Chapter 18 Tremors
Introduction
Tremor is a rhythmic, oscillatory movement produced by alternating or synchronous contractions of antagonist muscles. It is the most common form of involuntary movement, but only a small fraction of those who shake will seek medical attention. Indeed, in one epidemiologic study of normal controls, 96% were found to have clinically detectable postural tremor, and 28% had a postural tremor of “moderate amplitude” (Louis et al., 1998e).
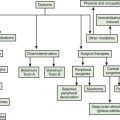
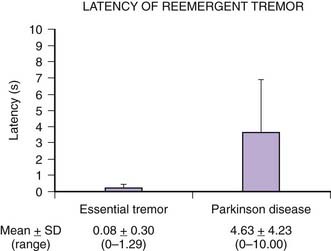
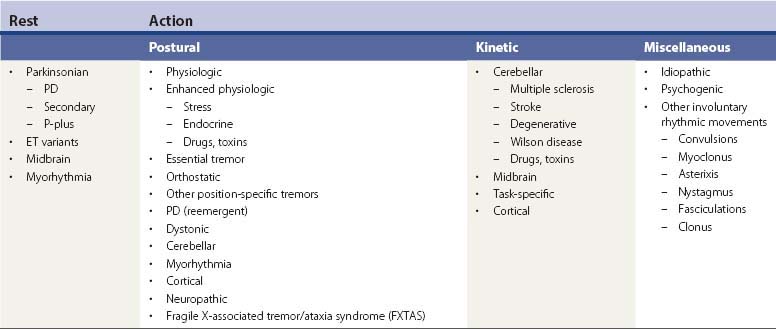
Tremors can be classified according to their phenomenology, distribution, frequency, or etiology (Hallett, 1991; Lou and Jankovic, 1991a; Bain, 1993; Findley, 1993; Deuschl et al., 1998a; Jankovic, 2000; Deuschl et al., 2001; Jankovic and Lang, 2008; Deuschl and Elble, 2009). Phenomenologically, tremors are divided into two major categories: rest tremors and action tremors (Table 18.1). Rest tremor is present when the affected body part is fully supported against gravity and not actively contracting; rest tremor is diminished or absent during voluntary muscle contraction and during movement. Action tremors occur with voluntary contraction of muscles, and they can be subdivided into postural, kinetic, task-specific or position-specific, and isometric tremors. Postural tremor is evident during maintenance of an antigravity posture, such as holding the arms in an outstretched horizontal position in front of the body. Some parkinsonian patients exhibit postural tremor that emerges after a latency of a few seconds. This tremor, referred to here as reemergent tremor, probably represents a rest tremor that has been “reset” during posture holding (Jankovic et al., 1999) (Fig. 18.1). The relationship of this reemergent tremor to the typical rest tremor is supported by the observation that this reemergent repose tremor shares many characteristics with the typical rest tremor; it has the same 3–6 Hz frequency, and it also responds to dopaminergic therapy (Video 18.1). Kinetic tremor can be seen when the voluntary movement starts (initial tremor), during the course of the movement (dynamic tremor), and as the affected body part approaches the target, such as while performing the finger-to-nose or the toe-to-finger maneuver (terminal tremor, also called intention tremor). Task-specific tremors occur only during, or are markedly exacerbated by, a certain task, such as while writing (primary handwriting tremor) (Video 18.2), while speaking or singing (voice tremor) (Rosenbaum and Jankovic, 1988; Soland et al., 1996b), or while smiling (Schwingenschuh et al., 2009). Besides writing, task-specific tremors may be triggered during other activities, such as while playing golf, particularly when putting (Video 18.3). Position-specific tremors occur while holding a certain posture (e.g., the “wing-beating” position or holding a spoon or a cup close to the mouth). One example of a task- or position-specific tremor is the tremor that occurs in performing the dot test (“dot approximation test”), during which the subject, seated at the desk with elbow elevated to a 90° shoulder abduction, is asked to hold the tip of the pen as close as possible to a dot on a horizontal paper without touching the dot. Patients with essential tremor (ET), to be discussed later, or other action tremors usually exhibit exacerbation of the tremor during this specific task. A variant of this tremor occurs in performing the modified finger–nose–finger test, during which the subject stands in front of a paper mounted on a wall and is asked to mark the center of the drawn target and to make a mark with a felt-tipped pen five times (Louis et al., 2005a). Isometric tremor occurs during a voluntary contraction of muscles that is not accompanied by a change in position of the body part, such as maintaining of a tightly squeezed fist or while standing (e.g., orthostatic tremor; see later). 
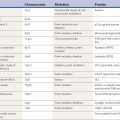
Table 18.1 Classification and differential diagnosis of tremors
| A. Rest tremors |
| 1. Parkinson disease (PD) |
| 2. Other parkinsonian syndromes |
Tremors can be also classified according to their anatomic distribution – for example, head, tongue, voice, and trunk. Orolingual tremors include physiologic, essential, task- and position-specific, dystonic, orthostatic, parkinsonian, palatal (also termed palatal myoclonus), drug-induced, hereditary, and psychogenic (Erer and Jankovic, 2007; Silverdale et al., 2008). Because of the complexity of limb tremors, it is best to describe them according to the joint about which the oscillation is most evident – for example, metacarpal-phalangeal joints, wrist, elbow, and ankle tremor. In most tremors, the frequency ranges between 4 and 10 Hz, but the cerebellar tremors may be slower, with a frequency of 2–3 Hz. The “slow” tremors (frequency: 1–3 Hz) are sometimes referred to as myorhythmia and are usually associated with brainstem pathology (Masucci et al., 1984; Cardoso and Jankovic, 1996; Tan et al., 2007a). The “fast” tremors (frequency: 11–20 Hz) may be distinct tremor disorders, such as orthostatic tremor, or may represent harmonics of other tremors. The clinical characteristics of tremors provide the most important clues to their etiology (Table 18.1).
Assessment of tremors
There have been many attempts to quantitate tremor, but it is not apparent whether electromyographic (EMG), accelerometric, or other methods of measuring tremor correlate with clinical rating scales. Indeed, one study suggested that assessments of spirography and handwriting correlate better with overall functional tremor-related disability than electrophysiologic methods (Bain et al., 1993). However, because the physiologic measurements and the clinical ratings were not performed simultaneously and because of other technical problems, interpretation of the study is difficult. Elble and colleagues (1996) described the use of a digitizing tablet in quantification of tremor during writing and drawing. Although relatively good inter-trial correlations were obtained with this method, the tablet does not capture the speed of writing or the amount of effort exerted by the patient in an attempt to control the tremor while writing. One study found high inter-observer reliability using a diagnostic protocol for ET (Louis et al., 1998a). The investigators also found high specificity and sensitivity of a screening questionnaire when compared to the physician’s examination in patients with definite and probable ET, but actual examination of the subjects is necessary to detect mild ET (Louis et al., 1998b). In another study, the authors concluded that when a limited number of tests are available in large epidemiologic surveys, a test such as the finger–nose maneuver may be used to screen populations for ET, whereas to exclude normal subjects, the spiral drawing test, water pouring test, or arm extension test may be utilized (Louis et al., 1999a). A performance-based test for ET has been validated and compared to other measures of tremor (Louis et al., 1999b). Although this performance-based test was thought to objectively assess functional capacity in patients with ET, the test seems somewhat cumbersome to perform because it requires a variety of props, such as a milk carton, a glass, a soup spoon, a bowl, a saucer, a wallet, coins, an electrical socket, a thread and needle, a strip of buttons, and a telephone. Using other instruments, the modified Klove–Matthews Motor Steadiness Battery and the Nine-Hole Steadiness Tester, Louis and colleagues (2000c) showed that these portable instruments provide a reliable and valid means of collecting objective quantitative data on tremor severity. A Tremor Disability Questionnaire has been developed and found to reliably correlate with multiple measures of tremor severity (Louis et al., 2000a). Another screening instrument for ET, consisting of seven items and a spiral drawing, has been found to have 70.5% sensitivity, 68.2% specificity, and 64.9% positive predictive value (Lorenz et al., 2008). A simple, user-friendly clinical tool, with even higher sensitivity, specificity and predictive value, is needed to assess tremors in the clinic and in the field. A teaching videotape for assessment of ET was developed to improve the uniform application of the Washington Heights–Inwood Genetic Study of Essential Tremor (Louis et al., 2001a). Using a clinical evaluation (interview and videotaped examination) and an electrophysiologic evaluation (quantitative computerized tremor analysis using accelerometry and EMG), Louis and Pullman (2001) found a very high concordance rate between the two methods in 51 of 54 (94%) subjects, suggesting that using either technique would arrive at a similar diagnosis. Although not yet validated, the Unified Tremor Rating Assessment developed by the Tremor Research Group has been used in a number of clinical, therapeutic trials (Bain, 1993; Jankovic et al., 1996). The other scale that has been used in several tremor studies is the Fahn–Tolosa–Marin Tremor Rating Scale (TRS) (Fahn et al., 1993). While the inter-rater reliability of this scale is relatively poor, there is a good consistency, with average Spearman correlation of 0.87, when the same rater repeatedly assesses the tremor (Stacy et al., 2007). Using the TRS, Putzke et al. (2006) showed that the total score increased by about 2 points during prospective follow-up of patients with ET over a mean of 3.6 years and that older age, longer duration of disease, and asymmetric onset of tremor were associated with increased tremor severity. Another tremor rating scale, the Tremor Research Group (TRG) Essential Tremor Rating Scale (TETRAS), is currently being validated against the TRS (Elble et al., 2008). The TETRAS has been found to correlate well with quantitative assessments using the Kinesia™ (CleveMed) system (Mostile et al., 2010). Any assessment of tremor must take into account minute-to-minute and hour-to-hour amplitude variability (Koller and Royse, 1985), and potential provocations, such as voluntary isometric contraction in the case of action tremors and walking and counting backwards in the case of rest tremor (Raethjen et al., 2008).
Rest tremors
Diagnosis
Rest tremor is most typically present in patients with Parkinson disease (PD). In one study, all 34 patients with pathologically proven cases of idiopathic (Lewy body) parkinsonism demonstrated typical rest tremor sometime during the course of their illness (Rajput et al., 1991). Although this study suggests that parkinsonian patients who do not exhibit rest tremor probably do not have idiopathic parkinsonism (PD), another study, involving 100 pathologically proven cases of PD, found that 32% of all patients apparently never manifested tremor during the course of their disease (Hughes et al., 1993).
Several studies have suggested that the natural course of PD is in part related to the presence or absence of tremor (Hughes et al., 1993). The tremor-dominant PD may be associated with earlier age at onset, less cognitive decline, and slower progression than the type of PD that is dominated by postural instability and gait difficulty (PIGD) (Jankovic et al., 1990). Clinical-pathologic correlations are needed to answer the question as to whether the tremor-dominant form and the PIGD-dominant form represent different diseases or merely variants of one disease, namely, PD. In support of the former is the finding that only 27% of patients with the PIGD form of idiopathic parkinsonism had Lewy bodies at autopsy (Rajput et al., 1993). In another clinical-pathologic study, Hirsch and colleagues (1992) demonstrated that patients with PD and prominent tremor have degeneration of a subgroup of midbrain (A8) neurons, whereas this area is spared in PD patients without tremor. This observation supports the hypothesis that differential damage of subpopulations of neuronal systems is responsible for the diversity of phenotypes seen in PD and other parkinsonian disorders. It is unclear whether the occasional patients with long-standing unilateral tremor and minimal or no other parkinsonian findings have a benign form of PD, as is suggested by positron emission tomography (PET) scans showing low fluorodopa uptake in the contralateral putamen (Brooks et al., 1992), or whether this condition represents a separate disease entity. In contrast to patients with the PIGD form of PD, patients with tremor-dominant PD have increased metabolic activity in the pons, thalamus, and motor association cortices (Antonini et al., 1998). When rest tremors involve the fingers, hands, lips, jaw, and tongue in the same individual, they share a common frequency, suggesting that they are of central origin (Hunker and Abbs, 1990). This pattern, however, changes during sleep in that non-rapid eye movement sleep transforms the alternating tremor that is typically seen in the awake patient into subclinical repetitive muscle contractions of variable frequency and duration during sleep stages I to IV, and the tremor disappears during rapid eye movement sleep (Askenasy and Yahr, 1990).
Rest tremor has other causes besides PD and related parkinsonian disorders (see Table 18.1). Patients with severe ET may have tremor at rest and prominent kinetic tremor. It is not known whether the ET patients with rest tremor have associated PD, whether they later develop other features of PD, or whether the rest tremor is a feature of ET (Jankovic, 1989; Shahed and Jankovic, 2007). Some patients with lesions in the cerebellar outflow pathways, particularly in the superior cerebellar peduncle near the red nucleus (cerebellar outflow, midbrain or “rubral” tremor, also referred to as Holmes tremor), also have tremor at rest, probably due to an interruption of the nigrostriatal pathway (Remy et al., 1995). This irregular, slow (2–5 Hz), predominantly unilateral tremor may be associated with other neurologic signs, such as ataxia, bradykinesia, and ophthalmoplegia. It is often associated with midbrain pathology, such as multiple sclerosis, stroke, tumor, or arteriovenous malformation (Lee et al., 2008). It rarely responds to any medical therapy, but wrist weights, levodopa, dopamine agonists, amantadine, propranolol, clonazepam, isoniazid, and levetiracetam (Ferlazzo et al., 2008) may be effective in reducing the amplitude of this, often disabling, tremor. The affected arm or leg may be also ataxic and may be associated with third nerve palsy (Benedikt syndrome) (Video 18.4). The cerebellar outflow tremor is most often caused by trauma, stroke, multiple sclerosis, and Wilson disease (Lou and Jankovic, 1993; Krauss et al., 1995; Miwa et al., 1996; Alarcon et al., 2004). Strokes involving the posterior circulation may involve the thalamus, producing slow (1–3 Hz) rest and postural tremors, sometimes referred to as myorhythmia (Masucci et al., 1984; Cardoso and Jankovic, 1996; Miwa et al., 1996). 
Myorhythmia is a slow (1–3 Hz) frequency, continuous or intermittent, relatively rhythmic movement that is present at rest but may persist during activity (Masucci et al., 1984; Cardoso and Jankovic, 1996). It may be associated with palatal myoclonus, and it disappears with sleep. Except for the slower frequency, the presence of flexion–extension rather than the typical supination–pronation pattern, and the absence of associated parkinsonian findings, myorhythmia resembles a parkinsonian tremor. In the cases that were examined at autopsy, the sites of maximum pathology involved chiefly the brainstem (particularly the substantia nigra and the inferior olive) and the cerebellum. The etiology for myorhythmia includes brainstem stroke, cerebellar degeneration, Wilson disease, and Whipple disease (Masucci et al., 1984; Tison et al., 1992; Cardoso and Jankovic, 1996).
Palatal myoclonus, sometimes referred to as palatal tremor, has some features of tremor, but in contrast to tremor which is produced by alternating or synchronous contractions of antagonist muscles, the palatal movement is produced by rhythmical contractions of agonist muscles, hence the term myoclonus is preferred despite the arguments raised against this nosology (Zadikoff et al., 2006). Palatal myoclonus, a form of segmental myoclonus, is manifested by rhythmical contractions of the soft palate resulting from acute or chronic lesions involving the Guillain–Mollaret triangle linking dentate nucleus with the red nucleus via the central tegmental tract to the inferior olivary nucleus. Symptomatic palatal myoclonus (SPM) usually persists during sleep, while essential palatal myoclonus (EPM), frequently associated with an ear-clicking sound, disappears with sleep. In EPM the muscle agonist is the tensor veli palatini, which opens the eustachian tube and is innervated by the trigeminal nerve. In SPM the palatal movement is due to contractions of the levator veli palatini, innervated by the facial nucleus and nucleus ambiguus. When the tensor muscle contracts, as in EPM, the entire soft palate moves, whereas only the edges of the soft palate move when the levator muscle contracts in SPM. Symptomatic, but not essential, palatal myoclonus is often associated with hypertrophy of the inferior olive (Goyal et al., 2000). SPM has been associated with a variety of lesions involving the brainstem as well as some neurodegenerative disorders such as Alexander disease (Pareyson et al., 2008).
Treatment with neuroleptics can also cause persistent tremor, referred to as tardive tremor (Stacy and Jankovic, 1992). This rest, postural, and kinetic tremor, with a frequency of 3–5 Hz, is aggravated by, and persists after, neuroleptic withdrawal and improves after treatment with the dopamine-depleting drug tetrabenazine. The tremor may be accompanied by other tardive movement disorders, including akathisia, chorea, dystonia, myoclonus, and stereotypy. There is usually no family history or other explanation for the tremor.
Spasmus nutans is characterized by the triad of nystagmus, abnormal head position, and irregular, multidirectional head nodding that disappears during sleep. This self-limited and often familial condition is first noted between the ages of 4 and 12 months, and it usually disappears within a year or two. Another oculomotor cause of head tremor is “head-shaking nystagmus” seen in patients with lateral medullary infarction (Choi et al., 2007). The 2–3 Hz horizontal head shaking has been postulated to be caused by unilateral impairment of nodulo-uvular inhibition of the velocity storage.
