy has allowed a reduction of the dose of craniospinal radiation therapy to 2340 cGy without any apparent deterioration in overall disease control in children with “average-risk” disease (see Figure 83-6) [Packer et al., 1999b, 2006]. This reduction was undertaken to reduce radiation-associated sequelae, and on-going prospective randomized studies are reducing the dose of craniospinal radiation therapy even further, to 1800 cGy [Goldwein et al., 1996]. An important caveat with these studies is that the staging of patients is of extreme importance. Upon central review in two international studies, approximately 20 percent of patients were inappropriately entered on these studies due to either misinterpretation of tumor extent or inadequate imaging [Oyharcabal-Bourden et al., 2005; Packer et al., 2006]. Patients who were placed on these reduced-dose craniospinal radiation studies with inadequate imaging or errors in interpretation had a significantly poorer outcome.
For children with poor-risk medulloblastoma, survival after craniospinal and local boost radiotherapy alone is only in the 30–40 percent range at 5 years [Evans et al., 1990; Hughes et al., 1988; Tait et al., 1990]. Studies that have added chemotherapy, once again primarily either during and/or after radiation therapy, have demonstrated survival rates ranging between 60 and 70 percent [Carrie et al., 2005; Gajjar et al., 2006; Gandola et al., 2009; Jakacki et al., 2004; Packer et al., 1994]. A variety of different chemotherapeutic approaches have been utilized and are still being studied, including the use of radiosensitization agents, such as carboplatin, during radiation therapy, and a variety of drugs after radiation therapy, in various combinations, including cisplatin, CCNU, cyclophosphamide, vincristine, and etoposide.
For infants and young children with medulloblastoma, treatment remains quite problematic. Due to concerns over the detrimental long-term effects of craniospinal radiation therapy, chemotherapeutic approaches have been used to delay, if not completely obviate, the need for radiotherapy [Ater et al., 1997; Duffner et al., 1993; Geyer et al., 2005]. With a variety of different regimens, including mechlorethamine, nitrogen mustard, procarbazine, and prednisone (MOPP), and cisplatin, cyclophosphamide, vincristine, and etoposide (augmented baby POG), approximately 20 percent of infants can be treated with chemotherapy alone [Ater et al., 1997; Duffner et al., 1993; Geyer et al., 2005]. These studies have now evolved into the use of postoperative chemotherapy and, for those patients who remain in remission or respond to the initial chemotherapy, consolidation with even higher doses of chemotherapy supported by autologous bone marrow transplantation or peripheral stem cell rescue [Dunkel and Finlay, 1996; Dupuis-Girod et al., 1996; Fisher et al., 1998; Mason et al., 1998]. Another approach to augmenting the efficacy of chemotherapy is to add methotrexate, in some studies intravenously at high dose, and in other trials both intravenously and intrathecally/intraventricularly, to improve survival and avoid the need for radiation [Rutkowski et al., 2005]. The infants most likely to survive are those with the nodular/desmoplastic form of the disease. Survival for children with non-nodular/desmoplastic disease is not as favorable, but still some of these children do survive after treatment with chemotherapy alone. The relative benefits of higher-dose chemotherapy with stem cell rescue or the addition of methotrexate have been difficult to discern, given that these treatments were not compared in prospective, randomized trials. In addition, there is renewed interest in reintroducing radiation therapy to the primary tumor site in these young children, especially for those patients without disseminated disease harboring tumors without desmoplastic/nodular histologic features. The added toxicities of methotrexate or focal radiation therapy have not been well studied. Treatment for infants and young children with disseminated disease remains suboptimal. Chemotherapeutic approaches alone have cured only a small group of those patients.
Relapsed medulloblastoma
Another extremely problematic group of patients to treat for medulloblastoma are those with relapsed disease. Given that the majority of older patients are now treated with both radiation therapy and multi-agent chemotherapy at time of initial diagnosis, most children over 3 years of age with relapsed disease are highly resistant to further therapy. Furthermore, approximately two-thirds of patients with medulloblastoma, be they high-risk, low-risk, or falling in the infant and young child category, will relapse with at least some component of disease outside the primary tumor site [Allen et al., 1987a; Friedman et al., 1986; Gaynon et al., 1990; Lefkowitz et al., 1990; Moghrabi et al., 1995]. For patients with disseminated disease at relapse, there are no proven effective retrieval regimens. For children who have relapsed only at the primary tumor site, possibly because they are biologically different, retrieval strategies have been somewhat more successful and have included attempts at re-resection followed by chemotherapy, predominantly with agents not previously seen or higher doses of previously utilized agents, followed by local radiotherapy. This approach has been most successful in young children who have not received prior radiation therapy.
Sequelae in medulloblastoma survivors
As more children survive medulloblastoma, it has become clearer that their quality of life is far from optimal [Armstrong et al., 2009; Gurney et al., 2003; Mulhern et al., 1989; Packer et al., 1989]. Although the dose of craniospinal radiation therapy has been reduced by almost one-third for patients with nondisseminated disease, it is yet unclear how beneficial such reductions will be. At the same time, while the radiotherapy dose has been decreased, there has been an increased use of chemotherapy, with its own inherent short- and long-term side effects [Bull et al., 2007]. In addition, the increased occurrence of the posterior fossa mutism syndrome negatively affects outcome [Rorke, 1983].
Neurocognitive sequelae are frequently seen in children of all ages treated for medulloblastoma [Kao et al., 1994; Mulhern et al., 1989, 1998; Ris et al., 2001]. It has been well documented that the majority of infant survivors are neurocognitively impaired at the time of diagnosis, probably due to multiple factors, including the tumor, associated hydrocephalus, and possibly the sequelae of surgery. Although radiation therapy has been avoided in some infants who survived medulloblastoma, follow-up testing demonstrates that the majority remain developmentally and, ultimately, neurocognitively impaired. The addition of potentially neurotoxic drugs, such as methotrexate, to the treatment of these children or the early use of focal radiotherapy may further cause neurocognitive issues.
In children with medulloblastoma who are between 3 and 7 years of age at the time of treatment, significant declines in overall intelligence have been demonstrated after 3600 cGy of craniospinal radiation [Mulhern et al., 1989, 1998; Ris et al., 2001]. This drop in intelligence can be seen as early as 1 year post treatment, is progressive, and may not plateau over time. Decline in intelligence in those children treated at full-dose radiation therapy has ranged between 20 and 30 IQ points within 3 years of treatment. Preliminary results suggest a lesser fall in overall intelligence in younger children who are treated with reduced doses of craniospinal radiation, but still a 10–20-point overall drop in global intelligence is likely. Older children have not demonstrated as severe a drop in global intelligence; however, these patients often have selective learning and attentional difficulties. Global intelligence is a crude estimate of intellectual function, and patients of all ages have been noted, in follow-up, to have selective sequelae, including memory difficulties, processing dysfunction, executive functioning abnormalities, visual-spatial difficulties, and attentional disorders. The attentional problems are often overlooked due to the lack of associated hyperactivity. Many of these children are hypoactive and studies are under way to determine if the use of stimulants or other agents will decrease fatigue and lethargy and improve attention.
Endocrinologic sequelae are the second most common long-term consequences of medulloblastoma treatment [Gurney et al., 2003; Packer et al., 2003; Shalet et al., 1987]. Growth hormone production is most commonly affected, but over time there can also be deficits in thyroid function, FSH-LH production, and adrenocortical function. Although there is hesitancy to utilize growth hormone replacement in children with cancer, studies in children with medulloblastoma have not shown an increased incidence of tumor relapse associated with its use [Packer et al., 2001]. Growth hormone insufficiency is exacerbated by the direct effects of radiation on vertebral growth, and many long-term survivors have disproportionate growth, with a decrease in truncal growth as compared to extremity growth. The effects on growth are also exacerbated by associated obesity, which is more common in long-term survivors.
Increasingly, secondary tumors have been reported in children surviving brain tumors, including medulloblastomas [Armstrong et al., 2009]. In a recent prospective Children’s Oncology Group study of 379 children, 14 secondary malignancies have occurred within 8 years of follow-up, including multiple malignant gliomas [Packer et al., 2006]. Since this patient population is less than 10 years from diagnosis, it is likely there will be an even greater rate of secondary tumor, especially the development of meningiomas over time.
Reports from the Children’s Cancer Survivor Study and others regarding children with medulloblastoma disclose that survivors are at higher risk, as compared to siblings, for focal neurologic deficits, strokes, neuropsychological deficits, and psychosocial deficits. They also had increased likelihood of early mortality [Armstrong et al., 2009; Neglia et al., 2006; Robison et al., 2005]. Survivors, at least those treated between 1975 and 1999 (the cohort initially studied in the 25-institution study), had limited abilities to live independently, have families, or be gainfully employed. Although the majority completed high school, often this was only possible with significant educational support for the majority, and only a minority attained higher degrees. It is hoped that, as the doses of radiation therapy are reduced for future patient populations, their quality of survival will improve. However, in one report, there was a suggestion that the addition of chemotherapy intensified the difficulties in some survivors [Bull et al., 2007]. All these results point to the need to develop less toxic treatments for children of all ages with medulloblastoma.
Supratentorial Primitive Neuroectodermal Tumors
Supratentorial primitive neuroectodermal tumors (PNETs) account for 2.5 percent or less of all childhood brain tumors [Cohen et al., 1995; Dirks et al., 1996; Yang et al., 1999]. They are reported to arise most commonly early in life, especially in infancy, although in a recent multicentered review, the median age at time of diagnosis (49.5 months) was found to be somewhat greater than in most series, and patients up to 15 years of age were diagnosed with the tumor [Johnston et al., 2008]. The male to female ratio has varied between studies, with a male predominance in most series.
Pathologically, supratentorial PNETs are usually relatively well circumscribed. Microscopically, they are composed of embryonal-appearing small round cells with hyperchromatic oval nuclei [Burger, 2007]. Tumors are mitotically active and frequently show areas of necrosis. Parts of the tumor may also show Homer–Wright rosettes or pseudorosettes; foci with various degrees of apparent cellular differentiation may also be present. When the tumor has a prominent connective tissue component or displays a nodular pattern, it has also been termed a central neuroblastoma.
The biology of supratentorial PNET is not as well elucidated as for medulloblastoma, although the tumor is biologically distinguishable from medulloblastoma [Pomeroy et al., 2002; Russo et al., 1999]. Loss of 14q and 19q is detected in up to 40 percent of supratentorial PNET. Unlike the situation in medulloblastoma, gains of 17q are uncommon [Inda et al., 2005; McCabe et al., 2006; Russo et al., 1999].
Clinical presentation and diagnosis
Clinical presentation tends to be relatively abrupt, with the majority of patients having focal neurologic deficits, including hemiparesis or visual field difficulties, at the time of diagnosis [Johnston et al., 2008; Yang et al., 1999]. Because of the proclivity of these tumors to arise early in life, it can be difficult to determine how long symptoms have been present, but most series suggest that the majority of patients have symptoms for less than 1 month prior to diagnosis. Overall, parent-perceived head pain, vomiting, and nausea are the most common symptoms early in illness, with one-quarter to one-third of patients having seizures prior to diagnosis.
Neuroimaging studies most commonly reveal a large mass with well-defined borders, which may arise cortically or in the deep periventricular white matter [Dai et al., 2003]. The tumor is heterogeneous on both CT and MRI, and two-thirds demonstrate cystic and/or necrotic foci (Figure 83-7). The solid portions are hyperdense on CT. Fifty percent of patients have some degree of calcification, and hemorrhage is seen in 20 percent. The solid component on MRI is usually isointense on T1-weighted images and hyperintense on T2-weighted images. Despite the large size of the tumor, minimal peritumoral edema often is seen. The primary difficulty in differential diagnosis is distinction from supratentorial atypical teratoid/rhabdoid tumors.
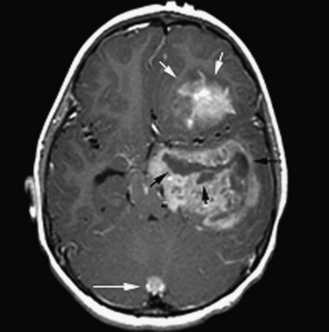
Management and outcome
As is the case for medulloblastoma, children with supratentorial PNETs are usually staged with neuroimaging of the entire neuroaxis prior to diagnosis [Albright et al., 1995; Cohen et al., 1995; Johnston et al., 2008]. Because of the cortical location of these tumors, lumbar cerebrospinal cytological examination may be impossible to obtain because of potential herniation. The incidence of dissemination is not well documented, but seems less than that seen in medulloblastoma, occurring in probably 10–15 percent of patients.
The initial step in treatment is surgical, and the degree of surgical resection has been related in some, but not all, studies to survival [Albright et al., 1995; Johnston et al., 2008; Pizer et al., 2006]. If possible, gross total surgical resection is the objective, but may not be possible in large vascular tumors invading functional areas of cortex.
Following surgery, radiation therapy is usually utilized [Chintagumpala et al., 2009; Gilheeney et al., 2008; McBride et al., 2008; Reddy et al., 2000; Taylor et al., 2009]. However, given the young age of the children and the cortical location of the tumor, such radiation therapy can be quite damaging. Treatment after radiation therapy alone provides long-term disease control in less than half of patients. There is some suggestion that the addition of chemotherapy to radiation therapy improves survival, and a variety of different chemotherapeutic approaches have been utilized, with survival rates ranging from 40 to 70 percent at 5 years [Gilheeney et al., 2008; Reddy et al., 2000]. In those patients without disseminated disease at the time of diagnosis, it seems that the craniospinal dose of radiation therapy can be reduced to 2400 cGy, as has been the experience for children with medulloblastoma. However, the local doses of radiation therapy often required will include large areas of the cortex at high doses. Infants with supratentorial PNETs have been treated with chemotherapy postoperatively – usually as part of clinical trials developed for infants with high-grade tumors, primarily medulloblastoma. In adjuvant chemotherapy studies, survival rates have ranged between 20 and 40 percent; the independent efficacy of chemotherapy may be difficult to determine as, in many of these trials, at least focal radiation therapy has been added following completion of the chemotherapy.
Atypical Teratoid/Rhabdoid Tumors
Atypical teratoid/rhabdoid tumors (AT/RTs) were first described clearly as a distinct entity by Rorke and colleagues in 1987 [Packer et al., 2002b; Rorke et al., 1995]. Initially diagnosed on the basis of distinct histologic and immunohistochemical appearances, cytogenetic and molecular genetic diagnosis is now the rule rather than the exception.
The tumor is defined by its distinctive combination of histological, cytogenetic, and molecular genetic features (Figure 83-8) [Burger et al., 1998; Rorke et al., 1995]. Although AT/RTs may exhibit considerable morphologic variability, the most common finding is a combination of rhabdoid cells intermixed with other regions of tumor resembling PNETs. The rhabdoid cells, which are large and have a “ground glass” appearance, express epithelial membrane antigen and vimentin and, somewhat less often, smooth muscle actin, neurofilament protein, glial fibrillary acidic protein, and keratin.
