Chapter 13 Treatment of dystonia
Despite the paucity of knowledge about the cause and pathogenesis of dystonic disorders, the symptomatic treatment of dystonia has markedly improved, particularly since the introduction of botulinum toxin. In most cases of dystonia, the treatment is merely symptomatic, designed to improve posture and function and to relieve associated pain. In rare patients, however, dystonia can be so severe that it can produce not only abnormal postures and disabling dystonic movements, sometimes compromising respiration, but also muscle breakdown and life-threatening hyperthermia, rhabdomyolysis, and myoglobinuria. In such cases of dystonic storm or status dystonicus, proper therapeutic intervention can be life-saving (Jankovic and Penn, 1982; Vaamonde et al., 1994; Manji et al., 1998; Dalvi et al., 1998; Jankovic, 2006, 2009a).
The assessment of various therapeutic interventions in dystonia is problematic for the following reasons: (1) Dystonia and its effects on function are difficult to quantitate; therefore, most trials utilize crude clinical rating scales, many of which have not been properly evaluated or validated; (2) dystonia is a syndrome with different etiologies, anatomic distributions, and heterogeneous clinical manifestations producing variable disability; (3) some patients, perhaps up to 15%, may have spontaneous, albeit transient, remissions; (4) many studies have used dosages that may have been insufficient or too short a duration to provide benefit; (5) the vast majority of therapeutic trials in dystonia are not double-blind, placebo-controlled; and (6) most studies, even those that have been otherwise well designed and controlled, have utilized small sample sizes, which makes the results difficult to interpret, particularly in view of a large placebo effect demonstrated in dystonia (Lindeboom et al., 1996). A variety of instruments have been used to assess the response in patients with cervical dystonia; the most frequently used is the Toronto Western Spasmodic Torticollis Rating Scale (TWSTRS) (Consky and Lang, 1994; Comella et al., 1997). TWSTRS (range: 0–87) consists of three subscales: severity (range: 0–35), disability (range: 0–23), and pain (range: 0–20). In addition, visual analog scale, global assessment of change, and pain analog assessments have been used in various clinical trials. Various scales, such as the Fahn–Marsden scale and the Unified Dystonia Rating Scale, have also been used to assess patients with generalized dystonia (Burke et al., 1985a; Ondo et al., 1998; Comella et al., 2003). Another scale designed to capture the burden of dystonia on patients is the Cervical Dystonia Impact Profile scale (Cano et al., 2004). The Jankovic Rating Scale was initially used in the original study of botulinum toxin (BTX) in patients with cranial dystonia which led to the approval of BTX by the Food and Drug Administration (Jankovic and Orman, 1987) and in subsequent studies (Roggenkamper et al., 2006) to assess the severity and frequency of involuntary eyelid contractions in patients with blepharospasm (Jankovic et al., 2009). Subsequent studies have refined the clinical rating scales and included quality-of-life scales (Lindeboom et al., 1996). Health-related quality-of-life instruments, such as the Craniocervical Dystonia Questionnaire (CDQ-24), are increasingly used to assess the function and the impact of treatment in patients with cranial, cervical, and other dystonias on activities of daily living and other measures of quality of life (Muller et al., 2004; Reimer et al., 2005; Hall et al., 2006).
The selection of a particular choice of therapy is guided largely by personal clinical experience and by empirical trials (Greene et al., 1988; Greene, 1995; Jankovic, 1997; Brin, 1998; Jankovic, 2004a, 2006; Albanese et al., 2006; Tinter and Jankovic, 2007) (Table 13.1). An evidence-based review concluded that except for BTX in cervical dystonia and high-dose trihexyphenidyl in young patients with segmental and generalized dystonia (level A, class I–II), none of the methods of pharmacologic intervention have been confirmed as being effective according to evidence-based criteria (Balash and Giladi, 2004). The patient’s age, the anatomic distribution of dystonia, and the potential risk of adverse effects are also important determinants of choice of therapy. The identification of a specific cause of dystonia, such as drug-induced dystonias or Wilson disease (Svetel et al., 2001), may lead to a treatment that is targeted to the particular etiology. It is therefore prudent to search for identifiable causes of dystonia, particularly when some atypical features are present. The diagnostic approach to patients with dystonia is based on being aware of the multiple causes of torsion dystonia, which was covered in Chapter 12 and in other reviews (Jankovic, 2007).
Table 13.1 Medical therapy of dystonia
Physical and supportive therapy
Before reviewing pharmacologic and surgical therapy of dystonia, it is important to emphasize the role of patient education and supportive care, which are integral components of a comprehensive approach to patients with dystonia. Various paramedical treatments reported to be useful in patients with primary dystonia have been systematically reviewed (Delnooz et al., 2009). Physical therapy and well-fitted braces are designed primarily to improve posture and to prevent contractures. Although braces are often poorly tolerated, particularly by children, they may be used in some cases as a substitute for a sensory trick. For example, some patients with cervical dystonia are able to construct neck–head braces that seem to provide sensory input by touching certain portions of the neck or head in a fashion similar to the patient’s own sensory trick, thus enabling the patient to maintain a desirable head position. Various hand devices have been developed in an attempt to help patients with writer’s cramp to use their hands more effectively and comfortably (Ranawaya and Lang, 1991; Tas et al., 2001). In one small study of five professional musicians with focal dystonia, Candia and colleagues (1999) reported success with immobilization by splints of one or more of the digits other than the dystonic finger followed by intensive repetitive exercises of the dystonic finger. It is not clear, however, whether this therapy provides lasting benefits. In one study of eight patients with idiopathic occupational focal dystonia of the upper limb, immobilization with a splint for 4–5 weeks resulted in a significant improvement at a 24-week follow-up visit, based on the Arm Dystonia Disability Scale (0 = normal; 3 = marked difficulty in playing) and the Tubiana and Champagne Score (0 = unable to play; 5 = returns to concert performances); the improvement was considered marked in four and moderate in three, but the initial improvement disappeared in one (Priori et al., 2001). The splint was applied for 24 hours every day except for 10 minutes once a week when it was removed for brief local hygiene. Immediately on full removal of the splint, all patients reported marked clumsiness and weakness, which resolved in 4 weeks. There was also some local subcutaneous and joint edema and pain in the immobilized joint, and nail growth stopped; none of the patients developed contractures. While the mechanisms of action of immobilization are unknown, the authors have postulated that removing all motor and sensory input to a limb might allow the cortical map to “reset” to the previous normal topography. One major concern about immobilization of a limb, particularly a dystonic limb, is that such immobilization can actually increase the risk of exacerbating or even precipitating dystonia, as has been well demonstrated in dystonia following casting or other peripheral causes of dystonia (Jankovic, 2001a). In one study of 21 patients with writer’s cramp, after 4 weeks of immbolization, “retraining” for 8 weeks using drawing and writing exercises was associated with significant improvement relative to baseline as assessed by the Writer’s Cramp Rating Scale (Zeuner et al., 2008). A variation of the immobilization therapy, constraint-induced movement therapy, has been used successfully in rehabilitation of patients after stroke and other brain insults, and the observed benefit has been attributed to cortical reorganization (Taub et al., 1999; Levy et al., 2001; Taub and Morris, 2001; Taub et al., 2002; Wolf et al., 2002).
Another technique, using a repetitive task during regional anesthesia of a weak arm in patients following stroke, was also associated with improved hand function (Muellbacher et al., 2002). Some patients find various muscle relaxation techniques and sensory feedback therapy useful adjuncts to medical or surgical treatments. Since some patients with dystonia have impaired sensory perception, it has been postulated that sensory training may relieve dystonia. In a study of 10 patients with focal hand dystonia, Zeuner and colleagues (2002) showed that reading Braille for 30–60 minutes daily for 8 weeks improved spatial acuity and dystonia that was sustained for up to 1 year in some patients (Zeuner and Hallett, 2003). Sensory training to restore sensory representation of the hand along with mirror imagery and mental practice techniques has also been reported to be useful in the treatment of focal hand dystonia (Byl and McKenzie, 2000; Byl et al., 2003).
Siebner and colleagues (1999) showed that using repetitive transcranial magnetic stimulation (rTMS) delivered at low frequencies (≤1 Hz) for 20 minutes can temporarily (8 of 16 patients reported improvement that lasted longer than 3 hours) improve handwriting impaired by dystonic writer’s cramp, presumably by increasing inhibition (and thus reducing excitability) of the underlying cortex. Despite some encouraging results from open-label pilot studies it remains to be seen whether rTMS or transcranial direct current stimulation (tDCS) will enter the mainstream of therapeutics of dystonia (Wu et al., 2008).
Other stimulation techniques are also being studied in patients with dystonia. Long-term neck muscle vibration of the contracting muscle might have a therapeutic value in patients with cervical dystonia. This is suggested by transient (minutes) improvement in head position in one patient who was treated for 15 minutes with muscle vibration (Karnath et al., 2000). Transcutaneous electrical stimulation has been found to improve dystonic writer’s cramp for at least 3 weeks in a double-blind, placebo-controlled trial (Tinazzi et al., 2005). Also, acoustic and galvanic vestibular stimulation was reported beneficial in one patient with cervical dystonia (Rosengren and Colebatch, 2006). Such observation is consistent with the notion that proprioceptive sensory input affects cervical dystonia.
Dopaminergic therapy
Pharmacologic treatment of dystonia is based largely on an empirical rationale rather than a scientific one. Unlike Parkinson disease, in which therapy with levodopa replacement is based on the finding of depletion of dopamine in the brains of parkinsonian animals and humans, our knowledge of biochemical alterations in idiopathic dystonia is very limited. One exception is dopa-responsive dystonia (DRD), in which the biochemical and genetic mechanisms have been elucidated by molecular DNA and biochemical studies in patients (Ichinose et al., 1994) and by studies of postmortem brains (Furukawa et al., 1999). Decreased neuromelanin in the substantia nigra with otherwise normal nigral cell count and morphology and normal tyrosine hydroxylase immunoreactivity were found in one brain of a patient with classic DRD (Rajput et al., 1994). There was a marked reduction in dopamine in the substantia nigra and the striatum. These findings suggested that in DRD, the primary abnormality was a defect in dopamine synthesis. This proposal is supported by the finding of a mutation in the GTP cyclohydrolase I gene on chromosome 14q that indirectly regulates the production of tetrahydrobiopterin, a cofactor for tyrosine hydroxylase, the rate-limiting enzyme in the synthesis of dopamine (Ichinose et al., 1994; Tanaka et al., 1995; Steinberger et al., 2000). Another form of DRD has been described in four cases belonging to two unrelated families of dopa-responsive cervical dystonia, presenting between 9 and 15 years of age and similar to classic DRD manifesting diurnal variation but no levodopa-induced dyskinesia (Schneider et al., 2006).
DRD usually presents in childhood with dystonia, mild parkinsonian features, and pseudopyramidal signs (hypertonicity and hyperreflexia) predominantly involving the legs. Many patients have a family history of dystonia or Parkinson disease. At least half of the patients have diurnal fluctuations, with marked progression of their symptoms toward the end of the day and a relief after sleep. Many patients with this form of dystonia are initially misdiagnosed as having cerebral palsy. Some patients with DRD are not diagnosed until adulthood, and family members of patients with typical DRD may present with adult-onset levodopa-responsive parkinsonism (Harwood et al., 1994). The take-home message from these reports is that a therapeutic trial of levodopa should be considered in all patients with childhood-onset dystonia, whether they have classic features of DRD or not.
Most patients with DRD improve dramatically even with small doses of levodopa (100 mg of levodopa with 25 mg of decarboxylase inhibitor), but some might require doses of levodopa as high as 1000 mg per day. In contrast to patients with juvenile Parkinson disease (Ishikawa and Miyatake, 1995), DRD patients usually do not develop levodopa-induced fluctuations or dyskinesias. If no clinically evident improvement is noted after 3 months of therapy, the diagnosis of DRD is probably an error, and levodopa can be discontinued. In addition to levodopa, patients with DRD also improve with dopamine agonists, anticholinergic drugs, and carbamazepine (Nygaard et al., 1991). In contrast to patients with DRD, patients with idiopathic or other types of dystonia rarely improve with dopaminergic therapy (Lang, 1988). While dopaminergic therapy is remarkably effective in treating DRD, this strategy is not useful in the treatment of primary dystonia. Apomorphine, however, may ameliorate dystonia, perhaps by decreasing dopamine as well as serotonin release (Zuddas and Cianchetti, 1996).
Antidopaminergic therapy
Although dopamine receptor-blocking drugs were used extensively in the past, most clinical trials have produced mixed results with these drugs. Because of the poor response and the possibility of undesirable side effects, particularly sedation, parkinsonism, and tardive dyskinesia, the use of these drugs in the treatment of dystonia should be discouraged (Jankovic, 1995b). Clozapine, an atypical neuroleptic, has been reported in a small open trial to be moderately effective in the treatment of segmental and generalized dystonia, but its usefulness was limited by potential side effects (Karp et al., 1999). Although antidopaminergic drugs have been reported to be beneficial in the treatment of dystonia, the potential clinical benefit is usually limited by the development of side effects. Dopamine-depleting drugs, however, such as tetrabenazine, have been found useful in some patients with dystonia, particularly those with tardive dystonia (Jankovic and Orman, 1988; Jankovic and Beach, 1997). Tetrabenazine, a vesicular monoamine transporter 2 (VMAT2) inhibitor, has the advantage over other antidopaminergic drugs in that it does not cause tardive dyskinesia, although it may cause transient acute dystonic reaction (Burke et al., 1985b; Jankovic and Beach, 1997; Kenney and Jankovic, 2006). Tetrabenazine is not readily available in the United States, but it is dispensed by prescription under the trade name Nitoman or Xenazine 25 in other countries, including the United Kingdom. It is possible that some of the new atypical neuroleptic drugs will be useful not only as antipsychotics but also in the treatment of hyperkinetic movement disorders. Risperidone, a D2 dopamine receptor-blocking drug with a high affinity for 5-HT2 receptors, has been reported to be useful in a 4-week trial of five patients with various forms of dystonia (Zuddas and Cianchetti, 1996). Clozapine, a D4 dopamine receptor blocker with relatively low affinity for the D2 receptors and high affinity for the 5-HT2A receptors, has been reported to ameliorate the symptoms of tardive dystonia (Trugman et al., 1994). The treatment of tardive dystonia and other tardive syndromes is discussed in Chapter 19 and in other reviews (Jankovic, 1995b).
Anticholinergic therapy
High-dosage anticholinergic medications, such as trihexyphenidyl, for dystonia were introduced by Fahn (1983) and confirmed by several groups (Marsden et al., 1984; Greene et al., 1988; Jabbari et al., 1989; Hoon et al., 2001), including a double-blind study (Burke et al., 1986). Although helpful in all types of dystonias, the supremacy of botulinum toxin therapy for focal dystonias has relegated anticholinergic therapy to be most useful in the treatment of generalized and segmental dystonia. In the experience of Greene and colleagues (1988), patients with blepharospasm, generalized dystonia, tonic (in contrast to clonic) dystonia, and onset of dystonia at age younger than 19 years seemed to respond better to anticholinergic drugs than other subgroups, but this difference did not reach statistical significance. Except for short duration of symptoms before onset of therapy, there was no other variable, such as gender or severity, that reliably predicted a favorable response. Treating patients within the first 5 years of disease onset was statistically significantly more successful than delaying treatment in both children and adults regardless of severity (Greene et al., 1988). Thus, starting treatment early is important.
This therapy is generally well tolerated when the dose is increased slowly. We recommend starting with a 5 mg preparation, half-tablet at bedtime, and adding a half-tablet a week, advancing up to 10 mg in four divided doses by the end of 4 weeks. Because there is a lag between reaching a dose and seeing a benefit, we hold this dose for a month, and then continue to increase at the same rate over the next 4 weeks until the dose is 20 mg in four divided doses. We continue to increase until there is adequate benefit or adverse effects that limit higher doses. Some patients require up to 60–140 mg/day, but may experience dose-related drowsiness, confusion, or memory difficulty that limit the dose. Children usually tolerate the very high dosages, whereas adults do not. In one study of 20 cognitively intact patients with dystonia, only 12 of whom could tolerate 15–74 mg of daily trihexyphenidyl, drug-induced impairments of recall and slowing of mentation were noted, particularly in the older patients (Taylor et al., 1991). Diphenhydramine, a histamine H1 antagonist with anticholinergic properties, has been reported to have an antidystonic effect in three of five patients (Truong et al., 1995). However, the drug was not effective in 10 other patients with cervical dystonia, and it was associated with sedation and other anticholinergic side effects in most patients. Pyridostigmine, a peripherally acting anticholinesterase, and eye drops of pilocarpine (a muscarinic agonist) often ameliorate many of the peripheral side effects, such as urinary retention and blurred vision. Pilocarpine (Salagen) 5 mg four times per day, Cevimeline (Evoxac) 30 mg three times per day, and synthetic saliva (Glandosane, Salagen, Salivart, Salix) have been found effective in the treatment of dry mouth.
Other pharmacologic therapies
Many patients with dystonia require a combination of several medications and treatments (Jankovic, 2004b, 2006). High-dosage oral baclofen appears to be the next most effective agent for dystonia, particularly in combination with high-dosage anticholinergic treatment. Baclofen is a GABAb autoreceptor agonist that is used to treat spasticity. It has been found to produce substantial and sustained improvement in 29% of children at a mean dose of 92 mg/day (range: 40–180 mg/day) (Greene, 1992; Greene and Fahn, 1992a). Although baclofen was initially effective in 28 of 60 (47%) of adults with cranial dystonia, only 18% continued baclofen at a mean dose of 105 mg/day after a mean of 30.6 months (Fahn et al., 1985a).