Treatment
The treatment of rest tremors is similar to that of parkinsonism (Jankovic and Marsden, 1998; also see Chapter 6). Secondary and potentially curable causes should be excluded, particularly when there are associated features to suggest disorders other than PD (see Table 18.1). Anticholinergic and dopaminergic drugs provide the most effective relief of rest tremors. Clozapine, an atypical neuroleptic that does not significantly exacerbate parkinsonism but can cause potentially serious side effects such as agranulocytosis, has been shown to be effective in the treatment of parkinsonian tremor (and ET) (Bonuccelli et al., 1997; Friedman et al., 1997; Ceravolo et al., 1999). Ethosuximide, an anticonvulsant that blocks low-threshold Ca2+ conductance in the thalamus, has been shown to reduce tremor in MPTP monkeys and to potentiate the effects of a D2 agonist (Gomez-Mancilla et al., 1992). However, ethosuximide was found ineffective in a pilot study of six PD patients with drug-resistant tremor (Pourcher et al., 1992). Mirtazapine (Remeron), a novel antidepressant that enhances noradrenergic and serotonergic transmission and acts as a presynaptic alpha-2, 5-HT2, and 5-HT3 receptor antagonist, has been reported to improve rest tremor (Pact and Giduz, 1999). Other drugs reported to have a possible beneficial effect in patients with ET include mirtazapine, clozapine, sodium oxybate, dimethoxymethyl-diphenyl-barbituric acid (T-2000), and carisbamate (Lyons and Pahwa, 2008). High-amplitude parkinsonian tremors and rest tremors caused by disorders other than PD usually do not improve with pharmacologic therapy. In some cases, botulinum toxin (BTX) injections in the involved muscles produce a satisfactory reduction in the tremor amplitude (Jankovic and Schwartz, 1991; Jankovic et al., 1996; Hou and Jankovic, 2002). A multicenter, randomized, double-blind, controlled trial confirmed the results of an earlier study (Jankovic et al., 1996) that BTX injections produce significant reduction in the postural hand tremor of ET and modest functional improvement (Brin et al., 2001). By avoiding injections of the forearm extensor muscles we prevent finger extensor weakness, a relatively frequent complication reported in the earlier studies (Pacchetti et al., 2000).
Ventral lateral thalamotomy, particularly involving the ventral intermediate nucleus of the thalamus (VIM), was considered the neurosurgical treatment of choice for disabling, drug-resistant tremors until the later 1980s when the ablative procedure was replaced by high-frequency deep brain (thalamic) stimulation (DBS) (Benabid et al., 1991; Fox et al., 1991; Jankovic et al., 1995b). Although effective in a majority of cases, the tremor recurs in about 20% of patients, and there is a considerable risk of contralateral hemiparesis, hemianesthesia, ataxia, speech disturbance, and other potential complications. These are compounded when the procedure is performed bilaterally. Thalamic DBS is now the surgical treatment of choice for patients with disabling tremors (Deiber et al., 1993; Benabid et al., 1996; Pahwa and Koller, 2001; Ondo et al., 2001a, 2001b; Pahwa et al., 2006) (see also Chapter 7). This technique has been proposed for chronic treatment of parkinsonian, essential, and other tremors. Using high-frequency (100 Hz) stimulation, with the tip of a monopolar electrode implanted stereotactically in the VIM contralateral to the disabling tremor, Benabid and colleagues (1991) noted “complete relief” of contralateral tremor in 27 of 43 (63%) thalami that were stimulated and “major improvement” in 11 (23%). The series included 26 patients with PD and 6 with ET, 7 of whom had previously been treated with thalamotomy. The benefit of thalamic stimulation was maintained for up to 29 months (mean follow-up: 13 months). The results were similar in their subsequent report of long-term effects of chronic VIM stimulation in 117 patients, 74 of whom had bilateral implantation (Benabid et al., 1996). The most robust tremor suppression was noted in patients with PD (n = 80); but patients with ET (n = 20) also benefited, although 18.5% deteriorated with time. Dysarthria and ataxia still occurred, but the patients were able to adjust the intensity of stimulation to ameliorate these side effects, though at the expense of increased tremor. Nevertheless, the investigators felt that the reversible nature of the side effects was the chief advantage of DBS over the permanent lesion produced by thalamotomy. To compare thalamic DBS with thalamotomy, Schuurman and colleagues (2000) conducted a prospective, randomized study of 68 patients with PD, 13 with ET, and 10 with multiple sclerosis. They found that the functional status improved more in the DBS group than in the thalamotomy group, and tremor was suppressed completely or almost completely in 30 of 33 (90.9%) patients in the DBS group and in 27 of 34 (79.4%) patients in the thalamotomy group. Although one patient in the DBS group died after an intracerebral hemorrhage, DBS was associated with significantly fewer complications than was thalamotomy. This procedure may be also advantageous in elderly patients and when bilateral effects are desirable (Blond et al., 1992). We found that bilateral thalamic DBS is more effective than unilateral DBS in controlling bilateral appendicular and midline tremors of ET and PD, and thalamic DBS does not seem to improve meaningfully any parkinsonian symptoms other than tremor (Ondo et al., 2001a). In addition, we found that VIM DBS produces modest improvement, rather than tremor augmentation as previously suggested, in ipsilateral tremor in patients with ET (Ondo et al., 2001b). A review of long-term efficacy of VIM DBS in 39 patients (20 with PD and 19 with ET) showed that the benefits may be maintained for at least 6 months (Rehncrona et al., 2003). In one of our patients, minimal foreign body reaction and gliosis around the electrodes was found 12 years after implantation, the longest reported follow-up with autopsy examination after DBS, supporting the long-term safety of DBS (DiLorenzo et al., 2010).
In addition to improving distal tremor associated with PD and ET, VIM DBS can effectively control ET head tremor, which usually does not respond to conventional therapy (Koller et al., 1999). Other midline tremors, such as voice, tongue, and face tremor, also may improve with unilateral VIM DBS, although additional benefit can be achieved with contralateral surgery (Obwegeser et al., 2000). The risk of local gliosis with chronic stimulation of the thalamus is minimal (Caparros-Lefebvre et al., 1994). Unfortunately, thalamic stimulation does not appear to be as effective in patients with predominantly kinetic and axial tremors, and it does not improve other parkinsonian features such as bradykinesia, rigidity, and levodopa-related motor complications. Furthermore, while VIM DBS is very effective in improving PD tremor, when performed bilaterally it is often associated with dysarthria and postural and gait abnormality (Pahwa et al., 2006) as a result of which the subthalamic nucleus (STN) has been suggested as a more appropriate target in PD patients with severe tremor (Limousin et al., 1998; Benabid et al., 2000; Diamond et al., 2007; Fishman, 2008). The mechanism of action of DBS is unknown, but “jamming” of low-frequency oscillatory inputs has been suggested as a possible mechanism for the antitremor effects of DBS. Regional cerebral blood flow, measured by PET scan, demonstrated that tremor suppression was associated with decreased cerebellar blood flow and, presumably, decreased synaptic activity in the cerebellum (Deiber et al., 1993).
In 1992, Laitinen of Stockholm, Sweden, reported the results of 90 pallidotomies in 86 patients with severe PD (Laitinen et al., 1992). The external, posteroventral portion of the medial globus pallidus interna (GPi) was the intended target for the stereotactically placed lesion. Nearly all patients had “marked improvement in tremor and akinesia.” In addition, some patients apparently also noted improvement in their gait, speech, and pain. Only two patients suffered permanent visual field defect, and one had “minor stroke with hemiparesis.” Several pallidotomy series have since confirmed the beneficial effects of pallidotomy on various parkinsonian symptoms, including tremor (Jankovic and Marsden, 1998). These results provide support for the notion that the GPi is “hyperactive” in PD and that surgical or chemical lesions of these structures may have a therapeutic value not only in controlling tremor but also in improving bradykinesia (Bergman et al., 1990; Aziz et al., 1991). Although some investigators (Subramanian et al., 1995) have suggested that posteroventral pallidotomy is as effective as thalamotomy in controlling parkinsonian tremor, others (Dogali et al., 1995) feel that pallidotomy provides only partial relief of tremor.
Postural tremors
Diagnosis and clinical features
Physiologic tremor
Normal and enhanced physiologic tremors are the most common forms of postural tremor, but they rarely require medical attention. Postural tremors are clinically similar despite different etiologies. In contrast to ET, the frequency of physiologic tremor can be slowed by mass loading (Elble and Koller, 1990). Indeed, there appear to be two components to physiologic tremor: variable frequency (peak: 8 Hz), which is dependent on loading, and consistent frequency (peak: 10 Hz), which is independent of peripheral influence. The latter suggests central origin of the tremor, as is presumed the case in ET. Thus the amplitude of ET is less dependent on the position of the tested limb than is the amplitude of other postural tremors, including physiologic tremors (Sanes and Hallett, 1990).
Essential tremor
Although the term “essential” implies necessary or desirable, it actually means that there is no known cause and the term is synonymous with “idiopathic.” The term “essential tremor” did not gain regular and widespread currency until a century or so after its initial use in 1874 by Pietro Burresi, a professor of medicine at the University of Siena, Italy (Louis et al., 2008b). He coined the term “tremore semplice essenziale” or “simple essential tremor” when he described the case of an 18-year-old man suffering from severe tremor of the arms when engaged in voluntary movement as well as head tremor. While the amplitude of ET tends to increase with age, the tremor frequency decreases with age (Elble et al.,1994; Elble, 2000b). The tremor of ET is typically a postural or kinetic tremor with frequency varying between 4 and 10 Hz. Although the frequency of the tremor is relatively constant in a particular individual, the amplitude may vary and in some cases may be even suppressed by mental concentration and distraction (Koller and Biary, 1989; Kenney et al., 2007).
Epidemiology of ET
In the past the modifier “benign” was used (“benign ET”) to indicate favorable prognosis of ET, even though it is now well accepted that ET can produce marked physical and psychosocial disability (Busenbark et al., 1991; Jankovic, 2000; Sullivan et al., 2004; Louis, 2005; Benito-León and Louis, 2006; Elble et al., 2006). Furthermore, in a longitudinal, prospective, population-based study, ET has been found to be associated with increased mortality at an estimated risk ratio of 1.59 (95% CI 1.11–2.27, P = 0.01) (Louis et al., 2007a). Meta-analysis of epidemiologic studies has found the prevalence of ET to range between 0.01% and 20.5%, but the pooled prevalence is 0.9%; the prevalence in people ≥65 years old is 4.6% and may be as high as 21.7% in people ≥95 years old; the prevalence is higher in males than females (Louis and Ferreira, 2010). There are many other estimates (Haerer et al., 1982; Louis et al., 1995, 1998d; Dogu et al., 2003) such as 5.5% in people over the age of 40 years (Rautakorpi et al., 1982) and 14% in people 65 years old or older (Moghal et al., 1994) (Table 18.2). In one epidemiologic study, 108 of 1056 (10%) nondemented individuals in upper Manhattan, aged 65 years or older, reported “shaking” (Louis et al., 1996). Neurologic examination confirmed rest tremor in 8.3% and action tremor in 17.6%, and the prevalence of PD and ET was estimated to be 3.2% and 10.2%, respectively. In a door-to-door survey of people aged 40 years or older in Mersin Province, Turkey, the prevalence of ET was found to be 4% (Dogu et al., 2003). In another population-based survey, involving 5278 subjects aged 65 years or older in central Spain who were followed for a median of 3.3 years, the adjusted annual incidence was determined to be 616 per 100 000 person-years; 64 of the 83 (77.1%) incident cases had not been previously diagnosed, and only 4 (4.8%) were taking antitremor medications (Benito-León et al., 2005). In yet another population-based study of northern Italian adults in which all participants were examined and classified by movement specialists using rigorous diagnostic criteria, tremors comprised the most common category of movement disorders, followed by restless legs syndrome (Wenning et al., 2005). These epidemiologic studies provide strong evidence that the prevalence and incidence of ET are higher than was previously recognized.
| Prevalence |
Diagnosis of ET
Although ET was described as early as the nineteenth century (Dana, 1887; Louis, 2010), there is still considerable controversy about the diagnostic criteria for ET (Chouinard et al., 1997; Louis et al., 1998c; Jankovic, 2000; Louis, 2010; Quinn et al., 2011). In one study of 71 patients, 37% diagnosed with ET based on the criteria for ET adapted from the consensus statement of the Movement Disorders Society (Deuschl et al., 1998a) were misdiagnosed, usually either as PD or dystonia (Jain et al., 2006). This is partly due to a lack of a disease-specific marker for ET. No specific pathologic changes indicative of PD were noted in 20 brains of ET patients that were examined at autopsy (Rajput et al., 2004). However, this study is fundamentally flawed, since all patients who were selected for the study had a diagnosis of ET at the time of death and patients who started with ET and later developed PD would have been excluded (Jankovic, 2004). It is of interest that one patient with severe ET that began at age 45 and no parkinsonian features, at the time of her death at age 91 years had Lewy bodies localized to the locus coeruleus, providing further evidence of a connection between ET and Lewy body disease (Louis et al., 2005b). The controversy about the possible association of ET and PD should be clarified once the genetic basis and pathophysiology of ET are understood. Until then, the operational diagnostic criteria must rely on the presence of typical clinical characteristics. The presence or absence of certain clinical characteristics may be used to categorize ET into “definite,” “probable,” and “possible” (Table 18.3). The diagnostic criteria may be used or modified according to specific needs. For example, for genetic linkage studies, only “definite” ET may be acceptable, whereas in studies that are designed to explore the clinical spectrum of ET, including associated features, the “possible” ET category might be more appropriate (Table 18.4). Family history, alcohol sensitivity, and propranolol responsiveness, while characteristic of ET, should not be considered necessary for the diagnosis. More recently, core and secondary criteria were proposed to facilitate a practical approach to the diagnosis of ET (Elble, 2000a). Core criteria include bilateral action tremor of the hands and forearms (but not rest tremor), absence of other neurologic signs, except for the Froment sign (a “cogwheel” phenomenon on passive movement of the affected limb with voluntary movement of the contralateral limb), and isolated head tremor without signs of dystonia, although the latter is rare. A recent analysis of ET patients from two large population-based studies and one large clinic-based cohort revealed no cases of pure head tremor in 583 patients (Louis and Dogu 2009). Therefore, patients with pure head tremor probably should not be regarded as definite ET. Head tremor is seen in about 18% of population-based cases of ET and in 37% of clinical samples of ET (Louis and Dogu, 2009). Secondary criteria include long duration (>3 years), a positive family history, and a beneficial response to alcohol (Mostile and Jankovic, 2010). Another feature, seen in about a third of patients with ET, is mirror movements (Louis et al., 2009d), more typically observed in patients with focal dystonia (Sitburana et al., 2009). There are red flags that indicate a diagnosis other than ET, such as unilateral tremor, present in only 4.4% of cases of ET (Phibbs et al., 2009), leg tremor, rigidity, bradykinesia, rest tremor, gait disturbance, focal tremor, isolated head tremor with abnormal posture (head tilt or turning), sudden or rapid onset, and drug treatment that may cause or exacerbate tremor. Thus head tremor is usually a manifestation of ET or cervical dystonia; it is almost never seen in PD unless there is coexistent ET (Roze et al., 2006; Gan et al., 2009).
| A. Definite essential tremor |
| 1. Inclusions |
Members of the Tremor Research and Investigation Group: M. Brin, C. Contant, R. Elble, L. Findley, J. Jankovic, W. Koller, P. LeWitt, A. Rajput. From Findley LJ, Koller WC. Definitions and behavioural classifications. In Findley LJ, Koller WC (eds): Handbook of Tremor Disorders. New York, Marcel Dekker, 1995, pp 1–5.
Table 18.4 NIH Essential Tremor Consortium diagnostic criteria for essential tremor
| Definite |
| Probable |
| Possible |
Tremor rating: 0, none perceived; 1, slight (barely noticeable); 2, moderate, noticeable, probably not disabling (<2 cm excursions); 3, marked, probably partially disabling (2–4 cm excursions); 4, severe, coarse, disabling (>4 cm excursions).
Participants in the July 1996 NIH meeting: J. Beach, S.B. Bressman, M.F. Brin, D. De Leon, L. Goldfarb, M. Hallett, J. Jankovic, W. Koller, D. Mirel, K. Wilhemsen. From Brin MF, Koller W. Epidemiology and genetics of essential tremor. Mov Disord 1998;13(Suppl. 3),55–63.
