[level-membership-for-pathology-category]
Sclerosing Bone Lesions
Sclerosing bone lesions represent a unique group of dysplastic anomalies with a wide range of clinical presentations, radiographic characteristics, and genetic backgrounds.7 They can be separated into two major groups with hereditary and nonhereditary clinical presentations.3 A comprehensive description of all conditions included in the group is beyond the scope of this book. In this chapter, we describe some of these conditions that may occasionally simulate a neoplasm and may require microscopic examination to confirm the diagnosis or to rule out a neoplastic process.
In the radiologic literature, these conditions are grouped into two major categories: those associated with abnormal enchondral ossification and those primarily affecting the sites of membranous bone formation. It is generally accepted that the common feature of the conditions described in this chapter is that they all arise from defects in bone resorption or formation during the process of skeletal development, maturation, and remodeling.1,5,8,10 Sometimes several such conditions can coexist, resulting in the so-called overlapping dysplastic syndromes, which further supports the concept of their unified pathogenesis.5,6,9 An abbreviated list of sclerosing bone dysplasias classified on the basis of the so-called target-site approach originally proposed by Norman and Greenspan5,10 (i.e., specific phase, site of skeletal development affected, or both) is provided in Table 21-1. A summary of clinical presentations, radiographic characteristics, and genetic alterations implicated in the development of hereditary bone dysplasia is provided in Table 21-2.7
TABLE 21-1
Classification of Sclerosing Dysplasias of Bone
| Dysplasias of Enchondral Bone Formation |
| Dysplasias of Membranous Bone Formation |

TABLE 21-2
Summary of the Hereditary Sclerosing Bone Dysplasias
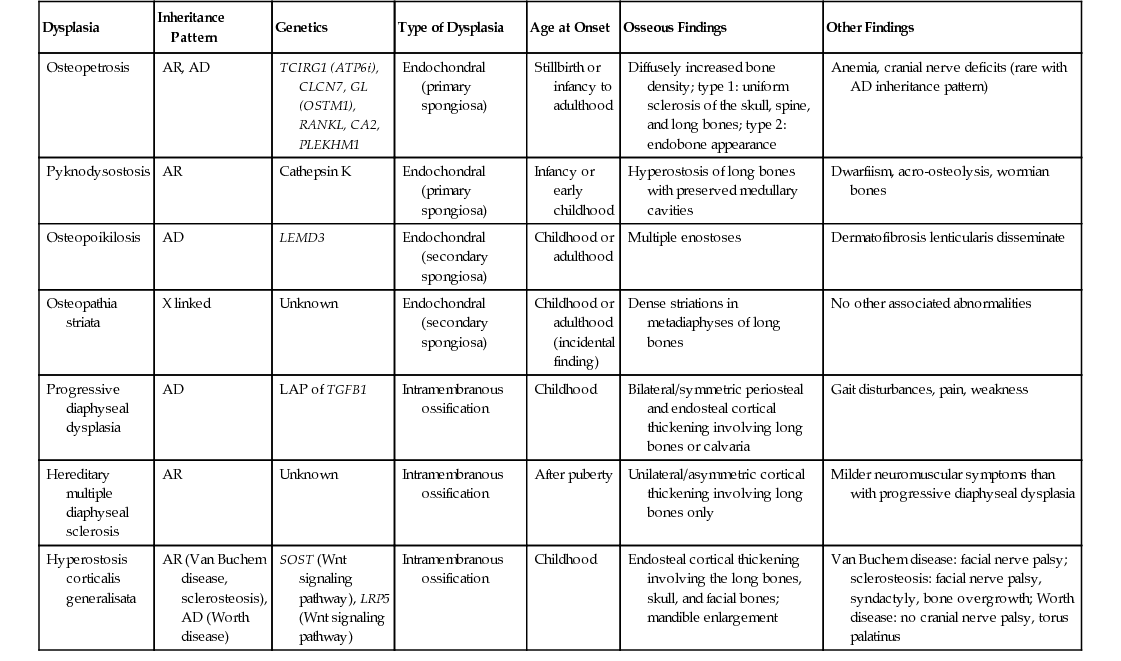
| Dysplasia | Inheritance Pattern | Genetics | Type of Dysplasia | Age at Onset | Osseous Findings | Other Findings |
| Osteopetrosis | AR, AD | TCIRG1 (ATP6i), CLCN7, GL (OSTM1), RANKL, CA2, PLEKHM1 | Endochondral (primary spongiosa) | Stillbirth or infancy to adulthood | Diffusely increased bone density; type 1: uniform sclerosis of the skull, spine, and long bones; type 2: endobone appearance | Anemia, cranial nerve deficits (rare with AD inheritance pattern) |
| Pyknodysostosis | AR | Cathepsin K | Endochondral (primary spongiosa) | Infancy or early childhood | Hyperostosis of long bones with preserved medullary cavities | Dwarfiism, acro-osteolysis, wormian bones |
| Osteopoikilosis | AD | LEMD3 | Endochondral (secondary spongiosa) | Childhood or adulthood | Multiple enostoses | Dermatofibrosis lenticularis disseminate |
| Osteopathia striata | X linked | Unknown | Endochondral (secondary spongiosa) | Childhood or adulthood (incidental finding) | Dense striations in metadiaphyses of long bones | No other associated abnormalities |
| Progressive diaphyseal dysplasia | AD | LAP of TGFB1 | Intramembranous ossification | Childhood | Bilateral/symmetric periosteal and endosteal cortical thickening involving long bones or calvaria | Gait disturbances, pain, weakness |
| Hereditary multiple diaphyseal sclerosis | AR | Unknown | Intramembranous ossification | After puberty | Unilateral/asymmetric cortical thickening involving long bones only | Milder neuromuscular symptoms than with progressive diaphyseal dysplasia |
| Hyperostosis corticalis generalisata | AR (Van Buchem disease, sclerosteosis), AD (Worth disease) | SOST (Wnt signaling pathway), LRP5 (Wnt signaling pathway) | Intramembranous ossification | Childhood | Endosteal cortical thickening involving the long bones, skull, and facial bones; mandible enlargement | Van Buchem disease: facial nerve palsy; sclerosteosis: facial nerve palsy, syndactyly, bone overgrowth; Worth disease: no cranial nerve palsy, torus palatinus |
Osteoma and Bone Island
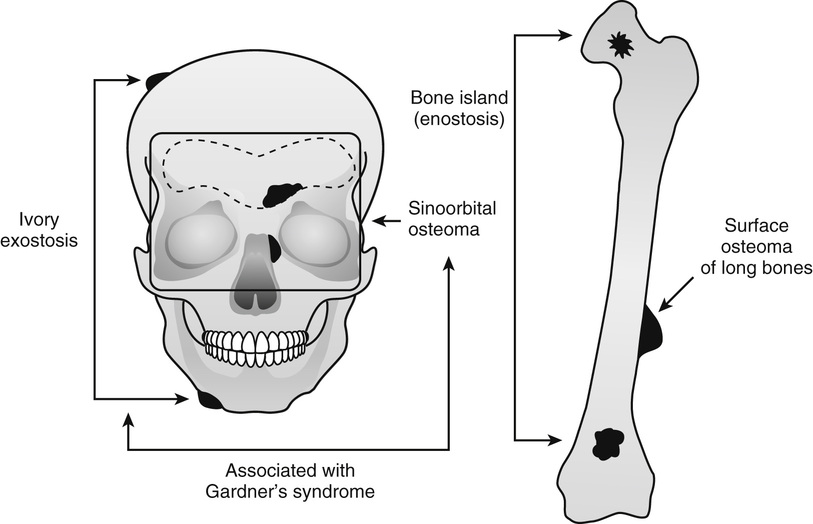
Several benign, slow-growing bony lesions are identified under the general heading of osteoma (Fig. 21-1). They can be divided into (1) calvarial and mandibular ivory exostoses; (2) osteomas of the paranasal sinuses, facial bones, and orbit (sino-orbital osteomas); (3) enostoses or bone islands; and (4) surface (juxtacortical) osteomas of long bones.
Some authors consider these lesions to be hamartomatous or dysplastic in nature, but there is a rationale for regarding those that affect the orbit, facial bones, and paranasal sinuses (sinoorbital osteoma) as benign bone-forming tumors.
Ivory Exostosis
Ivory exostoses represent a type of osteoma that affects bones formed by membranous ossification, chiefly affecting calvarial and mandibular surfaces2,4,6,10–12,20,22,28 (Fig. 21-2). Multiple osteomas of this type are frequently associated with colonic polyposis and fibrous soft tissue tumors, including mesenteric fibromatosis and epidermal inclusion cysts in the familial disorder known as Gardner’s syndrome.13,14,17,24,29 Some patients also had distinct hyperplastic changes of the retinal pigmented epithelium.22,32,33 The development of ocular lesions may precede the development of colonic and osseous manifestations. The polyps of the gastrointestinal tract are adenomatous and typically involve the colon but can also be present in the duodenum and stomach. The adenomatous polyps of the gastrointestinal tract have a high propensity for malignant transformation. In fact, colon cancer develops in all affected individuals unless prophylactic colectomy is performed.23 The disorder is transmitted by an autosomal dominant pattern of inheritance. Similar to other major familial colonic polyposis syndromes, such as flat adenoma and Turcot’s syndrome (colorectal polyposis with brain tumor), Gardner’s syndrome is linked to a malfunctioning adenomatous polyposis coli (APC) gene mapped to chromosome 5q21.26 Mutations involving different regions of APC are associated with the severity of the clinical syndrome and the extent of extracolonic manifestations. It has been shown that mutations involving the 38 end around codon 1444 of the APC gene are associated with particularly severe osseous manifestations.16 Patients with this type of APC alteration also have a higher incidence of soft tissue fibromatosis. The soft tissue fibrous lesions range from aggressive fibromatosis at one end of the spectrum to a lesion which is histologically similar to nuchal-type fibroma.15,19,27 These fibromas of low cellularity have been designated as Gardner fibromas, and their significance as early indicators of the development of other manifestations of Gardner’s syndrome such as desmoid-type fibromatosis has been proposed.15,34 Unlike typical nuchal-type fibromas, which most often affect patients in the third through fifth decades of life, Gardner-associated fibromas arise in younger patients and the majority of them present in the first decade of life. The most common location is in the back and paraspinal region followed by the head and neck and extremities. Immunohistochemically, these lesions stain positively for CD34 and CD99. Many of these lesions show positive nuclear immune-reactivity for β-catenin.15,19 Approximately 50% of patients with Gardner-associated fibromas eventually develop a desmoid-type fibromatosis at the same site. Overall, 10% to 15% of patients with Gardner’s syndrome develop aggressive desmoid-type fibromatosis. They typically develop intraabdominally spontaneously or more often after surgery. Similar to Gardner-associated fibromas, desmoid fibromatoses associated with this syndrome harbor the inactivating mutations of the APC gene. However, similar to sporadic fibromatoses, they are positive immunohistochemically for nuclear β-catenin.15,19 In addition to osteomas, which are often multiple, dental malformation or more diffuse sclerotic changes in the craniofacial region may develop in patients with Gardner’s syndrome.25 Osteomas in Gardner’s syndrome may increase in size over time; that is, progressively more severe malformation of the craniofacial bones may develop.31 Osseous lesions in Gardner’s syndrome do not behave as true neoplasms, and there is no evidence of their evolution into malignant lesions. Moreover, the presence of osteoblastoma-like areas, predominantly seen in sporadic sinoorbital osteomas, is not a feature of osseous lesions in Gardner’s syndrome.