Benzodiazepines (diazepam, lorazepam, or clonazepam) may provide additional benefit for patients whose response to anticholinergic drugs is unsatisfactory. Clonazepam might be particularly useful in patients with myoclonic dystonia. Muscle relaxants that are useful in the treatment of dystonia include cyclobenzaprine (Flexeril, 30–40 mg/day), metaxalone (Skelaxin, 800 mg two to three times per day), carisoprodol (Soma), methocarbamol (Robaxin oral and patch), orphenadrine (Norflex), and chlorzoxazone (Parafonforte). Structurally and pharmacologically similar to amitriptyline, cyclobenzaprine has been found at doses of 30–40 mg/day to be superior to placebo but equal to diazepam. Sodium oxybate, a salt of gamma-hydroxybutyrate, has been also reported to be effective in the myoclonus–dystonia syndrome, similar to alcohol (Frucht et al., 2005).
Narayan and colleagues (1991) first suggested that intrathecal baclofen (ITB) might be effective in the treatment of dystonia in 1991 in a report of an 18-year-old man with severe cervical and truncal dystonic spasms who was refractory to all forms of oral therapy and to large doses of paraspinal BTX injections. Muscle-paralyzing agents were necessary to relieve these spasms, which compromised his respiration. Within a few hours after the institution of ITB infusion, the patient’s dystonia markedly improved, and he was able to be discharged from the intensive care unit within 1–2 days. The subsequent experience with intrathecal infusions has been quite encouraging, and studies are currently in progress to further evaluate this form of therapy in patients with dystonia and other motor disorders (Penn et al., 1995; Dressler et al., 1997; van Hilten et al., 1999). In some patients who were treated with intrathecal baclofen for spasticity, the benefits persisted even after the infusion was stopped (Dressnandt and Conrad, 1996). Ford and colleagues (1996) reviewed the experience with ITB in 25 patients and concluded that this form of therapy may be “more effective when dystonia is associated with spasticity or pain.” ITB could have a role in selected patients with dystonic storm (Dalvi et al., 1998) and in secondary dystonias associated with pain and spasticity (Ford et al., 1998a). For example, Albright and colleagues (1998) found improvement in dystonia scores in 10 of 12 patients with cerebral palsy using an average daily dose of ITB of 575 µg; the improvement was sustained in 6 patients. In a subsequent study involving 86 patients aged 3–42 years (mean: 13 years) with generalized dystonia (secondary to cerebral palsy in 71% of patients), external infusion or bolus-dose screening was positive in approximately 90% of patients (Albright et al., 2001). Programmable pumps were implanted in 77 patients. Infusion began at 200 µg/day and increased by 10–20% per day until the best dose was achieved. The median duration of ITB therapy was 26 months. The mean dose increased over time, from 395 µg at 3 months to 610 µg at 24 months to 960 µg at 36 months. Patients and caregivers rated quality of life and ease of care as having improved in approximately 85% of patients. Seven patients, including four with cerebral palsy, lost their response to ITB during the study, usually during the first year. The most common side effects were increased constipation (19%), decreased neck and trunk control, and drowsiness. Surgical and device complications occurred in 38% of patients, including infections and catheter breakage and disconnection. Complication rates decreased over time. The authors conclude, “In our opinion, ITB is the treatment of choice for severe, generalized secondary dystonia after oral medications have been shown to be ineffective.” One potentially serious complication of ITB is life-threatening intermittent catheter leakage, which might not be detectable by standard noninvasive methods (Bardutzky et al., 2003).
In a long-term (6 years) follow-up of ITB in 14 patients, 5 patients were found to have improvement in their rating scale scores, although only 2 patients had sustained “clear clinical benefit” (Walker et al., 2000). Hou and colleagues (2001) provided a follow-up on long-term effects of ITB in 10 patients (2 males and 8 females; mean age: 43.2 years; range: 21–66 years) with severe segmental or generalized dystonia who responded unsatisfactorily to oral medications and BTX injections. Three patients had peripherally induced dystonia, all with reflex sympathetic dystrophy, two were idiopathic (one with reflex sympathetic dystrophy), two suffered brain injury, two had static encephalopathy, and one had an unknown neurodegenerative disorder. Anatomically, three patients suffered segmental dystonia involving the neck, arms, or abdominal muscles. Seven others had generalized dystonia. The average duration of dystonia prior to implantation was 11 years (range: 1.7–35 years). All patients received test doses of bolus ITB before implantations. Two had relatively poor initial response to the test dose, but still received implantations for continuous infusion. Neurologic assessments were conducted immediately after the implantations, 1 month later, and every 3 months thereafter. The average duration of follow-up was 4.7 years (range: 0.7–11.1 years). Only one patient abandoned the pump after 22 months. Initially, five improved markedly, two improved moderately, two improved mildly, and one had no improvement. Both patients who failed to respond to the initial test of ITB improved on continuous infusion with pump. In four patients, the improvement lessened over time, although the patients were still better than they had been preoperatively. All 10 patients needed to continue oral medications, and 8 continued BTX injections at similar doses. Hou and colleagues (2001) concluded that patients with secondary (spastic) dystonia involving primarily legs and trunk appeared to be the best candidates for continuous ITB infusion. Continuous ITB has been found to be safe and effective in other series of patients with reflex sympathetic dystrophy and dystonia (van Hilten et al., 2000). It is not yet clear whether ITB can induce lasting remissions in patients with dystonia. The American Academy for Cerebral Palsy and Developmental Medicine has published a systematic review of the use of ITB for spastic and dystonic cerebral palsy (Butler et al., 2000). The limited published data show that ITB reduced spasticity and dystonia, particularly in the lower extremities.
There are other medications that are used in the treatment of dystonia. For example, slow-release morphine sulfate has been shown to improve not only pain but also dystonic movement in some patients with primary and tardive dystonia (Berg et al., 2001). Besides clonazepam, gamma-hydroxybutyrate, used in the treatment of alcohol abuse, has been found to be beneficial in the treatment of myoclonus–dystonia syndrome (Priori et al., 2000). It is not known whether acamprosate, another drug that is used in the treatment of alcohol abuse, is useful in the treatment of myoclonus–dystonia. Anticonvulsants, such as levetiracetam (Keppra) and zonisamide (Zonegran), have been reported to be effective in the treatment of cortical myoclonus, but it is not clear whether these drugs play a role in the treatment of dystonia. An open-label trial in 10 patients with segmental and generalized dystonia showed no evidence of efficacy (Hering et al., 2007).
Peripheral deafferentiation with anesthetic was previously reported to improve tremor (Pozos and Iaizo, 1992), but this approach might also be useful in the treatment of focal dystonia, such as writer’s cramp (Kaji et al., 1995a) or oromandibular dystonia (Yoshida et al., 1998) that is unresponsive to other pharmacologic therapy. An injection of 5–10 mL of 0.5% lidocaine into the target muscle improved focal dystonia for up to 24 hours. This short effect can be extended for up to several weeks if ethanol is injected simultaneously. The observation that blocking muscle spindle afferents reduces dystonia suggests that somatosensory input is important in the pathogenesis of dystonia (Hallett, 1995; Kaji et al., 1995b). Mexiletine, an oral derivative of lidocaine, has been found to be effective in the treatment of cervical dystonia at doses ranging from 450 to 1200 mg/day (Ohara et al., 1998). Two-thirds of the patients, however, experienced adverse effects, including heartburn, drowsiness, ataxia, and tremor. On the basis of a review and a rating of videotapes by a “blind” rater, Lucetti and colleagues (2000) reported a significant improvement in six patients with cervical dystonia who had been treated with mexiletine.
Local electromyograph (EMG)-guided injection of phenol is currently being investigated as a potential treatment of cervical dystonia, but the results have not been very encouraging because of pain associated with the procedure and unpredictable response (Ruiz and Bernardos, 2000; Massey, 2002). Chemomyectomy with muscle-necrotizing drugs, such as doxorubicin, has been tried in some patients with blepharospasm and hemifacial spasm (Wirtschafter, 1991), but because of severe local irritation, it is doubtful that this approach will be adopted into clinical practice.
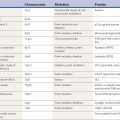
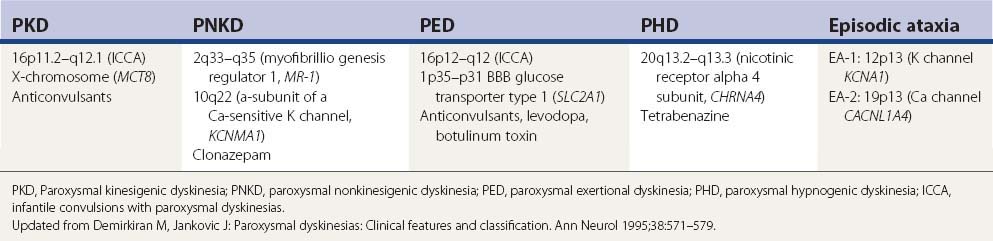
Attacks of kinesigenic paroxysmal dystonia may be controlled with anticonvulsants (e.g., carbamazepine, phenytoin) (Table 13.2). The nonkinesigenic forms of paroxysmal dystonia are less responsive to pharmacologic therapy, although clonazepam and acetazolamide may be beneficial. Treatment of paroxysmal dyskinesias is covered in Chapter 22 and in other reviews (Fahn, 1994; Demirkiran and Jankovic, 1995). Table 13.3 lists the common medications and procedures utilized for dystonia.
| Focal dystonias |
| Blepharospasm |
In addition to conventional forms of therapy described previously, many patients with dystonia seek complementary or alternative forms of therapy. In one survey of 180 members of the German Dystonia Group, 131 (73%) patients used some form of alternative treatments, such as acupuncture (56%), relaxation techniques (44%), homeopathy (27%), and massages (26%) (Junker et al., 2004).
The involuntary muscle contraction can lead to “status dystonicus” or “dystonic storm” resulting in breakdown of the muscle and a life-threatening myoglobinuria (Opal et al., 2002; Mariotti et al., 2007). In a review of 37 cases of status dystonicus or dystonic storm, the following treatment approach has been used with some success: early admission to the intensive care unit, monitor for evidence of myoglobinuria, avoid respiratory and renal compromise, sedation with intravenous midazolam (initially up to 10 µg/kg/min and subsequently at 30–100 µg/kg/hour), possible barbiturate anesthesia combined with endotracheal intubation and mechanical ventilation, continuous intrathecal baclofen, bilateral pallidotomy, or globus pallidus interna (GPi) deep brain stimulation (DBS) (Mariotti et al., 2007; Jech et al., 2009). Although administration of propofol has been also suggested, we would caution against it as this drug has been demonstrated to cause or exacerbate levodopa-related dyskinesia (Krauss et al., 1996).
Botulinum toxin
The introduction of BTX into clinical practice in the late 1980s revolutionized treatment of dystonia. The most potent biologic toxin, BTX has become a powerful therapeutic tool in the treatment of a variety of neurologic, ophthalmic, and other disorders that are manifested by abnormal, excessive, or inappropriate muscle contractions (Jankovic and Brin, 1991; Jankovic and Hallett, 1994; Brin et al., 2002; Thant and Tan, 2003; Jankovic, 2004a). In December 1989, after extensive laboratory and clinical testing, the Food and Drug Administration approved this biologic (BTX-A or Botox) as a therapeutic agent in patients with strabismus, blepharospasm, and other facial nerve disorders, including hemifacial spasm. In December 2000, the Food and Drug Administration approved Botox and BTX-B (Myobloc) as treatments for cervical dystonia. Although its widest application is still in the treatment of disorders manifested by abnormal, excessive, or inappropriate muscle contractions, the use of BTX is rapidly expanding to include treatment of a variety of ophthalmologic, gastrointestinal, urologic, orthopedic, dermatologic, secretory, painful, and cosmetic disorders (Tintner and Jankovic, 2001b; Jankovic and Brin, 2002; Thant and Tan, 2003; Jankovic, 2004a) (Fig. 13.1; Tables 13.4 and 13.5).
| Focal dystonia |
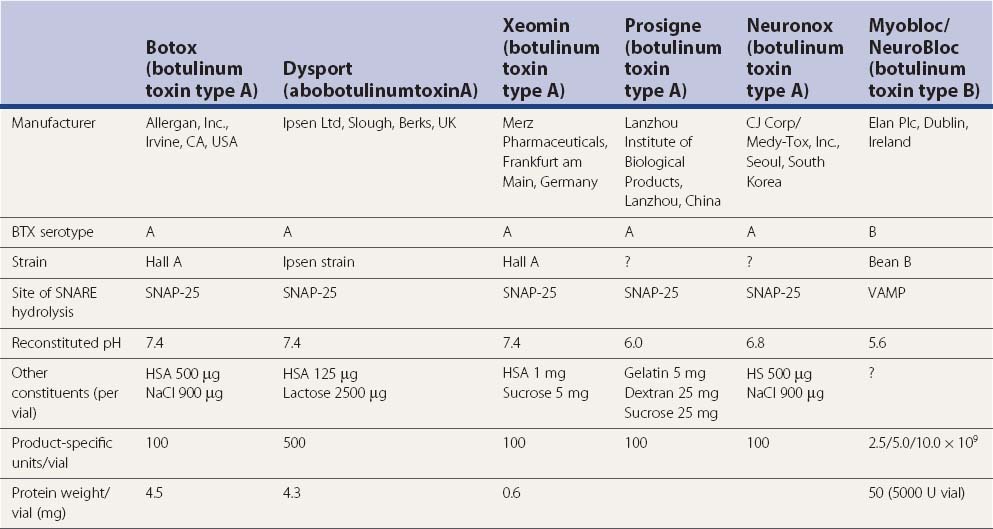
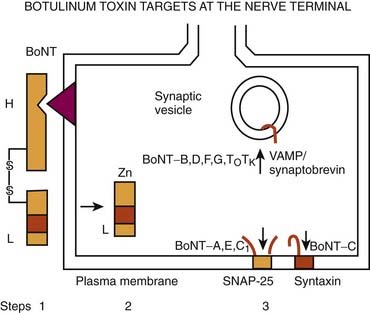
Few therapeutic agents have been better understood in terms of their mechanism of action before their clinical application or have had greater impact on patients’ functioning than BTX. The therapeutic value of BTX is due to its ability to cause chemodenervation and to produce local paralysis when injected into a muscle. There are seven immunologically distinct toxins that share structurally homologous subunits. Synthesized as single-chain polypeptides (molecular weight of 150 kDa), these toxin molecules have relatively little potency until they are cleaved by trypsin or bacterial enzymes into a heavy chain (100 kDa) and a light chain (50 kDa). The 150 kDa protein, the active portion of the molecule, complexes with one or more nontoxin proteins that support its structure and protect it from degradation. Type A is the only serotype that forms the 900 kDa complex. Types A, B, C1, and hemagglutinin-positive D form the 500 kDa complex and 300 kDa complex; types E, F, and hemagglutinin-negative D form only the 300 kDa complex (Melling et al., 1998). The three-dimensional structure of the BTX complex is known (Hanson and Stevens, 2002). When linked by a disulfide bond, these dichains exert their paralytic action by preventing the release of acetylcholine. BTX therefore does not affect the synthesis or storage of acetylcholine, but it interferes with the release of acetylcholine from the presynaptic terminal. This is a three-step process that involves binding to the acceptors on presynaptic membrane (heavy chain), internalization (endocytosis), and an enzymatic action (light chain). BTX serotypes bind to different acceptors, which contain both protein components and gangliosides with more than one neuraminic acid residue. BTX-A has been found to enter neurons by binding to the synaptic vesicle protein SV2 (isoform C) which acts as the BTX-A receptor (Dong et al., 2006; Mahrhold et al., 2006). The neural membrane proteins synaptotagmin I and II act as receptors for BTX-B and for BTX-G (Mahrhold et al., 2006). While the heavy chain of the toxin binds to the presynaptic cholinergic terminal, the light chain acts as a zinc-dependent protease that selectively cleaves proteins that are critical for fusion of the presynaptic vesicle with the presynaptic membrane. Thus, the light chains of BTX-A and BTX-E cleave SNAP-25 (synaptosome-associated protein), a protein that is needed for synaptic vesicle targeting and fusion with the presynaptic membrane. The light chains of BTX-B, BTX-D, and BTX-F prevent the quantal release of acetylcholine by proteolytically cleaving synaptobrevin-2, also known as VAMP (vesicle-associated membrane protein), an integral protein of the synaptic vesicle membrane. BTX-C cleaves syntaxin, another plasma membrane-associated protein. A three-dimensional study showed that syntaxin 1a forms a complex with neuronal-Sec1, forming a recognition site for the arriving vesicle through a Rab protein, and the subsequent formation of the syntaxin–VAMP–SNAP-25 complex promotes membrane fusion (Misura et al., 2000) (Fig. 13.1).