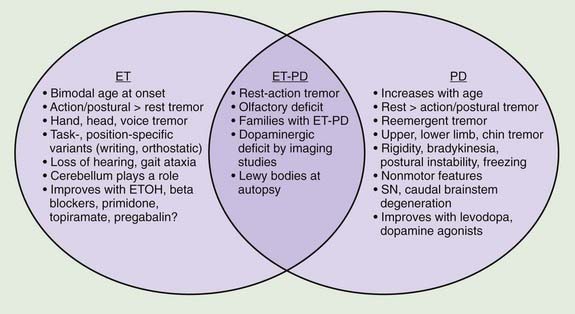
A review of the clinical features in 350 consecutive patients who were referred to the Movement Disorders Clinic at Baylor College of Medicine and diagnosed with ET has shown that although tremor is clearly the most troublesome symptom, it is not necessarily the only symptom in patients with ET (Lou and Jankovic, 1991b) (Tables 18.1 and 18.3). This is supported by the reports of well-studied families in which some members have typical ET, while others have dystonia, parkinsonism, or a combination of all three disorders (Jankovic et al., 1997; Farrer et al., 1999; Bertoli-Avella et al., 2003; Yahr et al., 2003; Spanaki and Plaitakis, 2009). One multigenerational family, 36 members in five generations, had an admixture of ET, PD, and dystonia (Yahr et al., 2003). Two twin brothers with ET and PD had the classic pathologic features of PD at autopsy. The authors concluded, “This unusual set of clinical and pathologic circumstances can hardly be attributed to chance occurrence and raises the question of a specific genetic mutation and/or clustering, which may link ET with PD.” In a study of the first-degree relatives of 303 PD probands and 249 controls from Crete, ET was present in the relatives of PD patients more often than in those of controls (OR: 3.64, P < 0.001) and the risk was even greater (OR: 4.48) when the affected proband had tremor-dominant or mixed PD (Spanaki and Plaitakis, 2009). Twelve subjects had both ET and PD phenotypes. The authors concluded that “in certain families ET and PD are genetically related probably sharing common hereditary predisposition.” Retrospective chart review at the Neurological Institute (NI) of New York showed that 56.7% of 210 PD patients versus 33.3% of 210 Parkinson-plus syndrome patients (P < 0.001) had kinetic tremor on examination and patients with PD were more likely to have a diagnosis of ET assigned by an NI neurologist (5.3% vs. 0.0%, OR 12.85, 95% CI 1.66–99.8, P = 0.001) (Louis and Frucht, 2007). Patients with PD were three to thirteen times more likely to have diagnoses of ET than patients with Parkinson-plus syndromes, thus confirming “the link between ET and PD, and possibly, between ET and Lewy body disease.” In another large family, originally from Cuba, manifested by parkinsonism and ET, the parkinsonism was linked to a marker on chromosome 19p13.3–q12, but it did not cosegregate with ET (Bertoli-Avella et al., 2003). Other gene mutations associated with PD have not been found in patients with ET alone (Deng et al., 2006a). Some, but not all (Adler et al., 2011) studies have suggested that there is an association between ET and dystonia and between ET and parkinsonism (Jankovic et al., 1997; Yahr et al., 2003; Shahed and Jankovic, 2007; Fekete and Jankovic, 2011) (Fig. 18.2). In addition to dystonic tremor, patients with dystonia frequently have postural ET-like tremor present in body parts distal to the dystonia, and they have a higher-than-expected family history of postural tremor (Chan et al., 1991; Jankovic et al., 1991; Deuschl et al., 1997; Jankovic and Mejia, 2005; Schneider et al., 2007). Asymmetric dystonic hand tremor may be initially misdiagnosed as PD-related tremor and, along with rest tremor associated with ET, may be responsible for scans without evidence of dopamine deficiency (SWEDDs) (Schneider et al., 2007; Bain, 2009; Schwingenschuh et al., 2010; Stoessl, 2010). When 25 tremulous SWEDDs patients were compared to 25 tremor-dominant PD patients, the former group lacked true bradykinesia, they had evidence of dystonia, and head tremor, whereas reemergent tremor, true fatiguing or decrement, good response to dopaminergic drugs, and presence of nonmotor symptoms favored PD (Schwingenschuh et al., 2010). Whether the hand tremor that is seen in about 25% (10–85%) of patients with cervical dystonia represents an enhanced physiologic tremor, ET, dystonic tremor, or some other form of postural tremor is unknown (Jankovic et al., 1991; Deuschl et al., 1997). Although the frequency of ET and the limb tremor in patients with cervical dystonia are similar, the tremor amplitude in patients with dystonia is smaller and the tremor is more irregular, suggesting that the two types of tremors, while similar, arise from different types of oscillators (Shaikh et al., 2008). The observation of significant overlap in association between variants in LINGO1 and ET and PD (see below) provides support for the genetic association between the two common movement disorders (Vilariño-Güell et al., 2010a).
The clinical heterogeneity of ET suggests that there may be different subtypes. Indeed, Louis and colleagues (2000b) found that patients with older onset (>60 years) and those without head tremor progressed more rapidly than did patients with young-onset tremor and those with head tremor. They also later found that head tremor was present four times more frequently in women than in men (Louis et al., 2003; Hardesty et al., 2004). Using the medical records linkage system of the Rochester Epidemiology Project, the authors identified ET patients who also had an autopsy report and found that women with ET were six times more likely to develop head tremor than men (Hardesty et al., 2004). The presence of jaw tremor, seen in 7.5–18.0% of patients with ET, has been found to be associated with older age at onset, more severe action tremor in arms, and the presence of head and voice tremor (Louis et al., 2006a). In a study of 34 patients with voice tremor, 93% were female and the voice tremor typically began in the seventh decade (62.9 ± 15.0 years) (Sulica and Louis, 2010). More than a third had a first-degree relative with tremor and more than a quarter reported a beneficial effect of ethanol. In this study only 11 (32.3%) were aware of an arm tremor and 10 (29.4%) had been misdiagnosed as spasmodic dysphonia. Only 56% of treated patients found botulinum toxin helpful and the response was often incomplete. Jaw tremor was also significantly associated with rest tremor, suggesting that some patients with ET and jaw tremor may convert to PD. Besides ET and PD, jaw tremor may be a manifestation of dystonia (Schneider and Bhatia, 2007). Lower extremity tremor is usually mild or asymptomatic (Poston et al., 2009).
The ongoing debate as to whether ET is a monosymptomatic or heterogeneous disorder or phenotypic manifestation of multiple entities will probably not be resolved until disease-specific physiologic, genetic, or other biologic markers are identified (Schrag et al., 2000; Elble, 2002; Jankovic, 2002; Elble and Tremor Research Group, 2006; Louis, 2009; Deuschl and Elble, 2009). One hypothesis is that the various nonmotor signs linked to ET could be secondary to “abnormal neuronal oscillation” (Deuschl and Elble, 2009), but his would not explain the heterogeneous presentation of ET. Also, the proposed classification of ET into “hereditary” (unequivocal family history), “sporadic” (no immediate family member with ET), and “senile” (onset after age 65) is too artificial and not easily applicable. Some studies have suggested that there is an association between ET and parkinsonism (Jankovic et al., 1997; Yahr et al., 2003; Shahed and Jankovic, 2007; Fekete and Jankovic, 2011), but other studies have not found a link (Adler et al., 2011) (Fig. 18.2). Differentiation between ET and PD is critical particularly in early stages since ET has been found to be erroneously treated with anti-PD drugs in 12.4% of 402 community cases re-evaluated by a movement disorder specialist (Meara et al., 1999). Postural tremor, similar to ET, has been reported to occur in as many as 93% of patients with PD and to correlate with the ipsilateral rest tremor but not with age at onset or disease duration (Louis et al., 2001e). Furthermore, in 22 patients with PD with family history of ET, 90% (20 of 22) had a tremor-predominant subtype of PD, suggesting that “these patients have inherited a genetic susceptibility factor for tremor, which affects the motor phenotype of PD” (Hedera et al., 2009). Phenomenologically similar to ET, the postural tremor of PD has been linked by some investigators to coexistent ET (Geraghty et al., 1985; Jankovic, 1989; Jankovic et al., 1997; Jankovic 2000). Others, however, believe that the coexistence of the two disorders simply “represents a chance occurrence of two common diseases” (Pahwa and Koller, 1993). On the basis of an analysis of 678 patients diagnosed as having ET, some by movement disorder specialists and others by private practice neurologists, 6.1% were found to have concomitant PD, and 6.9% had coexisting dystonia (Koller et al., 1994). The authors concluded that “the frequency of PD in ET is more than would be reported in the general population.” In the Neurological Disorders in Central Spain (NEDICES) study, a longitudinal, population-based study of 3813 people (mean age 73.4 ± 6.6 years), after a median of 3.3 years, 12 (5.8%) of 207 ET cases developed parkinsonism compared with 56 (1.6%) of 3606 controls, with adjusted relative risk (RR) of 3.47 (95% confidence interval 1.82–6.59; P < 0.001) (Benito-León et al., 2009b). Six (3.0%) of 201 ET cases developed incident PD versus 24 (0.7%) of 3574 controls, with adjusted RR of 4.27 (95% confidence interval 1.72–10.61; P = 0.002). The authors concluded that “Patients with ET were four times more likely than controls to develop incident PD during prospective follow-up.”
The coexistence of ET and PD may be difficult to recognize because once a patient develops PD, the postural tremor is usually attributed to the disease, and it is therefore difficult to diagnose ET in a patient who already has symptoms of PD (Shahed and Jankovic, 2007; Louis, 2009; Fekete and Jankovic, 2011) (Fig. 18.2). While rest tremor may be observed in patients with advanced ET, it may also be the initial manifestation of coexistent PD (Shahed and Jankovic, 2007; Fekete and Jankovic, 2010). The “postural tremor” that is seen in many patients with PD may represent an enhanced physiologic tremor (Forssberg et al., 2000), coexistent ET (Geraghty et al., 1985; Jankovic, 1995), or a reemergent classical rest tremor (Jankovic et al., 1999) with the same frequency and clinical characteristics as the typical rest tremor. This reemergent tremor is also often exacerbated during walking. In contrast to ET, which is seen immediately when patients outstretch their arms, the reemergent tremor of PD usually appears after a latency of several seconds (Jankovic et al., 1999). Furthermore, this PD-related tremor often responds to levodopa, whereas the postural tremor of ET does not (Kulisevsky et al., 1995). It is actually this action tremor that seems to correlate with motor disability rather than the typical rest tremor, which correlates chiefly with social handicap (Zimmermann et al., 1994). The clinical characteristics, however, may not always reliably differentiate between the two types of postural tremor (Henderson et al., 1995). We found a higher frequency of the 263 bp allele of the NACP-Rep1 polymorphism not only in patients with PD (odds ratio: 3.86) but also in patients with ET (odds ratio: 6.42), but not in patients with Huntington disease, supporting a genetic link between PD and ET (Tan et al., 2000). Further evidence that ET and PD may be related is the observation that patients with ET have an olfactory deficit that is similar, although milder, than that noted in patients with PD (Louis et al., 2002; Louis and Jurewicz, 2003; Djaldetti et al., 2008). In one study, however, there was no difference in results of olfactory testing between ET patients and controls (Shah et al., 2008). We and others (Gimenez-Roldan and Mateo, 1991) have noted that patients with ET seem to have a higher propensity toward neuroleptic-induced parkinsonism than do patients without ET, although a formal epidemiologic study is needed to confirm this clinical observation. To examine an ET–PD relationship, we described 22 patients with childhood-onset ET who later developed PD (Shahed and Jankovic, 2007; Fekete and Jankovic, 2011). Of 11 patients reporting asymmetric ET, PD symptoms began on the same side as the more severe ET tremor in 10 (90.9%, χ2 = 0.66, P = 0.024), with 68.2% reporting change in tremor as their first PD manifestation. These findings, supported by another study (Tan et al., 2006), suggest that in some patients, childhood ET evolves into adult tremor-dominant PD, explaining the coexistence of ET and PD within the same patient and family. It has been postulated that ET-related gene mutations may predispose some patients to subsequent development of PD. In a study of 53 patients with ET–PD combination, compared to 53 PD and 150 ET patients, the side of the greatest initial ET severity corresponded to the side of the greatest PD severity (Minen et al., 2008). In another study involving 13 patients who presented originally with asymmetrical postural tremor and no rest tremor for at least 10 years (mean: 19.2 years) and were initially diagnosed with ET, all patients subsequently developed evidence of PD (Chaudhuri et al., 2005). The onset of levodopa-responsive PD was manifested by rest tremor for a mean of 2.5 years before final presentation in the clinic. Furthermore, five patients who had β-CIT single photon emission computed tomography (SPECT) all showed reduced uptake in the contralateral striatum. It is not clear whether these patients with a long-standing history of asymmetrical postural tremor have PD at onset, whether patients with unilateral postural tremor (isolated tremor) or asymmetrical postural tremor (atypical ET) who later develop PD represent an overlap between ET and PD, or whether this type of postural tremor is an early marker for PD (Grosset and Lees, 2005).
In a population-based study (981 first-degree relatives of 162 patients with PD and of 838 first-degree relatives of 147 controls), the risk of ET was significantly increased for relatives of patients with onset of PD (P = 0.006) (Rocca et al., 2007). Also, in a referral-based sample (981 first-degree relatives of 162 patients with PD and of 838 first-degree relatives of 147 controls), the risk of ET among relatives increased with younger onset of PD in patients (P = 0.001) and was higher in relatives of PD patients with the tremor-predominant or mixed form when compared with relatives of patients with the akinetic-rigid form, and in men compared with women. The authors concluded that “These findings suggest that PD and ET may share familial susceptibility factors.” In a case-control study of 600 subjects evaluated for tremor, ET was significantly more frequent in patients with PD (12/204, 5.9%) compared to diseased controls (2/206, 1%) and healthy controls (1/190, 0.5%) (Tan et al., 2008). The authors concluded that “PD patients were 5–10 times more likely to have ET compared diseased and healthy controls.”
In addition to genetic factors, there may be environmental factors that determine the occurrence of ET and its relationship to PD. For example, heavy cigarette smoking has been associated with lower risk of PD and ET (Louis et al., 2008a). Although some studies have concluded dementia is more frequent in patients with ET than in controls (Bermejo-Pareja et al., 2007), and one study reported the adjusted odds ratios to vary between 1.64 and 1.84 (Thawani et al., 2009), a relationship between ET and Alzheimer disease has not been established (Elble et al., 2007a).
The possibility of additional cochlear involvement in ET is supported by the observation of high occurrence of partial or complete deafness in patients with ET (Ondo et al., 2003). Among 250 patients with ET, 42 (16.8%) patients wore hearing aids, compared to only 2 of 127 (1.6%) PD patients and 1 of 127 (0.8%) controls (P < 0.0001). Pure tone audiometry demonstrated age-dependent higher-frequency loss among patients with ET as compared to the general population. High risk of hearing loss among patients with ET has been confirmed by other studies (Benito-León et al., 2007). The combination of ET, sensorineuronal hearing loss, and early graying has been suggested to be a unique disorder, separate from Waardenburg syndrome (Karmody et al., 2005). Although mental functioning is usually intact in patients with ET, detailed testing of cognitive performance has found some subtle abnormalities on tests of verbal fluency, naming, mental set-shifting, verbal working memory, and other tests of cognitive function (Benito-León et al., 2006a) and elderly patients with ET may possibly have an increased risk of dementia compared with those without ET (Benito-León et al., 2006b). Furthermore, depression has been found to occur in about a third of the patients with ET, almost as frequently as in PD (Lombardi et al., 2001; Miller et al., 2007). These deficits have been interpreted as suggesting involvement of frontocerebellar circuits. In a cross-sectional study of personality, patients with ET were found to have a tendency to have increased levels of pessimism, fearfulness, shyness, anxiety, and easy fatigability, but none of these traits correlated with the severity of the tremor (Chatterjee et al., 2004).
ET appears to be a heterogeneous disorder, as is suggested by the multiple gene loci that have so far been identified and by the frequent association with other disorders, such as parkinsonism, dystonia, and myoclonus (Jankovic et al., 1997; Jankovic, 2002; Yahr et al., 2003; Deng et al., 2006b; Shahed and Jankovic, 2007) (Fig. 18.2). Postmortem studies of patients with ET have not provided evidence for nigrostriatal pathology in ET, but patients who had a diagnosis of PD at time of death (even though ET might have preceded the onset of PD) would have been excluded from these clinical-pathologic studies (Jankovic, 2004; Rajput et al., 2004). Furthermore, there is indirect evidence suggesting nigrostriatal impairment in some patients with ET. We found that relatives of patients with PD have at least a 2.5 times higher (those with the combination of ET–PD: 10 times higher) frequency of tremor than normal controls, providing additional support for the association of ET and PD (Jankovic et al., 1995a). Furthermore, about 20% of patients with ET have a rest tremor that has the clinical and physiologic characteristics of PD tremor (Cohen et al., 2003). However, when 9 brains of patients who exhibited advanced ET and upper extremity rest tremor (without any other evidence of parkinsonism) were examined with alpha-synclein staining, only two had Lewy bodies in the dorsal vagus nucleus and locus ceruleus, but none had Lewy body-containing neurons and/or Lewy neurites in the basal ganglia (Louis et al., 2011). Although this pathological study suggests that rest tremor in patients with ET is not associated with PD pathology, 19 of 24 (80%) patients with ET and rest tremor without other parkinsonian features had abnormal DAT uptake on DaTscan, particularly in the putamen (deVerdal et al., 2011). Similarly, a fourfold increase in prevalence of isolated tremor among relatives of patients with PD as compared to controls was found by Payami and colleagues (1994). Interestingly, among 196 twins with postural or kinetic tremors, Tanner and colleagues (2001) found that 137 had PD or had a twin with PD.