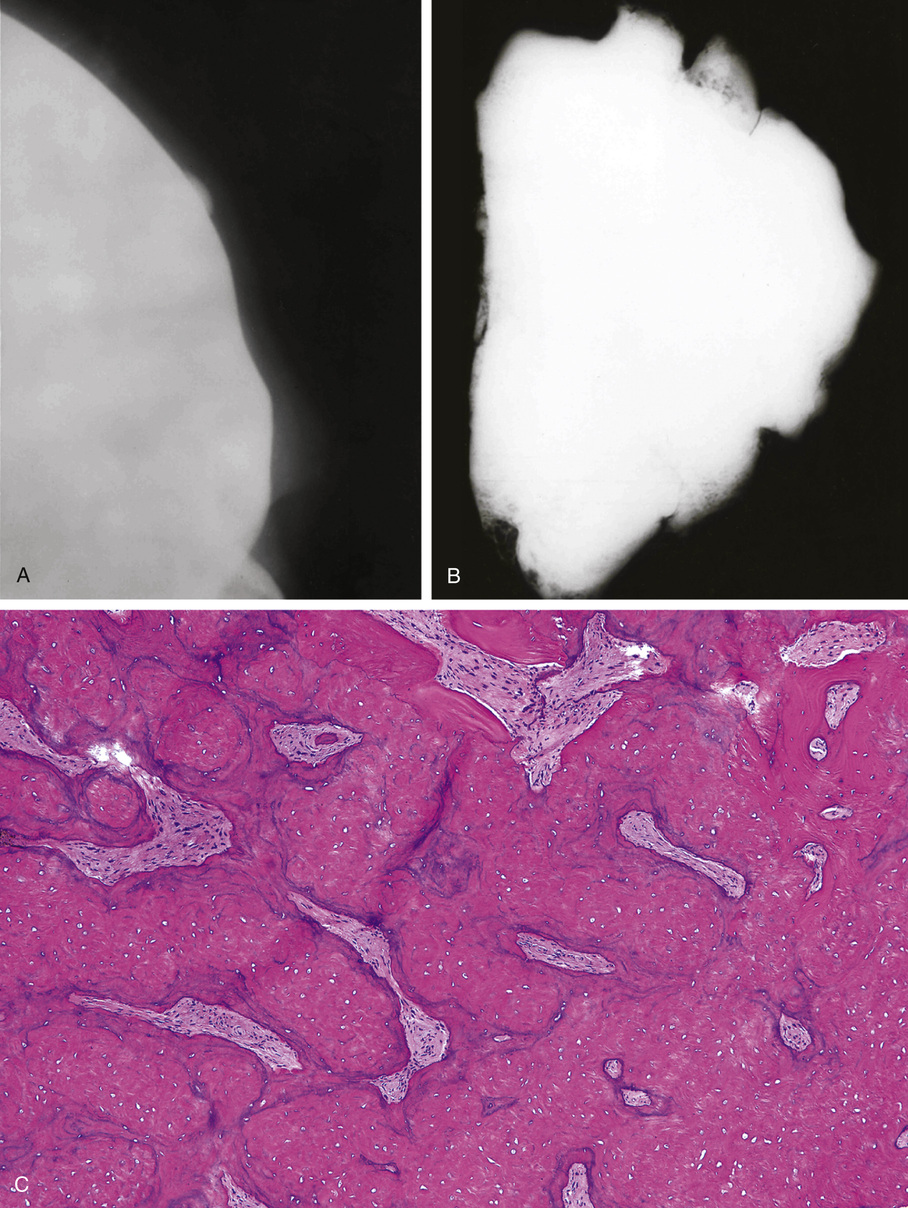
Microscopically, ivory exostoses are button-like excrescences of mature lamellar bone limited to the cortical surfaces and usually continuous with the outer table of the skull or the mandibular cortex. The dense cortical lamellar bone making up these exostoses contains osteons with haversian canals. Rare cases of compact surface osteoma have been reported in other flat bones as well as on long bones.44
Sinoorbital Osteoma
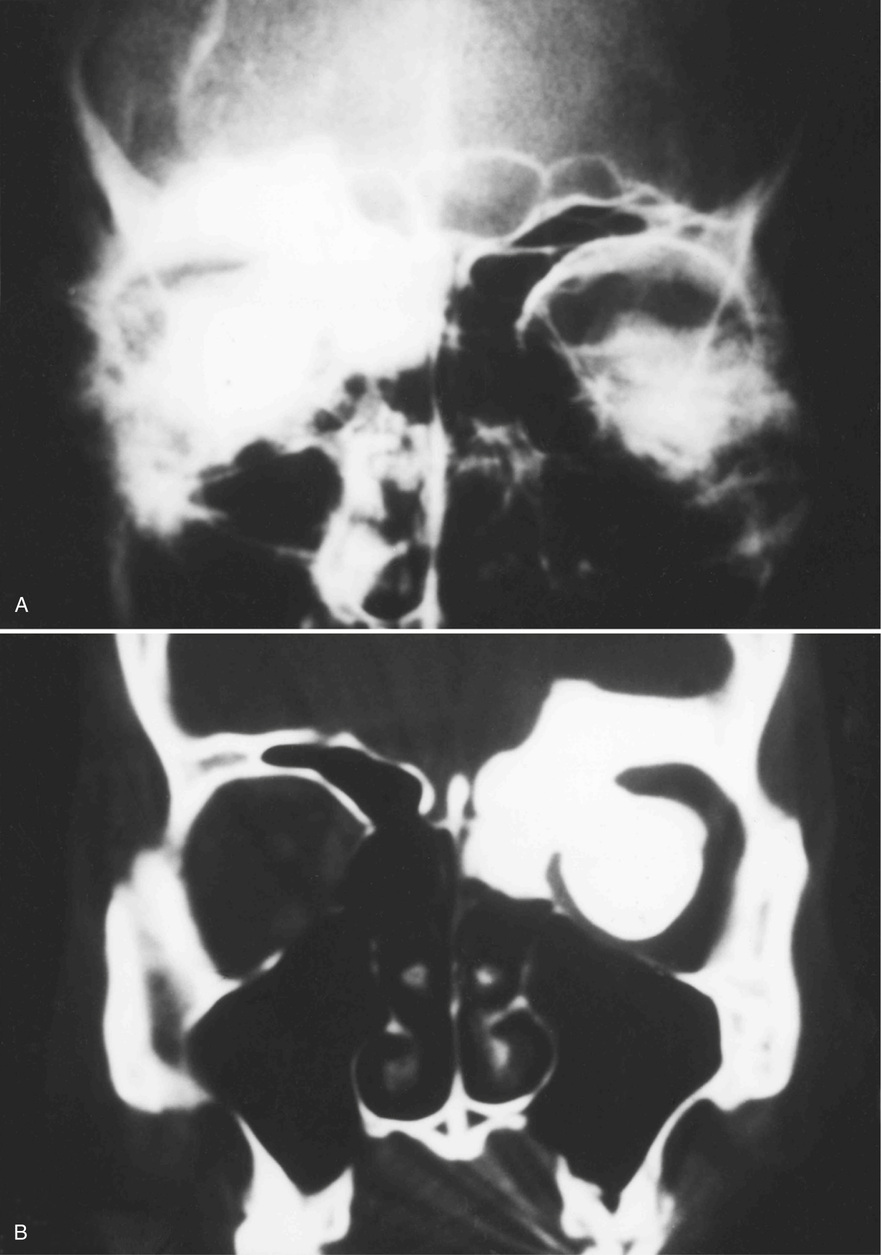
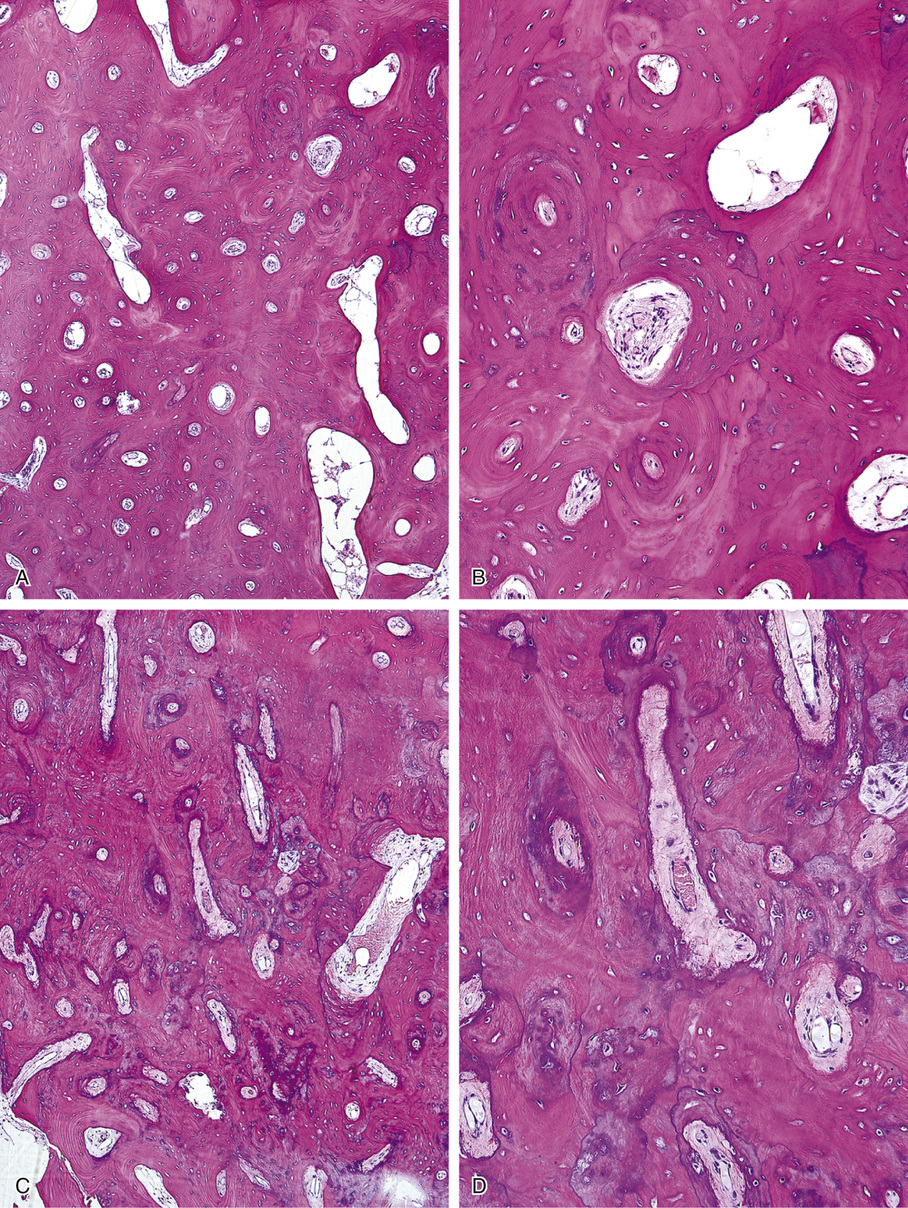
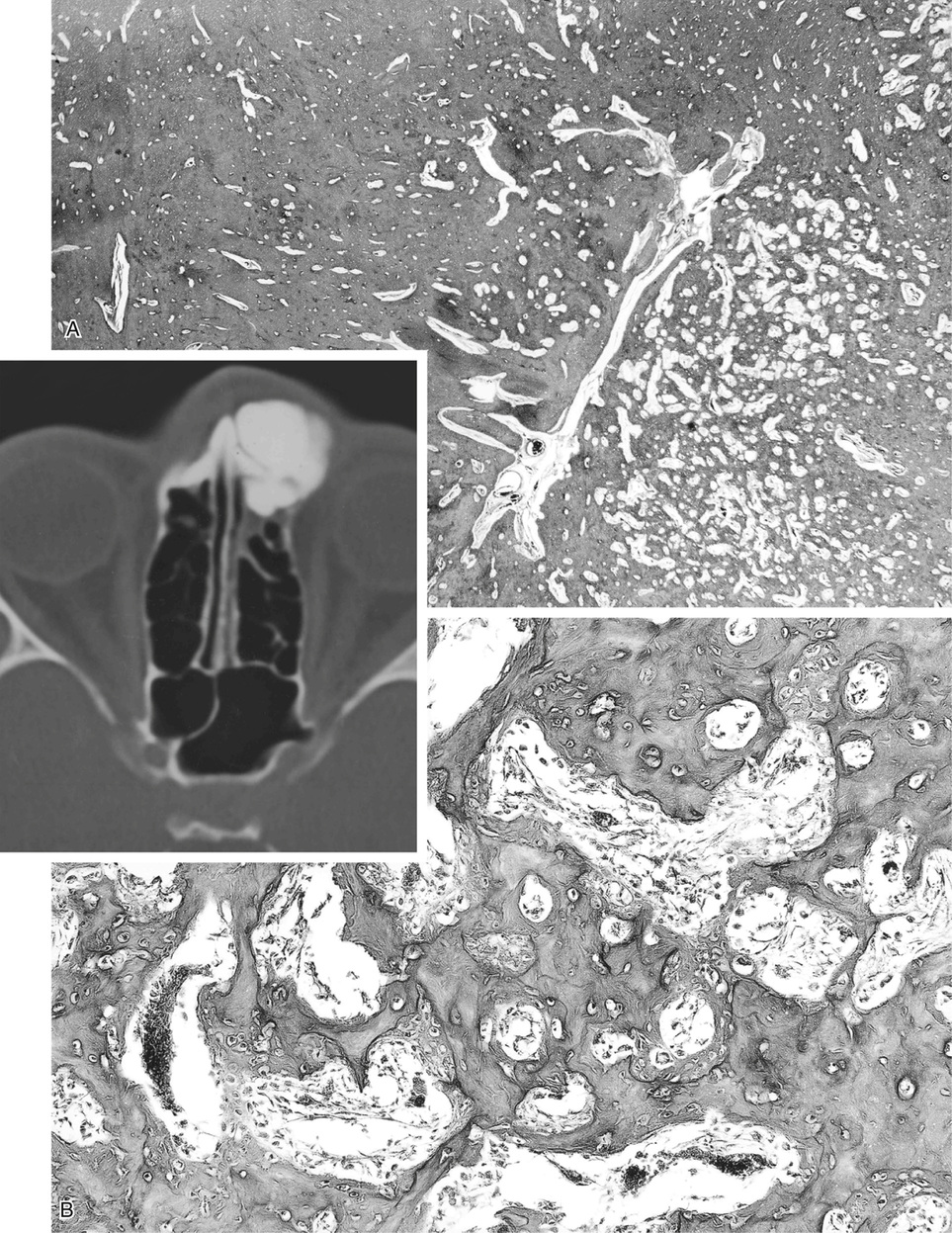
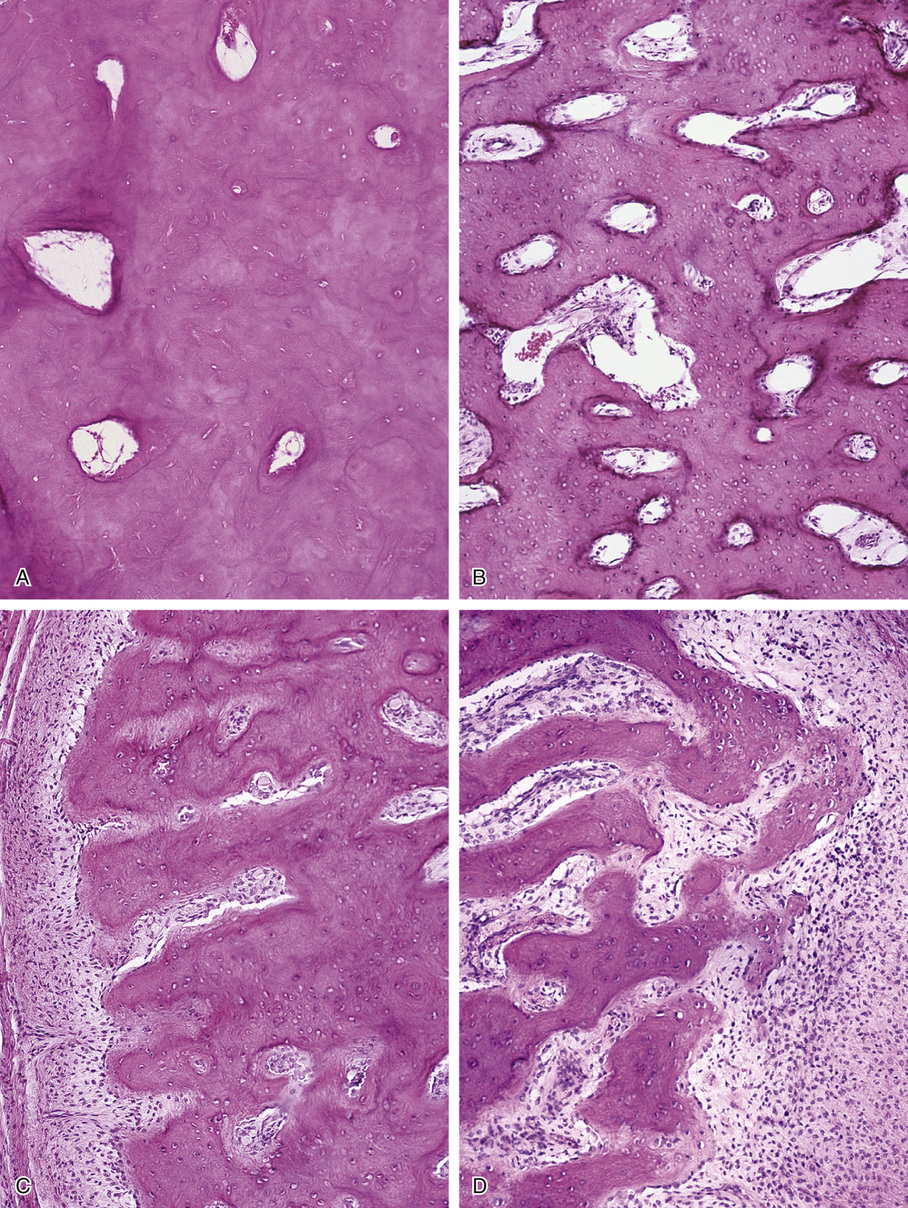
Osteomas involving paranasal sinuses, orbit, and facial bones tend to project into the sinus cavity from a broad attachment to the affected bone (Figs. 21-3 and 21-4).21,33,34,36,39,41,42,45,46 Some osteomas involving this region, especially those that are multifocal, are associated with Gardner’s syndrome. Microscopically, they are indistinguishable from ivory exostosis. In contrast, the so-called sporadic sinoorbital osteomas (not associated with Gardner’s syndrome) are usually composed of dense, immature bone and may have osteoblastic rimming as well as signs of bone resorption (Fig. 21-5). These lesions may contain central or peripheral foci of spongy or cancellous appearance (Figs. 21-6 and 21-7). Sometimes the latter areas show active remodeling, with cellular fibrous tissue filling the interstices, osteoblastic activity, and osteoclastic resorption.33 In ethmoid and frontal sinus osteomas, these lesions may occasionally