BTX-A has been studied most intensely and used most widely, but the clinical applications of other types of toxins, including BTX-B and BTX-F, are also expanding (Greene and Fahn, 1992b; Mezaki et al., 1995; Sheean and Lees, 1995; Figgit and Noble, 2002; Jankovic, 2004a). BTX-A is harvested from a culture medium after fermentation of a high-toxin-producing strain of Clostridium botulinum that lyses and liberates the toxin into the culture (Schantz and Johnson, 1992). The toxin is then extracted, precipitated, purified, and finally crystallized with ammonium sulfate. The crystalline toxin is diluted from milligram to nanogram concentrations, freeze-dried, and dispensed as a white powder in small vials containing 100 mouse units (U) of the toxin. When isolated from bacterial cultures, BTX is noncovalently associated with nontoxic macromolecules, such as hemagglutinin. These nontoxic proteins enhance toxicity by protecting the neurotoxin from proteolytic enzymes in the gut, but they apparently have no effect on the potency of the toxin if injected parenterally. Although the efficacy and duration of benefits of BTX-B or Myobloc, formerly NeuroBloc (Elan), are thought to be generally comparable to those of BTX-A (Botox and Dysport), no head-to-head comparisons between the various products have been performed. An in-vivo study using injections into the extensor digitorum brevis in healthy volunteers suggested that the muscle paralysis from BTX-B was not as complete or long-lasting as that from BTX-A (Sloop et al., 1997). Whether M-wave amplitude is a reliable measure of clinical response and whether the doses of BTX-A (7.5–10 U) and BTX-B (320–480 U), with a B/A ratio of about 45 : 1, are comparable are debatable. The apparently longer duration of action of BTX-A compared to that of BTX-B could possibly be explained by the observation that VAMP that is cleaved by BTX-B cannot form stable SNARE (soluble N-ethlymaleimide sensitive factor attachment protein receptor or SNAP receptor or neuronal synaptosome-associated proteins) complex and it turns over to form new VAMP, whereas SNAP-25, the substrate for BTX-A, forms a truncated SNAP-25A, which prevents degradation of SNARE, as a result of which the inhibition of exocytosis persists for 40–60 days. This correlates well with the reappearance of the original terminals (de Paiva et al., 1999). De Paiva and colleagues (1999) noted that a single intramuscular injection of BTX-A into the sternomastoid muscles of mice caused the formation of functional neuronal sprouts that connected with the muscle fiber. The primary BTX-A-intoxicated nerve terminal was incapable of neurotransmitter exocytosis; it produced new sprouts that were capable of exocytosis with subsequent upregulation of adjacent nicotinic receptors on the muscle fiber, thus forming a functional synapse. After a certain period of time, such as 3 months, consistent with return of clinical function of the muscle and wearing-off response from the previous injection, the original BTX-A-intoxicated terminal resumed exocytosis, and the sprouts regressed to return the neuromuscular junction to its original state. Other studies have also shown that the prolonged duration of action of BTX-A is due to persistence of catalytically active enzyme in the muscle cell (Adler et al., 2001). A physiologic study in monkeys showed that BTX-B diffuses less extensively than BTX-A into adjacent and remote muscles (Arezzo et al., 2002). In contrast to Botox, Myobloc is a solution that does not require reconstitution, and it may be stored in a refrigerator rather than a freezer. The commercial preparations are complex proteins that must be dissociated from the active neurotoxin molecule to exert the paralytic effect. This dissociation, which is pH dependent, unmasks the binding site on the heavy chain.
In addition to Botox, Myobloc, and Dysport (Truong et al., 2005), a new formulation of BTX-A, NT 201, has been recently introduced (Benecke et al., 2005). This formulation of BTX-A, which is free of complexing proteins, has been reported to be equivalent, in terms of efficacy and safety, to Botox in healthy volunteers and in patients with blepharospasm (Roggenkamper et al., 2006; Jankovic, 2009b) and cervical dystonia (Benecke et al., 2005). While low antigenicity has been predicted with this new formulation of BTX, no long-term data exist, and in view of the low frequency of blocking antibodies reported with the new formulation of Botox (see later) (Jankovic et al., 2003; Brin et al., 2008; Naumann et al., 2010) (Tables 13.6 and 13.7), it is not clear what role the new NT 201 BTX will play in the future treatment of dystonia. Patients resistant to BTX-A often initially respond to BTX-B, but a recent study showed that continued treatment with Myobloc is associated with high incidence of blocking antibodies (Jankovic et al., 2006).
Table 13.6 Risk factors for development of blocking antibodies (immunoresistance)
Table 13.7 Long-term effects of repeat botulinum toxin injections in cervical dystonia
From: Brin MF, Comella CL, Jankovic J, Lai F, Naumann M; CD-017 BoNTA Study Group. Long-term treatment with botulinum toxin type A in cervical dystonia has low immunogenicity by mouse protection assay. Mov Disord 2008;23:1353–1360.
The primary effect of BTX is to induce paralysis of injected skeletal muscles, especially the most actively contracting muscles. BTX paralyzes not only the extrafusal fibers but also the intrafusal fibers, thus decreasing the activity of Ib muscle afferents (Filippi et al., 1993). This might explain the effect of BTX on reciprocal inhibition. In untreated patients with dystonia, the second phase of reciprocal inhibition is usually decreased. BTX “corrects” the abnormal reciprocal inhibition by increasing the second phase, possibly through its effect on the muscle afferents (Priori et al., 1995). While the effect on intrafusal fibers might contribute in part to the beneficial action of BTX in patients with dystonia, this is not its main action because BTX is effective in facial dystonia, even though the facial muscles do not have spindles.
Measuring variations in fiber diameter and using acetylcholine-esterase staining as indexes of denervation, Borodic and colleagues (1994) showed that BTX diffuses up to 4.5 cm from the site of a single injection (10 U injected in the rabbit longissimus dorsi). Since the size of the denervation field is determined largely by the dose (and volume), multiple injections along the affected muscle rather than a single injection should therefore contain the biologic effects of the toxin in the targeted muscle (Borodic et al., 1992). Blackie and Lees (1990) also showed that frequency of dysphagia could be reduced by 50% when multiple rather than single injections are used.
A small percentage of patients receiving repeated injections develop antibodies against BTX, causing them to be completely resistant to the effects of subsequent BTX injections (Greene et al., 1994; Jankovic and Schwartz, 1995; Jankovic, 2002) (Tables 13.6 and 13.7). In one study, 24 of 559 (4.3%) patients treated for cervical dystonia developed BTX antibodies (Greene et al., 1994). The authors suggested that the true prevalence of antibodies might be more than 7%. In addition to patients with BTX antibodies, they studied 8 patients from a cohort of 76 (10.5%) who stopped responding to BTX treatments. These BTX-resistant patients had a shorter interval between injections, more booster injections, and a higher dose at the non-booster injection compared to nonresistant patients treated during the same period. As a result of this experience, clinicians are warned against using booster injections and are encouraged to extend the interval between treatments as long as possible, certainly at least 1 month, and to use the smallest possible dose. In addition to high dosages, young age is a potential risk factor for the development of immunoresistance to BTX-A (Jankovic and Schwartz, 1995; Hanna and Jankovic, 1998; Hanna et al., 1999). Some of the patients who developed BTX-A antibodies have benefited from injections by immunologically distinct preparations, such as BTX-F and BTX-B (Greene and Fahn, 1992b; Mezaki et al., 1994). After 1–3 years, some patients become antibody negative, and when reinjected with the same type of toxin, they may again experience transient benefit (Sankhla et al., 1998). The original preparation of Botox contained 25 ng of neurotoxin complex protein per 100 units, but in 1997, the Food and Drug Administration approved a new preparation that contains only 5 ng per 100 units, which should have lower antigenicity (Aoki et al., 1999). In fact, in a 3-year follow-up of patients treated with the current Botox, no evidence of blocking antibodies was found, compared to a 9.4% frequency of blocking antibodies in patients treated with the original Botox for the same period of time (Jankovic et al., 2003). The low antigenicity of the current Botox is also supported by the findings from a longitudinal, multicenter study in which de novo subjects were treated with Botox and only 4/326 (1.2%) subjects tested positive for antibodies by the mouse protective assay during the course of up to 15 cycles of treatment (Brin et al., 2008) (Table 13.7).
The preliminary data suggest that BTX-B provides clinical effects similar to those of BTX-A, but the duration of benefits is shorter (Comella et al., 2005) and Myobloc appears to have greater antigenicity than Botox, particularly in patients with prior resistance to BTX-A (Jankovic et al., 2007). To compare autonomic effects of BTX, Tintner and colleagues (2005) randomized patients with cervical dystonia to receive either BTX-A or BTX-B in a double-blind manner. Efficacy and physiologic questionnaire measures of autonomic function were assessed at baseline and 2 weeks after injection. Patients who were treated with BTX-B had significantly less saliva production (P < 0.01) and greater severity of constipation (P = 0.037) than those treated with BTX-A but did not differ with respect to other tests of autonomic function, including changes in blood pressure, heart rate, and ocular function. The autonomic effects of BTX-B were reviewed in several additional reports (Wan et al., 2005).
Many clinical studies have provided evidence not only that BTX is safe and effective, but also that this therapy leads to meaningful improvements in quality of life (Naumann and Jankovic, 2004; Jankovic et al., 2004), and the benefits are long-lasting (Mejia et al., 2005) (Video 13.1). Despite its proven therapeutic value, there are still many unresolved issues and concerns about BTX. These include lack of standardization of biologic activity of the different preparations of BTX, poor understanding of toxin antigenicity, variations in the methods of injection, and inadequate assays for BTX antibodies (Brin et al., 2008). Training guidelines for the use of botulinum toxin have been established (American Academy of Neurology, 1994). Clinicians who are interested in utilizing BTX chemodenervation in their practice must be aware of these concerns and must exercise proper precautions to minimize the potential risks associated with BTX. Most important, they should become thoroughly familiar with the movement disorders that they intend to treat and with the anatomy at the injection site. Possible contraindications to the use of BTX include the presence of myasthenia gravis (Emerson, 1994), Eaton–Lambert syndrome, motor neuron disease, aminoglycoside antibiotics, and pregnancy. Besides occasional complications, usually related to local weakness, a major limitation of BTX therapy is its high cost. Several studies analyzing the cost-effectiveness of BTX treatment, however, have demonstrated that the loss of productivity as a result of untreated dystonia and the cost of medications or surgery more than justify the financial expense of BTX treatments. It is likely that future research will result in the development of new, more effective neuromuscular blocking agents that provide therapeutic chemodenervation with long-term benefits and at lower cost. 
Blepharospasm
The effectiveness of BTX in blepharospasm was first demonstrated in double-blind, placebo-controlled trials (Fahn et al., 1985b; Jankovic and Orman, 1987). In a subsequent report of experience with BTX in 477 patients with various dystonias and hemifacial spasm, Jankovic and colleagues (1990) reviewed the results in 90 patients who had been injected with BTX for blepharospasm. Moderate or marked improvement was noted in 94% of the patients. The average latency from the time of the injection to the onset of improvement was 4.2 days; the average duration of maximum benefit was 12.4 weeks, but the total benefit lasted considerably longer: an average of 15.7 weeks. While 41% of all treatment sessions were followed by some side effects (ptosis, blurring of vision or diplopia, tearing, and local hematoma), only 2% of these affected patient’s functioning. Complications usually improved spontaneously in less than 2 weeks. These results are consistent with those of other studies (Elston, 1994). There is no apparent decline in benefit, and the frequency of complications actually decreases after repeat BTX treatments (Jankovic and Schwartz, 1993a).
The efficacy and safety of another type of BTX-A, NT-201 (or Xeomin), in the treatment of blepharospasm was also confirmed by a prospective, double-blind, placebo-controlled, randomized, multicenter study involving 109 patients (mean total dose of Xeomin per treatment visit was 64.8 U), in which the Jankovic Rating Scale severity subscore was significant reduced compared to placebo (P < 0.001). The most commonly reported adverse effects related to Xeomin versus placebo were eyelid ptosis (18.9 vs. 8.8%), dry eye (16.2 vs.11.8%), and dry mouth (16.2 vs. 2.9%) (Jankovic, 2009b; Jankovic et al., 2011). In a multicenter, clinical, fixed-dose, trial Dysport (40, 80, and 120 units/eye) or placebo (in a 3 : 1 randomization ratio) were administered to 119 patients with blepharospasm, 85 of whom completed the 16-week follow-up; several parameters showed robust improvement in Dysport arms compared to placebo (Truong et al., 2008).
In addition to the four different BTX-A preparations available on the market in one or more countries, Botox (Allergan Inc., Irvine, CA, USA), Dysport (Ipsen Ltd, Slough, UK), Xeomin (Merz Pharmaceuticals, Frankfurt am Main, Germany) (Albanese, 2011), and Prosigne (Lanzhou Biological Products, China), Meditoxin (Medy-Tox, Seoul, Korea; also known as Neuronox) was introduced for the treatment of blepharospasm and is currently available in Korea (Yoon et al., 2009). All these types were evaluated in blepharospasm and were found to be comparable with respect to clinical efficacy and adverse effects.
Reasons for the gradual enhancement in efficacy and reduction in the frequency of complications with repeat treatments include greater experience and improvements in the injection technique. For example, one controlled study showed that an injection into the pretarsal rather than preseptal portion of the orbicularis oculi is associated with significantly lower frequency of ptosis (Jankovic, 1996). Ptosis can be prevented also by injecting initially only the lateral and medial portions of the upper lid, thus avoiding the midline levator muscle. We usually initially inject 5 U in each site in the upper lid and 5 U in the lower lid laterally only. This finding has been confirmed by others (Cakmur et al., 2002).
The functional improvement, experienced by the vast majority of patients after BTX injection, is difficult to express numerically. Many patients could not work, drive, watch television, or read prior to the injections. As a result of reduced eyelid and eyebrow spasms, most can now function normally. In addition to the observed functional improvement, there is usually a meaningful amelioration of discomfort, and because of less embarrassment, the patients’ self-esteem also frequently improves. BTX injections are now considered by many to be the treatment of choice for blepharospasm (American Academy of Ophthalmology, 1989; American Academy of Neurology, 1990). In addition to idiopathic blepharospasm, BTX injections have been used effectively in the treatment of blepharospasm induced by drugs (e.g., levodopa in parkinsonian patients or neuroleptics in patients with tardive dystonia), dystonic eyelid and facial tics in patients with Tourette syndrome, and blepharospasm associated with apraxia of eyelid opening (Jankovic, 1994; Aramideh et al., 1995; Lepore et al., 1995; Jankovic, 1995a, 1996; Forget et al., 2002).
Oromandibular dystonia
Oromandibular dystonia is among the most challenging forms of focal dystonia to treat with BTX (Blitzer et al., 1989; Charous et al., 2011); it rarely improves with medications, there are no surgical treatments, and BTX therapy can be complicated by swallowing problems. The masseter muscles are usually injected in patients with jaw-closure dystonia; in patients with jaw-opening dystonia, either the submental muscle complex or the lateral pterygoid muscles are injected. In a total of 91 patients treated in 271 visits, the overall improvement was rated as 2.6 (0 = no response to 4 = marked improvement in spasms and function) (Jankovic and Schwartz, 1991b). A meaningful reduction in the oromandibular-lingual spasms and an improvement in chewing and speech were achieved in more than 70% of patients. The improvement was noted within an average of 5.5 days after injection and lasted an average of 11.5 weeks. Patients with dystonic jaw closure responded better than those with jaw-opening dystonia. A temporary swallowing problem, noted in fewer than one-third of patients and in only 17% of all treatment sessions, was the most frequent complication. Early treatment with BTX may prevent dental and other oral complications, including temporomandibular joint syndrome (Blitzer and Brin, 1991; Charous et al., 2011). BTX can provide lasting improvement not only in patients with primary (idiopathic) dystonia but also in orolingual-mandibular tardive dystonia. Clenching and bruxism are frequent manifestations of oromandibular dystonia, although nocturnal and diurnal bruxism can occur even without evident dystonia (Tan and Jankovic, 1999, 2000). Oromandibular involuntary movements caused by hemimasticatory spasms and other disorders such as Satoyoshi syndrome have been also successfully treated with BTX (Merello et al., 1994; Yaltho and Jankovic, 2011).
Laryngeal dystonia (spasmodic dysphonia)
Until the introduction of BTX, the therapy of spasmodic dysphonia was disappointing. The anticholinergic and benzodiazepine drugs only rarely provide meaningful improvement in voice quality. Unilateral transection of the recurrent laryngeal nerve, although effective in most patients, frequently causes unacceptable complications, and the voice symptoms often recur (Dedo and Izdebski, 1983). A surgical procedure that involves denervation of the adductor branch of the recurrent laryngeal nerve with reinnervation of the distal sumps with branches of the ansa cervicalis nerve was reported to produce marked improvement in patients with adductor spasmodic dysphonia (Berke et al., 1999), but further studies are needed to confirm this initial observation.
Several studies have established the efficacy and safety of BTX in the treatment of laryngeal dystonia, and this approach is considered by most to be the treatment of choice for spasmodic dysphonia (Brin et al., 1987; Blitzer et al., 1988; Jankovic et al., 1990; Brin et al., 1992; Ludlow, 1990; Brin et al., 1998; Blitzer et al., 2002). Before a patient can be considered a potential candidate for BTX injections, the diagnosis of spasmodic dysphonia must be confirmed by detailed neurologic, otolaryngologic, and voice assessment and documented by video and voice recordings.
Three approaches are currently used in the BTX treatment of spasmodic dysphonia: (1) unilateral EMG-guided injection of 5–30 U (Jankovic et al., 1990), (2) bilateral approach, injecting with EMG guidance 1.25–4 U in each vocal fold (Brin et al., 1992), and (3) an injection via indirect laryngoscopy without EMG (Ford et al., 1990). Irrespective of the technique, most investigators report about 75–95% improvement in voice symptoms. One controlled study, however, concluded that unilateral injections “may provide both superior and longer lasting benefits” than bilateral injections (Adams et al., 1993). The dosage can be adjusted depending on the severity of glottal spasms and the response to previous injections. Adverse experiences include transient breathy hypophonia, hoarseness, and rare dysphagia with aspiration.
