[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 19
Treatment of Type 1 Diabetes Mellitus in Adults
The discovery of insulin in the summer of 1921 by Frederick Banting, Charles Best, James Collip, and J. J. R. Macleod at the University of Toronto and its first therapeutic use on January 11, 1922, at the Toronto General Hospital, stands as one of the greatest achievements of contemporary medicine.1 By providing life-sustaining therapy for a previously fatal wasting condition, the discovery of insulin dramatically shifted the prognosis of type 1 diabetes to that of a chronic disease characterized by devastating long-term microvascular and macrovascular complications. Accordingly, the focus of treatment has changed significantly since 1922, with emphasis now directed toward the avoidance of complications and the optimization of patient quality of life. Given the incontrovertible evidence from the Diabetes Control and Complications Trial (DCCT) and other studies that optimization of glycemic control can significantly reduce the risk of vascular complications,2–5 the following overriding objective of modern diabetes management has emerged: the achievement of sustained euglycemia, with avoidance of hypoglycemia, in patients with type 1 diabetes through the physiologic replacement of insulin. Remarkably, however, despite significant advances in the past 80 years, this goal remains an elusive target.6 Modern management of type 1 diabetes involves a multifaceted, patient-centered approach, in which an interdisciplinary health care team works closely with the patient toward the goal of achieving sustained euglycemia. In this chapter, we review the essential elements of this approach to comprehensive care of patients with type 1 diabetes.
Goals of Management
The overriding goal in the management of type 1 diabetes is the optimization of the patient’s quality of life through the prevention and amelioration of the acute and chronic complications that are associated with this disease. In the modern era, a substantial body of epidemiologic and experimental evidence has established the relationship between diabetic complications and hyperglycemia (see Chapter 25). In particular, the findings of the DCCT have had an enormous impact on the management of type 1 diabetes and warrant review before discussion of specific therapeutic goals.
The DCCT was a multicenter, National Institutes of Health–funded randomized controlled clinical trial initiated in 1982 that was designed to assess whether intensive treatment with the goal of maintaining near-normal blood glucose concentrations could reduce the frequency and severity of diabetic complications.2 A total of 1441 patients with type 1 diabetes from the United States and Canada participated in the study and made up two cohorts: a primary prevention cohort of 726 subjects with no retinopathy at baseline and 1 to 5 years’ duration of diabetes, and a secondary intervention cohort of 715 individuals with mild retinopathy and duration of diabetes of up to 15 years. Participants were randomly assigned to receive either the conventional therapy of the day (one or two injections of insulin per day) or intensive therapy (either three or more insulin injections or continuous subcutaneous insulin infusion by external pump, with frequent blood glucose monitoring). After a mean 6.5 years of follow-up, mean glycosylated hemoglobin (A1c) was 7.2% in the intensive therapy arm and 9.0% in the conventional treatment arm. The intensive therapy group showed a relative risk reduction of 76% (95% confidence interval [CI]: 62% to 85%) for the development of retinopathy and a 54% relative risk reduction (95% CI: 39% to 66%) for progression of retinopathy. Similarly, intensive therapy was associated with relative risk reductions of 39% (95% CI: 21% to 52%) for the development of microalbuminuria (defined as urinary albumin excretion > 40 mg per 24 hours) and 54% (95% CI: 19% to 74%) for progression to albuminuria (urinary albumin excretion > 300 mg per 24 hours). The incidence of clinical neuropathy was also decreased in the intensive group, with a relative risk reduction of 60% (95% CI: 38% to 74%). Thus, the DCCT provided conclusive evidence that improved glycemic control with intensive therapy can reduce the incidence of microvascular complications (primary prevention) and can slow the progression of established microvascular disease (secondary intervention).
The benefits of near-normal glycemic control were found to persist for many years after the DCCT, according to data from the follow-up study, the Epidemiology of Diabetes Interventions and Complications (EDIC).7 In the ongoing EDIC observational study, a large cohort consisting of more than 95% of the DCCT participants (n = 1375) has been followed since the completion of the original trial and has been studied by using an intention-to-treat approach based on the original randomization in the DCCT. The mean A1c levels of the former intensive treatment cohort and the former conventional therapy group have converged (A1c ≈ 8%) over the course of the EDIC study and were no longer significantly different at 5 years post DCCT. Remarkably, however, despite similar glycemic control, the former intensive therapy group continued to display reduced risk for both retinopathy and nephropathy at 4 years and 7 to 8 years post DCCT, respectively, compared with the group of patients who were randomized originally to conventional therapy (Fig. 19-1).7,8 These persistent beneficial effects of previous near-normal glycemic control extend the results of the DCCT and demonstrate that intensive treatment should be initiated as early as is safely possible in the management of type 1 diabetes.
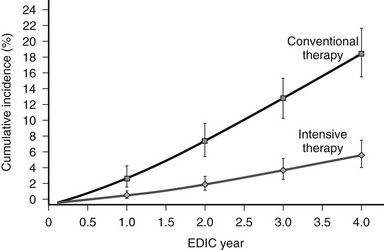
FIGURE 19-1 Cumulative incidence of further progression of retinopathy during the Epidemiology of Diabetes Interventions and Complications (EDIC) study in the former conventional therapy and intensive therapy cohorts from the Diabetes Control and Complications Trial (DCCT). Data are based on regression analysis adjusted for the level of retinopathy at the end of the DCCT, whether patients received therapy as primary prevention or as secondary intervention, and for the duration of diabetes and the A1c value on enrollment in the DCCT. (From DCCT/EDIC Research Group: Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. N Engl J Med 342[5]:387, 2000.)
Although the DCCT conclusively demonstrated that intensive therapy can reduce the risk for microvascular complications, the effect on macrovascular disease was initially less clear. By the end of the DCCT, the intensive therapy arm displayed a relative risk reduction of 34% (95% CI: 7% to 54%) for the development of elevated low-density lipoprotein (LDL) cholesterol levels. Nevertheless, even though intensive therapy was associated with a 41% relative risk reduction (95% CI: −10% to 68%) for macrovascular complications, this difference was not statistically significant.2 It is important to note, however, that the macrovascular benefits derived from near-normal glycemic control in the former intensive treatment arm of the DCCT have emerged over time in the EDIC study. Indeed, after mean follow-up of 17 years, it was observed that intensive therapy during the DCCT had reduced the subsequent risk for any cardiovascular disease (CVD) event by 42% (95% CI: 9% to 63%; P = .02) and the risk for nonfatal myocardial infarction (MI), stroke, or cardiovascular death by 57% (95% CI: 12% to 79%; P = .02) (Fig. 19-2).9 This remarkable risk reduction was largely explained by differences in the updated mean A1c during the DCCT. Furthermore, the reduction in CVD events was consistent with earlier observations that intensive therapy had reduced the progression of atherosclerosis (as measured by carotid artery intima media thickness [IMT]) and the prevalence of coronary artery calcification (CAC) during the DCCT/EDIC study, with both effects associated with the reduction in mean A1c achieved with intensive therapy.10,11 Thus, intensive therapy (with associated reduced glycemic exposure) during the mean 6.5 years of the DCCT first significantly reduced surrogate measures of atherosclerotic disease (carotid IMT, CAC) and later reduced clinical CVD events over the course of the EDIC study (Fig. 19-3). These data demonstrate that the long-term risk associated with past glycemic exposure extends to both microvascular and macrovascular disease.12 Indeed, it has been hypothesized that glycemic exposure early in the course of the disease may have an effect of metabolic imprinting (referred to as “metabolic memory”) that contributes to future risk for vascular complications.9 Taken together, these data further underscore the importance of initiating intensive therapy as early as safely possible in the management of type 1 diabetes.
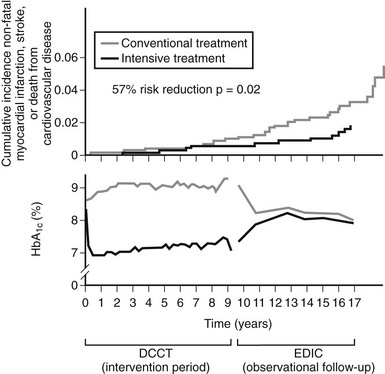
FIGURE 19-2 Differences in A1c (lower panel) and cardiovascular event rates (upper panel) over time between the intensive and conventional diabetes treatment arms of the Diabetes Control and Complications Trial (DCCT)/Epidemiology of Diabetes Interventions and Complications study (EDIC) cohort. (From Retnakaran R, Zinman B: Type 1 diabetes, hyperglycaemia and the heart. Lancet 371:1790–1799, 2008.)
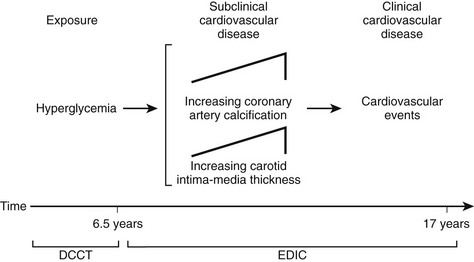
FIGURE 19-3 Pathophysiologic model of cardiovascular disease (CVD) in type 1 diabetes based on observations from the Diabetes Control and Complications Trial (DCCT)/Epidemiology of Diabetes Interventions and Complications study (EDIC). (From Retnakaran R, Zinman B: Type 1 diabetes, hyperglycaemia and the heart. Lancet 371:1790–1799, 2008.)
Overall, the DCCT helped to clarify the relationship between A1c and diabetic complications. For every 10% reduction in A1c, the risk of developing retinopathy was reduced by approximately 45%.13 Moreover, a continuous curvilinear relationship was observed between A1c and the incidence of retinopathy, the risk of progressive retinopathy being decreased substantially at A1c levels below 7%. Although the risk for progressive retinopathy was further reduced at A1c concentrations below 7% (i.e., no threshold effect), this benefit was achieved at the cost of a significantly increased risk for hypoglycemia, given an observed continuous and inverse relationship between hypoglycemic risk and A1c (Fig. 19-4). Thus, the DCCT firmly established the importance of glycemic control in the management of type 1 diabetes, demonstrated the feasibility of intensive therapy in improving glycemic control, and identified the limitations imposed on such control by hypoglycemia. In doing so, this study has provided a framework for current treatment goals.
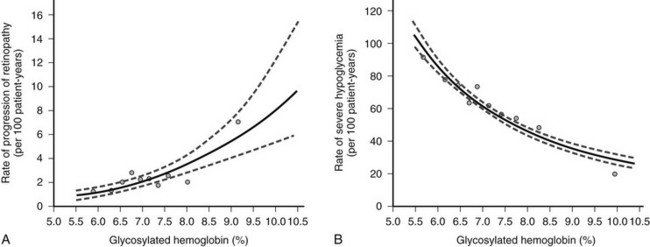
FIGURE 19-4 Risk for sustained progression of retinopathy (A) and rate of severe hypoglycemia (B) in patients receiving intensive therapy, in relation to mean A1c value during the Diabetes Control and Complications Trial (DCCT). (From the DCCT Research Group: The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 329[14]:984, 1993.)
Glycemic Goals
On the basis of the findings of the DCCT and other studies, the ultimate objective in the treatment of type 1 diabetes is metabolic normalization, that is, the achievement of sustained euglycemia and the avoidance of hypoglycemia. In this context, the 2008 American Diabetes Association (ADA) treatment guidelines suggest the following therapeutic targets for nonpregnant adults with type 1 or type 2 diabetes: (1) A1c less than 7.0% (referenced to nondiabetic range 4.0% to 6.0%); (2) preprandial plasma glucose 70 to 130 mg/dL; and (3) peak postprandial plasma glucose less than 180 mg/dL (Table 19-1).14 The evidence-based 2008 Canadian Diabetes Association Clinical Practice Guidelines recommend similar targets of A1c less than or equal to 7.0%, preprandial plasma glucose 72 to 126 mg/dL, and 2-hour postprandial plasma glucose 90 to 180 mg/dL.15 The American Association of Clinical Endocrinologists recommends slightly more stringent goals of A1c less than or equal to 6.5%, fasting glucose less than 110 mg/dL, and postprandial glucose less than 140 mg/dL.16
Table 19-1
American Diabetes Association Recommendations for Treatment Targets in Nonpregnant Adults With Diabetes

Goals might have to be individualized for the given patient.
HDL, High-density lipoprotein; LDL, low-density lipoprotein.
*For women, it has been suggested that the HDL goal should be increased by 10 mg/dL.
Adapted from American Diabetes Association: Standards of medical care in diabetes. Diabetes Care 31(Suppl 1):S12–S54, 2008.
Acute Complications
The major acute complications of type 1 diabetes are ketoacidosis and hypoglycemia. These topics are discussed in detail in Chapters 20 and 21, respectively. The risk for these acute complications can be reduced through the judicious use of insulin therapy. Specifically, the risk for ketoacidosis can be reduced through appreciation of the concept that patients with type 1 diabetes require insulin therapy at all times. Appropriate management of special situations, such as intercurrent illness, requires adherence to this principle (see the section entitled “Adjustment of Insulin Therapy in Special Situations”).
As was observed in the DCCT (see Fig. 19-4B), tight glycemic control typically is associated with an increased risk for hypoglycemia. Patients therefore must be educated about the recognition of hypoglycemic symptoms and the use of appropriate treatment measures when such symptoms arise. Patients are advised to keep rapidly absorbable sources of carbohydrate, such as glucose tablets, close at hand at all times. In general, 15 g of carbohydrate is needed to increase blood glucose levels by approximately 38 mg/dL within 20 minutes.17,18 Besides glucose tablets, other treatments that provide a similar amount of glucose include (1) 15 mL (1 tablespoon) or three packets of table sugar dissolved in water, (2) 175 mL ( cup) of juice, and (3) 15 mL (1 tablespoon) of honey. In addition, patients with type 1 diabetes should have an emergency glucagon injection kit. Both the patient and his or her family members should be trained in the use and intramuscular or subcutaneous injection of glucagon (0.5 to 1 mg) as a means of rapidly increasing blood glucose concentration when the patient is severely hypoglycemic. Finally, the avoidance of hypoglycemia is particularly important for patients who are unable to sense hypoglycemic symptoms (discussed in the section entitled “Complications of Insulin Therapy”).
cup) of juice, and (3) 15 mL (1 tablespoon) of honey. In addition, patients with type 1 diabetes should have an emergency glucagon injection kit. Both the patient and his or her family members should be trained in the use and intramuscular or subcutaneous injection of glucagon (0.5 to 1 mg) as a means of rapidly increasing blood glucose concentration when the patient is severely hypoglycemic. Finally, the avoidance of hypoglycemia is particularly important for patients who are unable to sense hypoglycemic symptoms (discussed in the section entitled “Complications of Insulin Therapy”).
Chronic Complications
The chronic microvascular complications of type 1 diabetes include retinopathy, neuropathy, and nephropathy (discussed in Chapters 26, 27, and 28, respectively). To reduce the impact of these complications, management strategies include regular surveillance for early detection of complications and tight glycemic control for both primary prevention and secondary intervention, as demonstrated in the DCCT.