Imaging studies have been helpful in providing insight into the relationship between ET and PD. A 10–13% reduction in 18F-dopa uptake in the striatum of patients with ET as compared to controls (Brooks et al., 1992) suggests a physiologically important compromise of the dopaminergic system in patients with ET (Jankovic et al., 1993). Furthermore, 18F-dopa uptake constants (Ki) in 5 of 32 asymptomatic relatives of patients with PD who had isolated postural tremor were reduced on average by 23% (P < 0.001) (Piccini et al., 1997). The mean Ki for the other 27 asymptomatic relatives was decreased by 17% (P < 0.001). Using 123I-IPT SPECT to image the striatal dopamine transporter, Lee and colleagues (1999) found the mean bilateral uptake in nine patients with isolated postural tremor (ET) to be slightly lower than that in normal control subjects (3.60 vs. 3.80), but this did not reach statistical significance. Six other patients in whom rest tremor developed 4–18 years (mean: 11.5 ± 6.7) after the onset of postural tremor without other parkinsonian features, however, had a significant reduction in the dopamine transporter compared to normal controls (2.61 vs. 3.83, P < 0.05) but lower than PD patients (1.97 contralateral and 2.35 ipsilateral). They concluded that some patients with postural tremor may acquire rest tremor in association with mild substantia nigra neuronal loss. Although the majority of ET patients have normal dopamine transporter (DAT) SPECT, some cases may start with isolated postural tremor, phenomenologically identical to ET, and later develop PD. In one study the mean latency between the onset of asymmetrical postural tremor and PD was 19.2 years whereas the mean latency between onset of rest tremor and PD was 2.5 years (Chaudhuri et al., 2005). In one study of 61 subjects presenting with “isolated atypical tremors defined as unilateral either postural, resting or mixed” followed at baseline and at mean 28.4 ± 7.2 months with 123I-FPCIT SPECT, those (n = 25) with normal baseline scan had only tremor at follow-up, and of the 36 with abnormal baseline scan, 23 (64%) developed PD, while the remaining patients had only tremor (presumably ET) (Ceravolo et al., 2008). They suggested that term “isolated tremor with dopaminergic presynaptic dysfunction” is used for the patients with unilateral or asymmetrical tremor with abnormal DAT SPECT. Whether the use of 123I-FPCIT SPECT in differentiating ET from PD is cost-effective is not clear, although one Italian study suggested some cost savings when using this diagnostic tool (Antonini et al., 2008). In one study FP-CIT SPECT showed that the pattern of dopaminergic loss over time is different between ET and PD, but both disorders exhibit impairment of DAT in the caudate nucleus (Isaias et al., 2010). Although this and other imaging studies reported by the same group (Isaias et al., 2008) provide important insights into the selective caudate dopaminergic deficit as a possible link between the two common disorders, some studies have reported that DAT SPECT remains normal over time in patients with mixed tremor (a combination of postural and rest tremor) (Arabia et al., 2010). Since medial substantia nigra (that predominantly projects to the caudate nucleus) is particularly involved in the tremor-dominant PD and is associated with more caudate loss of DAT, it is possible that tremor-dominant PD and ET share a selective dopaminergic loss in the caudate nucleus. This, in turn, may lead to a dysfunction of the caudate-thalamic pathway and disinhibition of the thalamic autorhythmic pacemakers, clinically expressed as tremor. Indeed, β-CIT SPECT, which mainly reflects serotonin transporters, is lower in the thalamus of patients with tremor-dominant PD as compared to those with non-tremor PD (Caretti et al., 2008). In addition to thalamus, the caudate also projects to the inferior olive and cerebellum, both implicated in the pathophysiology of ET, and supported by growing evidence or cerebellar pathology in ET. The clinical overlap between the two disorders undoubtedly contributes to the 10–15% frequency of patients diagnosed with mild PD who have SWEDDs (Schneider et al., 2007; Bain, 2009; Schwingenschuh et al., 2010; Stoessl, 2010).
A relationship between ET and nigral degeneration is supported by the finding of hyperechogenicity of substantia nigra on midbrain sonography in 16% of 44 ET patients as compared to 3% of 100 controls and 75% of 100 patients with PD (Stockner et al., 2007). Although not demonstrated by all studies (Doepp et al., 2008; Budisic et al., 2009), the slightly increased hyperechogenicity of the substantia nigra on midbrain sonography provides further support for the notion that some ET patients may later develop parkinsonism.
Although the relatively frequent coexistence of ET and dystonia supports the notions that there is a pathogenetic link between the two disorders, linkage analysis has excluded the dystonia (DYT1) gene on chromosome 9 in hereditary ET (Conway et al., 1993; Dürr et al., 1993). This suggests that the genes for these two disorders are on separate loci or that the relationship between the two disorders is physiologic rather than genetic. Münchau and colleagues (2001) studied 11 patients with classic ET and compared them to 19 patients with cervical dystonia and arm tremor. They found that the latency of the second agonist burst during ballistic wrist flexion movements was later in ET patients than in those with arm tremor associated with cervical dystonia. Furthermore, the latter group had a greater variability in reciprocal inhibition than the ET group. Patients with normal presynaptic inhibition had simultaneous onset of their arm tremor with onset of their cervical dystonia (mean age: 40 years), whereas patients with reduced or absent presynaptic inhibition had an earlier age at onset (mean 14 years), and the interval between the onset of the tremor and the onset of cervical dystonia was longer (mean: 21 years). This suggests that the mechanisms of arm tremor in patients with ET and cervical dystonia are different. The association between ET, dystonia, and PD is suggested by reports of families with manifestations of these three disorders in different or same members of the families (Jankovic et al., 1997; Yahr et al., 2003).
ET-like tremor has been described in patients with hereditary myoclonus and with hereditary motor-sensory neuropathy (sometimes referred to as Roussy–Levy syndrome) (Cardoso and Jankovic, 1993). ET-like tremor occurs in other genetic diseases, the study of which may provide important insights into possible genetic heterogeneity in families with clinically similar tremor. For example, postural tremor similar to that seen in ET has been reported in patients with Kennedy disease, also called X-linked recessive spinal and bulbar muscular atrophy, which is caused by a mutation characterized by expansion of CAG repeats in the gene on the X chromosome (Sperfeld et al., 2002). ET may also be associated with higher-than-expected frequency with restless legs syndrome (Ondo and Lai, 2006). The validity and meaning of such associations, however, are disputed, and the controversies are not likely to be resolved until a disease-specific marker (e.g., an ET-linked genetic locus) is identified. A diagnostic marker for ET would also help to resolve the question as to whether site-, position-, and task-specific tremors, such as primary handwriting tremor and orthostatic tremor, are distinct entities or whether these tremors represent clinical variants of ET (Rosenbaum and Jankovic, 1988; FitzGerald and Jankovic, 1991; Britton et al., 1992b; Danek, 1993; Soland et al., 1996b; Sander et al., 1998) (Table 18.5). It is still not clear whether primary writing tremor is a variant of ET, a type of focal dystonia such as writer’s cramp, or a separate nosological entity (Hai et al., 2010; Quinn et al., 2011).
Orthostatic tremor, first described by Heilman in 1984, is a fast (14–16 Hz) tremor, involving mainly the legs and trunk, but cranial muscles may be also involved (Koster et al., 1999) (Video 18.5). The latter observation suggests that supraspinal mechanisms play a role in the pathophysiology of orthostatic tremor. This is further supported by the finding of high intermuscular coherence between the two sides, providing evidence that the tremor originates from a common site (Lauk et al., 1999), and a high degree of EMG coherence between right and left muscle groups. This is in contrast to ET or PD tremors, in which there is no such left/right coherence, and these tremors are probably generated by more than one oscillator (Raethjen et al., 2000). Some authors have suggested that coherent high-frequency tremor in the legs may be a normal response to perceived unsteadiness when standing still and that orthostatic tremor may be an exaggeration of this response (Sharott et al., 2003). Others have postulated that orthostatic tremor merely unmasks 16 Hz central oscillators involved in postural tremor (McAuley et al., 2000). While there is robust evidence for a supraspinal origin of orthostatic tremor, the spinal cord may also serve as the generator of the tremor as suggested by the presence of a 16 Hz tremor in a man with complete paraplegia (Norton et al., 2004). Present chiefly on standing, orthostatic tremor may be precipitated also by isometric contraction of the upper limbs as well as facial and jaw muscles (Boroojerdi et al., 1999; Koster et al., 1999). This suggests that the generation of orthostatic tremor is more likely related to isometric force control rather than to regulation of stance. Orthostatic tremor is often associated with a feeling of unsteadiness and calf cramps, relieved by sitting or a supine position. Fung and colleagues (2001) postulated that “the sensation of unsteadiness arises from a tremulous disruption of proprioceptive afferent activity from the legs”. The leg cramps are presumably due to a high-frequency (tetanic) contraction of the calf muscles. The muscle contraction can be “heard” by auscultating over the thigh or calf and listening for the characteristic thumping sound (Brown, 1995). 
The pathophysiology of orthostatic tremor is not well understood, but some have suggested that it is a variant of ET. In support of the association between ET and orthostatic tremor is the relatively high occurrence of postural tremor, phenomenologically identical to ET, and the presence of family history of tremor in the majority of patients with orthostatic tremor (FitzGerald and Jankovic, 1991). Furthermore PET findings indicative of bilateral cerebellar (and contralateral lentiform and thalamic) dysfunction, similar to those observed in ET, have been also reported in patients with orthostatic tremor (Wills et al., 1996). Some studies have also suggested that there is a dopaminergic deficit in orthostatic tremor. Leg tremor, phenomenologically similar to orthostatic tremor, may be the initial manifestation of PD, particularly due to parkin mutation (Kim and Lee, 1993; Deng et al., 2006b). Some patients with orthostatic tremor respond to levodopa (Wills et al., 1999) and dopamine agonists (Finkel, 2000). Furthermore, [123I]-FP-CIT SPECT showed evidence of marked reduction of dopamine transporter in patients with orthostatic tremor (Katzenschlager et al., 2003).
Tremor that is present predominantly or only on standing, but usually of much lower frequency than the classic orthostatic tremor, can be also seen in other conditions, including parkinsonism, ET, head trauma, pontine lesions, and other disorders (Gabellini et al., 1990; Benito-León et al., 1997). In contrast to ET, orthostatic tremor does not respond to the conventional anti-ET medications, but usually improves with clonazepam and gabapentin (Rodrigues et al., 2006). In one study, five of nine patients with orthostatic tremor benefited from levodopa (Wills et al., 1999). In a review of 41 patients with orthostatic tremor, Gerschlager and colleagues (2004) found that 24 (58%) patients had associated postural arm tremor, and 10 (25%) had “orthostatic tremor plus”; 6 (15%) patients had parkinsonism. The response to medications was generally poor, but some, particularly those with associated parkinsonism, responded to dopaminergic therapy. Whether dopamine agonists and other antiparkinsonian treatments, including thalamotomy and VIM or STN/GPi DBS, will provide benefit to patients with orthostatic tremor remains to be determined. VIM DBS may be an effective treatment for patients with medically resistant orthostatic tremor (Guridi et al., 2008; Espay et al., 2008). Chronic spinal cord stimulation has been reported to be effective in two patients with medically intractable orthostatic tremor (Krauss et al., 2006).
The age at onset for ET showed a bimodal distribution with peaks in the second and sixth decades (Lou and Jankovic, 1991b). This was evident in both genders and in patients with and without dystonia and parkinsonism. Patients with early-onset (<30 years) ET had significantly more hand involvement, were more likely to have associated dystonia, and were more likely to improve with alcohol than were those with later onset (>40 years) ET (P < 0.05). There were no significant differences in any clinical variables between patients with and without a family history of tremor. Patients with older-onset ET, sometimes also referred to as “senile tremor,” tend to have more rapid progression and more degenerative pathology than the younger-onset patients (Louis et al., 2009b). The relative lack of important differences between subgroups (early versus late onset, familial versus sporadic, mild versus severe, low versus high frequency) suggests that ET represents a single disease entity with a variable clinical expression. This conclusion is supported by a recent study by Koller and colleagues (1992). In their clinical and physiologic study of 61 patients, they found a frequency below 7 Hz in 79% of the patients, a positive family history in 72%, an amelioration with alcohol in 75%, an amelioration with primidone in 71%, and an amelioration with propranolol in 46%. Since no significant correlations could be found to suggest any particular grouping, they concluded that “essential tremor cannot be classified into subtypes.”
Epidemiologic studies indicate that up to 5% of the adult population has ET, and 5–30% of adults with ET report symptom onset during childhood (Ferrara and Jankovic, 2009). Childhood-onset ET is usually hereditary, begins at a mean age of 6 years, and affects boys three times as often as girls. In a study of 39 patients with childhood-onset ET, a mean age at onset of 8.8 ± 5.0 years, and a mean age at evaluation of 20.3 ± 14.4 years, we found that some had their initial symptoms as early as infancy (Jankovic et al., 2004). A family history of tremor was noted for 79.5% of the patients. Eighteen (46.2%) patients had some neurologic comorbidity, such as dystonia, which was noted in 11 (28.2%) patients. Only 24 (61.5%) patients were treated with a specific antitremor medication; 5 of the 12 patients who were treated with propranolol experienced improvement. Other studies of childhood-onset ET also found male preponderance and paucity of head tremor (Louis et al., 2001b; Tan et al., 2006). Some investigators have suggested that “shuddering attacks” of infancy might be the initial manifestation of ET (Vanasse et al., 1976; Kanazawa, 2000).
Some isolated site-specific tremors, such as those involving the head and trunk (Rivest and Marsden, 1990) and some task- or position-specific tremors, might actually represent forms of dystonic tremor (Elble et al., 1990; Jedynak et al., 1991; Bain et al., 1995). Dystonic tremor is typically irregular and position-sensitive and when the patient is allowed to move the affected body part into the position of the maximal “pull”, the tremor often ceases, the so-called “null point” (Videos 18.6 and 18.7). Some patients with dystonic tremor present with asymmetric rest hand tremor and decreased armswing which may lead to initial misdiagnosis of PD (Jankovic and Mejia, 2005; Schneider et al., 2007; Bain, 2009). Although there is some overlap between primary writing tremor and dystonic writer’s cramp, the former is not usually associated with an excessive overflow of EMG activity into the proximal musculature, and the reciprocal inhibition of the median nerve H-reflex on radial nerve stimulation is normal (Bain et al., 1995; Modugno et al., 2002). The latter two features are typical of dystonia, and their presence in patients with task-specific tremors suggests that despite the absence of overt dystonia, these tremors represent forms of focal dystonia (Rosenbaum and Jankovic, 1988; Soland et al., 1996b). The overlap with primary handwriting tremor is supported by the observed activation of brain areas on functional magnetic resonance imaging (MRI) that are commonly activated in ET and dystonic writer’s cramp. Other causes of postural tremor include midbrain (rubral) lesions. In a study of six patients with midbrain tremors, PET studies indicated dopaminergic striatal denervation, supported by markedly decreased fluorodopa uptake in the ipsilateral striatum (Remy et al., 1995). This nigrostriatal denervation, however, was not accompanied by striatal dopamine receptor supersensitivity, and the density of striatal D2 receptors did not change. Furthermore, the density of dopamine transporter is the same as that in normal controls (Antonini et al., 2001). Another form of postural tremor with bilateral high-frequency (14 Hz) synchronous discharges was reported in a patient with sporadic olivopontocerebellar atrophy (Manto et al., 2003). 
