Tumor-Induced Osteomalacia and Rickets
Modern understanding of rachitic syndromes, originally referred to as vitamin D-resistant rickets, is based on the identification of a novel phosphate-regulating homeostatic system and its underlying diverse genetic background.7,10 Essential to this understanding were discoveries of two mutated genes involved in the development of somewhat overlapping yet distinct phosphaturic disorders. The first one involved the identification of phosphate regulating endopeptidase homolog on chromosome X (PHEX) as the mutated gene in X-linked phosphatemia.56 The second was the discovery that a mutant fibroblast growth factor 23 (FGF23) plays a role in the development of tumor induced osteomalacia and rickets as well as an autosomal dominant X-linked variant of hypophosphatemic rickets. A more complete list of phosphatemic disorders with their genetic background and biochemistry is provided in Table 22-1.7 Their detailed description is beyond the scope of this book, and this chapter focuses primarily on tumor-induced osteomalacia and rickets.
TABLE 22-1
Biochemistry and Genetics of Hypophosphatemic Disorders
| Calcium Metabolism | Phosphate Metabolism | Vitamin D Metabolism | Mutation | |||||
| SERUM CALCIUM | URINE CALCIUM | SERUM PTH | GI CALCIUM ABSORPTION | SERUM PHOSPHATE | TMP/GFR | SERUM 1,25(OH)2D | GENE | |
| FGF23-mediated | ||||||||
| XLH | N | ↓ | N, ↑ | ↓ | ↓ | ↓ | (↓) | PHEX |
| ADHR | N | ↓ | N | ↓ | ↓ | ↓ | (↓) | FGF23 |
| ARHR1 | N | N, ↓ | N, ↑ | ? | ↓ | ↓ | (↓) | DMP1 |
| TIO | N | N, ↓ | N, ↓, ↑ | ↓ | ↓ | ↓ | ↓ | FGF23 |
| ARHR2 | N | N, ↓ | N | ? | ↓ | ↓ | (↓) | ENPP1 |
| McCune-Albright | N, ↑ | N | N, ↑ | ? | N, ↓ | N, ↓ | N, ↓ | GNAS1 |
| ENS | N, ↑ | N, ↓ | N, ↑ | ? | N, ↓ | N, ↓ | (↓) | ? FGFR3* |
| NF | N, ↓ | N, ↓ | N | ? | N, ↓ | N, ↓ | N, (↓) | NF1 |
| OGD | N | N | N | ? | ↓ | ↓ | (↓) | FGFR1 |
| HRHPT | N, ↑ | N, ↑ | ↑ | ? | ↓ | ↓ | (↓) | KL (klotho) |
| Non-FGF23-mediated | ||||||||
| XLRH | N | ↑ | N, ↓, ↑ | N, ↑ | ↓ | ↓ | ↑ | CLCN5 |
| HHRH | N, ↑ | ↑ | N, ↓ | ↑ | ↓ | ↓ | ↑ | SLC34A3 |
| Fanconi | N, ↓, ↑ | N, ↑ | N | ? | N, ↓ | N, ↓ | N, ↓ | Various (SLC34A1) |
| NHERF1 | N | ? (stones) | N | ? | ↓ | ↓ | N, ↑ | NHERF1 |
The cause-and-effect relationship between certain mesenchymal tumors and severe osteomalacia or rickets was first recognized in 1959 by Prader et al.54 The original description of this peculiar paraneoplastic syndrome was provided in 1947 by McCance,41 who reported a case of resolution of vitamin D-resistant rickets after removal of “degenerated osteoid tissue.”
Tumor-Induced Osteomalacia
Tumor-induced osteomalacia (oncogenic osteomalacia) is a clinicopathologic entity in which vitamin D-resistant osteomalacia or rickets occurs in association with a bone or soft tissue tumor.1,64,72 These tumors have exhibited a wide spectrum of histologic features that have only recently been gathered under a unifying concept of phosphaturic mesenchymal tumors.20,82,83 Overlapping clinical presentation and biochemical abnormalities can be present in X-linked hypophosphatemia and autosomal dominant hyphosphatemia.7,37 A similar syndrome has also been reported in association with the linear sebaceous nevus syndrome and fibrous dysplasia.2,15,25,29
Clinical Data
The majority of patients with tumor-induced osteomalacia are adults; two thirds are age 30 years or older.36,45,55,58,63,74,89 The ages of reported patients range from 7 to 73 years, and the male-to-female ratio is 1.2 : 1. The typical presentation is a gradual onset of bone pain in weight bearing areas such as the legs, ankles, hips, and back. Some patients have pathologic fractures. The pain is frequently accompanied by generalized muscular weakness, leading to a severely debilitated state in which the patient is bedridden. Hence, in some patients an initial misdiagnosis of a primary muscular disorder is made. Symptoms are usually present anywhere from 3 months to 17 years before diagnosis. In some cases, a mass was noted up to 20 years before the onset of generalized symptoms. Occasionally an asymptomatic bone tumor is identified in radiographs in a patient with a diagnosis of vitamin-D resistant rickets.40 Children may initially have malaise, gait disturbance, swollen joints, and genu valgum or varum.