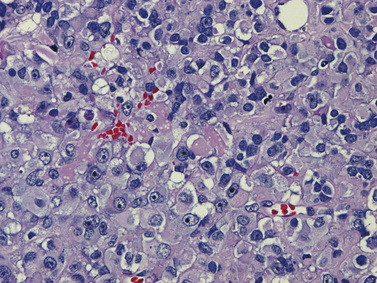
Work by Biegel and colleagues demonstrated that, in the majority of patients, the tumors show loss of chromosome 22 or partial deletion of 22q 11.2, which contains the INI1/hSNF5 (SMARCB1) gene; this gene encodes a component of the SWI/SFN (Switch/Sucrose nonfermentable) chromatin remodeling complex [Biegel et al., 2002b; Burger et al., 1998; Gessi et al., 2003; Judkins et al., 2004; Rickert and Paulus, 2004; Versteege et al., 1998; Wharton et al., 2003]. It is believed that this gene has multiple properties, including being a tumor suppressor gene and modifying the transcription of cellular genes implicated in tumorigenesis.
Germline mutations that cause inactivation of the INII/hSNFS gene in children cause multiple neoplasms, including rhabdoid neoplasms of the kidney [Biegel et al., 2000; Lynch et al., 1983; Sevenet et al., 1999a, b]. Chromosome 22 monosomy has also been reported in meningiomas and in acoustic neuromas in patients with neurofibromatosis type 2. Germline mutations were initially considered to be quite infrequent in patients with AT/RTs, but recent studies suggest that up to 50 percent of patients may have germline mutations. This raises the issue of whether parents or siblings of children with AT/RTs should be screened for the presence of a germline mutation and, if found, siblings should undergo screening for the presence or development of kidney or intracranial tumors.
Clinical presentation and diagnosis
Clinical presentation is not dramatically different from that of other embryonal tumors occurring in infants and young children [Lefkowitz et al., 1987; Rorke et al., 1995]. The tumor tends to present relatively explosively, and most patients are diagnosed within 1 month of onset of symptoms. There is a predilection for the cerebellopontine angle, and often AT/RTs cause multiple cranial nerve palsy, especially sixth and seventh cranial nerve palsies.
The radiologic features of AT/RTs are characteristic but are not absolutely diagnostic. AT/RTs tend to be hyperdense on CT and enhance intensely [Meyers et al., 2006; Rorke et al., 1995]. Calcifications may occur. On T1-weighted MRI there are often hyperintense foci within the lesion due to hemorrhage, and the tumors are heterogeneous on T2-weighted images. Most AT/RTs enhance with contrast agents (Figure 83-9). A helpful diagnostic feature is the predilection for AT/RT to present in the cerebellopontine angle, occurring in up to 50 percent of cases of posterior fossa AT/RTs. Intratumoral hemorrhage is greater in AT/RTs than in other posterior fossa tumors. Lack of or minimal enhancement is only evident in approximately 10 percent of tumors. Distinction between supratentorial PNET and AT/RTs is quite difficult.
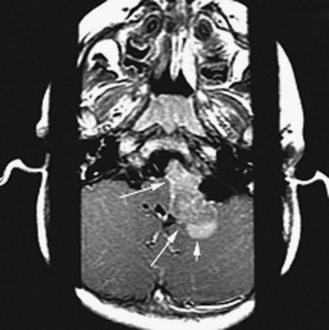
Management and outcome
The management of AT/RTs is evolving, but outcome is poor overall, especially in younger patients [Hilden et al., 2004; Meyers et al., 2006; Packer et al., 2002b; Rorke et al., 1995]. Initial studies in patients less than 2 years of age, especially less than 1 year of age, suggested that the tumor was uniformly fatal. This is not the case, but children less than 1 year of age fare very poorly.
Surgery remains the initial component of treatment for AT/RTs and gross total resections have been associated with a higher likelihood of long-term survival [Chi et al., 2009; Hilden et al., 2004; Packer et al., 2002b]. However, series have reported a low incidence of gross total resection, especially in infants with posterior fossa tumors.
A variety of different chemotherapeutic approaches have been utilized in children less than 3 years of age, primarily those that have been used for infants with medulloblastoma [Hilden et al., 2004; Packer et al., 2002b]. Some approaches now incorporate methotrexate and also utilize high-dose chemotherapy with peripheral stem cell rescue as consolidation. AT/RTs may also be sensitive to drug regimens used for children with sarcomas. An approach that has shown promise is essentially a combination of protocols used for infantile brain tumors with those used for children with sarcomas (which contain anthracyclines) [Chi et al., 2009; Tekautz et al., 2005]. Drug regimens are also now being coupled with earlier use of radiotherapy, especially local radiotherapy, for patients with nondisseminated disease. Survival rates as high as 70 percent have been reported in older children with AT/RTs after the use of aggressive chemotherapy and either local or local plus craniospinal radiation therapy [Chi et al., 2009; Squire et al., 2007; Tekautz et al., 2005].
Other Embryonal Tumors/Medulloepitheliomas and Ependymoblastomas
Both medulloepitheliomas and ependymoblastomas are extremely rare tumors that tend to occur in infants [Molloy et al., 1996]. The medulloepithelioma is characterized by a histologic appearance that mimics the embryonal neural tube [Molloy et al., 1996; Norris et al., 2005]. It is characterized by papillary or tubular neoplastic neuroepithelium with an external limiting membrane. Mitotic figures tend to be abundant and immunohistochemical studies demonstrate primitive staining for both nestin and vimentin. Medulloepithelioma comprises less than 1 percent of childhood brain tumors and may be congenital in origin. Despite its primitive nature, the mean age of diagnosis in series has been as high as 3–4 years. The tumor may occur in both the supratentorial and infratentorial spaces and tends to have a predilection for the cerebral hemispheres, especially periventricular regions. The tumor may also involve the orbit.
Ependymoblastomas are possibly even rarer than medulloepitheliomas, and recently it has been questioned whether they represent a distinct diagnostic entity [Mork and Rubinstein, 1985; Rubinstein, 1970]. Ependymoblastomas are characterized histologically by regions of ependymoblastic, multilayered rosettes, with sheets of small hyperchromatic cells [Judkins and Ellison, 2008; Lopez-Aguilar et al., 2007]. Histological features may overlap with those of other brain tumors, especially the recently reported “pediatric tumor with abundant neurophil and true rosettes.”
On neuroimaging, ependymoblastomas tend to be indistinguishable from other embryonal tumors [Dorsay et al., 1995]. They are commonly large and well circumscribed, with occasional cystic changes. Calcifications, necrosis, and hemorrhage may be present. Most tumors readily enhance with contrast and have associated peritumoral edema.
Management and outcome
The management of medulloepitheliomas and ependymoblastomas is similar to that of other infantile primitive embryonal tumors, and outcome is quite poor [Mork and Rubinstein, 1985; Norris et al., 2005]. Staging studies are usually undertaken, although, because of the rarity of these tumors, the incidence of dissemination is difficult to discern. In both tumors, there is a suggestion that total resections are related to a higher likelihood of long-term control. The use of radiotherapy is problematic in both tumor types because of the young age of the patients. Data concerning the efficacy of chemotherapy are essentially nonexistent, and the treatment protocols utilized for high-risk medulloblastomas are most often employed.
Glial-Astrocytic Tumors
Glial-astrocytic neoplasms comprise over 50 percent of all childhood brain tumors (Box 83-2). The majority are low grade – WHO 1 and 2 – and 20 percent or fewer are malignant [Qaddoumi et al., 2009]. Microscopically, juvenile pilocytic astrocytoma (JPA; WHO grade 1) demonstrates a biphasic pattern of fusiform astrocytes loosely interwoven with a fine fibrillary background and no, or rare, mitoses (Ki-67/MIB-1 labeling index of 0–4 percent) (Figure 83-10). A microcystic component and the presence of Rosenthal fibers, degenerative changes in astrocytes that appear as bright eosinophil bodies, are common. Vascular proliferation may be extensive, especially along the walls of the tumor cysts. Diffuse fibrillary astrocytomas (WHO grade 2) are composed of moderately increased numbers of infiltrating, neoplastic astrocytes with scant cytoplasm and nuclear atypia, but with little or no mitoses. By convention, pediatric glial tumors, especially low-grade tumors, are classified by location, as well as histology.













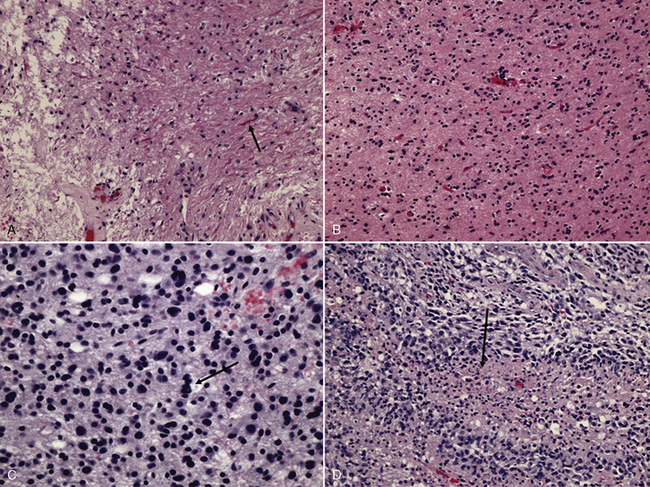
Cerebellar Astrocytomas
Cerebellar astrocytomas are the single most common tumor of the posterior fossa, comprising 40 percent of all posterior fossa tumors. The majority are JPAs, but up to 15 percent are diffuse [Qaddoumi et al., 2009]. Solid, midline tumors are more likely to be diffuse.
Clinical presentation and diagnosis
Cerebellar astrocytomas are commonly hypodense on CT, and hypointense on T1-weighted MRI and hyperintense on T2-weighted MRI (Figure 83-11). Calcification is a common finding. JPA frequently have a cystic component and, typically, the solid tumor portion or the mural nodule demonstrates prominent enhancement.
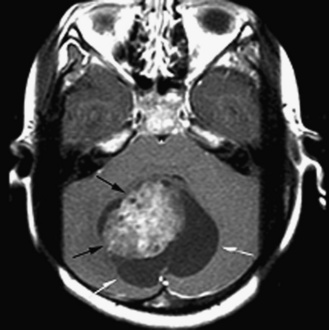
Management and outcome
Management of cerebellar astrocytomas is predominantly complete surgical excision [Sievert and Fisher, 2009]. Patients may be treated for pre-existing hydrocephalus with placement of a ventricular drain prior to or at the start of surgery. However, since more than 90 percent of astrocytomas are completely resectable, an attempt to defer shunt placement is often made. In the patient eventually requiring cerebrospinal fluid diversion, placing an endoscopic third ventriculostomy may avoid the need for long-term shunting. It is imperative to remove the solid tumor nodule, whereas removal of the cyst wall is not required. Postoperative care requires close observation for the development or progression of hydrocephalus, as well as supportive care for any post-resection morbidity involving brainstem or cranial nerves. Cerebellar mutism (posterior fossa mutism syndrome) has been reported in some patients following surgery, but is less common than after surgery for medulloblastoma. Most studies have shown close to 100 percent survival at 20 years after gross total resections.
Optic Pathway-Hypothalamic Gliomas
Optic pathway-hypothalamic gliomas (OPHG) constitute approximately 5 percent of all pediatric central nervous system tumors and invariably are either JPA or diffuse fibrillary astrocytomas; the majority are JPA, especially those localized to the optic nerve and chiasm [Qaddoumi et al., 2009]. Over 75 percent of isolated optic nerve gliomas occur in the first decade of life. Neurofibromatosis type 1 is present in 50–80 percent of patients with isolated optic nerve tumors, and in 15–20 percent of those with optic pathway tumors. Microscopically, these tumors have the appearance of JPA and other low-grade gliomas in different locations of the central nervous system.
Clinical prognosis and diagnosis
Generally, OPHG are homogeneously hypodense on CT, isointense on T1-weighted MRI, and hyperintense on T2-weighted MRI (Figure 83-12). Solid tumor portions commonly demonstrate diffuse contrast enhancement, although larger tumors may contain nonenhancing cystic elements, giving a more heterogeneous appearance.
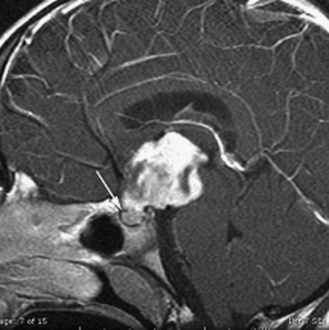
Management and outcome
Radiotherapy results in visual stabilization in the majority of patients with visual pathway gliomas; however, visual improvement after radiotherapy has been reported in a variable proportion of patients ranging between 9 and 44 percent [Jahraus and Tarbell, 2006]. Irradiation of large chiasmatic/hypothalamic lesions usually results in disease stabilization, with 5- and 10-year progression-free survival rates ranging between 70 and 90 percent. The doses of radiotherapy required (45–55 Gy) will result in significant endocrinologic deficits in the majority of patients and may have other long-term deleterious effects, including the risk for the development of vascular malformations and significant neurocognitive sequelae, especially in very young children [Jahraus and Tarbell, 2006]. Recent results using conformal radiation to lessen radiation-induced side effects are encouraging, although still not optimal in those less than 5 years of age [Merchant et al., 2009]. To delay, if not obviate, the need for radiotherapy, chemotherapy has been used in children with progressive optic pathway gliomas [Gnekow et al., 2004; Mahoney et al., 2000]. The combination of carboplatin and vincristine, or carboplatin alone, has been employed most commonly, and has been shown to result in disease stabilization in over 80–90 percent of patients with progressive lesions, and radiographic tumor shrinkage in 30–60 percent; however, approximately 60 percent of patients will eventually progress and require alternative therapy. Other chemotherapeutic regimens may be as effective; however, their use may cause more potential short- and long-term toxicities. Major concerns include potential mutagenesis, especially in children with neurofibromatosis type 1, and ototoxicity in children with already impaired vision if cisplatin is used. There is concern about the fact that infants less than 1 year of age tend to have more aggressive disease and may require more intensive therapy.
For children with neurofibromatosis type 1, the natural history of tumors of the optic nerve or visual pathway is erratic [Listernick et al., 1995]. The majority of asymptomatic children diagnosed on screening examinations do not require immediate treatment. Management includes careful neurologic, visual, endocrinologic, and neuroradiographic follow-up and treatment, only if there is clear evidence of tumor progression. Because of the potential risks of radiotherapy, patients with neurofibromatosis type 1 are initially treated with chemotherapy.
Cortical Gliomas
Up to one-half of cortical gliomas are located in the cerebral hemispheres and the remainder in deep midline structures of the diencephalon and basal ganglia [Pollack, 1999]. The majority of these are low-grade, but 20–25 percent will be malignant; malignant gliomas are somewhat more common in late childhood, but may also occur in infancy.