contain radiolucent areas that are histologically indistinguishable from conventional osteoblastoma and even aggressive osteoblastoma (Fig. 21-8).38,40,47 The latter cases sometimes resemble multifocal osteoblastomas arising within osteomas. The clinical behavior of these lesions is similar to that seen in osteoblastomas and aggressive osteoblastoma groups; that is, they exhibit a local destructive growth pattern and recurrences. For this reason, we prefer to regard the sporadic osteomas that arise in the orbit, paranasal sinuses, and facial bones, especially those that contain osteoblastoma-like foci or features of active bone production and remodeling, as benign bone-forming neoplasms rather than as hamartomas or dysplasias. Osteomas in this region typically do not exceed 2 cm in diameter. Occasionally, they attain a larger size (giant osteoma) that compresses adjacent structures and cause a disfiguring deformity.28,35,37,43
Bone Island (Enostosis)
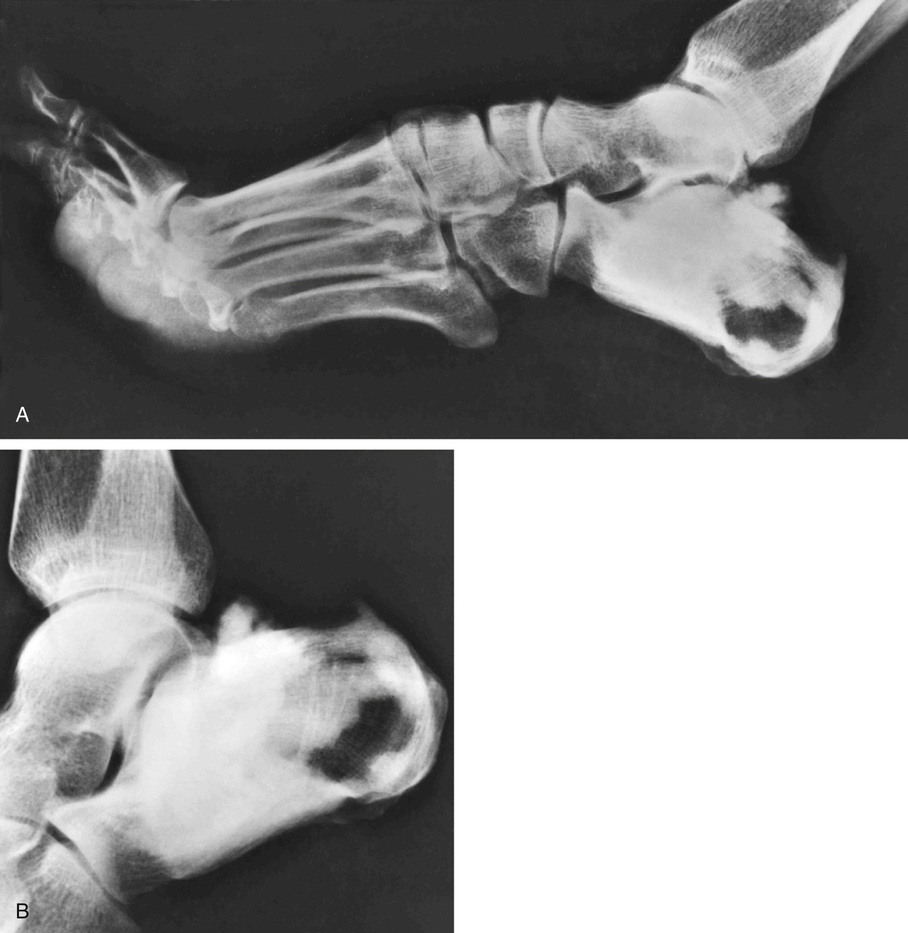
Bone islands are foci of dense, compact bone within the medullary cavity (i.e., within cancellous bone) (Fig. 21-9).48,50,51,54 Occasionally, they may be attached to the inner surface of the cortex. They are ovoid, with the long axis parallel to the cortex of the affected bone. Most bone islands are small (less than 1 cm in diameter), and the majority of lesions measure from 0.1 to 2.0 cm. Extremely rare examples of giant bone islands that measure more than 2.0 cm in diameter have been described (Figs. 21-10 and 21-11).18,30,49,52,57,60 The characteristic feature is the presence of so-called thorny spicules or pseudopodia at the periphery of the bone island.53,54,56