Characteristic metabolic abnormalities include a low serum phosphorus level (2.2 to 0.33 mg/dL), normal or slightly low serum calcium level, and variably elevated alkaline phosphatase activity (occasionally up to 1700 to 2300 IU). The urinary phosphate level is usually elevated, and most cases exhibit decreased tubular reabsorption of phosphate (23% to 70%). Urinary calcium levels tend to be low. Serum levels of 1,25-dihydroxyvitamin D3 are low or detectable, whereas serum levels of 25-hydroxyvitamin D3 are generally normal. In addition, aminoaciduria and glycosuria with normal serum glucose levels may be present.
Radiographic Imaging
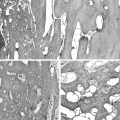
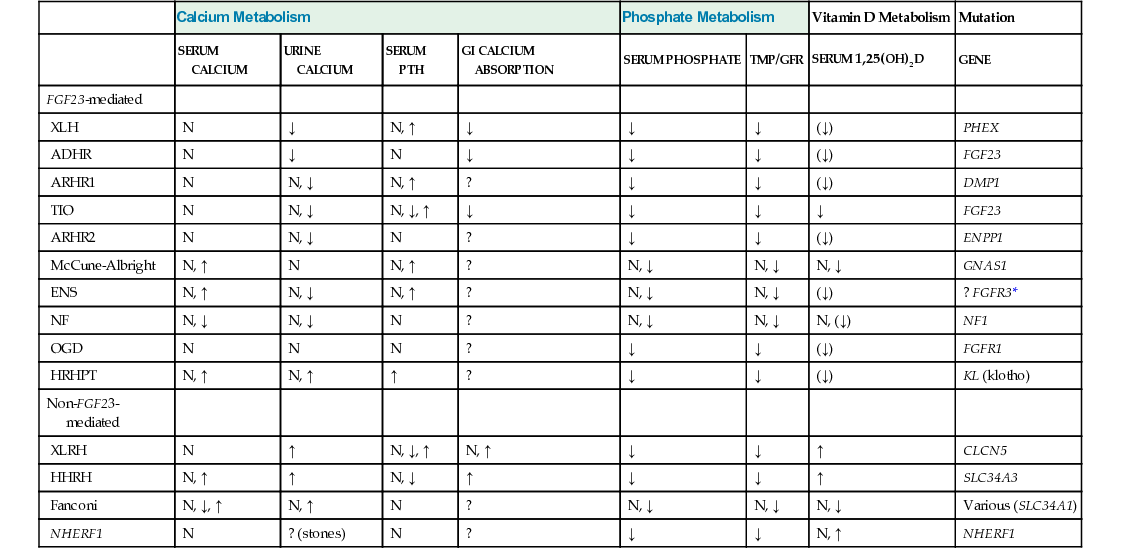
In skeletally immature patients, radiographs show widening of growth plates and radiolucent zones in the metaphyses consistent with rachitic changes (Fig. 22-1). Lower-extremity long bones may show bowing deformities. In skeletally mature patients, radiographic features consist of generalized osteopenia with Milkman’s pseudofractures or Looser’s lines, characteristic of osteomalacia. In addition, skeletal radiographs may reveal the presence of an asymptomatic or symptomatic tumor.11 Pathologic fractures in adults, particularly of the femoral neck, are relatively common.