Clinical presentation and diagnosis
The major clinical signs at diagnosis are nonspecific and nonlocalizing features related to increased intracranial pressure, occurring in up to 75 percent of patients regardless of tumor histology and location [Ciurea et al., 1997; Wisoff et al., 1998]. Seizures, most frequently grand mal, are present at diagnosis in at least 25 percent of patients. Although as many as one-third of patients with high-grade tumors have seizures, the frequency is higher in the more slowly evolving low-grade tumors, where they may precede diagnosis by months to years.
The more specific signs relate to tumor location. Midline-diencephalic tumors may present with the classic diencephalic syndrome [Gropman et al., 1998]. These tumors may also lead to rapid obstruction of CSF flow because of their location, and thus may present more acutely than hemispheric gliomas. Focal motor deficits and pyramidal tract findings, such as weakness, monoplegia, and hemiplegia, occur in up to 40 percent of patients. Basal ganglia tumors may be associated with dysmetria and chorea. Hypothalamic tumors may be associated with neuroendocrine abnormalities; growth hormone deficiency, diabetes insipidus, and precocious pubertal development in up to one-third of patients. These tumors may also impinge on the optic chiasm, resulting in optic atrophy and visual field loss.
Most cortical low-grade astrocytomas have a CT and MRI appearance similar to that observed in other locations of the CNS (Figure 83-13), while high-grade astrocytomas are more heterogeneous on CT, with more diffuse contrast enhancement, and more typically display mass effect. MRI shows similar findings but in general reveals associated peritumoral edema more clearly (Figure 83-14).
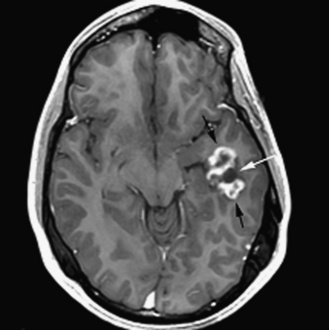
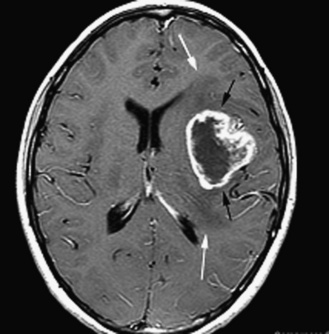
Management and outcome
High-Grade Glioma
Radical (>90 percent) surgical resection is the most important predictor of favorable outcome in high-grade glioma when followed by irradiation [Wisoff et al., 1998]. However, only 49 percent of high-grade glioma in the superficial hemisphere and 8 percent in the midline or deep cerebrum is amenable to radical resection [Wisoff et al., 1998]. Local (2- to 4-cm margin around the area of edema defined by imaging) or wide-field irradiation to 50–60 Gy is the mainstay of therapy. The addition of radiation therapy has improved 5-year survival rates (10–30 percent) compared to surgery alone (0 percent) [Marchese and Chang, 1990]. All patients are thus considered candidates for radiotherapy following surgery, with the exception of very young children, in whom attempts have been made to eliminate or delay the use of radiotherapy by utilizing chemotherapy because of concern regarding neurodevelopmental morbidity.
To date, no large randomized prospective clinical trial has clearly demonstrated a benefit of adjuvant chemotherapy. For infants with high-grade gliomas, treatment with chemotherapy alone has been successful in a small subset of patients and suggests that infantile malignant gliomas may be biologically different from those occurring later in childhood [Dufour et al., 2006]. Early phase II and III trials with single-agent and multi-agent chemotherapy demonstrated little or no impact on the overall survival, despite showing objective response rates as high as 45 percent [Finlay et al., 1995; MacDonald et al., 2005; Sposto et al., 1989]. Patients may have significant neurocognitive, neuroendocrine, and other neurologic deficits as a result of the tumor’s location and the secondary effects of surgery, irradiation, and chemotherapy.
Low-Grade Glioma
Complete surgical resection is curative for most low-grade gliomas, and even with incomplete excision, long-term, progression-free survival is common [Pollack et al., 1995]. If subsequent progression occurs, then re-resection is generally undertaken first. Surgical morbidity depends largely on tumor location and is highest in diencephalic tumors, in which the incidence of postoperative hemiparesis or visual field deficits may be 10–20 percent [Pollack et al., 1995]. Although gross total excisions are possible in up to 90 percent of hemispheric tumors, fewer than 40 percent of diencephalic tumors are similarly resectable [Pollack et al., 1995]. For patients with progressive disease not amenable to resection, local conformal irradiation to 50–55 Gy to the area of the tumor plus a 2-cm margin is warranted. The use of chemotherapy, as initial treatment in newly diagnosed low-grade glioma, has become a standard approach in young children and infants, in whom the goal is to avoid radiation neurotoxicity [Packer, 2000]. The use of such therapy in older patients is questionable since outcome is generally good with surgery and irradiation. The agents generally include an alkylating or platinating agent, such as carboplatin in combination with vincristine. The objective response rate with this combination is as high as 50–60 percent [Packer, 2000]. Overall 5-year survival for low-grade cortical glioma is 95 percent, while progression-free survival is 88 percent [Packer, 2000; Qaddoumi et al., 2009].
Brainstem Glioma
Brainstem gliomas comprise between 10 and 15 percent of all childhood primary central nervous system tumors [Qaddoumi et al., 2009]. Brainstem gliomas most commonly arise in the pons (diffuse intrinsic pontine glioma), in which location they typically act like adult glioblastomas multiforme and have an almost uniformly dismal prognosis [Frazier et al., 2009]. In contrast, those arising from midbrain or medulla are likely to be focal low-grade gliomas that have a more indolent course and improved outcome.
Clinical presentation and diagnosis
Diagnosis is by neuroradiographic imaging. On CT, appearance is variable, but most are isodense or hypodense, lack calcification, and enhance poorly. On MRI, the tumors are hypointense on T1- and hyperintense on T2-weighted imaging (Figure 83-15).
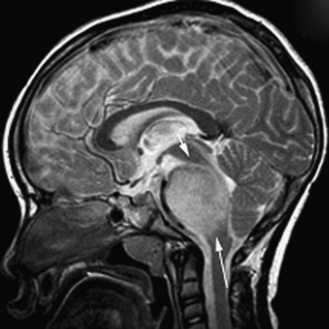
Management and outcome
Due to the severe neurologic compromise that patients with brainstem gliomas have at the time of diagnosis and during their disease, corticosteroids are often utilized in an attempt to improve neurologic function. Although corticosteroids may improve neurologic function temporarily, chronic use is associated with significant side effects and they should be tapered early in the course of treatment, if possible. For diffuse intrinsic pontine glioma, biopsy and partial resection are not warranted due to the risk of morbidity, because such interventions have not been shown to affect the natural history of the disease, and because the information obtained at the time of biopsy usually does not change management [Frazier et al., 2009]. The treatment for diffuse intrinsic pontine glioma is local irradiation to 55–60 Gy. Over 90 percent of patients with diffuse intrinsic pontine glioma respond transiently, and over half will have reduction in tumor size, but ultimately patients succumb to disease progression within 18 months of diagnosis [Frazier et al., 2009]. Neither hyperfractionated radiotherapy nor chemotherapy has been shown to add benefit.
Focal lesions within the pons, especially cystic ones presenting with isolated cranial nerve palsies, are often pilocytic astrocytomas (Figure 83-16) [Frazier et al., 2009]. Biopsy and cyst drainage, followed by focal radiotherapy, can result in prolonged disease control for many patients. Patients with tectal tumors may require no treatment other than cerebrospinal fluid diversion for many years after diagnosis (Figure 83-17). Biopsy is usually not required for diagnosis in these patients, and if there is progression, treatment with either chemotherapy (in very young children) or focal radiotherapy is usually effective. For patients with cervicomedullary lesions, especially those that are dorsally exophytic, usually the first treatment modality utilized is surgery. Although such lesions are often amenable to extensive resections as they are commonly pilocytic astrocytomas, such resections may cause significant neurologic morbidity. Alternatively, patients with cervicomedullary pilocytic astrocytomas may be treated successfully with partial surgical resection followed by either local radiotherapy or chemotherapy. The low-grade gliomas of the brainstem are treated with similar irradiation doses to other low-grade gliomas, but overall respond less favorably than their counterparts in other central nervous system locations.
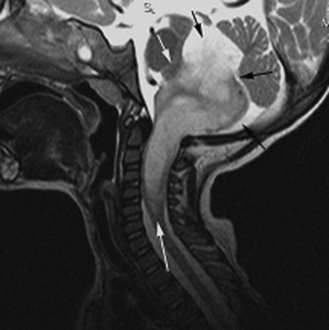
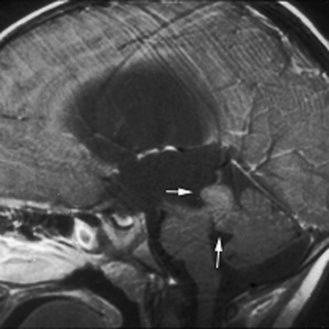
Gliomatosis Cerebri
Clinical presentation and diagnosis
Due to the diffuse infiltrating nature of the tumor and the various locations of the central nervous system that may be involved, signs and symptoms are quite variable and often remarkably subtle. Nonspecific symptoms, such as headaches, nausea, and lethargy, may be present early in disease [Jennings et al., 1995]. Later in illness, seizures, focal motor deficits, emotional disturbance, sensory abnormalities, and visual field deficits may occur. Neuroimaging usually demonstrates a diffuse infiltrating process in the lower-grade form of the disease. There is relatively minimal mass effect and the tumor, on MRI, may be isointense on T1-weighted images, and hyperintense on T2-weighted or FLAIR sequences. Areas of necrosis and enhancement are more common in higher-grade lesions, especially when the tumor meets glioblastoma multiforme criteria. Edema is more common in higher-grade gliomatosis cerebri.
Management and outcome
Given the diffuse infiltrative nature of the tumor, surgery is usually limited to confirm the diagnosis and grade the lesion. Sampling error may be an issue. Following surgery, radiation therapy is most commonly employed, with variable results. In low-grade forms of gliomatosis cerebri, radiation therapy may result in remission for months to years; however, malignant transformation is common. Few children with high-grade gliomatosis cerebri survive for longer than 18–24 months [Jennings et al., 1995]. Chemotherapy is often employed as an adjuvant to radiotherapy, but there is little evidence for increased efficacy.
Pleomorphic Xanthoastrocytoma
Pleomorphic xanthoastrocytomas are a rare form of astrocytic tumor that is most common in children and young adults [Giannini et al., 1999]. Two-thirds of patients with this tumor present before 18 years of age. The tumor tends to be superficial and attached to the meninges, with an infiltrating cortical component. There is a compact, fascicular or lobulated architecture with cellular pleomorphism, some degree of mitotic activity, and xanthomatous change. The histology can be quite variable and pleomorphic xanthoastrocytomas are usually classified as a WHO grade 2 lesion, but may act more aggressively. When there is increased mitotic activity, it is considered to be anaplastic.
Management and outcome
In the majority of patients, complete tumor resection is associated with an excellent outcome, with survival rates of 70–80 percent being reported at 5–10 years [Giannini et al., 1999; Kepes et al., 1979]. However, given the variability in histology and aggressivity, disease will recur in some patients. A benefit for radiation therapy or chemotherapy in patients with anaplastic lesions has not been well documented.
Astroblastoma
Astroblastomas are extremely rare glial neoplasms that tend to present in young adults but may also be diagnosed in children [Bonnin and Rubinstein, 1989; Thiessen et al., 1998]. The tumor is histologically marked by an atypical perivascular pseudorosette pattern of glial fibrillary acidic protein (GFAP)-positive cells with broad, nontapering processes radiating towards a central blood vessel. Similar histologic patterns can be seen in focal areas of low-grade and high-grade astrocytomas and in ependymomas.
Ependymoma
Ependymomas most frequently occur in young children, with more than half of the cases diagnosed before 5 years of age [Kun et al., 1988]. They constitute approximately 10 percent of all childhood brain tumors. Unlike the situation in adults, tumors in childhood are more likely to occur in the posterior fossa at a ratio of 3–4:1, as compared to supratentorial tumors. There is even a greater predilection for the posterior fossa in children less than 3 years of age. Ependymomas occur anywhere along the ventricular system and spinal cord. They are believed to arise from radial glial embryonic cells, which are neural progenitor cells. Ependymomas of the posterior fossa are genetically distinct from those of the cortex; spinal tumors are also likely molecularly different.
Clinical presentation and diagnosis
These tumors typically present with nonspecific and nonlocalizing signs and symptoms related to increased intracranial pressure. Infratentorial tumors may display cerebellar dysfunction and multiple lower cranial nerve findings, especially VI, VII, VIII, IX, and X [Kun et al., 1988]. Supratentorial tumors may present with seizures and focal cerebral deficits.
Although appearance varies, ependymomas are generally hyperdense and homogeneously enhance on CT. On MRI, tumors are iso- to hypodense on T1-weighted imaging and hyperdense on T2-weighted imaging (Figure 83-18).
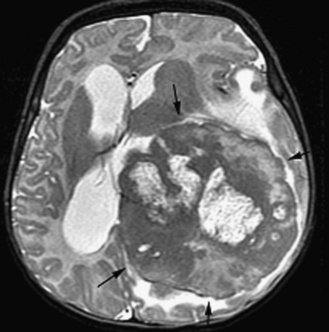
Management and outcome
Survival rates are clearly better for patients who have undergone complete surgical resections. However, complete resection is achieved in only 40–60 percent of patients [Bouffet et al., 1998a; Kun et al., 1988]. It is more difficult to attain a gross total or near-total resection with infratentorial tumors than in supratentorial tumors, as the former have a tendency to infiltrate the brainstem and entrap the lower cranial nerves. In addition, the 20–40 percent progression rate for patients without residual disease on postoperative imaging suggests that a significant number of tumors have extensive microscopic residual disease [Bouffet et al., 1998a]. Progression in almost all cases is local, with distant relapse occurring in less than 10 percent of cases [Bouffet et al., 1998a]. The possibility of local recurrence and success rates following complete resection have led to the concept of second-look surgery for tumors that have been incompletely resected.
Local radiotherapy to 5000–5500 cGy for supratentorial tumors and 5500–5900 cGy for infratentorial tumors remains the standard postoperative treatment for nondisseminated ependymomas. In older comparisons of patients treated with surgery alone or surgery plus radiation therapy, the survival rates were 17 and 40 percent, respectively [Garrett and Simpson, 1983]. More recent trials suggest a 70 percent or greater 5-year survival rate after gross total resections and conformal radiotherapy in patients with nonanaplastic tumors [Merchant et al., 2004b]. Craniospinal irradiation is recommended for those with tumor metastasis. Prophylactic craniospinal radiotherapy is no longer recommended, regardless of site and histology of the primary tumor, due to the relative rarity of distant relapse [Merchant et al., 1997].