Macrovascular disease, as manifested by cerebrovascular, peripheral vascular, and coronary artery disease, is a major chronic complication of type 1 diabetes (discussed in Chapter 25). Regular surveillance for vascular disease and management of cardiovascular risk factors is essential. In particular, modification of reversible cardiovascular risk factors, such as hypertension, dyslipidemia, and cigarette smoking, is an important management goal. The 2008 ADA treatment guidelines suggest a target blood pressure of less than 130/80 mm Hg in patients with diabetes. In addition, the following treatment targets for lipids are recommended: LDL less than 100 mg/dL, triglycerides less than 150 mg/dL, and HDL greater than 40 mg/dL in men and HDL greater than 50 mg/dL in women (see Table 19-1).14
Quality of Life
Type 1 diabetes is a chronic condition that requires a complex health care strategy that demands significant patient effort through frequent insulin injections, regular self-monitoring of blood glucose levels, and constant attention to nutrition and physical activity. As such, effective management requires that the patient take responsibility for his or her care. In this context, it should be noted that quality of life is an important feature that can affect a patient’s willingness to pursue a demanding treatment plan. Recognition of this concept at all times by the health care team will facilitate the success of treatment initiatives. Moreover, in recognition of the importance of quality of life, many research studies now utilize validated instruments such as the Diabetes Quality of Life Questionnaire to measure patient well-being as a therapeutic outcome.19
Monitoring
Glycemic Control
Acute Measurement of Glycemic Control
Patients with type 1 diabetes should monitor blood glucose frequently. In the DCCT, insulin doses for patients in the intensive treatment arm were adjusted according to the results of SMBG performed at least four times a day.20 Blood glucose typically is measured before administration of insulin prior to a meal and at bedtime. Meaningful data can be derived from measurements at other times as well. For instance, nocturnal hypoglycemia resulting from excessive bedtime insulin can be detected by blood glucose measurement in the early hours of the morning. Similarly, postprandial monitoring 2 hours after a meal can assess the adequacy of the preprandial insulin dose. Patients should check their blood glucose level before driving a motor vehicle, particularly if they have difficulty sensing hypoglycemic symptoms, as hypoglycemia (and hence neurologic impairment) while driving can be very dangerous for the patient and others. Patients should also consider SMBG at times when blood glucose might be changing rapidly, such as during the postexercise period or when experiencing symptoms of hypoglycemia.
Self-monitoring of blood glucose is of value only to the extent that the information it provides is used to guide treatment. With each measurement, patients are encouraged to use a logbook to record the blood glucose value, the time of day, and the temporal relationship to food intake. Data obtained from SMBG should be used in two ways: variable insulin dose scales and pattern management. Variable insulin dose scales refer to algorithms that guide the patient as to the appropriate dose adjustment of preprandial insulin based on the current degree of glycemia (discussed in the section entitled “Implementation of Intensive Insulin Therapy”). Pattern management describes the two-step process of using the glucose profile recorded over several days to identify glycemic patterns and then adjusting the daily insulin regimen for the coming days in a proactive fashion on the basis of these patterns. Therefore, both variable insulin dose scales and pattern management are essential components of patient education.
Given the value of blood glucose data, it is important to consider potential problems that are inherent in the process of self-monitoring. To ensure proper meter function, glucose meters should be calibrated by a laboratory reference standard. The patient’s use of the meter and self-monitoring technique should be reviewed periodically in the clinic.21 Inappropriate handling of the testing strips can lead to defective function and inaccurate glucose results. Finally, some patients find lancing of the fingertip to be painful and hence are unwilling to measure blood glucose as frequently as recommended.
In response to patient concerns regarding pain associated with lancing of the fingertip, alternative site glucose meters have recently been introduced. By sampling from alternative sites such as the forearm, these devices can provide accurate blood glucose measurements while causing less pain than fingertip testing.22 However, it should be noted that alternative site testing might not provide accurate results at times of rapid change in blood glucose concentration (e.g., immediately after a meal or after exercise).23,24 Therefore, traditional fingertip testing is recommended at these times.
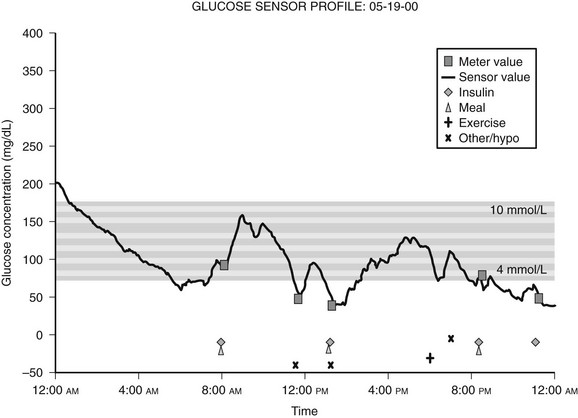
Because blood glucose concentrations are dynamic and fluctuate constantly, it is readily apparent that the limited data provided by even frequent SMBG might not be truly representative of an individual’s glucose excursions. In this context, continuous glucose monitoring (CGM) devices represent an important recent development. This technology utilizes the fact that glucose in interstitial fluid can reflect blood glucose levels.25 CGM systems typically consist of a pager-sized monitor worn by the patient that receives an electrical signal from a subcutaneous sensor that continuously measures the glucose concentration in interstitial fluid.26 Using this input, the monitor determines a blood glucose concentration every 5 minutes that can be downloaded to a computer to provide a graphic display of the patient’s glycemic profile over the period of time monitored (Fig. 19-5).27 During the period of monitoring (usually 3 days), the patient maintains a diary of meals, insulin doses, physical activity, and hypoglycemic symptoms to facilitate meaningful interpretation of the recorded data. Whereas older CGM systems yielded only retrospective glucose data, current models have the capability to provide patients with real-time access to their absolute blood glucose values and to relative trends in their glucose levels. Furthermore, these devices have alarms that can alert patients to either hypoglycemia or hyperglycemia, thereby precipitating appropriate corrective action. In addition, the technology exists wherein CGM systems have been combined with insulin pumps, although it should be noted that these systems currently require the input of the patient (since the insulin pump is not controlled by the CGM device).28 In this regard, it should be recognized (1) that CGM systems require calibration with SMBG readings, (2) that the accuracy of glucose measurements by these devices have been drawn into question in the past,29 and (3) that SMBG values remain the recommended data for guiding treatment decisions.14,30 Thus, the American Diabetes Association currently recommends that CGM be used as a supplemental tool to SMBG in selected patients with type 1 diabetes, particularly those with hypoglycemia unawareness.14 The latter consideration is a reflection of the fact that initial experience with CGM demonstrated higher rates of unrecognized nocturnal hypoglycemia than were previously suspected.31,32 Furthermore, several studies have since shown that CGM systems (particularly real-time devices) are associated with reductions in the total amount of time spent in hypoglycemia.30,33–35 To date, only a few studies have shown improvements in A1c.30,36,37 Although some challenges remain for this rapidly developing technology (including the acquisition of a broader evidence base from further clinical research), the role of CGM as a valuable tool in the management of type 1 diabetes is likely to grow in the near future.
Measurement of Chronic Glycemic Control
Glucose can attach to proteins in the blood through an irreversible, nonenzymatic process, resulting in the formation of glycated proteins.38 Because this process is irreversible, glucose remains attached to the protein until the latter is metabolized. The degree of glycation is a function of blood glucose concentration over time. Thus, measurement of glycated proteins provides a means of estimating chronic blood glucose concentration over a time period proportional to the half-life of the protein in question.
Erythrocytes are freely permeable to glucose. In the circulation, glucose attaches irreversibly to hemoglobin in red blood cells, leading to the formation of glycated hemoglobins called hemoglobin A1a, A1b, and A1c. Measurement of hemoglobin A1c (also known as A1c, glycohemoglobin, or glycosylated hemoglobin) has emerged as the most widely used test of chronic glycemic control. Specifically, the proportion of glycosylated hemoglobin to the total number of hemoglobin molecules reflects overall blood glucose concentration over the preceding 2 to 3 months.39 Therefore, regular measurement of A1c every 3 months is recommended as a method of determining overall glycemic control and evaluating the adequacy of the treatment regimen.21 For instance, a significantly elevated A1c value, consistent with suboptimal glycemic control, would suggest the need for a change in the treatment regimen. Analysis of daily glycemic patterns from the patient’s SMBG records can indicate the specific changes to be made.
Many different types of assays can be used for measurement of glycated hemoglobins.40 Some assays report A1c as a percentage of total hemoglobin, while other methods measure total glycated hemoglobin. Thus, nonstandardization of assays can complicate comparison of A1c measurements between laboratories. The National Glycohemoglobin Standardization Program, initiated in 1996, is an initiative that is designed to standardize laboratory A1c assays to the DCCT reference method.41
The DCCT provided insight into the correlation between A1c level and mean plasma glucose concentration (Fig. 19-6).42 This relationship has recently been studied with the use of CGM data.43 In practice, in some situations the measured A1c value may be discordant with the level that would be expected on the basis of the patient’s capillary blood glucose records. Such discrepancy might be due to glycemic excursions at unmonitored times of the day that are not reflected in SMBG records. Alternatively, the problem might be falsification of SMBG records by the patient.
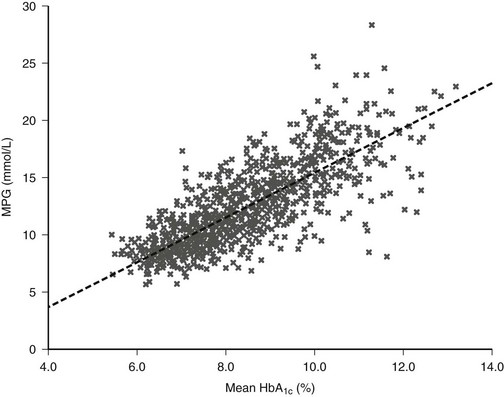
FIGURE 19-6 The relationship between mean plasma glucose and mean A1c in the Diabetes Control and Complications Trial (DCCT) (n = 1429; r = 0.82). (From Rohlfing CL, Wiedmeyer HM, Little RR, et al: Defining the relationship between plasma glucose and HbA1c: Analysis of glucose profiles and HbA1c in the Diabetes Control and Complication Trial. Diabetes Care 25[2]:276, 2002.)
Another possibility in certain clinical settings is that the measurement of A1c might not be accurate. For instance, rapid red blood cell turnover leads to a disproportionate representation of younger erythrocytes, whose collective exposure to ambient blood glucose levels has been shorter than that of other red blood cells. Accordingly, A1c levels tend to be reduced under such conditions.44 Pregnancy and hemolytic anemias are examples of settings characterized by rapid red blood turnover and hence lowered A1c levels. Similarly, A1c concentration may be lowered in hemoglobinopathies, such as thalassemia and sickle cell disease, because of a diminished propensity for glycation of the abnormal hemoglobin molecule. Conversely, depending on the assay used, A1c values may be falsely elevated in the presence of hemoglobin F, the carbamylated hemoglobin formed in uremia, or acetaldehyde-bound hemoglobin (seen in alcohol abuse).45
In situations in which measurement of A1c might be unreliable, an alternative approach to the estimation of glycemic control is the measurement of glycated serum proteins or albumin. The term fructosamine has been applied to the glycated proteins measured by such assays.46 Given the 14- to 21-day half-life of albumin, a serum fructosamine measurement reflects mean blood glucose concentration over the preceding 1 to 2 weeks. Generally, serum fructosamine values correlate well with A1c measurements.47 However, certain caveats should be noted in measuring fructosamine. First of all, fructosamine values can be affected by conditions that alter the synthesis or clearance of serum proteins. Indeed, the question of whether or not to correct fructosamine assays for serum protein or albumin concentration remains controversial.48 Second, serum fructosamine levels show greater intrasubject variability than A1c measurements do.49 Third, unlike A1c, fructosamine has not yet been shown to correlate with the risk for diabetic complications.
Ketone Testing
The presence of detectable levels of ketones in urine or blood may indicate impending or established ketoacidosis in patients with type 1 diabetes. Accordingly, patients should test for ketones in the following situations: when blood glucose concentrations are persistently elevated, during periods of acute illness or stress, or when the patient is experiencing symptoms compatible with ketoacidosis such as nausea, vomiting, and abdominal pain.48 Traditionally, patients have used urine dipsticks to test for ketonuria. In recent years, however, home monitors have been developed that can measure the capillary blood concentration of β-hydroxybutyrate, the most prevalent intermediate molecule in ketone formation.50 Blood ketone testing avoids some of the limitations of traditional urinary ketone testing, such as false-positive readings, the inability to detect β-hydroxybutyrate (urinary dipsticks measure different ketone bodies), and patient reluctance to perform urinary testing. In limited studies to date, home capillary blood ketone measurement appears to be more sensitive than urinary testing in the detection of ketosis.51,52 The use of blood ketone monitoring by patients is expected to increase in the future. In the clinical setting, serum ketone measurement is recommended over urinary testing.
Complication Surveillance
1. Retinopathy: Initial dilated and comprehensive eye examination should be performed within 5 years after diagnosis of type 1 diabetes and at least annually thereafter.14
2. Nephropathy: Patients with type 1 diabetes of 5 or more years’ duration should undergo an annual screening test for the presence of microalbuminuria.14 The preferred screening test is a measurement of the albumin-to-creatinine ratio in a random, spot urine sample.53
3. Neuropathy and foot care: A comprehensive foot examination should be performed annually.14 This examination includes evaluation of sensation by 10 g Semmes-Weinstein monofilament, testing of vibration sense by tuning fork, assessment of superficial pain sensation, palpation, and visual inspection. In screening for neuropathy, superficial pain sensation testing, monofilament examination, and vibration testing all show similar operating characteristics and may be used interchangeably.54
4. Cardiovascular disease: Regular evaluation of cardiovascular risk factors is recommended. Blood pressure should be measured at every diabetic clinic appointment. Lipid profile should be tested at least annually. Screening exercise stress testing may be considered in patients with typical or atypical cardiac symptoms; abnormal resting electrocardiogram; a history of peripheral or cerebrovascular disease; or sedentary lifestyle, age over 35 years, and plans to start a vigorous exercise program.14
5. Other: Other potential associated problems that warrant periodic screening include erectile dysfunction, depression, and thyroid disease.
Insulin Therapy
Principles of Insulin Replacement
Insulin replacement strategies in type 1 diabetes ideally aim to mimic the normal physiologic secretion of insulin by the pancreas.6 Therefore, to appreciate the rationale underlying the design of current insulin replacement regimens, an understanding of the basic structure and biochemistry of endogenous insulin is necessary (Fig. 19-7). This topic, discussed in detail in Chapter 6, is reviewed briefly here.
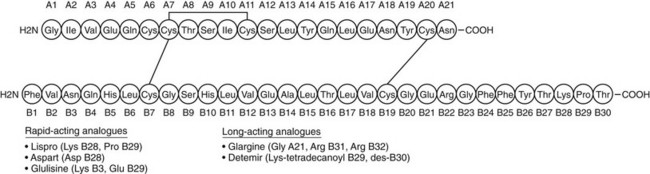
FIGURE 19-7 The amino acid sequence and structure of human insulin. The A and B chains are linked by two disulfide bonds. A third disulfide bridge is present within the A chain. (Adapted from Cheng AY, Zinman B: Insulin analogues and the treatment of diabetes. In Raz I, Skyler JS, Shafrir E [eds]: Diabetes: From Research to Diagnosis and Treatment. London, Martin Dunitz Ltd, 2002, pp 331–346.)
The pancreatic β cells secrete endogenous insulin into the portal venous system in response to physiologic demand for glucose homeostasis as determined by nutrient intake and energy expenditure. Despite broad fluctuations in these determinants, healthy individuals are able to maintain plasma glucose concentration within a narrow range of 3.5 to 7.0 mmol/L throughout the day (Fig. 19-8).55 This normal glucose homeostasis requires tightly regulated insulin secretion that consists of two components: a basal secretion rate and surges of markedly increased secretion following ingestion of a mixed meal. Accounting for approximately 40% of total 24 hour pancreatic insulin output, the basal secretion of ≈1 µ/hr serves to limit hepatic glucose production and adipocyte lipolysis in the postabsorptive (fasting) state such as between meals and overnight.56 Conversely, with ingestion of a mixed meal, dietary secretagogues (e.g., glucose, amino acids) and gastrointestinal hormones (e.g., glucagon-like peptide 1) stimulate abrupt pulses of insulin secretion up to five times the basal rate. These pulses regulate postprandial glycemia by inhibiting hepatic glucose production and increasing peripheral glucose uptake. Insulin secretion is also affected by energy expenditure. For instance, with moderate exercise, insulin secretion rapidly decreases to prevent hypoglycemia.57 Conversely, with strenuous exercise, catecholamine-mediated hyperglycemia may occur, leading to increased insulin secretion in the postexercise period.58 Thus, the tight coupling of insulin secretion to plasma glucose concentration in healthy individuals (see Fig. 19-8) requires the complex integration of multiple signals of nutrient availability and energy expenditure.