Although more complicated and less effective, BTX injections into the posterior cricoarytenoid muscle with the EMG needle placed posterior to the thyroid lamina may be used in the treatment of the abductor form of spasmodic dysphonia (Brin et al., 1989, 1992). Using a multidisciplinary team approach, consisting of an otolaryngologist who is experienced in laryngeal injections and a neurologist who is knowledgeable about motor disorders of speech and voice, BTX injections can provide effective relief for most patients with spasmodic dysphonia (American Academy of Neurology, 1990; American Academy of Otolaryngology, 1990). Outcome assessments clearly show that BTX injections for spasmodic dysphonia produce measurable improvements in the quality of life of patients with this disorder (Courey et al., 2000). BTX may be useful in the treatment of voice tremor and stuttering, but the results are less predictable (Ludlow, 1990; Brin et al., 1994; Warrick et al., 2000).
Cervical dystonia
The goal of therapy of cervical dystonia is not only to improve abnormal posture of the head and associated neck pain, but also to prevent the development of secondary complications such as contractures, cervical radiculopathy, and cervical myelopathy (Treves and Korczyn, 1986; Waterston et al., 1989).
With the use of TWSTRS and other scales, the efficacy and safety of BTX in the treatment of cervical dystonia have been demonstrated in several controlled and open trials (Greene et al., 1990; Jankovic and Schwartz, 1990; Poewe et al., 1992; Jankovic et al., 1994; Tintner and Jankovic, 2001a; Jankovic, 2002, 2004c; Truong et al., 2005; Brin et al., 2008) (Videos 13.2 and 13.3). In one double-blind, placebo-controlled study of 55 patients with cervical dystonia, 61% improved after BTX-A injection (Greene et al., 1990). BTX has been found to be superior not only to placebo but also to trihexyphenidyl (Brans et al., 1996). In a study of 66 consecutive patients with idiopathic cervical dystonia, Brans and colleagues (1996) compared the effectiveness of BTX-A (Dysport) with that of trihexyphenidyl in a prospective, randomized, double-blind design. Dysport or saline was injected under EMG guidance at study entry and again after 8 weeks. Patients were assessed for efficacy at baseline and after 12 weeks by different clinical rating scales. Sixty-four patients completed the study, 32 in each group. The mean dose of BTX-A was 292 U (first session) and 262 U (second session). The mean dose of trihexyphenidyl was 16.25 mg. TWSTRS (primary outcome) and other scales showed a significant improvement in favor of BTX-A, and adverse effects were significantly less frequent than in the group of patients randomized to trihexyphenidyl. In a multicenter, double-blind, randomized, controlled trial assessing the safety and efficacy of Dysport in cervical dystonia patients, 80 patients were randomly assigned to receive one treatment with Dysport (500 U) or placebo (Truong et al., 2005). Dysport was significantly more efficacious than placebo at weeks 4, 8, and 12 as assessed by TWSTRS (10 point vs. 3.8 point reduction in total score, respectively, at week 4; P ≤ 0.013). Of participants in the Dysport group, 38% showed positive treatment response, compared to 16% in the placebo group (95% confidence interval 0.02–0.41). The median duration of response to Dysport was 18.5 weeks. Side effects were generally similar in the two treatment groups; only blurred vision and weakness occurred significantly more often with Dysport. No participants in the Dysport group converted from negative to positive antibodies after treatment. These results confirm previous reports that Dysport (500 U) is safe, effective, and well tolerated in patients with cervical dystonia. 
Open-label studies generally report a more dramatic improvement, partly because of a placebo effect and, more important, because of greater flexibility in selecting the proper dosage and site of injection. Most trials report that about 90% of patients experience improvement in function and control of the head and neck and in pain. The average latency between injection and the onset of improvement (and muscle atrophy) is 1 week, and the average duration of maximum improvement is 3–4 months. On average, the injections are repeated every 4–6 months. Patients with long-duration dystonia have been found to respond less well than those who were treated relatively early, possibly because prolonged dystonia produced contractures (Jankovic and Schwartz, 1991b). In one study, 28% of patients experienced some complication, such as swallowing difficulties, neck weakness, and nausea, sometime during the course of their treatment (some patients had up to 12 visits in 5 years). Dysphagia, the most common complication, was encountered in 14% of all 659 visits, but in only five instances was this problem severe enough to require changing to a soft or liquid diet. Complications are usually related to focal weakness, although distant and systemic subclinical and clinical effects, such as generalized weakness and malaise, rarely occur, possibly as a result of blood distribution or retrograde axonal transport to the spinal motor neurons (Garner et al., 1993). Most complications resolve spontaneously, usually within 2 weeks. An injection into one or both sternocleidomastoid muscles was most frequently associated with dysphagia (Jankovic and Schwartz, 1991b; Comella et al., 1992b). One study showed that dosages as small as 20 units administered as a single injection into the sternocleidomastoid muscle completely eliminated muscle activity and could produce neck weakness and dysphagia (Buchman et al., 1994). In an analysis of patients who received five or more injections, the beneficial response was maintained and the frequency of complications with repeat injections actually declined, presumably as a result of improving skills (Jankovic and Schwartz, 1993a). Lindeboom and colleagues (1998) found that neurologic impairment and pain usually improve following BTX injections, but only functional status measures differentiate between patients who improve and those who have an insufficient response. There have been only a few long-term studies of BTX treatment in cervical dystonia. Brashear and colleagues (2000) showed that two-thirds of patients who received BTX reported the injections always helped. Another long-term study showed that 75% of patients continued to benefit for at least 5 years and only 7.5% developed secondary unresponsiveness; only 1.3% discontinued BTX therapy because of intolerable side effects (Hsiung et al., 2002).
Results similar to those obtained with BTX-A have been obtained in patients treated for cervical dystonia with BTX-B (Tsui et al., 1995; Lew et al., 1997; Figgitt and Noble, 2002). In a double-blind, controlled trial of 122 patients with cervical dystonia treated with BTX-B, there was a dose response effect, particularly at doses of 10 000 units (Lew et al., 1997). Using TWSTRS, 77% of the patients were found to respond at week 4. Other studies have subsequently confirmed the efficacy of BTX-B (Brashear et al., 1999), even in patients who are resistant to BTX-A (Brin et al., 1999). In a 16-week, randomized, multicenter, double-blind, placebo-controlled trial of BTX-B, 109 patients, who previously responded well to BTX-A, were randomized into one of the treatment groups: placebo, 5000 U, and 10 000 U administered into two to four cervical muscles (Brashear et al., 1999). At week 4, the total TWSTRS score improved by 4.3, 9.3 (P = 0.01), and 11.7 (P = 0.0004), respectively, when compared to baseline, and this was accompanied by significant improvements in pain, disability, and severity. The estimated median time until the total TWSTRS score returned to baseline was 63, 114, and 111 days, respectively. The most frequent side effects associated with BTX-B included dysphagia and dry mouth. An identical design was used in another study of BTX-B in cervical dystonia with one exception: the patients were resistant to BTX-A as determined by the frontalis-type A test (F-TAT) (Brin et al., 1999). A total of 77 patients were randomized to receive placebo or 10 000 U of BTX-B. At week 4, the total TWSTRS scores improved by 2 (placebo) and 11 (10 000 U) (P = 0.0001). There was also significant improvement in secondary and tertiary outcome measures, including global assessments and pain visual analog scores, as well as other measures of pain, disability, and severity. The estimated duration of effect, based on a Kaplan–Meier survival analysis, was 112 days (12–16 weeks). Subsequent studies have suggested that dosages as high as 45 000 U of BTX-B (Myobloc) per session may be effective and safe in patients with cervical dystonia.
The most important determinants of a favorable response to BTX treatments are a proper selection of the involved muscles and an appropriate dosage (Table 13.8). EMG may be helpful in some patients with obese necks or in whom the involved muscles are difficult to identify by palpation (Dubinsky et al., 1991; Gelb et al., 1991). One study attempted to determine the usefulness of EMG-assisted BTX injections and found that the percentage of patients who showed any improvement after BTX was similar whether the injections were assisted by EMG or not (Comella et al., 1992a). Comella et al. (1992a) also noted that “a significantly greater magnitude of improvement” was present in patients treated with the EMG-assisted method and that there was “a significantly greater number of patients with marked benefit” in the group that was randomly assigned to the EMG-assisted method of treatment. Since the majority (70–79%) of patients had previously been treated with BTX, some might have been experiencing residual effects from previous injections, making the interpretation of the results difficult. Furthermore, the patients who were treated without EMG assistance received a higher dose, indicating more severe dystonia, thus possibly explaining the lesser degree of observed improvement. The general consensus among most BTX users is that EMG is not needed in the vast majority of patients, except in rare instances when the muscles cannot be adequately palpated or the patient does not obtain adequate relief of symptoms with the conventional approach (Jankovic, 2001b). BTX treatment not only provides effective symptomatic relief in patients with cervical dystonia but has also dramatically changed the natural history of the disease as it also prevents contractures. Early introduction of BTX has been shown to prevent contractures even in patients with congenital torticollis (Collins and Jankovic, 2006).
|
1 Find the most uncompensated position
|
Writer’s cramp and other limb dystonias
Treatments of writer’s cramp with muscle relaxation techniques, physical and occupational therapy, and medical and surgical therapies have been disappointing. Several open (Rivest et al., 1990; Jankovic and Schwartz, 1993b; Karp et al., 1994; Priori et al., 1995; Pullman et al., 1996; Quirk et al., 1996; Wissel et al., 1996) and double-blind controlled (Yoshimura et al., 1992; Tsui et al., 1993; Cole et al., 1995) trials have concluded that BTX injections into selected hand and forearm muscles probably provide the most effective relief in patients with these task-specific occupational dystonias. In some studies, fine wire electrodes were used to localize bursts of muscle activation during the task, and the toxin was injected through a hollow EMG needle into the belly of the most active muscle (Cole et al., 1995). Similar beneficial results, however, were obtained in other studies without complex EMG studies (Rivest et al., 1990). Several lines of evidence support the notion that an intramuscular injection of BTX into the forearm muscles corrects the abnormal reciprocal inhibition (Priori et al., 1995).
Jankovic and Schwartz (1993b) studied the effects of BTX in 46 patients with hand dystonia who had injections into their forearm muscles in 130 treatment sessions. The average age was 49.4 years, and the dystonic symptoms had been present for an average of 8.6 years. After careful examination and palpation of the forearm muscles during writing, the toxin was injected into either the wrist flexors (116 injections) or the wrist extensors (52 injections). The average baseline severity of dystonia was 3.5 on a 0–4 rating scale. The average peak effect response for all treatment sessions was 2.3 (0 = no response to 4 = maximum benefit). The latency from injection to onset of effect averaged 5.6 days, and the benefit lasted an average of 9.2 weeks. Temporary hand weakness, the chief complication of this treatment, occurred in 54% of patients and in 34% of all treatment sessions. However, nearly all patients preferred the temporary weakness, which was usually mild, to the disabling writer’s cramps. Although one study showed that only 14 of 38 (37%) of needle placement attempts reached the proper hand muscles in the absence of EMG guidance, this does not mean that placement with EMG guidance correlates with better results, since the selection of the muscle involved in the hand dystonia is based on clinical examination, not on EMG (Molloy et al., 2002). One study showed that voluntary activity of the hand immediately after the treatment for 30 minutes enhanced the weakness produced by the injection which may, therefore, allow reduction in dosage and optimize the benefits (Chen et al., 1999).
In addition to improving writer’s cramp, BTX might provide relief in other task-specific disorders affecting typists, draftsmen, musicians, athletes, and other people who depend on skilled movements of their hands (Schuele et al., 2005). Other focal distal dystonias, besides those involving the hands, might be amenable to treatment with BTX. Patients with Parkinson disease, progressive supranuclear palsy, corticobasal degeneration, and other forms of parkinsonism or stroke-related hemiplegia occasionally develop secondary fixed dystonia of the hand (dystonic clenched fist), which might benefit in terms of pain and hygiene from local BTX injections (Cordivari et al., 2001). Patients with foot dystonia as a manifestation of primary (idiopathic) dystonia and patients with parkinsonism who experience foot dystonia as an early symptom of their disease or, more commonly, as a complication of levodopa therapy might benefit from local BTX infections (Pacchetti et al., 1995). BTX injections into the foot–toe flexors or extensors might not only alleviate the disability, pain, and discomfort that are often associated with such dystonia but also improve gait. Whether BTX injections will play an important role in the treatment of recurrent painful physiologic foot and calf cramps has yet to be determined.
Other indications for botulinum toxin
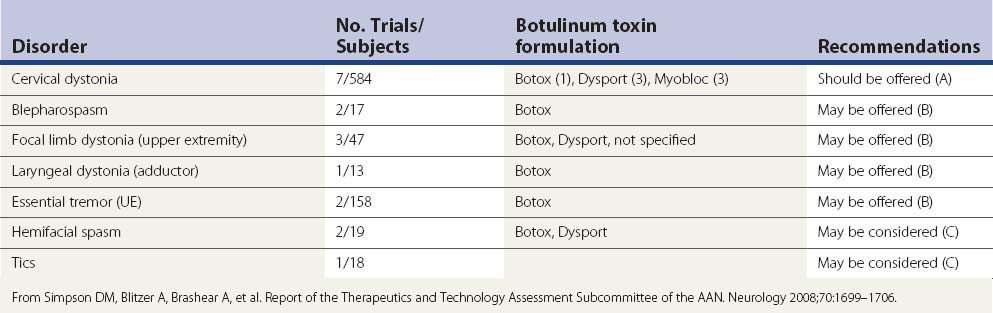
It is beyond the scope of this chapter to review the rapidly broadening indications for BTX therapy. The reader is referred to some recent reviews on this topic (Brin et al., 2002; Jankovic and Brin, 2002; Jankovic 2004a; Jankovic et al., 2009) and to Simpson et al. (2008) for evidence-based recommendations. Based on a thorough review of clinical trials with BTX, the American Academy of Neurology has made the following evidence-based recommendations: “BoNT should be offered as a treatment option for the treatment of cervical dystonia (Level A), may be offered for blepharospasm, focal upper extremity dystonia, adductor laryngeal dystonia, and upper extremity essential tremor (Level B), and may be considered for hemifacial spasm, focal lower limb dystonia and motor tics (Level C)” (Simpson et al., 2008) (Table 13.9).
Surgical treatment of dystonia
Although surgery has been used in the treatment of dystonia for a long time (Cooper 1965), there has been a recent resurgence in this approach, largely because of improvements in surgical and imaging techniques and as a result of surgical benefits observed in patients with tremor and Parkinson disease (Jankovic, 1998; Lang, 1998; Ondo et al., 2001).
Central ablative procedures and deep brain stimulation
Improved understanding of the functional anatomy of the basal ganglia and physiologic mechanisms underlying movement disorders, coupled with refinements in imaging and surgical techniques, has led to a resurgence of interest in thalamotomy in patients with disabling tremors, dystonia, and other hyperkinetic movement disorders (Grossman and Hamilton, 1993; Jankovic et al., 1996; Ondo et al., 2001). The observation, supported by both physiologic and positron emission tomographic studies, that there is a disruption of pallidothalamic-cortical projections in dystonia provides some rationale for treating dystonia by interrupting the abnormal outflow from the thalamus to the overactive prefrontal motor cortex (Lenz et al., 1990; Mitchel et al., 1990; Ceballos-Baumann et al., 1995).
In a longitudinal study of 17 patients with severe dystonia, 8 (47%) had a moderate to marked improvement in their abnormal postures and functional disability following thalamotomy (Cardoso et al., 1995). Patients with primary and secondary dystonia had similar responses, but 43% of the patients with primary dystonia deteriorated during a mean follow-up of 32.9 months, whereas only 30% of patients with secondary dystonia deteriorated during a mean follow-up of 41 months. Neurologic complications were observed in 6 of 17 patients (35%) immediately after surgery, but deficits (contralateral weakness, dysarthria, pseudobulbar palsy) persisted in only one subject. Mild weakness contralateral to the surgery was the most common complication, noted in 3 patients immediately following the procedure. The one patient who underwent bilateral procedures had no detectable dysarthria. These results are consistent with other studies that have reported improvement in 34–70% of patients with dystonia following thalamotomy (Cooper, 1976; Andrew et al., 1983; Tasker et al., 1988). While most studies concluded that distal dystonia responded more favorably to thalamotomy than axial dystonia, Andrew and colleagues (1983) felt that their patients with axial dystonia, including torticollis, also improved after thalamotomy. Most investigators, however, feel that the best candidates for thalamotomy are patients who have disabling, particularly unilateral, dystonia (hemidystonia) that is unresponsive to medical therapy. Patients with preexisting dysarthria are also good candidates because they have less to lose from thalamotomy-related speech disturbance. Patients with secondary (symptomatic) dystonia seem to respond better than those with idiopathic dystonia. Thalamotomy should not be recommended for patients with facial, laryngeal, and cervical or truncal dystonia.
The role of pallidotomy in the treatment of dystonia is currently being reevaluated in view of the emerging use of deep brain stimulation (Jankovic et al., 1997; Eltahawy et al., 2004). In one study of eight patients with severe generalized dystonia, there was 59% and 62.5% improvement in the Fahn–Marsden Dystonia Scale and in the Unified Dystonia Rating Scale, respectively, following pallidotomy (Ondo et al., 1998). The experience with pallidotomy continues to show favorable long-term effects, particularly in patients with primary dystonia (Ondo et al., 2001; Yoshor et al., 2001). Although the surgery does not slow or halt the progression of the underlying disease, most patients continue to benefit. The procedure is usually well tolerated, and the benefits have persisted for more than 10 years in most patients with primary dystonia. Pallidotomy has been also reported to be effective in a 47-year-old woman with paroxysmal dystonia induced by exercise (Bhatia et al., 1998).