Pathologic Findings
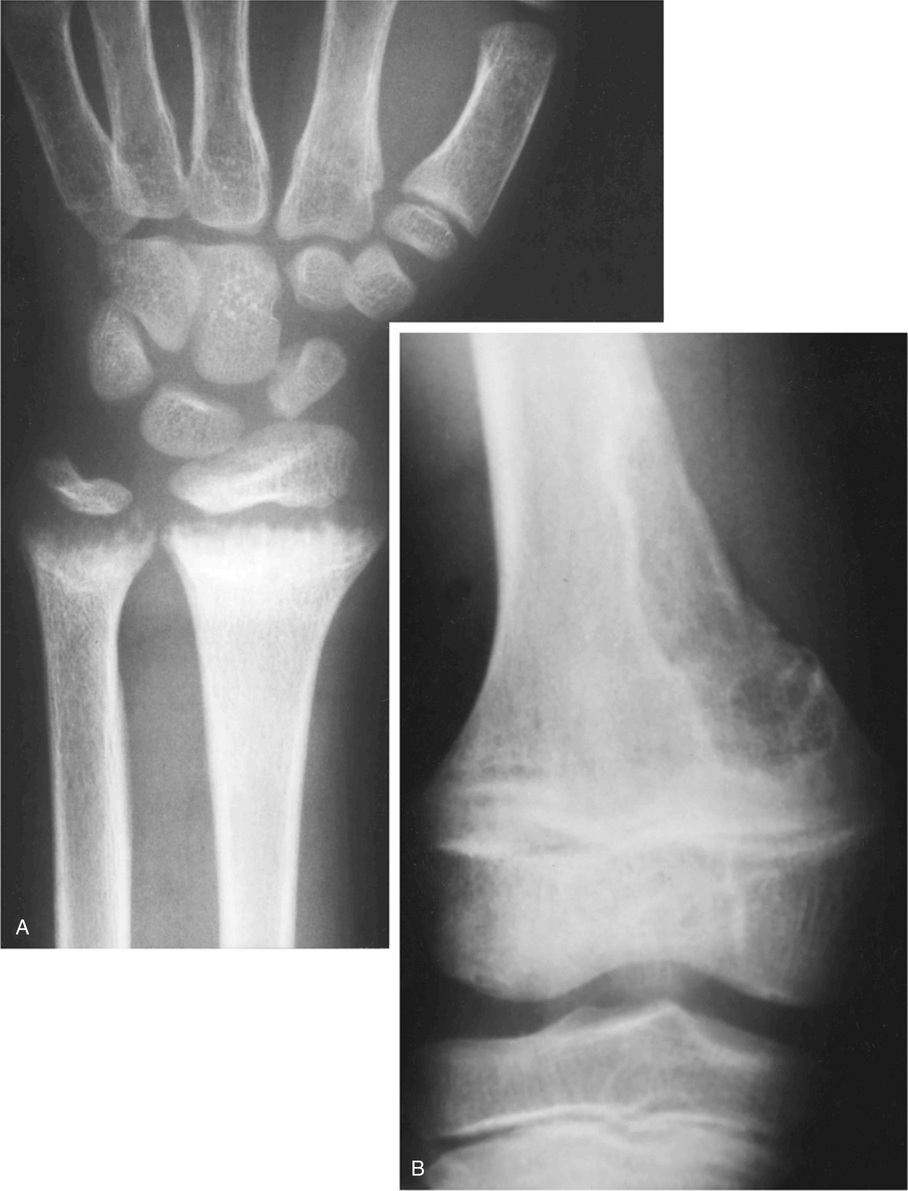
Histomorphometry on undecalcified bone sections shows hyperosteoidosis with increased osteoid volume and increased width of osteoid seams. In approximately 50% of cases, the tumor is primary in bone.49 Half of the cases involving bone occurred in the long bones of the lower extremities, particularly in the femur and tibia. The second most common location of osseous tumors is in the craniofacial bones.31,47 Soft tissue tumors comprise the other half of all cases and follow the same pattern of anatomic distribution as skeletal tumors—they occur in the soft tissues of the lower extremities and craniofacial region.
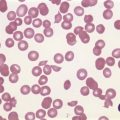
Histologically, 40% of reported cases are vascular tumors (Table 22-2). Of these, hemangiopericytoma is the most common, representing about half of the vascular tumors and approximately 20% of the total (Fig. 22-2).6,24,26,42,60,65,77,83,84,89 Primary hemangiopericytoma of bone is extremely rare and comprises 0.08% of all primary tumors of bone.79 Other vascular tumors include hemangioma, angiofibroma, low-grade hemangioendothelioma, and very rarely, high-grade angiosarcoma.1,13,39,43,63,81,89
TABLE 22-2
Types of Tumors Producing Osteomalacia and Rickets
| Histologic Type | No. of Cases |
| Vascular tumors | 31 |
| Hemangiopericytomas | 14 |
| Hemangiomas | 7 |
| “Fibrovascular” lesions | 6 |
| Angiosarcomas | 2 |
| Hemangioendothelioma | 1 |