Despite several trials demonstrating measurable disease responses, the role of chemotherapy in the treatment of ependymoma has not been established. In single-agent trials, platinum compounds have been the most active [Bouffet et al., 1998a]. To date, only one prospective randomized trial evaluating the role of chemotherapy in childhood ependymoma has been completed. The single prospective randomized trial compared postoperative craniospinal radiation to radiation in combination with chemotherapy consisting of vincristine, lomustine, and prednisone. The 5-year survival was 45 percent in patients receiving chemotherapy plus radiation therapy, and 35 percent in patients receiving radiation therapy alone; the difference was not statistically significant [Evans et al., 1996; Bouffet et al., 1998a]. Preliminary studies do support the potential use of chemotherapy in infants in an effort to delay radiation therapy, or in children with incompletely resected tumors as an adjunct to second-look surgery prior to radiation therapy. Objective response rates up to 48 percent and 2-year progression-free survival of 42 percent have been demonstrated with cyclophosphamide, cisplatin, etoposide, vincristine, and deferred irradiation in the infant population [Bouffet et al., 1998a].
Quality of life is variable for patients surviving ependymomas, although, after focal radiotherapy, little drop in overall intelligence has been noted [Merchant et al., 2004b]. For patients with infratentorial tumors, multiple lower cranial nerve palsies and long-term neurologic impairment may occur. At times, the degree of impairment in these patients can be so severe as to warrant the placement of a tracheostomy and gastric feeding tube. Patients with supratentorial tumors located in eloquent areas may sustain significant neurocognitive, neuroendocrinologic, and focal neurologic deficits as a result of surgery and radiotherapy.
Neuronal and Mixed Neuronal-Glial Tumors
Neuronal and mixed neuronal-glial tumors comprise a number of different tumors that may be diagnosed in childhood (Box 83-3). The most common neuronal-glial tumors are gangliogliomas, which may arise anywhere within the central nervous system but are most common in the temporal lobes. Other neuronal and mixed neuronal-glial tumors are rarer but may comprise a significant proportion of infantile tumors; these include the desmoplastic neuroepithelial tumor and the desmoplastic infantile astrocytoma/ganglioglioma. Dysplastic gangliomas may mimic more common tumors of the central nervous system. More recently, a term that has been utilized by some pathologists has been “neuroglial tumor,” for which there is limited outcome information. By definition, all neuronal-glial tumors have clear-cut glial and neuronal elements. Immunohistochemistry demonstrating avid staining with synaptophysin has greatly simplified diagnosis.






Dysembryoplastic Neuroepithelial Tumor
Dysembryoplastic neuroepithelial tumors (DNETs) are benign tumors that can occur in both children and adults [Daumas-Duport et al., 1988]. DNET is considered a glial-neuronal tumor, and some have suggested that it has a significant oligodendroglioma component. The tumors vary greatly in size and can be found in essentially any part of the cortex. DNETs have been most commonly described in the temporal lobe. Occasionally, deeper-seated DNETs have been seen. The tumor is characterized by columns that are formed by bundles of axons lined by small, possibly oligodendroglial, cells. These columns are oriented perpendicularly to the cortical surface. Between the columns, neurons are seen, and occasionally astrocytes. More complex variants can also be seen.
Clinical presentation and diagnosis
Although DNETs can be seen in patients of all ages, the majority present with seizures occurring in patients younger than 20 years of age [Daumas-Duport, 1993; Luyken et al., 2003]. Diagnosis has been made in infancy, although it is more common, in the pediatric patients, in the second decade of life. DNETs are increasingly diagnosed in epilepsy centers, where they comprise as many as 30 percent of all structural lesions operated upon for children with intractable seizures.
On neuroimaging, DNETs are usually seen as fairly well-defined nodules without mass effect or edema [Ostertun et al., 1996]. The cortical location of these tumors is better appreciated on MRI than on CT. DNETs are usually hypointense on T1-weighted images and hyperintense on T2-weighted images (Figure 83-19). The tumors enlarge the involved gyri and also often have a multicystic appearance. Scalloping of the overlying calvarium can also be seen. Contrast enhancement is variable, with one-third or less of tumor showing contrast enhancement.
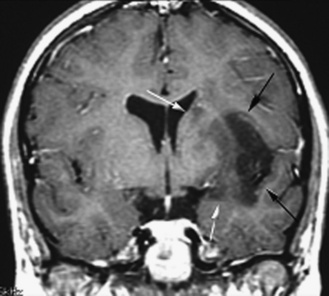
Giant Cell Astrocytoma/Subependymal Giant Cell Astrocytoma
Subependymal giant cell astrocytomas (SEGAs) occur primarily in patients with tuberous sclerosis [Shepherd et al., 1991]. By definition, they are slow-growing tumors arising in the wall of the lateral ventricles. SEGAs occur in approximately 6–16 percent of patients with tuberous sclerosis. They have been reported in patients without tuberous sclerosis, although it has been questioned whether those patients have a forme fruste of the disease. SEGAs are usually well-circumscribed, calcified tumors composed of large, plump cells resembling astrocytes. On immunohistochemical analysis, these tumor cells show both glial and neuronal immunoreactivity, and it has been suggested that the tumor is best labeled as a subependymal giant cell tumor rather than an astrocytoma.
Clinical presentation and diagnosis
The tumors are radiographically seen as bulky contrast-enhancing lesions with varying degrees of calcification [Shepherd et al., 1995]. There may be significant hydrocephalus at the time of diagnosis, or the hydrocephalus may develop over time.
Management and outcome
SEGAs have a variable growth rate, with some tumors growing steadily over time, while others are seemingly quiescent. In patients with hydrocephalus or those with growing tumors that are located in regions of brain where further growth is associated with a high likelihood of hydrocephalus, the treatment of choice is surgical excision. The tumor may be quite hemorrhagic and difficult to remove. Recent studies have shown that biologic therapy – specifically, treatment with mammalian target of rapamycin (mTOR) inhibitors – may be an effective treatment for those tumors that are not amenable to complete resection or that recur after apparent complete or incomplete resection [Franz et al., 2006].
Ganglioglioma
Ganglioglioma is a not uncommon brain tumor of childhood and, in some series, makes up as high as 5 percent of tumors in the pediatric age range [Chintagumpala et al., 1996]. In institutions that carry out a significant number of epilepsy surgeries, the incidence of gangliogliomas is relatively higher. Gangliogliomas are diagnosed in all ages and have a 1:1 male to female ratio [Johnson et al., 1997]. In children, the mean age at diagnosis is approximately 9–10 years.
Clinical presentation and diagnosis
For tumors that arise in the cerebrum, the most frequent presentation of gangliogliomas is seizures – usually complex partial seizures [Castillo et al., 1990]. The temporal lobe is most commonly involved and there are often few, if any other, symptoms. Symptom duration is typically long and distinction between a ganglioglioma and hamartomatous brain lesion can be difficult. For tumors of the brainstem or spinal cord, the mean duration of symptoms before diagnosis is shorter and patients usually present with focal brainstem symptoms or spinal cord dysfunction.
On CT, gangliogliomas are most commonly hypointense masses that are well circumscribed [Castillo et al., 1990]. There may be associated scalloping of the overlying calvarium in tumors that extend to the cortical surface. On MRI, gangliogliomas are hypointense on T1-weighted images and hyperintense on T2-weighted images. The tumor may enhance, but more commonly there is no enhancement. Peritumoral edema and mass effect are uncommon.
Management and outcome
For those patients with cortical tumors that are amenable to total resection, outcome is usually quite good, with the majority of patients being cured of their disease [Duffner et al., 1999; Luyken et al., 2004]. Surgery will also result in a marked diminution of seizures and many patients are seizure-free after surgery. Outcome for patients with subtotally resected tumors is less clear; many of these patients will be stable for a prolonged period of time after surgery, but will likely continue to have seizures. There are very little data to support the use of adjuvant therapies, such as chemotherapy or radiation therapy, in such circumstances. Similarly, for midline tumors such as those of the brainstem and spinal cord, the course of illness, after diagnosis, may be quite prolonged and the efficacy of additional therapy, after surgical confirmation, is essentially nonexistent.
Desmoplastic Infantile Astrocytoma/Ganglioglioma
The desmoplastic infantile astrocytoma, first described by Taratuto et al. in the early 1980s, is defined as a meningocerebral astrocytoma attached to dura with an associated desmoplastic reaction [Taratuto et al., 1984, 1995]. It was initially termed a desmoplastic cerebral astrocytoma of infancy, and then later investigators described similar tumors with divergent differentiation – the desmoplastic infantile gangliogliomas. These two tumors are now considered as representing the spectrum of one disease.
Clinical presentation and diagnosis
The majority of desmoplastic infantile astrocytomas/gangliogliomas present before 2 years of age, with a male to female ratio of 1.5:1 [Pommepuy et al., 2006; Sugiyama et al., 2002]. Most commonly, the tumor is diagnosed in the first year of life, although older patients have been reported. Symptoms, which are often short in duration, include increasing head circumference, bulging fontanels, and lethargy. Patients will also present with seizures and occasionally with focal motor signs.
Dysplastic Cerebellar Gangliocytoma/Lhermitte-Duclos Disease
Dysplastic cerebellar gangliocytoma is a rare form of cerebellar tumor that is often misdiagnosed. It is composed of cerebellar folia that are expanded by hypertrophic neurons of the internal granular layer. It is considered a WHO grade 1 tumor and is not neoplastic. In adults, it is associated with Cowden’s syndrome, an autosomal-dominant disorder caused by a germline mutation of the PTEN gene [Vital et al., 1994]. This syndrome includes multiple hamartomas involving tissue derived from all germ layers, a high risk of breast, nonmedullary thyroid, and endometrial cancer, and the presence of Lhermitte–Duclos disease/dysplastic gangliocytoma.
Clinical presentation and diagnosis
Although dysplastic gangliocytomas have been predominantly identified in adults, cases have been seen in children, including ones diagnosed in infancy [Vinchon et al., 1994]. PTEN mutations have been identified in all adult-onset cases but not in the childhood-onset cases. Clinical presentation in childhood is often subtle and consists of cerebellar deficits. Other children may present with signs of increased intracranial pressure due to compression of the fourth ventricle.
Neurocytoma
Central neurocytoma is a relatively recently classified tumor composed of uniform round cells with neuronal differentiation [Hassoun et al., 1982]. The tumor is usually benign, with a low mitotic index. This tumor is classically located in the lateral or third ventricle. Although primarily a tumor of adulthood, it can present in pediatrics as an intraventricular mass. Patients all along the pediatric age spectrum have been diagnosed with central neurocytomas; the tumor is more likely to occur in adolescence and in the teenage years.
Clinical presentation and diagnosis
The majority of children with central neurocytomas present with signs and symptoms of increased intracranial pressure secondary to hydrocephalus [Rades et al., 2005]. Neurocytomas can also be an incidental finding or may present abruptly due to acute hemorrhage.
Treatment of central neurocytomas is gross total resection, if possible [Rades et al., 2004]. After such resection, the majority of patients are cured of their disease. After subtotal resection, some patients can be observed for many years without any further treatment. There is little evidence for efficacy of either radiotherapy or chemotherapy.
Pineal Region Tumors
The WHO classification of tumors of the central nervous system recognizes four distinct subtypes of pineal tumors (Box 83-4) [Abay et al., 1981; Edwards et al., 1988; Wara et al., 1979]. The primary parenchymal tumor of the pineal is the pineocytoma. Pineoblastomas are classified within the pineal region tumor grouping, although in other classifications, this tumor has been considered a subtype of embryonal tumor. An intermediary tumor, the pineal parenchymal tumor of intermediate differentiation, first introduced as a separate entity in 1993, is a difficult tumor to diagnose, as it has components of both pineocytoma and pineoblastoma [Schild et al., 1993, 1996]. Depending on pathologic determinations, pineal parenchymal tumors of intermediate differentiation are diagnosed in anywhere between 0 and 60 percent of patients with pineal tumors, primarily in adulthood but also in childhood. Papillary tumors of the pineal region are even rarer and have also been diagnosed both in adults and children. These papillary tumors show immunopositivity for cytokeratin and have other features that can be seen in ependymomas.












Tumors of the pineal region account for 0.4–2 percent of all primary central nervous system tumors in children. Germ cell tumors, which also may occur in other regions of the brain, especially the suprasellar region, comprise the majority of pineal region tumors and will be discussed in the section on germ cell tumors [Jennings et al., 1985; Regis et al., 1996]. Overall, germ cell tumors make up 40–65 percent of all pineal region tumors, with the remaining 20 percent as pineal parenchymal tumors and 20 percent pineoblastomas. Astrocytomas may also occur in the pineal region.
Pineocytomas
Pineocytomas are rare tumors, accounting for 1 percent or less of all childhood primary central nervous system tumors [D’Andrea et al., 1987; Disclafani et al., 1989]. They occur in both adults and children, and, in childhood, tend to be diagnosed in the later portion of the second decade of life. They occur equally in males and females.
Clinical presentation and diagnosis
Neuroradiographically, pineocytomas are well marginated and may harbor cysts. Calcifications are not uncommon [Nakamura et al., 2000; Smirniotopoulos et al., 1992]. Pineocytomas usually have low signal on T1-weighted MRI sequences and are hyperintense on T2-weighted images. Contrast enhancement is usually intense and homogeneous. When there is an associated, more immature component (e.g., perhaps in pineal parenchymal tumors of intermediate differentiation), the tumor may be more heterogeneous in both signal intensity and contrast enhancement.
Management and outcome
In general, treatment of pineocytomas is surgical, and despite the difficult surgical location of the tumor, 5-year survival rates after gross total resection are excellent [D’Andrea et al., 1987; Disclafani et al., 1989; Edwards et al., 1988]. Local radiation therapy has been utilized for those tumors that are residual or progressive after surgery, although, given the rarity of the tumor, the efficacy of radiotherapy is unclear. There are very few data concerning the efficacy of chemotherapy.
Pineal Parenchymal Tumors of Intermediate Differentiation
As stated previously, this is a recently designated tumor that may occur in childhood, but is reported more frequently in adults [Mena et al., 2000; Schild et al., 1993; Trojanowski et al., 1982]. The tumor, by definition, has both features of pineocytomas and more aggressive areas, more consistent with pineoblastomas.
Management and outcome
Management varies, and, depending on histological appearance, the tumor has been managed as an aggressive pineocytoma, with surgery and often local radiotherapy, or as a pineoblastoma [Jouvet et al., 2000]. Survival rates have varied widely because of the difficulty in distinguishing this tumor clearly from other pineal region tumors. Overall survival is not as favorable as that which has been reported for pineocytomas. Leptomeningeal relapse, as well as local relapse, has been noted.
Pineoblastomas
Pineoblastomas comprise approximately 1 percent of all childhood brain tumors [Hinkes et al., 2007; Jakacki, 1999; Jakacki et al., 1995]. Although the tumor may occur at all ages, pineoblastomas are most likely to arise early in life. As is the situation with other embryonal tumors, there is a male predominance.
