[level-membership-for-neurology-category]
Chapter 74 Traumatic Brain Injury in Children
Introduction and Background
Epidemiology of Pediatric Traumatic Brain Injury
Traumatic brain injury (TBI) is a major cause of death and disability in the pediatric population and has been identified as a significant public health problem in the United States and worldwide [Engberg and Teasdale, 1998; Murgio et al., 1999; Thurman et al., 1999; Tsai et al., 2004; Weiner and Weinberg, 2000]. Age-related incidence rates for TBI in children have been estimated to be as high as 670 per 100,000 when head injuries of all severities are included [McCarthy et al., 2002]. When limited to TBI resulting in hospitalization, the reported incidence has declined over the past 15 years, most likely due to injury prevention measures and improved motor vehicle safety. None the less, TBI is responsible for the majority of trauma-related death and hospitalization. The incidence rate of TBI-related hospitalization remains significant, consistently around 75–80/100,000 [McCarthy et al., 2002; Reid et al., 2001]. Childhood TBI results in an estimated 3000 deaths, 29,000 hospitalizations, and 400,000 emergency department visits annually in the U.S. (children 0–14 years old) [Thurman et al., 1999]. TBI is six times more likely to cause death in childhood than AIDS/HIV infection, and 20 times more likely than asthma [Centers for Disease Control and Prevention, 2000]. Moderate and severe pediatric TBI has been associated with long-standing cognitive, neurological, and behavioral impairments [Fay et al., 1994; Massagli et al., 1996a], and the cost of these disabilities is often carried over the person’s lifetime. Fortunately, the majority of all TBI is considered mild in severity; however, recent studies suggest that even mild pediatric TBI may have adverse long-term functional consequences [Hawley et al., 2004]. There is also increasing evidence that repeated mild TBI, as commonly occurs in a sports-related setting, results in chronic cognitive impairment [Collins et al., 1999; Matser et al., 1999], and may even predispose to early memory disturbances and dementia in some individuals [Guskiewicz et al., 2005; McKee et al., 2009; Omalu et al., 2006]. Despite these facts, no specific treatment standards exist for pediatric TBI, either acutely or during recovery. A recent review of guidelines for pediatric TBI management concluded that acute supportive therapies are often administered inconsistently or simply extrapolated from adult TBI protocols, not taking into account the unique physiology of the immature brain [Adelson et al., 2003b].
The peak incidence of pediatric TBI occurs in the adolescent and young adult, with a secondary peak in infancy [Kraus and McArthur, 2000]. The etiology of TBI varies with age (Figure 74-1). Adolescents sustain most head injuries in motor vehicle accidents, as well as sports-related concussions and assaults. Pre-adolescent children are also frequent victims of motor vehicle accidents, but more often as a pedestrian or while riding a bicycle. Those under the age of 5 years are more prone to falls [Thurman, 2001], while infants are particularly vulnerable to repeated severe TBI in the form of abusive head trauma (AHT, previously inflicted TBI or nonaccidental trauma).
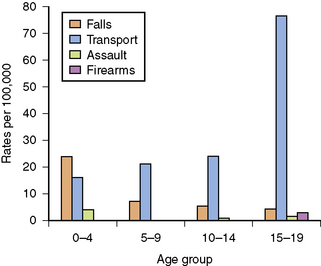
Fig. 74-1 Etiology of pediatric traumatic brain injury by age group.
(From Thurman D, for the Centers for Disease Control and Prevention. Traumatic brain injury in the U.S.: Assessing outcomes in children, Appendix B. Available at: http://www.cdc.gov.)
Traumatic brain injury is about twice as common in boys than girls overall, with this gender distinction becoming increasingly evident in the childhood and adolescent years [Kraus and McArthur, 2000; Rivara, 1994]. In the U.S. annually, it is estimated that about 30,000 pediatric patients incur permanent disability as a result of TBI. These sequelae include headaches, post-traumatic epilepsy, motor disturbances, learning disabilities, cognitive impairment, and behavioral problems. Furthermore, the vast majority of children suffering TBI, even those surviving moderate or severe injury, fail to receive adequate medical, rehabilitative, or psychosocial follow-up upon hospital discharge [Armstrong and Kerns, 2002; Hawley et al., 2004]. Finally, given the magnitude of recurrent head trauma as a problem of youth, it is increasingly noted that health-care providers should take every opportunity to provide important information and education to patients and their families, particularly with regard to potential sequelae of repeated injuries, as well as effective injury prevention.
Anatomy
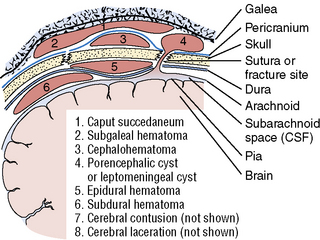
To understand the mechanisms and types of TBI best, knowledge of the basic anatomy of the brain and its coverings is essential (Figure 74-2). The scalp is the outermost covering and is highly vascular, tending to bleed profusely when lacerated. Under the scalp is a tendinous sheath, extending from the frontal to occipital regions, called the galea aponeurotic. The potential space beneath this is the subgaleal compartment, an occasional site of significant bleeding following head injury. The skull is next, with the periosteum covering its outer surface. The skull itself is composed of three layers, the bony outer and inner tables, separated by the diploic space, which is more vascular. Between the inner table of the skull and the dura mater is the epidural space, another potential space that is a significant site for arterial bleeding, particularly after skull fracture. The dura is the tough outer protective layer covering the brain itself. Below the dura is the subdural space, which is crossed by small veins that drain into the venous sinuses, which provides another site for post-traumatic hematomas. The next layer of brain coverings is the arachnoid, and under this, the subarachnoid space, which contains cerebrospinal fluid (CSF), and into which post-traumatic bleeding is also fairly common. The subarachnoid space is contiguous with the basal cisterns and the ventricular system, and CSF normally flows from its site of production at the choroid plexus within the ventricles, out into the basal cisterns, over the convexities of the cerebrum and into the venous drainage of the brain via the arachnoid villi. Subarachnoid blood can impair the flow of CSF after trauma and occasionally result in hydrocephalus. The last layer is the pia mater, which lies directly over the brain surface itself.
Biomechanics
Because the brain parenchyma is itself soft and deformable and brain regions are connected by fiber tracts, the brain is also prone to injury from rotational forces. These are the forces imparted when one part of the brain is twisted in relation to another part. In these circumstances, the underlying white-matter tracts are subjected to significant shearing forces that can result in stretch injury and microhemorrhages seen clinically [Tong et al., 2004] and in experimental models [Huh et al., 2008; Raghupathi and Margulies, 2002]. This type of damage following trauma is referred to as diffuse axonal injury (DAI), and can result in significant clinical disability. By its nature, DAI can occur throughout the brain, with a propensity for regions with major fiber tracts (corona radiata, corpus callosum, brainstem, etc.). While the developing brain has been shown to have remarkable resilience to focal injuries such as strokes or surgical resection, its ability to recover from diffuse injuries may be much more limited.
Penetrating trauma, while less common, often results in tremendous biomechanical forces, as well as physical disruption of tissue along the path of the foreign object. While the tissue destruction, with surrounding necrosis, hemorrhage, and edema, is a major factor in determining long-term disability, the transmitted forces from the passage of the projectile to more distant parts of the brain can result in immediate death, presumably by affecting respiratory and autonomic centers [Carey, 1995]. One large series reported that younger age was associated with worse outcome from gunshot wounds [Levy et al., 1993].
Injury Types
While specific injury types will be discussed in greater detail later in the chapter, an overview of the types of injuries commonly seen in the pediatric population ranges from superficial injuries to the scalp to severe diffuse cerebral edema with herniation. Scalp injuries are very common, and include contusions, lacerations, and hematomas. While many scalp injuries may occur with little, if any, evidence of underlying brain injury, careful history and examination should be undertaken in each case to rule out the possibility of concussion or, more rarely, intracranial injury. Concussion has been redefined as any trauma-induced transient disturbance of neurological function, even in the absence of unconsciousness [American Academy of Neurology, 1997]. A large scalp hematoma may conceal a skull fracture, and, in infants, can result in significant blood loss. Skull fractures are reported following 20 percent of childhood TBI [Harwood-Nash et al., 1971], and while simple linear fractures are generally benign, the presence of any fracture greatly increases the risk of intracranial hemorrhage [Lloyd et al., 1997]. Post-traumatic intracranial bleeding may occur in the form of cerebral contusion, cerebral laceration, and epidural, subdural, or subarachnoid hemorrhage, individually or often in combination. DAI represents stretch and shearing of white-matter tracts and can have profound clinical consequences, with prolonged unconsciousness and poor outcome. Traumatic injury, at its most severe, can lead to cerebral swelling and elevated ICP. While this problem may be accommodated to some degree by flexible sutures and open fontanels in infants, all ages are vulnerable to cerebral herniation, a displacement of cerebral contents within or outside of the intracranial space. Based on location of edema or focal lesions, distinct types of herniation can occur (Figure 74-3). Each of these injury types will be discussed in greater detail later in this chapter.
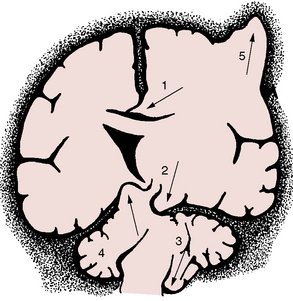
Fig. 74-3 Different forms of brain herniation.
1, Cingulate. 2, Uncal. 3, Cerebellar tonsillar. 4, Upward cerebellar. 5, Transcalvarial.
(From Fishman RA. Cerebrospinal fluid in diseases of the nervous system. Philadelphia: WB Saunders, 1980.)
While all of the above types of injury are associated with clinical syndromes of primary TBI, it is important to be aware that, inextricably intertwined with the primary injury, are the effects of so-called secondary injuries. Secondary injuries may be additional pathological insults independent of the original TBI, such as hypotension due to systemic hemorrhage, hyperthermia due to concomitant infection, or airway occlusion by a foreign body resulting in hypoxia or systemic hyper-/hypoglycemia. In many individuals, a secondary injury may be directly or indirectly related to the original brain injury, but occurs after the initial TBI. Thus, the spectrum of secondary injury encompasses entities such as cerebral edema with reduced brain perfusion, intracranial hemorrhage, early post-traumatic seizures, respiratory compromise of neurologic origin resulting in hypoxia, and post-traumatic hydrocephalus. More general etiologies of secondary injury include on-going excitotoxicity, free radical generation, oxidative stress, and inflammation. Clinical secondary injuries, such as hypotension, hypoxia, hydrocephalus, edema, hypoglycemia, and seizures, are of great medical significance, as they are generally treatable in the intensive care unit setting, and are often independent predictors of poor outcome [Chesnut et al., 1993a; Chiaretti et al., 2002].
Pathophysiology of Traumatic Brain Injury
Distinctions of Injury to the Developing Brain
Biomechanical Factors
Clearly, the physics of TBI in a child is different than in an adult. The relatively large head size, reduced muscular strength in the neck, and increased flexibility of the neck may promote a greater range of biomechanical force imparted to the head. On the other hand, there is less CSF space around the brain, and the prominent bony ridges of the anterior and middle cranial fossae are less developed. These factors may contribute to a lower occurrence of focal lesions in pediatric TBI [Berney et al., 1994; Bruce et al., 1979; Levi et al., 1991; Luerssen et al., 1988].
The physical properties of the developing skull also confer advantages and disadvantages. It is thinner, and thus more prone to diffuse deformation [Margulies and Thibault, 2000]. Some authors have reported a higher risk of skull fracture in children [Berney et al., 1994], although this finding is not universal [Levin et al., 1992]. However, the open fontanels and flexible sutures may serve to dampen traumatic forces, as well as to accommodate a greater degree of intracranial swelling. The brain itself, particularly in infants and young children, has a higher water content and is incompletely myelinated [Holland et al., 1986; Paus et al., 2001]. The higher water content tends to make the brain less compressible and less compliant than in the adult. Thus, the physical properties of the immature brain may provide both unique benefits and potential vulnerabilities to TBI, a theme that will be repeated when considering other developmental distinctions.
Changes in Cerebral Metabolism
It is well known that cerebral metabolism changes during maturation. The primary source of energy for the brain changes from lactate in the perinatal period, to ketone bodies while nursing, to glucose after weaning, and on through to adulthood [Nehlig, 2004; Vannucci and Vannucci, 2000]. The ability to use alternative fuels may have important consequences for acute energy metabolism in the injured brain at different ages [Prins et al., 2004].
Even after the brain has switched to predominantly glucose metabolism, the levels of this metabolic activity continue to change throughout the developing years. Basal glucose metabolism rates peak at around 6 years of age, correlating with the time of maximal synaptogenesis. As synaptic pruning and dendritic rearrangement occur throughout childhood and adolescence, cerebral glucose metabolism declines, although it still remains elevated compared to the adult brain [Chugani and Phelps, 1986]. Also, the brain does not mature uniformly, and cerebral metabolic rates for glucose and other substances have different time courses regionally.
Distinct Neurovascular Regulation
Neurovascular control may also differ in the young brain. Early studies suggested a propensity for diffuse cerebral swelling in children following closed-head injury, occurring 2–5 times more often than in adults [Aldrich et al., 1992; Lang et al., 1994]. Recent studies report that normal children have higher baseline cerebral blood flow (CBF) than adults, with significant variation across narrow age groups [Suzuki, 1990; Zwienenberg and Muizelaar, 1999]. At birth, CBF is lower than in adults, but increases rapidly to peak by age 5 years. CBF then appears to decline through adolescence to adult levels [Suzuki, 1990]. The effect of these developmental differences on injury-induced changes in CBF and the mechanism(s) of these differences are not yet well understood. Certainly, molecular mediators of neurovascular tone, such as nitric oxide synthase, have differential expression patterns in the immature brain [Keilhoff et al., 1996; Ohyu and Takashima, 1998], and these may play a role in age-dependent changes in blood flow and response to injury.
Increased Excitatory Neurotransmission
As mentioned earlier, the number of synapses in the human brain appears to peak in early childhood, and is associated with a peak in cerebral glucose metabolism. Levels of excitatory transmitter receptors are higher in the immature brain [Fosse et al., 1989; Insel et al., 1990; Miller et al., 1990a]. The occurrence of early post-traumatic seizures is also noticeably higher in children, and particularly in infants and younger children [Annegers et al., 1980; Berney et al., 1994].
Because excessive release of excitatory neurotransmitters can result in neuronal injury and death (excitotoxicity), the fact that the developing brain is more active might suggest that blocking excitatory transmission would be neuroprotective. However, early in development, neuronal activation also plays a critical role in both survival of immature neurons and in proper wiring of cerebral circuitry. In fact, in animal models, use of glutamate antagonists early in postnatal development (while protective against some degrees of excitotoxic insult) resulted in a large degree of programmed cell death (apoptosis) [Bittigau et al., 1999; Ikonomidou et al., 1999; Pohl et al., 1999]. Such evidence suggests that optimal recovery from brain injury may require a proper balance of excitation and inhibition, and may also account for the relative ineffectiveness of glutamate antagonists following human brain injury [Biegon et al., 2004; Hardingham et al., 2002; Ikonomidou and Turski, 2002].
On-Going Cerebral Maturation
In the past, the developing brain was generally felt to recover better after many types of brain injury, including stroke, TBI, and surgical resection. Developmental plasticity does confer some advantage when the brain must recover from a focal lesion [Kennard, 1942; Kolb et al., 2000; Villablanca and Hovda, 1999], if the lesion occurs at a specific age (see chapter 13). However, more recent work clearly indicates that the very young brain may be particularly vulnerable to diffuse injuries like TBI [Anderson et al., 2005; Babikian and Asarnow, 2009]. One mechanism for this vulnerability can be explained by post-traumatic impairment in experience-dependent neuroplasticity, which is the complex task of responding to environmental stimuli and rearranging neuronal network that results in enhanced function. In a mature brain, the consequences of such impairment might be overcome with time; in the young brain, it is likely that reduced responsiveness at a critical window of development will result in long-term dysfunction [Giza et al., 2009]. As a result, studies looking at post-TBI brain development, both in experimental animals and in children, must necessarily take into account the normal trajectory of brain maturation.
The Post-Traumatic Neurometabolic Cascade
Traumatic brain injury results in an immediate release of glutamate, widespread ionic changes, fluctuating cerebral glucose metabolism (initially elevated, then reduced), and dynamic changes in blood flow. Later, axonal damage and disconnection, reduced responsiveness to physiological stimuli, impaired neurotransmission, and apoptosis occur [Giza and Hovda, 2001]. For an overview of post-traumatic pathophysiology at the cellular level, see Figure 74-4. Specific components of the brain’s response to traumatic injury will be discussed below.
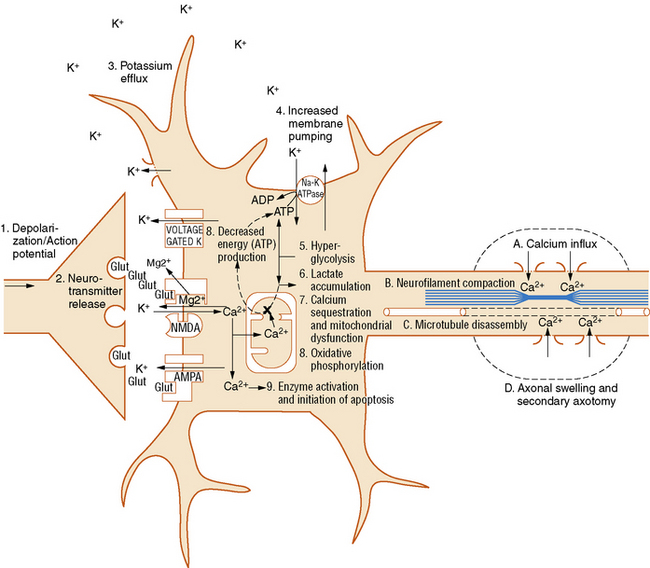
Fig. 74-4 Neurometabolic cascade after traumatic injury.