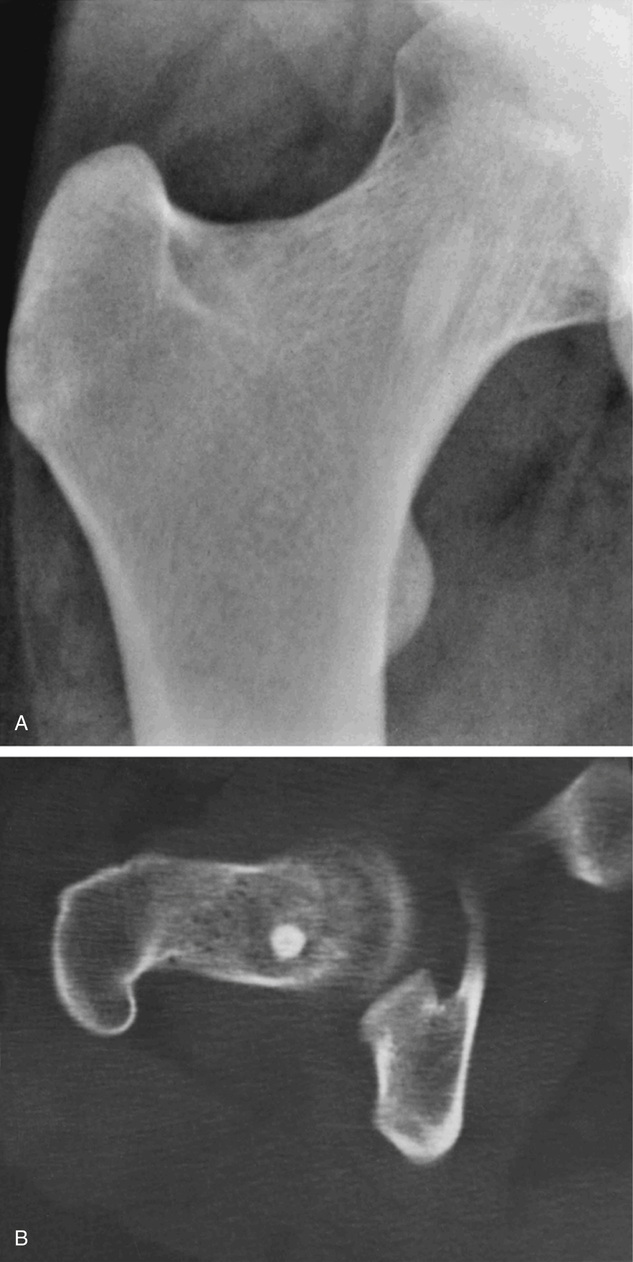
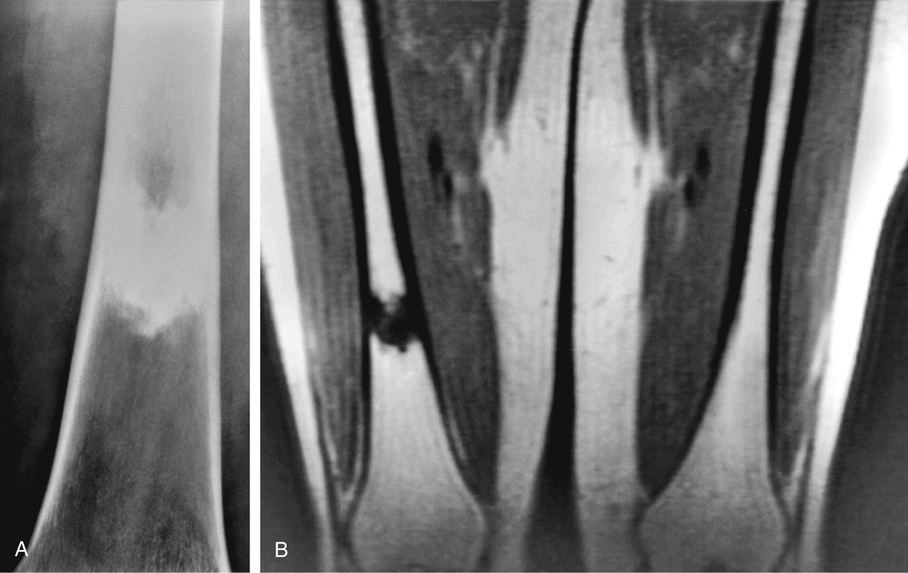
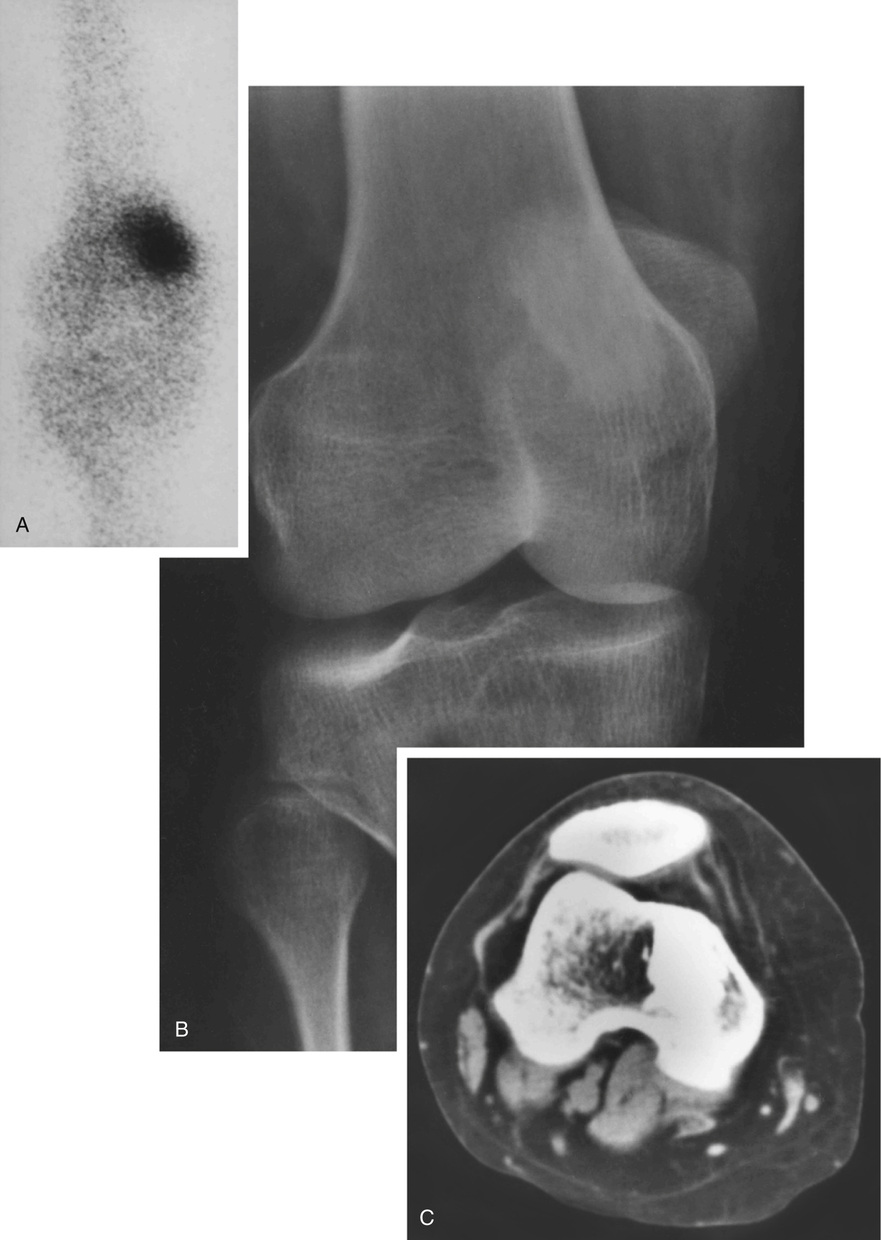
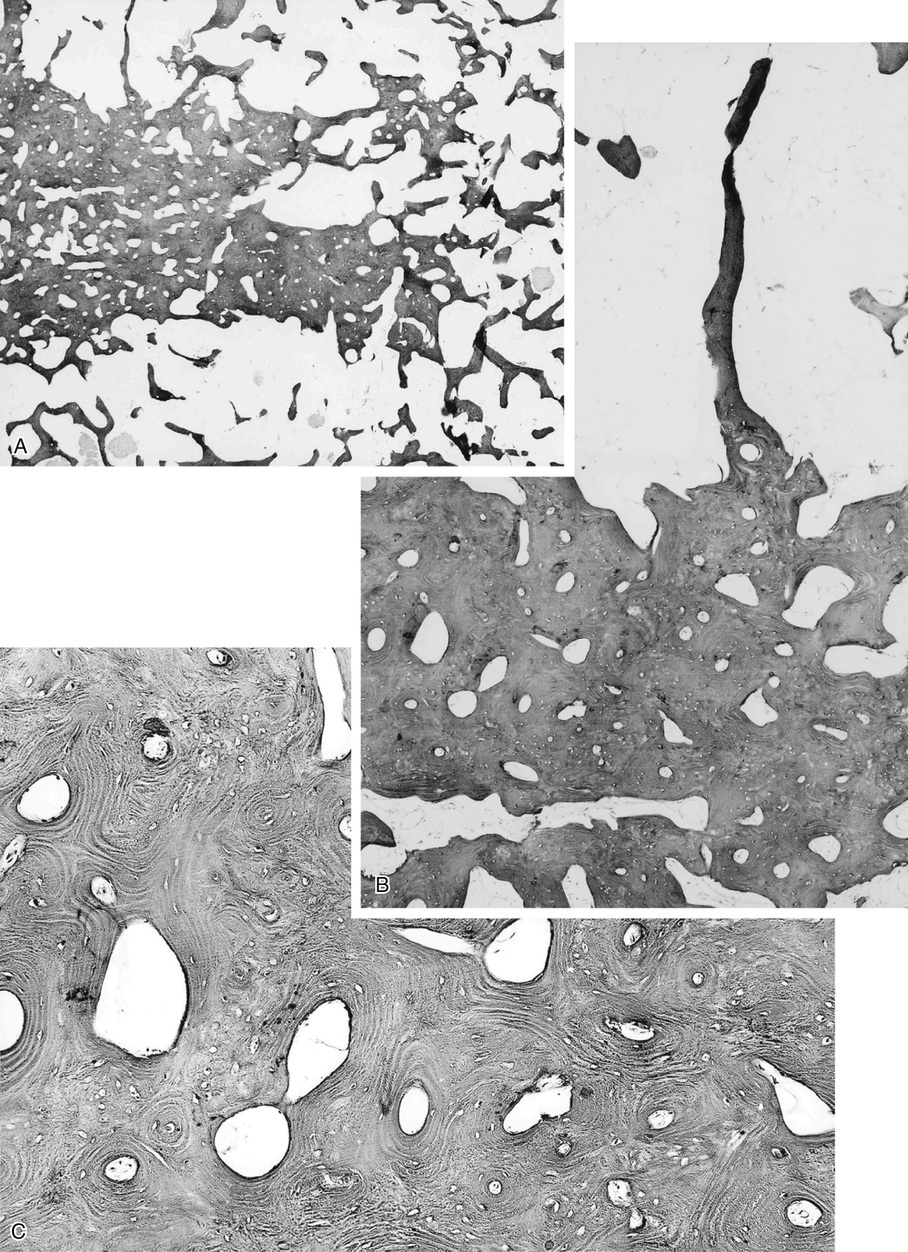
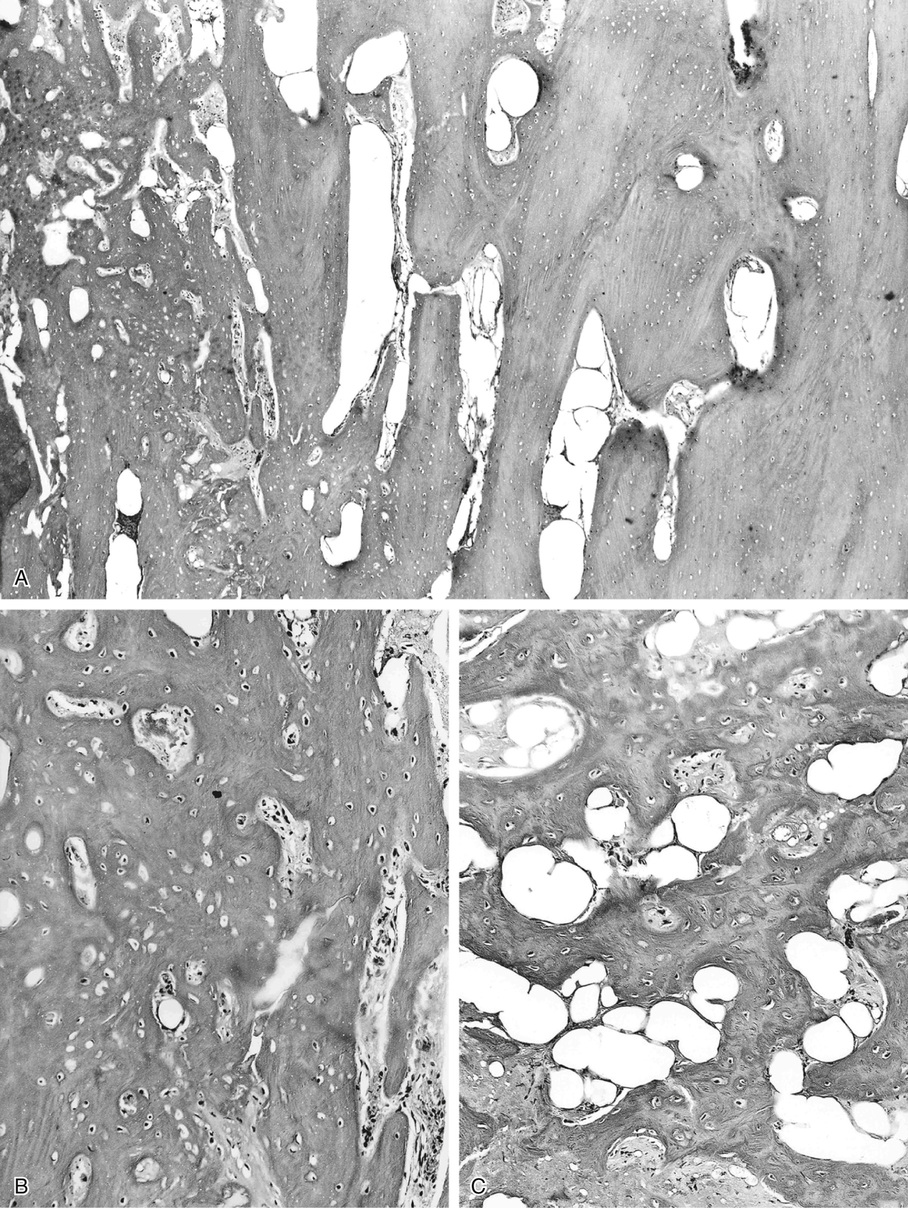
Microscopically, bone islands are composed of compact (cortical) lamellar bone. The thornlike projections represent thickened trabeculae radiating from the main mass and blending with the surrounding bone (Fig. 21-12). The lesions show prominent haversian canals that may contain osteoblasts and osteoclasts within Howship’s lacunae. Features of active bone deposition and remodeling are more often seen in larger lesions (Fig. 21-13). This explains the increased amount of bone turnover in bone islands, which can be demonstrated by skeletal scintigraphy.48,50,58 In fact, some bone islands may slowly increase in size (growing bone islands).48,50,55,59
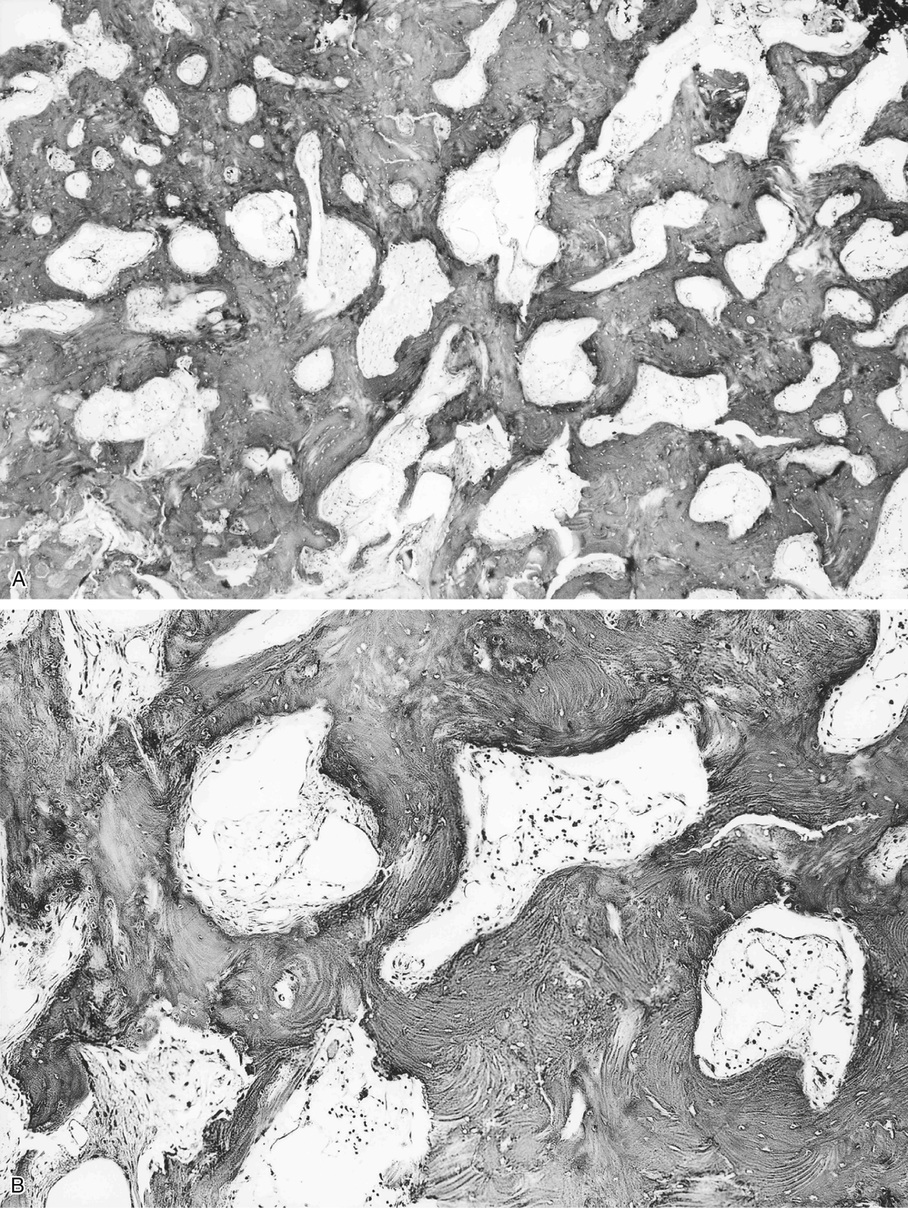
Biopsy specimens from small bone islands are rarely obtained, but larger lesions may raise the suspicion of a neoplasm, such as a sclerotic variant of osteosarcoma. We also have seen a giant bone island that was misinterpreted microscopically as a low-grade intraosseous fibroblastic osteosarcoma. Although some osteoblastic rimming may be present within dilated haversian channels of giant bone islands, the spindle-cell stroma and infiltrative pattern of growth characteristic of low-grade intraosseous fibroblastic osteosarcoma are not present in these lesions.