Insulin Preparations
For the first 60 years after the introduction of insulin therapy in 1922, the only commercially available preparations were animal insulins derived from bovine or porcine pancreatic extracts. Porcine and bovine insulin preparations differ from human insulin by one and three amino acids, respectively. The use of animal insulins, however, was inherently complicated by several issues, including potential supply limitations, incomplete purification, and a propensity to induce the formation of anti-insulin antibodies (which might affect the activity and absorption of the exogenous animal insulin and lead to unpredictable pharmacodynamics, although this relationship is controversial). These issues were reconciled with the introduction of recombinant human insulin in the 1980s.59 The past decade has seen further advances with the introduction of insulin analogues bearing molecular modifications that confer advantageous pharmacokinetics. Recombinant human insulin and the newer analogues are now the main preparations used in the treatment of type 1 diabetes.
In selecting types of insulin for use in a treatment regimen, the most important parameters to consider are the onset, peak, and duration of action of the given insulin preparation (Table 19-2). Commercially available human insulins and analogues include rapid-, short-, intermediate-, and long-acting preparations. Its pharmacokinetic properties will determine whether a preparation should be used as either meal insulin or basal insulin. Specifically, rapid- and short-acting preparations are used as meal (or bolus) insulins, and intermediate- and long-acting preparations provide basal insulins.
Table 19-2
Approximate Pharmacokinetic Properties of Human Insulin and Insulin Analogues Following Subcutaneous Injection
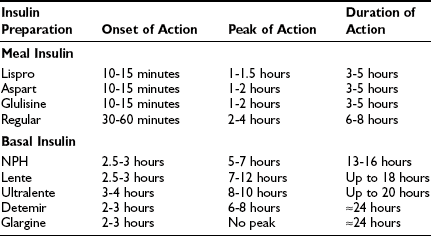
Human Insulins
Human insulin was the first medication to be commercially manufactured using recombinant DNA technology.60 Currently available human insulins include short-, intermediate-, and long-acting preparations. Each preparation has advantages and disadvantages, which will be considered in turn.
Short-Acting Human Insulin: Regular: Regular recombinant human insulin is identical to the endogenous insulin polypeptide. In solution, however, regular insulin tends to self-associate, first forming dimers and subsequently hexamers. After subcutaneous injection, the absorption of hexameric insulin molecules is delayed, pending dissociation into monomers and dimers, which can diffuse rapidly from subcutaneous tissue into the systemic circulation.61 This slowed absorption has important pharmacokinetic implications, leading to (1) a modestly delayed onset of biologic action in vivo (30 to 60 minutes after injection), (2) a relatively late peak effect (2 to 4 hours after injection), and (3) a prolonged duration of action (6 to 8 hours) (see Table 19-2).62 Clearly, this pharmacokinetic profile is quite different from the rapid and short-lived endogenous insulin response to a mixed meal that is seen in individuals without diabetes. Therefore, although regular insulin was widely used in the past, its role as meal insulin has declined since the introduction of rapid-acting analogues.
Intermediate-Acting Human Insulin: NPH and Lente: The development of intermediate-acting human insulins reflected the need for an exogenous insulin preparation that could mimic the basal insulin secretion that is observed in individuals who do not have diabetes. The principle underlying the development of these insulins is that the rate of absorption of exogenous insulin from subcutaneous tissue can be significantly reduced by manipulating its suspension. First introduced in 1946, Neutral Protamine Hagedorn (NPH) insulin is a suspension of insulin complexed with protamine and zinc.63 NPH is poorly absorbed from subcutaneous tissue, leading to delayed onset of action (2.5 to 3 hours), late peak effect (5 to 7 hours), and prolonged duration of action (13 to 16 hours) (see Table 19-2).62 A second intermediate-acting insulin preparation is lente, a crystalline suspension of insulin with zinc and acetate. It exhibits a pharmacokinetic profile similar to that of NPH, but with a slightly later peak and longer duration of action.
The intermediate-acting insulins show substantial variation in subcutaneous absorption both within an individual patient and between patients, resulting in variable glycemic excursions.64 Indeed, variability in the absorption of intermediate-acting insulin may account for as much as 80% of day-to-day variation in blood glucose concentrations.65 Other factors that contribute to the unpredictability of NPH and lente are dose-dependent changes in pharmacokinetics and the practical variability associated with resuspension of these insulin preparations by the patient before subcutaneous injection.
Given their underlying pharmacokinetic profiles (duration of action of 13 to 18 hours) and clinical variability in action, the intermediate-acting insulins generally do not provide adequate basal insulin coverage. To increase their utility as basal insulins, these preparations are given twice daily in at least 20% of patients with type 1 diabetes.66 Moreover, their peak effect is an undesirable quality in a basal insulin, insofar as it is clearly dissimilar to the relatively peakless profile of basal insulin secretion seen in an individual without diabetes.
Long-Acting Human Insulin: Ultralente: Ultralente, also a zinc insulin suspension, represents another attempt at providing appropriate basal insulin coverage. Its onset of action is 3 to 4 hours after injection with a peak effect at 8 to 10 hours and a duration of action of up to 20 hours.62 Clinically, its effects are similar to those of intermediate-acting insulin, but with possibly greater variability in absorption. Therefore, given the availability of intermediate-acting preparations and new long-acting analogues, its clinical utility is limited.
Insulin Analogues
The pharmacokinetic shortcomings of human insulin preparations have provided a strong impetus for the development of insulin analogues. Through specific modifications of the insulin molecule (see Fig. 19-7), researchers have designed analogues with tailored pharmacokinetic profiles that allow for better meal or basal coverage. Currently available analogues include rapid-acting and long-acting preparations.
Rapid-Acting Analogues: Lispro, Aspart, and Glulisine: Because the tendency to self-associate and resultant delayed absorption of regular insulin compromise its effectiveness in mimicking the normal prandial insulin surge, it follows that an analogue with enhanced absorption may provide a superior meal insulin. In this context, it was previously noted with interest that human insulin-like growth factor-1 (IGF-1), despite significant homology to insulin, exhibited a reduced tendency to form multimers. Identification of the salient structural differences between IGF-1 and insulin led to the development of lispro insulin, the first commercially available analogue.67 Lispro is identical to human insulin except for a reversal of the 28th and 29th amino acid residues (proline and lispro, respectively) of the normal insulin B chain (see Fig. 19-7). The resultant conformational change in the carboxyterminal of the B chain introduces steric hindrance at interfaces involved in dimerization. Thus, dimer formation with lispro is reduced by a factor of 300 compared with regular human insulin.68 It is important to note that apart from reduced self-aggregation, lispro displays biologic activity similar to that of regular insulin, with both comparable affinity for the insulin receptor and equivalent hypoglycemic potency.67 Thus, clinically, lispro acts similarly to monomeric human insulin. It is rapidly absorbed in 10 to 15 minutes, reaches peak activity in 60 to 90 minutes, and has a duration of action of 3 to 5 hours (see Table 19-2).69 With this pharmacokinetic profile, lispro mimics the normal prandial insulin surge in response to carbohydrate ingestion better than any of the exogenous meal insulins that preceded it do.
A second rapid-acting analogue is insulin aspart, engineered through replacement of the proline residue at position 28 of the B chain with aspartic acid. The negative charge of the aspartic acid residue causes repulsion from other negatively charged amino acids and thus leads to decreased self-association of insulin aspart monomers.70 Accordingly, insulin aspart has a similar pharmacokinetic profile to that of lispro and also is well suited for use as meal insulin.71 Finally, a third recently introduced rapid-acting analogue is insulin glulisine, in which the asparagine residue at position B3 of human insulin has been replaced by lysine and the lysine at position B29 has been replaced by glutamic acid.72
Given their pharmacokinetics, the rapid-acting insulin analogues have clear advantages over regular human insulin. Because patients can inject these analogues immediately before meals (rather than 30 minutes before meals, as is typically recommended with regular insulin), their use is associated with greater mealtime flexibility, easier application of carbohydrate estimation techniques (see the section entitled “Nutrition”), and improved quality of life.73 Both lispro and aspart provide improved postprandial glycemic control with less postprandial and nocturnal hypoglycemia than occurs with regular insulin.74–76 Despite these advantages, however, use of these rapid-acting analogues in intensive insulin regimens has not been uniformly associated with substantial improvement in A1c compared with regular insulin.73 This apparent discrepancy likely reflects the suboptimal postabsorptive coverage provided by the basal insulins that were used in the studies in question. Specifically, the shorter duration of action of the rapid-acting analogues is more liable to expose inadequate basal coverage between meals that might otherwise be masked by the longer duration of action of regular insulin when the latter is used for meal coverage. Nevertheless, even in the absence of an A1c benefit, the combination of improved postprandial glycemia with less hypoglycemia suggests an overall reduction in glycemic excursion and hence better glycemic control with rapid-acting analogues.
Long-Acting Analogues: Glargine and Detemir: An ideal basal insulin would have a peakless, 24 hour time-action profile. Attempts to develop long-acting analogues have focused on methods of achieving slow, prolonged absorption following subcutaneous injection. Insulin glargine, the first commercially available long-acting analogue, was introduced to the U.S. market in 2001. It has two modifications compared with human insulin: Two arginines have been added to the carboxy terminus of the B chain, and a glycine residue replaces an acid-sensitive asparagine at position A21.77 These changes have shifted the isoelectric point of glargine toward neutrality such that this analogue is completely soluble in its acidic injection solution at pH 4.0 but is much less soluble at the neutral physiologic pH of subcutaneous tissue. After injection into subcutaneous tissue, the acidic injection solution is neutralized, causing glargine to microprecipitate.78 Glargine subsequently is slowly absorbed into the systemic circulation from the subcutaneous microprecipitates, resulting in a smooth and gradual rise in serum concentration. Therefore, the pharmacokinetic profile of glargine shows an onset of action 2 to 3 hours after injection with a relatively peakless, 24 hour duration of action (see Table 19-2).79 Moreover, glargine is associated with less variability in absorption than is either NPH or lente.80,81 Furthermore, its in vivo hypoglycemic potency is equivalent to that of human insulin. The promise associated with these properties has been further supported by clinical trial evidence demonstrating lower fasting plasma glucose levels and less hypoglycemia (including nocturnal hypoglycemia) with the use of glargine, compared with NPH, as basal insulin in intensive therapy.82–84 Improved A1c with glargine compared with NPH has been demonstrated in a single study.85
The design of insulin detemir, a second long-acting analogue, reflects a different approach to achieve long-acting basal coverage. With detemir, the B30 amino acid has been removed, and a 14-carbon aliphatic fatty acid has been acylated to the B29 amino acid.86 This modification allows for reversible binding between albumin and the added fatty acid. After injection, an equilibrium develops between free and bound detemir, in which 98% of the analogue is bound to albumin. Because only the free analogue binds to the insulin receptor, the duration of action of detemir is prolonged owing to sustained release of the analogue that has been bound to circulating albumin.87 The time-action profile of insulin detemir is characterized by peak activity at 6 to 8 hours after injection and a prolonged 24 hour duration of action (see Table 19-2).87 Clinically, significant reductions in hypoglycemic risk have been demonstrated with detemir as basal insulin in intensive regimens compared with NPH.64 In addition, detemir has been associated with more predictable glycemic control than NPH, with significantly less within-subject variation in blood glucose levels.88,89 Moreover, its pharmacokinetic profile, unlike that of NPH, has been shown to be consistent in adults, adolescents, and children.90 It is interesting to note that this insulin formulation appears to be associated with less weight gain, which is a modest but consistent observation, the mechanism of which is uncertain.91
Intensive Insulin Therapy Regimens
Multiple Daily Injection Regimen
A multiple daily injection (MDI) regimen involves at least four injections of insulin per day, consisting of boluses of meal insulin before each meal and at least one injection of basal insulin, usually at bedtime. If needed, a second injection of basal insulin may be added at breakfast or before lunch. The meal insulin of choice is a rapid-acting analogue (lispro, aspart, glulisine). Use of rapid-acting analogues in MDI therapy has been associated with better postprandial glycemic control, a reduced incidence of hypoglycemia, and improvements in quality of life, compared with regular insulin.73 On the other hand, although some studies have shown a significant decrease in A1c with the use of rapid-acting analogues as compared with regular insulin in MDI therapy, other studies have not shown a significant difference in overall glycemic control.73 As was noted earlier, however, this phenomenon likely reflects suboptimal basal insulin replacement in the studies in question. Moreover, with similar A1c, the combination of reduced postprandial glycemic excursion with less hypoglycemia suggests reduced overall glycemic variability (and hence better control) with the use of rapid-acting analogues compared with regular insulin in MDI therapy.
In the past, basal insulin replacement in MDI usually has consisted of bedtime injection of NPH, lente, or ultralente. In clinical studies of MDI regimens using rapid-acting analogues as meal insulin, all three of these basal preparations have shown similar efficacy with regard to glycemic control and incidence of hypoglycemia.66,92 Given their pharmacokinetic advantages, however, the long-acting analogues have become the basal insulins of choice in MDI therapy. In studies comparing glargine and NPH as basal insulins in MDI regimens, glargine has been associated with improved fasting glucose levels and reduced rates of hypoglycemia.82–84 Moreover, a second, prebreakfast injection of basal insulin is far less likely to be required with glargine than with NPH, lente, or ultralente. Indeed, in studies using lispro as meal insulin in MDI regimens, glycemic control was similar with a single injection of glargine at breakfast, dinner, or bedtime.93 To date, few studies have compared glargine and NPH in MDI regimens using only rapid-acting analogues for meal insulin, although one such study demonstrated reduced A1c levels with a single dose of glargine compared with four daily injections of NPH.94 Detemir also has been shown to reduce A1c, compared with NPH, in patients on MDI using insulin aspart for meal coverage.95 Although large-scale studies evaluating the efficacy of MDI regimens that use rapid-acting analogues for meal insulin and long-acting analogues for basal coverage are pending, it is expected that this combination will provide the most physiologic insulin replacement to date among MDI regimens.