Pathologically, pineoblastomas are indistinguishable from medulloblastoma, which is why they have been classified by others as embryonal tumors [De Girolami et al., 2008]. The tumor consists of densely packed, small round cells with minimal cytoplasm and frequent mitoses. Immunohistochemically, there may be evidence of astrocytic or neuronal differentiation. The tumor may contain Flexner–Wintersteiner rosettes, suggesting differentiation towards retinoblastoma, and at times the tumor occurs in children with retinoblastoma. This combination of tumors has been called “trilateral retinoblastomas” [Bader et al., 1982]. Biologically, the tumor is different from medulloblastoma, and a variety of chromosomal deletions have been noted [Pomeroy and Sturla, 2003].
Clinical presentation and diagnosis
Pineoblastomas usually present with compression of the third ventricle and associated hydrocephalus, vomiting, headaches, and somnolence [Cuccia et al., 2006; Hinkes et al., 2007; Jakacki, 1999]. As they enlarge, they also frequently compress or infiltrate the tectum of the midbrain, resulting in Parinaud’s syndrome. Due the aggressive nature of the tumor, it may also infiltrate the thalamic area and present with motor or sensory abnormalities.
Pineoblastomas tend to be quite large and lobulated by the time of diagnosis [Nakamura et al., 2000; Smirniotopoulos et al., 1992]. On CT scan, they are characteristically hyperdense, with calcifications being seen in approximately one-third of cases. MRI discloses an isointense lesion on T1-weighted images, which demonstrates heterogeneity on the T2-weighted images (Figure 83-20). The tumor usually shows enhancement, either homogenous or heterogeneous.
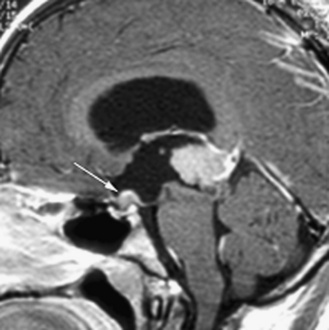
Management and outcome
Pineoblastomas are difficult-to-manage tumors [Cuccia et al., 2006; Hinkes et al., 2007; Jakacki, 1999]. Surgical resection is difficult and, since the majority of patients undergo an incomplete resection or biopsy, the relative benefits of the degree of resection have been difficult to determine.
Outcome has varied between series. In one prospective study, survival and progression-free survival rates for patients with pineoblastomas were reported to be higher than those for children with supratentorial PNETs [Jakacki et al., 1995]. However, in other studies the reverse has been found. Although survival rates as high as 70 percent have been noted after treatment with radiotherapy and chemotherapy, many reports have been much less favorable, with overall survival rates of 40 percent or less at 5 years [Hinkes et al., 2007.] For infants with pineoblastomas, outcome is even poorer, especially in those with disseminated disease at the time of diagnosis. Given the rarity of the tumor, there are essentially no data concerning long-term outcome of survivors. However, given that the boost radiotherapy often has to involve a significant portion of the cerebral cortex and that craniospinal radiation therapy at high doses is often required, long-term sequelae are likely.
Germ Cell Tumors
Germ cell tumors are a heterogeneous group of primary central nervous system tumors and diagnosis can be difficult [Jennings et al., 1985; Kretschmar, 1997; Matsutani et al., 1997]. The majority of tumors arise deep within the brain, in both the pineal and suprasellar region. Histological sampling is often limited. The WHO recognizes multiple different types of germ cell tumors (see Box 83-4), and these tumors range from mature lesions (the mature teratoma) to highly aggressive nongerminomatous germ cell lesions.
The WHO classification of germ cell tumor is a derivation from a schema initially proposed by Telium, suggesting that all germ cell tumors were derived from primordial germ cells that could either differentiate into a germinoma or be the origin of totipotential cells that could give rise to other types of germ cell tumors, although other theories exist [Sano and Matsutani, 1981; Takei and Pearl, 1981; Teilum, 1976]. If the neoplastic process involved differentiated embryonic tissue, such as ectoderm, mesoderm, or endoderm, the tumor became a malignant teratoma or a teratocarcinoma. Germinomas usually have a typical two-cell appearance, and are histopathologically indistinguishable from extracranial germ cell tumors [Mena et al., 2000]. Teratomas and mixed germ cell tumors harbor various mature and immature elements. Teratomas tend to be much better encapsulated.
Germ cell tumors can also be conceptualized as being either mature (teratomas) or malignant; within even a simplified classification, there are often important subvarieties. Still another way to distinguish between subtypes of germ cell tumors is based on secretion pattern, as germ cell tumors may secrete or produce a variety of different proteins that can be assayed in cerebrospinal fluid and, at times, in serum. Even in those tumors that secrete such markers, separation can be arbitrary, depending on the levels of these proteins in the cerebrospinal fluid. The two markers that are the most useful in diagnosis have been α-fetoprotein and human β-chorionic gonadotrophin (β-hCG) [Jennings et al., 1985; Matsutani et al., 1997; Packer et al., 1984].
Although germ cell tumors tend to arise predominantly in the pineal region, 20 percent will originate in the suprasellar region [Sung et al., 1978; Teilum, 1976]. The tumor may also arise in other areas of brain, especially in Asian series, and when this is the case, diagnosis is usually quite difficult [Teilum, 1976]. Nonpineal or suprasellar areas of involvement may include the ventricles, basal ganglia, thalamus, and diffuse periventricular regions. Germinomas may also present in both the suprasellar and the pineal regions at the time of diagnosis [Jennings et al., 1985; Lafay-Cousin et al., 2006; Matsutani et al., 1997; Sung et al., 1978; Teilum, 1976]. It is unclear whether this represents dissemination or true simultaneous presentation in both areas of brain; such dual presentations may occur in up to 10 percent of patients.
Clinical presentation and diagnosis
The diagnosis of germ cell tumors is notoriously delayed, despite the apparent malignant nature of most germ cell tumors. Symptoms may be present for months or, at times, years prior to diagnosis [Rushing et al., 2006; Teilum, 1976]. Suprasellar tumors causing diabetes insipidus or those tumors that are more widely infiltrated in the brain tend to have the longest period of time between onset of symptoms and diagnosis. Other endocrinologic sequelae seen in patients with suprasellar tumors include diabetes insipidus, delayed sexual development, panhypopituitarism, and isolated growth failure. Tumors in the pineal region present as other pineal region masses, with hydrocephalus, Parinaud’s syndrome, and obtundation. Patients with more aggressive nongerminomatous germ cell tumors tend to have more neurologic compromise and focal deficits at the time of diagnosis, usually secondary to infiltration of surrounding brain. Precocious puberty has been reported in children with both suprasellar and pineal region tumors. Although it has been postulated that the precocious puberty may be caused by secretion of β-hCG, it is also quite conceivable that many patients diagnosed with pineal region tumors who have precocious puberty may also have subtle suprasellar or hypothalamic involvement. Infants with teratomas tend to have extremely large tumors at the time of diagnosis, with resultant hydrocephalus, macrocephaly, and marked neurologic impairments.
The neuroimaging of germ cell tumors is also variable and there is considerable overlap between different germ cell tumors [Nakamura et al., 2000; Packer et al., 1984; Smirniotopoulos et al., 1992]. Germinomas tend to be relatively homogeneous, although small cysts can be seen in up to 50 percent of patients. On CT, these tumors are isodense to hyperdense. On MRI, the solid portions of the tumor are isointense compared to gray matter on T1-weighted images, and hypointense to isointense on T2-weighted images; calcifications may also be seen (Figure 83-21). Nongerminomatous germ cell tumors tend to be more heterogeneous and frequently have cysts and regions of hemorrhage. Choriocarcinomas may also be hemorrhagic and tend to be hypervascular. Teratomas are more likely to be heterogeneous lesions with multilocular microcystic and solid elements; lipomatous elements may also been seen (Figure 83-22).
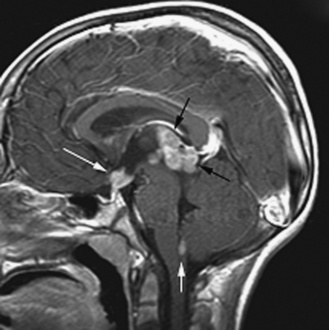
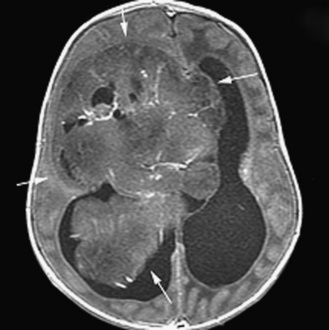
Although neuroimaging findings are relatively characteristic for germ cell tumors, these lesions cannot be clearly distinguished from pineoblastomas and, at times, pineal parenchymal tumors of intermediate differentiation [Abay et al., 1981; Calaminus et al., 2005; Jennings et al., 1985; Matsutani et al., 1997; Packer et al., 1984]. Measurements of cerebrospinal fluid tumor markers are an accepted means of diagnosis of secreting germ cell tumors. Pure germinomas usually do not have elevated markers, though some tumors have a moderate elevation of β-hCG. In contradistinction, mixed germ cell tumors are associated with elevated α-fetoprotein and β-hCG levels in cerebrospinal fluid, especially lumbar fluid, and – depending on the degree of elevation – also in blood. Choriocarcinomas will secrete high levels of β-hCG. Distinction between germinomas that secrete small amounts of β-hCG, syncytiotrophoblastic germinomas that secrete higher levels of β-hCG, and nongerminomatous germ cell tumors that may secrete even higher levels is often arbitrary, and classifications based on the levels of β-hCG vary between series.
Management and outcome
Most patients with germ cell tumors, independent of their location in the nervous system, undergo subtotal resections, at best, prior to diagnosis. Patients with secreting pineal region tumors are often treated without any surgical intervention [Abay et al., 1981]. The exceptions are those patients, especially infants, with what are believed to be teratomas. After total resections, patients with teratomas tend to have an extremely favorable outcome.
For patients with pure germinomas, radiotherapy alone is often curative; however, there have been significant concerns raised as regards the volume and doses of radiotherapy required for cure [Calaminus et al., 2005; Jennings et al., 1985; Lafay-Cousin et al., 2006; Matsutani et al., 1997; Merchant et al., 2000; Rich et al., 1985; Wolden et al., 1995]. Treatment with craniospinal and local radiotherapy results in event-free and overall survival rates of well over 90 percent, even in patients with disseminated disease. It is unclear whether patients with nondisseminated germinomas require craniospinal radiation therapy. Excellent survival rates have been reported after the use of whole ventricular radiotherapy supplemented with “boost” radiotherapy to the primary tumor site [Linstadt et al., 1988; Shibamoto et al., 2001; Shirato et al., 2004]. In studies utilizing whole ventricular radiation, there is variability in the inclusion of the fourth ventricle in the whole ventricular volume. There are no clear-cut comparative studies demonstrating which dose of cranial or local boost radiotherapy is required, if radiation is used alone. Between 2100 and 2700 cGy of radiation therapy to the whole ventricle or craniospinal axis is conventionally utilized, and 4000–4500 cGy to the primary tumor site. Higher doses are utilized in patients with leptomeningeal disease at the time of diagnosis.
Chemotherapy has been increasingly utilized in children with germinomas in attempts to reduce the volume and overall dose of radiotherapy [Allen et al., 1987b, 1994; Bouffet et al., 1999; Nguyen et al., 2006; Ogawa et al., 2004; Raggi et al., 2008; Robertson et al., 1997; Shibamoto et al., 1994; Shikama et al., 2005]. As is the case in extracranial germ cell tumors, germ cell tumors of the central nervous system are quite chemosensitive. A variety of different agents have been utilized, including cyclophosphamide, ifosfamide, etoposide, cisplatin, carboplatin, and bleomycin. Studies have suggested that, in patients with a complete response to preradiotherapy chemotherapy, the volume and doses of radiotherapy can even be safely reduced. Chemotherapy alone has not been shown to be an effective strategy for patients with germinomas, as there is a significantly higher rate of disease relapse after approaches based on chemotherapy alone and overall poorer survival [Balmaceda et al., 1996].
Controversy exists concerning the treatment of germinomas that secrete low to moderate levels of β-hCG [Calaminus et al., 2005; Matsutani et al., 1990; Paulino et al., 1999]. Survival rates for these patients, whether they are classified as having secreting germinomas or syncytiotrophoblastic germinomas, tend to be favorable, and there is no consensus on the appropriate dose or volume of radiotherapy and the need for chemotherapy.
The outcome of patients with nongerminomatous germ cell tumors is less favorable. Results of studies utilizing radiotherapy alone, including doses of radiotherapy of 3600 cGy to the craniospinal axis and 5400 cGy or higher to the local tumor site, have demonstrated long-term disease control of 50 percent or less of patients [Bouffet et al., 1999; Calaminus et al., 1994, 2004, 2008; Kobayashi et al., 1989; Matsutani, 2008; Schild et al., 1996]. The addition of multiagent chemotherapy (utilizing drugs similar to those used for germinomas) prior to radiotherapy, followed by craniospinal and local boost radiotherapy, results in a better overall progression-free survival, ranging as high as 70 percent for patients in recent series. There is no consensus as to which chemotherapeutic regimen is optimal for disease control.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 83 Tumors of the Brain and Spine
Introduction
Primary central nervous system tumors comprise, in total, the second most common form of childhood cancer, exceeded only by the leukemias [CBTRUS, 2008; Jemal et al., 2008; Louis et al., 2007a; Preston-Martin, 1996; Ries et al., 1997]. Although relatively rare compared to other pediatric neurological conditions, these neoplasms cause significant neurologic morbidity and mortality. Unlike the situation in adults, where metastatic brain tumors outnumber primary central nervous system neoplasms by a ratio as high as 10 to 1, in the pediatric years, metastatic lesions are much less common and primary brain and spine tumors predominate [Bleyer, 1999; CBTRUS, 2008; Jemal et al., 2008; Louis et al., 2007b; Ries et al., 1997; Smith et al., 1998].
Tumors of the brain and spine constitute approximately one-quarter of all childhood cancers. In the United States, depending on what age is utilized to distinguish between adult and pediatric patients, between 3000 and 4000 children each year are diagnosed. Concepts and management of pediatric brain tumors are constantly evolving [Bleyer, 1999; Jemal et al., 2008; Louis et al., 2007a; Smith et al., 1998]. The classification of these neoplasms, on which treatment is based, is also changing, primarily due to the incorporation of molecular diagnostics.
As in adults, the majority of primary central nervous system tumors in children are glial in origin; however, a significant proportion of tumors are embryonal (Figure 83-1) [CBTRUS, 2008; Jemal et al., 2008]. Embryonal, and mixed neuronal and glial tumors in infants are notoriously difficult to diagnose. The routine utilization of immunohistochemical analysis and, more recently, molecular analysis of tumors have altered concepts of diagnosis and classification, but their incorporation has lagged behind those in other childhood cancers, such as leukemia and neuroblastoma, and has led to the continued subjectivity of diagnosis.