(From Giza CC, Hovda DA. The neurometabolic cascade of concussion. J Athl Train 2001;36:230).
Glutamate Release and Ionic Flux
Following a traumatic brain injury, there is an immediate and indiscriminate release of the excitatory neurotransmitter, glutamate [Bullock et al., 1998; Katayama et al., 1990]. This can occur due to widespread triggering of action potentials, synaptic neurotransmitter release, and membrane disruption. This flood of glutamate results in a massive efflux of potassium into the extracellular space [Katayama et al., 1990; Nilsson et al., 1993], and an influx of sodium and calcium [Nilsson et al., 1996; Osteen et al., 2001]. While hyperacute measures of extracellular glutamate are obviously unobtainable in human patients, microdialysis or CSF samples from severely injured patients (including children) in the days after injury have demonstrated glutamate elevations and have been associated with secondary injury and poorer outcomes [Bullock et al., 1998; Ruppel et al., 2001; Vespa et al., 1998].
Dynamic Changes in Cerebral Metabolism
As the injured cells attempt to restore ionic equilibrium post-trauma, membrane pumps such as the Na+-K+ ATPase are activated. An increase in cerebral glucose uptake, termed hyperglycolysis, is seen early after experimental TBI and has been postulated as a mechanism of providing additional substrate to generate the energy necessary to drive these membrane ionic pumps [Kawamata et al., 1992; Yoshino et al., 1991]. Acute studies of cerebral glucose metabolism using positron emission tomography (PET) in adult patients have also shown evidence for increased cerebral glucose uptake [Bergsneider et al., 1997]. Interestingly, there is increasing laboratory evidence that injured neurons are capable of utilizing alternative fuels to glucose [Magistretti et al., 1999; Pellerin and Magistretti, 2004; Prins et al., 2004], and it is well known that the capacity to use alternative fuels in uninjured neurons is age-specific [Vannucci and Vannucci, 2000].
After the acute period of hyperglycolysis, there is a prolonged period of diminished glucose uptake by the brain. This hypometabolism is seen in both adult and immature rats, although it lasts longer in adults (7–10 days) than in rat pups (3 days) [Yoshino et al., 1991; Thomas et al., 2000]. In traumatically injured adult patients, the period of reduced glucose metabolism has been shown to last weeks or months [Bergsneider et al., 2001].
Cerebral Blood Flow: Hyperemia? Hypoperfusion?
Blood flow also undergoes dynamic changes after injury and the pattern these changes take may depend on the type of injury and its severity. A pattern of diffuse cerebral swelling has been reported more often in children than adults [Aldrich et al., 1992; Lang et al., 1994]. This phenomenon was originally attributed to hyperemia [Bruce et al., 1981], but subsequent studies suggest that the incidence of post-traumatic hyperemia has been overestimated [Muizelaar et al., 1989]. In the earlier studies of post-TBI CBF in children, flow values were compared with normal values from a young adult control group. When CBF has been measured in normal children, however, it appears that blood flow values change dramatically across development, and are generally much higher in young children than in adults [Chiron et al., 1992; Suzuki, 1990; Zwienenberg and Muizelaar, 1999]. Subsequent studies of CBF in children suffering TBI, when compared to age-appropriate control values, have failed to show clear hyperemia [Adelson et al., 1997a; Sharples et al., 1995b]. In an experimental model of TBI, there is evidence for dramatically increased cerebral blood flow adjacent to a traumatic contusion, and this focal hyperemia was most pronounced in immature animals [Biagas et al., 1996]. While clinically it is important to recognize that diffuse cerebral swelling is more common in children, the mechanism of this swelling does not appear to be solely attributable to hyperemia.
The relation between CBF and outcome appears to depend upon the state of cerebral autoregulation. In the presence of intact autoregulation, increased CBF has been associated with better perfusion and improved outcome in children and young adults [Kelly et al., 1996; Vavilala et al., 2004]. However, after very severe injuries, in which autoregulation is dysfunctional and cerebral vasculature is pressure-passive, increased CBF can result in intractable elevations of ICP and worse outcome.
Beyond the acute phase of head injury, CBF generally declines. Post-TBI CBF can decline below normal, and it is important to monitor for ischemia, as this is associated with worse outcome [Bouma et al., 1991; Vespa et al., 2003]. Investigations are still under way to determine whether neurovascular coupling is intact during this period of reduced flow.
Altered Neurotransmission
Reductions in excitatory neurotransmitter systems have been reported after TBI in experimental animals, including impairments in glutamatergic [Giza et al., 2006; Miller et al., 1990b; Osteen et al., 2004; Sihver et al., 2001], noradrenergic [Fujinaka et al., 2003; Krobert et al., 1994], and cholinergic transmission [Gorman et al., 1996]. These changes may correlate with the injury-induced reduction in cerebral metabolism described above. Furthermore, these changes may be an underlying mechanism for post-TBI deficits in neuronal activation [Dietrich et al., 1994; Ip et al., 2003; Sanders et al., 2000], diminished activity-dependent molecular responsiveness [Giza et al., 2002; Griesbach et al., 2004], and impaired experience-dependent plasticity in developing animals [Fineman et al., 2000; Giza et al., 2005; Ip et al., 2002]. Activation studies using functional magnetic resonance imaging (fMRI), even after relatively mild TBI, have shown reduced or aberrant activation patterns in traumatically injured teenagers and young adults [Jantzen et al., 2004; McAllister et al., 1999].
Axonal Disconnection
Mechanical stretch can disrupt axonal membranes, resulting in calcium influx and microtubule and neurofilament disruption. Damage to these important cytoskeletal components impairs normal axonal transport, endangering distal axonal segments and synapses. Over time, disrupted axonal transport leads to an accumulation of transported proteins and organelles at the injury site, causing axonal blebs and, eventually, disconnection [Pettus and Povlishock, 1996; Povlishock and Christman, 1995; Povlishock and Pettus, 1996]. This type of damage is seen following experimental TBI in both immature [Raghupathi and Margulies, 2002] and adult animals [Povlishock and Pettus, 1996]. In postmortem human specimens, evidence of axonal blebbing and damage has been reported long after injury [Maxwell et al., 1997], and MRI can clearly show abnormalities of diffuse axonal injury, in pediatric and adult TBI patients [Tong et al., 2004]. Newer imaging modalities, such as diffusion tensor imaging (DTI) and MR spectroscopy, have shown white-matter abnormalities that correlate with functional impairments after pediatric TBI, even in brain regions without overt structural lesions [Ashwal et al., 2006; Babikian et al., 2009a; Ewing-Cobbs et al., 2008; Wilde et al., 2008].
Cell Death: Necrosis and Apoptosis
Apoptosis is characterized by nuclear condensation, DNA fragmentation, and preservation of the cell membrane. Apoptosis requires energy and protein synthesis, and triggers an acute inflammatory response that may be distinct from that seen with necrosis (see chapter 14). Usually, apoptotic cell death takes longer to evolve after injury, and studies have demonstrated that apoptosis can continue to occur long after experimental trauma [Conti et al., 1998; Wilson et al., 2004]. While apoptosis is seen after TBI, it is also part of the brain’s normal maturational program. Thus apoptotic pathways are more active in the immature brain, and apoptotic cell death appears more prominently after experimental injury to the developing brain [Bittigau et al., 1999; Pohl et al., 1999].
Impaired Plasticity
An important aspect of injury in the young brain is the effect of this injury on normal neuronal responsiveness and on developmental plasticity (see chapter 13). As mentioned earlier, increased plasticity in the immature brain results in significantly better recovery than in the adult following a focal lesion, and this has been well described in rats [Kolb and Tomie, 1988], cats [Burgess and Villablanca, 1986; Villablanca and Hovda, 1999], primates [Kennard, 1938], and humans [Hogan et al., 2000; Trauner et al., 1993]. However, the benefits of youth are less apparent when the injury is more diffuse and/or occurs at a critical window of brain development. Environment enrichment (EE) is an experimental model of enhancing brain development, and animals reared in EE grow up to have larger brains [Bennett et al., 1964; Diamond et al., 1964; Rosenzweig and Bennett, 1996], increased dendritic arborization [Faherty et al., 2003; Greenough et al., 1973; Juraska, 1984; Volkmar and Greenough, 1972], and superior performance on neurobehavioral tasks [Tees et al., 1990; Venable et al., 1988; Williams et al., 2001]. While injured rat pups show less overt cell death and less behavioral impairment than adults [Gurkoff et al., 2006; Prins et al., 1996; Prins and Hovda, 2003], they do lose the ability to benefit from EE rearing. Specifically, they show a loss of EE-induced experience-dependent plasticity; cortical thickening is blocked [Fineman et al., 2000], expansion of dendritic arbors is inhibited [Ip et al., 2002], and EE-induced cognitive enhancements are absent [Giza et al., 2005]. These studies of altered developmental plasticity are also supported by the findings of Prins and colleagues using a different model [Prins et al., 2003]. Following a lesion, regrowth of entorhinal cortical axons in a nontraumatically injured brain occurs in a well-characterized fashion. In juvenile rats subjected to experimental TBI, this normal pattern of axonal regrowth was markedly disturbed, with evidence of disruption down to the synaptic level.
Studies of developmental plasticity in traumatically injured children are difficult, as the effects of both age at injury and age at assessment must be considered in the control groups. However, there is growing evidence that pediatric TBI results in severity-dependent altered trajectories of brain development [Babikian and Asarnow, 2009; Catroppa et al., 2008]. First, although children generally have better outcome from TBI than adults, those injured at the earliest ages (i.e., in infancy) actually have worse outcomes [Anderson et al., 2005; Levin et al., 1992; Luerssen et al., 1988]. Some of this may be due to different mechanisms of injury, including nonaccidental trauma, which has a very poor developmental prognosis and is predominantly an etiology seen in infancy. It is also possible that the very young brain is decidedly more vulnerable to injury, as suggested by experimental studies using hypoxia-ischemia [Ikonomidou et al., 1989] and TBI [Bittigau et al., 1999; Pohl et al., 1999]. Second, there are rare anecdotal reports of neonates with head trauma that appear to have resulted in abnormal cortical development that was subsequently confirmed pathologically after epilepsy surgery for intractable seizures [Lombroso, 2000; Marin-Padilla et al., 2002]. Third, studies of severe pediatric TBI show more persistent cognitive deficits when the injury was diffuse rather than focal [Levin et al., 2000]. Lastly, in studies of repeated, mild sports-related TBI sustained during adolescence or early adulthood, subtle but significant cognitive impairments are detected [Collins et al., 1999; Matser et al., 1999].
Examination
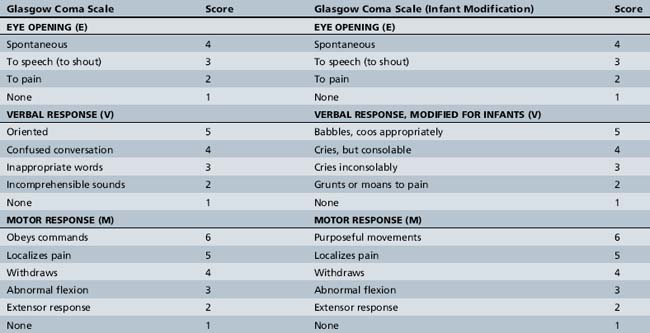
The patient’s Glasgow Coma Scale (GCS) score should be determined. The GCS is a quick, reproducible means of rating the severity of the patient’s neurological injury (Table 74-1) [Teasdale and Jennett, 1976]. There are three components to the GCS score:
The total GCS score ranges from 3 (worst) to 15 (normal). There are obvious limitations to this scale in the setting of pediatric trauma, such as how to determine verbal score in infants and young children. Several GCS modifications have been proposed to make it more usable in this age group, and one modified scale is shown in Table 74-1. By convention, mild TBI is defined by a GCS score of 13–15, moderate by 9–12, and severe by 8 or less. A patient with a GCS of 13–15 but having an intracranial lesion would generally be classified as having a moderate TBI [Malec et al., 2007] (see chapter 73).
Sensorimotor examination can also be done quickly, starting with the best motor response elicited on GCS testing. Limb posture and muscle tone should be examined, and particular note should be made of any asymmetry. If the patient is unable to follow commands, then sensorimotor responses may be assessed by application of a peripheral noxious stimulus (e.g., nail bed pressure) in each limb. If no response occurs, then the site of noxious stimuli can be moved centrally (e.g., sternal rub, supraorbital pressure) to distinguish between afferent versus efferent impairment. Application of painful stimulus centrally also allows one to observe more easily whether the child can localize pain (distinction between a 4 and 5 for best motor score). If the child is awake, coordination should be assessed by observation of posture, spontaneous movements, and directed movements when the child reaches for an object or mimics the examiner. Deep tendon reflexes should be checked for asymmetry, hyperreflexia/clonus or areflexia. Flaccid paresis with no reflexes should immediately raise suspicion of a spinal injury. An outline of the rapid trauma examination is provided in Box 74-1.
Box 74-1 Rapid Pediatric Trauma Examination
In the mildly injured child who is conscious at the time of initial evaluation, the neurological examination can be conducted less urgently and more completely. In instances, mental status testing can be more extensive, starting with observation and possibly digit span recall to assess the child’s attention. This can be followed by evaluation of orientation, language, memory (both anterograde – for new items, and retrograde – for past events, including events surrounding the injury), and behavior. Cranial nerve, motor, coordination, and sensory examinations can include voluntary responses to the examiner’s commands, as well as subjective reports of sensory input (visual fields, tactile stimulation). Gait should be observed for signs of unsteadiness or ataxia. As with the more severely injured child, deep tendon reflexes and plantar responses should be checked. Aspects of the neurological examination are reviewed in detail in Chapters 1–4 and 73.
Immediate Management
Initial management of the child suffering from moderate or severe TBI centers on implementation of the ABCs and avoidance of secondary insults (see chapter 76). Early management will occur concomitantly with initial evaluation and examination. The airway must be secured and supplemental oxygen provided. In circumstances where the child is unable to maintain his/her airway adequately, then endotracheal intubation should be performed. Appropriate noninvasive monitors should be placed, including cardiac, respiratory, temperature, blood pressure, and those for pulse oximetry. Intubated patients should also have an end tidal CO2 monitor. Intravenous access must be established immediately and fluid resuscitation begun. Blood should be drawn for serum electrolytes, glucose, renal and hepatic function, complete blood count, prothrombin time (PT)/partial thromboplastin time (PTT), type and cross for blood transfusion, and, if appropriate, alcohol or drug screens.
Acute Clinical Syndromes
Herniation Syndromes
Herniation is the displacement of brain tissue from one intracranial compartment to another. This can occur as a result of a space-occupying lesion in one region, diffuse swelling of both cerebral hemispheres, or hydrocephalus (see Figure 74-3 and chapter 73) [Fishman, 1980; Plum and Posner, 1980]. The Cushing response is a paradoxical bradycardia with hypertension and slow irregular respiration that can be seen in the setting of elevated ICP and impending herniation. Definitive treatment for herniation requires surgical removal of the offending mass, if present. In cases where herniation is triggered by edema, positioning the head so it is slightly elevated and in the midline is indicated (to facilitate venous drainage), as well as immediately initiating first-tier measures (see the section on management below) to control elevated ICP: sedation, hyperventilation, and hyperosmolar therapy. Ventriculostomy with CSF drainage should be instituted as soon as possible. Infants can tolerate some elevation of ICP better due to unfused sutures and an open fontanel. These properties also allow for additional physical examination clues to the presence of increased ICP, such as a tense or bulging fontanel or a steadily increasing head circumference (if chronic).
Diffuse Cerebral Swelling
Since trauma, particularly closed head trauma, is by nature a diffuse process, bilateral swelling can occur even in the absence of discrete focal lesions. In fact, diffuse cerebral swelling is more common in children [Aldrich et al., 1992; Lang et al., 1994], while focal cerebral damage, such as contusions, is more common in older adolescents and adults. The etiology of this swelling was originally postulated to be due to hyperemia [Bruce et al., 1981]. However, as previously discussed, original reports of post-traumatic hyperemia in children overestimated the frequency of this phenomenon by comparing CBF values with those of uninjured young adult controls rather than uninjured children [Muizelaar et al., 1989; Zwienenberg and Muizelaar, 1999]. It is more likely that severe degrees of cerebral edema are related to the severity and nature of the injury, and particularly to the degree of cytotoxic and vasogenic edema formation. Cytotoxic edema occurs as a result of injured neurons undergoing energy failure, with loss of membrane ionic gradients and a rapid increase in intracellular water. In contrast, vasogenic edema results from fluid seeping into the extracellular space across a damaged or dysfunctional blood–brain barrier. Cytotoxic edema is felt to predominate after traumatic injury. Diffuse edema can occur as a direct response to trauma, but can also be due to a post-traumatic secondary insult, such as hypotension or hypoxia. At its extreme, diffuse edema can result in central transtentorial herniation and death.
Diffuse Axonal Injury
A clinical correlate to the pathophysiological process of axonal injury and disconnection, DAI is thought to result from shearing and rotational forces on white-matter fiber tracts [Adams et al., 1982]. These forces are usually considerable, and DAI is most commonly seen after a high-impact injury, such as a motor vehicle accident or a high fall.
DAI can occur in the presence or absence of hypoxia-ischemia or elevated ICP, but is a discrete process. The pathology of these lesions shows tearing and necrosis of nerve fibers, often with microhemorrhages. Blebs and swellings occur at points of axonal injury, locations that may subsequently become sites of axonal disconnection [Maxwell and Graham, 1997; Pettus and Povlishock, 1996; Povlishock and Christman, 1995]. These lesions are often seen at the gray/white junction of the frontal and temporal lobes, in the corpus callosum, and in the rostral brainstem. DAI is rarely seen on CT, and then only if hemorrhage is present. MRI is much more sensitive in detecting DAI, which can be seen as hypointensity on T2*-weighted gradient recall echo (GRE) imaging (Figure 74-5). Recently, MRI using susceptibility-weighted imaging (SWI) has more clearly demonstrated and allowed quantification of hemorrhagic DAI lesions [Tong et al., 2004].