Continuous Subcutaneous Insulin Infusion
The external insulin infusion pump was first developed in the late 1970s, providing the basis for modern continuous subcutaneous insulin infusion (CSII) therapy.96 With CSII, an external infusion pump delivers a continuous infusion of rapid- or short-acting insulin through a catheter inserted into the subcutaneous tissue of the abdominal wall. The pump is preprogrammed by the patient to deliver insulin continuously at a specified rate that is designed to meet the individual’s basal insulin demands. For meal coverage, patients use the pump to deliver a specified bolus of insulin before eating. The rapid-acting insulin analogues are the insulins of choice in CSII, as both lispro and aspart have been shown to reduce postprandial glycemia, A1c concentration, and the incidence of hypoglycemia compared with regular insulin in this setting.97–99
The major limitation with CSII is the significant cost associated with the pump and necessary supplies, such as the tubing, which must be changed every 48 to 72 hours. Another disadvantage is the risk for infection at the insertion site of the catheter. Catheter site infections occur at an estimated rate of 7.3 to 11.3 events per 100 years of patient follow-up.20,100 These infections usually are readily treatable with antibiotics and a change of insertion site. In rare cases, a subcutaneous abscess requiring surgical drainage can develop. Finally, interruption of basal insulin delivery due to pump malfunction or catheter disruption can rapidly lead to hyperglycemia or even ketoacidosis, as patients will quickly become markedly insulinopenic owing to the use of only rapid- or short-acting insulin in CSII.101
Clinical Efficacy of CSII versus MDI
In clinical trials comparing optimized MDI therapy with CSII, glycemic control has been found to be similar with both regimens. A meta-analysis of trials comparing CSII and MDI regimens reported a difference in A1c of 0.51% favoring CSII, although all but 1 of the 12 studies included in this analysis were older studies using suboptimal, non-analogue meal insulins.102 Furthermore, given improvements in pump technology and greater clinical experience with intensified regimens, the current applicability of these studies is unclear. To date, the few adult studies comparing CSII and optimized MDI therapy with both regimens using rapid-acting analogues have shown slightly improved glycemic control with insulin pump therapy, with no significant difference in hypoglycemia, although all but one study used NPH as the basal insulin for MDI therapy.103–107 A pooled analysis of three of these NPH-based MDI studies revealed that the glycemic advantage of CSII over MDI may be related to baseline A1c, such that patients with the poorest initial glycemic control enjoy the greatest benefit with insulin pump therapy.108,109 The lone adult study comparing CSII with MDI using analogues for both meal and basal insulin (aspart and glargine, respectively) used a 10-week crossover design and demonstrated that insulin pump therapy was associated with lower glycemic exposure, as measured by fructosamine and area under the glucose curve.107 At present, additional studies comparing CSII versus optimized MDI regimens using long-acting analogues for basal insulin and rapid-acting analogues for prandial coverage are eagerly awaited.
Implementation of Intensive Insulin Therapy
Implementation of an intensive insulin regimen using MDI or CSII can be accomplished in many ways. In general, starting doses are estimated by using an algorithm such as the method shown in Figure 19-9. The initial total daily insulin (TDI) requirement is estimated at 0.5 unit/kg of body weight, although most patients with type 1 diabetes ultimately require 0.6 to 0.7 unit/kg. Alternatively, for patients who are already on an insulin regimen, TDI is simply the sum of all current insulin doses. When CSII is implemented in such a patient, this TDI dose must be reduced by 20% to determine the total daily amount of insulin to be delivered by the pump.
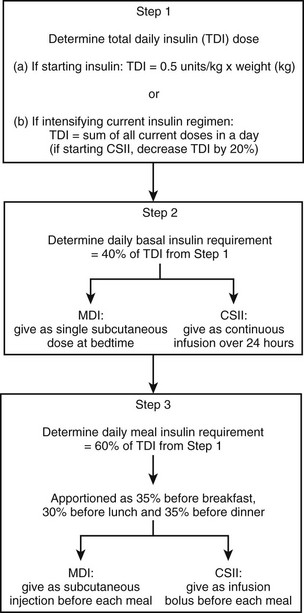
FIGURE 19-9 One algorithm for the determination of initial insulin doses when intensive insulin therapy using multiple daily injection (MDI) or continuous subcutaneous insulin infusion (CSII) is implemented. (Adapted from Shah BR, Zinman B: Insulin regimens for type 1 diabetes. In Sperling MA [ed]: Contemporary Endocrinology: Type 1 Diabetes: Etiology and Treatment. Totowa NJ, Humana Press, 2003, p 205.)
As an example, consider the following case of a patient who is currently on a nonintensified regimen of 12 units of NPH at both breakfast and bedtime and 6 units of regular insulin at breakfast and dinner. To convert this patient’s insulin therapy to an intensified MDI regimen, the algorithm in Fig. 19-9 would be applied as follows:
• The TDI would be 12 + 12 + 6 + 6 = 36 units of insulin per day.
• The basal requirement would be estimated at 40% × 36 units = 14 units per day.
• The daily meal insulin requirement would be estimated at 60% × 36 units = 22 units per day. This meal insulin could be divided such that the patient receives 35% × 22 units = 8 units at breakfast, 30% × 22 units = 6 units at lunch, and 35% × 22 units = 8 units at dinner. These meal insulin doses ideally would be provided in the form of preprandial subcutaneous injections of a rapid-acting analogue.
In practice, the insulin doses calculated in Fig. 19-9 are adjusted continually by the patient on the basis of the following three considerations: preprandial capillary blood glucose concentration, anticipated carbohydrate intake, and upcoming physical activity. By determining the preprandial blood glucose level using capillary monitoring, the patient can adjust prandial insulin dosage using a variable insulin dose scale (Table 19-3). Using this approach, the patient is directed to take a higher dose of prandial bolus insulin when the blood glucose concentration is above a specified range. The second consideration in dose adjustment is anticipated carbohydrate intake. By estimating the amount of carbohydrate to be ingested in the upcoming meal (a practice that is referred to as carbohydrate counting), the patient can adjust the prandial insulin dose accordingly. Although most patients require an additional 1 unit of insulin per 10 to 15 grams of carbohydrate ingested at a meal, the actual ratio must be determined individually for each meal in any given patient. Finally, patients must consider the effects of upcoming physical activity when adjusting insulin dosage. For instance, with MDI therapy, prandial insulin administered at the meal before moderate exercise might have to be reduced, depending on the individual’s glycemic response to exercise.110 Patients using CSII have the additional flexibility of being able to adjust their basal insulin rate to accommodate for exercise.
Table 19-3
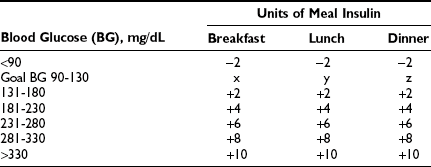
Adapted from Leadership Sinai Centre for Diabetes, Mount Sinai Hospital, Toronto, Ontario, Canada.
Insulin Delivery
A newer, alternative delivery method is provided by pen-and-cartridge devices. In this case, replaceable cartridges containing insulin are placed into a pen-shaped delivery device that is referred to as an insulin pen, which then is used to inject the insulin subcutaneously. Disposable insulin pens with prefilled cartridges are also available and provide the added convenience of being ready to use. Insulin pens offer the advantages of more accurate dosing and easier administration, particularly for patients with visual or motor impairment.111,112 One disadvantage, however, is that basal and meal insulins that are being taken at the same time cannot be mixed with the pen and hence require separate injections. Nevertheless, insulin pens have become the delivery mode of choice for most patients using MDI therapy.
Many aspects of subcutaneous insulin administration can affect the absorption of the medication and hence its clinical efficacy. One key factor is the site of insulin injection. Insulin absorption varies inversely with the thickness of subcutaneous fat at the site of injection.113 Thus, insulin absorption is fastest from the subcutaneous fat of the abdominal wall, the preferred site of injection, and slower from other sites that have more subcutaneous fat such as the upper arm, anterior thigh, and buttocks.114 Another factor is depth of injection, as shallow insertion of the needle can lead to poorly absorbed intradermal delivery of insulin. Conversely, if one is injecting into a lean site with little subcutaneous fat, intramuscular injection may occur, which can be painful and can lead to rapid systemic absorption. After injection, alterations in subcutaneous blood flow may affect insulin absorption. For instance, after insulin injection into an extremity, physical exercise involving the affected limb can increase local blood flow and enhance insulin absorption.115 Other factors that can increase absorption include increased skin temperature and local massage.116,117
Alternative Routes of Insulin Delivery
Despite best efforts, the faithful imitation of normal physiologic insulin secretion generally cannot be achieved with the subcutaneous delivery of current insulin preparations. Factors that limit the success of current treatment approaches include the pharmacokinetics of exogenous insulin preparations following subcutaneous injection, the systemic dissemination of this injected insulin via the peripheral venous system (as opposed to normal pancreatic secretion into the portal venous system), and patient dissatisfaction with the demands and inconvenience of complex treatment regimens. Therefore, alternative routes for delivery of insulin have long been of interest. Options considered include nasal, pulmonary, oral, transdermal, and peritoneal delivery of insulin,118 although these attempts generally have met with limited success to date.
Intraperitoneal insulin delivery offers an approach that could simulate normal pancreatic secretion of insulin into the portal venous system. Indeed, intraperitoneal insulin infusion has been shown to reproduce a positive portosystemic insulin gradient and has been associated with a more rapid onset of action and reduced duration of activity compared with subcutaneous insulin.119 These findings have led to the development of implantable insulin pumps, that is, disk-shaped infusion systems that are implanted surgically in the abdominal subcutaneous tissue.120 The pump reservoir holds a 2 to 3 month supply of insulin and is connected to a free-moving peritoneal catheter. Rates of insulin delivery are adjusted by the patient, based on SMBG results. The patient uses an external programmer that sends radiowave signals to the electronic command unit of the pump to control the rate of insulin infusion. Studies to date suggest that implantable insulin pumps are associated with comparable A1c levels and a reduced incidence of severe hypoglycemia compared with subcutaneous insulin delivery.120,121 The main problem with implantable pumps is the potential for underdelivery of insulin due to catheter occlusion, an event that has been estimated to occur at a rate of 15 per 100 patient-years.122 Nevertheless, intraperitoneal delivery may be an option for insulin administration in the future.
Attempts to utilize other routes for insulin administration have been largely unsuccessful. Although inhaled insulin has attracted considerable interest in the past, concerns regarding delivery systems and long-term safety recently have led several manufacturers to withdraw from the development of this product. Oral insulin has not proved to be a viable option owing to enzymatic degradation of the insulin polypeptide in the gastrointestinal tract and poor absorption (<1% of oral dose).123 Strategies to overcome these problems have included the coadministration of enzyme inhibitors and attempts to improve the chemical stability of oral insulin through the use of liposomes and polymer-based systems.118 Similarly, intranasal insulin delivery has been hampered by low and unpredictable bioavailability that can fluctuate significantly with even minor changes in the nasal mucosa.124 Finally, transdermal insulin delivery has been unsuccessful owing to the relative impermeability of the skin to large, hydrophilic polypeptides such as insulin.118 Strategies that are being studied for improving transdermal drug delivery include iontophoresis, low-frequency ultrasound, and the use of drug carrier agents.
Complications of Insulin Therapy
The most feared complication of intensive insulin therapy is hypoglycemia. In the DCCT, the risk for hypoglycemia was approximately threefold higher in the intensive treatment group than in the conventional therapy arm.125 Because insulin analogues were not available at the time of the DCCT, it is not clear whether this finding reflects current practice. It is surprising that despite the high rates of hypoglycemia with intensive therapy in the DCCT, no evidence of dectectable long-term decline in cognitive function was noted in study participants after a mean 18 years of follow-up in the EDIC study.126 Nevertheless, hypoglycemia is an important factor that might limit the ability to achieve optimal glycemic control. Hypoglycemia generally reflects an imbalance between carbohydrate intake, physical activity, and the dose of exogenous insulin. In addition, patients with type 1 diabetes have a deficient counterregulatory response to hypoglycemia.127 Previous episodes of severe hypoglycemia and physiologic inability to detect the attendant symptoms (hypoglycemia unawareness) are other factors that are associated with increased hypoglycemic risk. Thus, a patient’s propensity for hypoglycemia must be considered in establishing therapeutic goals, as excessive hypoglycemia is best managed by raising glycemic targets. Similarly, in patients with hypoglycemia unawareness, raising glycemic targets is utilized as a means of restoring physiologic awareness of hypoglycemic symptoms.128 Specifically, with increased overall glycemia, the careful avoidance of further hypoglycemia can help to restore awareness and improve defective counterregulation.
Weight gain is another adverse effect that is associated with insulin therapy. In the DCCT, patients receiving intensive therapy gained 4.75 kg more than participants in the conventional treatment arm over the course of the study.129 This effect reflects both insulin’s anabolic properties and the decrease in glycosuria observed with improved glycemic control. Specifically, for patients with poor glycemic control, the elimination of glycosuria with insulin therapy will lead to a positive caloric balance and weight gain. In addition, intermittent hypoglycemia associated with intensive insulin therapy can lead to hunger and increased caloric intake. Concerns regarding weight gain may cause some patients to be reluctant to initiate intensive therapy and should be addressed by the health care team.
Rapid improvement in glycemic control following the initiation of intensive insulin therapy can cause transient exacerbation of underlying retinopathy.130 Patients with proliferative retinopathy and baseline A1c greater than 10% are at highest risk for this complication.131 Baseline ophthalmologic evaluation with frequent surveillance should be considered for such patients when intensive therapy is started.130 In addition, more gradual reduction in A1c may be prudent in such cases.
Allergic reactions to insulin are rare, particularly because human insulin is much less immunogenic than earlier animal insulins were.92 Most allergic reactions to insulin reflect local hypersensitivity at the site of injection. Generalized allergic reactions can occur rarely. Protocols for desensitization have been developed for both local and systemic hypersensitivity to insulin.92
Lipohypertrophy refers to localized swelling at a site of repeated insulin injection. This complication is related to the lipogenic effects of insulin. Continued injection of insulin into sites of lipohypertrophy is discouraged, as absorption of the insulin can be erratic, which may lead to unpredictable glycemic effects. Lipoatrophy reflects a different pathophysiology, in which an immune-mediated response to exogenous insulin leads to atrophy of subcutaneous tissue at the site of injection.132 With the current use of recombinant human insulin preparations and analogues, the problem of insulin-induced lipoatrophy is infrequent.
Adjustment of Insulin Therapy in Special Situations
Intercurrent Illness
Even with continued insulin therapy, patients with type 1 diabetes are at risk for developing ketoacidosis during times of illness owing to the effects of the counterregulatory hormones in antagonizing insulin action and promoting ketogenesis. Frequent testing of blood ketone levels using a home capillary blood ketone monitor is recommended at such times. Alternatively, if such testing is unavailable at home, then traditional urinary ketone monitoring should be performed (see the section entitled “Ketone Testing,” earlier in the chapter).
Nutrition
The nutritional recommendations associated with healthy eating habits for the general public are also applicable to patients with type 1 diabetes.133 The important difference in patients with type 1 diabetes is that dietary intake must be coordinated appropriately with insulin therapy and physical activity to achieve regulation of blood glucose levels. In particular, attention must be paid to the sources and amount of carbohydrate in the diet. Carbohydrate is the main dietary constituent that affects postprandial glycemic excursion and hence prandial insulin requirements.134 This concept has led to the practice of “carbohydrate counting.”