| Angiolipoma | 1 |
| Giant cell lesions | 9 |
| GCT of bone | 2 |
| Soft tissue GCT | 2 |
| Other | 5 |
| Malignant GCT | |
| PVNS | |
| GCRG | |
| Giant cell variant of soft-part chondroma | |
| Giant cell MFH | |
| Nonossifying fibromas | 8 |
| “Mesenchymal” tumors (malignant) | 9 |
| Osteoblastomas | 4 |
| Cartilaginous | 2 |
| Chondroma | 1 |
| Mesenchymal chondrosarcoma | 1 |
| Odontogenic | 2 |
| Undifferentiated sarcomas | 2 |
| Other | 5 |
| “Degenerate osteoid tissue” | |
| Neuroma | |
| Osteosarcoma | |
| Fibrous xanthoma | |
| “Vascular” fibrous histiocytoma |
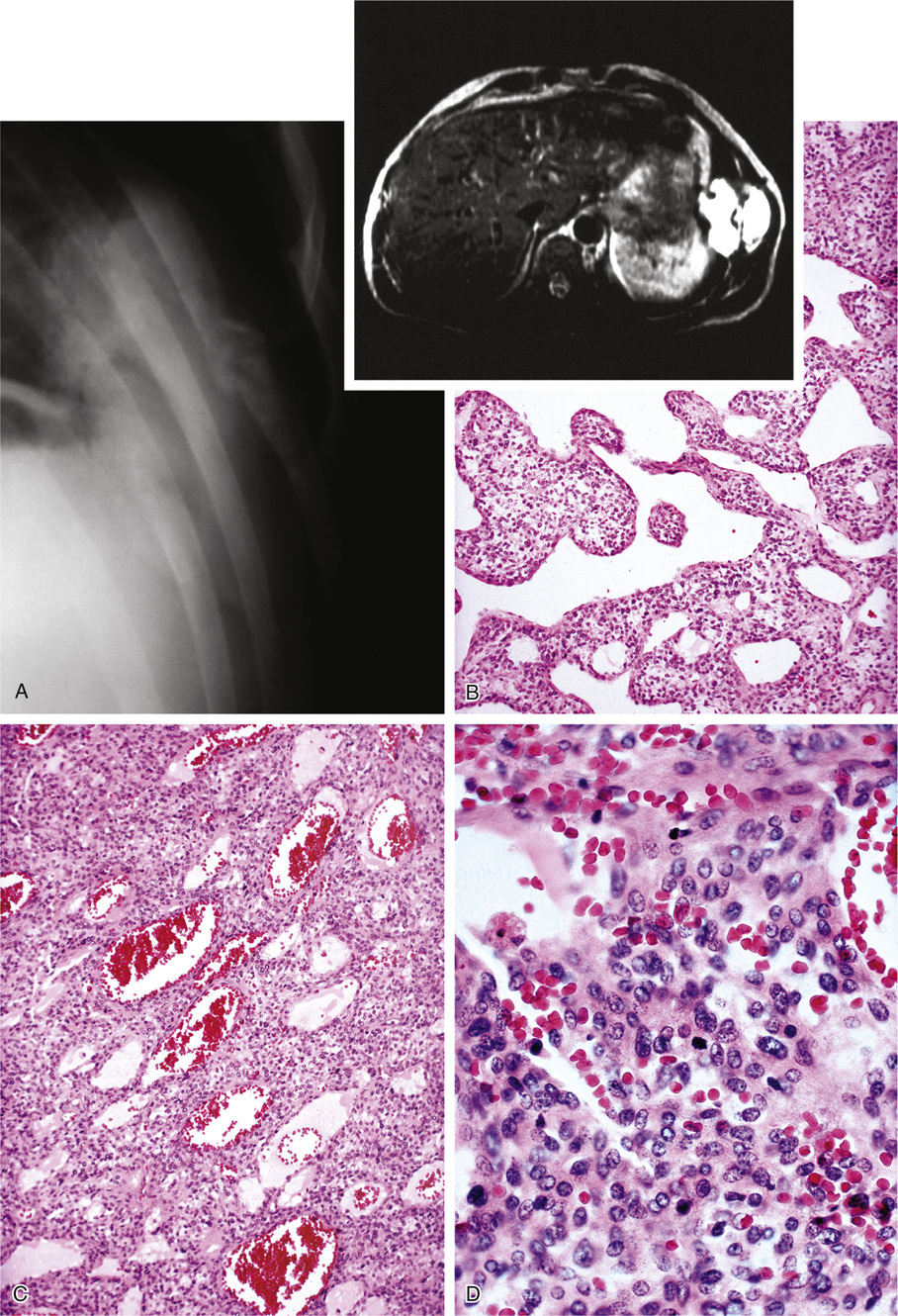
The second group of tumors associated with rickets or osteomalacia can be histologically classified as having features of fibrohistiocytic and giant cell lesions exhibiting prominent vasculature (Fig. 22-3). The specific lesions of this group are nonossifying fibromas and giant cell reparative granuloma, giant cell tumor, and pigmented villonodular synovitis.3 A small proportion of reported tumors represent malignant lesions, which include osteosarcoma and mesenchymal chondrosarcoma.28,46,48,75,76,87 Some of the tumors defy clear-cut classifications and are descriptively designated as fibrovascular or mesenchymal. The majority of tumors are relatively small—within the range of 1 to 4 cm—but occasionally measure 7 cm or larger. Rarely malignant tumors such as osteosarcoma are associated with rickets or osteomalacia (Fig. 22-4).