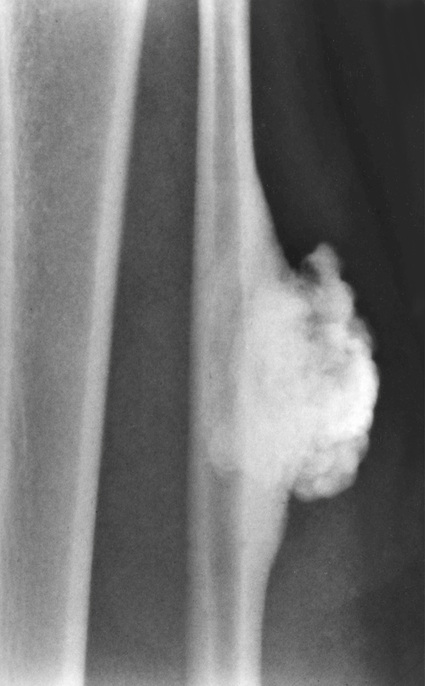
Surface Osteoma of Long Bones
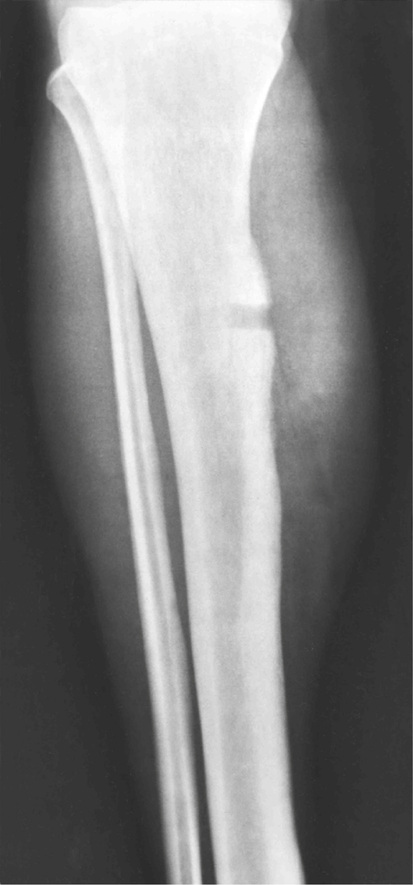
Surface osteoma that involves the clavicle, pelvic, or long bones of the extremities is the rarest form of osteoma.61–69 On radiographs, it appears to resemble ivory exostosis (surface osteoma) of the craniofacial bones (Fig. 21-14). Similar to other osteomas, this lesion is composed of mature lamellar bone organized into osteons and haversian canals. The outer surface is covered by a thin layer of fibrous tissue. There is no cartilaginous cap covering the lesion. The underlying cortex may be thickened but is otherwise intact. No continuity between the lesion and the underlying medullary cavity can be seen grossly or on radiographs. Osteoma that involves the surface of a long bone must be radiographically and microscopically differentiated from parosteal osteosarcoma, melorheostosis, and osteochondroma. The spindle-cell fibroblastic stroma that is characteristic of parosteal osteosarcoma is not present in osteoma. Moreover, the bone in osteoma is mature and forms solid areas with recognizable haversian canals, which are typically not present in parosteal osteosarcoma. The distinction from melorheostosis is based mainly on the radiographic pattern of the bone surface lesion. Multifocality and the so-called wax-dripping patterns are not present in surface osteoma. In contrast to osteochondroma, parosteal osteoma is not covered by a cartilaginous cap, and there is no continuation between the lesion and the underlying medullary cavity.
Osteopoikilosis (Spotted Bone Disease)
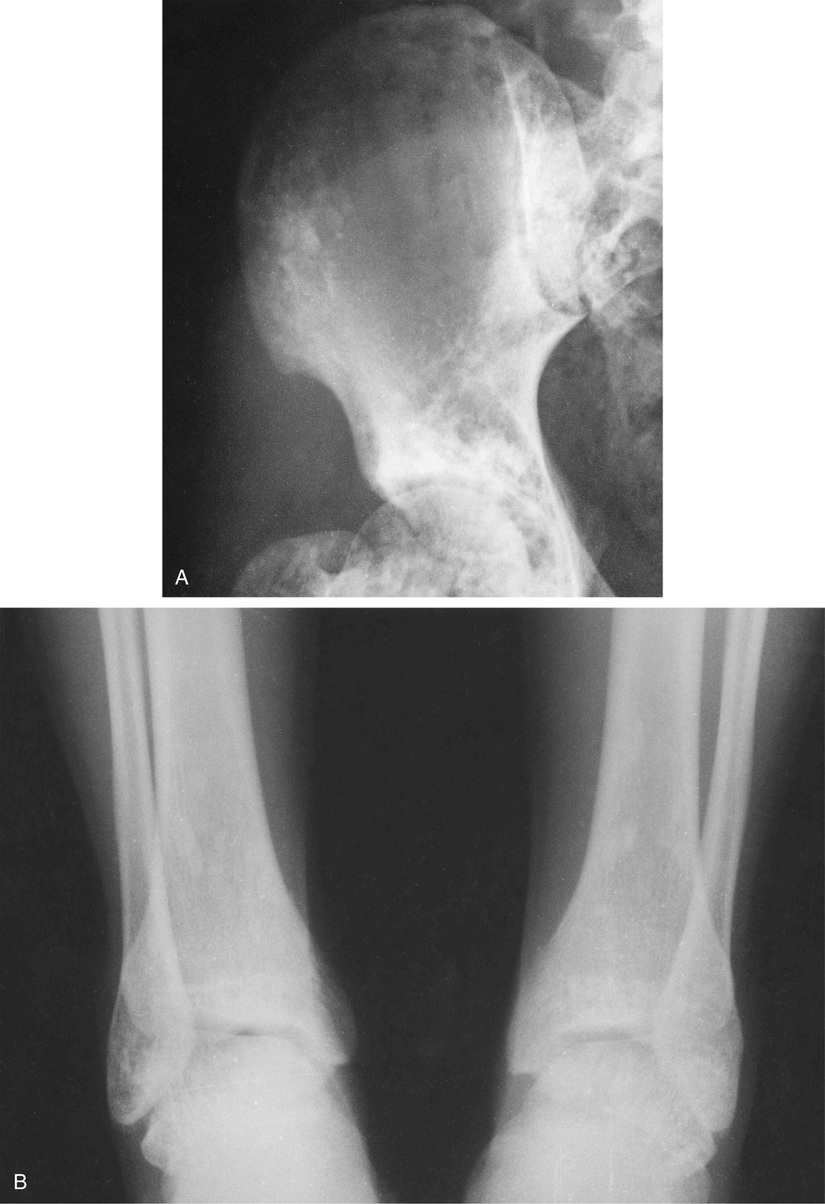
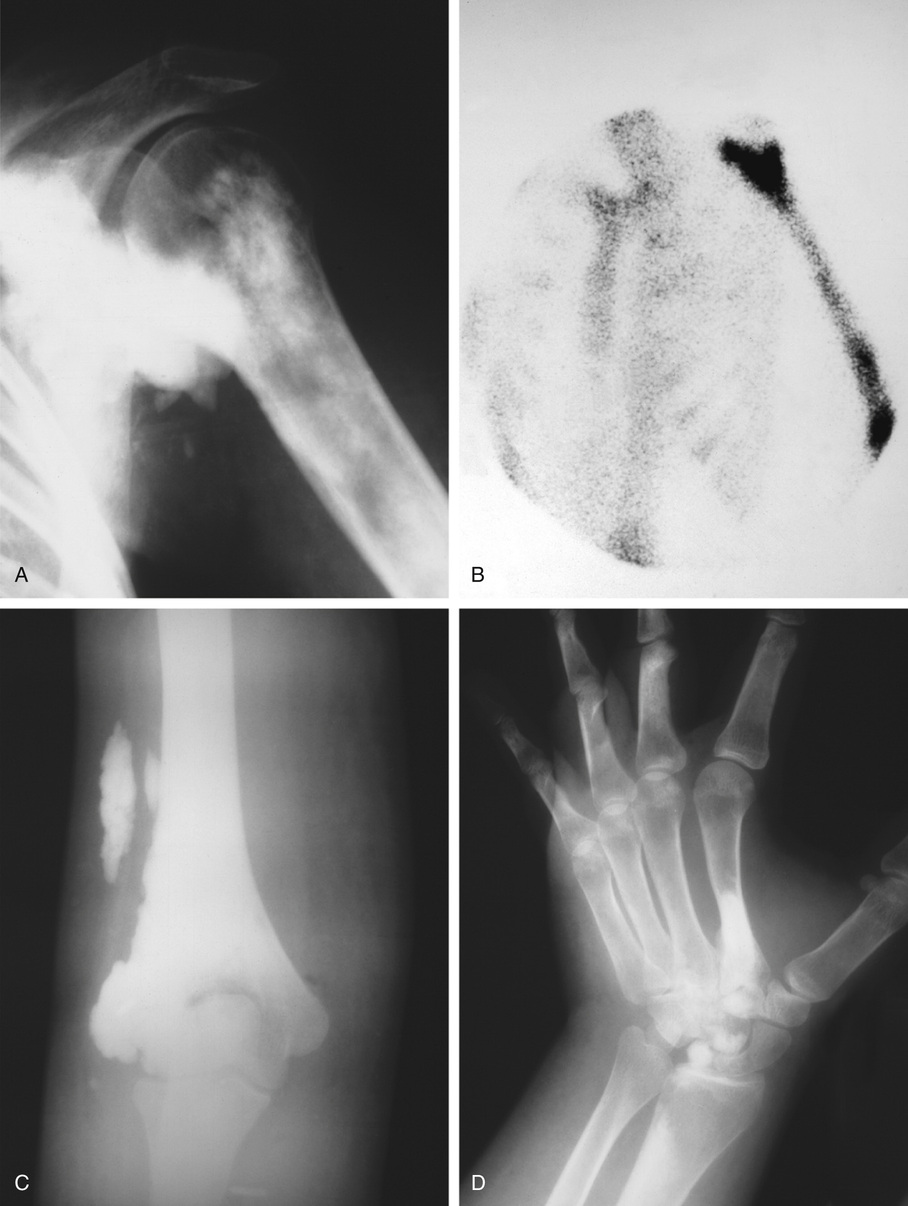
Osteopoikilosis is closely related to osteomas and bone islands. The difference is most likely related to the degree of phenotypic penetration of the genetic defect. Some investigators have suggested that a bone island represents a unifocal minor form of osteopoikilosis.6,10 Osteopoikilosis is inherited in an autosomal dominant pattern by inactivating mutations involving the LEM domain-containing protein 3 gene. LEMD3 interacts with transforming growth factor TGF-β and bone morphogenetic protein signaling, causing focal deposit of compact lamellar bone.7 On radiographs, osteopoikilosis presents as multiple bone islands clustered at the ends of the metaphyses.70–72 It is rare, with only about 400 cases described in the literature. Osteopoikilosis can be associated with the hereditary dermatologic condition known as dermatofibrosis lenticularis disseminata (Buschke-Ollendorff syndrome), which is characterized by multiple papular fibromas on the back, arms, and thighs.5,71
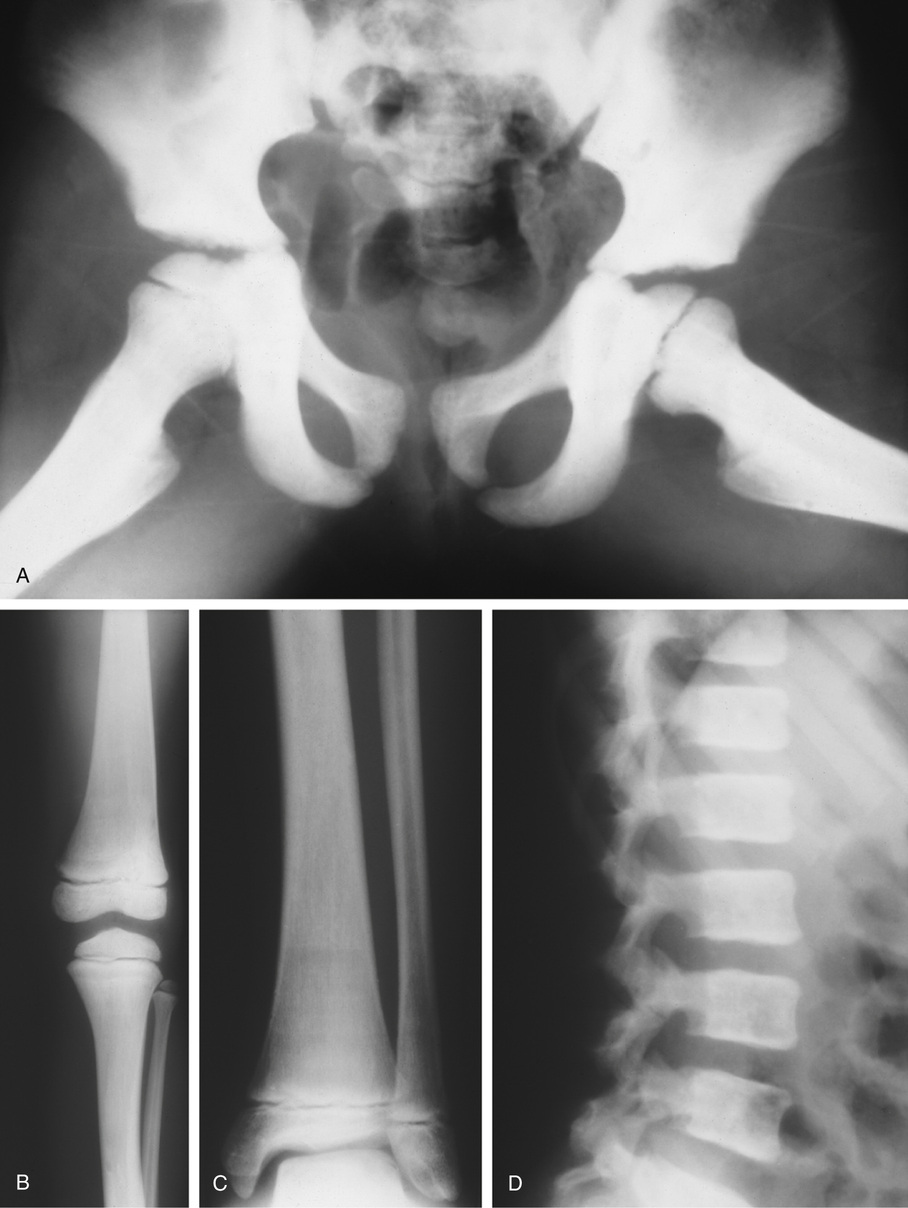
The symmetric distribution of multiple bone islands in the metaphyseal ends of the long tubular bones and in the carpal or tarsal bones is a characteristic radiographic feature (Figs. 21-15 and 21-16). Less frequently the lesions may also be found in other parts of the skeleton in the vicinity of joints (e.g., around the acetabulum or glenoid fossa). The skull, spine, and ribs are extremely rare sites of involvement.
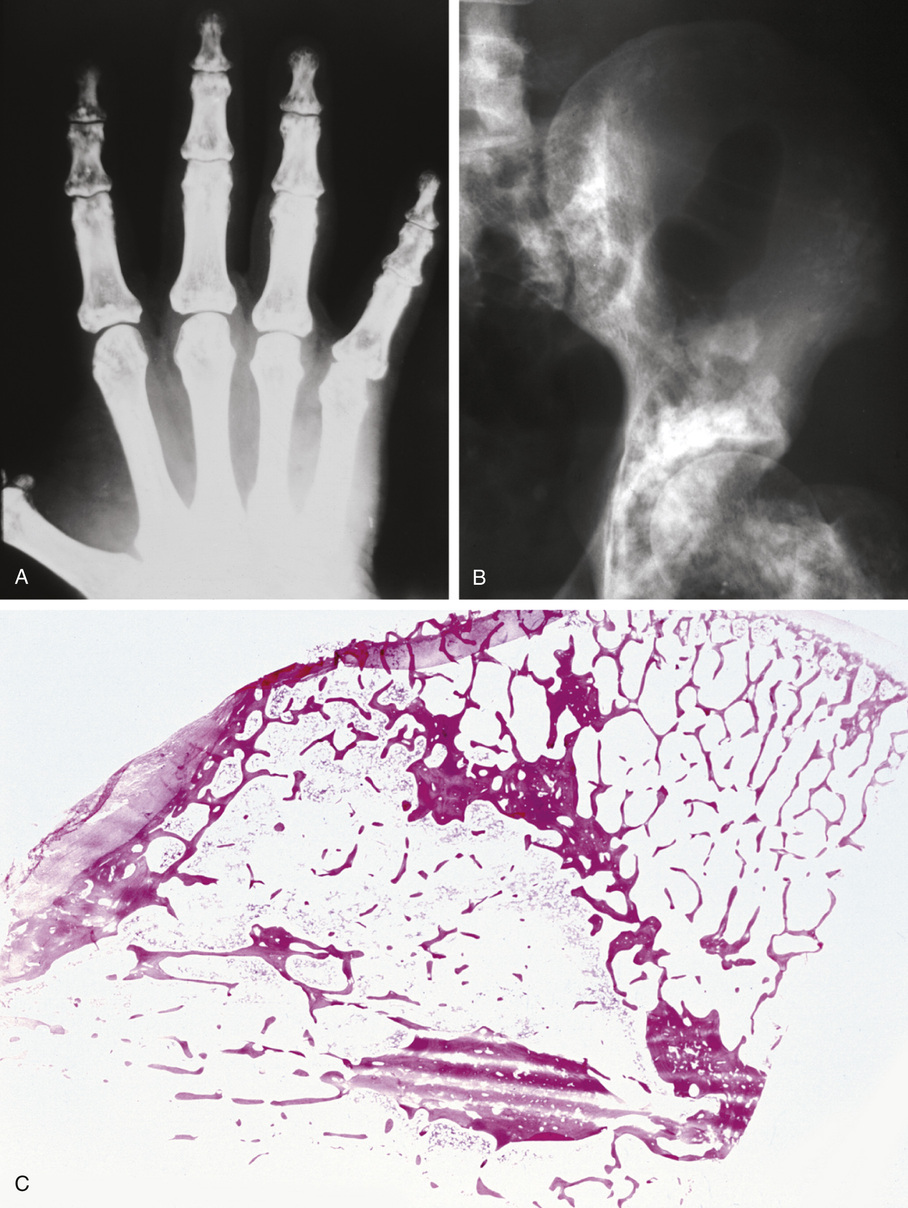