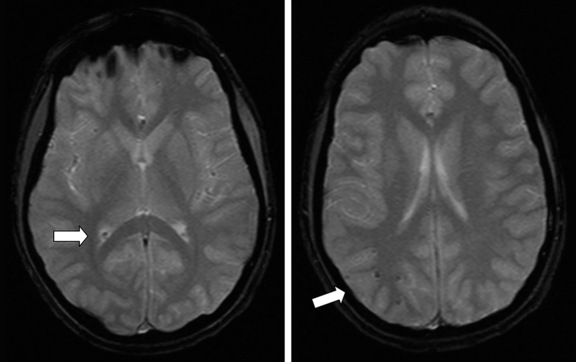
In general, patients with DAI will have lasting neurological impairments, with good outcome at 3 months ranging from 65 percent of mild DAI patients to only 15 percent of severely injured [LeRoux et al., 2000]. The aforementioned autonomic impairments may be the etiology of significant mortality in patients with DAI.
Paroxysmal Dysautonomia
Autonomic disturbances are well known to accompany CNS injuries. These disturbances are referred to by a large number of terms, including dysautonomia, thalamic or sympathetic storms, acute midbrain syndrome, and diencephalic seizures [Baguley et al., 2008]. Within the first few weeks following more severe TBI, a subset of patients appear to develop persistent and paroxysmal manifestations of dysautonomia, such as tachycardia, tachypnea, hyperthermia, hypertension, posturing/dystonia, and diaphoresis. In a case-control cohort study, patients with dysautonomia had worse functional outcomes than those without such symptoms, both groups having had comparable injury severities [Baguley et al., 1999]. It has been postulated that dysautonomia may be a potentially treatable cause of post-TBI morbidity. Interestingly, the average age of patients with dysautonomia was lower than the age of the entire TBI cohort, and post-traumatic dysautonomia has been reported in patients younger than 18 years of age. Treatment to mitigate autonomic disturbances is directed towards minimizing external triggers while also using medications that are thought to reduce hyperactive autonomic neurotransmission. Agents that have been used include morphine, benzodiazepines, propranolol, bromocriptine, and intrathecal baclofen. A recent open-label trial reported that gabapentin may have efficacy for this syndrome [Baguley et al., 2007; Baguley et al., 2008]. Further investigations are necessary to determine whether this syndrome is more common after TBI in pediatric patients than in adults, and if early recognition and treatment can improve long-term outcome.
Abusive Head Trauma
Child abuse has previously been termed “shaken baby syndrome” because the etiology of the cerebral injuries was thought to be rapid shaking of the infant. Biomechanical studies attempting to model these injuries found that shaking alone may not be capable of generating the forces necessary to cause the severe and diffuse intracranial injuries typically seen [Prange et al., 2003], and it has been proposed that most cases involve not only shaking, but also a final impact [Duhaime et al., 1987]. Many different terms, including “nonaccidental trauma,” “inflicted TBI,” or “shaking impact syndrome,” have been used to describe this clinical entity. Most recently, the American Academy of Pediatrics has officially adopted the term “abusive head trauma” (AHT) [Christian and Block, 2009].
The clinical presentation is rarely straightforward, as clinical signs in infants are often nonspecific and parents may not be forthcoming as to the circumstances of the injury. These infants can present with lethargy, irritability, seizures, or vomiting. Physical signs include a tense or bulging fontanel, split sutures, enlarged head circumference, retinal hemorrhages, and often evidence of other inflicted bodily injuries, such as fractures, bruising in unusual locations (back, upper legs), or burns. CT scanning typically reveals subdural hematomas, contusions, or intracranial hemorrhage. Subdural hematomas of differing ages are particularly suspicious for AHT, and are frequently seen over the cerebral convexities or along the falx (Figure 74-6B). The inconsistency between the reported history and the severity of these injuries is another indication that should lead the physician to suspect child abuse.
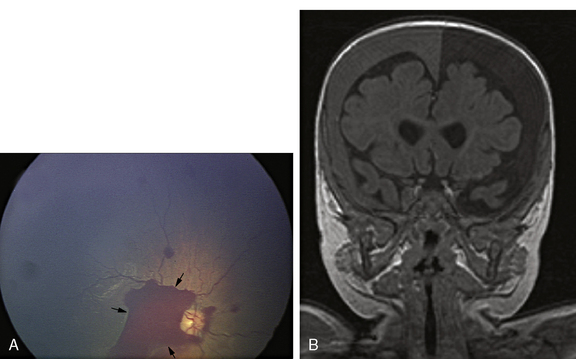
Infants with AHT may often need vigorous resuscitation, controlled ventilation, anticonvulsant treatment, and intensive care unit management. Beyond CT neuroimaging, diagnostic work-up may include whole-body x-rays to look for other acute or partially healing fractures. Ophthalmoscopic examination is essential to identify retinal hemorrhages (Figure 74-6A), as is a careful inspection of the skin for burns or excessive bruising, which are often in unusual locations and in various stages of resolution. Laboratory tests should be sent to evaluate for a coagulopathy. Two rare genetic metabolic disorders (glutaric aciduria and Menkes’ disease) can present with bilateral subdural fluid collections/hematomas and may mimic AHT.
Recent advances in neuroimaging have offered additional insight into the pathophysiology of AHT [Ashwal et al., 2010, 2010]. There is substantial evidence that AHT is associated with an increased incidence of concomitant hypoxic-ischemic injury. In cases of AHT, MR spectroscopy has been reported to show low N-acetylaspartate/phosphocreatinine (NAA/Cre) and the presence of lactate [Makoroff et al., 2005], and diffusion weighted imaging (DWI) has demonstrated areas of restricted diffusion [Suh et al., 2001; Ichord et al., 2007], both findings consistent with ischemic injury. Importantly, these abnormalities can be seen in areas of the brain that appear normal on standard structural imaging, and those showing these imaging patterns are reported to have worse long-term outcomes.
The outcome of infants suffering AHT is exceedingly poor. Mortality rates range from 7 to 30 percent, and severe neurocognitive sequelae are reported in one-third to one-half [Weiner and Weinberg, 2000]. Up to 80 percent of deaths due to trauma under the age of 2 years are attributed to nonaccidental injury [Bruce and Zimmerman, 1989]. It is believed that these children have a poorer prognosis reported than that usually reported for young infants, which is different than the general trend of improved outcome seen in younger infants. Several explanations may account for this observation. First, the very young brain may be selectively vulnerable; it is well known to be particularly sensitive to excitotoxicity, but also to depend upon excitatory glutamatergic stimulation for neuronal survival and development of connectivity. Second, the nature of AHT is such that repeated, fairly severe, injuries are sustained, and the cumulative effect of this injury pattern may result in the poor outcomes. Third, biomechanical properties of the infantile brain, including incomplete myelination and increased water content, may predispose it to shearing- and disconnection-type injuries, and disconnection at this maturational stage may result in developmental failure. Lastly, medical care for these children is often delayed, perhaps setting the stage for secondary injuries due to hypoperfusion, hypoxia-ischemia, and recurrent seizures. For a more detailed discussion of AHT, see Chapters 75 and 76.
Subarachnoid Hemorrhage
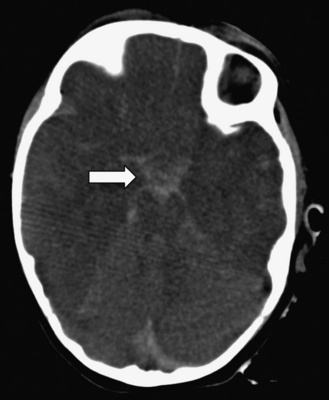
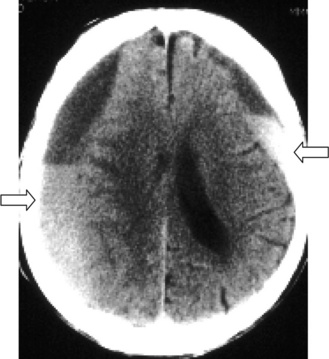
Subarachnoid hemorrhage (SAH) occurs commonly after TBI. Clinical symptoms of SAH are fairly nonspecific in infants (irritability, lethargy, poor feeding), but older children present with persistent headache, nausea, vomiting, neck stiffness, and altered mental status. Blood products are irritating to the brain itself and to the cerebral vasculature. Cortical irritation can result in seizures, and subarachnoid blood can provoke vasospasm, usually after several days. Vasospasm can be severe enough to cause cerebral ischemia or infarction. SAH is easily seen on CT, with a hyperdense signal of acute blood visible in the sulci and fissures, and often in the basal cisterns (Figure 74-7). Occasionally, subarachnoid blood can be seen in the ventricular system and is associated with an increased risk of post-traumatic hydrocephalus.
Subdural Hematoma
The appearance of SDH on CT scan is characteristic. An acute SDH appears as a crescent-shaped hyperdensity along the inner surface of the skull. Large lesions may track across suture lines (Figure 74-8). SDHs can also occur over the cerebral convexities, along the falx or tentorium, or more rarely, in the posterior fossa. Often there is associated focal edema in the vicinity of the SDH, and midline shift can lead to subfalcine or transtentorial herniation.
Epidural Hematoma
Epidural hematomas (EDHs) are acute collections of blood that occur in the potential space over the dura but beneath the calvarium. In adults, and to a certain degree in children, they are caused by arterial bleeding, particularly underlying the pterion, where they are often due to skull fracture with tearing of the middle meningeal artery. On CT, an EDH classically appears as a convex lens-shaped hyperdensity adjacent to the skull (Figure 74-9). EDHs generally are constrained by dural attachment to the skull and do not cross suture lines.
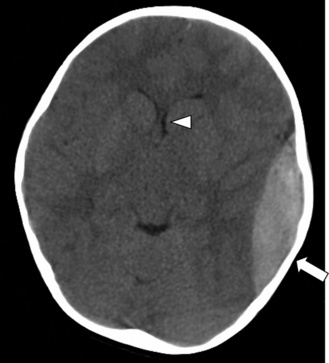
Although these characteristics are typical for EDH, in children there are significant distinctions. First, EDHs occur less frequently in children than in adults. Second, EDHs are more likely to be due to bleeding from bridging veins or dural sinuses, and thus less commonly associated with skull fracture. Those associated with fractures in infants can occasionally push through the fracture line of the thin skull to decompress, masking symptoms and allowing the EDH to accumulate to the point where anemia may be noted. Because they are more often venous in origin, the clinical presentation of EDHs in children may be more gradual. Some reports indicate that over 50 percent of children and over 80 percent of infants show no alteration in mental state at the time of injury [Choux et al., 1975]. Third, although the majority of EDHs in adults or children are supratentorial, the percentage of posterior fossa EDHs is higher in the pediatric population [Wright, 1966]. These are more often associated with occipital trauma, are venous in origin, and have delayed symptomatology (ataxia, vomiting, and headache), occurring a few days after the injury. Because of the risk of compression of critical brainstem structures and potential cardiorespiratory collapse, surgical removal is preferred.
Cerebral Contusion and Laceration
Cerebral contusion and laceration represent types of direct, focal damage to the brain. These injuries are less common in children than in adults. Contusions tend to be frontal or temporal, usually along the surface of the gyri (Figure 74-10). Their appearance may be as a subtle hypodensity on CT. Some contusions evolve into hemorrhagic lesions and become hyperdense on CT (see Figure 74-10), and in adults, acquiring serial CT scans for evaluation has been recommended. In severe cases with edema and mass effect, surgical excision of necrotic material can be performed.
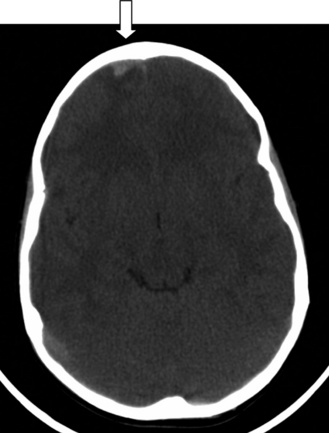
Lacerations are also fairly rare and involve physical tearing of brain tissue. These lesions can be associated with a depressed fracture or penetrating injury. Dura and vascular structures can be similarly torn. In infants, tearing of white matter can occur, even in the setting of blunt head trauma [Calder et al., 1984]. This is due to the lack of myelin and increased water content that are present in children.
Traumatic Arterial Dissection and Traumatic Aneurysms
There is increasing awareness that traumatic arterial dissection may be a more common cause of acute ischemic stroke in children than previously thought, occurring in approximately 5 percent of cases [Wraige et al., 2005]. In a recent review of 132 pediatric arterial dissections, only one-third were deemed “spontaneous,” and over 50 percent were related to some form of trauma [Fullerton et al., 2001]. Unlike in adults, headache and/or neck pain are rarely presenting symptoms in children. Traumatic dissections of the craniocervical arteries were almost never diagnosed prior to onset of focal neurological symptoms, suggesting that a high degree of suspicion needs be maintained in order to identify this etiology promptly. Arterial dissections appear to occur more often in boys, even independently of risk of trauma, and the onset of symptoms most commonly occurs between 1 and 7 days post-injury [Fullerton et al., 2001]. There are no evidence-based guidelines for treatment of pediatric craniocerebral dissections, although there is an increasing tendency to use antiplatelet agents or anticoagulation [Fullerton et al., 2001; Chamoun et al., 2008]. While traumatic dissections occur more commonly in the extracranial vessels, intracranial dissections do occur, and appear to be associated with intracranial aneurysms. Presenting signs and symptoms of traumatic intracranial aneurysms may include focal neurological signs related to ischemia or a delayed occurrence of subarachnoid hemorrhage, several weeks after the initial TBI [Kneyber et al., 2005]. Onset of focal symptoms in the first days after TBI or unexplained subarachnoid hemorrhage later after TBI should prompt additional diagnostic testing. MR angiography may be the preferred initial diagnostic modality due to its noninvasive nature.
Concussion
Concussion has been defined as any traumatically induced disturbance of neurological function and mental state, occurring with or without actual loss of consciousness [American Academy of Neurology, 1997]. Concussion in isolation is often referred to as mild TBI, and it is important to be aware that concussive-type injury can occur in almost every setting of head trauma, although it may be overshadowed by greater symptomatology, e.g., from a skull fracture or mass lesion. Mild TBI is the most common form of TBI in all age groups, accounting for over 75–85 percent of all head injuries [Kraus and McArthur, 2000].
Because there is little evidence of structural damage, the etiology of concussive symptoms is often ascribed to neural dysfunction that recovers over time. Several points regarding concussion are particularly relevant in children. First, there is much concern about the proper evaluation of concussion/mild TBI so as not to miss any associated intracranial injury, albeit exceedingly rare. This will be addressed in the section discussing general management of the mildly injured child. Second, duration of postacute physiological derangement is uncertain, but has significant implications for returning to normal activity. For example, a child returning to normal activity prior to complete physiological recovery may have subtle cognitive or fine motor deficits that need be considered in order to integrate them best into their pre-injury educational settings. Also, there is some concern that premature return to physical activity, such as contact sports, may place the individual at greater risk for a second injury. Third, while mild TBI in adults is well recognized to result in persistent neurobehavioral problems in a subset of patients, studies of a single mild TBI in children differ with regard to the significance of long-term sequelae [Asarnow et al., 1995; Hawley et al., 2004]. Finally, there is increasing evidence that repeated mild TBI, clinically encountered most commonly in a setting of sports-related concussion, may result in a chronic accumulation of subtle deficits [Collins et al., 1999; Matser et al., 1999] or even an early onset of memory disturbances and dementia in middle age [Guskiewicz et al., 2005; Omalu et al., 2006]. Many of these recurrent sports concussions occur in the late childhood and adolescent age groups, developmental periods where demonstrable brain maturation continues to occur, particularly in the frontal lobes [Sowell et al., 1999; Sowell et al., 2001]. Because of the unique nature of sports concussion, which has a mild, repetitive character and often occurs in brains still undergoing maturation, these issues will be discussed separately.
Acute Post-Concussive Syndromes
Certain clinical syndromes are characteristic of pediatric concussion and deserve particular mention. Acute behavioral changes commonly occur following mild pediatric TBI, including lethargy, irritability, disorientation, and listlessness. These symptoms can be worrisome for worsening intracranial injury, but in the presence of a normal CT scan, must be attributed to some as yet undetermined physiological dysfunction. Spreading depression, seizure activity, changes in blood flow, and even migraine have all been proposed [Ryan and Edmonds, 1988; Sanford, 1988].
Vomiting is very common following childhood head injury, occurring in up to 50 percent of individuals [Hugenholtz et al., 1987]. Some report that frequent vomiting is actually rare in more severe head injury, but characteristic of mild TBI [Luerssen, 1991]. However, the presence of vomiting is nonspecific, and if it is persistent or worsening, CT scanning should be performed.
Acute post-traumatic migraine is predominantly a behavioral syndrome and is diagnosed only after more serious lesions have been excluded [Haas and Lourie, 1988]. After a mild injury, with normal postinjury behavior, the child becomes progressively more agitated, aggressive, and combative. This is followed by tiredness and sleep, sometimes with no recollection of the earlier events upon awakening. Headache may occur but is not invariable. A prior history or family history of migraine is not uncommon.
Cortical blindness has also been reported following mild pediatric TBI, in the absence of intracranial pathology [Kaye and Herskowitz, 1986; Yamamoto and Bart, 1988]. Pupillary responses are preserved, and malingering may be suspected. Complete recovery is the rule. Migraine, spreading depression, or other neurovascular abnormalities may contribute to this phenomenon.
Sports Concussion/Repeated Concussion
Sports concussion is an increasingly common problem in childhood, occurring during team play in contact sports such as football and soccer, but also in basketball, wrestling, hockey, and others [Cohen et al., 2009]. Children also frequently experience mild TBI from accidents involving bicycles, rollerblades, skateboards, and scooters. Because of its association with particular recreational activities, the occurrence of sports-related concussion is more predictable than other forms of head trauma. This provides the opportunity to understand this injury type better by premorbid testing of at-risk individuals, as well as by instituting postinjury clinical assessments, beginning immediately after the concussion and re-assessing symptoms over the range of minutes to days. Another important opportunity is the possibility of more effective prevention strategies, by using approved protective gear when engaging in these activities and also by avoiding return to at-risk sports activity until recovery from injury is complete.