Although there are no data from healthy humans, microelectrode recordings from patients with generalized dystonia indicate that the mean discharge rates (about 50 Hz) in the GPi are lower than those in patients with Parkinson disease (80–85 Hz) (Lozano et al., 1997) or parkinsonian primates (70–75 Hz) (Filion and Tremblay, 1991) but higher than those in hemiballism (Vitek et al., 1998, 1999) and that the proportion of GPi cells that respond to stimulation is higher in patients with dystonia than in those with hemiballism (Lenz et al., 1998). In comparison to normal or parkinsonian primates, the GPi discharges in patients with dystonia seem to be more irregular, with more bursting and pauses, and the receptive fields to passive and active movements seem to be widened (Kumar et al., 1999). The intraoperative neurophysiologic recordings in 15 patients with dystonia were compared with those of 78 patients with Parkinson disease undergoing pallidotomy; the discharge rates were lower in frequency and more irregular in the GPi and GPe of patients with dystonia (Sanghera et al., 2003).
Physiologic studies in patients with dystonia, based on lower mean discharge rates in GPe and GPi (Vitek et al., 1998, 1999), suggest overactivity of both direct (striatum–GPi) and indirect (striatum–GPe–GPi) pathways; the dopamine receptor ligand PET studies (Perlmutter et al., 1997; Asanuma et al., 2005) suggest increased activity in the direct and decreased activity in the indirect pathway. This is compatible with FDG PET studies supportive of excessive activity in the direct pathway (Eidelberg et al., 1995). In multiple reports, pallidotomy (Lozano et al., 1997; Ondo et al., 1998; Vitek et al., 1999) and GPi DBS (Coubes et al., 2000; Tronnier and Fogel, 2000; Vercueil et al., 2001; Bereznai et al., 2002; Vidailhet et al., 2005; Diamond et al., 2006; Isaias et al., 2008, 2009; Sensi et al., 2009) appear to be effective procedures for patients with dystonia (Fig. 13.2) (Videos 13.4, 13.5, and 13.6). Both procedures may provide benefit by disrupting the abnormal GPi pattern and thus reduce cortical overactivation, characteristic of dystonia (Eidelberg et al., 1995). Primary dystonia clearly responds better than secondary dystonia to either pallidotomy or GPi DBS, although patients with pantothenate kinase-associated neurodegeneration also experience marked improvement (Coubes et al., 2000; Eltahawy et al., 2004). 
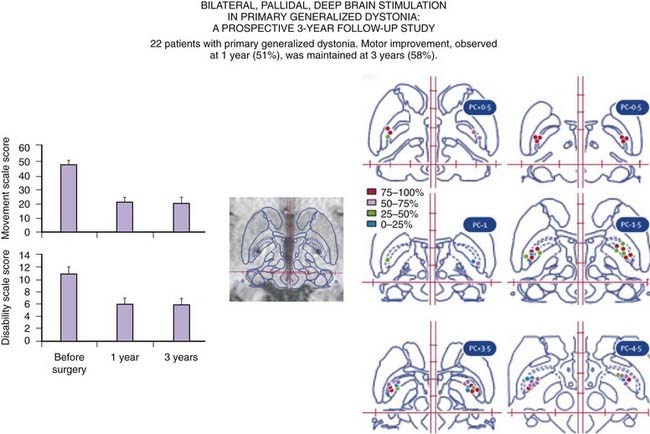
Figure 13.2 Long-term experience with GPi DBS in dystonia.
Redrawn from Vidailhet M, Vercueil L, Houeto JL, et al., French SPIDY Study Group. Bilateral, pallidal, deep-brain stimulation in primary generalised dystonia: a prospective 3 year follow-up study. Lancet Neurol 2007;6:223–229.
DBS targeting GPi has generally replaced ablative surgery in patients with disabling dystonia (Ostrem and Starr, 2008). Since an ablative lesion or high-frequency stimulation of the GPi can both produce and improve dystonia, this suggests it is the pattern of discharge in the basal ganglia rather than the actual location or frequency of discharge that is pathophysiologically relevant to dystonia (Münchau et al., 2000). In a prospective, multicenter study of 22 patients with generalized dystonia, seven of whom had a DYT1 mutation, the Fahn–Marsden Dystonia Scale score (FMDRS) improved after bilateral GPi DBS from a mean of 46.3 ± 21.3 to 21.0 ± 14.1 at 12 months (P < 0.001) (Vidailhet et al., 2005). A “blinded” review of the videos at 3 months showed improvement with stimulation from a mean of 34.63 ± 12.3 to 24.6 ± 17.7. The improvement in mean dystonia motor scores was 51%, and one-third of the patients improved more than 75% compared to preoperative scores. In addition, there was a significant improvement in health-related quality of life as measured by the SF-36, but there was no change in cognition or mood. Although the sample was rather small, the authors were not able to find any predictors of response such as DYT1 gene status, anatomic distribution of the dystonia, or location of the electrodes. Patients with the phasic form of dystonia improved more than those with tonic contractions and posturing. The maximum benefit was not achieved in some patients until 3–6 months after surgery. Vercueil and colleagues (2001) described 10 patients with bilateral GPi DBS; 5 had a major improvement, 2 had a moderated improvement, and 1 had a minor improvement after 14 months. They concluded that GPi DBS is much more effective than thalamic DBS for the treatment of dystonia. In a 2-year follow-up of 31 patients with primary generalized dystonia, Coubes and colleagues (2004) noted a mean improvement in clinical and functional FMDRS of 79% and 65%, respectively. There was no difference in response between DYT1-positive and DYT1-negative patients, but the magnitude of improvement was greater in children than in adults. Kupsch et al. (2006) compared GPi DBS with sham stimulation in a randomized, controlled clinical trial of 40 patients with primary segmental or generalized dystonia. The primary endpoint was the change from baseline to 3 months in the severity of symptoms, according to the movement subscore on the FMDRS. Two investigators who were unaware of treatment status assessed the severity of dystonia by reviewing videotaped sessions. Three months after randomization, the change from baseline in the mean (± SD) movement score was significantly greater in the neurostimulation group (−15.8 ± 14.1 points) than in the sham-stimulation group (−1.4 ± 3.8 points, P < 0.001). There was other evidence of benefit from DBS in this study. Several studies have demonstrated a gradual deterioration of dystonia over 48 hours or longer after GPi DBS is interrupted but this is rapidly reversible once the DBS is turned on (Grabli et al., 2009). In a study of 39 patients with dystonia undergoing GPi DBS, short duration of disease was identified as the only predictor of good outcome (among 32 patients with mobile dystonia the FMDRS score improved 87.8%) but the presence of fixed axial skeletal deformities was associated with poor outcome (Isaias et al., 2008). In a long-term follow-up of 30 patients with primary and generalized dystonia followed for up to 8 years, the hardware and stimulation adverse events were rare and the implantable pulse generators were replaced on the average every 24 months (Isaias et al., 2009).
In addition to primary dystonia, GPi DBS has been reported to be effective in patients with tardive dystonia (Trottenberg et al., 2005), cranial-cervical dystonia (Foote et al., 2005; Hebb et al., 2007; Ostrem et al., 2007) and other forms of segmental dystonia (Krauss et al., 2002). In addition, bilateral GPi DBS has been reported to be effective in patients with neurodegeneration with brain iron accumulation (NBIA) (Timmermann et al., 2010). Further studies are needed before surgery is recommended for these forms of dystonia. For example, blepharospasm associated with craniocervical dystonia has been reported to be exacerbated by bilateral GPi DBS (Vagefi et al., 2008). Although DBS has not been used frequently in the treatment of cranial-cervical dystonia, both GPi and STN have been targeted in the DBS treatment of this form of dystonia (Ostrem et al., 2011). GPi DBS has also been found helpful in the treatment of dystonic or parkinsonian camptocormia, an extreme flexion of the trunk (Jankovic, 2010).
Peripheral surgery
Peripheral denervation procedures were used extensively for cervical dystonia prior to the advent of BTX therapy. In one series of patients with cervical dystonia seen before 1990, 40 of 300 (13%) patients elected to have this type of surgery (Jankovic et al., 1991). While 10% of the patients noted worsening after the surgery, 38% experienced a noticeable improvement in the ability to control their head position or in reduction of the neck pain.
Three procedures have been used in the treatment of cervical dystonia: (1) extradural selective sectioning of posterior (dorsal) rami (posterior ramisectomy) with or without myotomy, (2) intradural sectioning of anterior cervical roots (anterior cervical rhizotomy), and (3) microvascular decompression of the spinal accessory nerve. Although the first procedure, championed by Bertrand (Bertrand and Molina-Negro, 1988), is considered by many clinicians to be the procedure of choice, no study has compared the different surgical approaches. Bertrand and Molina-Negro (1988) reported that 97 of 111 (87%) patients had “excellent” or “very good” results. In a smaller series, five of nine (56%) patients had moderate benefit that was sustained during up to 21 months of follow-up (Davis et al., 1991). The procedure is performed under general anesthesia without a paralyzing agent so that intraoperative nerve root stimulation can be used to identify the innervation to the dystonic muscles. This information, coupled with preoperative EMG, is used to avulse selected nerve roots, usually the branches of the spinal accessory nerve and the posterior rami of C1 through C6. Thorough avulsion of the peripheral branches is believed to be essential in preventing recurrences. Pain seems to improve more than the abnormal posture following the cervical muscle denervation, although some patients complain of stiff neck, sometimes lasting several months, after the surgery. Other complications may include local numbness, neck weakness, and rarely dysphagia. The chief disadvantage of anterior rhizotomy compared to posterior primary ramisectomy is that the former procedure causes denervation of both involved and uninvolved muscles, and it cannot be carried out at or below the C4 level because of the potential for involvement of the roots to the phrenic nerve, leading to paralysis of the diaphragm. The posterior ramisectomy (C2 to C6) allows more selective denervation of the involved muscles. Krauss and colleagues (1997) reported the effects of 70 intradural or extradural approaches in 46 patients with severe cervical dystonia. During a mean duration of follow-up of 6.5 years, 21 (46%) of the patients reported excellent or marked improvement on a global outcome scale. There was no difference in the distribution of outcome when patients who still responded to BTX were compared with the BTX nonresponders. Using a modified TWSTRS scale, there was statistically significant improvements not only in the severity of dystonia but also in occupational and domestic work as well as in various activities of daily living. The results of this study are comparable with those of Ford and colleagues (1998b), who reported on an open-label, retrospective study of selective denervation for severe cervical dystonia (torticollis) in 16 patients who were refractory to injections with BTX-A. Using functional capacity scales, they concluded that 6 (37.5%) patients had “a moderate or complete return of normal neck function.” Despite some improvement in 12 of 14 (85.7%) patients on the TWSTRS dystonia rating scale applied to “blinded” ratings of videotaped examinations, the surgery failed to return patients to their occupations. On the basis of the reported experience, we conclude that surgical treatment tailored to the specific pattern of dystonic activity in the individual patient is a valuable alternative in the long-term management of cervical dystonia, particularly for patients who are anxious to avoid having the intracranial surgical procedure of DBS targeting the GPi.
Surgical treatments, such as facial nerve lysis and orbicularis oculi myectomy, once used extensively in the treatment of blepharospasm, have been essentially abolished because BTX treatment is usually very effective and because postoperative complications, such as ectropion, exposure keratitis, facial droop, and postoperative swelling and scarring, are common (Chapman et al., 1999). Similarly, recurrent laryngeal nerve section, once used in the treatment of spasmodic dysphonia (Dedo and Izdebski, 1983), is used rarely and only when BTX fails to provide a satisfactory relief. Another once popular procedure, spinal cord stimulation for cervical dystonia, has been shown to be ineffective by a controlled trial (Goetz et al., 1988).
Medical, surgical, and other therapeutic approaches to dystonia have been described in several recent reviews (Jankovic 2006; Jankovic, 2009a) (Fig. 13.3).
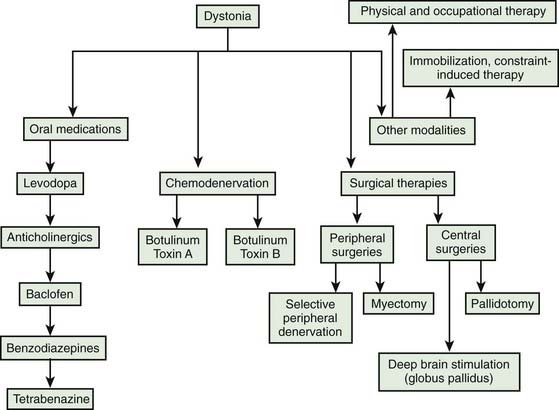
Other therapies
Inhibiting expression of mutant torsinA could be potentially a powerful therapeutic strategy in DYT1 dystonia. Using small interfering RNA (siRNA) to silence the expression of mutant torsinA, Gonzalez-Alegre and colleagues (2003) successfully suppressed expression of this abnormal protein in transfected cells that was allele-specific. Novel and experimental therapies are needed in order to make incremental advances in the treatment of dystonia (Jinnah and Hess, 2008).
Therapeutic guidelines
In summary, patients with segmental or generalized dystonia beginning in childhood or adolescence should be initially tried on levodopa/carbidopa up to 1000 mg of levodopa per day. If this therapy is successful, it should be maintained at a lowest possible dose. If it is ineffective after 3 months, then high-dose anticholinergic (e.g., trihexyphenidyl) therapy should be instituted, and the dosage should be increased to the highest tolerated level. If the results are poor, then baclofen, benzodiazepines, carbamazepine, and tetrabenazine should be tried. Some patients may require triple therapy consisting of an anticholinergic agent (e.g., trihexyphenidyl), a monoamine-depleting drug (e.g., tetrabenazine), and a dopamine receptor-blocking drug (e.g., fluphenazine, pimozide, risperidol, or clozapine) (Marsden et al., 1984). Tetrabenazine alone or with anticholinergic drugs is particularly useful in the treatment of tardive dystonia. In some patients, BTX injections may be helpful to control the most disabling symptom of the segmental or generalized dystonia. In most patients with adult-onset dystonia, the distribution is usually focal; therefore, BTX injections are usually considered the treatment of choice. Besides the abnormal movement and posture, pain, associated depression, anxiety, and other psychological comorbidities can have an important impact on the quality of life of patients with cervical dystonia and must be appropriately treated (Ben-Shlomo et al., 2002). In some patients, this treatment might need to be supplemented by other drugs noted previously or by surgical peripheral denervation. DBS should be reserved only for patients whose symptoms continue to be disabling despite optimal medical therapy. Any form of therapy should, of course, be preceded by a thorough evaluation designed to rule out secondary causes of dystonia. Finally, it is important to emphasize that patient education and counseling are essential components of a comprehensive therapeutic approach to all patients with dystonia (see the Appendix).
Adams S.G., Hunt E.J., Charles D.A., Lang A.E. Unilateral versus bilateral botulinum toxin injections in spasmodic dysphonia: Acoustic and perceptual results. J Otolaryngol. 1993;22:171-175.
Adler M., Keller J.E., Sheridan R.E., Deshpande S.S. Persistence of botulinum neurotoxin A demonstrated by sequential administration of serotypes A and E in rat EDL muscle. Toxicon. 2001;39:233-243.
Albanese A. Terminology for preparations of botulinum neurotoxins: what a difference a name makes. JAMA. 2011;305(1):89-90.
Albanese A., Barnes M.P., Bhatia K.P., et al. A systematic review on the diagnosis and treatment of primary (idiopathic) dystonia and dystonia plus syndromes: report of an EFNS/MDS-ES Task Force. Eur J Neurol. 2006;13:433-444.
Albright A.L., Barry M.J., Painter M.J., Shultz B. Infusion of intrathecal baclofen for generalized dystonia in cerebral palsy. J Neurosurg. 1998;88:73-76.
Albright A.L., Barry M.J., Shafron D.H., Ferson S.F. Intrathecal baclofen for generalized dystonia. Dev Med Child Neurol. 2001;43:652-657.
American Academy of Neurology. Assessment: The clinical usefulness of botulinum toxin-A in treating neurologic disorders: Report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 1990;40:1332-1336.
American Academy of Neurology. Training guidelines for the use of botulinum toxin for the treatment of neurologic disorders: Report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 1994:2401-2403.
American Academy of Ophthalmology. Botulinum toxin therapy of eye muscle disorders: Safety and effectiveness. Ophthalmology. 1989;96(2):37-41.
American Academy of Otolaryngology. Position statement on the clinical usefulness of botulinum toxin in the treatment of spasmodic dysphonia. Arch Otolaryngol Head Neck Surg Bull. 1990;9:8.
Andrew J., Fowler C.J., Harrison M.J.G. Stereotaxic thalamotomy in 55 cases of dystonia. Brain. 1983;106:981-1000.
Aramideh M., Ongerboer de Visser B.W., et al. Motor persistence of orbicularis oculi muscle in eyelid-opening disorders. Neurology. 1995;45:897-902.
Arezzo J.C., Litwak M.S., Caputo F.A., et al. Spread of paralysis to nearby and distant noninjected muscles in a monkey hand model: Comparison of BoNT-B and BoNT-A. In: Brin M.F., Hallett M., Jankovic J., editors. Scientific and Therapeutic Aspects of Botulinum Toxin. Philadelphia: Lippincott Williams & Wilkins; 2002:123-134.
Aoki K.R., Merlino G., Spanoyannis A.F., Wheeler L.A. BOTOX (Botulinum Toxin Type A) Purified Neurotoxin Complex prepared from the new bulk toxin retains the same preclinical efficacy as the original but with reduced antigenicity. Neurology. 1999;52(Suppl. 2):A521-A522.
Asanuma K., Ma Y., Okulski J., et al. Decreased striatal D2 receptor binding in non-manifesting carriers of the DYT1 dystonia mutation. Neurology. 2005;64(2):347-349.
Balash Y., Giladi N. Efficacy of pharmacological treatment of dystonia: Evidence-based review including meta-analysis of the effect of botulinum toxin and other cure options. Eur J Neurol. 2004;11:361-370.