Advances and refinements in therapy, to date, have resulted in a modest improvement in overall survival rate of patients. It has been estimated that there has been a 15–20 percent improvement in survival in patients diagnosed between 1975 and 1979, compared to those diagnosed and treated between 1996 and 2003 [CBTRUS, 2008; Jemal et al., 2008]. The complexities involved in the treatment of childhood brain and spine tumors have resulted in the development of regional centers focusing on the comprehensive management of children with these neoplasms. It is widely accepted that, for optimum management, a multidisciplinary approach is required, involving many specialties, including neurosurgery, neurology, oncology, neuropathology, neuroradiology, radiation oncology, ophthalmology, and rehabilitative specialists. In addition, given the relative infrequency of specific subtypes of childhood brain tumors, for progress to be made expeditiously, prospective multicentered, cooperative group-based trials are optimal. National and, in selected cases, international collaborations have become the cornerstone of modern management of childhood brain tumors.
Incidence
The incidence of childhood nervous system tumors is best estimated from population-based registries. Institution-based registries are potentially flawed by referral patterns; the tendency for larger programs to receive referrals of selective patients, skewing patient number; and the increasing mobility of patients, allowing individuals to seek second opinions and thus being included in statistics at multiple centers. The reported incidence rates of brain tumors for patients between 0 and 19 years ranges from 3.3 to 4.5 cases per 100,000 person years [CBTRUS, 2008]. This reported incidence is higher than the 2.7 cases per 100,000 person years reported in the late 1970s and early 1980s. It is believed that this higher incidence is partially due to the availability of magnetic resonance imaging (MRI), identifying more brain tumors earlier, and possibly the use of stereotactic biopsies to confirm the presence of primary central nervous system tumors [Bleyer, 1999; Smith et al., 1998]. Better reporting, the inclusion of lower-grade tumors in registries, and the wider utilization of epilepsy surgery confirming the diagnosis of low-grade neoplasms may also contribute to this increased incidence, rather than it representing a true increase in the number of brain and spine tumors. However, some studies have suggested that there is indeed a true increase in incidence. Among the major histological groupings, rates are highest for tumors of neuroepithelial tissue, with pilocytic astrocytomas and medulloblastomas being the most common individual subtypes of tumors.
Primary central nervous system tumors are more common in the first decade of life (Figure 83-2) [CBTRUS, 2008]. The reported incidence of brain tumors is higher in whites (4.7 per 100,000 person years) than in blacks (3.0 per 100,000 person years). The overall incidence for all brain tumors is highest among those between 0 and 4 years of age (5.2 per 100,000 person years), and lowest among those between 10 and 14 years of age (4.1 per 100,000 person years) [CBTRUS, 2008]. However, incidence does vary depending on tumor type, with ependymomas and medulloblastomas decreasing with age, and pilocytic astrocytomas peaking at 5–9 years of age. Germ cell tumors increase with age. In the 0–19 years age group, brain tumors are more common in boys, especially tumors of neuroepithelial origin and pineal germ cell tumors.
Overall, gliomas of multiple different subtypes, including pilocytic astrocytomas, astrocytomas not otherwise specified, and gliomas, are the most common type of childhood brain tumors, accounting for over 50 percent of all tumors in children between 0 and 14 years of age, and nearly 75 percent of all malignant tumors [CBTRUS, 2008]. Embryonal tumors, including medulloblastoma, comprise 15–20 percent of all tumors in children between 0 and 14 years, occurring in approximately 5 percent of those between 15 and 19 (see Figure 83-2) [CBTRUS, 2008].
Etiology
The causation of the majority of all childhood brain tumors is unknown [Bleyer, 1999; Bunin, 2000]. Despite this, there are genetic conditions and, to a lesser extent, environmental exposures that predispose to the development of tumors [Bleyer, 1999; Bunin, 2000]. Genetic syndromes related to increased development of specific tumor types have provided molecular genetic insights into childhood brain tumors that have been partially translatable to the study of apparently nongenetically inherited tumors. The known syndromes that are associated with the development of childhood brain tumors usually have an autosomal-dominant pattern of inheritance. At times, for unclear reasons, brain tumors will arise in multiple family members [Draper et al., 1977; Fitzgerald, 2000; Kuijten et al., 1993; Malmer et al., 2007; Moschovi et al., 1998].
Neurofibromatosis
Probably the most common genetic syndrome associated with development of primary central nervous system tumors is neurofibromatosis type 1, which is an autosomal-dominant disorder due to mutation of the NF1 gene [Burger et al., 1987; Cawthon et al., 1990; Listernick et al., 1999; Wallace et al., 1990]. The NF1 gene on the long arm of chromosome 17 encodes the neurofibromin protein, which, among other actions, is a tumor suppressor gene [Burger et al., 1987; Listernick et al., 1999; Pollack and Mulvihill, 1997]. Its absence allows ras activation and subsequent downstream intracellular signaling through the RAS/MAPK (MAP kinase) pathway. The gene is large, spanning 350 kb, and a mutation in the initially unaffected (wild-type) NF1 gene is thought to result in the development of a host of tumors. In the central nervous system, the most common are low-grade, most often pilocytic astrocytomas, which early in life have a marked predilection for the optic nerves and other parts of the visual pathway. Later in childhood, gliomas may develop in other central nervous system sites, especially midline areas, including the brainstem and corpus callosum.
Separation of low-grade tumors from areas of brain dysplasia, also common in neurofibromatosis type 1, can be difficult [Listernick et al., 1997]. These areas of abnormal signal on T2-weighted MRI are at least partially developmental, and many disappear as the child enters into puberty. Other central nervous system tumors, such as ependymomas and possibly medulloblastomas, are more common in patients with neurofibromatosis type 1. Neurofibromatosis type 1 occurs in as many as 1 in 3500 patients at risk, and brain tumors may occur in as high as 20 percent of patients, although, for unclear reasons, many affected patients do not require specific treatment.
Children with neurofibromatosis type 1 are also prone to development of neurofibromas, of which the most problematic in prepubertal patients are plexiform neurofibromas [Listernick et al., 1999; Packer and Rosser, 2002; Packer et al., 2002a]. Plexiform neurofibromas are congenital and may cause significant cosmetic disfigurement, organ dysfunction, and, dependent on location, neurologic compromise, including spinal cord compression. A percentage of plexiform neurofibromas, possibly as high as 10 percent, will undergo malignant transformation, although in the vast majority of patients this occurs after puberty, especially in the early adult years. Since patients with neurofibromatosis type 1 have an increased predilection for the development of other malignancies, including myelogenous leukemia and sarcomas, treatment of patients with neurofibromatosis type 1-associated brain tumors can be quite challenging. Chemotherapeutic alkylating drugs and radiation therapy may increase the risk of developing secondary tumors.
Neurofibromatosis type 2, which is also an autosomal-dominant disorder affecting approximately 1 in 25,000 to 1 in 400,000, is due to a gene abnormality on the short arm of chromosome 22 [Pollack and Mulvihill, 1997; Rouleau et al., 1993; Trofatter et al., 1993]. Patients with this condition are predisposed to developing schwannomas of the cranial nerves, spinal nerves, and peripheral nervous system. Bilateral acoustic schwannomas are essentially pathognomonic of the illness and are usually present by early adulthood. Gliomas and ependymomas, especially spinal ependymomas, also occur with increased frequency.
Tuberous Sclerosis and Other Autosomal-Dominant Conditions
Other autosomal-dominant neurogenetic diseases are related to specific tumor types. Tuberous sclerosis is associated with the development of subependymal giant cell astrocytomas, which tend to occur in the subependymal ventricular zone and most commonly cause obstructive hydrocephalus early in illness [Kandt et al., 1992; van Slegtenhorst et al., 1997]. Cerebellar gangliocytomas are associated with Cowden’s syndrome, caused by a germline mutation of the PTEN gene [Liaw et al., 1997]. The occurrence of dysplastic gangliocytoma of the cerebellum is also known as Lhermitte–Duclos disease. Caused by a mutation of the VHL gene, von Hippel–Landau syndrome is associated with the development of hemangioblastomas in the cerebellum, spinal cord, and retina [Duan et al., 1995; Latif et al., 1993].
Li–Fraumeni Syndrome
Li–Fraumeni syndrome is caused by a germline mutation in the multifunctional TP53 gene [Malkin et al., 1990; Srivastava et al., 1990]. The protein encoded by the gene has a role in cell-cycle control and in DNA integrity and repair. Gliomas, both high-grade and low-grade, are most common in patients with Li–Fraumeni syndrome, and such tumors may be multifocal. Other central nervous system tumors that occur in increased incidence in this syndrome include medulloblastomas, supratentorial neuroectodermal tumors, and choroid plexus tumors.
Gorlin’s Syndrome
Gorlin’s syndrome, also known as the nevoid basal cell carcinoma syndrome, accounts for probably less than 1 percent of all patients with medulloblastoma, but the relationship between this condition and the development of medulloblastoma has led to a better understanding of the molecular pathogenesis of a larger subset of patients with medulloblastoma [Amlashi et al., 2003; Stavrou et al., 2001; Taylor et al., 2000]. It has been estimated that 3–5 percent of patients with Gorlin’s syndrome will develop medulloblastoma. Gorlin’s syndrome is caused by an inherited germline mutation of the PATCHED 1 (PTCH1) gene on chromosome 9, which encodes the sonic hedgehog (SHH) receptor. PTCH1 normally suppresses sonic hedgehog signaling [Taylor et al., 2000]. The syndrome may be diagnosed early in life by characteristic dermatological and skeletal features, including odontogenic keratocysts of the jaw, bifid or fused ribs, macrocephaly, and calcification of the falx. The basal cell carcinomas tend to occur later in life. Mutations of the PTCH1 gene can be demonstrated in 20–40 percent of medulloblastomas, especially in the desmoplastic variant in infants. Since patients with Gorlin’s syndrome are predisposed to the development of basal cell carcinomas later in life, the use of craniospinal radiotherapy, which is the backbone of treatment for most patients with medulloblastoma, is problematic, as basal cell carcinomas may occur in large numbers in the radiation fields. The diagnosis of Gorlin’s syndrome is often made later in life, when the basal cell tumors become evident.
Turcot’s Syndrome
Turcot’s syndrome is another autosomal-dominant disorder due to mutations in the adenomatous polyposis coli (APC) gene (type 2), or secondary to mutations in DNA mismatch repair genes HPS2 and MLH1 (type 1) [Attard et al., 2007; Foulkes, 1995; Hamilton et al., 1995; Ng et al., 2005]. Patients with mutation of a mismatch repair gene are prone to developing colorectal carcinomas and gliomas. Patients with type 2 disease are at increased risk for developing medulloblastomas. Mutations in the APC gene result in abnormalities of the Wnt molecular pathway. This pathway has been noted to be abnormal in up to 15 percent of patients over 3 years of age with medulloblastoma, especially older patients, and may constitute a different, more favorable form of the disease.
Environmental Exposures, Including Radiation Therapy
Of the environment exposures that predispose to the development of brain tumors, the best documented is ionizing radiation [Bakhshi et al., 2003; Kimball Dalton et al., 1998; Rimm et al., 1987; Ron et al., 1988]. This association was demonstrated in children treated with radiation for tinea capitis during the 1940s and 1950s, who were found to have an increased incidence, later in life, of meningiomas, gliomas, and nerve sheath tumors [Ron et al., 1988]. Secondary brain tumors have been reported after cranial irradiation for leukemia, head and neck tumors, and primary brain tumors [Kimball Dalton et al., 1998; Kleinerman, 2009; Neglia et al., 1991; Rimm et al., 1987; Thierry-Chef et al., 2008]. The most common form of malignant brain tumor occurring after radiation is malignant gliomas, which tend to appear 5–15 years after radiation and are highly resistant to therapy. Meningiomas are the most common radiation-induced tumors, increase in frequency with time after radiotherapy, and do not begin to peak until ten years after exposure [Neglia et al., 1991; Rimm et al., 1987]. Other tumor types that have been reported include lower-grade gliomas, medulloblastomas, and ependymomas.
The effects of a multitude of environmental exposures, including maternal diet, on the occurrence of childhood brain tumors have not been conclusively shown [Bunin, 2000; Preston-Martin, 1996]. Factors that have been associated with a higher incidence of medulloblastoma in some, but not all, studies include maternal farm residence during pregnancy and pesticide exposure [Connelly and Malkin, 2007; Norman et al., 1996; Preston-Martin, 1996; Robison et al., 1995]. In some studies, mothers’ multivitamin use and food consumption during pregnancy have been found to be potentially protective against of the development of tumors, especially medulloblastoma. Initial studies suggested an association between significant exposure to electromagnetic waves or the use of cell phones in the development of gliomas, but other studies have not been able to confirm this relationship [Ali Kahn et al., 2003; Connelly and Malkin, 2007; Hardell et al., 2002]. Similarly, initial studies suggested that patients exposed to simian virus 40 (SV40), through immunizations, may have a higher incidence of developing medulloblastoma [Hayashi et al., 2001; Kim et al., 2002]. Subsequent studies could not confirm this association. Immunosuppression, either due to an underlying disorder of the immune system or after treatment with chronic immunosuppressive agents in situations such as organ transplantation, has been associated with an increased incidence of central nervous system lymphoma [Kuijten and Bunin, 1993; Penn, 1998].
Pathology and Classification
The present classification of primary central nervous system tumors dates back to the 1920s and, as devised by Bailey and Cushing, was based on a cell of origin concept and the theory that tumor cells developed from cells arrested in various stages of development [Bailey, 1925; Louis et al., 2007b]. For each developmental stage of a cell, a corresponding tumor could be identified. In the Bailey and Cushing classification, tumors were also classified on the basis of the morphology of the tumor [Bailey, 1925]. This classification was modified by histologic grading of predominantly glial tumors on a 1–4 scale, with 1 being the most benign and 4 being the most malignant. Over time, other systems have been devised, essentially to score tumors to connote their relative aggressivity and to link the nomenclature to prognosis.
At present, the most widely accepted classification schema is that of the World Health Organization (WHO) [Louis et al., 2007a]. This system, since its first publication in 1979, has included a grading scheme that basically attempts to be a malignancy scale. Generically, grade 1 tumors are those with low proliferative potential and the possibility of cure following surgical resection alone. Grade 2 lesions are believed to be more infiltrative in nature and may have a higher likelihood of recurring. Dependent on the tumor type, grade 2 tumors are also those considered to more likely to progress to higher grades, especially in the case of gliomas. Grade 3 is generally reserved for tumors with histological evidence of malignancy, which are less likely to be cured by surgery alone. Grade 4 tumors are those that are considered the most cytologically malignant and mitotically active. There have been attempts to modify the WHO classification for use in pediatrics. Although the WHO classification is applicable to pediatrics, the congenital nature of many tumors, often with elements that seem to show mixed lineage, such as neuronal and glial elements, can make classification difficult. In addition, in the pediatric modifications, which have not been fully accepted, tumor location is also a component of classification.