Epidemiology
It is estimated that over 300,000 sports-related head injuries occur annually in the United States, the majority of them mild [Thurman et al., 1998]. In high school athletes, 5–6 percent of all injuries were head injuries, the majority occurring during football play, but with almost every sport encountering some number of concussions [Guskiewicz et al., 2000; Powell and Barber-Foss, 1999]. Interestingly, after sustaining a concussion, there was a three-fold risk of a subsequent concussion during the same sports season [Guskiewicz et al., 2000]. This may be due to a concussion-induced physiological vulnerability, a manifestation of a particularly risky style of play in certain individuals, or perhaps an indication of subtle coordination and reaction time impairments in concussed individuals, setting them up for repeated injuries when they return to practice or play prior to full recovery. The vast majority of sports concussions (91 percent) do not involve loss of consciousness, and headache is the most common complaint (86 percent). Perhaps because of the mild nature of these injuries, almost one-third of these players returned to play the same day [Guskiewicz et al., 2000]. There is a common mentality among both players and coaches that one can “shake it off” and “take one for the team,” which sets the stage for recurrent injuries.
Pathophysiology
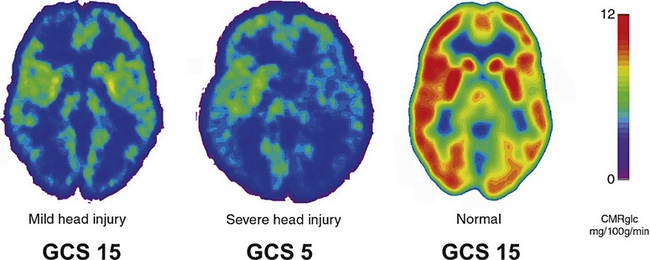
Physiological studies of sports concussion have demonstrated abnormalities in brain metabolism and activation in the absence of anatomical lesions [Bergsneider et al., 2000; McAllister et al., 1999; Vagnozzi et al., 2008] (Figure 74-11). PET scanning of mildly injured young adults has demonstrated dramatic reductions in cerebral glucose metabolism in walking, talking players that resemble abnormalities seen in comatose patients [Bergsneider et al., 2000]. Experimental studies of diffuse concussive injury in immature rats show periods of reduced glucose metabolism lasting for 2–3 days [Thomas et al., 2000].
More recently, fMRI has been used to study brain activation in mildly injured patients. Even while overall performance on particular cognitive tasks was acceptable, the task-associated activation patterns seen in these patients were decidedly abnormal [McAllister et al., 1999; McAllister et al., 2001]. A general theme of fMRI testing in concussed patients is that task-specific activation patterns become less focused and more diffuse in the injured brain [Jantzen et al., 2004]. This has been interpreted as a compensatory strategy to bypass dysfunctional circuits by utilizing less efficient alternative routes.
Evidence of postconcussive axonal dysfunction also has been reported using DTI, a newer MRI technique that quantifies the direction of diffusion along white-matter tracts. Abnormalities in DTI signal that correlate with clinical symptoms have been reported following even mild TBI in children and adolescents [Wilde et al., 2008; Wozniak et al., 2007].
Symptomatology
Symptomatology in sports concussions is similar to that described above regarding concussions. One consideration with subjective reporting of symptoms in a sports setting is that the player, being motivated to return to play, may attempt to minimize symptoms. Many athletic teams and physicians are utilizing a concussion checklist to semiquantitatively assess symptoms [McCrea et al., 2003]. This can be administered pre-injury and repeated at intervals during the recovery period. There is also increasing use of computerized neuropsychological tests that can be administered by an athletic trainer using a laptop, shortly after injury and again repeatedly during recovery. Interestingly, there is some evidence that recovery from sports concussion is prolonged in high-school students compared to college athletes [Field et al., 2003]. This raises legitimate concerns about blanket return-to-play guidelines and suggests that age-specific guidelines may be more appropriate [McCrory et al., 2009].
Sequelae
While the long-term significance of a single mild pediatric TBI is uncertain, several large studies have demonstrated multiple neurocognitive deficits in athletes who experienced an accumulation of lifetime concussions. Collins and colleagues reported impairments in executive functions and information-processing speed in college football players who experienced two or more lifetime concussions [Collins et al., 1999]. Other studies similarly showed neuropsychological deficits in amateur soccer players when compared with control athletes participating in noncontact sports [Matser et al., 1999]. Studies in adult rats have modeled this effect, demonstrating no significant deficit after a single mild injury, but definite impairment in spatial learning tasks after multiple repeated mild injuries [DeFord et al., 2002]. Individuals with previous concussions have shown a significantly longer duration of symptoms and length of recovery [Bruce and Echemendia, 2004; Guskiewicz et al., 2003]. More recent studies of retired professional football players suggest an increased risk of earlier-onset memory problems and even dementia. There also are anecdotal case reports of substantial tau protein deposition in the brains of ex-athletes who died of unrelated causes [Guskiewicz et al., 2005; McKee et al., 2009; Omalu et al., 2006]. Clearly, further work is needed to delineate the risk of chronic cognitive impairment and its relation to timing, number, and age at injury.
Second Impact Syndrome
Some controversy surrounds the existence of so-called “second impact syndrome” [Cantu, 1998; McCrory and Berkovic, 1998]. In general, it was invoked in circumstances where a mild TBI, often in a child or adolescent, resulted in catastrophic brain swelling (Figure 74-12) and often death. On some occasions, an earlier head injury was reported, and it has been suggested that the initial injury somehow primed the brain for the dramatic and seemingly disproportionate response occurring after the second injury [Kelly and Rosenberg, 1997]. A careful review of the literature raised concerns regarding poor documentation of a “first impact” in many cases [McCrory and Berkovic, 1998] and the terminology “malignant brain swelling” was proposed. None the less, much of the effort to devise return-to-play guidelines is aimed at reducing the likelihood of this syndrome, as well as the chronic, apparently cumulative effects of repeated injuries.
Skull Fractures
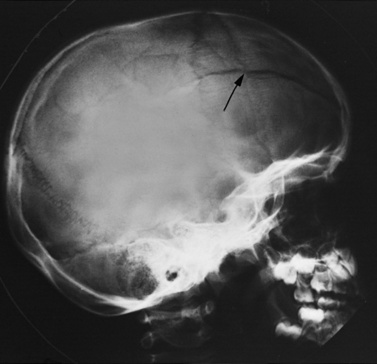
Linear fractures account for 66–75 percent of all pediatric skull fractures (Figure 74-13). These are most commonly temporoparietal in location, and can be associated with an overlying scalp injury. Although the fractures themselves rarely require intervention, CT scanning should always be performed to determine whether there is underlying pathology.
Depressed skull fractures involve displacement of the bony skull and increase the risk of underlying cerebral contusion or laceration. In neonates, the “ping-pong” fracture is a type of depressed fracture where the softer infant skull is simply indented rather than broken. Scalp injuries can be a sign of an underlying depressed fracture, with pain, swelling, or contusion at the site of injury. Indications for surgical repair of depressed fractures vary, but treatment is certainly indicated in patients with bony fragments displaced into brain parenchyma, dural tear, an underlying large hematoma, associated focal neurological deficits or seizures, and overt wound contamination. Depression >5 mm or in cosmetically unappealing locations has been considered a relative indication. Surgical repair has not been shown to alter neurological outcome or later risk of epilepsy [Braakman, 1972; van den Heever and van der Merwe, 1989], presumably due to the fact that any cerebral injury was likely sustained at the moment of impact.
Basal skull fractures tend to occur frontally or in the area of the petrous bone. While these fractures can be seen on radiographs, a number of clinical signs are highly indicative of their presence. Periorbital or retroauricular ecchymosis (raccoon eyes or Battle sign), rhinorrhea, otorrhea, and hemotympanum all point to a fracture of the skull base. Traumatic cranial neuropathies are associated with these fractures, typically involving CN I, II, VII, or VIII. The presence of CSF leakage (rhinorrhea or otorrhea) theoretically increases the risk of CNS infection, although studies have reported conflicting results [Langley et al., 1993], and the prophylactic use of antibiotics in these patients is not universally practiced [Einhorn and Mizrahi, 1978; Francel and Honeycutt, 2004]. The majority of CSF leaks resolve spontaneously; however, a longer duration of the leak increases the risk of meningitis. Persistent leaks require CSF diversion with a lumbar drain; if that fails, surgical repair is indicated.
Diagnostic Evaluation
Neuroimaging
Skull X-Rays
Skull x-rays are still the most sensitive method to detect fractures. The presence of a skull fracture increases the likelihood of an underlying intracranial abnormality 21- to 80-fold [Quayle et al., 1997; Teasdale et al., 1990]. However, the absence of a fracture on skull x-ray does not preclude intracranial pathology. The frequency of intracranial lesions discovered on CT in the absence of skull fracture on x-ray ranges from 16 to 39 percent [Levi et al., 1991; Lloyd et al., 1997], but these results are skewed toward more severe injuries, as CT scans were obtained only in patients with “clinical indications” or who were hospitalized. Of all pediatric TBI patients with CT scans positive for an intracranial lesion, 35–50 percent occurred in the absence of a skull fracture [Lloyd et al., 1997; Quayle et al., 1997; Teasdale et al., 1990]. In patients where underlying brain injury is already suspected, the value of screening skull radiography has been questioned [Bell and Loop, 1971; Lloyd et al., 1997].
Skull fracture does occur much more frequently in infants less than 1–2 years old and is associated with intracranial hematomas, although less so than in older children [Lloyd et al., 1997; Quayle et al., 1997; Schutzman et al., 2001]. None the less, identification of isolated skull fractures in infants is still important, with the implications of diagnosing nonaccidental trauma. Also, significant scalp injuries or hematomas may overlie a skull fracture. Given this information, skull x-rays may be warranted in evaluation of TBI under the following circumstances: in settings where there is a suspicion of nonaccidental trauma, in head-injured infants below 1–2 years of age, particularly if there is scalp swelling or hematoma, or as a screening tool in older children only in areas where CT scanning is not readily available.
Computed Tomography
Noncontrast CT scanning is the neurodiagnostic test of choice in the evaluation of acute head trauma. Many studies have been performed to determine clinical indications for CT following pediatric TBI [Dunning et al., 2004; Halley et al., 2004; Homer and Kleinman, 1999; Kuppermann et al., 2009; Maguire et al., 2009; Murgio et al., 2001; Quayle et al., 1997]. The proper use of CT scanning depends upon the severity of the injury sustained and the age of the child. The rationale behind use of CT is to identify intracranial complications, particularly those that would warrant neurosurgical or medical intervention to achieve optimal outcome.
In a large, prospective series (n = 653) of children with TBI of all severities admitted to a neurosurgical department, 34.6 percent demonstrated significant intracranial abnormalities [Levi et al., 1991]. Fractures were excluded, but nonsurgical pathology, such as DAI and contusions, was included, along with acute hematomas. If only acute hematomas were tabulated, 23.7 percent of severely injured (GCS 3–8) children had a lesion, as compared with 12.4 percent of moderate (GCS 9–12) and 5 percent of mild (GCS 13–15) patients. A consecutive, prospective study of children (n = 322) seen in an urban emergency room for “nontrivial” head injury reported an 8.3 percent rate of intracranial pathology. In this case, “nontrivial” was defined as head injury with symptoms or physical findings, except for superficial scalp injuries, which were only deemed “nontrivial” in infants less than 12 months of age [Quayle et al., 1997]. Differences in these studies may be attributed to geographic area, inclusion criteria (neurosurgical admissions versus emergency department visits), distribution of injury severity, and age range. In ten studies published since 1990, each with n >100 patients, the frequency of significant intracranial pathology in all pediatric TBI ranged from 1.3 percent [Chan et al., 1990; Teasdale et al., 1990] to 34.6 percent [Levi et al., 1991]. A recent meta-analysis of 16 published papers examining variables predictive of intracranial hemorrhage in pediatric TBI reported that skull fracture was associated with a relative risk (RR) of 6.1 (that is, the presence of a skull fracture carried a 6.1-fold increased risk of intracranial bleed), focal neurological deficits (RR, 9.4), GCS score less than 15 (RR, 5.5), and loss of consciousness (RR, 2.2) [Dunning et al., 2004]. Seizure activity had a relative risk of 2.8; however, due to variability of this parameter between studies, it did not reach statistical significance. Headache and vomiting were not predictive. Given the high frequency of intracranial lesions in children with moderate or severe TBI, cranial CT is warranted in patients with a GCS <13. While the presence of the risk factors listed above should further raise suspicion of an intracranial process, the absence of a factor does not necessarily mean the risk of a lesion is reduced. Imaging recommendations are summarized in Box 74-2 and Box 74-3.
Box 74-2 Pediatric Traumatic Brain Injury >2 Years Old: Indications for CT Scanning











Box 74-3 Pediatric Traumatic Brain Injury <2 Years Old: Indications for CT Scanning

















The indication for CT is more controversial when only mild TBI pediatric patients are considered. In this population, the true prevalence of intracranial injury is difficult to determine. Review of 12 studies published since 1986, each with n >100 patients, shows that from 0 percent [Immordino, 1986] to 19.1 percent [Wang et al., 2000] of mildly injured children will have a significant CT abnormality. Davis and colleagues reported intracranial bleeding in 7 percent of pediatric patients (n = 168) who suffered a loss of consciousness after mild TBI; however, in a subset of 49 patients with normal neurological examination, 0 percent were found to have CT abnormalities [Davis et al., 1994]. Other studies have shown lower utility of clinical variables and normal neurological examination in predicting the absence of intracranial problems in mildly injured children [Halley et al., 2004; Quayle et al., 1997]. A formal review of this question [Homer and Kleinman, 1999] concluded that it is clear that only a very small number of mildly injured children will have significant intracranial damage, but that clinical history and examination are relatively insensitive predictors of those who will. Intracranial lesions requiring surgical intervention do occur, albeit very rarely, even when the neurological examination is normal. Therefore, the American Academy of Pediatrics consensus guideline for mild head injury states that CT imaging (in conjunction with observation – see management, below) is a reasonable management option, but is not required in children older than 2 years of age who experience brief (<1 minute) loss of consciousness and who have no abnormal physical or neurological findings at the time of medical evaluation [American Academy of Pediatrics, 1999]. While there are currently no definitive guidelines for children with mildly abnormal GCS (13–14) or normal GCS (15) with loss of consciousness lasting over 1 minute, careful observation in all and liberal use of CT is recommended (see Box 74-2) [Dietrich et al., 1993; Halley et al., 2004; Savitsky and Votey, 2000]. A recently developed and validated clinical prediction rule shows promise in identifying the subset of children with mild TBI who do not require CT scanning [Kuppermann et al., 2009]; the specifics of this rule are discussed later (and see Figure 74-16 below).
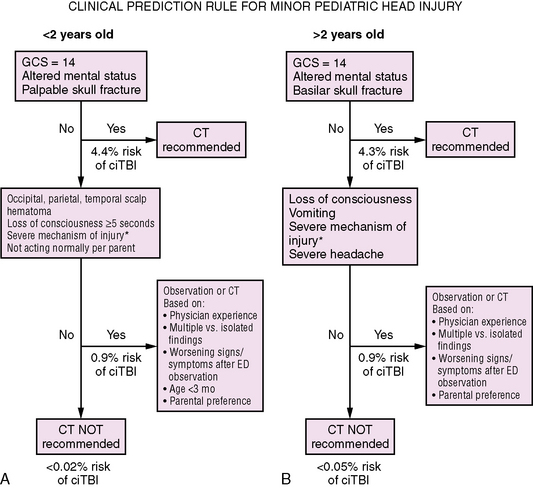
Fig. 74-16 Clinical algorithm for CT scanning in children with minor head injury (GCS 14-15).
(From Kuppermann N et al., Identification of children at very low risk of clinically-important brain injuries after head trauma: a prospective cohort study. Lancet 2009; 374: 1160–1170.)
Recommendations for CT scanning are even broader when children under the age of 2 years are concerned. In this age group, skull fractures are more common, and abnormalities on neurological examination may not be as easily detected as in older children [Berney et al., 1994; Dietrich et al., 1993]. An expert review of the literature has resulted in a proposed guideline for stratification of risk in head-injured infants younger than 2 years of age [Schutzman et al., 2001]. Indicators of high and intermediate risk stratification are presented in Box 74-3. In general, this algorithm recognizes that low-risk patients (low energy mechanism of injury, no signs or symptoms, more than 2 hours since injury, and age over 12 months) may be discharged from the emergency department without CT scanning, but with continued observation by reliable caretakers. Kupperman and colleagues have also devised and validated a CT rule for children less than 2 years of age that shows very good negative predictive value for clinically significant TBI [Kuppermann et al., 2009] (see Management of mild TBI and Figure 74-16, below).
Magnetic Resonance Imaging
MRI has limited usefulness in the acute management of pediatric TBI, primarily because of the longer time needed for scanning. MRI is also less able to visualize bony structures and fractures, and confers no advantage over CT in diagnosing large (surgical) hematomas. MRI is better able to delineate structures in the posterior fossa, as well as subacute hematomas that may appear isodense on CT. The ability of MRI to distinguish the approximate age of an intracranial hemorrhage makes it useful in cases of nonaccidental trauma (Table 74-2; see Subacute and chronic SDH). DWI has been used to demonstrate areas of restricted diffusion related to post-traumatic secondary hypoxic-ischemic injury [Suh et al., 2001]. Particular MRI sequences (T2* GRE, and more recently, SWI) are especially good at detecting microhemorrhages that indicate foci of DAI [Tong et al., 2004] and might not be seen on conventional CT. MR spectroscopy performed subacutely (within the first week) in patients with persistent coma has been highly accurate at predicting outcome [Ashwal et al., 2000; Holshouser et al., 2000], and is discussed later in this chapter (see Figure 74-17 below), under the section on prognosis.