Bardutzky J., Tronnier V., Schwab S., Meinck H.M. Intrathecal baclofen for stiff-person syndrome: Life-threatening intermittent catheter leakage. Neurology. 2003;60:1976-1978.
Benecke R., Jost W.H., Kanovsky P., et al. A new botulinum toxin type A free of complexing proteins for treatment of cervical dystonia. Neurology. 2005;64:1949-1951.
Ben-Shlomo Y., Camfield L., Warner T. What are the determinants of quality of life in people with cervical dystonia? J Neurol Neurosurg Psychiatry. 2002;72:608-614.
Bereznai B., Steude U., Seelos K., Botzel K. Chronic high-frequency globus pallidus internus stimulation in different types of dystonia: A clinical, video, and MRI report of six patients presenting with segmental, cervical, and generalized dystonia. Mov Disord. 2002;17:138-144.
Berg D., Becker G., Naumann M., Reiners K. Morphine in tardive and idiopathic dystonia. J Neural Transm. 2001;108:1035-1041.
Berke G.S., Blackwell K.E., Gerratt B.R., et al. Selective laryngeal adductor denervation-reinnervation: A new surgical treatment for adductor spasmodic dysphonia. Ann Otol Rhinol Laryngology. 1999;108:227-237.
Bertrand C.M., Molina-Negro P. Selective peripheral denervation in 111 cases of spasmodic torticollis: Rationale and results. In: Fahn S., Marsden C.D., Calne D.B., editors. Dystonia: Advances in Neurology, vol. 50. New York: Raven Press; 1988:637-643.
Bhatia K.P., Marsden C.D., Thomas D.G.T. Posteroventral pallidotomy can ameliorate attacks of paroxysmal dystonia induced by exercise. J Neurol Neurosurgy Psychiatry. 1998;65:604-615.
Blackie J.D., Lees A.J. Botulinum toxin treatment in spasmodic torticollis. J Neurol Neurosurg Psychiatry. 1990;53:640-643.
Blitzer, Brin M.F. Layngeal dystonia: A series with botulinum toxin therapy. Ann Otol Rhinol Layngol. 1991;100:85-90.
Blitzer A., Brin M.F., Fahn S., Lovelace R.E. The use of botulinum toxin for the treatment of focal laryngeal dystonia (spastic dysphonia). Laryngoscope. 1988;98:193-197.
Blitzer A., Brin M.F., Greene P.E., Fahn S. Botulinum toxin injection for the treatment of oromandibular dystonia. Ann Otol Rhinol Laryngol. 1989;98:93-97.
Blitzer A., Zalvan C., Gonzalez-Yanez O., Brin M.F. Botulinum toxin type A injections for the management of hyperfunctional larynx. In: Brin M.F., Hallett M., Jankovic J., editors. Scientific and Therapeutic Aspects of Botulinum Toxin. Philadelphia: Lippincott Williams & Wilkins; 2002:207-216.
Borodic G.E., Ferrante R., Pearce L.B., Smith K. Histologic assessment of dose-related diffusion and muscle fiber response after therapeutic botulinum A toxin injections. Mov Disord. 1994;9:31-39.
Borodic G.E., Pearce L.B., Smith K., Joseph M. Botulinum A toxin for spasmodic torticollis: Multiple vs single injection points per muscle. Head Neck. 1992;14:33-37.
Brans J.W.M., Lindeboom R., Snoek J.W., et al. Botulinum toxin versus trihexyphenidyl in cervical dystonia: A prospective, randomized, double-blind controlled trial. Neurology. 1996;46:1066-1072.
Brashear A., Lew M.F., Dykstra D.D., et al. Safety and efficacy of Neurobloc (botulinum toxin type B) in type A-responsive cervical dystonia. Neurology. 1999;53:1439-1446.
Brashear A., Bergan K., Wojcieszek J., et al. Patients’ perception of stopping or continuing treatment of cervical dystonia with botulinum toxin type A. Mov Disord. 2000;15:150-153.
Brin M.F. Treatment of Dystonia. In: Jankovic J., Tolosa E., editors. Parkinson’s Disease and Movement Disorders. third ed. Baltimore: Williams & Wilkins; 1998:553-578.
Brin M.F., Blitzer A., Fahn S., et al. Adductor laryngeal dystonia (spastic dysphonia): Treatment with local injections of Botulinum toxin (Botox). Mov Disord. 1989;4:287-296.
Brin M.F., Blitzer A., Stewart C. Laryngeal dystonia (spasmodic dysphonia) observations of 901 patients and treatment with botulinum toxin. Adv Neurol. 1998;78:1-10.
Brin M.F., Blitzer A., Stewart C., Fahn S. Treatment of spasmodic dysphonia (laryngeal dystonia) with local injections of botulinum toxin: Review and technical aspects. In: Blitzer A., Brin M.F., Sasaki C.T., et al, editors. Neurologic Disorders of the Larynx. New York: Thieme Medical Publishers; 1992:214-228.
Brin M.F., Comella C.L., Jankovic J., et alCD-017 BoNTA Study Group. Long-term treatment with botulinum toxin type A in cervical dystonia has low immunogenicity by mouse protection assay. Mov Disord. 2008;23:1353-1360.
Brin M.F., Fahn S., Moskowitz C., et al. Localized injections of botulinum toxin for the treatment of focal dystonia and hemifacial spasm. Mov Disord. 1987;2:237-254.
Brin M.F., Lew M.F., Adler C.H., et al. Safety and efficacy of NeuroBloc (botulinum toxin type B) in type A-resistant cervical dystonia. Neurology. 1999;53:1431-1438.
Brin M.F., Hallett M., Jankovic J., editors. Scientific and Therapeutic Aspects of Botulinum Toxin. Philadelphia: Lippincott Williams & Wilkins. 2002:1-507.
Brin M.F., Stewart C., Blitzer A., et al. Laryngeal botulinum toxin injections for disabling stuttering in adults. Neurology. 1994;44:2262-2266.
Buchman A.S., Comella C.L., Stebbins G.T., et al. Determining a dose-effect curve for botulinum toxin in the sternocleidomastoid muscle in cervical dystonia. Clin Neuropharmacol. 1994;17:188-193.
Burke R.E., Fahn S., Marsden C.D., et al. Validity and reliability of a rating scale for the primary torsion dystonias. Neurology. 1985;35:73-77.
Burke R.E., Reches A., Traub M.M., et al. Tetrabenazine induces acute dystonic reactions. Ann Neurol. 1985;17:200-202.
Burke R.E., Fahn S., Marsden C.D. Torsion dystonia: A double-blind, prospective trial of high-dosage trihexyphenidyl. Neurology. 1986;36:160-164.
Butler C., Campbell S. AACPDM Treatment Outcomes Committee Review Panel: Evidence of the effects of intrathecal baclofen for spastic and dystonic cerebral palsy. Dev Med Child Neurol. 2000;42:634-645.
Byl N.N., McKenzie A. Treatment effectiveness for patients with a history of repetitive hand use and focal hand dystonia: A planned, prospective follow-up study. J Hand Ther. 2000;13:289-301.
Byl N.N., Nagajaran S., McKenzie A.L. Effect of sensory discrimination training on structure and function in patients with focal hand dystonia: A case series. Arch Phys Med Rehab. 2003;84:1505-1514.
Cakmur R., Ozturk V., Uzunel F., et al. Comparison or preseptal and pretarsal injections of botulinum toxin in the treatment of blepharospasm and hemifacial spasm. J Neurol. 2002;249:64-68.
Candia V., Elbert T., Altenmüller E., et al. Constraint-induced movement therapy for focal hand dystonia in musicians. Lancet. 1999;53:42.
Cano S.J., Warner T.T., Linacre J.M., et al. Capturing the true burden of dystonia on patients: The Cervical Dystonia Impact Profile (CDIP-58). Neurology. 2004;63:1629-1633.
Cardoso F., Jankovic J., Grossman R., Hamilton W. Outcome after stereotactic thalamotomy for dystonia and hemiballismus. Neurosurgery. 1995;36:501-508.
Ceballos-Baumann A.O., Passingham R.E., Warner T., et al. Overactive prefrontal and underactive motor cortical areas in idiopathic dystonia. Ann Neurol. 1995;37:363-372.
Chapman K.L., Bartley G.B., Waller R.R., Hodge D.O. Follow-up of patients with essential blepharospasm who underwent eyelid protractor myectomy at the Mayo Clinic from 1980 through 1995. Ophthal Plast Reconstr Surg. 1999;15:106-110.
Charous S.J., Comella C.L., Fan W. Jaw-opening dystonia: Quality of life after botulinum toxin injections. Ear Nose Throat J. 2011;90(2):E9-E12.
Chen R., Karp B.I., Goldstein S.R., et al. Effect of muscle activity immediately after botulinum toxin injection for writer’s cramp. Mov Disord. 1999;14:307-312.
Cole R., Hallett M., Cohen L.G. Double-blind trial of botulinum toxin for treatment of focal hand dystonia. Mov Disord. 1995;10:466-471.
Collins A., Jankovic J. Botulinum toxin injection for congenital muscular torticollis presenting in children and adults. Neurology. 2006;67:1083-1085.
Comella C.L., Buchman A.S., Tanner C.M., et al. Botulinum toxin injection for spasmodic torticollis: Increased magnitude of benefit with electromyographic asssistance. Neurology. 1992;42:878-882.
Comella C.L., Jankovic J., Shannon K.M., et alDystonia Study Group. Comparison of botulinum toxin serotypes A and B for the treatment of cervical dystonia. Neurology. 2005;65:1423-1429.
Comella C.L., Leurgans S., Wuu J., et al. Rating scales for dystonia: A multicenter assessment. Mov Disord. 2003;18:303-312.
Comella C.L., Stebbins G.T., Goetz C.G., et al. Teaching tape for the motor section of the Toronto Western Spasmodic Torticollis Scale. Mov Disord. 1997;12:570-575.
Comella C.l., Tanner C.M., DeFoor-Hill L., Smith C. Dysphagia after botulinum toxin injections for spasmodic torticollis. Clinical and radiologic findings. Neurology. 1992;42:1307-1310.
Consky E.S., Lang A.E. Clinical assessments of patients with cervical dystonia. In: Jankovic J., Hallett M., editors. Therapy with botulinum toxin. New York: Marcel Dekker; 1994:211-237.
Cooper I.S. Clinical and physiologic implications of thalamic surgery for disorders of sensory communication: II. Intention tremor, dystonia, Wilson’s disease and torticollis. J Neurol Sci. 1965;2:520-533.
Cooper I.S. 20-year followup study of the neurosurgical treatment of dystonia musculorum deformans. In: Eldridge R., Fahn S., editors. Dystonia Advances in Neurology, vol. 14. New York: Raven Press; 1976:423-452.
Cordivari C., Misra P., Catania S., Lees A.J. Treatment of dystonic clenched fist with botulinum toxin. Mov Disord. 2001;16:907-913.
Coubes P., Cif L., El Fertit H., et al. Electrical stimulation of the globus pallidus internus in patients with primary generalized dystonia: Long-term results. J Neurosurg. 2004;101:189-194.
Coubes P., Roubertie A., Vayssiere N., et al. Treatment of DYT1-generalised dystonia by stimulation of the internal globus pallidus. Lancet. 2000;355:2220-2221.
Courey M.S., Garrett C.G., Billante C.R., et al. Outcomes assessment following treatment of spasmodic dysphonia with botulinum toxin. Ann Otol Rhinol Laryngol. 2000;109:819-822.
Dalvi A., Fahn S., Ford B. Intrathecal baclofen in the treatment of dystonic storm. Mov Disord. 1998;13:611-612.
Davis D.H., Ahlskog J.E., Litchy W.J., Root L.M. Selective peripheral denervation for torticollis: Preliminary results. Mayo Clin Proc. 1991;66:365-371.
de Paiva A., Meunier F.A., Molgó J., et al. Functional repair of motor endplates after botulinum neurotoxin A poisoning: Bi-phasic switch of synaptic activity between nerve sprouts and their parent terminals. Proc Natl Acad Sci USA. 1999;96:3200-3205.
Dedo H.H., Izdebski K. Intermediate results of 306 recurrent laryngeal nerve sections for spastic dysphonia. Laryngoscope. 1983;93:9-15.
Delnooz C.C., Horstink M.W., Tijssen M.A., van de Warrenburg B.P. Paramedical treatment in primary dystonia: a systematic review. Mov Disord. 2009;24(15):2187-2198.
Demirkiran M., Jankovic J. Paroxysmal dyskinesias: Clinical features and classification. Ann Neurol. 1995;38:571-579.
Diamond A., Shahed J., Azher S., et al. Globus pallidus deep brain stimulation in dystonia. Mov Disord. 2006;21:692-695.
Dong M., Yeh F., Tepp W.H., et al. SV2 is the protein receptor for botulinum neurotoxin A. Science. 2006;312:592-596.
Dressler D., Oeljeschlager R.-O., Ruther E. Severe tardive dystonia: Treatment with continuous intrathecal balcofen administration. Mov Disord. 1997;12:585-587.
Dressnandt J., Conrad B. Lasting reduction of severe spasticity after ending chronic treatment with intrathecal baclofen. J Neurol Neurosurg Psychiatry. 1996;60:168-173.
Dubinsky R.M., Gray C.S., Vetere-Overfield B., Koller W.C. Electromyographic guidance of botulinum toxin treatment in cervical dystonia. Clin Neuropharmacol. 1991;14:262-267.
Eidelberg D., Moeller J.R., Ishikawa T., et al. The metabolic topography of idiopathic torsion dystonia. Brain. 1995;118:1473-1484.
Elston J.S. Botulinum toxin for blepharospasm. In: Jankovic J., Hallett M., editors. Therapy with Botulinum Toxin. New York: Marcel Dekker; 1994:191-198.
Eltahawy H.A., Saint-Cyr J., Giladi N., et al. Primary dystonia is more responsive than secondary dystonia to pallidal interventions: Outcome after pallidotomy or pallidal deep brain stimulation. Neurosurgery. 2004;54:613-619.
Emerson J. Botulinum toxin for spasmodic torticollis in a patient with myasthenia gravis. Mov Disord. 1994;9:367.
Fahn S. High dosage anticholinergic therapy in dystonia. Neurology. 1983;33:1255-1261.
Fahn S. Paroxysmal dyskinesias. In: Marsden C.D., Fahn S., editors. Movement Disorders 3. Oxford: Butterworth-Heinemann; 1994:310-345.
Fahn S., Henning W.A., Bressman S., et al. Long-term usefulness of baclofen in the treatment of essential blepharospasm. Adv Ophthalm Plast Reconstr Surg. 1985;4:219-226.
Fahn S., List T., Moskowitz C., et al. Double-blind controlled study of botulinum toxin for blepharospasm. Neurology. 1985;35(Suppl. 1):271-272.
Figgit D.P., Noble S. Botulinum toxin B: A review of its therapeutic potential in the management of cervical dystonia. Drugs. 2002;62:705-755.
Filion M., Tremblay L. Abnormal spontaneous activity of globus pallidus neurons in monkeys with MPTP-induced parkinsonism. Brain Res. 1991;547:142-151.
Filippi G.M., Errico P., Samtarelli R., et al. Botulinum A toxin effects on rat jaw muscle spindles. Acta Otolaryngol (Stockh). 1993;113:400-404.
Foote K.D., Sanchez J.C., Okun M.S. Staged deep brain stimulation for refractory craniofacial dystonia with blepharospasm: case report and physiology. Neurosurgery. 2005;56(2):E415.
Ford B., Greene P., Louis E.D., et al. Use of intrathecal balcofen in the treatment of patients with dystonia. Arch Neurol. 1996;53:1241-1246.
Ford B., Greene P.E., Louis E.D., et al. Intrathecal baclofen in the treatment of dystonia. Adv Neurol. 1998;78:199-210.
Ford B., Louis E.D., Greene P., Fahn S. Outcome of selective ramisectomy for botulinum toxin resistant torticollis. J Neurol Neurosurg Psychiatry. 1998;65:472-478.
Ford C.N., Bless D.M., Lowery J.D. Indirect laryngoscopic apprach for injection of botulinum toxin in spasmodic dysphonia. Otolaryngol Head Neck Surg. 1990;103:752-758.
Forget R., Tozlovanu V., Iancu A., Boghen D. Botulinum toxin improves lid opening in blepharospasm-associated apraxia of lid opening. Neurology. 2002;58:1843-1846.
Frucht S.J., Bordelon Y., Houghton W.H., Reardam D. A pilot tolerability and efficacy trial of sodium oxybate in ethanol-responsive movement disorders. Mov Disord. 2005;20:1330-1337.
Furukawa Y., Nygaard T.G., Gutlich M., et al. Striatal biopterin and tyrosine hydroxylase protein reduction in dopa-responsive dystonia. Neurology. 1999;53:1032-1041.
Garner C.G., Straube A., Witt T.N., et al. Time course effects of local injections of botulinum toxin. Mov Disord. 1993;8:33-37.
Gelb D.J., Yoshimura D.M., Olney R.K., et al. Change in pattern of muscle activity following botulinum toxin injections for torticollis. Ann Neurol. 1991;29:370-376.
Goetz C.G., Penn R.D., Tanner C.M. Efficacy of cervical cord stimulation in dystonia. In: Fahn S., Marsden C.D., Calne D.B., editors. Dystonia: Advances in Neurology, vol. 50. New York: Raven Press; 1988:645-649.
Gonzalez-Alegre P., Miller V.M., Davidson B.L., Paulson H.L. Toward therapy for DYT1 dystonia: Allele-specific silencing of mutant TorsinA. Ann Neurol. 2003;53:781-787.