Carbohydrate counting is a meal-planning approach in which patients are taught to estimate the carbohydrate content of a meal preprandially and to adjust their dose of meal insulin accordingly.135 Most patients require 1 extra unit of insulin for each 10 to 15 g of carbohydrate ingested, although this ratio must be determined individually on the basis of personal food preferences and individual physiologic response. As such, carbohydrate counting is an integral component of patient education, and it is a skill that patients generally develop with the assistance of a clinical dietitian. Although the concept of carbohydrate counting was described shortly after the discovery of insulin,136 its recent resurgence is linked in part to its inclusion as one of the dietary interventions in the intensive treatment arm of the DCCT. In the DCCT, the practice of adjusting food and/or insulin dosage in response to hyperglycemia emerged as a dietary behavior associated with improved glycemic control.137 Moreover, carbohydrate counting allows for significant flexibility in food choices. Alternatively, for patients on fixed insulin doses who are unwilling to adjust prandial insulin dosage on the basis of dietary content, consistency of carbohydrate intake is advised.133
Another important consideration that affects food choices is hypoglycemia. For instance, patients must recognize that ingested alcohol may reduce hepatic glucose production and mask the symptoms of hypoglycemia. Indeed, moderate alcohol consumption (one to two standard drinks) 2 to 3 hours after the evening meal can lead to delayed hypoglycemia the next morning after breakfast.138 Conversely, patients are advised to avoid the overtreatment of hypoglycemia by excessive food intake, as this practice may lead to weight gain and significant glycemic excursion.17
Other Therapies for Type 1 Diabetes
The two most important alternative therapies currently available for type 1 diabetes are islet cell and pancreatic transplantation. These treatment modalities are discussed in detail in Chapter 24. A recent adjunctive therapy to be used in conjunction with insulin treatment is pre-meal subcutaneous injection of pramlintide, an analogue of the pancreatic β cell hormone amylin that is otherwise deficient in patients with type 1 diabetes. Amylin affects glucose homeostasis through multiple mechanisms, including slowing of gastric emptying, regulation of postprandial glucagon, and reduction in food intake.139 Pramlintide can be used in the treatment of type 1 diabetes in conjunction with insulin therapy (although it should be noted that pramlintide and insulin cannot be mixed in the same syringe). Overall, its effect on glycemic control is modest, but pramlintide may help to limit weight gain and reduce meal insulin requirements.140
Enhanced understanding of the natural history and immunogenetics of type 1 diabetes has led to the ability to identify individuals who are at high risk for developing this condition and recognition of the existence of a long, asymptomatic prediabetic period that might be amenable to intervention (see Chapter 14).141 Therefore, significant recent research interest has focused on the concept of prevention of type 1 diabetes in high-risk subjects who have been identified on the basis of genetic and antibody markers.142 Although several strategies have been studied, it is important to recognize that no intervention to date has proved to be consistently successful in preventing type 1 diabetes. Nevertheless, research efforts aimed at the prevention of type 1 diabetes are expected to increase in the coming years and are likely to ultimately yield novel therapeutic approaches.
Preventive strategies that have been considered thus far have aimed to prevent β cell loss in high-risk individuals either by modifying the immune response (immunomodulation) or by suppressing it (immunosuppression). Immunoregulatory strategies have included the administration of nicotinamide, insulin injections, and oral insulin to high-risk individuals.28,143 To date, however, large trials with these agents have not demonstrated efficacy. Support for an immunosuppressive approach was provided initially by the observation that drugs such as azathioprine and cyclosporine can temporarily decrease insulin requirements in patients with recently diagnosed type 1 diabetes.144,145 Unfortunately, the remission that these medications induce appears to be only temporary.146,147 More recently, studies using anti-CD3 monoclonal antibodies at the onset of type 1 diabetes have shown significant preservation of C-peptide secretion for at least 1 year.148,149 Nevertheless, the duration of this effect and the long-term safety of this therapy are important questions that remain to be addressed.
Future Perspectives
One such area is glucose sensing. Several approaches to noninvasive glucose sensing are currently being developed and could provide convenient, patient-friendly means for self-monitoring in the future.150 These initiatives utilize various technologies, including near-infrared (700 to 1300 nm) spectroscopy, in which an optical sensor determines glucose concentration based on the absorption pattern of light passing through body tissue, and reverse iontophoresis, in which a low-level electrical current is applied to the skin to generate a signal that is proportional to the interstitial fluid glucose concentration.120
Implantable intravenous glucose sensing devices could offer a more physiologic alternative for real-time blood glucose measurement. Although earlier prototypes were plagued by problems such as in situ thrombosis, recent intravenous sensors might hold promise for safe, reliable function.120 Such technology could ultimately lead to the successful development of the long-awaited artificial β cell. With such a system, an intravenous sensor would transmit real-time blood glucose measurements to a responsive intraperitoneal insulin pump. Indeed, the resultant closed-loop system theoretically could mimic physiologic insulin secretion by linking appropriate portal insulin secretion to real-time blood glucose levels. Such a system holds promise for achieving sustained euglycemia.
Similarly exciting is the concept of β cell bioengineering. The isolation of pluripotent pancreatic ductal epithelial stem cells and the identification of islet cell growth factors have raised the possibility of generating islet cells in vitro. Such efforts could provide an unlimited source of islet cells for transplantation. Alternatively, a gene therapy approach can be used to confer transgenic insulin secretory capacity to an appropriate target cell. For instance, this approach has been applied to create a liver-targeted insulin transgene driven by a glucose-responsive promoter in a rodent model of diabetes.151 Gene therapy could also be applied to reprogram liver stem cells into insulin-producing cells. In a murine model, adenovirus-mediated gene transfer of pancreatic and duodenal homeobox gene 1, a key developmental factor in pancreatic organogenesis, has been shown to reprogram hepatocytes to create a β cell phenotype.152 Although still many years away from clinical application, these initiatives might eventually lead to stable β cell regeneration and a potential cure for type 1 diabetes at some point in the future.
References
1. Banting, FG, Best, CH, Collip, JB, et al. Pancreatic extracts in the treatment of diabetes mellitus. Can Med Assoc J. 1922;12:141–146.
2. The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:977–986.
3. Reichard, P, Nilsson, B-Y, Rosenqvist, U. The effect of long-term intensified insulin treatment on the development of microvascular complications of diabetes mellitus. N Engl J Med. 1993;329:304–309.
4. Lauritzen, T, Rost-Larsen, K, Larsen, H-W, et al. Two-year experience with continuous subcutaneous insulin infusion in relation to retinopathy and neuropathy. Diabetes. 1985;34(Suppl 1):74–79.
5. Brinchmann-Hansen, O, Dahl-Jorgensen, K, Hanssen, KF, et al. The response of diabetic retinopathy to 41 months of multiple insulin injectins, insulin pumps, and conventional insulin therapy. Arch Ophthalmol. 1988;106:1242–1246.
6. Zinman, B. The physiologic replacement of insulin. N Engl J Med. 1989;321:363–370.
7. Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. N Engl J Med. 2000;342:381–389.
8. Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. Sustained effect of intensive treatment of type 1 diabetes mellitus on development and progression of diabetic nephropathy. JAMA. 2003;290:2159–2167.
9. Nathan, DM, Cleary, PA, Backlund, JY, et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. 2005;353:2643–2653.
10. Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. Intensive therapy and carotid intima-media thickness in type 1 diabetes mellitus. N Engl J Med. 2003;348(23):2294–2303.
11. Cleary, PA, Orchard, TJ, Genuth, S, et al. The effect of intensive glycemic treatment on coronary artery calcification in type 1 diabetic participants of the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study. Diabetes. 2006;55:3556–3565.
12. Retnakaran, R, Zinman, B. Type 1 diabetes, hyperglycaemia, and the heart. Lancet. 2008;371:1790–1799.
13. The Diabetes Control and Complications Trial Research Group. The relationship of glycemic exposure (HbA1c) to the risk of development and progression of retinopathy in the Diabetes Control and Complications Trial. Diabetes. 1995;44(8):968–983.
14. American Diabetes Association. Standards of medical care in diabetes. Diabetes Care. 2008;31(Suppl 1):S12–S54.
15. Canadian Diabetes Association Clinical Practice Guidelines Expert Committee. Targets for glycemic control. Can J Diabetes. 2008;32(Suppl 1):S29–S31.
16. American Association of Clinical Endocrinologists (AACE). medical guidelines for clinical practice for the management of diabetes mellitus. Endocr Pract. (Suppl May/June):2007.
17. Canadian Diabetes Association Clinical Practice Guidelines Expert Committee. Hypoglycemia. Can J Diabetes. 2008;32(Suppl 1):S62–S64.
18. Slama, G, Traynard, P–Y, Desplanque, N, et al. The search for an optimized treatment of hypoglycemia. Carbohydrates in tablets, solution, or gel for the correction of insulin reactions. Arch Intern Med. 1990;150:589–593.
19. The Diabetes Control and Complications Trial Research Group. Reliability and validity of a diabetes quality-of-life measure for the Diabetes Control and Complications Trial. Diabetes Care. 1988;11:725–732.
20. The Diabetes Control and Complications Trial Research Group. Implementation of treatment protocols in the Diabetes Control and Complications Trial. Diabetes Care. 1995;18(3):361–376.
21. American Diabetes Association. Test of glycemia in diabetes. Diabetes Care. 2004;27(Suppl 1):S91–S93.
22. Reynolds, LR, Karounos, DG. Emerging technology in diabetes mellitus: Glucose monitoring and new insulins. South Med J. 2002;95(8):914–918.
23. Ellison, JM, Stegmann, JM, Colner, SL, et al. Rapid changes in postprandial blood glucose produce concentration differences at finger, forearm, and thigh sampling sites. Diabetes Care. 2002;25(6):961–964.
24. Bina, DM, Anderson, RL, Johnson, ML, et al. Clinical impact of prandial state, exercise and site preparation on the equivalence of alternative-site blood glucose testing. Diabetes Care. 2003;26(4):981–985.
25. Rebrin, K, Steil, GM, van Antwerp, WP, et al. Subcutaneous glucose predicts plasma glucose independent of insulin: Implications for continuous monitoring. Am J Physiol. 1999;277:E561–E571.
26. Mastrototaro, JJ. The MiniMed Continuous Glucose Monitoring System (CGMS). J Pediatr Endocrinol Metab. 1999;12(Suppl 3):S751–S758.
27. Cheyne, E, Kerr, D. Making “sense” of diabetes: Using a continuous glucose sensor in clinical practice. Diabetes Metab Res Rev. 2002;18(Suppl 1):S43–S48.
28. Eisenbarth, GS. Update in type 1 diabetes. J Clin Endocrinol Metab. 2007;92:2403–2407.
29. Metzger, M, Leibowitz, G, Wainstein, J, et al. Reproducibility of glucose measurements using the glucose sensor. Diabetes Care. 2002;25:1185–1191.
30. Reach, G. Continuous glucose monitoring and diabetes health outcomes: a critical appraisal. Diabetes Technology & Therapeutics. 2008;10:69–80.
31. Chico, A, Vidal-Rios, P, Subira, M, et al. The continuous glucose monitoring system is useful for detecting unrecognized hypoglycemias in patients with type 1 and type 2 diabetes but is not better than frequent capillary glucose measurements for improving metabolic control. Diabetes Care. 2003;26(4):1153–1157.
32. Kaufman, FR, Austin, J, Neinstein, A, et al. Nocturnal hypoglycemia detected with the Continuous Glucose Monitoring System in pediatric patients with type 1 diabetes. J Pediatr. 2002;141(5):625–630.
33. Tanenberg, R, Bode, B, Lane, W, et al. Use of the Continuous Glucose Monitoring System to guide therapy in patients with insulin-treated diabetes: a randomized controlled trial. Mayo Clin Proc. 2004;79(12):1521–1526.
34. Garg, SK, Schwartz, S, Edelman, SV. Improved glucose excursions using an implantable real-time continuous glucose sensor in adults with type 1 diabetes. Diabetes Care. 2004;27(3):734–738.
35. Garg, S, Zisser, H, Schwartz, S, et al. Improvement in glycemic excursions with a transcutaneous, real-time continuous glucose sensor: a randomized controlled trial. Diabetes Care. 2006;29(1):44–50.
36. Ludvigsson, J, Hanas, R. Continuous subcutaneous glucose monitoring improved metabolic control in pediatric patients with type 1 diabetes: a controlled crossover study. Pediatrics. 2003;111(5 Pt 1):933–938.
37. Deiss, D, Bolinder, J, Riveline, JP, et al. Improved glycemic control in poorly controlled patients with type 1 diabetes using real-time continuous glucose monitoring. Diabetes Care. 2006;29(12):2730–2732.
38. Bunn, HF, Haney, DN, Gabbay, KH, et al. Further identification of the nature and linkage of the carbohydrate in hemoglobin A1c. Biochem Biophys Res Commun. 1975;67:103–109.
39. Nathan, DM, Singer, DE, Hurxthal, K, et al. The clinical information value of the glycosylated hemoglobin assay. N Engl J Med. 1984;310:341–346.
40. Little, RR, Wiedmeyer, HM, England, JD, et al. Interlaboratory comparison of glycohemoglobin results: College of American Pathologists survey data. Clin Chem. 1991;37:1725–1729.
41. Little, RR, Rohlfing, CL, Widemeyer, HM, et al. The National Glycohemoglobin Standardization Program (NGSP): A five-year progress report. Clin Chem. 2001;47:1985–1992.
42. Rohlfing, CL, Wiedmeyer, HM, Little, RR, et al. Defining the relationship between plasma glucose and HbA1c: Analysis of glucose profiles and HbA1c in the Diabetes Control and Complication Trial. Diabetes Care. 2002;25:275–278.
43. Nathan, DM, Kuenen, J, Borg, R, et al. A1c-Derived Average Glucose Study Group. Translating the A1C assay into estimated average glucose values. Diabetes Care. 2008;31(8):1473–1478.
44. Panzer, S, Kronik, G, Lechner, K, et al. Glycosylated hemoglobins (GHb): An index of red cell survival. Blood. 1982;59:1348–1350.
45. Paisey, R, Banks, R, Holton, R, et al. Glycosylated haemoglobin in uremia. Diabet Med. 1986;3:445–448.
46. Armbruster, DA. Fructosamine: Structure, analysis and clinical usefulness. Clin Chem. 1987;33:2153–2163.
47. Baker, JR, Mf, PA, Holdaway, IM, et al. Serum fructosamine concentration as a measure of blood glucose control in type 1 (insulin-dependent) diabetes mellitus. Br Med J. 1985;290:352–355.
48. American Diabetes Association. Test of glycemia in diabetes. Diabetes Care. 2004;27(Suppl 1):S91–S93.
49. Howey, JE, Bennet, WM, Browning, MC, et al. Clinical utility of assays of glycosylated hemoglobin and serum fructosamine compared: Use of data on biologic variation. Diabet Med. 1989;6:793–796.
50. Byrne, HA, Tieszen, KL, Hollis, S, et al. Evaluation of an electrochemical sensor for measuring blood ketones. Diabetes Care. 2000;23:500–503.
51. Guerci, B, Benichout, M, Floriot, M, et al. Accuracy of an electrochemical sensor for measuring capillary blood ketones by fingerstick samples during metabolic deterioration after continuous subcutaneous insulin infusion interruption in type 1 diabetic patients. Diabetes Care. 2003;26(4):1137–1141.
52. Wallace, TM, Meston, NM, Gardner, SG, et al. The hospital and home-use of a 30 second hand-held blood ketone meter: Guidelines for clinical practice. Diabet Med. 2001;18(8):640–645.
53. Eknoyan, G, Hostetter, T, Bakris, GL, et al. Proteinuria and other markers of chronic kidney disease: A position statement of the National Kidney Foundation (NKF) and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). Am J Kidney Dis. 2003;42(4):617–622.
54. Perkins, BA, Olaleye, D, Zinman, B, et al. Simple screening tests for peripheral neuropathy in the diabetes clinic. Diabetes Care. 2001;24(2):250–256.
55. Owens, DR, Zinman, B, Bolli, GB. Insulins today and beyond. Lancet. 2001;358:739–746.
56. Shah, BR, Zinman, B. Insulin regimens for type 1 diabetes. In: Sperling MA, ed. Type 1 Diabetes: Etiology and Treatment. Totowa, NJ: Humana Press; 2003:199–214.
57. Zinman, B, Murray, FT, Vranic, M, et al. Glucoregulation during moderate exercise in insulin treated diabetes. J Clin Endocrinol Metab. 1977;45:641–652.
58. Mitchell, TH, Abraham, G, Schiffrin, A, et al. Hyperglycemia after intense exercise in IDDM subjects during continuous subcutaneous insulin infusion. Diabetes Care. 1988;11:311–317.
59. Chance, RE, Kroeff, EP, Hoffmann, JA, et al. Chemical, physical and biologic properties of biosynthetic human insulin. Diabetes Care. 1981;4:147–154.