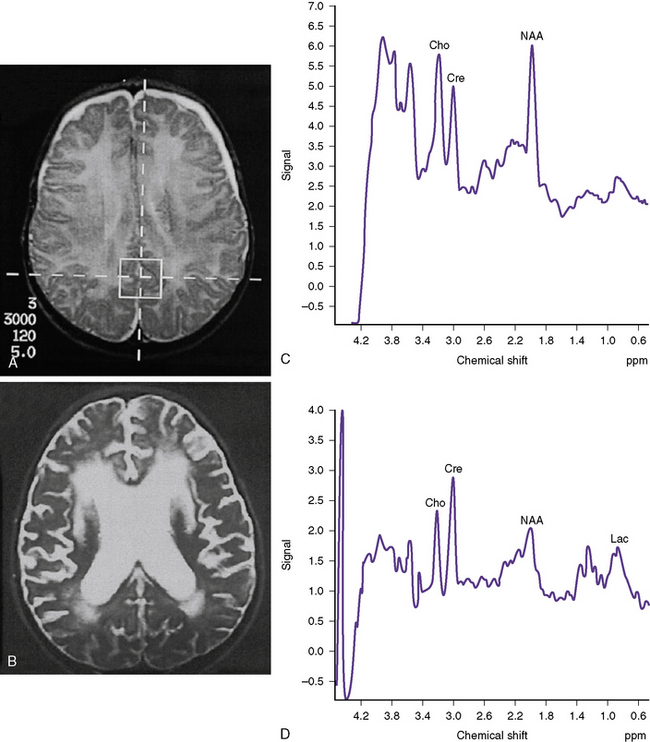
Fig. 74-17 Examples of children after traumatic brain injury with good or bad outcomes.
(From Ashwal S et al. Predictive value of proton magnetic resonance spectroscopy in pediatric closed head injury. Pediatr Neurol 2000;23:119–120.)
Advanced MRI techniques have shown promise in characterizing pediatric TBI and recovery from injury, but are currently used primarily for investigational purposes [Ashwal et al., 2006; Suskauer and Huisman, 2009; Difiori and Giza, 2010]. fMRI (altered patterns of network activation [Lovell et al., 2007; McAllister et al., 1999]), MR spectroscopy (abnormal cerebral metabolites [Babikian et al., 2006; Babikian et al., 2009a]), DWI (restricted diffusion [Babikian et al., 2009b]), and DTI (perturbations in diffusion along white-matter tracts [Ewing-Cobbs et al., 2008; Wozniak et al., 2007]) have all been reported to show changes after TBI, including pediatric TBI.
Angiography
Angiography is useful for diagnosing traumatic vascular lesions, particularly dissections of the great cervical arteries, traumatic intracranial aneurysms, and cavernous carotid fistulas. In some cases, MR angiography is being recommended as a screening test for suspected dissection in children, in preference to conventional angiography [Chamoun et al., 2008].
Ultrasound
An open fontanel in younger infants provides a window for ultrasound examination. While able to visualize more centrally located intracranial lesions, ultrasound is less sensitive at detecting epidural or subdural hematomas, and is not generally utilized in an acute trauma setting. Use of ultrasound is reviewed in more detail in Chapter 11.
Neurophysiological Testing
EEG may be help in the evaluation of the persistently comatose patient whose mental status is altered disproportionately to the documented injury. In some cases, subclinical seizures may underlie prolonged unresponsiveness after TBI [Vespa et al., 1999]. Continuous EEG (cEEG) monitoring is one method of measuring cerebral function that may play a particularly important role in patients with TBI. While there is little written specifically about cEEG for pediatric TBI patients [Liesemer et al., 2011], there is a growing literature evaluating the adult neurointensive care unit population.
Subclinical seizures are not infrequently seen in the intensive care unit setting. A retrospective review of 570 consecutive adult intensive care unit patients undergoing cEEG found seizures in 19 percent, 92 percent of which were subclinical [Claassen et al., 2004]. A very high percentage (36 percent) of the children included in the study had seizures. It was also demonstrated in an adult intensive care unit population that 24 hours of EEG monitoring was sufficient to capture 98 percent of seizures in patients without altered mental status. In patients with altered mental status, 48 hours was necessary to capture 96 percent of seizures [Hirsch, 2004].
In adults with severe TBI, 22 percent had early post-traumatic seizures, half of which were nonconvulsive and would not have been detected without the cEEG monitoring [Vespa et al., 1999]. To demonstrate the potential of seizures to contribute to secondary injury after TBI, another study by the same group evaluated physiologic changes in ten adults with post-TBI seizures [Vespa et al., 2007]. During subclinical post-traumatic seizures, episodic and long-lasting elevations in ICP and in the microdialysis lactate/pyruvate ratio were seen, indicative of on-going cerebral metabolic stress. In addition to seizures, other studies have demonstrated frequent abnormal interictal findings after TBI, including slowing, spike discharges, and fast activity [Ronne-Engstrom and Winkler, 2006].
Continuous EEG monitoring may be even more important in children [Liesemer et al., 2011]. It is often quite challenging to assess the mental status in young children and it is often very difficult to determine the cause of paroxysmal events in children. In children, early post-traumatic seizures are more common than late. In addition, studies have shown that younger age is a risk factor for early post-traumatic seizures [Chiaretti et al., 2000]. The incidence of subclinical seizures in this population is not known, as most of the studies to date have not included continuous EEG monitoring. However, subclinical seizures after pediatric TBI have recently been reported [Lerner et al., 2009]. Out of 10 patients in this on-going prospective study, clinical seizures occurred in 3 and subclinical seizures were demonstrated in 2 (aged 4 months and 5 months), one of whom was in subclinical status epilepticus.
General Management Principles for Severe Pediatric TBI
An expert review (which is currently being revised and updated) summarizes the existing published literature regarding medical management of severe pediatric TBI [Adelson et al., 2003b]. An evidence-based approach was utilized; however, the literature did not support any treatment standards. Only five items achieved the level of treatment guidelines:
Multiple treatment options and their rationale were provided and the following sections summarize these findings and synthesize them with traditional management strategies.
Stabilization and Prevention of Secondary Injury
While it is clear that hypoxemia results in poorer outcome following TBI [Ong et al., 1996; Pigula et al., 1993], studies aimed at prehospital endotracheal intubation for children have failed to show any significant benefit in outcome [Cooper et al., 2001; Gausche et al., 2000]. After initial assessment and resuscitation, acute management is aimed primarily at avoiding or preventing secondary injuries until more definitive therapy can be instituted. Airway maintenance and provision of supplemental oxygen is crucial, and upon arriving at the hospital or the intensive care unit, intubation and ventilation are often necessary for the more severely injured child.
Fluid resuscitation to avoid hypotension is also essential [Luerssen et al., 1988; Pigula et al., 1993]. It is rare for pediatric head injury alone to cause systemic hypoperfusion; an alternative cause must be sought, and may include internal or external bleeding or spinal cord injury. Signs of hypoperfusion and shock include diminished pulses, prolonged capillary refill, tachycardia, and reduced urine output. Hypotension is a late sign in pediatric shock. Fluid restriction out of concern about exacerbation of cerebral edema is to be avoided in the shocky, head-injured child; perfusion must be maintained.
First-Tier Intracranial Pressure Management
Sedation and Neuromuscular Blockade
Sedatives and analgesics may be useful to reduce struggling and exacerbation of elevated ICP, promote optimal ventilation, alleviate pain, and allow for adequate nursing care (suctioning, catheter placement, etc.). A theoretical benefit of sedatives is to minimize increases in cerebral metabolic activity provoked by pain or stress. In addition, some sedatives possess anticonvulsant or antiemetic properties. A potential adverse effect of sedation is hypotension, which can exacerbate poor cerebral perfusion if not addressed promptly. A recent expert literature review found insufficient evidence to recommend a specific choice of sedative medications in the setting of pediatric TBI [Adelson et al., 2003d]. Sedatives with published reports of use in pediatric TBI include fentanyl, morphine, sufentanil, remifentanil, diazepam, ketamine, propofol, etomidate, and barbiturates. However, the majority of these reports were small series, often with mixed adult and adolescent patients. There is anecdotal evidence of ICP reduction with remifentanil [Tipps et al., 2000], ketamine [Albanese et al., 1997], propofol [Farling et al., 1989; Spitzfaden et al., 1999], and barbiturates. However, the use of a continuous propofol infusion in children has been associated with a lethal syndrome of metabolic acidosis [Bray, 1998; Canivet et al., 1994; Cray et al., 1998; Parke et al., 1992], and has not been approved for pediatric sedation by the FDA.
Neuromuscular blockade may aid in the management of TBI patients by promoting ease of ventilation, reducing ICP, and stopping metabolic demands from skeletal muscle. Complications of neuromuscular blockade include an increased risk of pneumonia, cardiovascular depression, immobilization stress (if used with inadequate sedation), unnoticed seizure activity, and prolonged paralysis/myopathy. Myopathy appears to be associated with the combination of nondepolarizing agents and corticosteroids [Martin et al., 2001; Rudis et al., 1996]. When using iatrogenic paralysis, it is important to monitor the depth of blockade periodically using a “train of four” stimulation. Vecuronium, pancuronium, and doxacurium have all been used for pediatric neuromuscular blockade in the setting of TBI.
Hyperventilation
However, excessive hyperventilation has been shown, in both adults [Kiening et al., 1997; von Helden et al., 1993] and children [Skippen et al., 1997], to result in reductions in cerebral blood flow that may reach ischemic levels. The routine use of prophylactic hyperventilation (PCO2 <35 mmHg) in severe pediatric TBI is no longer recommended.
Controlled hyperventilation (PCO2 30–35 mmHg) may be useful to control elevated ICP that is refractory to sedation, neuromuscular blockade, CSF drainage, and hyperosmolar therapy. More aggressive hyperventilation (PCO2 <30 mmHg) should be considered a second-tier therapy for intractably increased ICP. The risk of potential ischemic complications must be considered, and the patient monitored appropriately for these problems. Jugular venous oxygen saturation measurements via a jugular bulb catheter should be utilized. This methodology provides a more global measure of cerebral tissue perfusion and can be monitored frequently as the patient’s condition and treatment change. Alternative methods of measuring for ischemia include CBF studies and brain-tissue oxygen monitoring. CBF studies have the advantage of providing some regional data, but are usually limited to the actual time of testing. Brain-tissue oxygen levels can be monitored continuously over time but has the disadvantage of being limited to measurements in the vicinity of the probe [Figaji et al., 2009].
Hyperosmolar Therapy
Two agents have been recommended for hyperosmolar therapy in pediatric TBI: 3% saline and mannitol. The theory behind hyperosmolar therapy is to raise the solute concentration of the serum, thus creating an osmotic gradient whereby free water is drawn out from the brain’s extracellular space. One drawback of the evidence-based medicine approach is made apparent when reviewing this therapy. Which agent is preferable, one with a long history of effectiveness but little in terms of blinded, placebo-controlled clinical trials, or a newer intervention with demonstrated efficacy in small but appropriately designed clinical trials? Mannitol, an agent whose clinical use for ICP management has become almost ubiquitous, appears clearly effective in many older adult studies [James, 1980b; Marshall et al., 1978; McGraw et al., 1978; Mendelow et al., 1985; Muizelaar et al., 1984; Schwartz et al., 1984], as well as those with mixed populations of children and adults [Nara et al., 1998; Rosner et al., 1995; Smith et al., 1986]. Unfortunately, the pediatric component of the mixed studies was not separately defined. Hypertonic saline has been more recently investigated [Peterson et al., 2000], and several studies used prospective data collection specifically in pediatric patients to document the effectiveness of this agent in controlling refractory ICP [Simma et al., 1998], yet this therapy lacks widespread clinical implementation.
Mannitol exerts its beneficial effects through several mechanisms. First, it reduces blood viscosity, improving blood flow and allowing for a reduction in cerebral blood volume [Levin et al., 1979; Muizelaar et al., 1983; Muizelaar et al., 1986]. Second is the osmotic effect described above, drawing free water out of the brain (across an intact blood–brain barrier) back into the bloodstream [James, 1980a]. A less well understood effect of mannitol is its ability to act as an antioxidant [Kontos and Hess, 1983]. Mannitol is administered with a bolus infusion, at 0.25–1.5 g/kg up to every 4 hours. Overall, mannitol begins to exert its ICP-reducing effects within 10–20 minutes, and these effects can last for several hours.
A caveat to remember when using mannitol is that, in the presence of a leaky or damaged blood–brain barrier, mannitol can potentially enter the injured area and cause a rebound osmotic effect [Kaieda et al., 1989; McManus and Soriano, 1998]. Partially for this reason, bolus rather than continuous administration of mannitol is recommended. Hypertonic saline exerts its effect through several mechanisms, including reducing blood viscosity, osmotically drawing free water out of brain parenchyma, restoring cellular membrane potential and volume [McManus and Soriano, 1998; Nakayama et al., 1985], reducing inflammation [Qureshi and Suarez, 2000], and improving cardiovascular function. The effectiveness of hypertonic saline in pediatric patients has been demonstrated in several recent studies [Khanna et al., 2000; Peterson et al., 2000; Simma et al., 1998]. Despite reducing ICP, none of these studies investigated whether the therapy resulted in better outcomes. Dosing of hypertonic (3%) saline was on a sliding scale titrated to ICP and serum sodium, but the infusion rates ranged from 0.1 to 1.0 mL/kg/hr.
ICP Monitoring – Indications and Treatment Threshold
Outcome from pediatric and adult TBI depends on avoiding secondary insults, such as ischemia and hypoperfusion [Chesnut et al., 1993a; Narayan et al., 1981]. Elevated ICP can lead to global hypoperfusion, as well as precipitating herniation, which can cause tissue ischemia via compression and, in some situations, by impinging upon cerebral arteries. Many of the specific therapies for severe TBI are directed at preventing excessive ICP; thus, it logically follows that monitoring ICP is an important component of TBI management [Becker et al., 1977; Marshall et al., 1979; Narayan et al., 1982]. Although there has been no randomized clinical trial specifically validating the benefits of ICP monitoring in children, many studies have demonstrated benefits of aggressive management of elevated ICP in head-injured children [Bruce et al., 1981; Cho et al., 1995; Peterson et al., 2000; Taylor et al., 2001].
A pediatric study comparing Camino catheters (a tip-mounted fiberoptic sensor) with ventricular catheters found that they similarly measured ICP, although there were slight differences between the measured ICP from the two devices, dependent upon the patient’s GCS score [Gambardella et al., 1993]. Several adult studies addressing these issues have been published and summarized in published adult TBI management guidelines [Brain Trauma Foundation et al., 2000]. A ventricular catheter with an external sensor is considered the simplest and most reliable method to measure ICP and has the added benefit of allowing therapeutic removal of CSF. Catheter-tip mounted devices (pressure transducers or fiberoptic) are more expensive, but similarly accurate when placed within the ventricular catheter. Catheter-tip devices used for intraparenchymal placement may experience measurement drift, and external transducers coupled to subarachnoid, epidural, or subdural devices were felt to be less reliable; these modalities may be useful when ventricular access is problematic. ICP monitoring is an invasive procedure that requires a burr hole at a minimum, although suture lines may be used in infants/neonates. It is thus important to have standard indications for use of monitors in those children who are at greatest risk of having elevated ICP following TBI. In one series, 19 of 22 (86 percent) children with a GCS score ≤8 (i.e., severely injured), who received ICP monitoring, had ICP values >20 mmHg [Shapiro and Marmarou, 1982]. In a large series of severely injured patients (n = 654) from the National Traumatic Coma Data Bank, 72 percent had ICP >20 mmHg during the course of their monitoring [Marmarou et al., 1991].
These studies suggest that clinical determination of severe injury (GCS ≤8) delineates a group of patients at high risk of developing elevated ICP. Examination of less severely injured patients can provide greater sensitivity in determining clinical deterioration. However, particularly in young children and infants, clinical examination and GCS are less reliable than in older children or adults [Humphreys et al., 1990; Shapiro and Marmarou, 1982]. Using CT findings to predict elevated ICP can also be problematic in children, who are less likely to present with mass lesions than adults. Despite the presence of open fontanels and flexible sutures, infants can still develop elevated ICP [Cho et al., 1995]. Given this information, ICP monitoring is recommended in children with a GCS ≤8. In addition, ICP monitoring may be reasonable in selected patients with moderate TBI, particularly those whose clinical examination may be clouded by concomitant sedation, paralysis, or anesthesia. ICP monitoring can also still be beneficial in infants, particularly those with severe injuries due to abusive head trauma.
Normative values for ICP are generally felt to be lower in children, but the consequences of exceeding particular ICP levels have only been incompletely examined. Several retrospective studies in children have shown worse outcome in children with ICP elevations above either 20 mmHg [Esparza et al., 1985; Pfenninger et al., 1983] or 30 mmHg [Cho et al., 1995]. Other studies have shown cerebral compliance and CBF to be inversely proportional to ICP [Shapiro and Marmarou, 1982; Sharples et al., 1995a]. In general, it is recommended to institute ICP-lowering therapy in pediatric patients whose ICP exceeds 20 mmHg [Adelson et al., 2003c].
Complication rates for fiberoptic monitoring in children (n = 98) have been reported, with 7 percent of catheters becoming colonized (all with Staphylococcus aureus; average duration of catheter placement >7 days) and 13 percent having mechanical failure (on average by 9.5 days) [Jensen et al., 1997]. The complication rate was significantly different for catheters placed in the emergency department or in the intensive care unit. No pediatric reports of significant brain injury, hemorrhage, or seizure due to an ICP monitor placement or monitoring were found, suggesting a very low complication rate.