Probably the most controversial aspect of the classification of pediatric brain tumors has been the nomenclature utilized for central nervous system embryonal tumors [Rorke, 1983]. The most common embryonal tumor is the medulloblastoma, a term that was coined over 80 years ago and still exists, although it is now accepted that there is no such tumor cell as a medulloblast. The primary controversy centered on whether all histologically similar, primitive neuroectodermal tumors should be classified as one tumor type and then subclassified on the basis of other histological features, such as histological differentiation along one or more cellular lineages, or whether the tumor should be diagnosed based on both histological features and tumor location. The introduction of immunohistochemical techniques demonstrated the variable cell types that occur in embryonal tumors, although the majority of cells are small hyperchromatic round or oval cells (small blue cells) [Gould et al., 1990; Molenaar et al., 1989]. The availability of molecular genetic analysis helped settle this controversy, as it was shown that primitive embryonal tumors occurring in the cerebellum were molecularly distinct from those arising in the pineal region and from those arising in the cortex [Ellison, 2002; Haberler et al., 2006; Pomeroy and Sturla, 2003; Rorke et al., 1995].
The WHO classification does not separate tumor types on the basis of tumor location [Louis et al., 2007a]. However, by convention, childhood central nervous system gliomas are often discussed by location, and cerebellar astrocytomas, intrinsic brainstem gliomas, and visual pathway gliomas, especially optic nerve and/or chiasmatic tumors, are considered relatively distinct entities. There are few biologic data to support such separations, and there is probably little, if any, prognostic difference between a diffuse intrinsic brainstem glioma and a similar tumor involving the thalamus, or, for that matter, a juvenile pilocytic astrocytoma of the cerebrum versus juvenile pilocytic astrocytoma of the cerebellum.
Medulloblastoma and Supratentorial PNET
Isochromosome 17q, often in combination with loss of 17p, is the most common cytogenetic alteration in medulloblastoma, occurring in up to 50 percent of tumors [Biegel, 1999]. A clear association with clinical outcome has not been confirmed; however, recent evidence suggests that isochromosome 17q is an unfavorable prognostic indicator [Pan et al., 2005]. Supratentorial primitive neuroectodermal tumors (sPNET) display a unique set of genomic alterations that rarely include isochromosome 17q, indicating that sPNET are molecularly distinct from medulloblastoma, despite sharing a similar histological appearance [Bayani et al., 2000; Pfister et al., 2007].
High expression of the neurotrophin-3 receptor, TRKC, was the first molecular alteration identified as an independent predictor of favorable outcome in medulloblastoma [Grotzer et al., 2000; Segal et al., 1994]. Increased expression of ErbB2, a member of the epidermal growth factor receptor (EGFR) family, and MYCC oncogene independently correlates with a poor outcome [Gilbertson et al., 1995; Grotzer et al., 2001]. High-level amplifications of MYC family members are also seen in 5–15 percent of medulloblastomas, and are associated with the aggressive anaplastic medulloblastoma variant and unfavorable outcome [Herms et al., 2000].
Aberrant signaling through the developmental pathways, Hedgehog (Hh), Notch, and Wnt, has been linked to medulloblastoma formation and growth (Figure 83-3). The relationship between Hh and medulloblastoma was deduced from the observation that medulloblastomas arise in patients with nevoid basal cell carcinoma syndrome (Gorlin’s syndrome), a condition caused by a somatic mutation of the PATCHED (PTCH) gene [Wicking and Bale, 1997]. PTCH is an important regulator of the Hh mitogenic signaling pathway. Gorlin’s syndrome accounts for 1–2 percent of all medulloblastomas, most commonly of the desmoplastic subtype [Amlashi et al., 2003]. Mutations in Hh pathway genes have been identified in approximately 25 percent of sporadically occurring medulloblastomas, predominantly of the desmoplastic subtype [Dong et al., 2000; Pomeroy et al., 2002; Raffel et al., 1997].
Increased copy numbers of Notch and mutations in the Wnt signaling pathway have each been detected in up to 15 percent of medulloblastomas. Expression of the Notch pathway target gene Hes1 is associated with shorter survival time, while nuclear immunoreactivity for beta-catenin, a marker of Wnt pathway activation, is associated with a highly favorable outcome [Ellison et al., 2005; Fan et al., 2004]. A more recent integrative genomic study identified five distinct molecular subtypes of medulloblastoma, including a Wnt signaling group associated with classic histology tumors in older children, and an Hh signaling group associated with desmoplastic tumors in younger children [Kool et al., 2008]. Both tumor groups shared increased expression of Notch and platelet-derived growth factor (PDGF) pathway genes.
Astrocytoma
Perhaps the most significant discovery to date for this group of tumors is the identification of BRAF oncogene-activating mutations in association with juvenile pilocytic astrocytoma (JPA) [Jones et al., 2008]. BRAF mutations appear to be rare in other types of pediatric low-grade glial tumors, and are associated primarily with JPA of the cerebellum and optic pathway [Jacob et al., 2009]. Low-grade gliomas, usually JPA of the optic nerves and chiasm, also occur in about 15 percent of children with neurofibromatosis type 1; however, BRAF mutations have not been identified in neurofibromatosis type 1-associated JPA [Jacob et al., 2009].
Evidence shows that childhood and adult malignant gliomas are molecularly distinct. Overexpression of EGFR is seen in 60–80 percent of high-grade gliomas in both children and adults; however, amplification of EGFR is found in 40 percent of adult tumors, but only rarely seen in childhood gliomas [Bredel et al., 1999]. TP53 is mutated in 12 percent of malignant gliomas in children aged less than 3 years, compared to 40 percent of the tumors in children older than 3 years [Pollack et al., 2001]. The 5-year survival is 17 percent in patients with high TP53 expression, and 44 percent in those with tumors that have low TP53 expression [Pollack et al., 2002]. PTEN mutations occur in only 8 percent of high-grade lesions in children, compared to 30 percent of adult high-grade tumors; however, expression of activated PDGF receptors and PTEN deficiency are associated with increasing malignant histology and decreased survival, respectively, among children with high-grade gliomas [Cheng et al., 1999; Raffel et al., 1999; Thorarinsdottir et al., 2008].
Other Childhood Brain Tumors
One of the most important molecular genetic alterations in childhood brain tumors is the discovery of hSNF5/INI1 gene mutations in association with atypical teratoid/rhabdoid tumors (AT/RT) [Biegel et al., 2002a]. AT/RT has sometimes been confused with medulloblastoma and PNET, but now screening for hSNF5/INI1 mutations can confirm the molecular diagnosis of AT/RT, which has important prognostic and therapeutic implications.
In ependymoma, chromosome 17p deletions have been found in 50–80 percent of tumors, but TP53 mutations have not been found to date [von Haken et al., 1996]. An integrative genomic study of ependymoma also revealed that supratentorial, posterior fossa, and spinal tumors have distinguishable molecular signatures, and that genes of supratentorial and spinal tumors are similarly expressed in mouse radial glial cells in the corresponding region of the CNS [Taylor et al., 2005].
Neuroradiology
The ability to stage tumor extent – especially to detect the presence of subarachnoid metastatic dissemination – is crucial. Many pediatric brain tumors have a propensity to disseminate throughout the subarachnoid spaces, often at the time of presentation. High-quality screening MRI studies of the entire neural axis are needed; clear outcome differences have been demonstrated with regard to pediatric brain tumors, depending on the presence or absence of dissemination [Packer et al., 2006]. Finally, repeated follow-up at appropriate time intervals is the best method for detecting early tumor recurrence; detection of smaller, presymptomatic recurrent tumor leads to improved outcomes [Saunders et al., 2003; Shaw et al., 1997].
Diffusion Imaging
Diffusion imaging (DI) investigates molecular translational movements (i.e., Brownian motion) of water molecules. Brownian motion produces signal loss proportional to the degree of molecular translation/diffusion. Within the cerebral milieu, cellular membranes and other macromolecular structures restrict water diffusion, which results in increased signal on DI. (Less diffusion means less signal loss secondary to spin dephasing.) Thus, signal on diffusion images can reflect cellular density and microarchitecture. The apparent diffusion coefficient (ADC) is a measure of the ability of tissue to restrict water diffusion. ADC adversely correlates with the grade of tumor and the cellular proliferation Ki-67 labeling index [Higano et al., 2006]. Increase in ADC, reflecting decrease in cellularity, has been observed as tumors respond to treatment.
In the intracellular and extracellular spaces of the brain, macromolecular proteins, intracellular organelles, cell walls, and myelin sheaths restrict or slow diffusion in certain directions. The most significant determinant of the preferred diffusion direction is the myelin sheath within cerebral white matter [Miller et al., 2003]. DI can therefore assess the microscopic structural integrity of white-matter tracts as the movement of water occurs preferentially along the long axis of the myelin sheath surrounding the axon. Anisotropy is an aggregate measure of the magnitude of displacement across all three dimensions; using information gleaned from the size, shape, orientation, and distribution (or spatial pattern) of diffusion ellipsoids within an imaged volume, one can characterize features of water diffusion within a voxel – the diffusion tensor. A voxel by voxel analysis of anisotropy can be performed, resulting in diffusion tensor imaging (DTI). Connectivity maps (i.e., tractography) can also be generated, which demonstrate the long association (axonal) pathways that convey cortical connections as tracklike structures.
DTI tractography is used to plan the surgical approach to a focal tumor. Important association pathways (e.g., pyramidal, optic tracts) can be localized in relation to a tumor; this helps the surgeon to avoid injury to these white-matter tracts. DTI also shows promise as a tool to evaluate for the presence and extent of tumoral invasion; whereas focal (well-marginated) tumors should displace but not disrupt axons outside of their borders, the microscopic tumor extension of infiltrative tumors is expected to disrupt local axonal structures [Helton et al., 2006; Talos et al., 2007].
Magnetic Resonance Spectroscopy
Spectroscopy can be performed with single-voxel or multivoxel techniques. Tissue composition within a voxel must be homogeneous, otherwise partial volume averaging of tissue of various compositions (e.g., normal, tumor, and necrotic tissue) occurs and the spectrum has little value. Single-voxel studies have improved spectral resolution over multivoxel techniques, though minimal voxel size for good signal to noise is about 4 cm3; further, in single-voxel imaging, the prescription (i.e., the determination of the voxel location) of the voxel is totally operator-dependent – and therefore not very reproducible. Spectroscopic analysis of a large volume of cerebral tissue is feasible with multivoxel techniques (i.e., two- or three-dimensional chemical shift imaging [CSI]), in which several subcentimeter voxels can be examined simultaneously. CSI is very helpful in the evaluation of small or heterogeneous tumors; in the evaluation of the extent infiltrative tumors where the tumor/normal brain boundary may be difficult to determine with conventional techniques; and to distinguish recurrent tumor from radiation necrosis. MR spectroscopy may be of help to monitor tumor response versus progression [Tzika et al., 2004]; prognosticate overall survival [Warren et al., 2000]; and improve preoperative identification of tumor type [Panigrahy et al., 2006].
Perfusion Imaging
Perfusion imaging can be performed with techniques based on dynamic susceptibility contrast (DSC) or on vascular permeability. DSC techniques assess cerebral tissue perfusion following a dynamic injection of gadolinium. During the first-pass transit of contrast through cerebral capillaries, which lasts anywhere from 5 to 15 seconds, gadolinium is (normally) restricted to the intravascular space. This intravascular restriction gives rise to signal inhomogeneity within image voxels (i.e., a susceptibility effect), which is reflected as signal loss on susceptibility-sensitive T2 (T2*) imaging techniques. A curve showing concentration of gadolinium over time can be generated. This enables a time versus signal intensity curve to be drawn, from which a variety of parameters can be inferred. These include mean transit time (MTT); time-to-peak; area under the curve, a measure of relative cerebral blood volume (rCBV); and relative cerebral blood flow (rCBF). rCBV corresponds to the volume of blood within brain tissue and can reflect the neovascularization associated with tumor growth (tumor angiogenesis). Perfusion imaging is therefore useful for the study of neovascularization and angiogenesis inhibition; for evaluation of tumor grade [Schmainda et al., 2004]; for targeting a lesion for biopsy; and to differentiate hypervascular tumors from tissue necrosis. DSC perfusion imaging may also be able to identify low-grade tumors that progress rapidly, and low-grade gliomas that have a propensity for malignant transformation [Law et al., 2006].
DSC perfusion studies do not consistently generate usable data. First, a larger-bore intravenous catheter must be placed for safe delivery of the injection of 3–4 cc/sec of gadolinium necessary to generate a well-defined first-pass curve; that is not always easy, especially in children with poor intravenous access. (Recent commercial introduction of ports and central lines that are safe to use with power injectors may alleviate this issue.) Second, susceptibility artifacts are not negligible, as DSC perfusion utilizes an echo planar technique (echo planar images are prone to image distortion by susceptibility artifacts); studies performed on children with dental braces or in the presence of significant amounts of intracranial/intratumoral hemorrhage are often useless. Finally, gadolinium contrast can leak through tumor capillaries; such leakage can result in an underestimation of rCBV, as the computations performed to process the susceptibility T2* signal assume that the contrast remains confined to the capillary [Cha et al., 2002].
Permeability-based perfusion techniques take advantage of leakage of gadolinium through tumor vessel walls. In healthy cerebral capillaries, the blood–brain barrier prevents gadolinium leakage into the interstitium. Tumor capillaries have disrupted or absent blood–brain barriers, which allows contrast to leak outside the vessel. The rate of leakage is proportional to the permeability of the capillary wall. Permeability imaging, also known as relaxivity imaging, is a T1-weighted technique in which a curve of dynamic signal intensity (i.e., enhancement) versus time is generated. The slope of enhancement reflects the degree of permeability of capillaries. The intensity of initial enhancement reflects tissue vascularity; fractional blood volume (fBV) can be extrapolated [Roberts et al., 2000]. fBV is a fairly good estimate of the rCBV data obtained with susceptibility perfusion [Haroon et al., 2007].
Positron Emission Tomography
Positron emission tomography (PET), with use of (18)F-fluorodeoxyglucose (FDG), allows for the evaluation of regional cerebral glucose metabolism. PET is useful in the determination of tumor metabolism/grade and of tumor extent, and to distinguish persistent or recurrent tumor from radiation necrosis [Pirotte et al., 2007]. Metabolic changes can be measured with a spatial resolution approaching 2–3 mm in the newer scanners. Images can be co-acquired with a CT scan (CT-PET), or can be co-registered with MR images using fusion software. Metabolic activity of tumor sites can be determined with quantitative, as well as qualitative (comparing FDG uptake in a tumor to uptake of normal gray or white matter), assessment. Higher-grade tumors tend to be more glucose-avid than lower-grade tumors, except for juvenile pilocytic astrocytomas, which show very high glucose uptake. For lesions that do not show abnormal glucose uptake, of (11)C-methionine PET scanning is helpful to differentiate neoplastic from non-neoplastic types. Thallium-201 single-photon emission CT (SPECT) now has a decreasing role in evaluation of pediatric brain tumors, given its more limited anatomic resolution.