60. Skyler, JS. Symposium on human insulin of recombinant DNA origin. Diabetes Care. 1982;5(Suppl 2):1–186.
61. Kang, S, Brange, J, Burch, A, et al. Subcutaneous insulin absorption explained by insulin’s physicochemical properties: Evidence from absorption studies of soluble human insulin and insulin analogues in humans. Diabetes Care. 1991;14:942–948.
62. Heinemann, L, Richter, B. Clinical pharmacology of human insulin. Diabetes Care. 1993;16(Suppl 3):90–100.
63. Krayenbuhl, C, Rosenberg, T. Crystalline protamine insulin. Rep Steno Hosp. 1946;1:60–73.
64. Barnett, AH. A review of basal insulins. Diabet Med. 2003;20:873–885.
65. Lauritzen, T, Faber, OK, Binder, C. Variation in 125 I-insulin absorption and blood glucose conception. Diabetologia. 1979;17:291–295.
66. Zinman, B, Ross, S, Campos, R, et al. Effectiveness of human ultralente versus NPH insulin in providing basal insulin replacement for an insulin lispro multiple daily injection regimen. Diabetes Care. 1999;22:603–608.
67. Holleman, F, Hoekstra, JBL. Insulin lispro. New Engl J Med. 1997;337:176–183.
68. Brems, DN, Alter, LA, Beckage, MJ, et al. Altering the association properties of insulin by amino acid replacement. Protein Eng. 1992;5:527–532.
69. Howey, DC, Bowsher, RR, Brunelle, RF, et al. [Lys(B28), Pro(B29)]-human insulin: A rapidly absorbed analogue of human insulin. Diabetes. 1994;43:396–402.
70. Lee, W, Zinman, B. From insulin to insulin analogs: Progress in the treatment of type 1 diabetes. Diabetes Rev. 1998;6:73–88.
71. Mudaliar, SR, Lindberg, FA, Joyce, M, et al. Insulin aspart (B28 Asp-insulin): A fast-acting analog of human insulin. Diabetes Care. 1999;22:1501–1506.
72. Becker, RH, Frick, AD. Clinical pharmacokinetics and pharmacodynamics of insulin glulisine. Clin Pharmacokinet. 2008;47(1):7–20.
73. Cheng, AY, Zinman, B. Insulin analogues and the treatment of diabetes. In: Raz I, Skyler JS, Shafrir E, eds. Diabetes: From Research to Diagnosis and Treatment. London: Martin Dunitz Ltd; 2002:331–346.
74. Brunelle, RL, Llewelyn, J, Anderson, JH, et al. Meta-analysis of the effect of insulin lispro on severe hypoglycemia in paitents with type 1 diabetes. Diabetes Care. 1998;21:1726–1731.
75. Anderson, JH, Brunelle, RL, Koivisto, VA, et al. Reduction of postprandial hyperglycemia and frequency of hypoglycemia in IDDM patients on insulin-analog treatment. Diabetes. 1997;46:265–270.
76. Raskin, P, Guthrie, RA, Leiter, L, et al. Use of insulin aspart, a fast-acting insulin analog, as the mealtime insulin in the management of patients with type 1 diabetes. Diabetes Care. 2000;23:583–588.
77. Bolli, GB, Owens, DR. Insulin glargine. Lancet. 2000;356:443–444.
78. Buse, J. Insulin glargine (HOE901). Diabetes Care. 2000;23:576–578.
79. Heinemann, L, Linkeschova, R, Rave, K, et al. Time-action profile of the long-acting insulin analog insulin glargine (HOE901) in comparison with those of NPH insulin and placebo. Diabetes Care. 2000;23:644–649.
80. Owens, DR, Coates, PA, Luzio, SD, et al. Pharmacokinetics of 125I-labelled insulin glargine (HOE901) in healthy men: Comparison with NPH insulin and the influence of different subcutaneous injection sites. Diabetes Care. 2000;23:813–819.
81. Lepore, M, Pampanelli, S, Fanelli, C, et al. Pharmacokinetics and pharmacodynamics of subcutaneous injection of long-acting insulin analog glargine, NPH insulin, and ultralente human insulin and continuous subcutaneous infusion of insulin lispro. Diabetes. 2000;49:2142–2148.
82. Ratner, RE, Hirsch, IB, Neifing, JL, et al. Less hypoglycemia with insulin glargine in intensive therapy for type 1 diabetes. Diabetes Care. 2000;23:639–643.
83. Raskin, P, Klaff, L, Berenstal, R, et al. A 16-week comparison of the novel insulin analog insulin glargine (HOE 901) and NPH human insulin used with lispro in patients with type 1 diabetes. Diabetes Care. 2000;23:1666–1671.
84. Rosenstock, J, Park, G. Zimmerman J for the US Insulin Glargine (HOE 901) Type 1 Diabetes Investigator Group: Basal insulin glargine (HOE 901) versus NPH insulin in patients with type 1 diabetes on multiple daily insulin regimens. Diabetes Care. 2000;23:1137–1142.
85. Pieber, TR, Eugene-Jolchine, I, Derobert, E. Efficacy and safety of HOE 901 versus NPH insulin in patients with type 1 diabetes. Diabetes Care. 2000;23:157–162.
86. Markussen, J, Havelund, S, Kurtzhals, P, et al. Soluble, fatty acid acylated insulins bind to albumin and shown protracted action in pigs. Diabetologia. 1996;39:281–288.
87. Heinemann, L, Sinha, K, Weyer, C, et al. Time-action profile of the soluble, fatty acid acylated, long-acting insulin analogue NN304. Diabet Med. 1999;16:332–338.
88. Hermansen, K, Madsbad, S, Perrild, H, et al. Comparison of the soluble basal insulin analog insulin detemir with NPH insulin. Diabetes Care. 2001;24:296–301.
89. Vague, P, Selam, JL, Skeie, S, et al. Insulin detemir is associated with more predictable glycemic control and reduced risk of hypoglycemia than NPH insulin in patients with type 1 diabetes on a basal-bolus regimen with premeal insulin aspart. Diabetes Care. 2003;26:590–596.
90. Danne, T, Lupke, K, Walte, K, et al. Insulin detemir is characterized by a consistent pharmacokinetic profile across age-groups in children, adolescents, and adults with type 1 diabetes. Diabetes Care. 2003;26:3087–3092.
91. Hermansen, K, Davies, M. Does insulin detemir have a role in reducing risk of insulin-associated weight gain? Diabetes Obes Metab. 2007;9(3):209–217.
92. Cheng, AY, Zinman, B. Insulin for treating type 1 and type 2 diabetes. In: Gerstein HC, Haynes RB, eds. Evidence-Based Diabetes Care. Hamilton, Ontario: BC Decker; 2001:323–343.
93. Hamann, A, Matthaei, S, Rosak, C, et al. HOE901/4007 Study Group: A randomized clinical trial comparing breakfast, dinner or bedtime administration of insulin glargine in patients with type 1 diabetes. Diabetes Care. 2003;26(6):1738–1744.
94. Rossetti, P, Pampanelli, S, Fanelli, C, et al. Intensive replacement of basal insulin in patients with type 1 diabetes given rapid-acting insulin analog at mealtime: A 3-month comparison between administration of NPH insulin four times daily and glargine insulin at dinner or bedtime. Diabetes Care. 2003;26(5):1490–1496.
95. Bartley, PC, Bogoev, M, Larsen, J, et al. Long-term efficacy and safety of insulin detemir compared to Neutral Protamine Hagedorn insulin in patients with Type 1 diabetes using a treat-to-target basal-bolus regimen with insulin aspart at meals: a 2-year, randomized, controlled trial. Diabet Med. 2008;25(4):442–449.
96. Pickup, J, Keen, H. Continuous subcutaneous insulin infusion at 25 years: Evidence base for the expanding use of insulin pump therapy in type 1 diabetes. Diabetes Care. 2002;25:593–598.
97. Zinman, B, Tildesley, H, Chiasson, JL, et al. Insulin lispro in CSII: Results of a double-blind crossover study. Diabetes. 1997;46:440–443.
98. Melki, V, Renard, E, Lassmann-Vague, V, et al. Improvement of HbA1c and blood glucose stability in IDDM patients treated with lispro insulin analog in external pumps. Diabetes Care. 1998;21:977–982.
99. Renner, R, Pfutzner, A, Trautmann, M, et al. Use of insulin lispro in continuous subcutaneous insulin infusion treatment: results of a multicenter trial: German Humalog-CSII Study Group. Diabetes Care. 1999;22:784–788.
100. Lenhard, MJ. Continuous subcutaneous insulin infusion: A comprehensive review of insulin pump therapy. Arch Intern Med. 2001;161(19):2293–2300.
101. Zinman, B. Insulin pump therapy and rapid acting insulin: What have we learned? Int J Clin Pract Suppl. 2001;123:47–50.
102. Pickup, J, Mattock, M, Kerry, S. Glycaemic control with continuous subcutaneous insulin infusion compared with intensive insulin injections in patients with type 1 diabetes: Meta-analysis of randomized controlled trials. Br Med J. 2002;324:1–6.
103. Tsui, E, Barnie, A, Ross, S, et al. Intensive insulin therapy with insulin lispro: A randomized trial of continuous subcutaneous insulin infusion versus multiple daily insulin injection. Diabetes Care. 2001;24:1722–1727.
104. DeVries, JH, Snoek, FS, Kostense, PJ, et al. A randomized trial of continuous subcutaneous insulin infusion and intensive injection therapy in type 1 diabetes for patients with long-standing poor glycemic control. Diabetes Care. 2002;25:2074–2080.
105. Hanaire-Broutin, H, Melki, V, Bessieres-Lacombe, S, et al. Comparison of continuous subcutaneous insulin infusion and multiple daily injection regimens using insulin lispro in type 1 diabetic patients on intensified treatment: A randomized study. Diabetes Care. 2000;23:1232–1235.
106. Hoogma, RP, Hammond, PJ, Gomis, R, et al. Comparison of the effects of continuous subcutaneous insulin infusion (CSII) and NPH-based multiple daily insulin injections (MDI) on glycaemic control and quality of life: results of the 5-nations trial. Diabet Med. 2006;23(2):141–147.
107. Hirsch, IB, Bode, BW, Garg, S, et al. Continuous subcutaneous insulin infusion (CSII) of insulin aspart versus multiple daily injection of insulin aspart/insulin glargine in type 1 diabetic patients previously treated with CSII. Diabetes Care. 2005;28(3):533–538.
108. Retnakaran, R, Hochman, J, DeVries, JH, et al. Continuous subcutaneous insulin infusion versus multiple daily injections: the impact of baseline A1c. Diabetes Care. 2004;27(11):2590–2596.
109. Retnakaran, R, DeVries, JH, Hanaire-Broutin, H, et al. Continuous subcutaneous insulin infusion versus multiple daily injections: modeling predicted benefits in relationship to baseline A1c. Diabetes Care. 2005;28(7):1835–1836.
110. Rabasa-Lhoret, R, Bourque, J, Ducros, F, et al. Guidelines for premeal insulin dose reduction for postprandial exercise of different intensities and durations in type 1 diabetic subjects treated intensively with a basal-bolus insulin regimen (ultralente-lispro). Diabetes Care. 2001;24(4):625–630.
111. Lteif, AN, Schwenk, WF. Accuracy of pen injectors versus insulin syringes in children with type 1 diabetes. Diabetes Care. 1999;22:137–140.
112. Graff, MR, McClanahan, MA. Assessment by patients with diabetes mellitus of two insulin pen delivery systems versus a vial and syringe. Clin Ther. 1998;20:486–496.
113. Sindelka, G, Heinemann, L, Berger, M, et al. Effect of insulin concentration, subcutaneous fat thickness and skin temperature on subcutaneous insulin absorption in healthy subjects. Diabetologia. 1994;37:377.
114. Koivisto, VA, Felig, P. Alterations in insulin absorption and in blood glucose control associated with varying insulin injection sites in diabetic patients. Ann Intern Med. 1980;92:59.
115. Koivisto, VA, Felig, P. Effects of leg exercise on insulin absorption in diabetic patients. N Engl J Med. 1978;298:79–83.
116. Koivisto, VA. Sauna-induced acceleration in insulin absorption from subcutaneous injection site. Br Med J. 1980;280:1411.
117. Linde, B. Dissociation of insulin absorption and blood flow during massage of a subcutaneous injection site. Diabetes Care. 1986;9:570.
118. Owens, DR, Zinman, B, Bolli, G. Alternative routes of insulin delivery. Diabet Med. 2003;20:886–898.
119. Nelson, JA, Stephen, R, Landau, ST, et al. Intraperitoneal insulin administration produces a positive portal-systemic blood insulin gradient in unanesthetized, unrestrained swine. Metabolism. 1982;31:969–972.
120. Renard, E. Implantable closed-loop glucose-sensing and insulin delivery: The future for insulin pump therapy. Curr Opin Pharmacol. 2002;2:708–716.
121. Gin, H, Renard, E, Melki, V, et al. Combined improvements in implantable pump technology and insulin stability allow safe and effective long term intraperitoneal insulin delivery in type 1 diabetic patients: The EVADIAC experience. Diabetes Metab. 2003;29(6):602–607.
122. Selam, JL. External and implantable insulin pumps: Current place in the treatment of diabetes. Exp Clin Endocrinol Diabetes. 2001;109(Suppl 2):S333–S340.
123. Carino, GP, Mathiowitz, E. Oral insulin delivery. Adv Drug Deliv Rev. 1999;35:249–257.
124. Saudek, CD. Novel forms of insulin delivery. Endocrinol Metab Clin North Am. 1997;26:599–610.
125. The Diabetes Control and Complications Trial Research Group. Adverse events and their association with treatment regimens in the Diabetes Control and Complications Trial. Diabetes Care. 1995;18:1415–1427.
126. Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Study Research GroupJacobson, AM, Musen, G, Ryan, CM, et al. Long-term effect of diabetes and its treatment on cognitive function. N Engl J Med. 2007;356(18):1842–1852.
127. Cryer, PE. Managing diabetes: Lessons from type 1 diabetes mellitus. Diabet Med. 1998;15(Suppl 4):S8–S12.
128. Bolli, GB, Pampanelli, S, Porcellati, F, et al. Recovery and prevention of hypoglycemia unawareness in type 1 diabetes mellitus. Diabetes Nutr Metab. 2002;15(6):402–409.
129. The Diabetes Control and Complications Trial Research Group. Influence of intensive diabetes treatment on body weight and composition of adults with type 1 diabetes in the Diabetes Control and Complications Trial. Diabetes Care. 2001;24(10):1711–1721.
130. DeWitt, DE, Hirsch, IB. Outpatient insulin therapy in type 1 and type 2 diabetes mellitus. JAMA. 2003;289:2254–2264.
131. Chantelau, E, Kohmer, EM. Why some cases of retinopathy worsen when diabetic control improves. Br Med J. 1997;315:1105–1106.
132. Reeves, W, Allen, B, Tatersall, R. Insulin-induced lipoatrophy: Evidence for an immune pathogenesis. Br Med J. 1980;280:1500.
133. American Diabetes Association. Nutrition recommendations and interventions for diabetes. Diabetes Care. 2008;31(Suppl 1):S61–S78.
134. Nuttal, FQ. Carbohydrate and dietary management of clients with insulin-requiring diabetes. Diabetes Care. 1993;16:1039–1042.
135. Gillespie, SJ, Kulkarni, KD, Daly, AE. Using carbohydrate counting in diabetes clinical practice. J Am Diet Assoc. 1998;98:897–905.
136. Joslin, EP. The diabetic diet. J Am Diet Assoc. 1927;3:89–92.
137. Delahanty, LM, Halford, BN. The role of diet behaviours in achieving improved glycemic control in intensively treated patients in the Diabetic Control and Complications Trial. Diabetes Care. 1993;16:1453–1458.