Genetics
A family history of tremor has been reported in 17–100% of patients with ET (Busenbark et al., 1996; Louis and Ottman, 1996). The reason for such a large discrepancy is that unless all the symptomatic and asymptomatic members of the family are examined, the number of affected relatives will be under-ascertained (Jankovic et al., 1997; Louis et al., 1999b). Tremor in relatives is often wrongly attributed to aging, stress, nervousness, PD, alcoholism, or an associated illness or medications. In a study of 169 relatives of 46 ET patients, 12 (7.5%) were diagnosed as having probable or definite ET, but only 2 were reported by probands to have tremor (sensitivity: 16.7%); only 1 of 136 normal relatives were reported to have tremor (specificity: 99.3%) (Louis et al., 1999b). In other studies, the investigators found that 23% of elderly individuals had ET, and relatives of ET patients were five times more likely to develop the disease than a control population (Louis et al., 2001c), and first-degree relatives were more likely to have tremor compared to relatives of controls than were second-degree relatives (Louis et al., 2001d). Factors that were associated with more accurate reporting were female informant, increased tremor severity, sibling relationship, and higher level of education. In a comprehensive study of 20 index patients with hereditary ET and their 93 first-degree relatives and 38 more distant relatives, Bain and colleagues (1994) examined 53 definite and 18 possible cases. Similar to the findings of Lou and Jankovic (1991a), the investigators found a bimodal distribution and autosomal dominant inheritance with nearly complete penetrance by the age of 65 years. In contrast to some previous studies, they found no cases of dystonia, PD, task-specific tremors, or primary orthostatic tremors, but migraine headaches occurred with a higher-than-expected frequency of 26%. About 50% were alcohol-responsive, but there was marked heterogeneity of responsiveness between and within families (Mostile and Jankovic, 2010). In contrast, in a study of 252 members in four large kindreds with ET, three of the kindreds had a total of 41 members with the combination of ET and dystonia, and two had associated parkinsonism (Jankovic et al., 1997). Besides the one kindred with “pure” ET without any associated disorders (Jankovic et al., 1997), we subsequently studied 216 individuals of another large kindred with “pure” ET. The observation of earlier age at onset in successive generations suggests the phenomenon of anticipation, although the relatively small number of subjects and the possibility of ascertainment bias preclude any definite conclusions. Since ET is so common in the general population, bilineal transmission is not rare, and the amplitude of tremor appears to be greater in the children than in either affected parent (Rajput and Rajput, 2006). The younger the age at onset of ET, the higher the frequency of positive family history. In one study, 91% of cases with onset before the age of 20 years had a family history of tremor (Louis and Ottman, 2006).
Although the genetic origin of ET is widely recognized, the gene or genes responsible for ET have eluded intensive search by several groups of investigators (Deng et al., 2007) (Table 18.6). A genome scan of 16 ET Icelandic families containing 75 affected relatives with “definite” ET identified a marker for the familial ET gene, FET1 or ETM1, on chromosome 3q13.1 (Gulcher et al., 1997). An analysis of a large Czech-American family established linkage to a locus ETM2 on chromosome 2p22–p25 (Higgins et al., 1997). Two of our families with “pure” ET and one with ET–parkinsonism–dystonia also mapped to the same locus (Higgins et al., 1998). On the basis of studies of genetically diverse populations of ET, the locus has been narrowed to 2p24.1 with a candidate interval to a 192-kilobase interval between the loci etm1231 and APOB (Higgins et al., 2004). More recently, a missense mutation (828C→G) in the HS1-BP3 gene was identified in two American families with ET and was absent in 150 control samples (300 chromosomes) (Higgins et al., 2005, 2006). The 828C→G mutation causes a substitution of a glycine for an alanine residue in the HS1-BP3 protein (A265G), which is normally highly expressed in motor neurons and Purkinje cells and regulates the Ca2+/calmodulin-dependent protein kinase activation of tyrosine and tryptophan hydroxylase. Studies in our own population of patients with ET and suitable controls, however, have led us to conclude that the variant might not be pathogenic for ET and might simply represent a polymorphism in the HS1-BP3 gene (Deng et al., 2005). A linkage to 6p23 with a LOD score ranging from 1.265 to 2.983 was identified in two ET families, but further studies are needed to determine whether this merely represents a susceptibility locus or whether this gene region contains a causative gene (Shatunov et al., 2006). Variants in the coding region of the D3 receptor gene (DRD3), localized on 3q13.3, have been found to be associated with ET in some families and in a case-control study (Lucotte et al., 2006). In a genome-wide scan involving families from France and North America, Ser9Gly variant in the DRD3 gene was found to increase dopamine affinity 4–5-fold and produce a gain-of-function abnormality as demonstrated by increased cAMP dopamine-mediated response and prolonged mitogen-associated protein kinase (MAPK) signal (Jeanneteau et al., 2006). The DRD3Gly was found to be associated with an increased risk for and age at onset of ET in Spanish (García-Martín et al., 2009) and French populations (Lorenz et al., 2009). This variant, however, has not been found in the majority of Asian (Tan et al., 2007b) or Italian (Vitale et al., 2008) patients with ET, and some have argued against the role of DRD3 gene in the pathogenesis of ET (Blair et al., 2008).
When the genome-wide scan of 452 ET patients was compared to that of 14 378 controls, a marker in intron 3 of LINGO1 gene on chromosome 15q24.3 was found to be significantly associated with ET (Stefansson et al., 2009). In several large European and American populations, the odds ratios for carriers of one G allele averaged 1.55, with P = 10−9. Odds increased to 2.4 for carrying two G alleles. Based on this and the prevalence of the allele, the population attributable risk of the variant was 20%. Another study involving a North American population demonstrated a significant association between LINGO1 rs9652490 and essential tremor (P = 0.014) and PD (P = 0.0003), again suggesting that variations in LINGO1 can increase risk of ET and provide the first evidence of a genetic link between ET and PD (Vilariño-Güell et al., 2010a, 2010b). Furthermore, a slightly higher frequency of the allele G of LINGO1 marker rs9652490 was found in ET patients of Asian origin (Tan et al., 2009). LINGO1 may be potentially pathogenic as it has been implicated in axon regeneration, and when mutated it may be responsible for fusiform swellings of Purkinje cell axons, similar to those found in autopsied brains of patients with ET.
Another marker for ET has been mapped to chromosome 4p14–p16.3 (MIM 168601) in a family with autosomal dominant PD (Farrer et al., 1999, 2004). This family, however, was later found to have α-synuclein gene (SNCA) triplication, and this SNCA triplication segregated with parkinsonism but not the postural tremor (Singleton et al., 2003). This suggests that the postural tremor is a coincidental finding. Since not all families map to the three known loci (ETM1, ETM2, or the 4p locus) (Kovach et al., 2001), it is likely that familial tremor has not only marked phenotypic but also genetic heterogeneity and that additional gene loci will be identified in the near future. Furthermore, models other than autosomal dominant, including interaction between susceptibility genes and environmental risk factors, should be considered (Ma et al., 2006).
In 1935, Minor (1935), a Russian neurologist, not only stated that “the older one is, the more likely one displays tremor (ET),” but also suggested “that a factor for longevity was also contained in the tremor gamete.” In support of the latter hypothesis, he offered the description of 51 cases of “hereditary tremor” associated with longevity. The longevity was based on patients’ parents and grandparents usually being older than 70 years. His “control group” consisted of 11 cases of parkinsonism in which “no example of longevity was found out.” However, the author did not state who in the ET families had tremor, and he did not provide any details on his parkinsonian patients (clinical features, ages of the subjects and relatives), and normal controls were not examined. Supporting the clinical observation that patients with ET live longer than a controlled population, Jankovic, Beach and colleagues (Jankovic, 1995) found that parents of patients with ET who had tremor (presumably ET) lived on the average 9.2 years longer than did parents without tremor. The association of familial tremor with significantly increased longevity suggests that familial tremor confers some anti-aging influence. Alternatively, patients with ET might have an underlying personality trait that encourages dietary, occupational, and physical habits that promote longevity. Furthermore, the small amounts of alcohol to calm the tremor might prolong life; and finally, the tremor itself might be viewed as a form of exercise that would have long-standing beneficial effects on general health (Mostile and Jankovic, 2010). Despite the overwhelming evidence that ET is of genetic origin, some researchers have suggested that environmental factors might also play a role, particularly since concordance among monozygotic twins is only 60% (Louis, 2001; Tanner et al., 2001). However, in another study involving 92 twins with ET from the Danish twin registry, the concordance rate was 93% among monozygotic twins and 29% among dizygotic twins when the Tremor Research and Investigation Group consensus criteria were used (Lorenz et al., 2004).
Treatment
In designing protocols to study the effects of a therapeutic intervention on tremor-related functional impairment and on the mean amplitude (and frequency) of tremor, it is important to take into account the marked intraindividual and interindividual and diurnal variations in physiologic and pathologic tremors (van Hilten et al., 1991; Deuschl et al., 2011). Factors such as anxiety, caffeine, or alcohol intake, drugs, and even temperature (Lakie et al., 1994) can affect hour-to-hour variations in the amplitude of tremor. Caffeine, a nonselective adenosine receptor antagonist, can trigger or exacerbate tremor and the observed marked increase in the DBS-induced release of ATP from local astrocytes, which metabolizes to adenosine, has been suggested as one possible mechanism of DBS in suppression of tremor (Bekar et al., 2008).
Even without specific triggers or exacerbating factors, the ET amplitude may vary by 30–50% from hour to hour (Koller and Royse, 1985). The antitremor drugs exert their ameliorating effects by reducing tremor amplitude without any effect on tremor frequency. Reduction of tremor amplitude, however, does not always translate into improvement in function. There are currently no uniformly accepted ET rating scales, but the Unified Tremor Rating Assessment developed by the Tremor Research and Investigation Group (Jankovic et al., 1996; Ondo et al., 2000) and the Tremor Rating Scale developed by the Tremor Research Group (Tintner and Tremor Research Group, 2004) have been used in several studies.
The treatment of postural tremor depends largely on its severity; many patients require nothing more than simple reassurance. Most patients who are referred to a neurologist, however, have troublesome tremors that require pharmacologic or surgical treatments (Ondo and Jankovic, 1996; Bain, 1997; Lyons et al., 2003) (Fig. 18.3). The large-amplitude and slow-frequency postural tremors usually do not respond to any pharmacologic therapy. Since alcohol reduces the amplitude of ET in about two-thirds of patients, some use it regularly for its calming effect, and some use it prophylactically – for example, before an important engagement at which the presence of tremor could be a source of embarrassment. Although evidence concerning the risk of alcoholism among ET patients is contradictory, regular use of alcohol to treat ET is inadvisable (Mostile and Jankovic, 2010). One epidemiologic, prospective, population-based study in central Spain involving 3285 elderly participants, 76 of whom developed incident ET, concluded that baseline alcohol consumption was associated with substantially higher risk of developing ET (Louis et al., 2009a). They suggested that the alcohol may be in part responsible for the cerebellar toxicity associated with ET. Although the authors dismiss the possibility that the participants who developed incident ET between the baseline and follow-up evaluations had preclinical ET at baseline, because they answered “no” when asked at baseline whether their arms or legs shook, many patients deny having tremor before the onset of alcohol consumption but validation by other family members often confirms that they had tremor prior to alcohol. Furthermore, family members of ET patients sometimes consume alcohol without being aware that it “calms” their tremor. Arguing against alcohol as contributing to the cause of ET is also the observation that many patients with ET never drink alcohol (Mostile and Jankovic, 2010).
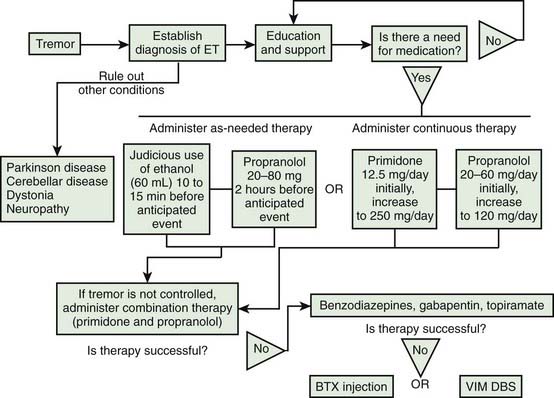
The mechanism by which alcohol reduces tremor (effective blood level is only 300 mg/L (0.03%)) is unknown, but it is thought to act centrally, since infusion of alcohol into the brachial artery of a tremulous arm is ineffective (Growdon et al., 1975). Furthermore, the amplitude of the central but not peripheral components of ET is decreased after alcohol consumption (Zeuner et al., 2003a). Alcohol is known to affect multiple neurotransmitters, and it might stabilize neuronal membranes by potentiating gamma-aminobutyric acid (GABA) receptor-mediated chloride influx. A pilot trial (n = 12) of 1-octanol, a food additive approved by the Food and Drug Administration that was previously demonstrated to suppress harmaline tremor (see below), found that it significantly decreased amplitude of ET for about 90 minutes after oral dose of 1 mg/kg (Bushara et al., 2004). The benefits and tolerance (except for unusual taste) of this drug were demonstrated in an open-label trial at doses up to 64 mg/kg (Shill et al., 2004).
Treatment of ET and other action tremors is not optimal. In a survey of 261 patients with ET about a third who had been prescribed medication for tremor had discontinued the therapy and another third of cases with severe tremor had stopped medication (Louis et al., 2010). Propranolol, a β-adrenergic blocker, remains the most effective drug for the treatment of ET and enhanced physiologic tremors, but other beta-blockers may also ameliorate postural tremor (Caccia et al., 1989; Calzetti et al., 1990; Koller et al., 2000) (Table 18.7). Propranolol is less effective in head tremor than hand tremor (Calzetti et al., 1992). Although a central mechanism of action has been suggested for this group of drugs, some beta-blockers exert potent antitremor activity even though they are not lipid soluble and hence do not cross the blood–brain barrier. This suggests that the therapeutic effect of beta-blockers may be mediated, at least in part, by the peripheral β-adrenergic receptors (Guan and Peroutka, 1990). The major side effects of propranolol and, to a lesser degree, other beta-blockers include fatigability, sedation, depression, and sexual impotence. A critical review of 42 articles, based on controlled trials, concluded that despite the conventional wisdom, “there is no significant increased risk of depressive symptoms and only small increased risk of fatigue and sexual dysfunction” associated with beta-blocker therapy (Ko et al., 2002). These drugs are contraindicated in the presence of asthma, second-degree atrioventricular block, and insulin-dependent diabetes. Contrary to traditional recommendations, they may be used safely in patients with stable congestive heart failure due to left ventricular systolic dysfunction (Packer et al., 1999). The efficacy of sotalol, a nonselective β-antagonist, in reducing ET is comparable to that of propranolol and atenolol, even though both atenolol and sotalol have very low lipid solubility and therefore act mainly through peripheral mechanisms (Leigh et al., 1983). Arotinolol, an alpha- and beta-blocker that is used as an antiobesity drug, was found to be as effective as propranolol in a randomized crossover study (Lee et al., 2003).
Table 18.7 Evidence-based recommendations for treatment of ET (Quality Standards Subcommittee of the AAN)
The antitremor effect of primidone has been confirmed by several open trials and placebo-controlled studies (Koller and Royse, 1986). By starting primidone at a very low dose (25 mg at bedtime), the occasional idiosyncratic acute, toxic side effects (nausea, vomiting, sedation, confusion, and ataxia) can be prevented. Since there is little or no correlation between blood levels and tremolytic effects, the daily dosage should be gradually increased over a period of several weeks until the optimal therapeutic response is achieved. Dosages above 250 mg/day are only rarely necessary. The antitremor effect of primidone is largely attributed to the parent compound rather than to its metabolites, phenylethyl-malonamide (PEMA) or phenobarbital (Sasso et al., 1991). In one study (Koller and Royse, 1986), primidone alone was found to decrease tremor more than propranolol alone, but a combination of the two drugs might be more efficacious than either drug alone. In a double-blind, placebo-controlled, crossover study, Gorman and colleagues (1986) found that both propranolol and primidone significantly reduced tremor compared to placebo. There is no evidence that either one of these primary anti-ET drugs is more efficacious than the other, but acute adverse effects with primidone and chronic side effects of propranolol limit therapy. The combination of the two drugs might be more efficacious than monotherapy (Koller and Royse, 1986). Although the benefits are usually maintained, the dosages might have to be increased after the first year to sustain the antitremor effectiveness. A double-blind study of 87 patients with ET, however, showed that low doses of primidone (250 mg/day) were as effective as, or more effective than, high doses (750 mg/day) (Serrano-Duenas, 2003).
In addition to beta-blockers and primidone, the benzodiazepine drugs, such as diazepam, lorazepam, clonazepam, and alprazolam (Gunal et al., 2000), as well as barbiturates, also may have some ameliorating effects on ET or its variants (Ondo and Jankovic, 1996). In a double-blind, crossover, placebo-controlled study, 22 patients with ET received in random order alprazolam, acetazolamide, primidone, and placebo for 4 weeks, each separated by a 2-week washout period. The study demonstrated that alprazolam was superior to placebo and equipotent to primidone, whereas there was no statistically significant difference between acetazolamide and placebo (Gunal et al., 2000). The mean effective daily dose of alprazolam was 0.75 mg, and no troublesome side effects were reported by the patients who took alprazolam. Clonazepam was shown to be ineffective in controlling ET in one double-blind study, but at a mean dose of 2.2 mg/day, it improved kinetic tremor (Biary and Koller, 1987). Methazolamide (Neptazane), a sulfonamide that is used in the treatment of glaucoma, was reported to be effective in the treatment of ET (Muenter et al., 1991). Ten of 28 patients apparently achieved moderate to complete relief of their tremor. The average maintenance dose was 129 mg/day, and reported side effects included sedation, nausea, epigastric discomfort, and parasthesias. Aplastic anemia, the most feared complication, did not occur during the 6-month (10 weeks to 29 months) follow-up. The beneficial effects of methazolamide suggested by this open trial, however, could not be confirmed by a double-blind controlled study (Busenbark et al., 1993). Although our personal experience with this drug has been disappointing, we have found that up to 10–20% of patients who were previously unresponsive to other antitremor treatments note a marked improvement in their tremor with methazolamide. Flunarizine, a calcium channel blocker, was reported to improve ET in 13 of 15 patients (Biary and Deeb, 1991). This drug, however, is not available for use in the United States, and it can produce parkinsonism and tardive dyskinesia. Another calcium channel blocker, nimodipine at 120 mg/day, was found to improve ET in 8 of 15 patients who completed a double-blind, placebo-controlled trial (Biary et al., 1995). Anecdotally, fenofibric acid (Trilipix), a lipid regulating agent, has been reported to also improve ET.