The lesions in osteopoikilosis are intramedullary but may also be attached to the inner surface of the cortex. Microscopically, they are composed of lamellar bone identical to that seen in bone islands (Fig. 21-16).
Osteopetrosis
Osteopetrosis (Albers-Schönberg disease) occurs in two basic clinical settings: as an autosomal recessive condition with early manifestation and as an autosomal dominant condition with delayed manifestation.77,81,82 These two forms of osteopetrosis are referred to as congenital or malignant osteopetrosis and adult osteopetrosis or osteopetrosis tarda. The autosomal recessive subtype, also referred to as osteopetrosis type 1, has been linked to several genetic defects, including inactivating mutations of T-cell immune regulator 1 (TCIRG1); homozygous mutations in the chloride 7 channel (CLCN7); a defect in the gray-lethal or osteopetrosis-associated transmembrane protein gene (GL); and mutations of the receptor activator of nuclear factor κ-B ligand (RANKL).7,86,88,89 The autosomal dominant subtype, referred to as osteopetrosis type 2, was linked to mutations inactivating one allele of the chloride 7 channel gene (CLCN7), and mutations involving carbonic anhydrase II (CA2), T-cell immune regulator 1 (TCIRG1), osteopetrosis-associated transmembrane protein 1 (OSTM1), and pleckstrin homology domain-containing family M member 1 (PLEKHM1).7,76,86,88,89 At least eight subtypes of osteopetrosis, each with unique clinical presentation and mode of transmission, have been described in humans.73,74,77 Deficiency of the carbonic anhydrase II isoenzyme is associated with autosomal recessive osteopetrosis. The majority of malignant autosomal recessive osteopetrosis is caused by mutations in the TCIRG1 gene, whereas mutations in the gene encoding the CLCN7 chlorine channel pump are responsible for the majority of the most common autosomal dominant osteopetrosis.75,78,80,83,84,85,86,88,89
The adult form occurs more frequently than the congenital form. Under normal conditions, the primary spongiosa is resorbed by osteoclasts. Focal ineffective resorption of primary spongiosa results in accumulated masses of calcified cartilage in the medullary cavity. Congenital osteopetrosis is typically fatal and in many instances results in stillbirth. When a child is born alive, the disease rapidly progresses, and most patients die in early infancy after manifesting profound anemia and other sequelae of bone marrow compromise. Adults with osteopetrosis usually live a long time. Many patients are asymptomatic, and the diagnosis is based on incidental findings on radiographs made for other reasons or as a result of pathologic fractures.
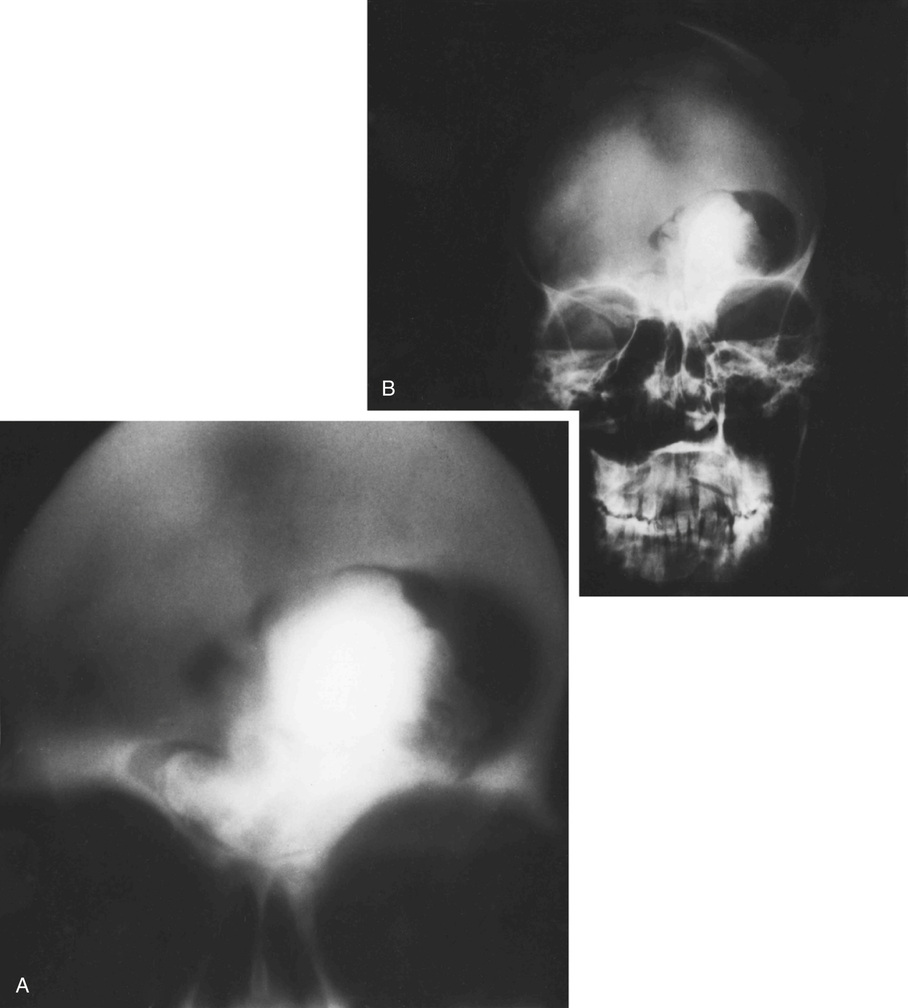
The radiologic hallmark of all variants of osteopetrosis is generalized opacification of bone (Figs. 21-17 and 21-18). The skeletal sites that are formed by membranous ossification are usually unaffected. Thus in the skull, sclerosis is more prominent at the base. In the long bones, the metaphyseal parts typically show prominent sclerosis. In the spine, the sclerosis of the end plates gives the vertebrae a “sandwiched” appearance. The metaphyseal regions of long bones appear clublike without the normal flare (Erlenmeyer flask deformity). The metaphyses may also show horizontal striations of lucent and sclerotic areas.