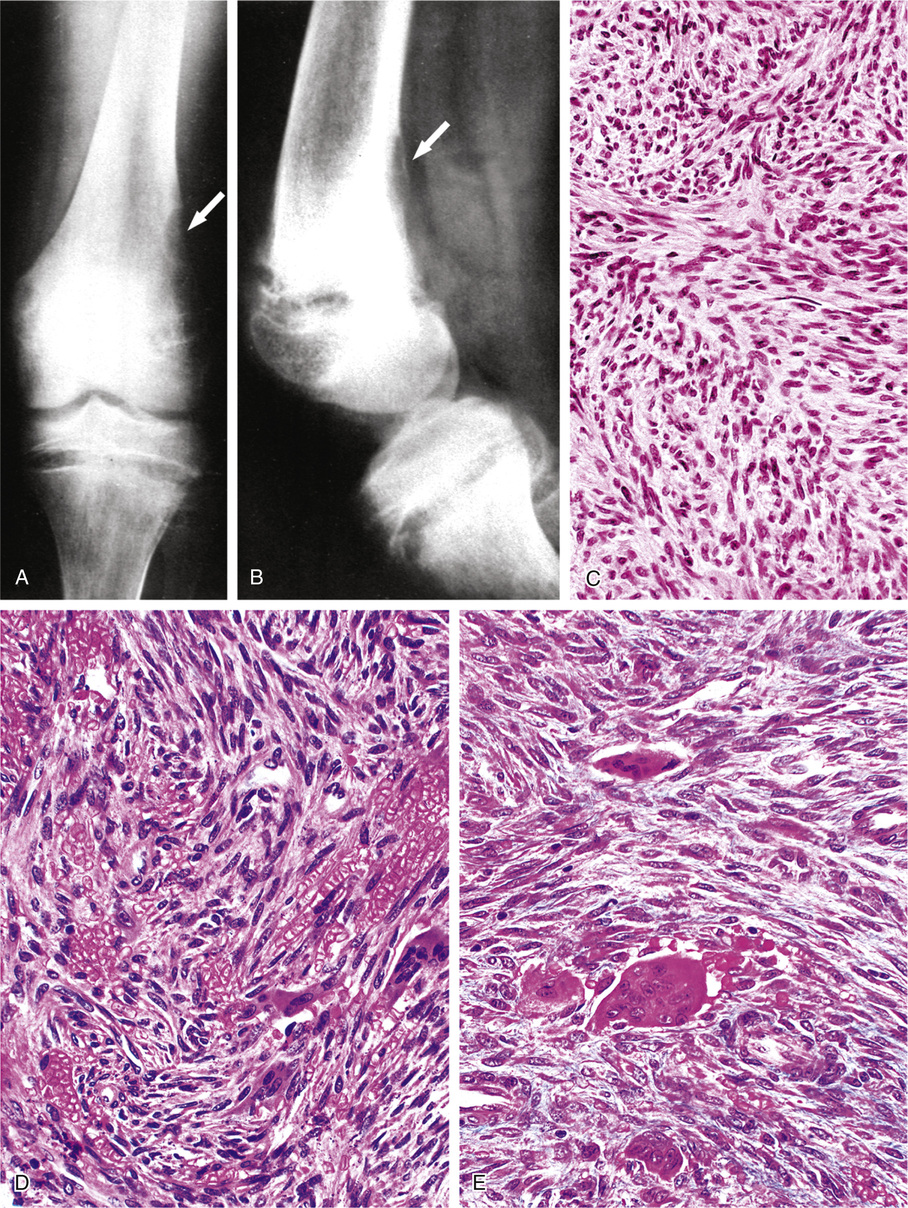
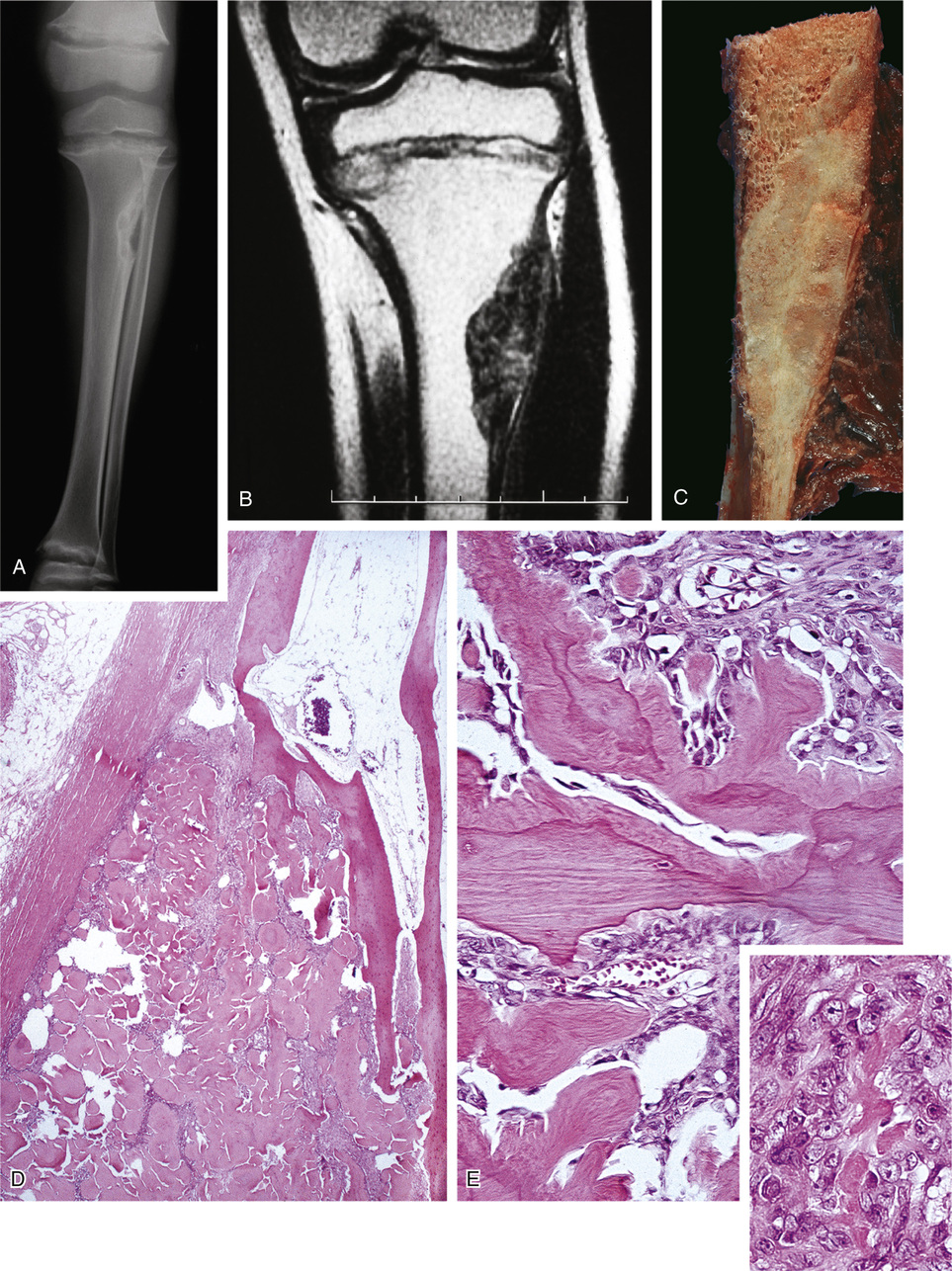
Ultrastructurally or immunohistochemically, the tumors inducing rickets or osteomalacia do not differ histologically from similar lesions not associated with this syndrome (see Fig. 22-2).
It was recognized by several authors that many of these tumors show a distinctive morphology and have microscopic features in common.8,18,50,63 In 1987, Weidner and Santa Cruz first introduced the term phosphaturic mesenchymal tumor, mixed connective tissue variant, proposing that these tumors of wide-ranging histologic patterns had common features of spindle cells, prominent blood vessels, osteoclast-like giant cells, cystic spaces, metaplastic bone, and cartilage-like matrix, which indicated that they represent a distinct clinicopathologic entity.83
A collaborative multiinstitutional study of 32 examples of phosphaturic mesenchymal tumors in which reverse transcription polymerase chain reaction (RT-PCR) demonstration of FGF23 gene expression in two cases and immunohistochemical demonstration of FGF23 protein expression in 17 cases has strengthened the concept of a recognizable clinicopathologic entity with a demonstrable phosphaturic factor in the tumor cells. These authors pointed out that wider recognition of the histologic features of phosphaturic mesenchymal tumors could occasionally identify patients with previously undiagnosed osteomalacia.22
Treatment and Behavior
Conventional treatment for rickets or osteomalacia consisting of increasing doses of vitamin D or phosphate provides only transient improvement in approximately 15% of patients with oncogenic osteomalacia.3,4,12,51,52 Removal of the tumor is required and results