138. Turner, BC, Jenkins, E, Kerr, D, et al. The effect of evening alcohol consumption on next-morning glucose control in type 1 diabetes. Diabetes Care. 2001;24:1888–1893.
139. Pullman, J, Darsow, T, Frias, JP. Pramlintide in the management of insulin-using patients with type 2 and type 1 diabetes. Vasc Health Risk Manag. 2006;2(3):203–212.
140. Marrero, DG, Crean, J, Zhang, B, et al. Effect of adjunctive pramlintide treatment on treatment satisfaction in patients with type 1 diabetes. Diabetes Care. 2007;30(2):210–216.
141. Atkinson, MA, Eisenbarth, GS. Type 1 diabetes: New perspectives on disease pathogenesis and treatment. Lancet. 2001;358:221–229.
142. Thivolet, C. New therapeutic approaches to type 1 diabetes: From prevention to cellular or gene therapies. Clin Endocrinol. 2001;55:565–574.
143. Diabetes Prevention Trial/Type 1 Diabetes Study Group. Effects of insulin in relatives of patients with type 1 diabetes mellitus. N Engl J Med. 2002;346(22):1685–1691.
144. Assan, R, Feutren, G, Debray-Sachs, M, et al. Metabolic and immunological effects of cyclosporin in recently-diagnosed type 1 diabetes mellitus. Lancet. 1985;1:67–71.
145. Silverstein, J, Maclean, N, Riley, W, et al. Immunosuppression with azathioprine and prednisone in recent-onset insulin-dependent diabetes mellitus. N Engl J Med. 1988;319:599.
146. Carel, JC, Boitard, C, Eisenbarth, G, et al. Cyclosporine delays but does not prevent clinical onset in glucose intolerant pre-type 1 diabetic children. J Autoimmun. 1996;9:739–745.
147. Cook, JJ, Hudson, I, Harrison, LC, et al. A double-blind controlled trial of azathioprine in children with newly-diagnosed type 1 diabetes. Diabetes. 1989;38:779.
148. Herold, KC, Gitelman, SE, Masharani, U, et al. A single course of anti-CD3 monoclonal antibody hOKT3gamma1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes. 2005;54(6):1763–1769.
149. Keymeulen, B, Vandemeulebroucke, E, Ziegler, AG, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med. 2005;352(25):2598–2608.
150. Rohrscheib, M, Robinson, R, Eaton, RP. Non-invasive glucose sensors and improved informatics: The future of diabetes management. Diabetes Obes Metab. 2003;5:280–284.
151. Thule, PM, Liu, JM. Regulated hepatic insulin gene therapy of STZ-diabetic rats. Gene Therapy. 2000;7:1744–1752.
152. Ferber, S, Halkin, A, Cohen, H, et al. Pancreatic and duodenal homeobox gene 1 induces expression of insulin genes I liver and ameliorates streptozotocin-induced hyperglycemia. Nat Med. 2000;6:568–572.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 19
Treatment of Type 1 Diabetes Mellitus in Adults
The discovery of insulin in the summer of 1921 by Frederick Banting, Charles Best, James Collip, and J. J. R. Macleod at the University of Toronto and its first therapeutic use on January 11, 1922, at the Toronto General Hospital, stands as one of the greatest achievements of contemporary medicine.1 By providing life-sustaining therapy for a previously fatal wasting condition, the discovery of insulin dramatically shifted the prognosis of type 1 diabetes to that of a chronic disease characterized by devastating long-term microvascular and macrovascular complications. Accordingly, the focus of treatment has changed significantly since 1922, with emphasis now directed toward the avoidance of complications and the optimization of patient quality of life. Given the incontrovertible evidence from the Diabetes Control and Complications Trial (DCCT) and other studies that optimization of glycemic control can significantly reduce the risk of vascular complications,2–5 the following overriding objective of modern diabetes management has emerged: the achievement of sustained euglycemia, with avoidance of hypoglycemia, in patients with type 1 diabetes through the physiologic replacement of insulin. Remarkably, however, despite significant advances in the past 80 years, this goal remains an elusive target.6 Modern management of type 1 diabetes involves a multifaceted, patient-centered approach, in which an interdisciplinary health care team works closely with the patient toward the goal of achieving sustained euglycemia. In this chapter, we review the essential elements of this approach to comprehensive care of patients with type 1 diabetes.
Goals of Management
The overriding goal in the management of type 1 diabetes is the optimization of the patient’s quality of life through the prevention and amelioration of the acute and chronic complications that are associated with this disease. In the modern era, a substantial body of epidemiologic and experimental evidence has established the relationship between diabetic complications and hyperglycemia (see Chapter 25). In particular, the findings of the DCCT have had an enormous impact on the management of type 1 diabetes and warrant review before discussion of specific therapeutic goals.
The DCCT was a multicenter, National Institutes of Health–funded randomized controlled clinical trial initiated in 1982 that was designed to assess whether intensive treatment with the goal of maintaining near-normal blood glucose concentrations could reduce the frequency and severity of diabetic complications.2 A total of 1441 patients with type 1 diabetes from the United States and Canada participated in the study and made up two cohorts: a primary prevention cohort of 726 subjects with no retinopathy at baseline and 1 to 5 years’ duration of diabetes, and a secondary intervention cohort of 715 individuals with mild retinopathy and duration of diabetes of up to 15 years. Participants were randomly assigned to receive either the conventional therapy of the day (one or two injections of insulin per day) or intensive therapy (either three or more insulin injections or continuous subcutaneous insulin infusion by external pump, with frequent blood glucose monitoring). After a mean 6.5 years of follow-up, mean glycosylated hemoglobin (A1c) was 7.2% in the intensive therapy arm and 9.0% in the conventional treatment arm. The intensive therapy group showed a relative risk reduction of 76% (95% confidence interval [CI]: 62% to 85%) for the development of retinopathy and a 54% relative risk reduction (95% CI: 39% to 66%) for progression of retinopathy. Similarly, intensive therapy was associated with relative risk reductions of 39% (95% CI: 21% to 52%) for the development of microalbuminuria (defined as urinary albumin excretion > 40 mg per 24 hours) and 54% (95% CI: 19% to 74%) for progression to albuminuria (urinary albumin excretion > 300 mg per 24 hours). The incidence of clinical neuropathy was also decreased in the intensive group, with a relative risk reduction of 60% (95% CI: 38% to 74%). Thus, the DCCT provided conclusive evidence that improved glycemic control with intensive therapy can reduce the incidence of microvascular complications (primary prevention) and can slow the progression of established microvascular disease (secondary intervention).
The benefits of near-normal glycemic control were found to persist for many years after the DCCT, according to data from the follow-up study, the Epidemiology of Diabetes Interventions and Complications (EDIC).7 In the ongoing EDIC observational study, a large cohort consisting of more than 95% of the DCCT participants (n = 1375) has been followed since the completion of the original trial and has been studied by using an intention-to-treat approach based on the original randomization in the DCCT. The mean A1c levels of the former intensive treatment cohort and the former conventional therapy group have converged (A1c ≈ 8%) over the course of the EDIC study and were no longer significantly different at 5 years post DCCT. Remarkably, however, despite similar glycemic control, the former intensive therapy group continued to display reduced risk for both retinopathy and nephropathy at 4 years and 7 to 8 years post DCCT, respectively, compared with the group of patients who were randomized originally to conventional therapy (Fig. 19-1).7,8 These persistent beneficial effects of previous near-normal glycemic control extend the results of the DCCT and demonstrate that intensive treatment should be initiated as early as is safely possible in the management of type 1 diabetes.
FIGURE 19-1 Cumulative incidence of further progression of retinopathy during the Epidemiology of Diabetes Interventions and Complications (EDIC) study in the former conventional therapy and intensive therapy cohorts from the Diabetes Control and Complications Trial (DCCT). Data are based on regression analysis adjusted for the level of retinopathy at the end of the DCCT, whether patients received therapy as primary prevention or as secondary intervention, and for the duration of diabetes and the A1c value on enrollment in the DCCT. (From DCCT/EDIC Research Group: Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. N Engl J Med 342[5]:387, 2000.)
Although the DCCT conclusively demonstrated that intensive therapy can reduce the risk for microvascular complications, the effect on macrovascular disease was initially less clear. By the end of the DCCT, the intensive therapy arm displayed a relative risk reduction of 34% (95% CI: 7% to 54%) for the development of elevated low-density lipoprotein (LDL) cholesterol levels. Nevertheless, even though intensive therapy was associated with a 41% relative risk reduction (95% CI: −10% to 68%) for macrovascular complications, this difference was not statistically significant.2 It is important to note, however, that the macrovascular benefits derived from near-normal glycemic control in the former intensive treatment arm of the DCCT have emerged over time in the EDIC study. Indeed, after mean follow-up of 17 years, it was observed that intensive therapy during the DCCT had reduced the subsequent risk for any cardiovascular disease (CVD) event by 42% (95% CI: 9% to 63%; P = .02) and the risk for nonfatal myocardial infarction (MI), stroke, or cardiovascular death by 57% (95% CI: 12% to 79%; P = .02) (Fig. 19-2).9 This remarkable risk reduction was largely explained by differences in the updated mean A1c during the DCCT. Furthermore, the reduction in CVD events was consistent with earlier observations that intensive therapy had reduced the progression of atherosclerosis (as measured by carotid artery intima media thickness [IMT]) and the prevalence of coronary artery calcification (CAC) during the DCCT/EDIC study, with both effects associated with the reduction in mean A1c achieved with intensive therapy.10,11 Thus, intensive therapy (with associated reduced glycemic exposure) during the mean 6.5 years of the DCCT first significantly reduced surrogate measures of atherosclerotic disease (carotid IMT, CAC) and later reduced clinical CVD events over the course of the EDIC study (Fig. 19-3). These data demonstrate that the long-term risk associated with past glycemic exposure extends to both microvascular and macrovascular disease.12 Indeed, it has been hypothesized that glycemic exposure early in the course of the disease may have an effect of metabolic imprinting (referred to as “metabolic memory”) that contributes to future risk for vascular complications.9 Taken together, these data further underscore the importance of initiating intensive therapy as early as safely possible in the management of type 1 diabetes.
FIGURE 19-2 Differences in A1c (lower panel) and cardiovascular event rates (upper panel) over time between the intensive and conventional diabetes treatment arms of the Diabetes Control and Complications Trial (DCCT)/Epidemiology of Diabetes Interventions and Complications study (EDIC) cohort. (From Retnakaran R, Zinman B: Type 1 diabetes, hyperglycaemia and the heart. Lancet 371:1790–1799, 2008.)
FIGURE 19-3 Pathophysiologic model of cardiovascular disease (CVD) in type 1 diabetes based on observations from the Diabetes Control and Complications Trial (DCCT)/Epidemiology of Diabetes Interventions and Complications study (EDIC). (From Retnakaran R, Zinman B: Type 1 diabetes, hyperglycaemia and the heart. Lancet 371:1790–1799, 2008.)
Overall, the DCCT helped to clarify the relationship between A1c and diabetic complications. For every 10% reduction in A1c, the risk of developing retinopathy was reduced by approximately 45%.13 Moreover, a continuous curvilinear relationship was observed between A1c and the incidence of retinopathy, the risk of progressive retinopathy being decreased substantially at A1c levels below 7%. Although the risk for progressive retinopathy was further reduced at A1c concentrations below 7% (i.e., no threshold effect), this benefit was achieved at the cost of a significantly increased risk for hypoglycemia, given an observed continuous and inverse relationship between hypoglycemic risk and A1c (Fig. 19-4). Thus, the DCCT firmly established the importance of glycemic control in the management of type 1 diabetes, demonstrated the feasibility of intensive therapy in improving glycemic control, and identified the limitations imposed on such control by hypoglycemia. In doing so, this study has provided a framework for current treatment goals.
FIGURE 19-4 Risk for sustained progression of retinopathy (A) and rate of severe hypoglycemia (B) in patients receiving intensive therapy, in relation to mean A1c value during the Diabetes Control and Complications Trial (DCCT). (From the DCCT Research Group: The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 329[14]:984, 1993.)
Glycemic Goals
On the basis of the findings of the DCCT and other studies, the ultimate objective in the treatment of type 1 diabetes is metabolic normalization, that is, the achievement of sustained euglycemia and the avoidance of hypoglycemia. In this context, the 2008 American Diabetes Association (ADA) treatment guidelines suggest the following therapeutic targets for nonpregnant adults with type 1 or type 2 diabetes: (1) A1c less than 7.0% (referenced to nondiabetic range 4.0% to 6.0%); (2) preprandial plasma glucose 70 to 130 mg/dL; and (3) peak postprandial plasma glucose less than 180 mg/dL (Table 19-1).14 The evidence-based 2008 Canadian Diabetes Association Clinical Practice Guidelines recommend similar targets of A1c less than or equal to 7.0%, preprandial plasma glucose 72 to 126 mg/dL, and 2-hour postprandial plasma glucose 90 to 180 mg/dL.15 The American Association of Clinical Endocrinologists recommends slightly more stringent goals of A1c less than or equal to 6.5%, fasting glucose less than 110 mg/dL, and postprandial glucose less than 140 mg/dL.16
Table 19-1
American Diabetes Association Recommendations for Treatment Targets in Nonpregnant Adults With Diabetes
Goals might have to be individualized for the given patient.
HDL, High-density lipoprotein; LDL, low-density lipoprotein.
*For women, it has been suggested that the HDL goal should be increased by 10 mg/dL.
Adapted from American Diabetes Association: Standards of medical care in diabetes. Diabetes Care 31(Suppl 1):S12–S54, 2008.
Acute Complications
The major acute complications of type 1 diabetes are ketoacidosis and hypoglycemia. These topics are discussed in detail in Chapters 20 and 21, respectively. The risk for these acute complications can be reduced through the judicious use of insulin therapy. Specifically, the risk for ketoacidosis can be reduced through appreciation of the concept that patients with type 1 diabetes require insulin therapy at all times. Appropriate management of special situations, such as intercurrent illness, requires adherence to this principle (see the section entitled “Adjustment of Insulin Therapy in Special Situations”).
As was observed in the DCCT (see Fig. 19-4B), tight glycemic control typically is associated with an increased risk for hypoglycemia. Patients therefore must be educated about the recognition of hypoglycemic symptoms and the use of appropriate treatment measures when such symptoms arise. Patients are advised to keep rapidly absorbable sources of carbohydrate, such as glucose tablets, close at hand at all times. In general, 15 g of carbohydrate is needed to increase blood glucose levels by approximately 38 mg/dL within 20 minutes.17,18 Besides glucose tablets, other treatments that provide a similar amount of glucose include (1) 15 mL (1 tablespoon) or three packets of table sugar dissolved in water, (2) 175 mL ( cup) of juice, and (3) 15 mL (1 tablespoon) of honey. In addition, patients with type 1 diabetes should have an emergency glucagon injection kit. Both the patient and his or her family members should be trained in the use and intramuscular or subcutaneous injection of glucagon (0.5 to 1 mg) as a means of rapidly increasing blood glucose concentration when the patient is severely hypoglycemic. Finally, the avoidance of hypoglycemia is particularly important for patients who are unable to sense hypoglycemic symptoms (discussed in the section entitled “Complications of Insulin Therapy”).
Chronic Complications
The chronic microvascular complications of type 1 diabetes include retinopathy, neuropathy, and nephropathy (discussed in Chapters 26, 27, and 28, respectively). To reduce the impact of these complications, management strategies include regular surveillance for early detection of complications and tight glycemic control for both primary prevention and secondary intervention, as demonstrated in the DCCT.