Grabli D., Ewenczyk C., Coelho-Braga M.C., et al. Interruption of deep brain stimulation of the globus pallidus in primary generalized dystonia. Mov Disord. 2009;24:2363-2369.
Greene P. Baclofen in the treatment of dystonia. Clin Neuropharmacol. 1992;15:276-288.
Greene P. Medical and surgical therapy of idiopathic torsion dystonia. In: Kurlan R., editor. Treatment of Movement Disorders. Philadelphia: J B Lippincott; 1995:153-181.
Greene P.E., Fahn S. Baclofen in the treatment of idiopathic dystonia in children. Mov Disord. 1992;7:48-52.
Greene P., Fahn S. Treatment of torticollis with injections of botulinum toxin type F in patients with antibodies to botulinum toxin type A. Mov Disord. 1992;7(Suppl. 1):134.
Greene P., Fahn S., Diamond B. Development of resistance to botulinum toxin type A in patients with torticollis. Mov Disord. 1994;9:213-217.
Greene P., Kang U., Fahn S., et al. Double-blind, placebo controlled trial of botulinum toxin injection for the treatment of spasmodic torticollis. Neurology. 1990;40:1213-1218.
Greene P., Shale H., Fahn S. Analysis of open-label trials in torsion dystonia using high dosage of anticholinergics and other drugs. Mov Disord. 1988;3:46-60.
Grossman R.G., Hamilton W.J. Surgery for movement disorders. In: Jankovic J., Tolosa E., editors. Parkinson’s Disease and Movement Disorders. second ed. Baltimore: Williams & Wilkins; 1993:531-548.
Hall T.A., McGwin G.Jr., Searcey K., et al. Health-related quality of life and psychosocial characteristics of patients with benign essential blepharospasm. Arch Ophthalmol. 2006;124:116-119.
Hallett M. Is dystonia a sensory disorder? Ann Neurol. 1995;38:139-140.
Hanna P.A., Jankovic J. Mouse bioassay versus Western blot assay for botulinum toxin antibodies: Correlation with clinical response. Neurology. 1998;50:1624-1629.
Hanna P.A., Jankovic J., Vincent A. Comparison of mouse bioassay and immunoprecipitation assay for botulinum toxin antibodies. J Neurol Neurosurg Psychiatry. 1999;66:612-616.
Hanson M.A., Stevens R.C. Structural view of botulinum neurotoxin in numerous functional states. In: Brin M.F., Hallett M., Jankovic J., editors. Scientific and Therapeutic Aspects of Botulinum Toxin. Philadelphia: Lippincott Williams & Wilkins; 2002:11-28.
Harwood G., Hierons R., Fletcher N.A., Marsden C.D. Lessons from a remarkable family with dopa-responsive dystonia. J Neurol Neurosurg Psychiatry. 1994;57:460-463.
Hebb M.O., Chiasson P., Lang A.E., et al. Sustained relief of dystonia following cessation of deep brain stimulation. Mov Disord. 2007;22:1958-1962.
Hering S., Wenning G.K., Seppi K., et al. An open trial of levetiracetam for segmental and generalized dystonia. Mov Disord. 2007;22:1649-1651.
Hoon A.H.Jr., Freese P.O., Reinhardt E.M., et al. Age-dependent effects of trihexyphenidyl in extrapyramidal cerebral palsy. Pediatr Neurol. 2001;25:55-58.
Hou J.G., Ondo W., Jankovic J. Intrathecal baclofen for dystonia. Mov Disord. 2001;16:1201-1202.
Hsiung G.-Y.R., Das S.K., Ranawaya R., et al. Long-term efficacy of botulinum toxin A in treatment of various movement disorders over a 10-year period. Mov Disord. 2002;17:1288-1293.
Ichinose H., Ohye T., Takahi E., et al. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat Genet. 1994;8:236-242.
Isaias I.U., Alterman R.L., Tagliati M. Outcome predictors of pallidal stimulation in patients with primary dystonia: the role of disease duration. Brain. 2008;131:1895-1902.
Isaias I.U., Alterman R.L., Tagliati M. Deep brain stimulation for primary generalized dystonia. Long-term outcomes. Arch Neurol. 2009;66:465-470.
Ishikawa A., Miyatake T. A family with hereditary juvenile dystonia-parkinsonism. Mov Disord. 1995;10:482-488.
Jabbari B., Scherokman B., Gunderson C.H., et al. Treatment of movement disorders with trihexyphenidyl. Mov Disord. 1989;4:202-212.
Jankovic J. Botulinum toxin in the treatment of dystonic tics. Mov Disord. 1994;9:347-349.
Jankovic J. Apraxia of eyelid opening [Letter to the editor]. Mov Disord. 1995;10:686-687.
Jankovic J. Tardive syndromes and other drug-induced movement disorders. Clin Neuropharmacol. 1995;18:197-214.
Jankovic J. Pretarsal injection of botulinum toxin for blepharospasm and apraxia of eyelid opening. J Neurol Neurosurg Psychiatry. 1996;60:704.
Jankovic J. Treatment of dystonia. In: Watts R.L., Koller W.C., editors. Movement Disorders: Neurologic Principles and Practice. New York: McGraw Hill; 1997:443-454.
Jankovic J. Re-emergence of surgery for dystonia [Editorial Commentary]. J Neurol Neurosurg Psychiatry. 1998;65:434.
Jankovic J. Can peripheral trauma induce dystonia and other movement disorders? Yes!. Mov Disord. 2001;16:7-12.
Jankovic J. Needle EMG guidance is rarely required. Muscle Nerve. 2001;24:1568-1570.
Jankovic J. Botulinum toxin: Clinical implications of antigenicity and immunoresistance. In: Brin M.F., Hallett M., Jankovic J., editors. Scientific and Therapeutic Aspects of Botulinum Toxin. Philadelphia: Lippincott Williams & Wilkins; 2002:409-416.
Jankovic J. Botulinum toxin in clinical practice. J Neurol Neurosurg Psychiatry. 2004;75:951-957.
Jankovic J. Dystonia: Medical therapy and botulinum toxin. In: Fahn S., Hallett M., DeLong D.R., editors. Dystonia 4 Advances in Neurology, vol. 94. Philadelphia: Lippincott Williams & Wilkins; 2004:275-863.
Jankovic J. Treatment of cervical dystonia with botulinum toxin. Mov Disord. 2004;19(Suppl. 8):S109-S115.
Jankovic J. Treatment of dystonia. Lancet Neurol. 2006;5:864-872.
Jankovic J. Dystonic disorders. In: Jankovic J., Tolosa E., editors. Parkinson’s Disease and Movement Disorders. fifth ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2007:321-347.
Jankovic J. Treatment of hyperkinetic movement disorders. Lancet Neurol. 2009;8(9):844-856.
Jankovic J. Clinical efficacy and tolerability of Xeomin in the treatment of blepharospasm. Eur J Neurol. 2009;16(Suppl. 2):14-18.
Jankovic J. Camptocormia, head drop and other bent spine syndromes: heterogeneous etiology and pathogenesis of parkinsonian deformities. Mov Disord. 2010;25:527-528.
Jankovic J., Albanese A., Atassi M.Z., et al. Botulinum Toxin. Therapeutic Clinical Practice and Science. Philadelphia, PA: Saunders (Elsevier); 2009.
Jankovic J., Beach J. Long-term effects of tetrabenazine in hyperkinetic movement disorders. Neurology. 1997;48:358-362.
Jankovic J., Brin M.F. Therapeutic uses of botulinum toxin. N Engl J Med. 1991;324:1186-1194.
Jankovic J., Brin M. Botulinum toxin: Historical perspective and potential new indications. In: Mayer N.H., Simpson D.M., editors. Spasticity: Etiology, Evaluation, Management and the Role of Botulinum Toxin. New York: WEMOVE; 2002:100-109.
Jankovic J., Brin M.F., Comella C. Handbook of botulinum toxin treatment for cervical dystonia. New York: Churchill Livingstone; 1994.
Jankovic, J., Comella, C., Hanschmann, A., Grafe, S., 2011. Efficacy and safety of NT 201 (botulinum neurotoxin type A free from complexing proteins) in the treatment of blepharospasm (in press).
Jankovic J., Esquenazi A., Fehling D., et al. Evidence-based review of patient reported outcomes with botulinum toxin type A. Clin Neuropharmacol. 2004;27:234-244.
Jankovic J., Hallett M., editors. Therapy with botulinum toxin. New York: Marcel Dekker, 1994.
Jankovic J., Hamilton W., Grossman R.G. Thalamic surgery for movement disorders. In: Obeso J.A., DeLong M., Ohye C., Marsden C.D., editors. Advances in Understanding the Basal Ganglia and New Surgical Approaches for Parkinson’s Disease Advances in Neurology, vol. 74. New York: Raven Press; 1997:221-233.
Jankovic J., Hunter C., Dolimbek B.Z., et al. Clinico-immunologic aspects of botulinum toxin type B treatment of cervical dystonia. Neurology. 2006;67:2233-2235.
Jankovic J., Kenney C., Grafe S., et al. Relationship between various clinical outcome assessments in patients with blepharospasm. Mov Disord. 2009;24:407-413.
Jankovic J., Leder S., Warner D., Schwartz K. Cervical dystonia: Clinical findings and associated movement disorders. Neurology. 1991;41:1088-1091.
Jankovic J., Orman J. Botulinum A toxin for cranial-cervical dystonia: A double-blind, placebo-controlled study. Neurology. 1987;37:616-623.
Jankovic J., Orman J. Tetrabenazine treatment in dystonia, chorea, tics and other dyskinesias. Neurology. 1988;38:391-394.
Jankovic J., Penn A. Severe dystonia and myoglobinuria. Neurology. 1982;32:1195-1197.
Jankovic J., Schwartz K. Botulinum toxin injections for cervical dystonia. Neurology. 1990;41:277-280.
Jankovic J., Schwartz K. Clinical correlates of response to botulinum toxin injections. Arch Neurol. 1991;48:1253-1256.
Jankovic J., Schwartz K. Longitudinal follow-up of botulinum toxin injections for treatment of blepharospasm and cervical dystonia. Neurology. 1993;43:834-836.
Jankovic J., Schwartz K. The use of botulinum toxin in the treatment of hand dystonias. J Hand Surg. 1993;18A:883-887.
Jankovic J., Schwartz K. Response and immunoresistance to botulinum toxin injections. Neurology. 1995;45:1743-1746.
Jankovic J., Schwartz K., Clemence W., et al. A randomized, double-blind, placebo-controlled study to evaluate botulinum toxin type A in essential hand tremor. Mov Disord. 1996;11:250-256.
Jankovic J., Schwartz K., Donovan D.T. Botulinum toxin treatment of cranial-cervical dystonia, spasmodic dysphonia, other focal dystonias and hemifacial spasm. J Neurol Neurosurg Psychiatry. 1990;53:633-639.
Jankovic J., Vuong K.D., Ahsan J. Comparison of efficacy and immunogenicity of original versus current botulinum toxin in cervical dystonia. Neurology. 2003;60:1186-1188.
Jech R., Bares M., Urgosík D., et al. Deep brain stimulation in acute management of status dystonicus. Mov Disord. 2009;24:2291-2292.
Jinnah H.A., Hess E.J. Experimental therapeutics for dystonia. Neurotherapeutics. 2008;5:198-209.
Junker J., Oberwittler C., Jackson D., Berger K. Utilization and perceived effectiveness of complementary and alternative medicine in patients with dystonia. Mov Disord. 2004;19:158-161.
Kaji R., Kohara N., Katayama M., et al. Muscle afferent block by intramuscular injection of lidocaine for the treatment of writer’s cramp. Muscle Nerve. 1995;18:234-235.
Kaji R., Rothwell J.C., Katayama M., et al. Tonic vibration reflex and muscle afferent block in writer’s cramp. Ann Neurol. 1995;38:155-162.
Karnath H.-O., Konczak J., Dichgans J. Effect of prolonged neck muscle vibration on lateral head tilt in severe spasmodic torticollis. J Neurol Neurosurg Psychiatry. 2000;69:658-660.
Karp B.I., Cole R.A., Cohen L.G., et al. Long-term botulinum toxin treatment of focal hand dystonia. Neurology. 1994;44:70-76.
Karp B.I., Goldstein S.R., Chen R., et al. An open trial of clozapine for dystonia. Mov Disord. 1999;14:652-657.
Kenney C., Jankovic J. Tetrabenazine in the treatment of hyperkinetic movement disorders. Expert Rev Neurother. 2006;6:7-17.
Krauss J.K., Akeyson E.W., Giam P., Jankovic J. Propofol-induced dyskinesias in Parkinson’s disease. Anesth Analg. 1996;83:420-422.
Krauss J.K., Loher T.J., Pohle T., et al. Pallidal deep brain stimulation in patients with cervical dystonia and severe cervical dyskinesias with cervical myelopathy. J Neurol Neurosurg Psychiatry. 2002;72:249-256.
Krauss J.K., Toops E.G., Jankovic J., Grossman R.G. Symptomatic and functional outcome of surgical treatment of cervical dystonia. J Neurol Neurosurg Psychiatry. 1997;63:642-648.
Kumar R., Dagher A., Hutchison W.D., et al. Globus pallidus deep brain stimulation for generalized dystonia: clinical and PET investigation. Neurology. 1999;53:871-874.
Kupsch A., Benecke R., Muller J., et al. Pallidal deep-brain stimulation in primary generalized or segmental dystonia. N Engl J Med. 2006;355:1978-1990.
Lang A.E. Dopamine agonists and antagonists in the treatment of idiopathic dystonia. In: Fahn S., Marsden C.D., Calne D.B., editors. Dystonia: Advances in Neurology, vol. 50. New York: Raven Press; 1988:561-570.
Lang A.E. Surgical treatment of dystonia. Adv Neurol. 1998;78:185-198.
Lenz F.A., Martin R., Kwan H.C., et al. Thalamic single-unit activity occurring in patients with hemidystonia. Stereotact Funct Neurosurg. 1990;55:159-162.
Lenz F.A., Suarez J.I., Verhagen Metman L., et al. Pallidal activity during dystonia: Somatosensory reorganization and changes with severity. J Neurol Neurosurg Psychiatry. 1998;65:767-770.
Lepore V., Defazio G., Acquistapance D., et al. Botulinum A toxin for the so-called apraxia of lid opening. Mov Disord. 1995;10:525-526.
Levy C.E., Nichols D.S., Schmalbrock P.M., et al. Functional MRI evidence of cortical reorganization in upper-limb stroke hemiplegia treated with constraint-induced movement therapy. Am J Phys Med Rehabil. 2001;80:4-12.
Lew M.F., Adomato B.T., Duane D.D., et al. Botulinum toxin type B: A double-blind, placebo-controlled, safety and efficacy study in cervical dystonia. Neurology. 1997;49:701-707.
Lindeboom R., Brans J.W.M., Aramindeh M., et al. Treatment of cervical dystonia: A comparison of measures for outcome assessment. Mov Disord. 1998;13:706-712.
Lindeboom R., de Haan R.J., Brans J.W.M., Speelman J.D. Treatment outcomes in cervical dystonia: A clinimetric study. Mov Disord. 1996;11:371-376.
Lozano A.M., Kumar R., Gross R.E., et al. Globus pallidus internus pallidotomy for generalized dystonia. Mov Disord. 1997;12:865-870.
Lucetti C., Nuti A., Gambaccini G., et al. Mexiletine in the treatment of torticollis and generalized dystonia. Clin Neuropharmacol. 2000;23:186-189.
Ludlow C.L. Treatment of speech and voice disorders with botulinum toxin. JAMA. 1990;264:2671-2675.
Manji H., Howard R.S., Miller D.H., et al. Status dystonicus: The syndrome and its management. Brain. 1998;121:243-252.
Mahrhold S., Rummel A., Bigalke H., et al. The synaptic vesicle protein 2C mediates the uptake of botulinum neurotoxin A into phrenic nerves. FEBS Lett. 2006;580:2011-2014.
Mariotti P., Fasano A., Contarino M.F., et al. Management of status dystonicus: our experience and review of the literature. Mov Disord. 2007;22:963-968.
Marsden C.D., Marion M.-H., Quinn N. The treatment of severe dystonia in children and adults. J Neurol Neurosurg Psychiatry. 1984;47:1166-1173.
Massey J.M. EMG-Guided chemodenervation with phenol in cervical dystonia (spasmodic torticollis. In: Brin M.F., Hallett M., Jankovic J., editors. Scientific and Therapeutic Aspects of Botulinum Toxin. Philadelphia: Lippincott Williams & Wilkins; 2002:459-462.
Mejia N.I., Vuong K.D., Jankovic J. Long-term botulinum toxin efficacy, safety and immunogenicity. Mov Disord. 2005;20:592-597.
Melling J., Hambleton P., Shone C.C. Clostridium botulinum toxins: nature and preparation for clinical use. Eye. 1998;2:16-23.
Merello M., Garcia H., Nogues M., Leiguarda R. Masticatory muscle spasm in non-Japanese patient with Satoyoshi syndrome successfully treated with botulinum toxin. Mov Disord. 1994;9:104-105.
Mezaki T., Kaji R., Hamano T., et al. Optimisation of botulinum treatment for cervical and axial dystonias: Experience with Japanese type A toxin. J Neurol Neurosurg Psychiatry. 1994;57:1535-1537.
Mezaki T., Kaji R., Kohara N., et al. Comparison of therapeutic efficacies of type A and F botulinum toxins for blepharospasm: A double-blind, controlled study. Neurology. 1995;45:506-508.
Misura K.M.S., Scheller R.H., Weis W.I. Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature. 2000;404:355-362.