in complete cure of osteomalacia or rickets in approximately 80% of patients.14,84 If the tumor cannot be completely removed because of its location, partial removal is necessary to support the medical treatment to reverse symptoms and skeletal changes. Recurrence of the original tumor (benign or malignant) results in the reappearance of the chemical findings with or without accompanying clinical symptoms or skeletal changes. In fact, chemical monitoring can be used for identification of local recurrence or metastasis.
Correction of the laboratory abnormalities usually occurs from 1 day to 20 months after surgery. Symptoms may disappear from the first day to 1 year after resection.43,55,67,71 Continuation of medical treatment (vitamin D, phosphate) is required in patients with advanced skeletal changes and persistent symptoms.13,44 In some cases the persistence of symptoms, chemical abnormalities, and skeletal changes can be attributed to incomplete removal of the tumor.18,57 In malignant tumors, local recurrence and metastases are associated with reappearance of chemical changes, skeletal abnormalities, and clinical symptoms of rickets or osteomalacia.38,61,62
Follow-up of reported cases varies from 3 days to 30 years. In eight cases, the asymptomatic patients were followed for more than 5 years. Among the 10 histologically malignant tumors, only 1 recurred and 2 metastasized, leading to death as a result of generalized sarcomatosis.38,59,83 Another death occurred in the case of nasal hemangiopericytoma and was attributed to persistent bleeding.9 A reported complication of medical therapy with calcitriol and phosphates is the development of parathyroid autonomy (tertiary hyperparathyroidism).