Macrovascular disease, as manifested by cerebrovascular, peripheral vascular, and coronary artery disease, is a major chronic complication of type 1 diabetes (discussed in Chapter 25). Regular surveillance for vascular disease and management of cardiovascular risk factors is essential. In particular, modification of reversible cardiovascular risk factors, such as hypertension, dyslipidemia, and cigarette smoking, is an important management goal. The 2008 ADA treatment guidelines suggest a target blood pressure of less than 130/80 mm Hg in patients with diabetes. In addition, the following treatment targets for lipids are recommended: LDL less than 100 mg/dL, triglycerides less than 150 mg/dL, and HDL greater than 40 mg/dL in men and HDL greater than 50 mg/dL in women (see Table 19-1).14
Quality of Life
Type 1 diabetes is a chronic condition that requires a complex health care strategy that demands significant patient effort through frequent insulin injections, regular self-monitoring of blood glucose levels, and constant attention to nutrition and physical activity. As such, effective management requires that the patient take responsibility for his or her care. In this context, it should be noted that quality of life is an important feature that can affect a patient’s willingness to pursue a demanding treatment plan. Recognition of this concept at all times by the health care team will facilitate the success of treatment initiatives. Moreover, in recognition of the importance of quality of life, many research studies now utilize validated instruments such as the Diabetes Quality of Life Questionnaire to measure patient well-being as a therapeutic outcome.19
Monitoring
Glycemic Control
Acute Measurement of Glycemic Control
Patients with type 1 diabetes should monitor blood glucose frequently. In the DCCT, insulin doses for patients in the intensive treatment arm were adjusted according to the results of SMBG performed at least four times a day.20 Blood glucose typically is measured before administration of insulin prior to a meal and at bedtime. Meaningful data can be derived from measurements at other times as well. For instance, nocturnal hypoglycemia resulting from excessive bedtime insulin can be detected by blood glucose measurement in the early hours of the morning. Similarly, postprandial monitoring 2 hours after a meal can assess the adequacy of the preprandial insulin dose. Patients should check their blood glucose level before driving a motor vehicle, particularly if they have difficulty sensing hypoglycemic symptoms, as hypoglycemia (and hence neurologic impairment) while driving can be very dangerous for the patient and others. Patients should also consider SMBG at times when blood glucose might be changing rapidly, such as during the postexercise period or when experiencing symptoms of hypoglycemia.
Self-monitoring of blood glucose is of value only to the extent that the information it provides is used to guide treatment. With each measurement, patients are encouraged to use a logbook to record the blood glucose value, the time of day, and the temporal relationship to food intake. Data obtained from SMBG should be used in two ways: variable insulin dose scales and pattern management. Variable insulin dose scales refer to algorithms that guide the patient as to the appropriate dose adjustment of preprandial insulin based on the current degree of glycemia (discussed in the section entitled “Implementation of Intensive Insulin Therapy”). Pattern management describes the two-step process of using the glucose profile recorded over several days to identify glycemic patterns and then adjusting the daily insulin regimen for the coming days in a proactive fashion on the basis of these patterns. Therefore, both variable insulin dose scales and pattern management are essential components of patient education.
Given the value of blood glucose data, it is important to consider potential problems that are inherent in the process of self-monitoring. To ensure proper meter function, glucose meters should be calibrated by a laboratory reference standard. The patient’s use of the meter and self-monitoring technique should be reviewed periodically in the clinic.21 Inappropriate handling of the testing strips can lead to defective function and inaccurate glucose results. Finally, some patients find lancing of the fingertip to be painful and hence are unwilling to measure blood glucose as frequently as recommended.
In response to patient concerns regarding pain associated with lancing of the fingertip, alternative site glucose meters have recently been introduced. By sampling from alternative sites such as the forearm, these devices can provide accurate blood glucose measurements while causing less pain than fingertip testing.22 However, it should be noted that alternative site testing might not provide accurate results at times of rapid change in blood glucose concentration (e.g., immediately after a meal or after exercise).23,24 Therefore, traditional fingertip testing is recommended at these times.
Because blood glucose concentrations are dynamic and fluctuate constantly, it is readily apparent that the limited data provided by even frequent SMBG might not be truly representative of an individual’s glucose excursions. In this context, continuous glucose monitoring (CGM) devices represent an important recent development. This technology utilizes the fact that glucose in interstitial fluid can reflect blood glucose levels.25 CGM systems typically consist of a pager-sized monitor worn by the patient that receives an electrical signal from a subcutaneous sensor that continuously measures the glucose concentration in interstitial fluid.26 Using this input, the monitor determines a blood glucose concentration every 5 minutes that can be downloaded to a computer to provide a graphic display of the patient’s glycemic profile over the period of time monitored (Fig. 19-5).27 During the period of monitoring (usually 3 days), the patient maintains a diary of meals, insulin doses, physical activity, and hypoglycemic symptoms to facilitate meaningful interpretation of the recorded data. Whereas older CGM systems yielded only retrospective glucose data, current models have the capability to provide patients with real-time access to their absolute blood glucose values and to relative trends in their glucose levels. Furthermore, these devices have alarms that can alert patients to either hypoglycemia or hyperglycemia, thereby precipitating appropriate corrective action. In addition, the technology exists wherein CGM systems have been combined with insulin pumps, although it should be noted that these systems currently require the input of the patient (since the insulin pump is not controlled by the CGM device).28 In this regard, it should be recognized (1) that CGM systems require calibration with SMBG readings, (2) that the accuracy of glucose measurements by these devices have been drawn into question in the past,29 and (3) that SMBG values remain the recommended data for guiding treatment decisions.14,30 Thus, the American Diabetes Association currently recommends that CGM be used as a supplemental tool to SMBG in selected patients with type 1 diabetes, particularly those with hypoglycemia unawareness.14 The latter consideration is a reflection of the fact that initial experience with CGM demonstrated higher rates of unrecognized nocturnal hypoglycemia than were previously suspected.31,32 Furthermore, several studies have since shown that CGM systems (particularly real-time devices) are associated with reductions in the total amount of time spent in hypoglycemia.30,33–35 To date, only a few studies have shown improvements in A1c.30,36,37 Although some challenges remain for this rapidly developing technology (including the acquisition of a broader evidence base from further clinical research), the role of CGM as a valuable tool in the management of type 1 diabetes is likely to grow in the near future.
Measurement of Chronic Glycemic Control
Glucose can attach to proteins in the blood through an irreversible, nonenzymatic process, resulting in the formation of glycated proteins.38 Because this process is irreversible, glucose remains attached to the protein until the latter is metabolized. The degree of glycation is a function of blood glucose concentration over time. Thus, measurement of glycated proteins provides a means of estimating chronic blood glucose concentration over a time period proportional to the half-life of the protein in question.
Erythrocytes are freely permeable to glucose. In the circulation, glucose attaches irreversibly to hemoglobin in red blood cells, leading to the formation of glycated hemoglobins called hemoglobin A1a, A1b, and A1c. Measurement of hemoglobin A1c (also known as A1c, glycohemoglobin, or glycosylated hemoglobin) has emerged as the most widely used test of chronic glycemic control. Specifically, the proportion of glycosylated hemoglobin to the total number of hemoglobin molecules reflects overall blood glucose concentration over the preceding 2 to 3 months.39 Therefore, regular measurement of A1c every 3 months is recommended as a method of determining overall glycemic control and evaluating the adequacy of the treatment regimen.21 For instance, a significantly elevated A1c value, consistent with suboptimal glycemic control, would suggest the need for a change in the treatment regimen. Analysis of daily glycemic patterns from the patient’s SMBG records can indicate the specific changes to be made.
Many different types of assays can be used for measurement of glycated hemoglobins.40 Some assays report A1c as a percentage of total hemoglobin, while other methods measure total glycated hemoglobin. Thus, nonstandardization of assays can complicate comparison of A1c measurements between laboratories. The National Glycohemoglobin Standardization Program, initiated in 1996, is an initiative that is designed to standardize laboratory A1c assays to the DCCT reference method.41
The DCCT provided insight into the correlation between A1c level and mean plasma glucose concentration (Fig. 19-6).42 This relationship has recently been studied with the use of CGM data.43 In practice, in some situations the measured A1c value may be discordant with the level that would be expected on the basis of the patient’s capillary blood glucose records. Such discrepancy might be due to glycemic excursions at unmonitored times of the day that are not reflected in SMBG records. Alternatively, the problem might be falsification of SMBG records by the patient.
FIGURE 19-6 The relationship between mean plasma glucose and mean A1c in the Diabetes Control and Complications Trial (DCCT) (n = 1429; r = 0.82). (From Rohlfing CL, Wiedmeyer HM, Little RR, et al: Defining the relationship between plasma glucose and HbA1c: Analysis of glucose profiles and HbA1c in the Diabetes Control and Complication Trial. Diabetes Care 25[2]:276, 2002.)
Another possibility in certain clinical settings is that the measurement of A1c might not be accurate. For instance, rapid red blood cell turnover leads to a disproportionate representation of younger erythrocytes, whose collective exposure to ambient blood glucose levels has been shorter than that of other red blood cells. Accordingly, A1c levels tend to be reduced under such conditions.44 Pregnancy and hemolytic anemias are examples of settings characterized by rapid red blood turnover and hence lowered A1c levels. Similarly, A1c concentration may be lowered in hemoglobinopathies, such as thalassemia and sickle cell disease, because of a diminished propensity for glycation of the abnormal hemoglobin molecule. Conversely, depending on the assay used, A1c values may be falsely elevated in the presence of hemoglobin F, the carbamylated hemoglobin formed in uremia, or acetaldehyde-bound hemoglobin (seen in alcohol abuse).45
In situations in which measurement of A1c might be unreliable, an alternative approach to the estimation of glycemic control is the measurement of glycated serum proteins or albumin. The term fructosamine has been applied to the glycated proteins measured by such assays.46 Given the 14- to 21-day half-life of albumin, a serum fructosamine measurement reflects mean blood glucose concentration over the preceding 1 to 2 weeks. Generally, serum fructosamine values correlate well with A1c measurements.47 However, certain caveats should be noted in measuring fructosamine. First of all, fructosamine values can be affected by conditions that alter the synthesis or clearance of serum proteins. Indeed, the question of whether or not to correct fructosamine assays for serum protein or albumin concentration remains controversial.48 Second, serum fructosamine levels show greater intrasubject variability than A1c measurements do.49 Third, unlike A1c, fructosamine has not yet been shown to correlate with the risk for diabetic complications.
Ketone Testing
The presence of detectable levels of ketones in urine or blood may indicate impending or established ketoacidosis in patients with type 1 diabetes. Accordingly, patients should test for ketones in the following situations: when blood glucose concentrations are persistently elevated, during periods of acute illness or stress, or when the patient is experiencing symptoms compatible with ketoacidosis such as nausea, vomiting, and abdominal pain.48 Traditionally, patients have used urine dipsticks to test for ketonuria. In recent years, however, home monitors have been developed that can measure the capillary blood concentration of β-hydroxybutyrate, the most prevalent intermediate molecule in ketone formation.50 Blood ketone testing avoids some of the limitations of traditional urinary ketone testing, such as false-positive readings, the inability to detect β-hydroxybutyrate (urinary dipsticks measure different ketone bodies), and patient reluctance to perform urinary testing. In limited studies to date, home capillary blood ketone measurement appears to be more sensitive than urinary testing in the detection of ketosis.51,52 The use of blood ketone monitoring by patients is expected to increase in the future. In the clinical setting, serum ketone measurement is recommended over urinary testing.
Complication Surveillance
1. Retinopathy: Initial dilated and comprehensive eye examination should be performed within 5 years after diagnosis of type 1 diabetes and at least annually thereafter.14
2. Nephropathy: Patients with type 1 diabetes of 5 or more years’ duration should undergo an annual screening test for the presence of microalbuminuria.14 The preferred screening test is a measurement of the albumin-to-creatinine ratio in a random, spot urine sample.53
3. Neuropathy and foot care: A comprehensive foot examination should be performed annually.14 This examination includes evaluation of sensation by 10 g Semmes-Weinstein monofilament, testing of vibration sense by tuning fork, assessment of superficial pain sensation, palpation, and visual inspection. In screening for neuropathy, superficial pain sensation testing, monofilament examination, and vibration testing all show similar operating characteristics and may be used interchangeably.54
4. Cardiovascular disease: Regular evaluation of cardiovascular risk factors is recommended. Blood pressure should be measured at every diabetic clinic appointment. Lipid profile should be tested at least annually. Screening exercise stress testing may be considered in patients with typical or atypical cardiac symptoms; abnormal resting electrocardiogram; a history of peripheral or cerebrovascular disease; or sedentary lifestyle, age over 35 years, and plans to start a vigorous exercise program.14
5. Other: Other potential associated problems that warrant periodic screening include erectile dysfunction, depression, and thyroid disease.
Insulin Therapy
Principles of Insulin Replacement
Insulin replacement strategies in type 1 diabetes ideally aim to mimic the normal physiologic secretion of insulin by the pancreas.6 Therefore, to appreciate the rationale underlying the design of current insulin replacement regimens, an understanding of the basic structure and biochemistry of endogenous insulin is necessary (Fig. 19-7). This topic, discussed in detail in Chapter 6, is reviewed briefly here.
FIGURE 19-7 The amino acid sequence and structure of human insulin. The A and B chains are linked by two disulfide bonds. A third disulfide bridge is present within the A chain. (Adapted from Cheng AY, Zinman B: Insulin analogues and the treatment of diabetes. In Raz I, Skyler JS, Shafrir E [eds]: Diabetes: From Research to Diagnosis and Treatment. London, Martin Dunitz Ltd, 2002, pp 331–346.)
The pancreatic β cells secrete endogenous insulin into the portal venous system in response to physiologic demand for glucose homeostasis as determined by nutrient intake and energy expenditure. Despite broad fluctuations in these determinants, healthy individuals are able to maintain plasma glucose concentration within a narrow range of 3.5 to 7.0 mmol/L throughout the day (Fig. 19-8).55 This normal glucose homeostasis requires tightly regulated insulin secretion that consists of two components: a basal secretion rate and surges of markedly increased secretion following ingestion of a mixed meal. Accounting for approximately 40% of total 24 hour pancreatic insulin output, the basal secretion of ≈1 µ/hr serves to limit hepatic glucose production and adipocyte lipolysis in the postabsorptive (fasting) state such as between meals and overnight.56 Conversely, with ingestion of a mixed meal, dietary secretagogues (e.g., glucose, amino acids) and gastrointestinal hormones (e.g., glucagon-like peptide 1) stimulate abrupt pulses of insulin secretion up to five times the basal rate. These pulses regulate postprandial glycemia by inhibiting hepatic glucose production and increasing peripheral glucose uptake. Insulin secretion is also affected by energy expenditure. For instance, with moderate exercise, insulin secretion rapidly decreases to prevent hypoglycemia.57 Conversely, with strenuous exercise, catecholamine-mediated hyperglycemia may occur, leading to increased insulin secretion in the postexercise period.58 Thus, the tight coupling of insulin secretion to plasma glucose concentration in healthy individuals (see Fig. 19-8) requires the complex integration of multiple signals of nutrient availability and energy expenditure.
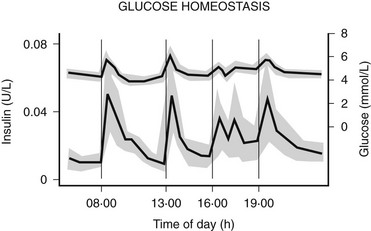
FIGURE 19-8 Twenty-four-hour plasma glucose and insulin profiles in healthy individuals (n = 12) (shown as mean values with 95% confidence interval). (From Owens DR, Zinman B, Bolli GB: Insulins today and beyond. Lancet 358:739, 2001.)
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