Mitchel I.J., Luquin R., Boyce S. Neural mechanisms of dystonia: Evidence from a 2-deoxyglucose uptake study in a primate model of dopamine agonist-induced dystonia. Mov Disord. 1990;5:49-54.
Molloy F.M., Shill H.A., Kaelin-Lang A., Karp B.I. Accuracy of muscle localization without EMG: Implications of limb dystonia. Neurology. 2002;58:805-807.
Muellbacher W., Richards C., Ziemann U., et al. Improving hand function in chronic stroke. Arch Neurol. 2002;59:1278-1282.
Muller J., Wissel J., Kemmler G., et al. Craniocervical dystonia questionnaire (CDQ-24): development and validation of a disease-specific quality of life instrument. J Neurol Neurosurg Psychiatry. 2004;75:749-753.
Münchau A., Mathen D., Cox T., et al. Unilateral lesions of the globus pallidus: Report of four patients presenting with focal or segmental dystonia. J Neurol Neurosurg Psychiatry. 2000;69:494-498.
Narayan R.K., Loubser P.G., Jankovic J., et al. Intrathecal baclofen for intractable axial dystonia. Neurology. 1991;41:1141-1142.
Naumann M., Jankovic J. Safety of botulinum toxin type A: A systematic review and meta-analysis. Curr Med Res Opin. 2004;20:981-990.
Naumann M., Carruthers A., Carruthers J., et al. Meta-analysis of neutralizing antibody conversion with onabotulinumtoxinA (BOTOX®) across multiple indications. Mov Disord. 2010;25:2211-2218.
Nygaard T.G., Marsden C.D., Fahn S. Dopa-responsive dystonia: Long-term treatment response and prognosis. Neurology. 1991;41:174-181.
Ohara S., Hayashi R., Momoi H., et al. Mexiletine in the treatment of spasmodic torticollis. Mov Disord. 1998;13:934-940.
Ondo W.G., Desaloms J.M., Jankovic J., Grossman R. Surgical pallidotomy for the treatment of generalized dystonia. Mov Disord. 1998;13:693-698.
Ondo W.G., Desalom J.M., Jankovic J., Grossman R.G. Pallidotomy and thalamotomy for dystonia. In: Krauss J.K., Jankovic J., Grossman R.G., editors. Surgery for Parkinson’s Disease and Movement Disorders. Philadelphia: Lippincott Williams & Wilkins; 2001:299-306.
Opal P., Tintner R., Jankovic J., et al. Intrafamilial phenotypic variability of the DYT1 dystonia: From asymptomatic TOR1A gene carrier status to dystonic storm. Mov Disord. 2002;17:339-345.
Ostrem J.L., Starr P.A. Treatment of dystonia with deep brain stimulation. Neurotherapeutics. 2008;5:320-330.
Ostrem J.L., Marks W.J.Jr., Volz M.M., et al. Pallidal deep brain stimulation in patients with cranial-cervical dystonia (Meige syndrome). Mov Disord. 2007;22:1885-1891.
Ostrem J.L., Racine C.A., Glass G.A., et al. Subthalamic nucleus deep brain stimulation in primary cervical dystonia. Neurology. 2011;76:870-878.
Pacchetti C., Albani G., Martignoni E., et al. “Off” painful dystonia in Parkinson’s disease treated with botulinum toxin. Mov Disord. 1995;10:333-336.
Penn R.D., Gianino J.M., York M.M. Intrathecal baclofen for motor disorders. Mov Disord. 1995;10:675-677.
Perlmutter J.S., Stambuk M.K., Markham J., et al. Decreased [18F]spiperone binding in putamen in idiopathic focal dystonia. J Neurosci. 1997;17:843-850.
Poewe W., Schlosky L., Kleedorfer B., et al. Treatment of spasmodic torticollis with local injections of botulinum toxin. J Neurol. 1992;239:21-25.
Pozos R.S., Iaizo P.A. Effects of topical anesthesia on essential tremor. Electromyogr Clin Neurophysiol. 1992;32:369-372.
Priori A., Berardelli A., Mercuri B., Mafredi M. Physiological effects produced by botulinum toxin treatment of upper limb dystonia: Changes in reciprocal inhibition between forearm muscles. Brain. 1995;118:801-807.
Priori A., Bertolasi L., Pesenti A., et al. Gamma-hydroxyburinic acid for alcohol-sensitive myoclonus in dystonia. Neurology. 2000;54:1706.
Priori A., Pesenti A., Cappellari A., et al. Limb immobilization for the treatment of focal occupational dystonia. Neurology. 2001;57:405-409.
Pullman S.L., Greene P., Fahn S., Pederson S.F. Approach to the treatment of limb disorders with botulinum toxin. Arch Neurol. 1996;53:617-624.
Quirk J.A., Sheean G.L., Marsden C.D., Lees A.J. Treatment of nonoccupational limb and trunk dystonia with botulinum toxin. Mov Disord. 1996;11:377-383.
Rajput A.H., Gibb W.R.G., Zhong X.H., et al. DOPA-responsive dystonia: Pathologiucal and biochemical observations in a case. Ann Neurol. 1994;35:396-402.
Ranawaya R., Lang A. Usefulness of a writing device in writer’s cramp. Neurology. 1991;41:1136-1138.
Reimer J., Gilg K., Karow A., et al. Health-related quality of life in blepharospasm or hemifacial spasm. Acta Neurol Scand. 2005;111:64-70.
Rivest J., Lees A.J., Marsden C.D. Writer’s cramp: Treatment with botulinum toxin injections. Mov Disord. 1990;6:55-59.
Roggenkamper P., Jost W.H., Bihari K., et alfor the NT 201 Blepharospasm Study Team. Efficacy and safety of a new Botulinum Toxin Type A free of complexing proteins in the treatment of blepharospasm. J Neural Transm. 2006;113:303-312.
Rosengren S.M., Colebatch J.G. Cervical dystonia responsive to acoustic and galvanic vestibular stimulation. Mov Disord. 2006;21:1495-1499.
Ruiz P.J.G., Bernardos V.S. Intramuscular phenol injection for severe cervical dystonia. J Neurol. 2000:247146-247147.
Sanghera M., Grossman R.G., Kalhorn C.G., et al. Basal ganglia neuronal discharge in primary and secondary dystonia in patients undergoing pallidotomy. Neurosurgery. 2003;52:1358-1373.
Sankhla C., Jankovic J., Duane D. Variability of the immunologic and clinical response in dystonic patients immunoresistant to botulinum toxin injections. Mov Disord. 1998;13:150-154.
Schantz E.J., Johnson E.A. Properties and use of botulinum toxin and other microbial neurotoxins in medicine. Microbiol Rev. 1992;56:80-99.
Schneider S.A., Mohire M.D., Trender-Gerhard I., et al. Familial dopa-responsive cervical dystonia. Neurology. 2006;66:599-601.
Schuele S., Jabusch H.C., Lederman R.J., Altenmuller E. Botulinum toxin injections in the treatment of musician’s dystonia. Neurology. 2005;64:341-343.
Sensi M., Cavallo M.A., Quatrale R., et al. Pallidal stimulation for segmental dystonia: long term follow up of 11 consecutive patients. Mov Disord. 2009;24:1829-1835.
Sheean G.L., Lees A.J. Botulinum toxin F in the treatment of torticollis clinically resistant to botulinum toxin A. J Neurol Neurosurg Psychiatry. 1995;59:601-607.
Siebner H.R., Tormos J.M., Ceballos-Baumann A.O., et al. Low-frequency repetitive transcranial magnetic stimulation of the motor cortex in writer’s cramp. Neurology. 1999;52:529-537.
Simpson D.M., Blitzer A., Brashear A., et alTherapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Assessment: Botulinum neurotoxin for the treatment of movement disorders (an evidence-based review): report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2008;70:1699-1706.
Sloop R.R., Cole B.A., Escutin R.O. Human response to botulinum toxin injection: Type B compared with type A. Neurology. 1997;49:189-194.
Steinberger D., Korinthenber R., Topka H., et al. Dopa-responsive dystonia: Mutation analysis of GCH1 and analysis of therapeutic doses of L-dopa. Neurology. 2000;55:1735-1737.
Svetel M., Kozic D., Stefanoval E., et al. Dystonia in Wilson’s disease. Mov Disord. 2001;16:719-723.
Tan E.-K., Jankovic J. Botulinum toxin A in patients with oromandibular dystonia: Long-term follow-up. Neurology. 1999;53:2102-2105.
Tan E.-K., Jankovic J. Treating severe bruxism with botulinum toxin. J Am Dent Assoc. 2000;131:211-216.
Tanaka H., Endo K., Tsuji S., et al. The gene for hereditary progressive dystonia with marked diurnal fluctuation maps to chromosome 14q. Ann Neurol. 1995;37:405-408.
Tas N., Karatas K., Sepici V. Hand orthosis as a writing aid in writer’s cramp. Mov Disord. 2001;16:1185-1189.
Tasker R.R., Doorly T., Yamashiro K. Thalamotomy in generalized dystonia. In: Fahn S., Marsden C.D., Calne D.B., editors. Dystonia 2: Advances in Neurology, vol. 50. New York: Raven Press; 1988:615-631.
Taub E., Morris D.M. Constraint-induced movement therapy to enhance recovery after stroke. Curr Atheroscler Rep. 2001;3:279-286.
Taub E., Uswatte G., Elbert T. New treatments in neurorehabilitation founded on basic research. Nat Rev Neurosci. 2002;3:226-236.
Taub E., Uswatte G., Pidikiti R. Constraint-induced movement therapy: A new family of techniques with broad application to physical rehabilitation – a clinical review. J Rehabil Res Dev. 1999;36:237-251.
Taylor A.E., Lang A.E., Saint-Cyr J.A., et al. Cognitive processes in idiopathic dystonia treated with high-dose anticholinergic therapy: Implications for treatment strategies. Clin Neuropharmacol. 1991;14:62-77.
Thant Z.S., Tan E.K. Emerging therapeutic applications of botulinum toxin. Med Sci Monit. 2003;9:40-48.
Timmermann L., Pauls K.A., Wieland K., et al. Dystonia in neurodegeneration with brain iron accumulation: outcome of bilateral pallidal stimulation. Brain. 2010;133:701-712.
Tinazzi M., Farina S., Bhatia K., et al. TENS for the treatment of writer’s cramp dystonia: A randomized, placebo-controlled study. Neurology. 2005;64:1946-1948.
Tintner R., Gross R., Winzer U.F., et al. Autonomic function after botulinum toxin type A or B: A double-blind, randomized trial. Neurology. 2005;65:765-767.
Tintner R., Jankovic J. Botulinum toxin for the treatment of cervical dystonia. Expert Opin Pharmacother. 2001;2:1985-1994.
Tintner R., Jankovic J. Focal dystonia: The role of botulinum toxin. Curr Neurol Neurosci. 2001;1:337-345.
Tintner R., Jankovic J. Botulinum toxin. In: Warner T.T., Bressman S., editors. Clinical Diagnosis and Management of Dystonia. London, UK: Informa Healthcare; 2007:189-207.
Treves T., Korczyn A.D. Progressive dystonia and paraparesis in cerebral palsy. Eur Neurol. 1986;25:148-153.
Tronnier V.M., Fogel W. Pallidal stimulation for generalized dystonia: Report of three cases. J Neurosurg. 2000;92:453-456.
Trottenberg T., Volkmann J., Deuschl G., et al. Treatment of severe tardive dystonia with pallidal deep brain stimulation. Neurology. 2005;64:344-346.
Trugman J.M., Leadbetter R., Zalis M., et al. Treatment of severe axial tardive dystonia with clozapine: Case report and hypothesis. Mov Disord. 1994;9:441-446.
Truong D., Comella C., Fernandez H.H., Ondo W.G., on behalf of the Dysport Benign Essential Blepharospasm Study Group. Efficacy and safety of purified botulinum toxin type A (Dysport®) for the treatment of benign essential blepharospasm: A randomized, placebo-controlled, phase II trial. Parkinsonism Relat Disord. 2008;14:407-414.
Truong D., Duane D.D., Jankovic J., et al. Efficacy and safety of botulinum type A toxin (Dysport) in cervical dystonia: Results of the first US randomized, double-blind, placebo-controlled study. Mov Disord. 2005;20:783-791.
Truong D.D., Sandromi P., van der Noort S., Matsumoto R.R. Diphenhydramine is effective in the treatment of idiopathic dystonia. Arch Neurol. 1995;52:405-407.
Tsui J.K.C., Bhatt M., Calne S., Calne D.B. Botulinum toxin in treatment of writer’s cramp: A double-blind study. Neurology. 1993;43:183-185.
Tsui J.K.C., Hayward M., Mak E.K.M., Schulzer M. Botulinum toxin type B in the treatment of cervical dystonia: A pilot study. Neurology. 1995;45:2109-2110.
Vaamonde J., Narbona J., Weiser R., et al. Dystonic storms: A practical management problem. Clin Neuropharmacol. 1994;17:344-347.
Vagefi M.R., Lin C.C., McCann J.D., Anderson R.L. Exacerbation of blepharospasm associated with craniocervical dystonia after placement of bilateral globus pallidus internus deep brain stimulator. Mov Disord. 2008;23:454-456.
van Hilten B.J., van de Beek W.J., Hoff J.I., et al. Intrathecal baclofen for the treatment of dystonia in patients with reflex sympathetic dystrophy. N Engl J Med. 2000;343:625-630.
van Hilten J.J., Hoff J.I., Thang M.C., et al. Clinimetric issues of screening for responsiveness to intrathecal baclofen in dystonia. J Neural Transm. 1999:106931-106941.
Vercueil L., Pollak P., Fraix V., et al. Deep brain stimulation in the treatment of severe dystonia. J Neurol. 2001;248:695-700.
Vidailhet M., Vercueil L., Houeto J.L., et al. French Stimulation du Pallidum Interne dans la Dystonie (SPIDY) Study Group. Bilateral deep-brain stimulation of the globus pallidus in primary generalized dystonia. N Engl J Med. 2005;352:459-467.
Vidailhet M., Vercueil L., Houeto J.L., et al. French SPIDY Study Group. Bilateral, pallidal, deep-brain stimulation in primary generalised dystonia: a prospective 3 year follow-up study. Lancet Neurol. 2007;6:223-229.
Vitek J.L., Chockkan V., Zhang J.-Y., et al. Neuronal activity in the basal ganglia in patients with generalized dystonia and hemiballism. Ann Neurol. 1999;46:22-35.
Vitek J.L., Zhang J., Evatt M., et al. GPi pallidotomy for dystonia: Clinical outcome and neuronal activity. In: Fahn S., Marsden C.D., DeLong D.R., editors. Dystonia 3: Advances in Neurology, vol. 78. Philadelphia: Lippincott-Raven; 1998:211-219.
Walker R.H., Danisi F.O., Swope D.M., et al. Intrathecal baclofen for dystonia: Benefits and complications during six year of experience. Mov Disord. 2000;15:1242-1247.
Wan X.H., Vuong K.D., Jankovic J. Clinical application of botulinum toxin type B in autonomic symptoms. Chin Med Sci J. 2005;20:44-47.
Warrick P., Dromey C., Irish J.C., et al. Botulinum toxin for essential tremor of the voice with multiple anatomical sites of tremor: A crossover design study of unilateral versus bilateral injection. Laryngoscope. 2000;110:1366-1374.
Waterston J.A., Swash M., Watkins E.S. Idiopathic dystonia and cervical spondylotic myelopathy. J Neurol Neurosurg Psychiatry. 1989;52:1424-1426.
Wirtschafter J.D. Clinical doxorubicin chemomyectomy: An experimental treatment for benign essential blepharospasm and hemifacial spasm. Ophthalmology. 1991;98:357-366.
Wissel J., Kabus C., Wenzel R. Botulinum toxin in writer’s cramp: Objective response evaluation in 31 patients. J Neurol Neurosurg Psychiatry. 1996;61:172-175.
Wolf S.L., Blanton S., Baer H., et al. Repetitive task practice: A critical review of constraint-induced movement therapy in stroke. Neurology. 2002;8:325-338.
Wu A.D., Fregni F., Simon D.K., et al. Noninvasive brain stimulation for Parkinson’s disease and dystonia. Neurotherapeutics. 2008;5:345-361.
Yaltho T.C., Jankovic J. The many faces of hemifacial spasm: Differential diagnosis of unilateral facial spasms. Mov Disord. 2011. (in press)
Yoon J.S., Kim J.C., Lee S.Y. Double-blind, randomized, comparative study of Meditoxin versus Botox in the treatment of essential blepharospasm. Korean J Ophthalmol. 2009;23:137-141.
Yoshida K., Kaji R., Kubori T., et al. Muscle afferent block for the treatment of oromandibular dystonia. Mov Disord. 1998;13:699-705.
Yoshimura D.M., Aminoff M.J., Olney R.K. Botulinum toxin therapy for limb dystonias. Neurology. 1992;42:627-630.
Yoshor D., Hamilton W.J., Ondo W., et al. Comparison of thalamotomy and pallidotomy for the treatment of dystonia. Neurosurgery. 2001;48:818-824.
Zeuner K.E., Bara-Jimenez W., Noguchi P.S., et al. Sensory training for patients with focal hand dystonia. Ann Neurol. 2002;51:593-598.
Zeuner K.E., Hallett M. Sensory training as treatment for focal hand dystonia: A 1-year follow-up. Mov Disord. 2003;18:1044-1047.
Zeuner K.E., Peller M., Knutzen A., et al. Motor re-training does not need to be task specific to improve writer’s cramp. Mov Disord. 2008;23:2319-2327.
Zuddas A., Cianchetti C. Efficacy of risperidone in idiopathic segmental dystonia. Lancet. 1996;347:127-128.