Despite some encouraging results with gabapentin based on pilot studies, subsequent double-blind controlled studies showed mixed results (Louis, 1999), ranging from no benefit (Pahwa et al., 1998) to modest improvement (Ondo et al., 2000) to a marked benefit comparable to that obtained with propranolol (Gironell et al., 1999). Topiramate, a broad-spectrum anticonvulsant, has been also reported to reduce ET in a double-blind, placebo-controlled trial at 400 mg/day dose (Connor, 2002). Although well tolerated, topiramate may cause parasthesias and weight loss and may adversely affect cognition (Thompson et al., 2000). Some patients who are unresponsive to conventional treatments do improve even with low-dose (50 mg/day) topiramate (Gatto et al., 2003). In a multicenter, double-blind, placebo-controlled trial involving 208 patients (topiramate, 108; placebo, 100) the final visit score (last observation carried forward) was lower in the topiramate group than with placebo (P < 0.001) (Ondo et al., 2006). Mean percentage improvement in overall TRS scores was 29% with topiramate at a mean final dose of 292 mg/day and 16% with placebo (P < 0.001) and topiramate was associated with greater improvement in function and disability (P = 0.001). The most common adverse effects were parasthesias (28%), weight loss (22%), and taste perversion (19%), but only the following adverse effects resulted in discontinuation of the drug: paresthesia (5%), nausea (3%), concentration/attention difficulty (3%), and somnolence (3%). Overall, adverse events were treatment limiting in 31.9% of topiramate patients and 9.5% of placebo patients. Thus, this multicenter study showed that topiramate was effective in the treatment of ET with acceptable tolerability profile. In a pilot, open-label, crossover trial designed to compare zonisamide and arotinolol in 14 patients with ET using the Fahn–Tolosa–Marin clinical rating scale at baseline and 2 weeks after administration of each drug, both drugs were found to have significant and equal antitremor effect, but zonisamide was more effective for voice, face, tongue, and head tremors (Morita et al., 2005). In a subsequent double-blind, placebo-controlled trial involving 20 patients with ET, zonisamide failed to provide any improvement in clinical rating scales, although it reduced tremor by accelerometry (Zesiewicz et al., 2007a). Zonisamide was, however, found to be more effective than propranolol in the treatment of isolated head tremor (Song et al., 2008). One study showed that 200 mg/day seemed to the optimal dose for zonisamide in the treatment of ET (Handforth et al., 2009). Levetiracetam, another antiepileptic, was shown to have a significant antitremor effect in one double-blind, placebo-controlled trial at 1000 mg as a single dose (Bushara et al., 2005), but not in other studies (Handforth and Martin, 2004; Ondo et al., 2004; Elble et al., 2007b). Pregabalin has been found effective in a pilot, double-blind, placebo-controlled trial (Zesiewicz et al., 2007b), but not in a double-blind, placebo-controlled trial (Ferrara et al., 2009b).
Mirtazapine has been reported to improve rest tremor (Pact and Giduz, 1999), but in a double-blind, placebo-controlled trial, the drug was not found to exert a significant benefit in ET (Pahwa and Lyons, 2003). Finally, clozapine has been found to effective in selected drug-resistant patients with ET (Ceravolo et al., 1999). Gabapentin (Onofrj et al., 1998), levodopa (Wills et al., 1999), primidone, clonazepam, and phenobarbital seem to be particularly useful in patients with orthostatic tremor (Cabrera-Valdivia et al., 1991; FitzGerald and Jankovic, 1991). Despite earlier reports, amantadine has not been found effective in patients with ET in a randomized, placebo-controlled trial, and in some patients actually exacerbated the postural tremor (Gironell et al., 2006). Sodium oxybate, a salt of gamma-hydroxybutyrate (GHB), may suppress tremor in a manner similar to alcohol (Frucht et al., 2005).
Other treatment modalities for postural tremors include injections of BTX into muscles that are involved in the production of the oscillatory movement and various surgical approaches. In one open trial of BTX treatment, 67% of 51 patients with various disabling tremors noted at least some improvement (Jankovic and Schwartz, 1991). The average duration of improvement was 10.5 weeks, and side effects were chiefly related to local muscle weakness; 40% of 42 patients who were injected in the neck muscles to control head tremor and 60% of 10 patients who were injected in the forearm muscles to control hand tremor improved. Other studies have demonstrated the usefulness of BTX in the treatment of hand tremor (Trosch and Pullman, 1994). A double-blind, placebo-controlled trial has demonstrated a mild to moderate efficacy of BTX injections in patients with severe hand ET (Jankovic et al., 1996) and in patients with ET involving the head (Pahwa et al., 1995; Wissel et al., 1997). Although in one study, only 20–30% of patients with voice tremor were found to benefit from vocal cord injections of BTX, the majority of patients benefited from a subjective reduction in vocal effort that might have been attributable to reduced laryngeal airway resistance (Warrick et al., 2000). In another study involving 27 patients with adductor spasmodic dysphonia and vocal tremor and in four patients with severe vocal tremor alone, a significant improvement in various acoustic measures was observed after thyroarytenoid and interarytenoid BTX injections and less tremor was demonstrated in 73% of the paired comparisons (Kendall and Leonard, 2010). Although 67% of the patients with spasmodic dysphonia and vocal tremor wished to continue to receive BTX injections, only one patient with severe vocal tremor wished to continue with injections. BTX may also be effective in primary writing tremor, although a specially designed writing device might be a simpler and at least as effective treatment (Espay et al., 2005).
Peripheral deafferentation with anesthetic is currently being re-explored as a potential treatment of focal dystonia and tremor (Rondot et al., 1968; Pozos and Iaizzo, 1992; Kaji et al., 1995). An injection of 5–10 mL of 0.5% lidocaine into the target muscle not only improved focal dystonia, but also reduced the amplitude of the postural tremor. This short effect (<24 hours) can be extended for up to several weeks if ethanol is injected simultaneously (Kaji, personal communication). In a study of 10 patients with ET, Gironell and colleagues (2002) reported a transient (<1 hour) improvement in tremor after transient magnetic stimulation of the cerebellum.
The neurosurgical treatments, including DBS, that were discussed in the section on rest tremors may be also efficacious in patients with action hand tremors (Benabid et al., 1991; Blond et al., 1992; Deiber et al., 1993; Jankovic et al., 1995b; Koller et al., 1997), head tremors (Koller et al., 1999), and even task-specific tremors (Racette et al., 2001). It is of interest that cerebellar lesions can abolish ET, but it is unlikely that this observation will lead to surgically induced cerebellar lesions as a therapeutic modality in patients with ET (Dupuis et al., 1989). On the other hand, chronic cortical stimulation has been reported to improve contralateral action tremor (Nguyen et al., 1998). The observation that injection of muscimol, a GABAA agonist, into the VIM thalamus improved tremor in patients with ET suggests that GABA agonists might be useful in the treatment of ET (Pahapill et al., 1999). On the basis of the observation that vagus nerve stimulation has a nonspecific calming effect in treated epileptic patients and that it suppresses harmaline-induced tremor (see below) in rats (Handforth and Krahl, 2001), a multicenter trial was conducted to study the effects of vagus nerve stimulation in patients with essential and parkinsonian tremor, but no meaningful benefit was demonstrated (Handforth et al., 2003).
Kinetic tremors
Diagnosis
Kinetic tremor is typically associated with lesions or diseases that involve the cerebellum or its outflow pathways. The term kinetic tremor more accurately describes the oscillation occurring with limb movement than the classic term intention tremor, which is ambiguous because it implies tremors that are present when “contemplating, initiating, performing, or completing a movement” (Lou and Jankovic, 1993). It is important to recognize that kinetic tremor is not simply a consequence of cerebellar ataxia (Diener and Dichgans, 1992), hypotonia, dysmetria, or dysdiadochokinesia but that the different motor disorders may coexist, often causing severe functional disability (Sabra and Hallett, 1984). Although kinetic tremor was traditionally considered a predominantly proximal tremor, using wrist (distal) or whole-arm (proximal) visually guided tracking in patients with multiple sclerosis showed a major frequency component at 4–5 Hz, most of the action tremor being distal rather than proximal (Liu et al., 1999).
In addition to this action, kinetic tremor patients with cerebellar lesions often exhibit postural tremors and titubation. The term titubation simply refers to a rhythmic oscillation of the head or trunk, presumably caused by hypotonia of the axial muscles. Kinetic cerebellar outflow tremor might be a component of the thalamic ataxia syndrome characterized in addition to the tremor by contralateral ataxia, hemisensory loss, and transient hemiparesis caused by a lesion in the mid to posterior thalamus involving the dentatorubrothalamic and ascending sensory pathways (Solomon et al., 1994). Kinetic terminal and postural tremors have been reported to result from repetitive transcranial magnetic stimulation, possibly by interfering with cerebellar inflow to the motor cortex (Topka et al., 1999). Although kinetic tremors are more difficult to assess than ET or PD tremors, by using handwriting, spirals, and other tests, objective assessments of kinetic tremors, such as those seen in multiple sclerosis, can be reliably obtained (Alusi et al., 2000). In a study of 100 patients with definite multiple sclerosis, Alusi and colleagues (2001b) found 58 with tremor, but it was symptomatic in only 38 and incapacitating in 10. The authors concluded that multiple sclerosis tremors are usually related to involvement of the cerebellum.
Treatment
Some causes of tremor, such as alcohol withdrawal, phenytoin, lithium, amiodarone, and valproate toxicity (Nouzeilles et al., 1999) and cerebellar tumors and abscesses are specifically treatable. Tremor and ataxia in such cases can resolve after the underlying cause is removed. Some kinetic tremors can be reduced by attaching weights to the wrist, but this method provides only limited improvement in function (Aisen et al., 1993).
No drugs have been shown to reduce cerebellar tremor satisfactorily and reproducibly, but the application of wrist weights before eating may enable to patient feed himself. Isoniazid was initially thought to improve cerebellar, postural tremor more than kinetic tremor (Sabra and Hallett, 1984), but the results of a double-blind trial were disappointing (Hallett et al., 1991). Sechi and colleagues (1989) reported that carbamazepine was effective in the treatment of cerebellar tremor, possibly by reducing hyperactivity in thalamic neurons. Ten patients, seven with multiple sclerosis and three with cerebrovascular disease, were followed for 2–24 months. All patients improved on a clinical rating scale and by accelerometric recording when given carbamazepine, 400–600 mg daily. There was no improvement with placebo. Trelles and colleagues (1984) reported that cerebellar kinetic tremor secondary to multiple sclerosis and to olivopontocerebellar degeneration responded to clonazepam treatment at a daily dose of 8–15 mg. Alcohol not only improves ET but also may improve tremor associated with multiple sclerosis (Hammond and Kerr, 2008). Glutethimide, a piperidinedione derivative with sedative and anticholinergic effects, has recently been reported to be effective at doses of 750–1250 mg/day in action tremors, including ET, cerebellar tremors, and midbrain tremors (Aisen et al., 1991). The encouraging results from this open trial await confirmation by properly designed controlled studies. Even if it is found to be effective, however, the potential side effects of glutethimide, including respiratory depression, aplastic anemia, and physical and psychological dependence, will limit its usefulness. Buspirone, a serotonin (5-HT1A) agonist was reported to be useful in some patients with mild cerebellar ataxia, but placebo-controlled study is needed before it can be concluded that this drug is effective in cerebellar ataxia or tremor (Lou et al., 1995). Baker and colleagues (2000) found that cannabinoids control tremor in a mouse model of multiple sclerosis, but the effects of tetrahydrocannabinol in patients with cerebellar outflow tremor are unknown. Levetiracetam has been also found to be effective in rare cases (Ferlazzo et al., 2008).
Stereotactic thalamotomy has been reported to successfully relieve kinetic tremor in patients with multiple sclerosis and other etiologies (Andrew, 1984). Radiofrequency lesions in the VIM seem to provide the best control of tremor with the lowest risk of neurologic deficit (Speelman and Manen, 1984; Nagaseki et al., 1986; Alusi et al., 2001a), although thalamic DBS has been also reported to be useful in selected patients with cerebellar-outflow tremor due to multiple sclerosis or other causes (Whittle et al., 1998; Montgomery et al., 1999). A recent report of the Quality Standards Subcommittee of the American Academy of Neurology, based on a review of evidence-based publications, published the following practice parameters related to therapies for ET. Propranolol and primidone reduce limb tremor (Level A); alprazolam, atenolol, gabapentin (monotherapy), sotalol, and topiramate are probably effective in reducing limb tremor (Level B); propranolol reduces head tremor (Level B); clonazepam, clozapine, nadolol, and nimodipine possibly reduce limb tremor (Level C); BTX type A may reduce limb, head, and voice tremor, but this treatment is limited by potential adverse effects (Level C); chronic DBS and thalamotomy are highly efficacious but potentially risky procedures (Level C); and there is insufficient evidence regarding surgical treatment of head and voice tremor and the use of gamma knife thalamotomy (Level U) (Zesiewicz et al., 2005) (Table 18.7).
Pathophysiologic mechanisms of rest and action tremors
Our understanding of mechanisms that are involved in generation of tremors has been facilitated by the development of neurophysiologic and other quantitative techniques such as EMG recordings and uniaxial and triaxial accelerometers and by the application of computer technology to analyze the frequency spectra and other tremor-related physiologic variables (Elble and Koller, 1990; Gresty and Buckwell, 1990; Elble et al., 1994; Louis et al., 2001b). A frequency of 6 Hz is usually the maximum rate of oscillation produced by a voluntary effort. During a voluntary contraction, motor units usually start firing at 8 Hz, and tetanic fusion frequency is reached at 15–20 Hz. Different parts of the human limb have mechanical characteristics (inertia and stiffness) that determine its natural resonant frequency. Such frequency is inversely related to the mass of the body part: finger, 25 Hz; wrist, 9 Hz; and elbow, 2 Hz.
There is a growing body of evidence supporting the notion that central oscillators are important in the generation of physiologic and pathologic tremors (Pare et al., 1990; Plenz and Kitai, 1999; McAuley and Marsden, 2000; Brown, 2003; Gatev et al., 2006). Using microelectrode recordings in awake or decerebrate monkeys who have been curarized and whose limbs were deafferented by sectioning C2 to T4 dorsal roots, Lamarre (1984) demonstrated spontaneous 3–6 Hz rhythmic discharges in the ventral thalamus and 7–12 Hz activity in the olivocerebellar system. Deafferenting the thalamus by a lesion in the ventromedial tegmentum of the midbrain facilitates synchronization of thalamic neurons, which is ultimately expressed as a spontaneous, 3–6 Hz, parkinsonian rest tremor. The amplitude of these discharges and associated tremors is markedly enhanced by the tremorgenic drug harmaline. Harmaline-induced tremor is currently considered the most reliable animal model of ET as it generates both postural and kinetic tremors. It has been postulated that harmaline induces release of glutamate and acts as a potent reversible monoamine oxidase inhibitor. These and possibly other pharmacologic effects of harmaline result in facilitating low-threshold calcium conductance with resultant oscillatory activity of the olivocerebellar system, and eventual coupling and synchrony in the inferior olivary nucleus. Ethanol reduced harmaline-induced tremor, and possibly ET, by desynchronizing the olivary discharges, in part by decreasing glutamate (Manto and Laute, 2008). There are two types of harmaline-induced tremors, both of which appear to originate in the inferior olivary nucleus: an 8–12 Hz tremor in normal animals (analogous to physiologic tremor) and a 6–8 Hz tremor in monkeys with lesions in the dentate nucleus. Single-unit recordings from patients with parkinsonian tremors showed that neurons in the thalamic ventral nuclear group show rhythmic activity that correlated with EMG activity. Some, but not all, of these cells responded to somatosensory activity, indicating the importance of peripheral modulation (Lenz et al., 1994). In addition to the thalamus, there are other subcortical nuclei – particularly the subthalamic nucleus (STN) and external globus pallidus (GPe) – that contain neuronal populations that produce synchronized oscillating bursts at 0.4, 0.8, and 1.8 Hz in a cell-culture environment (Plenz and Kitai, 1999). In addition to the low-frequency (<10 Hz) oscillations, there are 11–30 Hz and >60 Hz oscillatory activities within the subthalamopallidal-thalamocortical circuit (Brown, 2003). In PD, synchronized oscillatory activity in the 10–50 Hz band (often termed “β-band”) prevalent in the basal ganglia thalamocortical circuit may be important in mediating certain parkinsonian features, including bradykinesia and tremor, and can be reduced by dopaminergic treatments (Gatev et al., 2006). Furthermore, there is evidence that as a result of dopaminergic deficiency, the normal independent firing of pallidal neurons changes to both low-frequency (4–7 Hz) and high-frequency (10–16 Hz) oscillatory activity, which can be also recorded from the STN, and these oscillations often correlate with arm tremor (Bergman et al., 1998). This pacemaker could be responsible for the generation of tremor under pathologic conditions such as dopaminergic deficiency in PD. Therefore, surgical treatments of PD, such as lesion or stimulation of the GPi or STN, might act by desynchronizing the oscillatory basal ganglia–thalamocortical network activity. Such microelectrode-guided stereotactic operations in PD allow for single-cell recordings in the globus pallidus, thalamic nuclei, and other subcortical structures, leading to hypotheses about the normal and abnormal function of the basal ganglia. One such study, for example, found tremor-locked cells in the center median-parafascicular complex of the thalamus with low-threshold calcium spike type bursts in central lateral nucleus and the paralamellar division of mediodorsal nucleus of the thalamus (Magnin et al., 2000).