Pathogenesis
A wide variety of causes may give rise to osteomalacia or rickets, and their description is beyond the scope of this book. In developed countries, however, given that inadequate nutrition is infrequent, some of the rare causes are X-linked hypophosphatemia and autosomal dominant hypophosphatemia.7,10,17 Oncogenic osteomalacia presents with analogous symptoms and chemical abnormalities requiring large doses of vitamin D and phosphate for mild, transitory improvement.21 However, serum calcium levels usually are normal in contrast to other forms of osteomalacia.
After the tumor is excised, a dramatic reversion to normal bone metabolism implies that the tumor is directly responsible for skeletal changes. Immunoassays performed on saline tumor extracts show no evidence of parathyroid hormone or calcitonin.61,62 Elaboration by the tumor of a “phosphaturic substance” that would act on the proximal renal tubule and cause a renal phosphate leak has been proposed.35,43,50,61,62 Some investigators were able to induce phosphaturia in experimental animals by injection of saline tumor extract that did not contain parathyroid hormone or calcitonin.2,34,53 A substance affecting phosphaturia alone would not explain the low or undetectable levels of 1,25-dihydroxyvitamin D3.16 It has been postulated that the decreased levels of 1,25 dihydroxyvitamin D3 are due to elaboration and secretion by the tumor of a factor that would inhibit the hydroxylase, which acts on the mitochondria of the proximal renal tubule. Phosphaturia would be a direct effect of 1,25-dihydroxyvitamin D3 deficiency, because it has been proved that the latter increases renal phosphate resorption.32 Transplantation of soft tissue hemangiopericytoma causing osteomalacia into athymic mice resulted in hyperphosphatemia, high serum alkaline phosphate levels, and increased urinary phosphorus excretion.45 On the other hand, decreased levels of the metabolically active form of vitamin D3 alone do not fully account for the phosphate depletion because increasing the dosage does not correct the defect.23,62,63,73,80,86
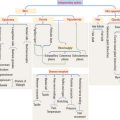
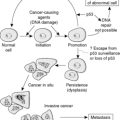
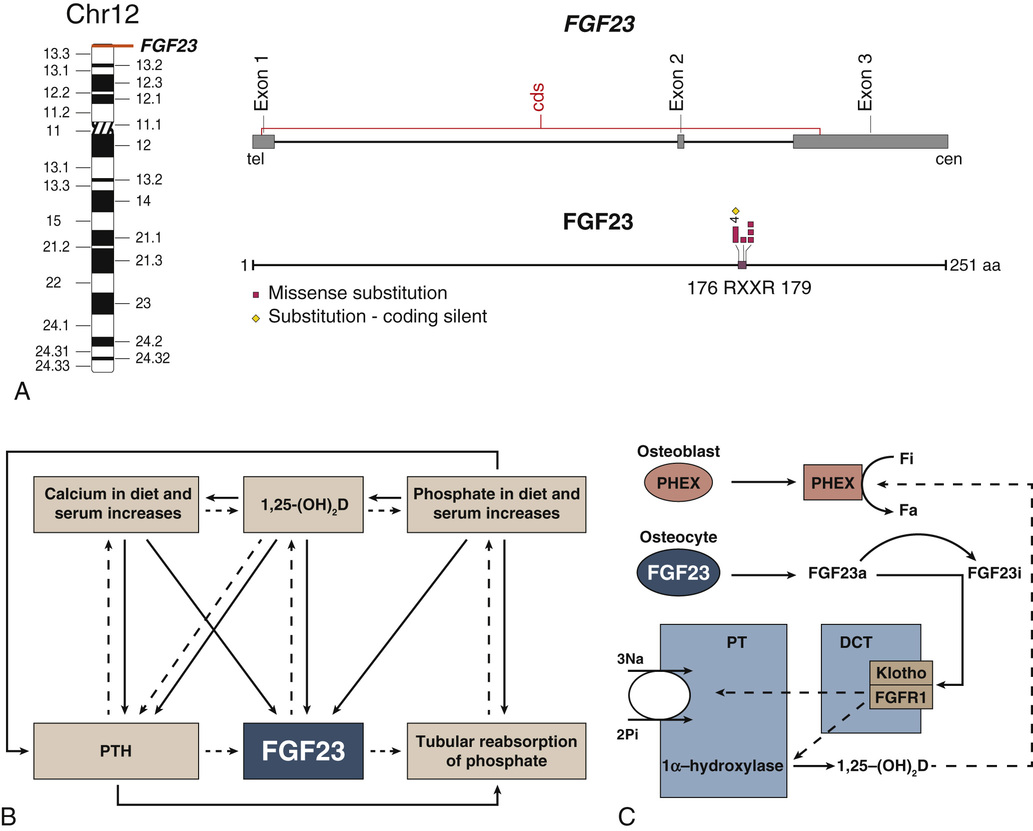