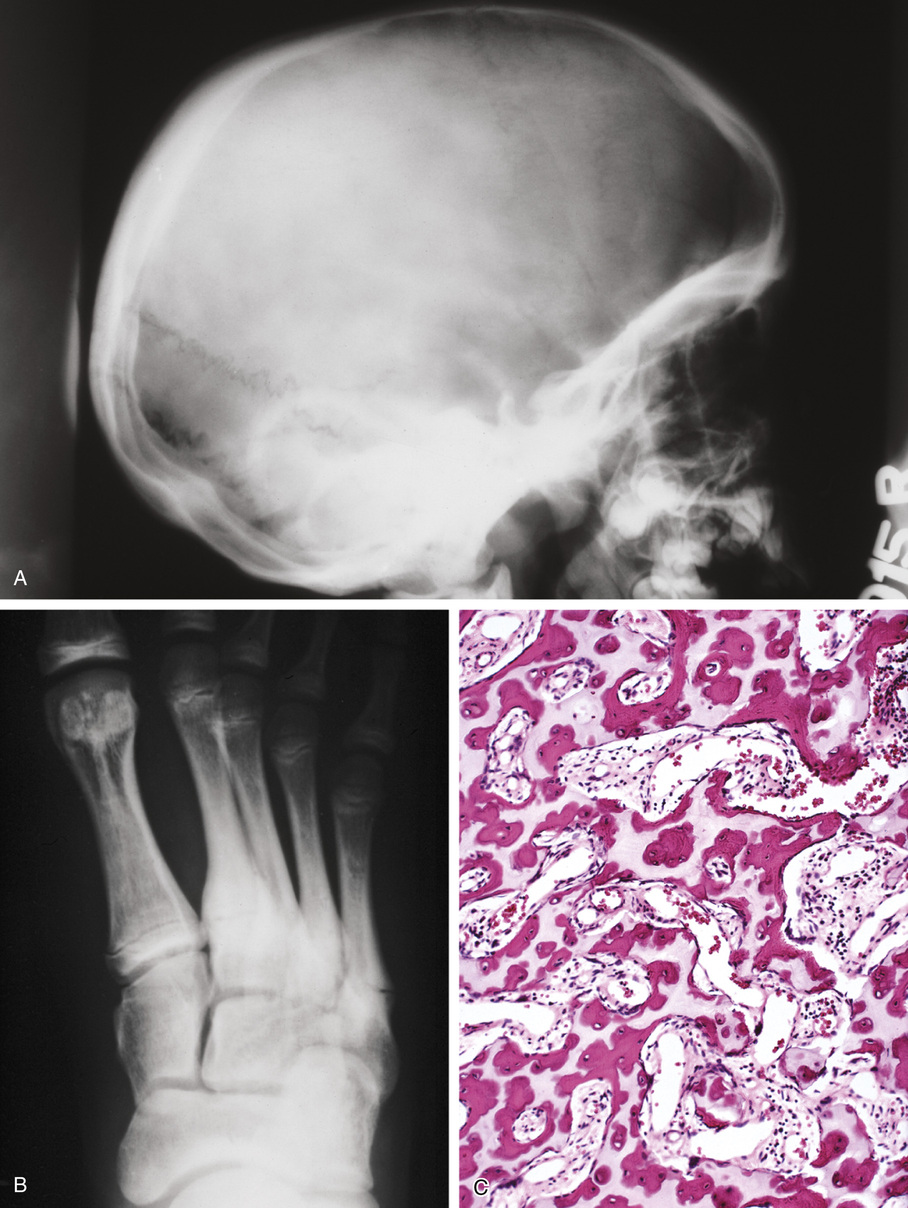
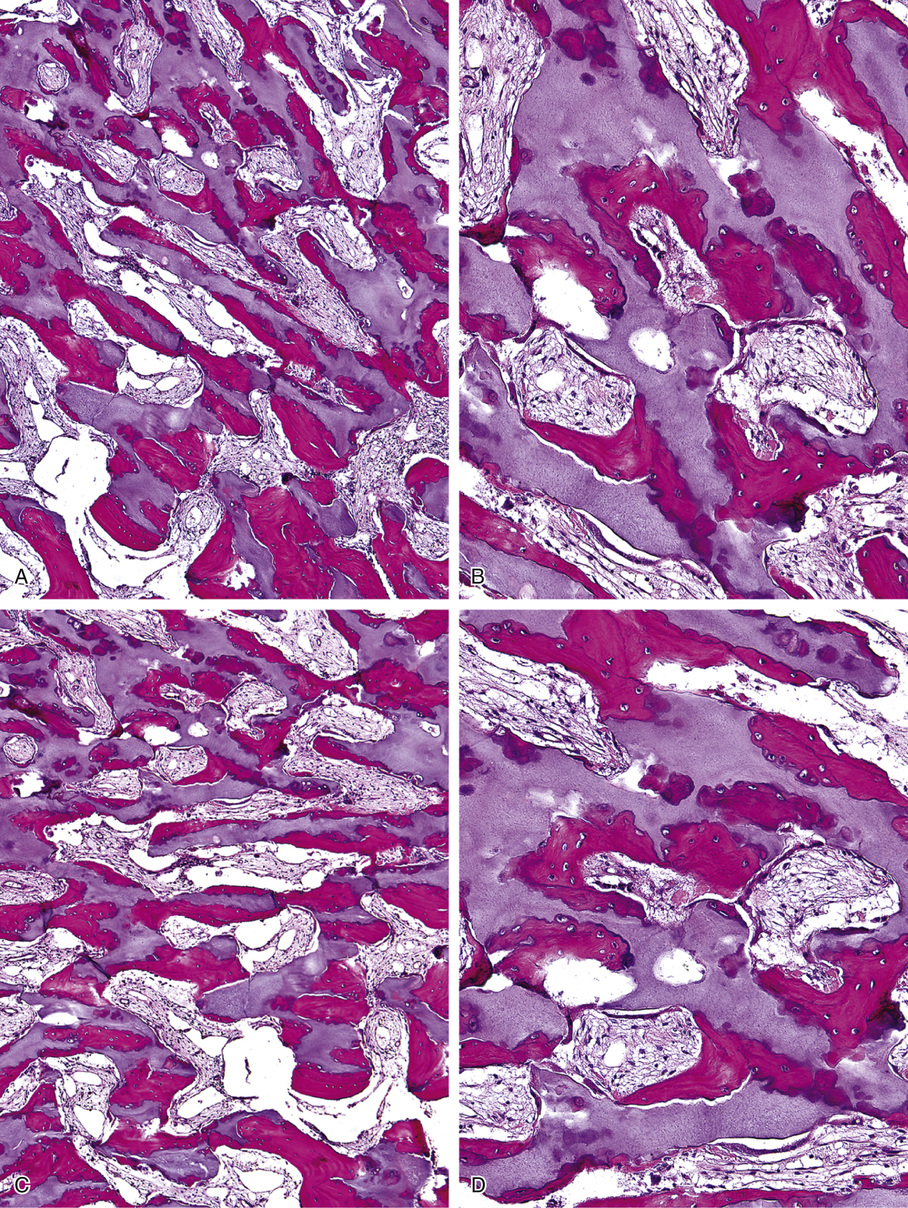
Microscopically, the sclerotic areas of bone show an accumulation of cartilage with an interconnecting network of bone trabeculae87 (Figs. 21-18 and 21-19). Osteoclasts may be present on bone surfaces, but they do not show Howship’s lacunae and lack ruffled borders. These features indicate that the basic defect in this disorder involves osteoclastic cells that cannot resorb bone. This is consistent with the observation that the metaphyseal region closest to the growth plate is packed with thickened trabeculae of woven bone that has a central core of calcified cartilage (i.e., lack of resorption of primary spongiosa). This concept is further supported by the significant normalization of skeletal density and the reversal of bone opacification that can be induced by bone marrow transplantation in patients with congenital osteopetrosis.79,89
Melorheostosis
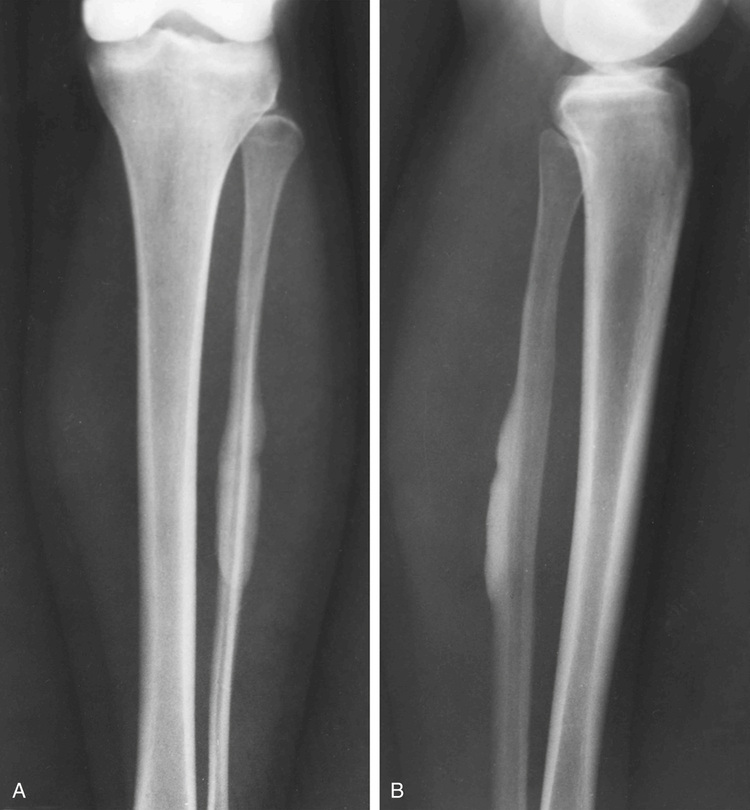
Melorheostosis is a rare condition characterized by a localized, diffuse thickening of the cortical bone (Figs. 21-20 through 21-23). The lesions frequently are described radiographically as resembling wax dripping down the side of a candle (guttering).90,92,93,96,102 The condition may involve one bone (monostotic form) or may affect multiple bones in one extremity (monomelic form). Rarely, it presents as a disseminated skeletal disorder that involves more than one limb (polyostotic polymelic form) (Fig. 21-23).91 The most prominent hyperostosis is usually at the site developmentally considered to be formed by membranous ossification (shafts in the long tubular bones). In the polyostotic involvement of one limb, the distribution of lesions in melorheostosis seems to correlate with the anatomic distribution of sclerotomes.101 It can also be associated with other lesions such as arteriovenous aneurysms and scleroderma.99,100,103 Microscopically, these lesions represent hyperostosis of compact cortical bone similar to that seen in osteomas, bone islands, and osteopoikilosis (Fig. 21-24). Features of immaturity (absence of well-organized osteons and a woven bone appearance) can be present. Melorheostosis typically involves the surface of bone but in rare cases can also involve the medullary cavity. It is believed that hyperostosis tends to progress through childhood into adult life. It is characterized by periods of exacerbation and arrest.95,98 Overlying soft tissues and nearby joints may be involved with fibrous thickening and contractures. Radioisotopic bone scans are usually positive in melorheostosis and show increased uptake at the sites of lesions. Mineralized soft tissue masses may occur in as many as 27% of patients with melorheostosis.101 These may be found adjacent to areas of cortical hyperostosis or at a distance, usually in paraarticular location.97 Histologically, these bony and cartilaginous soft tissue masses often contain a core of mature osseous tissue with a peripheral cap of hyaline cartilage.94
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
Sclerosing Bone Lesions
Sclerosing bone lesions represent a unique group of dysplastic anomalies with a wide range of clinical presentations, radiographic characteristics, and genetic backgrounds.7 They can be separated into two major groups with hereditary and nonhereditary clinical presentations.3 A comprehensive description of all conditions included in the group is beyond the scope of this book. In this chapter, we describe some of these conditions that may occasionally simulate a neoplasm and may require microscopic examination to confirm the diagnosis or to rule out a neoplastic process.