Intracellular recordings from brainstem sections have demonstrated that neurons in the inferior olive have spontaneous oscillatory activity (Llinas, 1988; Kepler et al., 1990; Bevan et al., 2002). The autorhythmic properties of these neurons make this brainstem nucleus a prime candidate as a neuronal generator for tremor. In the olivary neurons, low-threshold Ca2+ conductance at the somatic membrane enables these neurons to generate action potentials (low-threshold spike) even at subthreshold depolarization. A fast-action potential, generated by Na+ current into the cell body, is followed by a slow, high-threshold Ca2+ spike that activates prolonged (80–100 ms), K+-mediated hyperpolarization. This is followed by an abrupt rebound response, generated by low-threshold Ca2+ conductance, often large enough to generate a second, Na+-dependent, action potential, and the cycle repeats itself. In addition to the olivary nucleus, there are other areas of the brain, particularly the STN and globus pallidus, that display spontaneous rhythmic activities. In the STN neurons, voltage-gated Na+ channels inactivate slowly during the depolarizing phase of the oscillation. In addition, Ca2+-dependent K+ current is activated by Ca2+ entry through high-voltage Ca2+ channels that open briefly during each Na+ action potential (Bevan et al., 2002). Harmaline enhances normal hyperpolarization, which is terminated by Ca2+ influx and rebound excitation, leading to rhythmic activation of the neurons. Besides harmaline, there are many toxins that can produce tremors in animals and provide useful models for the study of tremors (Wilms et al., 1999). Microelectrode-guided single-cell recordings in patients with PD showed that the average firing rate in the GPi was 91 ± 52 Hz, and that in the globus pallidus externa was 60 ± 21 Hz (Magnin et al., 2000). In addition, rhythmic, low-threshold calcium spike bursts are often recorded in the pallidum and medial thalamus; some, but not all, are synchronous (in phase) with the typical rest tremor. It has been postulated that the low-threshold calcium spike bursts contribute to rigidity and dystonia by activating the supplementary motor area.
Physiologic tremor is present in all humans. It is an asymptomatic oscillation of a body part, resulting from a complex interaction between local mechanical-reflex mechanisms and central oscillators (McAuley and Marsden, 2000) (Fig. 18.4). The mechanical-reflex component is determined partly by the ballistocardiogram, mechanical properties of the muscle, motor neuron firing characteristics, stretch reflex and muscle spindle feedback, supraspinal influences, and state of activation of the muscle beta receptors (Marsden, 1984). The frequency of this component is inversely proportional to the mass and stiffness of the limb; thus, increasing the external mass load decreases the tremor frequency (Elble and Koller, 1990). Besides this variable frequency component, which is determined largely by the mechanical properties of the oscillating body part, spectral analyses of normal physiologic tremor show another, generally smaller, component with a relatively consistent frequency peak at about 10 (8–12) Hz. This 10 Hz frequency component is independent of peripheral influence, and it persists even after deafferentation, suggesting that this component of physiologic tremor is centrally generated. Supraspinal influence on physiologic tremor is supported by the observation that the loss of visual input when the eyes are closed reduces or abolishes this tremor. The amplitude of physiologic tremor is determined largely by the degree of synchronization of motor unit discharges, modulated by muscle spindle Ia afferents. This process is exaggerated during anxiety, exercise, fatigue, and other conditions that are known to enhance peripheral β-adrenergic activity, and it may result in a visible tremor called enhanced physiologic tremor (Table 18.1).
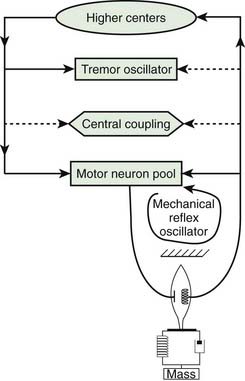
In most pathologic tremors, such as the rest or reemergent tremor of PD (Jankovic et al., 1999), the central oscillators are thought to drive the tremor, and the peripheral mechanisms are thought to merely modify or modulate its amplitude (Marsden, 1984; Britton et al., 1992a). Magnetic brain stimulation can also modify certain pathologic tremors, providing further evidence that these tremors are centrally generated (Britton et al., 1993). The motor pathways that are involved in transmission of signals from these central generators are not well understood. Inhibition of tremor by lesions in the basal ganglia outflow nuclei and in the thalamus suggests that these subcortical nuclei are involved in the pathophysiology of tremors. In parkinsonian states, as a result of nigrostriatal dopamine depletion, excessive GABA-mediated inhibition of the GPe within the indirect striatopallidal pathway leads to a disinhibition of the STN and enhanced glutamate-mediated excitatory drive to the basal ganglia output nucleus (GPi) (DeLong, 1990). This increased drive from the output nuclei is reinforced by reduced inhibitory input to the GPi through the direct pathway. The typical 3–6 Hz rest tremor of PD possibly results from increased inhibition of the thalamic neurons that normally suppress the oscillators or from hyperpolarization of the oscillator neurons. In PD, the increased inhibitory output from the GPi leads to deafferentation of the thalamus, particularly the reticular nucleus, and synchronization of local neuronal networks with intrinsic properties to spontaneously discharge (Lamarre, 1984; Huntsman et al., 1999). This spontaneous bursting is transmitted via the thalamocortical-spinal circuitry to the motor neurons in the spinal cord, causing synchronization of motor unit discharges, ultimately expressed as an oscillatory movement.
Pathophysiology of Parkinsonian tremor
In support of the role of the thalamus in the generation of parkinsonian rest tremor is the observation that the ventral thalamus of patients with PD contains a population of neurons that fire spontaneously and rhythmically and that their firing frequency correlates relatively well with the frequency of tremor, recorded by EMG in the contralateral limb (Lenz et al., 1988). Furthermore, a lesion in the nucleus ventralis intermedius (VIM) or electrical stimulation of the nucleus, so-called deep brain stimulation (DBS), abolishes contralateral rest tremor (Kelly et al., 1987; Benabid et al., 1991, 1996; Hubble et al., 1997; Koller et al., 1997; Ondo et al., 1998; Limousin et al., 1999). That parkinsonian tremor is centrally driven is supported by the observation that complete paralysis of extensor forearm muscles as a result of radial nerve palsy actually increased the amplitude of the tremor without changing its frequency (Pullman et al., 1994). Volkmann and colleagues (1996) used magnetoencephalography to study parkinsonian resting tremor, and on the basis of these studies, they concluded that the typical 3–6 Hz parkinsonian tremor is generated by the same central mechanisms that are involved in voluntary rapid alternating movements. They proposed the following sequence of events for typical parkinsonian rest tremor: increased inhibitory output from GPi → hyperpolarization and activation of low-threshold calcium conductance in anterior ventrolateral nucleus (VLa) → 5–10 ms → activation of lateral premotor and motor cortex → 25–30 ms → EMG activity → 100 ms → activation of the central sulcus (reafferent input from muscle spindle to area 3a) → second activation of the somatosensory cortex by the antagonist contraction. The study of pacemaker neuronal networks as well as fast and slow oscillations within the basal ganglia circuitry will undoubtedly provide important insights into the pathophysiology of rhythmic involuntary movements such as tremors (Plenz and Kitai, 1999; Ruskin et al., 1999).
Pathophysiology of essential tremor
It has been suggested that the postural tremor of ET arises from spontaneous firing of the inferior olivary nucleus, which drives the cerebellum and its outflow pathways via the thalamus to the cerebral cortex and then to the spinal cord (Deuschl and Elble, 2000). In contrast to physiologic tremor, the frequency of ET does not decrease with mass loading, indicating a primary role of central mechanisms in ET (Elble and Koller, 1990). On the other hand, local cooling may significantly reduce the amplitude of ET, without changing its frequency (Lakie et al., 1994). Abnormal triphasic pattern with a delayed second agonist burst and other kinematic problems in response to a ballistic movement further support impaired cerebellar function in patients with ET (Koster et al., 2002). In addition, impaired eyeblink conditioning in patients with ET has been used as evidence of functional disturbance in the olivocerebellar circuits (Kronenbuerger et al., 2007). MRI scans, including diffusion-weighted imaging, have found no abnormalities associated with ET (Martinelli et al., 2007). Using a 3-tesla MRI in 19 ET patients (mean age 69.8 ± 9.4 years) and 20 age- and gender-matched controls, Benito-León et al. (2009a) found that ET patients with severe tremor had significantly more white matter changes in the midbrain, both occipital lobes, and right frontal lobe, and gray matter changes bilaterally in the cerebellum. Diffusion tensor imaging (DTI) MRI showed microstructural abnormalities in the dentate nucleus and superior cerebellar peduncle in patients with ET, providing further support for neurodegenerative pathology of cerebellar structures in ET (Nicoletti et al., 2010).
The involvement of the olivocerebellar-thalamocortical circuitry in ET is supported by changes in the cerebellar blood flow as measured by PET during inhalation of C15-labeled O2 (Jenkins et al., 1993). Using this technique, the authors compared the cerebellar and cerebral blood flow in patients with ET and normal controls. They found that when the control subjects were holding one arm outstretched (in the absence of tremor), there was increased blood flow in the contralateral sensorimotor cortex and the supplementary motor area. In contrast, unilateral postural arm tremor in patients with ET was associated with markedly increased blood flow to both cerebellar hemispheres; in both premotor cortices; and to the contralateral striatal, thalamic, and sensorimotor cortex (but not the supplementary motor area). Passive and voluntary flexion–extension movements of the wrist significantly increased the blood flow only in the ipsilateral cerebellum. The investigators suggested that the increased flow in the cerebellum reflected increased activity of neurons involved in generation of tremor and that ET was due to oscillation within the olivocerebellar pathways, relayed by way of the thalamus and motor cortex to the spinal cord. Using a higher-resolution camera, the investigators were additionally able to demonstrate bilateral midbrain activation in the region of the red nuclei during tremor, but there was no change in the activity of the inferior olive either at rest or during postural tremor (Wills et al., 1994). Additional evidence of increased activation of the cerebellum and red nucleus in ET has been provided by functional MRI studies (Bucher et al., 1997). The preponderance of evidence, based on PET and functional MRI studies, indicates that the inferior olive probably does not play an important role in the generation of ET. This is in contrast to one previous study that indicated the involvement of the inferior olive in ET, suggested by the finding of glucose hypermetabolism during activation (finger-to-nose) of tremor in patients with ET (Dubinsky and Hallett, 1987). The absence of activity in the inferior olive and the bilateral overactivity of cerebellar-rubral-thalamic connections suggest that the tremor generator for ET is located in the cerebellum rather than the inferior olive. This conclusion is further supported by the finding of abnormal bilateral overactivity of cerebellar connections, as measured by regional cerebral blood flow determined by PET with 15O-labeled water, in patients with primary writing tremor and primary orthostatic tremor (Wills et al., 1995, 1996). Furthermore, alcohol has been found to suppress cerebellar synaptic overactivity in patients with ET, which in turn increases afferent input to the inferior olive (Boecker et al., 1996). In one study, 25% of ET patients were found to have moderate or severe kinetic tremor and other elements of cerebellar dysfunction (Deuschl et al., 2000). The demonstration by magnetic resonance spectroscopy of reduced N-acetyl-L-aspartate/creatine and N-acetyl-L-aspartate/choline ratios in the cerebellum of patients with ET compared to controls provides additional evidence for cerebellar dysfunction in ET (Pagan et al., 2003).
Cerebellar pathology, including loss of Purkinje cells, has been also described in some brains of patients with ET (Louis et al., 2006b; Axelrad et al., 2008; Louis and Vonsattel, 2008) but this finding has not been replicated by others (Rajput et al., 2011). In one of the largest clinical-pathologic studies, involving 33 ET and 21 control brains, 8 (24.2%) brains were found to have Lewy bodies in the brainstem, mainly in the locus coeruleus, and these patients were older than those ET cases without Lewy bodies (Louis et al., 2007b). The major pathologic changes, however, were observed in the cerebellum, particularly marked reduction in the number of Purkinje cells and 7-fold increase in Purkinje cell torpedoes (Louis et al., 2009c). Other findings included degeneration of the dentate nucleus, Purkinje cell heterotopias and dendrite swellings. Lewy body ET cases were older than ET cases without Lewy bodies. Increased cerebellar gliosis and depletion of pigmented neurons in the locus coeruleus and substantia nigra (of which 3 had Lewy bodies) were found in a prospective study of 24 subjects enrolled in the Sun Health Research Institute and Body Donation Program (Shill et al., 2008). In a subsequent study, presumably based on the same pathologic cases, they found no difference in the putaminal tyrosine hydroxylase concentration between the ET cases (91.7 ± 113.2 ng/mg) and controls (96.4 ± 102.7 ng/mg) and concluded that “This study provides neurochemical support that ET does not appear to be pre-clinical PD.” The data, however, do not support the conclusion for several reasons. First, the mean age in the pathologic series was 86.2 years and mean duration of tremor was 11.1 years; and 19/24 (79%) had a duration of ≤8 years. Although the authors do not provide the mean age at onset, based on their demographic data, the mean age at onset of ET in their population was about 81 years, which is 50 years later than the usual mean age at onset of ET of 31 years. It is, therefore, likely that most of the patients in the Shill et al. (2008) pathologic series had “senile tremor” rather than typical ET. Furthermore, patients with prior history of a movement disorder, including those with parkinsonian features, were excluded; thus, by definition, clinical, pathologic, or biochemical evidence of PD could not be demonstrated in this selected ET population.
Cerebellar dysfunction in ET is also suggested by abnormalities in tandem gait, noted in 50% of ET patients (Singer et al., 1994; Stolze et al., 2001), and by mild postural instability (Henderson et al., 1996; Overby et al., 1996). Although ataxic gait is rare, some ET patients exhibit a slightly slow or cautious pattern of walking (Kronenbuerger et al., 2009). This gait ataxia improves with alcohol at blood level of 0.45% (Klebe et al., 2005). Posturographic analysis in patients with ET showed that balance control is only minimally impaired in ET, but patients with head tremor and longer disease duration may have slightly reduced postural stability (Bove et al., 2006). ET patients also show eye movement abnormalities indicative of cerebellar dysfunction, such as impaired smooth pursuit initiation and pathologic suppression of the vestibular-ocular reflex time constant by head tilts (otolith dumping) (Helmchen et al., 2003). Further evidence of cerebellar involvement in ET is suggested by a delay in second agonist EMG burst during rapid wrist movements (Britton et al., 1994). That the cerebellum might be involved in ET is also supported by the report of a 71-year-old man with ET in whom postural tremor disappeared on the right side after an ipsilateral cerebellar infarct (Dupuis et al., 1989). Since the amplitude of ET can be substantially reduced by thalamic lesions or thalamic stimulation (Benabid et al., 1991) and close correlation has been demonstrated between thalamic neuronal activity and forearm EMG in patients with ET (Hua et al., 1998), it is likely that the thalamus also plays an important role in the generation or transmission of ET. Despite the reported evidence of cerebellar dysfunction in patients with ET, voxel-based morphometry, however, shows no reduction in cerebellar gray matter volume (Daniels et al., 2006).