Staging and Stratification
Pre- and postoperative staging is the cornerstone of therapy for most embryonal childhood central nervous system tumors and, to a lesser extent, glial neoplasms [Gajjar et al., 2004; Packer et al., 1999a]. The utility of staging for both determination of prognosis and decisions concerning most appropriate therapy has been best shown for medulloblastoma, as staging has been an accepted component of management since the mid-1980s. The first widely accepted staging system was the “Chang” classification schema, which has been extensively modified over time. All these schemas share the property of being based on degree of surgical resection or, more specifically, the amount of tumor left after surgery and the extent of tumor spread outside the primary tumor site. The latter is determined by a combination of results from MRI of the entire brain and spine, and cerebrospinal fluid cytological examination, preferably from the lumbar space, if deemed medically safe [Deutsch and Reigel, 1980; Gajjar et al., 2004; Packer, 1999; Packer et al., 1999a]. Embryonal tumors and glial neoplasms rarely spread to non-neural sites; thus, evaluation for disease outside the primary nervous system, at the time of diagnosis, is usually not indicated. The classical TNM (tumor, nodes, metastasis) staging system used for non-central nervous system tumors is replaced by a TM system for brain and spine tumors. Although the need for lumbar cerebrospinal fluid cytological examination has been debated, studies have shown that it provides complementary information to imaging results [Packer et al., 1999a; Zeltzer et al., 1999].
A complicating factor has been the fact that the performance and interpretation of imaging results have been variable in national and international studies [Packer et al., 2006]. In the most recent medulloblastoma studies, approximately 10 percent of studies were inadequately performed, primarily due to movement artifact or incomplete imaging (primarily of the spine), and another 5–10 percent of studies are considered, upon central review, to be incorrectly interpreted. Tumors that are nonenhancing at the primary site may result in nonenhancing dissemination along the neuroaxis, which is difficult to assess and leads to under-assessment of the extent of disease. In addition, normal superficial spinal cord vascularity can be misinterpreted as leptomeningeal enhancement.
Histology has been incorporated into risk stratification for many tumor types. For embryonal tumors, such as medulloblastoma, histological features, including desmoplasia/nodularity in infants and anaplasia in infants and older children, are now increasingly accepted as important components of staging and risk-stratification classifications, although the independent significance of such features has been related variably to outcome [Eberhart et al., 2004; Giangaspero et al., 1999, 2006; Grotzer et al., 2007]. For glial tumors or ependymomas, histological features that have been translated into grades are an accepted component of classification and are used both for risk stratification and for determinations of treatment.
It has become clear for the embryonal tumors, especially medulloblastoma, that molecular genetic parameters provide extremely important information [Ellison et al., 2005; Fernandez-Teijeiro et al., 2004; Gajjar et al., 2004; Grotzer et al., 2007; Pfister et al., 2009; Ray et al., 2004]. In retrospective studies, a host of chromosomal and molecular genetic findings had been related to outcome. In some cases, these findings show a strong correlation with other parameters, such as the tendency to disseminate or features of anaplasia, but in other studies they are of independent prognostic value.
Clinical Presentation
Nonlocalizing Signs and Symptoms
Nonlocalizing signs and symptoms predominate early in the course of illness in almost all types of childhood nervous system tumors. This is due to a variety of factors, including the predilection of childhood brain tumors to arise in the posterior fossa and cause obstruction of cerebrospinal fluid early in the course of illness, resulting in the nonspecific symptoms of headaches, nausea, vomiting, and lethargy. Separation of headaches caused by neoplasms, as opposed to the more common headaches children develop, can be extremely difficult [Glaser et al., 1971; Honig and Charney, 1982]. The majority of children with brain tumors, especially those with posterior fossa tumors and large supratentorial tumors, will have headaches by the time of diagnosis. However, headaches are notoriously nonspecific early in the course of illness. A change from a nonspecific headache pattern to morning headaches should raise concern that a mass lesion is present. Headaches that wake a child from sleep also suggest the possibility of increased intracranial pressure. Although this is true in general terms, recognizing such changes, in practicality, can be quite difficult for the child or family, and migraine headaches can wake a child from sleep. Headaches are especially difficult to characterize in very young children.
Localizing Symptoms
Supratentorial Tumors
The diagnosis of primary central nervous system tumors is usually easier when there are associated localizing signs and symptoms (Table 83-1). Supratentorial tumors present with symptoms and signs similar to those seen in older patients and adults. Headaches are the most common initial complaint and are often nonlocalizing early in the course of illness [Hirsch et al., 1989]. Seizures are the second most common symptom encountered and approximately one-quarter of children with supratentorial tumors will have seizures as their initial symptom. This is especially true with tumors of the frontal or temporal lesion. Slower-growing cortical tumors, especially gliomas, are more likely to result in convulsions early in the course of illness. Although patients with more aggressive cortical gliomas may have seizures, focal neurologic deficits, such as hemiparesis or, in a patient amenable to careful examination, hemisensory loss or visual field loss, are more common.
Posterior Fossa Tumors
Five tumor types are most likely to occur in the posterior fossa, as noted in Table 83-2. Since cerebellar astrocytomas are more laterally placed and are slower-growing tumors, their symptoms tend to be more indolent than those of medulloblastomas, which fill the fourth ventricle early in the course of illness and result in obstructive hydrocephalus. However, midline cerebellar astrocytomas may mimic medulloblastomas [Packer et al., 1999a]. Brainstem gliomas are an insidious lesion and are likely to cause cranial nerve neuropathies associated with motor, cerebellar, and sensory deficits. Ependymomas are the great mimickers of other tumor types in the posterior fossa and may present as slow-growing, medulloblastoma-like tumors or, if the tumor involves the cerebellopontine angle, can mimic brainstem gliomas and result in multiple cranial neuropathies, especially sixth, seventh, and eighth nerve palsies.
Midline Tumors
The infiltrating nature of midline tumors may also make diagnosis difficult [Albright et al., 1984; Glaser et al., 1971]. Many of these neoplasms are low-grade and classically result in insidious, nonspecific, and often nonlocalizing symptoms early in the course of illness. In the school-age child, infiltrating tumors and occasionally midline compressive masses, such as craniopharyngiomas, may also result in the nonspecific finding of declining academic performance, associated with fatigue and personality change.
General Aspects of Treatment
Surgery
Surgery remains the initial step in the treatment for the overwhelming majority of childhood central nervous system tumors, and the degree of resection, even for malignant neoplasms that require other forms of adjuvant therapy, is often an important factor in determining outcome [Albright et al., 1995; Campbell et al., 1996; Chen et al., 2007; Horn et al., 1999; Packer et al., 1999a; Roujeau et al., 2007]. As regards diagnosis, attainment of a representative portion of the neoplasm for histological and increasingly biological analysis is crucial. Exceptions to the need for tissue for diagnosis include neuroradiographic characteristic diffuse intrinsic brainstem tumors; chiasmatic/hypothalamic infiltrating gliomas in children with neurofibromatosis type 1 and possibly for those without; and germ cell tumors, predominantly mixed germ cell tumors, which secrete diagnostic proteins into cerebrospinal fluid and blood.
For some tumor types, surgery is curative and complete resection obviates the need for alternative therapies [Schneider et al., 1992]. This is primarily true for low-grade gliomas, especially pilocytic astrocytomas of the cerebellum or cerebrum, gangliogliomas, other mixed neuronal-glial tumors, and possibly supratentorial low-grade ependymomas [Poussaint and Rodriguez, 2006]. For other, more aggressive tumor types, surgery may be a crucial component of multimodality treatment, as outcome is statistically better for patients with nondisseminated medulloblastomas, posterior fossa ependymomas, and cortical malignant gliomas if total or near-total resection can be obtained [Albright et al., 1989; Packer et al., 1992; Wisoff et al., 1998]. For those tumors that located in eloquent areas of brain or are disseminated, the preoperative assessment of whether “total” resection or an attempt at total resection provides a survival benefit is usually the prime determinant of whether aggressive tumor removal is tried or less aggressive approaches, including open or stereotactic needle biopsy, will suffice. Stereotactic biopsy techniques, be they CT- or MRI-guided, are highly accurate in targeting deep-seated lesions and are associated with morbidities of less than 5 percent and mortalities of less than 1 percent [Broggi et al., 1983]. The advent of biologic therapies, often requiring fresh frozen tissue for molecular analysis, has resulted in the need, in certain circumstances, for additional tissue that cannot be obtained by needle biopsy alone.
Intraoperative Techniques
For those tumors in which extent of resection is a major determinant of outcome, improvements in neurosurgical technology have made “total resections” more likely and possibly safer. Progressive improvements in the operative microscope, which facilitates the visualization of the interface between the neoplasm and normal brain, have allowed for more extensive resections. Frame-based and frameless stereotactic guidance systems are now routinely used for improved preoperative targeting of the tumor and to decrease injury to the surrounding normal brain [Barnett et al., 1993; Kelly, 1990]. Similarly, ultrasonic guidance has allowed for better intraoperative localization. A recent addition to the neurosurgical armamentarium has been the availability of intraoperative MRI, which allows real-time evaluation for extent of surgical resection [Martin et al., 2000]. Intraoperative MRI is performed both with low-field units, which require interruption of the operation for set-up and visualization, and with high-field units, which utilize MRI-compatible surgical equipment and allow the operation to be carried out essentially within the MRI unit. These techniques are most useful for circumscribed benign tumors; however, for infiltrating neoplasms the extent of resection of the neoplasm continues to be difficult to determine.
In functionally critical areas of the brain, intraoperative monitoring of the visual, auditory, and somatosensory pathways can be used to improve the safety of tumor resection [Schneider et al., 1993]. Similarly, for intrinsic and extrinsic brainstem lesions, intraoperative monitoring of cranial nerve function is an important adjunct [Berger et al., 1990]. Maintenance of function intraoperatively does not assure normal function postoperatively, as edema and apparent delayed damage may occur. Awake craniotomy can be used in older children, but it is usually of limited utility in the majority of pediatric brain tumor cases.
Perioperative Considerations
It has been suggested by many that improvements in preoperative and postoperative care have resulted in lower surgical morbidity and mortality. However, what constitutes acceptable morbidity is extremely difficult to define, and obviously, one of the goals of surgery is to have no mortality and limited morbidity. Preoperatively, in children with increased intracranial pressure, every attempt is made to try to decrease pressure and stabilize the patient. In patients with hydrocephalus, this often includes the placement of a temporary external ventricular drain [Dias and Albright, 1989]. In most centers, in patients with posterior fossa tumors and hydrocephalus, a temporary drain is utilized and, within 24–72 hours, the tumor is removed. Removal may restore adequate cerebrospinal fluid flow to avoid and the need for permanent ventriculoperitoneal diversion (ventriculoperitoneal shunting). The minority of patients with posterior fossa lesions, usually fewer than 40 percent, require permanent external drainage. Third ventriculostomy has been proven to be a useful adjunct in reducing the need for permanent ventriculoperitoneal drainage, but the effect of this technique on long-term outcome has not yet been shown [Hopf et al., 1999; Jones et al., 1990].
In children with cortical tumors, prophylactic anticonvulsants are often utilized, even if there is no history of seizures. The need for such treatment and the duration of treatment, postoperatively, are unclear [Foy et al., 1992]. Phenytoin is often used preoperatively due to its lack of significant sedation in most patients, its ability to be given and loaded intravenously, and the ease of switching from intravenous to oral dosing. In patients on steroids, due to drug metabolism interactions, maintenance of therapeutic levels of phenytoin may be difficult, especially when patients are switched to oral dosing. In children who may require postoperative chemotherapy, levetiracetam is an effective alternative, since it also can be used orally or intravenously, and has the added advantage that it does not undergo hepatic metabolism and, thus, does not interact with the metabolism of most antineoplastic chemotherapeutic agents.
Postoperative Morbidity
Surgery in the suprasellar area is frequently associated with increased postoperative visual impairments, which may include decreases in visual acuity and increased deficits in visual fields [Sanford, 1994]. Hypothalamic abnormalities and significant hormonal deficits secondary to disruption of the hypothalamic–pituitary axis also frequently occur. In large tumors that extend to the subfrontal area, removal of the tumor may cause additional damage and both early and late emotional/personality changes. Hypothalamic injury may also result in abnormalities in satiety control and excessive weight gain, primarily in children with craniopharyngiomas.
Surgery for posterior fossa tumors may result in direct brainstem damage, with resultant cranial nerve deficits and damage to the cerebellum or cerebellar peduncles, with increased ataxia and dysmetria. Such direct brainstem damage may be difficult to separate from the increasingly recognized posterior fossa mutism syndrome, which is a constellation of the delayed onset of mutism (characteristically, a few hours to a day after surgery), hypotonia, cerebellar deficits, supranuclear palsies, emotional lability, and severe irritability [Doxey et al., 1999; Packer et al., 1992; Pollack, 1997]. The exact pathogenesis of the posterior fossa mutism syndrome is unknown. Seen primarily, but not exclusively, after medulloblastoma resection, it is thought to be secondary to unilateral or bilateral cerebellar dentate nuclei damage and/or disruption of critical pathways between the cerebellum and the cortex, especially the premotor and supplementary motor cortices. This syndrome results in permanent sequelae in approximately 50 percent of initially symptomatic patients, including persistent abnormalities in tone, coordination, speech, and cognitive abilities [Robertson et al., 2006; Wells et al., 2008]. The presence of lower motor neuron cranial nerve deficits or MRI features of immediate brainstem injury or cerebellar damage makes direct brainstem injury more likely. Postoperative brainstem edema has been reported to be more common in patients with medulloblastoma who develop posterior fossa mutism syndrome compared to patients who do not.
Radiation Therapy
Radiation therapy is an integral component of treatment for many forms of childhood brain tumors. For children with benign tumors, its use is usually limited to those tumors that are not amenable to gross total resections. For children with more aggressive or malignant tumors, radiation therapy has been the backbone of most curative and palliative therapies, even in those patients whose tumors are totally resected. For medulloblastoma, other embryonal tumors, and germ cell tumors, delivery of radiotherapy to the entire neuroaxis (craniospinal radiotherapy), supplemented with boost radiotherapy to the primary tumor site, is routinely employed to treat neuroaxis sanctuary disease and prevent exoprimary disease relapse, even in patients without evidence of tumor dissemination [Packer et al., 1999b]. Craniospinal radiotherapy has been associated with improved survival, but also significant long-term endocrinologic, growth, and cognitive sequelae. In very young children, especially infants, radiotherapy may result in even more severe sequelae and attempts have been made to delay, if not obviate, the need for radiotherapy by the use of chemotherapy. Chemotherapy is often utilized with radiotherapy in older patients, not only to increase survival, but also to decrease the dose or volume of radiation therapy required for disease control or cure.
[/not-level-membership-for-neurology-category]