In the radiologic literature, these conditions are grouped into two major categories: those associated with abnormal enchondral ossification and those primarily affecting the sites of membranous bone formation. It is generally accepted that the common feature of the conditions described in this chapter is that they all arise from defects in bone resorption or formation during the process of skeletal development, maturation, and remodeling.1,5,8,10 Sometimes several such conditions can coexist, resulting in the so-called overlapping dysplastic syndromes, which further supports the concept of their unified pathogenesis.5,6,9 An abbreviated list of sclerosing bone dysplasias classified on the basis of the so-called target-site approach originally proposed by Norman and Greenspan5,10 (i.e., specific phase, site of skeletal development affected, or both) is provided in Table 21-1. A summary of clinical presentations, radiographic characteristics, and genetic alterations implicated in the development of hereditary bone dysplasia is provided in Table 21-2.7
TABLE 21-1
Classification of Sclerosing Dysplasias of Bone
| Dysplasias of Enchondral Bone Formation |
| Dysplasias of Membranous Bone Formation |
TABLE 21-2
Summary of the Hereditary Sclerosing Bone Dysplasias
| Dysplasia | Inheritance Pattern | Genetics | Type of Dysplasia | Age at Onset | Osseous Findings | Other Findings |
| Osteopetrosis | AR, AD | TCIRG1 (ATP6i), CLCN7, GL (OSTM1), RANKL, CA2, PLEKHM1 | Endochondral (primary spongiosa) | Stillbirth or infancy to adulthood | Diffusely increased bone density; type 1: uniform sclerosis of the skull, spine, and long bones; type 2: endobone appearance | Anemia, cranial nerve deficits (rare with AD inheritance pattern) |
| Pyknodysostosis | AR | Cathepsin K | Endochondral (primary spongiosa) | Infancy or early childhood | Hyperostosis of long bones with preserved medullary cavities | Dwarfiism, acro-osteolysis, wormian bones |
| Osteopoikilosis | AD | LEMD3 | Endochondral (secondary spongiosa) | Childhood or adulthood | Multiple enostoses | Dermatofibrosis lenticularis disseminate |
| Osteopathia striata | X linked | Unknown | Endochondral (secondary spongiosa) | Childhood or adulthood (incidental finding) | Dense striations in metadiaphyses of long bones | No other associated abnormalities |
| Progressive diaphyseal dysplasia | AD | LAP of TGFB1 | Intramembranous ossification | Childhood | Bilateral/symmetric periosteal and endosteal cortical thickening involving long bones or calvaria | Gait disturbances, pain, weakness |
| Hereditary multiple diaphyseal sclerosis | AR | Unknown | Intramembranous ossification | After puberty | Unilateral/asymmetric cortical thickening involving long bones only | Milder neuromuscular symptoms than with progressive diaphyseal dysplasia |
| Hyperostosis corticalis generalisata | AR (Van Buchem disease, sclerosteosis), AD (Worth disease) | SOST (Wnt signaling pathway), LRP5 (Wnt signaling pathway) | Intramembranous ossification | Childhood | Endosteal cortical thickening involving the long bones, skull, and facial bones; mandible enlargement | Van Buchem disease: facial nerve palsy; sclerosteosis: facial nerve palsy, syndactyly, bone overgrowth; Worth disease: no cranial nerve palsy, torus palatinus |
Osteoma and Bone Island
Several benign, slow-growing bony lesions are identified under the general heading of osteoma (Fig. 21-1). They can be divided into (1) calvarial and mandibular ivory exostoses; (2) osteomas of the paranasal sinuses, facial bones, and orbit (sino-orbital osteomas); (3) enostoses or bone islands; and (4) surface (juxtacortical) osteomas of long bones.
Some authors consider these lesions to be hamartomatous or dysplastic in nature, but there is a rationale for regarding those that affect the orbit, facial bones, and paranasal sinuses (sinoorbital osteoma) as benign bone-forming tumors.
Ivory Exostosis
Ivory exostoses represent a type of osteoma that affects bones formed by membranous ossification, chiefly affecting calvarial and mandibular surfaces2,4,6,10–12,20,22,28 (Fig. 21-2). Multiple osteomas of this type are frequently associated with colonic polyposis and fibrous soft tissue tumors, including mesenteric fibromatosis and epidermal inclusion cysts in the familial disorder known as Gardner’s syndrome.13,14,17,24,29 Some patients also had distinct hyperplastic changes of the retinal pigmented epithelium.22,32,33 The development of ocular lesions may precede the development of colonic and osseous manifestations. The polyps of the gastrointestinal tract are adenomatous and typically involve the colon but can also be present in the duodenum and stomach. The adenomatous polyps of the gastrointestinal tract have a high propensity for malignant transformation. In fact, colon cancer develops in all affected individuals unless prophylactic colectomy is performed.23 The disorder is transmitted by an autosomal dominant pattern of inheritance. Similar to other major familial colonic polyposis syndromes, such as flat adenoma and Turcot’s syndrome (colorectal polyposis with brain tumor), Gardner’s syndrome is linked to a malfunctioning adenomatous polyposis coli (APC) gene mapped to chromosome 5q21.26 Mutations involving different regions of APC are associated with the severity of the clinical syndrome and the extent of extracolonic manifestations. It has been shown that mutations involving the 38 end around codon 1444 of the APC gene are associated with particularly severe osseous manifestations.16 Patients with this type of APC alteration also have a higher incidence of soft tissue fibromatosis. The soft tissue fibrous lesions range from aggressive fibromatosis at one end of the spectrum to a lesion which is histologically similar to nuchal-type fibroma.15,19,27 These fibromas of low cellularity have been designated as Gardner fibromas, and their significance as early indicators of the development of other manifestations of Gardner’s syndrome such as desmoid-type fibromatosis has been proposed.15,34 Unlike typical nuchal-type fibromas, which most often affect patients in the third through fifth decades of life, Gardner-associated fibromas arise in younger patients and the majority of them present in the first decade of life. The most common location is in the back and paraspinal region followed by the head and neck and extremities. Immunohistochemically, these lesions stain positively for CD34 and CD99. Many of these lesions show positive nuclear immune-reactivity for β-catenin.15,19 Approximately 50% of patients with Gardner-associated fibromas eventually develop a desmoid-type fibromatosis at the same site. Overall, 10% to 15% of patients with Gardner’s syndrome develop aggressive desmoid-type fibromatosis. They typically develop intraabdominally spontaneously or more often after surgery. Similar to Gardner-associated fibromas, desmoid fibromatoses associated with this syndrome harbor the inactivating mutations of the APC gene. However, similar to sporadic fibromatoses, they are positive immunohistochemically for nuclear β-catenin.15,19 In addition to osteomas, which are often multiple, dental malformation or more diffuse sclerotic changes in the craniofacial region may develop in patients with Gardner’s syndrome.25 Osteomas in Gardner’s syndrome may increase in size over time; that is, progressively more severe malformation of the craniofacial bones may develop.31 Osseous lesions in Gardner’s syndrome do not behave as true neoplasms, and there is no evidence of their evolution into malignant lesions. Moreover, the presence of osteoblastoma-like areas, predominantly seen in sporadic sinoorbital osteomas, is not a feature of osseous lesions in Gardner’s syndrome.
Microscopically, ivory exostoses are button-like excrescences of mature lamellar bone limited to the cortical surfaces and usually continuous with the outer table of the skull or the mandibular cortex. The dense cortical lamellar bone making up these exostoses contains osteons with haversian canals. Rare cases of compact surface osteoma have been reported in other flat bones as well as on long bones.44
Sinoorbital Osteoma
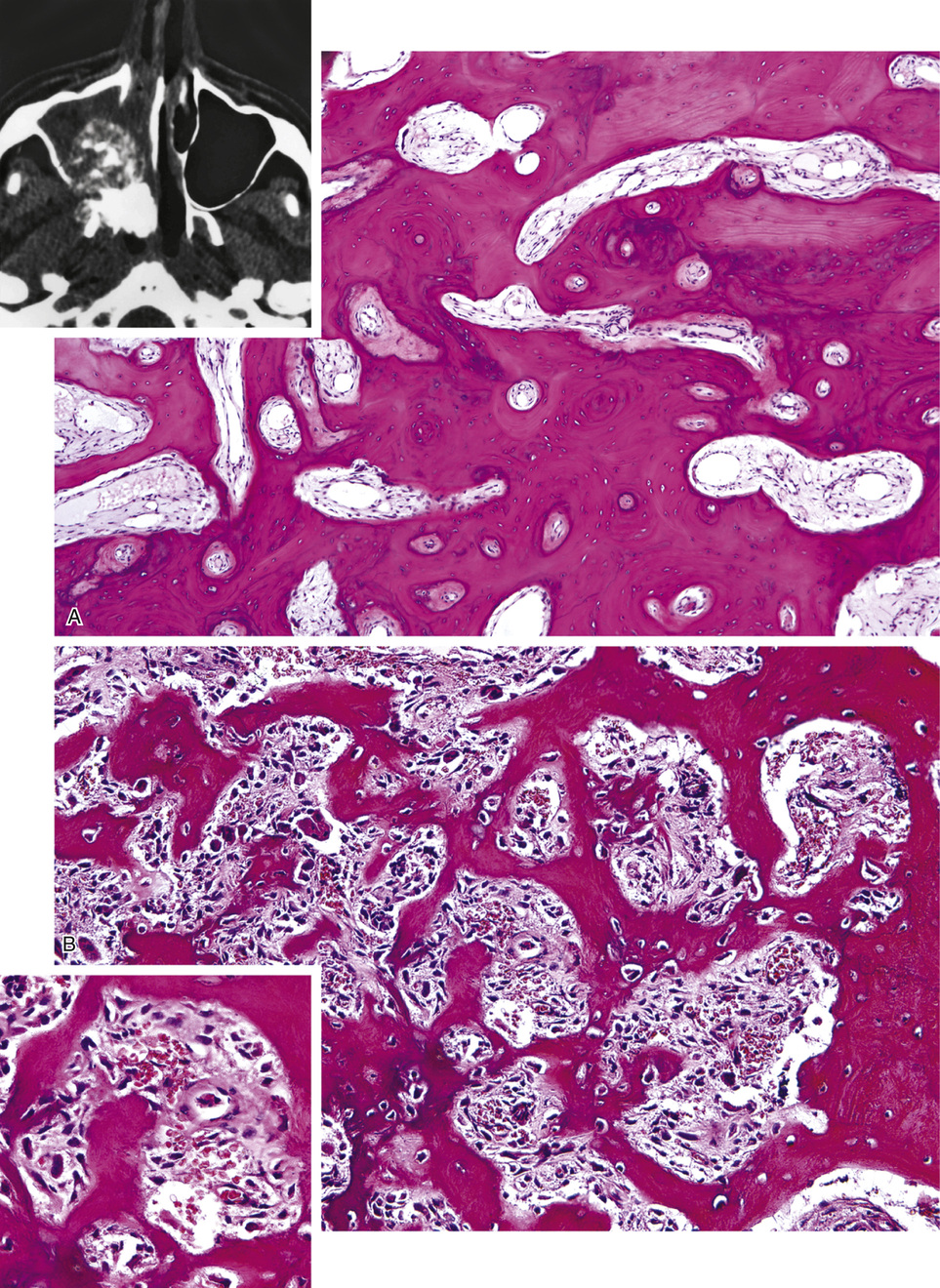
Osteomas involving paranasal sinuses, orbit, and facial bones tend to project into the sinus cavity from a broad attachment to the affected bone (Figs. 21-3 and 21-4).21,33,34,36,39,41,42,45,46 Some osteomas involving this region, especially those that are multifocal, are associated with Gardner’s syndrome. Microscopically, they are indistinguishable from ivory exostosis. In contrast, the so-called sporadic sinoorbital osteomas (not associated with Gardner’s syndrome) are usually composed of dense, immature bone and may have osteoblastic rimming as well as signs of bone resorption (Fig. 21-5). These lesions may contain central or peripheral foci of spongy or cancellous appearance (Figs. 21-6 and 21-7). Sometimes the latter areas show active remodeling, with cellular fibrous tissue filling the interstices, osteoblastic activity, and osteoclastic resorption.33 In ethmoid and frontal sinus osteomas, these lesions may occasionally contain radiolucent areas that are histologically indistinguishable from conventional osteoblastoma and even aggressive osteoblastoma (Fig. 21-8).38,40,47 The latter cases sometimes resemble multifocal osteoblastomas arising within osteomas. The clinical behavior of these lesions is similar to that seen in osteoblastomas and aggressive osteoblastoma groups; that is, they exhibit a local destructive growth pattern and recurrences. For this reason, we prefer to regard the sporadic osteomas that arise in the orbit, paranasal sinuses, and facial bones, especially those that contain osteoblastoma-like foci or features of active bone production and remodeling, as benign bone-forming neoplasms rather than as hamartomas or dysplasias. Osteomas in this region typically do not exceed 2 cm in diameter. Occasionally, they attain a larger size (giant osteoma) that compresses adjacent structures and cause a disfiguring deformity.28,35,37,43
[/not-level-membership-for-pathology-category]