Recently, some debate over the utility of ICP monitoring following TBI has arisen after publication of two retrospective observational studies [Cremer et al., 2005; Salim et al., 2008]. In the pediatric study, results were compared between two groups (ICP monitor versus no ICP monitor) managed in one center between 1996 and 2006 [Salim et al., 2008]. The ICP-monitored group had significantly more severe overall injuries (Injury Severity Score [ISS] 25 versus 18, p = 0.001) and more severe head injuries (head Abbreviated Injury Score [head AIS] >3, 81 percent versus 55 percent, p = 0.01), as well as more frequent use of central venous catheter placement and use of mechanical ventilation. This group also had longer intensive care unit and hospital stays, and more systemic medical complications. After statistically correcting for the difference in TBI severity, there was no difference in survival between the two groups. No subgroup analysis was done for different types of TBI or age at injury. The adult study was performed at two centers with different TBI management protocols, including ICP monitoring [Cremer et al., 2005]. This study also found no survival benefit but did not perform subgroup analysis related to the type of TBI. These studies have suggested that a controlled clinical trial of ICP monitoring be considered. However, due to the limitations mentioned above, as well as the substantial existing physiological data suggesting the importance of ICP monitoring, there has been no consensus to limit the use of ICP monitoring in adult or pediatric TBI.
Cerebral Perfusion Pressure
Cerebral perfusion pressure (CPP) is defined as the difference between the mean arterial pressure (MAP) and ICP. Reduced CPP can occur during hypotension, intracranial hypertension, or both. Retrospective pediatric studies have shown a strong correlation between CPP <40 mmHg and death [Downard et al., 2000; Elias-Jones et al., 1992]. In one study of 118 children with ICP monitors placed within 24 hours of TBI, outcome was compared to the mean CPP over the first 48 hours of monitoring. None of the 22 children with a mean CPP <40 mmHg survived [Downard et al., 2000]. When outcome was assessed in comparison with CPP decile (i.e., 40–50 mmHg, 50–60 mmHg, etc.), no further relationship was found. Mean CPP <40 mmHg was thus interpreted as a limit of survivability in severe pediatric TBI [Downard et al., 2000]. More recently, using receiver operating curve (ROC) analysis and a novel pressure–time index (PTI), critical CPP thresholds of 48, 54, and 58 mmHg have been proposed. These values demarcate absolute CPP levels for the 2–6-year, 7–10-year, and 11–16-year age groups respectively, below which secondary brain injury contributes significantly to worsened global outcome [Chambers et al., 2006].
While low CPP is associated with poor outcome in children and adults, the benefits of CPP-directed therapy are uncertain. In adults, CPP of >80 mmHg has been associated with better outcome in two large, retrospective studies [Changaris et al., 1987; McGraw, 1989]. In one study, improved neurological outcome at 6 months was observed in patients managed for CPP and cerebral oxygen extraction (using jugular venous oxygen saturation monitoring) [Cruz, 1998]. However, a large randomized adult trial (n = 189) directly comparing CPP-directed therapy with ICP-directed therapy failed to show an outcome benefit of the former, and also reported an increased risk of acute respiratory distress syndrome in the CPP group [Robertson et al., 1999].
Cerebrospinal Fluid Drainage
Physical drainage of CSF is the most direct means of reducing ICP, and has been done via intraventricular or lumbar catheters. In most studies, it is difficult to separate CSF drainage from other first- and second-tier means of ICP reduction. However, in one prospective adult TBI study, which compared patients with medical management only versus those with medical management with ventriculostomy (after initial removal of surgical lesions), it was found that the group without ICP monitoring had a four-fold higher mortality rate (53 percent versus 12 percent), and a one-third lower rate of independent living (20 percent versus 59 percent) [Ghajar et al., 1993]. In a pediatric study, risk of sudden increases in ICP was assessed using the pressure–volume index (PVI) technique [Shapiro and Marmarou, 1982]. This method relies upon the observation that brain compliance does not change linearly as fluid volume is added to the intracranial space. As the threshold for brain compliance is neared, addition of even small volumes of fluid results in dramatic increases in ICP. Reduced PVI (less than 80 percent of predicted normal) was shown to predict intracranial hypertension accurately. All patients were managed with CSF drainage, which was found to reduce ICP and increase PVI. Based on such findings, CSF drainage can be considered a reasonable treatment option to reduce elevated ICP in children.
Second-Tier Intracranial Pressure Management
When elevated ICP is refractory to first-tier interventions, more intensive therapies are often instituted. These “second-tier” strategies involve methods to reduce brain metabolism and activity (barbiturates, hypothermia) or physical opening of the intracranial space to relieve pressure (craniectomy). In addition, more aggressive hyperventilation, in the setting of perfusion monitoring to avoid ischemia, remains an option, as does lumbar CSF drainage, provided a ventriculostomy is in place and the basal cisterns are open. Second-tier therapies are generally less proven or have an increased risk of potential side effects. Despite their apparent efficacy in spinal cord injury, corticosteroids have been associated with increased mortality in a very large, international, multicenter, randomized, placebo-controlled trial of severe traumatic brain injury, and are not recommended [Roberts et al., 2004].
Barbiturates
Barbiturate coma has benefit in cases of intractable ICP elevation by several potential mechanisms. Barbiturates are well known to suppress cerebral metabolism [Piatt, and Schiff, 1984]. They can reduce cerebral blood flow and volume by coupling with reduced cerebral metabolism [Kassell et al., 1980]. There is also some evidence that barbiturates decrease markers of free radical-induced damage in neonates with perinatal asphyxia [Singh et al., 2004].
Efficacy for barbiturates in refractory intracranial hypertension comes primarily from a study that included patients from 15 to 50 years of age [Eisenberg et al., 1988]. In this study, ICP control was achieved in 32 percent of the barbiturate group, which was about twice the response rate in the conventional treatment group. The 1-month survival rate was 92 percent among barbiturate responders, compared to only 17 percent of nonresponders.
The use of barbiturates in children with TBI has been described in several case series. One study reported cardiovascular complications and a high rate of requiring pressor support (91 percent), but did not mention whether there was any effect on cerebral hemodynamics or ICP [Kasoff et al., 1988]. In a different series of children with refractory ICP, addition of pentobarbital therapy achieved ICP control (<20 mmHg) in 52 percent [Pittman et al., 1989]. Dosing of pentobarbital is best titrated to burst-suppression on the EEG rather than serum levels, so continuous EEG monitoring should be used. Intravenous loading doses of pentobarbital range from 5 to 15 mg/kg, followed by either slow continuous infusion 1 mg/kg/hr, or 5 mg/kg intermittent doses every 4–6 hours.
The major complications of barbiturate therapy are myocardial depression and hypotension. Infants are perhaps particularly prone to these adverse effects. There is experimental evidence in neonatal rodents that even brief blockade of synaptic activity can result in a significant increase in neuronal apoptosis [Bittigau et al., 2002; Ikonomidou et al., 1999; Olney et al., 2002; Pohl et al., 1999].
Temperature Control and Hypothermia
Temperature control can be used either to avoid hyperthermia or to institute hypothermia. There is substantial experimental and adult clinical literature that suggests hyperthermia can exacerbate brain injury by increasing metabolism, triggering seizures, elevating levels of excitotoxins, promoting inflammation, and so on [Busto et al., 1989; Dietrich et al., 1996; Ginsberg and Busto, 1998; Wass et al., 1995]. It is recommended that fever/hyperthermia be avoided in brain-injured children.
Since the original report of hypothermia to treat TBI in children [Hendrick, 1959], interest in reducing temperature therapeutically has periodically been considered. Hypothermia can substantially reduce cerebral activity, and has been anecdotally associated with neurological preservation after prolonged drowning [Young et al., 1980]. In adults, several single-center studies showed either physiological benefits (improved CPP, reduced ICP) or trends toward better neurological outcomes [Clifton et al., 1993; Marion et al., 1993; Marion et al., 1997; Shiozaki et al., 1993], as did a recent meta-analysis [Polderman, 2008]. However, a large multicenter hypothermia trial that excluded children under the age of 16 years failed to show efficacy in improving outcome [Clifton et al., 2001]. There was a trend toward better outcomes in patients who were hypothermic at presentation and in younger patients, but there was no overall benefit.
More recently, a prospective randomized multicenter trial of therapeutic hypothermia for severe pediatric TBI was completed [Hutchison et al., 2008]. This study found a trend toward higher mortality (21 percent versus 12 percent, p = 0.06) and morbidity (unfavorable outcome, 31 percent versus 22 percent, p = 0.14) in the hypothermia group compared to the normothermia group. The hypothermia group also showed more systemic hypotension and use of vasopressors during the rewarming period than the normothermic patients. While this study utilized a 24-hour period of hypothermia with rapid rewarming, other investigators have reported that the short-duration/rapid-rewarming hypothermia protocol is associated with rebound increases in ICP [Adelson et al., 2005]. Unfortunately, a second multi-center hypothermia trial for pediatric TBI (‘cool kids’ trial) that uses a longer period of hypothermia with a more gradual rewarming period [Adelson, 2009] was halted in 2011.
Surgical Management of ICP
Decompressive craniectomy has been used to alleviate intractable intracranial hypertension and malignant cerebral edema. This procedure has been reported to lower ICP on average by 9–47 mmHg in children and adolescents [Cho et al., 1995; Polin et al., 1997; Ruf et al., 2003; Taylor et al., 2001].
One study divided 23 pediatric victims of nonaccidental trauma into a surgical group (with ICP >30 mmHg) and two medical groups (ICP >30 mmHg and <30 mmHg). Children in the surgical group and the medical group with less severe ICP elevation had significantly better outcome than the medically managed patients with ICP >30 mmHg [Cho et al., 1995].
Taylor et al. prospectively studied 27 children and randomly assigned them to best medical management with ventricular drainage (n = 14) or bilateral decompressive craniectomy with best medical management (n = 13) [Taylor et al., 2001]. Mean ICP was reduced by 9 mmHg postoperatively. Over a similar 48-hour period, the decompressed group had fewer episodes of ICP >30 mmHg (9 versus 59), fewer episodes of ICP >20 mmHg (107 versus 223), and a narrower range of ICP (11–25 mmHg versus 11–44 mmHg) when compared to the medical-only group. The surgically managed patients also tended towards a better Glasgow Outcome Scale score, although follow-up times were not standardized and mild pre-existing cognitive impairments were identified in two of the medically managed patients. Other studies reporting outcome after decompressive craniectomy were case series without a control/medical-only group.
The most commonly used surgical approach for decompressive craniectomy is a combination of bilateral craniectomy and expansion duraplasty [Cho et al., 1995; Dam et al., 1996; Guerra et al., 1999; Polin et al., 1997]. The studies reported above had another feature in common: surgery was undertaken less than 48 hours post-injury. Expert review of the literature suggested that severely injured children with intractably elevated ICP who are more likely to benefit from decompressive craniectomy could be selected on the basis of meeting at least some of the criteria listed in Box 74-4. In clinical practice, unilateral craniectomy has been performed ipsilateral to the hemisphere with greater edema or a surgical lesion, although there are no definitive published studies comparing bilateral with unilateral decompression in pediatric TBI.
Early Post-Traumatic Seizures and Seizure Prophylaxis
Seizures are not infrequent following TBI. Acutely, seizures may result from stretch-induced depolarization of neurons, post-traumatic glutamate release [Vespa et al., 1998; Vespa et al., 2007], toxic effects of blood products [D’Alessandro et al., 1988; Willmore et al., 1978], focal necrotic damage due to contusion or skull fracture, compression of neurons secondary to mass effect or cerebral edema [Chiaretti et al., 2000], infection secondary to penetrating injury or skull fracture, and/or hypoxia-ischemia [Ichord et al., 2007].
Post-traumatic epilepsy (recurrent unprovoked seizures) may arise due to neuron death/gliosis/scarring (such as mesial temporal sclerosis), development of recurrent excitatory neuronal pathways (sprouting, kindling), and loss of inhibitory gamma-aminobutyric acid (GABA)ergic interneurons. In rare circumstances, neonatal head trauma has been shown to result in abnormal neuronal migration, with generation of dysplastic cortical regions [Lombroso, 2000; Marin-Padilla et al., 2002].
Acute Anticonvulsant Therapy
Early after TBI, an acute seizure may act as a secondary injury, increasing cerebral metabolic demand, depleting neuronal energy reserves, causing release of excitotoxins, inducing hypoxia by impairing respiration or in association with aspiration, or, particularly in generalized tonic-clonic seizures, increasing body temperature and raising ICP. The incidence of early post-traumatic seizures (PTS) is higher in children than in adults, and, following severe pediatric TBI, early seizure rates of 20–39 percent have been reported [Annegers et al., 1980; Lewis et al., 1993; Ratan et al., 1999]. This risk is increased with lower GCS and younger age [Hahn et al., 1988a; Hahn et al., 1988b; Hendrick and Harris, 1968; Lewis et al., 1993; Ratan et al., 1999]. Most early PTS in children occur within the first 24 hours.
In adults, prophylactic phenytoin and carbamazepine reduce the risk of early PTS [Glotzner et al., 1983; Temkin et al., 1990; Chang and Lowenstein, 2003]. In children, one study determined early seizure prophylaxis based on caregiver preference [Lewis et al., 1993]. Fifty-three percent of untreated children and only 15 percent of the treated children experienced early seizures. A larger pediatric study retrospectively described 128 severely injured children (out of a total of 477 of all severities) and compared treatment differences between centers. The use of prophylactic anticonvulsants for early seizures ranged from 10 to 35 percent, and the overall risk of an early PTS in the entire cohort was 9 percent. However, the risk of early seizure by treatment group or severity group was not reported. It was noted that the group treated for early seizures had an improved survival rate [Tilford et al., 2001].
More recently, levetiracetam, a newer antiepileptic medication, has been evaluated as a possible alternative to phenytoin. The availability of an intravenous form, good side-effect profile, and lack of hepatic metabolism all make it an attractive medication. Two small head-to-head trials have shown no difference in the incidence of seizures between levetiracetam and phenytoin. One of these studies showed an increase in interictal EEG abnormalities with levetiracetam [Jones et al., 2008; Szaflarski et al., 2009].
Prevention of Post-Traumatic Epilepsy
Despite the effectiveness of early prevention, several prospective, randomized adult studies have revealed the inability of anticonvulsant prophylaxis to prevent subsequent development of late post-traumatic epilepsy [Temkin et al., 1990; Young et al., 1983]. Given the potentially different mechanisms underlying the genesis of early and late PTS, this finding is not surprising.
In a trial of anticonvulsant prophylaxis for late PTS, 41 children were randomized in a double-blind, placebo-controlled study [Young et al., 1983]. Phenytoin or placebo was administered; those with phenytoin sensitivity were treated with phenobarbital. Patients were then followed for 18 months, and 12 percent of the treated group had a late PTS, as opposed to 6 percent of the untreated group (not significant). Importantly, compliance with treatment was poor. Therapeutic levels were present in only 50 percent of treated patients at 3 months’ follow-up, and by 1 year only 10 percent were therapeutic.
In addition to the above clinical data showing no efficacy for late PTS/pulmonary thromboembolism prophylaxis, there are also substantial animal data demonstrating that the immature brain is particularly sensitive to anti-excitatory pharmacological agents like anticonvulsants and anesthetics. Excitatory activity is necessary for survival of developing neurons and for on-going experience-dependent plasticity. Immature rodents show major increases in neuronal apoptosis and development of learning deficits when treated with many common anesthetics and anticonvulsants [Bittigau et al., 2002; Jevtovic-Todorovic et al., 2003; Olney et al., 2004]. (Topiramate and levetiracetam were notable exceptions [Kaindl et al., 2006].) These animal data are supported by human studies showing reductions in IQ for children treated with phenobarbital for febrile seizures [Farwell et al., 1990], and more recently showing an apparent dose-dependent increased risk for learning disabilities in children treated with anesthetics [Wilder et al., 2009].
Given the available data in adult and pediatric patients with late PTS and the potential concerns around use of agents that block neurotransmission in the developing brain, anticonvulsants cannot be recommended routinely for long-term prophylaxis after pediatric TBI. After the acute period, anticonvulsants can be gradually weaned. If a late PTS occurs, it should be evaluated and treated as a new-onset seizure [Adelson et al., 2003f].
Supportive Care
Management of severe TBI includes general supportive measures to prevent complications of intensive care unit hospitalization and potentially prolonged immobilization. Aspiration precautions should be maintained, including nasogastric tube placement acutely (orogastric placement in the case of frontobasal skull fractures). Particular care should be taken shortly after a recovering patient is extubated. Meticulous skin care is necessary to prevent breakdown, decubiti, and potential infection in sedated/paralyzed, critically ill children, as well as recovering but debilitated children following severe TBI. Consideration should be given to prophylaxis against deep venous thromboembolism, although anticoagulants should be used cautiously, if at all, in a setting of acute or chronic intracranial hemorrhage. Children with hypothalamic, brainstem or spinal cord injury are at risk for autonomic instability and should be carefully monitored and treated as needed. Pain is a common problem following traumatic injury, including head and bodily pain due to musculoskeletal injury. While analgesia should not be withheld, care must be taken not to sedate patients overly when neurological examination is necessary to observe for signs of deterioration (e.g., a more moderately injured patient without an ICP monitor). Sedation and analgesia may be lightened daily to allow for clinical assessment of mental status. Nutritional status must not be neglected. While enteral feedings are preferred, total parenteral nutrition should be provided for individuals in whom the gut is nonfunctioning. Because of the catabolic state existing after severe injury, nutritional calculations should attempt to provide 130–160 percent of resting metabolic needs [Adelson et al., 2003a]. Psychosocial support is also necessary for the patient’s family, and for the patients themselves as they begin to recover from injury. When nonaccidental trauma is suspected, a report should be filed to social services for full investigation.
Treatment Algorithm for Increased Intracranial Pressure in Severe Pediatric Traumatic Brain Injury
As previously noted, a comprehensive expert review and consensus parameter, titled “Guidelines for the Acute Medical Management of Severe Traumatic Brain Injury in Infants, Children, and Adolescents,” was published [Adelson et al., 2003b]. Many of the recommendations included in this chapter are drawn from these guidelines which are being revised. These guidelines provided critical pathways for treatment of elevated ICP in pediatric brain injury that is divided into first-tier (Figure 74-14) and second-tier therapies (Figure 74-15) [Adelson et al., 2003e].