In addition to proposed cerebellar involvement in ET, there is some evidence that cerebral cortex plays a role in the generation of ET (McAuley, 2001). Using simultaneous electroencephalogram (EEG)-EMG recordings, Hellwig and colleagues (2001) showed significant corticomuscular coherences in five of nine patients with ET. This is in contrast to a previous study that used magnetoencephalography (Halliday et al., 2000). That study found that in contrast to PD rest tremor, which is associated with marked disruption to the organization of the descending motor signals, there was no coherence between the magnetoencephalogram and EMG in patients with ET. However, the interpretation of the study that primary motor cortex does not contribute to the generation or maintenance of ET has been challenged because of possible insensitivity of the method (Elble, 2000c). The limitations of the various methods that were used in these studies have been pointed out by McAuley (2001), who also noted that EEG-EMG coherence does not necessarily establish cortical origin of the tremor (McAuley, 2001). Some patients with ET have abnormal vibration-induced illusion of movement, suggesting abnormal sensorimotor processing (Frima and Grunewald, 2005).
Rarely, postural tremor that occurs during sustained isometric muscle contraction can be a manifestation of cortical reflex myoclonus (Toro et al., 1993). This “cortical tremor,” with or without seizures, usually starts between 19 and 30 years of age as tremulous finger movements, with a benign course and marked response to anticonvulsants, possibly cognitive decline, and a family history suggestive of ET (Okuma et al., 1998; van Rootselaar et al., 2005). It is characterized physiologically by an 8–15 Hz, synchronous, agonist-antagonist, EMG burst lasting less than 50 ms (10–50 ms). The condition shares neurophysiologic features with myoclonus, including giant somatosensory evoked responses and C-reflexes, and it responds well to anticonvulsants such as clonazepam, primidone, and valproate but not to beta-blockers (Okuma et al., 1998). Since the EMG pattern can be influenced by transcranial magnetic and electrical stimulation but not by peripheral nerve stimulation, the rhythmic movement is thought to arise from central, possibly cortical, generators. Jerk-locked back-averaging revealed a positive EEG wave 15 ms preceding the EMG burst of the wrist extensor muscle (Okuma et al., 1998). In contrast to ET, strong cortico- and intermuscular coherence in the 8–30 Hz range was demonstrated in patients with familial cortical myoclonic tremor with epilepsy (van Rootselaar et al., 2006).The syndrome of autosomal dominant cortical tremor, myoclonus, and epilepsy has been linked to three genetic markers, on 8q24 and 2p11.1–q12.2 (Striano et al., 2005) and 5p15.31–p15 (Depienne et al., 2010). The term familial cortical myoclonic tremor with epilepsy has been suggested for this disorder, which has been reported in over 50 Japanese and European families (van Rootselaar et al., 2005) and in patients with spinocerebellar ataxia type 6 (Bour et al., 2008).
Other abnormalities that are found in patients with ET include high blood levels of β-carboline alkaloids. Interestingly, these endogenous compounds, also found in plant-derived foods, increase the synchrony of neuronal firing in the inferior olive and cause tremor in experimental animals and humans. Some investigators have suggested that a loss of GABAA receptors may impair signaling by cerebellar Purkinje cells and cause tremor, as has been demonstrated in mice in which the gene coding for the GABAA receptor α1 subunit has been knocked out (Jankovic and Noebels, 2005; Kralic et al., 2005). Mutations in the coding region of the GABRA1 gene, however, do not seem to be a major genetic cause of ET (Deng et al., 2006c).
Clinical-anatomic correlations indicate that kinetic tremor is usually associated with lesions in the cerebellar outflow pathways (Lou and Jankovic, 1993). Lesions of the dentate nucleus or the superior cerebellar peduncle proximal to the decussation correlate with kinetic tremor ipsilateral to the side of the lesion, whereas lesions that are distal to the decussation result in kinetic tremor contralateral to the side of the lesion (Carrea and Mettler, 1955). Lesions in the cerebellar hemisphere tend to produce kinetic tremors with a variable frequency ranging from 5 to 11 Hz; high brainstem lesions are usually associated with tremor frequency in the range of 5–7 Hz; and lower brainstem lesions produce faster tremors, ranging from 8 to 11 Hz (Cole et al., 1988). In contrast, the frequency of classical midbrain (red nucleus) tremor is relatively slow, at 2–3 Hz. In one study of patients with multiple sclerosis, two types of action tremors were identified. One group of patients had pure kinetic tremor with a frequency of 5–8 Hz and an EMG burst duration of 75–100 ms; the other group had coexisting kinetic and postural tremor. In contrast to kinetic tremor, postural tremor had a slower (2.5–4 Hz) frequency and a higher amplitude with a longer burst duration (125–200 ms) (Sabra and Hallett, 1984). The postural tremor was thought to result from a lesion in the cerebellar outflow pathway. Cerebellar kinetic tremor is associated with errors in timing and amplitude of the EMG activities of agonists and antagonists (Flament and Hore, 1986; Hore and Flament, 1986). The important role of peripheral reflex mechanisms in the pathogenesis of cerebellar kinetic tremor is supported by the observation that the character of the tremor can be altered by mechanical loading to the tremulous limb (Sanes et al., 1988) and by improvement in writing after 3–5 min of compressive ischemia of the affected arm (Dash, 1995). Lakie and colleagues (2004) showed that reduction of tremor by ischemia may be due to an accumulation of interstitial potassium in the muscle. The frequency of the tremor can be increased by increasing the spring stiffness and by decreasing the mass.
Other tremors
Fragile X-associated tremor/ataxia syndrome
Other causes of kinetic tremor, phenomenologically similar to ET, include premutation in the fragile X gene (FMR1) (Hagerman et al., 2001; Berry-Kravis et al., 2003; Jacquemont et al., 2003, 2004; Leehey et al., 2003; Hagerman and Hagerman, 2004, 2008; Amiri et al., 2008). While healthy individuals have about 30 CGG repeats in the FMR1 gene, the carriers of this premutation syndrome have 55–200 repeats. The FMR1 CGG repeat length has been found to predict motor dysfunction in premutation carriers (Leehey et al., 2008). In addition to the ET-like tremor, frequently affecting the head, and ataxia, this fragile X-associated tremor/ataxia syndrome (FXTAS) is also associated with mild cognitive impairment with frontal executive deficit, dementia, parkinsonism, dysautonomia, erectile dysfunction, peripheral neuropathy, and generalized brain atrophy, regardless of family history (Berry-Kravis et al., 2007) (Table 18.8). FXTAS may also, rarely, occur in females with a similar phenotype but in early menopause due to ovarian failure. One of our patients with severe FXTAS-associated tremor improved markedly with thalamic DBS (Ferrara et al., 2009a). Unusual bilateral T2 middle cerebellar hyperintensities have been identified on magnetic resonance imaging in some cases (Leehey et al., 2003). Overexpression and CNS toxicity of the FMR1 mRNA has been thought to cause this fragile-X-associated tremor/ataxia syndrome (Berry-Kravis et al., 2007; Jacquemont et al., 2007; Hagerman and Hagerman, 2008).
Table 18.8 Clinical features of fragile X-associated tremor/ataxia syndrome (FXTAS)
Although some features overlap with those of multiple system atrophy, FXTAS is a rare cause of multiple system atrophy (Biancalana et al., 2005). Patients with FXTAS and parkinsonism have been found to have normal dopamine transporter as determined by CIT-SPECT, indicating a postsynaptic dopaminergic deficit (Ceravolo et al., 2005). Postmortem studies on brains of four elderly premutation carriers showed eosinophilic intranuclear inclusions in both neuronal and astrocytic nuclei throughout the brain, particularly involving the hippocampus (Greco et al., 2002). Subsequent neuropathologic studies of patients with FXTAS showed: (1) cerebral and cerebellar white matter disease with spongiosis in the middle cerebellar peduncles, (2) astrocytic pathology with enlarged inclusion-bearing astrocytes, and (3) intranuclear inclusions in the brain and spinal cord, including the cranial nerve nucleus XII (Greco et al., 2006). The number of inclusions seem to correlate with the number of CGG repeats (Cohen et al., 2006). The inclusions contain a variety of proteins, such as RNA binding proteins, but ubiquinated proteins represent only a small portion of all the proteins, suggesting that breakdown of proteasomal degradation is not the main mechanism of the inclusions (Iwahashi et al., 2006).It has been postulated that premutation RNA is responsible for the observed neurodegeneration (Orr, 2004). Marked loss of Purkinje cells in the cerebellum, Purkinje axonal torpedoes, and Bergmann gliosis were also found. SCA12, a rare autosomal-dominant ataxia, may also present with ET-like tremor before other signs of ataxia become evident. This form of ataxia has been associated with CAG repeat expansion in the PPP2R2B gene (5q31–q33) coding for protein phosphatase PP2A (Fujigasaki et al., 2001). The frequency of fragile X premutation, even in patients with tremor, parkinsonism, and ataxia, is so low that it is not cost-effective to routinely test for it even in an enriched movement disorders population (Arocena et al., 2004; Deng et al., 2004; Tan et al., 2004). Interestingly, the location of the CAG repeat expansion in the 5′ region is similar to that of the CGG repeat in FMR1, in which an expansion results in CpG hypermethylation and disruption of transcription, resulting in the fragile X phenotype.
Neuropathic tremors
Tremor has been described in patients with various types of peripheral neuropathy (Said et al., 1982; Shahani, 1984). Distal postural tremor was noted in patients with chronic relapsing and dysgammaglobulinemic polyneuropathies (Bain et al., 1996). The amplitude was not related to the severity of weakness or proprioceptive sensory loss, and propranolol improved the tremor in some patients. Hereditary motor and sensory neuropathy is associated with tremor, clinically similar to ET, in nearly half of cases (Cardoso and Jankovic, 1993). The frequent association between tremor caused by peripheral nerve injury and reflex sympathetic dystrophy suggests that the sympathetic nervous system contributes to the pathogenesis of peripherally induced tremor (Jankovic and Van der Linden, 1988; Deuschl et al., 1991; Cardoso and Jankovic, 1995; Jankovic, 1994; van Rooijen et al., 2011).
Tremors caused by trauma or stroke
Tremor can also occur after severe or even minimal head trauma (Biary et al., 1989; Goetz and Pappert, 1992; Jankovic, 1994; Krauss et al., 1995). Biary and colleagues (1989) described seven patients who developed postural and kinetic tremors involving various body parts up to several weeks after mild head injury without loss of consciousness. Ipsilateral predecussational dentatothalamic lesion was involved in the majority (56%) of 25 instances of severe post-traumatic tremor in 19 patients, followed by involvement of the contralateral predecussational dentatothalamic pathway (28%), dentate nucleus (4%), and thalamus (4%); no lesion was identified by neuroimaging in 2 (8%) patients (Krauss et al., 1995). Clonazepam was effective in reducing tremor in 3 patients, and propranolol was effective in 1. Post-traumatic midbrain tremor may improve with anticholinergic and dopaminergic drugs (Samie et al., 1990), but in general, this tremor only rarely responds satisfactorily to drug therapy. Many of our patients with post-traumatic tremors have benefited from VIM thalamotomy; additional patients may benefit from thalamic stimulation (Broggi et al., 1993). In addition to trauma, a variety of tremors can occur following ischemic or hemorrhagic strokes (Ferbert and Gerwig, 1993; Miwa et al., 1996).
Trauma can also cause injury to the peripheral nerves, producing a form of neuropathic tremor, which was discussed earlier. Traumatic neck injury may cause cervical radiculopathy, which may be associated with a postural tremor in the ipsilateral arm (Hashimoto et al., 2002). Loading and ischemic nerve block reduced the frequency of the tremor, suggesting a role of the mechanical reflex mechanism and stretch reflex loop in this peripherally induced tremor.
Psychogenic tremor
One of the most challenging tasks in the diagnosis of tremors is differentiating between psychogenic and neurogenic tremors (Kenney et al., 2007) (Table 18.9). Koller and colleagues (1989) provided the following diagnostic criteria useful in the diagnosis of psychogenic tremors: (1) abrupt onset, (2) static course, (3) spontaneous remission, (4) difficulty in classifying (various combinations of rest, postural, and kinetic tremors), (5) selective disability, (6) changing amplitude and frequency, (7) unresponsiveness to antitremor drugs, (8) increasing of tremor with attention, (9) lessening of tremor with distractibility, (10) responsiveness to placebo, (11) absence of other neurologic signs, and (12) remission with psychotherapy (Video 18.8). Deuschl and colleagues (1998b) found the following features particularly characteristic of psychogenic tremor: sudden onset and variable (rarely remitting) course, coactivation sign (fluctuations in muscle tone and in tremor during passive movements), and absent finger tremor. Although overt signs of hysteria were often lacking, evidence of depression and psychosomatic conditions were common. In another study of 70 patients with psychogenic tremor, 73% had an abrupt onset, and 46% had their maximal disability at onset (Kim et al., 1999). Additional features that were found helpful in confirming the diagnosis of psychogenic tremor included spread from a focal onset to generalized tremor, variability, and entrainment. Electrophysiologic studies are rarely helpful in differentiating organic tremor from psychogenic tremor (McAuley et al., 1998). Using accelerometry, Zeuner and colleagues (2003b) demonstrated that in contrast to essential and parkinsonian tremor, patients with PT showed larger tremor frequency changes and higher individual variability while tapping. Longitudinal studies are needed to determine the natural history of psychogenic tremors. In one study, a 6-year follow-up of 64 patients with unexplained motor disorders, only 3 were later found to have an organic diagnosis (Crimlisk et al., 1998). In the Parkinson’s Disease Center and Movement Disorders Clinic at the Baylor College of Medicine, psychogenic tremor is the most common of all psychogenic movement disorders, accounting for 4.1% of all patients (Jankovic and Thomas, 2006). Clinical information was obtained on 228 of 517 (44.1%) patients, who were followed for a mean of 3.4 ± 2.8 years. Among the 127 patients who were diagnosed with psychogenic tremor, 92 (72.4%) were female, the mean age at initial evaluation was 43.7 ± 14.1 years, and the mean duration of symptoms was 4.6 ± 7.6 years. The following clinical features were considered to be characteristic of psychogenic tremor: abrupt onset (78.7%), distractibility (72.4%), variable amplitude and frequency (62.2%), intermittent occurrence (35.4%), inconsistent movement (29.9%), and variable direction (17.3%). In the majority of patients, some precipitating event could be identified prior to the onset of tremor, including personal life stress (33.9%), physical trauma (23.6%), major illness (13.4%), surgery (9.4%), or reaction to a medical treatment or procedure (8.7%). Coexistent organic neurologic disorder was present in 37% of patients with psychogenic tremor; psychiatric comorbidities included depression in 50.7% and anxiety in 30.7%. Evidence of secondary gain was present in 32.3%, including maintenance of a disability status in 21.3%, pending compensation in 10.2%, and litigation in 9.4%. Improvement in tremor, reported on a global rating scale at last follow-up by 55.1%, was attributed chiefly to “effective treatment by physician” and “elimination of stressors.” On the basis of the analysis of data in this largest longitudinal study of patients with psychogenic tremor, we have concluded that accurate diagnosis is not only based on exclusion of other causes but also dependent on positive clinical criteria, the presence of which should avoid unnecessary investigation. 
Data from Kenney C, Diamond A, Mejia N, et al. Distinguishing psychogenic and essential tremor. J Neurol Sci 2007;263:94–99.
Miscellaneous tremors
There are many other causes of tremors. One of the most common causes of tremors are certain drugs, such as antipsychotics, amiodarone, cyclosporine, lamotrigine, lithium, tricyclic antidepressants, selective serotonin reuptake inhibitors (SSRIs), valproate, and other drugs (Morgan and Sethi, 2005). In one study of 707 patients treated with amiodarone, 2.8% had some neurotoxic effects, including de novo tremor and exacerbation of essential tremor, gait ataxia, peripheral neuropathy, and cognitive impairment (Orr and Ahlskog, 2009). These side effects correlated with duration of exposure and were not always reversible. The bobble-head doll syndrome is a form of slow (2–30 Hz) oscillatory movement that is usually associated with cystic enlargement of the third ventricle, suprasellar arachnoid cysts, aqueductal stenosis, and other lesions involving the third ventricle (Goikhman et al., 1998; Bhattacharyya et al., 2003). Hereditary chin tremor, or hereditary geniospasm, is an autosomal dominant disorder characterized by recurrent episodes of chin tremor (Soland et al., 1996a; Erer and Jankovic, 2007) (Video 18.9). Usually a benign condition with a peak in the mid-twenties (although it may be persistent in men), this condition has been mapped to a marker on chromosome 9q13–q21 (Jarman et al., 1997). Some investigators have suggested that “isolated generalized polymyoclonus” may present as “whole body tremulousness” and that some of the cases are drug-induced (e.g., SSRI) or due to an autoimmune disorder (McKeon et al., 2007). Genetic phenocopies of ET include FXTAS, Kennedy syndrome, SCA12, hereditary neuropathies, Klinefelter (XXY) syndrome (Harlow et al., 2008), and the 48,XXYY syndrome, which is also associated with gait ataxia, dysarthria, and nystagmus (Tartaglia et al., 2009).