The circulating phosphaturic factor secreted by the tumor cells was originally termed phosphatonin but is now recognized to represent fibroblast growth factor-23 (FGF23).17,22,32,33,70 Missense mutations in the gene encoding FGF23 are associated with the development of tumor-induced osteomalacia and rickets in the majority of cases. They also play a role in autosomal dominant hypophosphatemic rickets, which has clinical manifestations virtually identical to those of X-linked hypophosphatemic rickets and tumor-induced osteomalacia. Mutations of the PHEX gene are believed to play a role in the development of X-linked hypophosphatemia. The FGF23 gene, located on human chromosome 12p1,3 is one of 23 known members of the FGF family.68,70 The full-length FGF23 is an 26-kDa (251 amino acids) protein with an NH2-terminal region containing the FGF homology domain and a novel 71-amino acid COOH terminus.70 Cleavage of FGF23 at the 176-RXXR-179 motif generates biologically inactive NH2- and COOH-terminal fragments.76 The basic physiologic defects in tumor-induced osteomalacia and rickets depends on increased activity of circulating mutant FGF23 produced by tumor cells that cannot be cleaved into inactive fragments.69 The increased FGF23 activity reduces the expression of NaPi-2α and β (type 2 sodium phosphate cotransporters) on the apical surface of renal proximal tubules.37 The signal transduction of FGF23 is accomplished by forming a complex with FGF receptors (FGFR1c and FGFR3c) interacting with the klotho protein.27 In physiologic states, FGF23 plays a central role in phosphate homeostasis, interacting with 1,25-(OH)2D, parathyroid hormone, and tubular reabsorption of phosphate (Fig. 22-5). It is also a PHEX substrate that provides a functional link between FGF23-mediated phosphaturic syndrome and PHEX mutations in X-linked phosphatemia.5 Its primary hyperactivation by mutations that prevent degradation of the active protein or by overexpression by the upstream regulatory factors decreases the mineralization of bone and inhibits renal tubular reabsorption of phosphate, causing the phosphate waste related syndromes (Fig. 22-6).
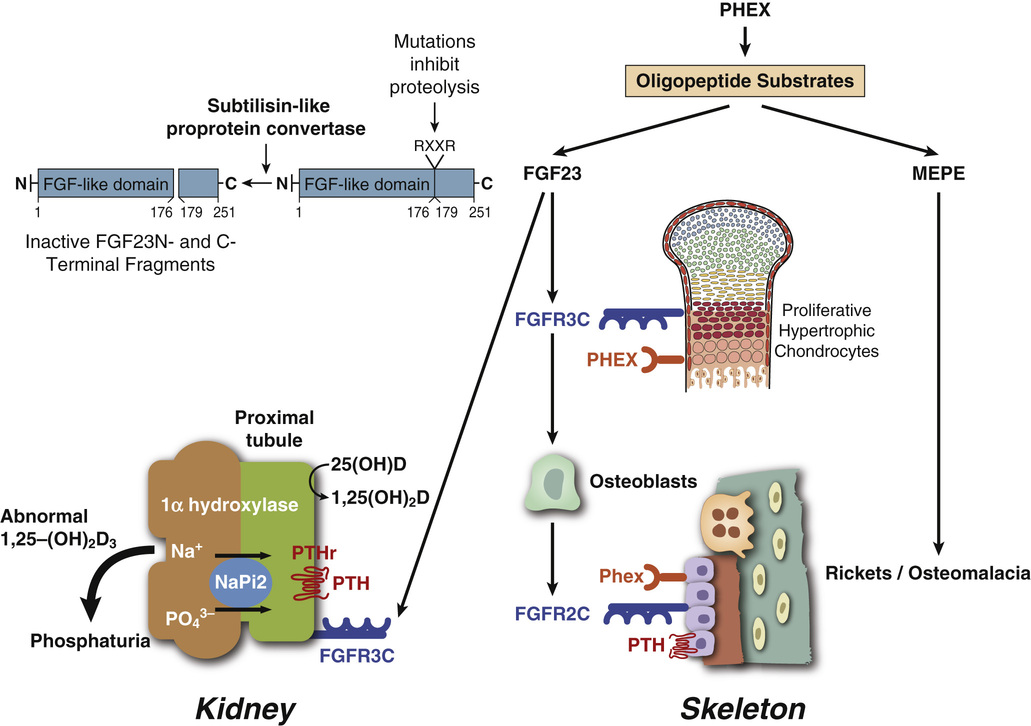
The RXXR motif of FGF23 is similar to the consensus recognition site for precursor convertases, which belong to the family of serine proteases of the subtilisin/kexin type. The increased level of uncleaved FGF23 in peripheral blood can be used to confirm preoperative diagnosis of tumor-induced osteomalacia and to monitor the treatment.78,85,88 The demonstration that some of the tumors involved in the syndrome may express somatostatin receptors has led to the use of octreotide imaging using labeling with indium-111 to detect occult phosphaturic mesenchymal tumors and in some cases to treat overproduction of FGF23 secretion by these tumors.19,22,30,66