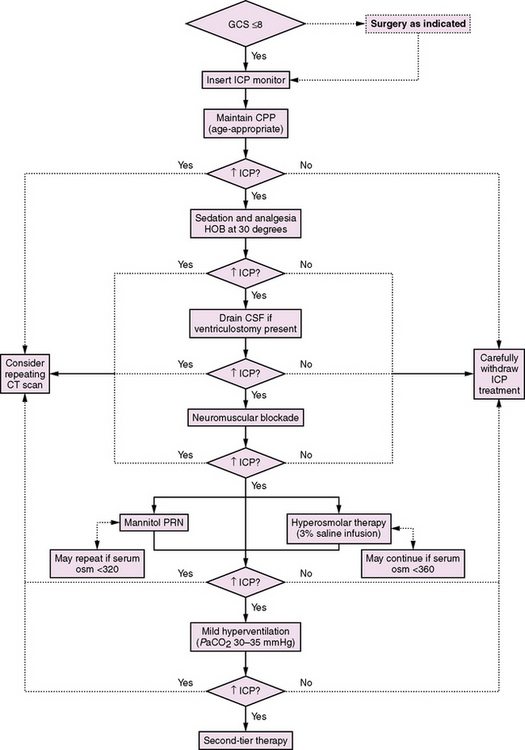
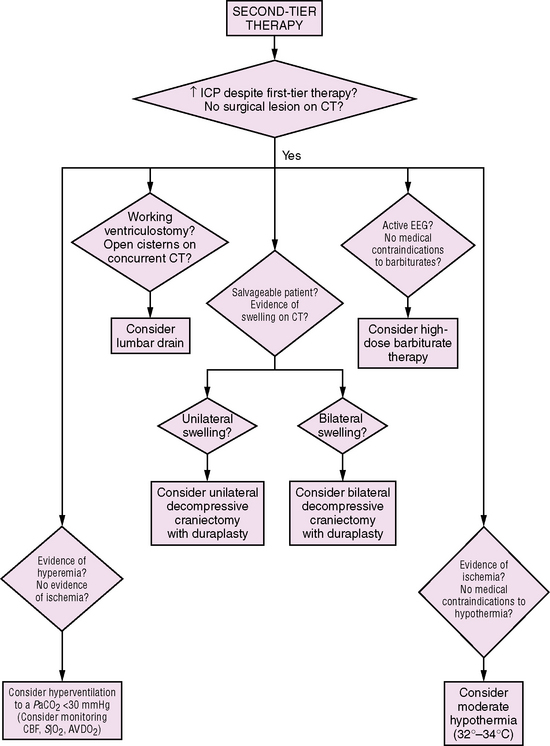
General Management of Mild Traumatic Brain Injury/Concussion
Two sets of management guidelines are provided. The first set represents the expert consensus guidelines developed by the American Academy of Pediatrics for the evaluation and management of the mildly head-injured child [American Academy of Pediatrics, 1999], with some added clinical prediction rules for CT scanning based upon the recent literature [Kuppermann et al., 2009; Maguire et al., 2009]. The second set of guidelines has been developed specifically for sports-related concussion by the International Conference on Concussion in Sport Group, with on-going revisions [Aubry et al., 2002; McCrory et al., 2005; McCrory et al., 2009]. It is important to realize that the majority of current sports concussion guidelines do not specifically take into account potential age-dependent differences in symptomatology or recovery; however, the most recent revision to the Concussion in Sport Group recommendations does articulate the need for special considerations for children and adolescents, albeit without specifying what they should be [McCrory et al., 2009].
Given the rapidly growing use of CT scanning and the radiosensitivity of the developing brain, legitimate concerns about the risk–benefit ratio of brain CT scanning for minor TBI in pediatric patients have been articulated [Brenner and Hall, 2007]. A systematic review of clinical prediction rules for CT scanning in the setting of pediatric head injury concluded that existing rules showed considerable variation and none was validated. The authors called for future development of high-quality, high-performance predictors, including prediction rules specifically for the patient population under the age of 2 years [Maguire et al., 2009]. Subsequently, a large multicenter study appears to have provided precisely the type of clinical rules required to fill this gap [Kuppermann et al., 2009]. The approach to mild TBI is more comprehensively reviewed in a forthcoming publication [Kirkwood and Yeates, 2012].
Mild Traumatic Brain Injury without Loss of Consciousness in Child Over 2 Years Old
Assuming the physical and neurological examinations do not reveal any worrisome findings, careful observation is the appropriate management [American Academy of Pediatrics, 1999; Homer and Kleinman, 1999]. Observation may take place in the clinic, in the emergency department, or at home under the supervision of a competent caregiver. Observation should be for a duration of at least 24 hours. Signs and symptoms to be watched for include deteriorating mental status, seizures, focal neurological signs, and worsening headache, nausea, or vomiting. Post-head injury instructions should be verbally explained to the caregiver, who should also be provided with written instructions and a means of returning to medical care should deterioration occur.
Neuroimaging is not generally recommended, as the likelihood of a significant intracranial abnormality in this setting is exceedingly small. A recently developed, high-quality, high-performance, validated clinical prediction rule for identifying pediatric minor head-injury patients (GCS 14–15) in whom CT scans are unnecessary showed a 96.7 percent sensitivity and a 99.9 percent negative predictive value for children over 2 years old (Figure 74-16). For this report, “clinically important TBI” (ciTBI) was defined as death from TBI, neurosurgical intervention, intubation for more than 24 hours for TBI, or hospital admission for more than 2 nights for TBI. Using this rule, CT is recommended if the patient has:
Patients meeting one of these three criteria represented 14 percent of the minor head injury population in this study (3585 of 25,608), with a 4.3 percent risk of ciTBI (154 of 3585). If none of these conditions was met, the second branch point was determining whether there was a history of loss of consciousness (of any duration), vomiting, severe mechanisms of injury, or severe headache. Patients with “no” answers to all of these questions represented 58.3 percent of the population (14,929 of 25,608), had a <0.05 percent risk of ciTBI (7 of 14,929), and CT scanning was not recommended. Patients with a “yes” answer to one of these questions represented an intermediate group (27.7 percent of the population, 0.9 percent risk of ciTBI), for whom observation or CT could be utilized based upon clinician experience. In the same report, this rule was validated in a separate population (n = 6411), showing a 96.8 percent sensitivity and 99.9 percent negative predictive value [Kuppermann et al., 2009].
Mild Traumatic Brain Injury with Brief Loss of Consciousness in Child Over 2 Years Old
This management algorithm does not require immediate neuroimaging of affected children. This is compatible with the new clinical predictor rule developed and validated by Kupperman et al., as patients with LOC <1 min would fall into the intermediate group, for whom either observation or CT imaging would be appropriate [Kuppermann et al., 2009] (see above and Figure 74-16). While clinical signs such as headache, nausea, and vomiting are probably more common in children with intracranial injury, they are not sensitive, and the majority of children with these symptoms will not have an intracranial lesion. However, there are children with documented normal neurological examination who eventually require neurosurgical intervention. Rather than acquiring an immediate CT scan in every patient, a reasonable strategy is to evaluate and observe the patient, with neuroimaging reserved for those individuals who clinically do not recover or who worsen. If neuroimaging is warranted, CT scanning is the examination of choice (see Box 74-2). While skull radiographs are adequate to visualize many types of skull fractures, fractures may be present without intracranial injury, and vice versa. In settings where CT scanning is not available, skull radiographs may provide useful additional information for triaging and assessing the risk of intracranial hemorrhage and the need for CT. When the CT scan and examination are normal, the likelihood of clinically significant late deterioration is very low so children in this category can be discharged home under parental observation.
Mild Traumatic Brain Injury in Children Under 2 Years Old
Special consideration needs to be given to the evaluation of mild TBI in children below 2 years of age, as clinical assessment can be difficult, the risk of skull fracture is particularly high [Schutzman et al., 2001], and the youngest children are the most radiosensitive to excessive CT scanning [Brenner and Hall, 2007]. In this age group, based upon the American Academy of Pediatrics recommendations, more weight may be placed on neuroimaging results, depending upon assessment of the degree of risk of intracranial injury (low, intermediate or high – see Box 74-3). The lowest-risk group under 2 years is made up of those children with low-energy mechanisms of injury (i.e., short falls) and no signs or symptoms, who have been observed for more than 2 hours. The older child is even more reassuring under these circumstances.
Patients identified as high risk have one or more of the following clinical characteristics:
These patients should undergo noncontrast CT scanning.
A recently published and validated clinical decision rule to identify children below 2 years of age who have a very low risk for ciTBI may substantially simplify the identification of those young children for whom CT scanning is not necessary [Kuppermann et al., 2009] (see Figure 74-16). Following this rule, CT scanning was recommended for children with mild TBI if they had one of the following: GCS <15, altered mental status, or palpable skull fracture. The second branch point in this rule seeks to identify individuals: with nonfrontal scalp hematomas, loss of consciousness for longer than 5 seconds, a severe mechanism of injury, or not acting normally per parent. For those who answer “no” to all of the second-tier questions (4549 of 8502; 53.5 percent of the study population), the risk of ciTBI was <0.02 percent (1 of 4549) and CT scanning was not recommended. Patients with a “yes” answer to any of the second-branch questions, however, would be managed by observation or neuroimaging, depending upon clinical judgment. This rule showed similarly high levels of performance when validated in a separate group of patients (n = 2216) with 100 percent sensitivity and a 100 percent negative predictive value.
Guidelines for Return to Play Following Sports Concussion
There is growing awareness that grading concussion “severity” based upon early symptomatology is difficult. Acute factors, such as loss of consciousness, amnesia, impact force, and headache, have all been investigated as measures of concussion severity, but existing data remain inconclusive [Asplund et al., 2004; Collins et al., 2003; Dischinger et al., 2009; Guskiewicz et al., 2007; Lovell et al., 1999; Meares et al., 2008]. More recently, sports concussion guidelines have moved away from grading concussion severity, and earlier recommendations have been updated [Aubry et al., 2002; McCrory et al., 2005; McCrory et al., 2009] or are currently under revision [American Academy of Neurology, 1997].
There is some general agreement on return-to-play recommendations across different guidelines. Most guidelines require the athlete to be symptom-free, at rest and with exertion, before returning to physical activity – persistent symptoms that are likely reflect on-going cerebral dysfunction and have been interpreted as indicating incomplete recovery. Second, following symptom resolution, a gradual, supervised return to physical activity is suggested (Table 74-3).
| Rehabilitation Stage | Functional Exercise | Objective |
|---|---|---|
| 1. No activity | Physical and cognitive rest | Recovery |
| 2. Light aerobic exercise | Moderate-intensity walking, swimming, stationary cycling; no resistance training | Increase heart rate |
| 3. Sport-specific exercise | Skating drills in hockey, running drills in soccer; no head impact activities | Add sport-specific movement |
| 4. Noncontact training drills | Progression to more complex training drills (passing drills in football and hockey, etc.); start resistance training | Exercise, coordination, and cognitive load |
| 5. Full-contact practice | Following medical clearance, participate in normal training activities | Restore confidence and assess functional skills by coaching staff |
| 6. Return to play | Normal competitive game play |
(From McCrory P et al. Consensus statement on Concussion in Sport, 3rd International Conference on Concussion in Sport held in Zurich, November 2008. Clin J Sport Med 2009;19:185.)
It is important to remember that longer symptom duration has been linked to younger patients [Field et al., 1986] and patients with multiple concussions [Guskiewicz et al., 2003]. The pediatric practitioner must also recognize that none of the existing guidelines specifically addresses pediatric or adolescent populations, although some of the more recent ones indicate that age-specific modifications need to be considered [McCrory et al., 2004; McCrory et al., 2009].
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 74 Traumatic Brain Injury in Children
Introduction and Background
Epidemiology of Pediatric Traumatic Brain Injury
Traumatic brain injury (TBI) is a major cause of death and disability in the pediatric population and has been identified as a significant public health problem in the United States and worldwide [Engberg and Teasdale, 1998; Murgio et al., 1999; Thurman et al., 1999; Tsai et al., 2004; Weiner and Weinberg, 2000]. Age-related incidence rates for TBI in children have been estimated to be as high as 670 per 100,000 when head injuries of all severities are included [McCarthy et al., 2002]. When limited to TBI resulting in hospitalization, the reported incidence has declined over the past 15 years, most likely due to injury prevention measures and improved motor vehicle safety. None the less, TBI is responsible for the majority of trauma-related death and hospitalization. The incidence rate of TBI-related hospitalization remains significant, consistently around 75–80/100,000 [McCarthy et al., 2002; Reid et al., 2001]. Childhood TBI results in an estimated 3000 deaths, 29,000 hospitalizations, and 400,000 emergency department visits annually in the U.S. (children 0–14 years old) [Thurman et al., 1999]. TBI is six times more likely to cause death in childhood than AIDS/HIV infection, and 20 times more likely than asthma [Centers for Disease Control and Prevention, 2000]. Moderate and severe pediatric TBI has been associated with long-standing cognitive, neurological, and behavioral impairments [Fay et al., 1994; Massagli et al., 1996a], and the cost of these disabilities is often carried over the person’s lifetime. Fortunately, the majority of all TBI is considered mild in severity; however, recent studies suggest that even mild pediatric TBI may have adverse long-term functional consequences [Hawley et al., 2004]. There is also increasing evidence that repeated mild TBI, as commonly occurs in a sports-related setting, results in chronic cognitive impairment [Collins et al., 1999; Matser et al., 1999], and may even predispose to early memory disturbances and dementia in some individuals [Guskiewicz et al., 2005; McKee et al., 2009; Omalu et al., 2006]. Despite these facts, no specific treatment standards exist for pediatric TBI, either acutely or during recovery. A recent review of guidelines for pediatric TBI management concluded that acute supportive therapies are often administered inconsistently or simply extrapolated from adult TBI protocols, not taking into account the unique physiology of the immature brain [Adelson et al., 2003b].
The peak incidence of pediatric TBI occurs in the adolescent and young adult, with a secondary peak in infancy [Kraus and McArthur, 2000]. The etiology of TBI varies with age (Figure 74-1). Adolescents sustain most head injuries in motor vehicle accidents, as well as sports-related concussions and assaults. Pre-adolescent children are also frequent victims of motor vehicle accidents, but more often as a pedestrian or while riding a bicycle. Those under the age of 5 years are more prone to falls [Thurman, 2001], while infants are particularly vulnerable to repeated severe TBI in the form of abusive head trauma (AHT, previously inflicted TBI or nonaccidental trauma).
Fig. 74-1 Etiology of pediatric traumatic brain injury by age group.
(From Thurman D, for the Centers for Disease Control and Prevention. Traumatic brain injury in the U.S.: Assessing outcomes in children, Appendix B. Available at: http://www.cdc.gov.)
Traumatic brain injury is about twice as common in boys than girls overall, with this gender distinction becoming increasingly evident in the childhood and adolescent years [Kraus and McArthur, 2000; Rivara, 1994]. In the U.S. annually, it is estimated that about 30,000 pediatric patients incur permanent disability as a result of TBI. These sequelae include headaches, post-traumatic epilepsy, motor disturbances, learning disabilities, cognitive impairment, and behavioral problems. Furthermore, the vast majority of children suffering TBI, even those surviving moderate or severe injury, fail to receive adequate medical, rehabilitative, or psychosocial follow-up upon hospital discharge [Armstrong and Kerns, 2002; Hawley et al., 2004]. Finally, given the magnitude of recurrent head trauma as a problem of youth, it is increasingly noted that health-care providers should take every opportunity to provide important information and education to patients and their families, particularly with regard to potential sequelae of repeated injuries, as well as effective injury prevention.
Anatomy
To understand the mechanisms and types of TBI best, knowledge of the basic anatomy of the brain and its coverings is essential (Figure 74-2). The scalp is the outermost covering and is highly vascular, tending to bleed profusely when lacerated. Under the scalp is a tendinous sheath, extending from the frontal to occipital regions, called the galea aponeurotic. The potential space beneath this is the subgaleal compartment, an occasional site of significant bleeding following head injury. The skull is next, with the periosteum covering its outer surface. The skull itself is composed of three layers, the bony outer and inner tables, separated by the diploic space, which is more vascular. Between the inner table of the skull and the dura mater is the epidural space, another potential space that is a significant site for arterial bleeding, particularly after skull fracture. The dura is the tough outer protective layer covering the brain itself. Below the dura is the subdural space, which is crossed by small veins that drain into the venous sinuses, which provides another site for post-traumatic hematomas. The next layer of brain coverings is the arachnoid, and under this, the subarachnoid space, which contains cerebrospinal fluid (CSF), and into which post-traumatic bleeding is also fairly common. The subarachnoid space is contiguous with the basal cisterns and the ventricular system, and CSF normally flows from its site of production at the choroid plexus within the ventricles, out into the basal cisterns, over the convexities of the cerebrum and into the venous drainage of the brain via the arachnoid villi. Subarachnoid blood can impair the flow of CSF after trauma and occasionally result in hydrocephalus. The last layer is the pia mater, which lies directly over the brain surface itself.
Biomechanics
Because the brain parenchyma is itself soft and deformable and brain regions are connected by fiber tracts, the brain is also prone to injury from rotational forces. These are the forces imparted when one part of the brain is twisted in relation to another part. In these circumstances, the underlying white-matter tracts are subjected to significant shearing forces that can result in stretch injury and microhemorrhages seen clinically [Tong et al., 2004] and in experimental models [Huh et al., 2008; Raghupathi and Margulies, 2002]. This type of damage following trauma is referred to as diffuse axonal injury (DAI), and can result in significant clinical disability. By its nature, DAI can occur throughout the brain, with a propensity for regions with major fiber tracts (corona radiata, corpus callosum, brainstem, etc.). While the developing brain has been shown to have remarkable resilience to focal injuries such as strokes or surgical resection, its ability to recover from diffuse injuries may be much more limited.
Penetrating trauma, while less common, often results in tremendous biomechanical forces, as well as physical disruption of tissue along the path of the foreign object. While the tissue destruction, with surrounding necrosis, hemorrhage, and edema, is a major factor in determining long-term disability, the transmitted forces from the passage of the projectile to more distant parts of the brain can result in immediate death, presumably by affecting respiratory and autonomic centers [Carey, 1995]. One large series reported that younger age was associated with worse outcome from gunshot wounds [Levy et al., 1993].
Injury Types
While specific injury types will be discussed in greater detail later in the chapter, an overview of the types of injuries commonly seen in the pediatric population ranges from superficial injuries to the scalp to severe diffuse cerebral edema with herniation. Scalp injuries are very common, and include contusions, lacerations, and hematomas. While many scalp injuries may occur with little, if any, evidence of underlying brain injury, careful history and examination should be undertaken in each case to rule out the possibility of concussion or, more rarely, intracranial injury. Concussion has been redefined as any trauma-induced transient disturbance of neurological function, even in the absence of unconsciousness [American Academy of Neurology, 1997]. A large scalp hematoma may conceal a skull fracture, and, in infants, can result in significant blood loss. Skull fractures are reported following 20 percent of childhood TBI [Harwood-Nash et al., 1971], and while simple linear fractures are generally benign, the presence of any fracture greatly increases the risk of intracranial hemorrhage [Lloyd et al., 1997]. Post-traumatic intracranial bleeding may occur in the form of cerebral contusion, cerebral laceration, and epidural, subdural, or subarachnoid hemorrhage, individually or often in combination. DAI represents stretch and shearing of white-matter tracts and can have profound clinical consequences, with prolonged unconsciousness and poor outcome. Traumatic injury, at its most severe, can lead to cerebral swelling and elevated ICP. While this problem may be accommodated to some degree by flexible sutures and open fontanels in infants, all ages are vulnerable to cerebral herniation, a displacement of cerebral contents within or outside of the intracranial space. Based on location of edema or focal lesions, distinct types of herniation can occur (Figure 74-3). Each of these injury types will be discussed in greater detail later in this chapter.
Fig. 74-3 Different forms of brain herniation.
1, Cingulate. 2, Uncal. 3, Cerebellar tonsillar. 4, Upward cerebellar. 5, Transcalvarial.
(From Fishman RA. Cerebrospinal fluid in diseases of the nervous system. Philadelphia: WB Saunders, 1980.)
While all of the above types of injury are associated with clinical syndromes of primary TBI, it is important to be aware that, inextricably intertwined with the primary injury, are the effects of so-called secondary injuries. Secondary injuries may be additional pathological insults independent of the original TBI, such as hypotension due to systemic hemorrhage, hyperthermia due to concomitant infection, or airway occlusion by a foreign body resulting in hypoxia or systemic hyper-/hypoglycemia. In many individuals, a secondary injury may be directly or indirectly related to the original brain injury, but occurs after the initial TBI. Thus, the spectrum of secondary injury encompasses entities such as cerebral edema with reduced brain perfusion, intracranial hemorrhage, early post-traumatic seizures, respiratory compromise of neurologic origin resulting in hypoxia, and post-traumatic hydrocephalus. More general etiologies of secondary injury include on-going excitotoxicity, free radical generation, oxidative stress, and inflammation. Clinical secondary injuries, such as hypotension, hypoxia, hydrocephalus, edema, hypoglycemia, and seizures, are of great medical significance, as they are generally treatable in the intensive care unit setting, and are often independent predictors of poor outcome [Chesnut et al., 1993a; Chiaretti et al., 2002].
Pathophysiology of Traumatic Brain Injury
Distinctions of Injury to the Developing Brain
Biomechanical Factors
Clearly, the physics of TBI in a child is different than in an adult. The relatively large head size, reduced muscular strength in the neck, and increased flexibility of the neck may promote a greater range of biomechanical force imparted to the head. On the other hand, there is less CSF space around the brain, and the prominent bony ridges of the anterior and middle cranial fossae are less developed. These factors may contribute to a lower occurrence of focal lesions in pediatric TBI [Berney et al., 1994; Bruce et al., 1979; Levi et al., 1991; Luerssen et al., 1988].
The physical properties of the developing skull also confer advantages and disadvantages. It is thinner, and thus more prone to diffuse deformation [Margulies and Thibault, 2000]. Some authors have reported a higher risk of skull fracture in children [Berney et al., 1994], although this finding is not universal [Levin et al., 1992]. However, the open fontanels and flexible sutures may serve to dampen traumatic forces, as well as to accommodate a greater degree of intracranial swelling. The brain itself, particularly in infants and young children, has a higher water content and is incompletely myelinated [Holland et al., 1986; Paus et al., 2001]. The higher water content tends to make the brain less compressible and less compliant than in the adult. Thus, the physical properties of the immature brain may provide both unique benefits and potential vulnerabilities to TBI, a theme that will be repeated when considering other developmental distinctions.
Changes in Cerebral Metabolism
It is well known that cerebral metabolism changes during maturation. The primary source of energy for the brain changes from lactate in the perinatal period, to ketone bodies while nursing, to glucose after weaning, and on through to adulthood [Nehlig, 2004; Vannucci and Vannucci, 2000]. The ability to use alternative fuels may have important consequences for acute energy metabolism in the injured brain at different ages [Prins et al., 2004].
Even after the brain has switched to predominantly glucose metabolism, the levels of this metabolic activity continue to change throughout the developing years. Basal glucose metabolism rates peak at around 6 years of age, correlating with the time of maximal synaptogenesis. As synaptic pruning and dendritic rearrangement occur throughout childhood and adolescence, cerebral glucose metabolism declines, although it still remains elevated compared to the adult brain [Chugani and Phelps, 1986]. Also, the brain does not mature uniformly, and cerebral metabolic rates for glucose and other substances have different time courses regionally.
Distinct Neurovascular Regulation
Neurovascular control may also differ in the young brain. Early studies suggested a propensity for diffuse cerebral swelling in children following closed-head injury, occurring 2–5 times more often than in adults [Aldrich et al., 1992; Lang et al., 1994]. Recent studies report that normal children have higher baseline cerebral blood flow (CBF) than adults, with significant variation across narrow age groups [Suzuki, 1990; Zwienenberg and Muizelaar, 1999]. At birth, CBF is lower than in adults, but increases rapidly to peak by age 5 years. CBF then appears to decline through adolescence to adult levels [Suzuki, 1990]. The effect of these developmental differences on injury-induced changes in CBF and the mechanism(s) of these differences are not yet well understood. Certainly, molecular mediators of neurovascular tone, such as nitric oxide synthase, have differential expression patterns in the immature brain [Keilhoff et al., 1996; Ohyu and Takashima, 1998], and these may play a role in age-dependent changes in blood flow and response to injury.
Increased Excitatory Neurotransmission
As mentioned earlier, the number of synapses in the human brain appears to peak in early childhood, and is associated with a peak in cerebral glucose metabolism. Levels of excitatory transmitter receptors are higher in the immature brain [Fosse et al., 1989; Insel et al., 1990; Miller et al., 1990a]. The occurrence of early post-traumatic seizures is also noticeably higher in children, and particularly in infants and younger children [Annegers et al., 1980; Berney et al., 1994].
Because excessive release of excitatory neurotransmitters can result in neuronal injury and death (excitotoxicity), the fact that the developing brain is more active might suggest that blocking excitatory transmission would be neuroprotective. However, early in development, neuronal activation also plays a critical role in both survival of immature neurons and in proper wiring of cerebral circuitry. In fact, in animal models, use of glutamate antagonists early in postnatal development (while protective against some degrees of excitotoxic insult) resulted in a large degree of programmed cell death (apoptosis) [Bittigau et al., 1999; Ikonomidou et al., 1999; Pohl et al., 1999]. Such evidence suggests that optimal recovery from brain injury may require a proper balance of excitation and inhibition, and may also account for the relative ineffectiveness of glutamate antagonists following human brain injury [Biegon et al., 2004; Hardingham et al., 2002; Ikonomidou and Turski, 2002].
On-Going Cerebral Maturation
In the past, the developing brain was generally felt to recover better after many types of brain injury, including stroke, TBI, and surgical resection. Developmental plasticity does confer some advantage when the brain must recover from a focal lesion [Kennard, 1942; Kolb et al., 2000; Villablanca and Hovda, 1999], if the lesion occurs at a specific age (see chapter 13). However, more recent work clearly indicates that the very young brain may be particularly vulnerable to diffuse injuries like TBI [Anderson et al., 2005; Babikian and Asarnow, 2009]. One mechanism for this vulnerability can be explained by post-traumatic impairment in experience-dependent neuroplasticity, which is the complex task of responding to environmental stimuli and rearranging neuronal network that results in enhanced function. In a mature brain, the consequences of such impairment might be overcome with time; in the young brain, it is likely that reduced responsiveness at a critical window of development will result in long-term dysfunction [Giza et al., 2009]. As a result, studies looking at post-TBI brain development, both in experimental animals and in children, must necessarily take into account the normal trajectory of brain maturation.
The Post-Traumatic Neurometabolic Cascade
Traumatic brain injury results in an immediate release of glutamate, widespread ionic changes, fluctuating cerebral glucose metabolism (initially elevated, then reduced), and dynamic changes in blood flow. Later, axonal damage and disconnection, reduced responsiveness to physiological stimuli, impaired neurotransmission, and apoptosis occur [Giza and Hovda, 2001]. For an overview of post-traumatic pathophysiology at the cellular level, see Figure 74-4. Specific components of the brain’s response to traumatic injury will be discussed below.
Fig. 74-4 Neurometabolic cascade after traumatic injury.
(From Giza CC, Hovda DA. The neurometabolic cascade of concussion. J Athl Train 2001;36:230).
Glutamate Release and Ionic Flux
Following a traumatic brain injury, there is an immediate and indiscriminate release of the excitatory neurotransmitter, glutamate [Bullock et al., 1998; Katayama et al., 1990]. This can occur due to widespread triggering of action potentials, synaptic neurotransmitter release, and membrane disruption. This flood of glutamate results in a massive efflux of potassium into the extracellular space [Katayama et al., 1990; Nilsson et al., 1993], and an influx of sodium and calcium [Nilsson et al., 1996; Osteen et al., 2001]. While hyperacute measures of extracellular glutamate are obviously unobtainable in human patients, microdialysis or CSF samples from severely injured patients (including children) in the days after injury have demonstrated glutamate elevations and have been associated with secondary injury and poorer outcomes [Bullock et al., 1998; Ruppel et al., 2001; Vespa et al., 1998].
Dynamic Changes in Cerebral Metabolism
As the injured cells attempt to restore ionic equilibrium post-trauma, membrane pumps such as the Na+-K+ ATPase are activated. An increase in cerebral glucose uptake, termed hyperglycolysis, is seen early after experimental TBI and has been postulated as a mechanism of providing additional substrate to generate the energy necessary to drive these membrane ionic pumps [Kawamata et al., 1992; Yoshino et al., 1991]. Acute studies of cerebral glucose metabolism using positron emission tomography (PET) in adult patients have also shown evidence for increased cerebral glucose uptake [Bergsneider et al., 1997]. Interestingly, there is increasing laboratory evidence that injured neurons are capable of utilizing alternative fuels to glucose [Magistretti et al., 1999; Pellerin and Magistretti, 2004; Prins et al., 2004], and it is well known that the capacity to use alternative fuels in uninjured neurons is age-specific [Vannucci and Vannucci, 2000].
After the acute period of hyperglycolysis, there is a prolonged period of diminished glucose uptake by the brain. This hypometabolism is seen in both adult and immature rats, although it lasts longer in adults (7–10 days) than in rat pups (3 days) [Yoshino et al., 1991; Thomas et al., 2000]. In traumatically injured adult patients, the period of reduced glucose metabolism has been shown to last weeks or months [Bergsneider et al., 2001].
Cerebral Blood Flow: Hyperemia? Hypoperfusion?
Blood flow also undergoes dynamic changes after injury and the pattern these changes take may depend on the type of injury and its severity. A pattern of diffuse cerebral swelling has been reported more often in children than adults [Aldrich et al., 1992; Lang et al., 1994]. This phenomenon was originally attributed to hyperemia [Bruce et al., 1981], but subsequent studies suggest that the incidence of post-traumatic hyperemia has been overestimated [Muizelaar et al., 1989]. In the earlier studies of post-TBI CBF in children, flow values were compared with normal values from a young adult control group. When CBF has been measured in normal children, however, it appears that blood flow values change dramatically across development, and are generally much higher in young children than in adults [Chiron et al., 1992; Suzuki, 1990; Zwienenberg and Muizelaar, 1999]. Subsequent studies of CBF in children suffering TBI, when compared to age-appropriate control values, have failed to show clear hyperemia [Adelson et al., 1997a; Sharples et al., 1995b]. In an experimental model of TBI, there is evidence for dramatically increased cerebral blood flow adjacent to a traumatic contusion, and this focal hyperemia was most pronounced in immature animals [Biagas et al., 1996]. While clinically it is important to recognize that diffuse cerebral swelling is more common in children, the mechanism of this swelling does not appear to be solely attributable to hyperemia.
The relation between CBF and outcome appears to depend upon the state of cerebral autoregulation. In the presence of intact autoregulation, increased CBF has been associated with better perfusion and improved outcome in children and young adults [Kelly et al., 1996; Vavilala et al., 2004]. However, after very severe injuries, in which autoregulation is dysfunctional and cerebral vasculature is pressure-passive, increased CBF can result in intractable elevations of ICP and worse outcome.
Beyond the acute phase of head injury, CBF generally declines. Post-TBI CBF can decline below normal, and it is important to monitor for ischemia, as this is associated with worse outcome [Bouma et al., 1991; Vespa et al., 2003]. Investigations are still under way to determine whether neurovascular coupling is intact during this period of reduced flow.
Altered Neurotransmission
Reductions in excitatory neurotransmitter systems have been reported after TBI in experimental animals, including impairments in glutamatergic [Giza et al., 2006; Miller et al., 1990b; Osteen et al., 2004; Sihver et al., 2001], noradrenergic [Fujinaka et al., 2003; Krobert et al., 1994], and cholinergic transmission [Gorman et al., 1996]. These changes may correlate with the injury-induced reduction in cerebral metabolism described above. Furthermore, these changes may be an underlying mechanism for post-TBI deficits in neuronal activation [Dietrich et al., 1994; Ip et al., 2003; Sanders et al., 2000], diminished activity-dependent molecular responsiveness [Giza et al., 2002; Griesbach et al., 2004], and impaired experience-dependent plasticity in developing animals [Fineman et al., 2000; Giza et al., 2005; Ip et al., 2002]. Activation studies using functional magnetic resonance imaging (fMRI), even after relatively mild TBI, have shown reduced or aberrant activation patterns in traumatically injured teenagers and young adults [Jantzen et al., 2004; McAllister et al., 1999].
Axonal Disconnection
Mechanical stretch can disrupt axonal membranes, resulting in calcium influx and microtubule and neurofilament disruption. Damage to these important cytoskeletal components impairs normal axonal transport, endangering distal axonal segments and synapses. Over time, disrupted axonal transport leads to an accumulation of transported proteins and organelles at the injury site, causing axonal blebs and, eventually, disconnection [Pettus and Povlishock, 1996; Povlishock and Christman, 1995; Povlishock and Pettus, 1996]. This type of damage is seen following experimental TBI in both immature [Raghupathi and Margulies, 2002] and adult animals [Povlishock and Pettus, 1996]. In postmortem human specimens, evidence of axonal blebbing and damage has been reported long after injury [Maxwell et al., 1997], and MRI can clearly show abnormalities of diffuse axonal injury, in pediatric and adult TBI patients [Tong et al., 2004]. Newer imaging modalities, such as diffusion tensor imaging (DTI) and MR spectroscopy, have shown white-matter abnormalities that correlate with functional impairments after pediatric TBI, even in brain regions without overt structural lesions [Ashwal et al., 2006; Babikian et al., 2009a; Ewing-Cobbs et al., 2008; Wilde et al., 2008].
Cell Death: Necrosis and Apoptosis
Apoptosis is characterized by nuclear condensation, DNA fragmentation, and preservation of the cell membrane. Apoptosis requires energy and protein synthesis, and triggers an acute inflammatory response that may be distinct from that seen with necrosis (see chapter 14). Usually, apoptotic cell death takes longer to evolve after injury, and studies have demonstrated that apoptosis can continue to occur long after experimental trauma [Conti et al., 1998; Wilson et al., 2004]. While apoptosis is seen after TBI, it is also part of the brain’s normal maturational program. Thus apoptotic pathways are more active in the immature brain, and apoptotic cell death appears more prominently after experimental injury to the developing brain [Bittigau et al., 1999; Pohl et al., 1999].
Impaired Plasticity
An important aspect of injury in the young brain is the effect of this injury on normal neuronal responsiveness and on developmental plasticity (see chapter 13). As mentioned earlier, increased plasticity in the immature brain results in significantly better recovery than in the adult following a focal lesion, and this has been well described in rats [Kolb and Tomie, 1988], cats [Burgess and Villablanca, 1986; Villablanca and Hovda, 1999], primates [Kennard, 1938], and humans [Hogan et al., 2000; Trauner et al., 1993]. However, the benefits of youth are less apparent when the injury is more diffuse and/or occurs at a critical window of brain development. Environment enrichment (EE) is an experimental model of enhancing brain development, and animals reared in EE grow up to have larger brains [Bennett et al., 1964; Diamond et al., 1964; Rosenzweig and Bennett, 1996], increased dendritic arborization [Faherty et al., 2003; Greenough et al., 1973; Juraska, 1984; Volkmar and Greenough, 1972], and superior performance on neurobehavioral tasks [Tees et al., 1990; Venable et al., 1988; Williams et al., 2001]. While injured rat pups show less overt cell death and less behavioral impairment than adults [Gurkoff et al., 2006; Prins et al., 1996; Prins and Hovda, 2003], they do lose the ability to benefit from EE rearing. Specifically, they show a loss of EE-induced experience-dependent plasticity; cortical thickening is blocked [Fineman et al., 2000], expansion of dendritic arbors is inhibited [Ip et al., 2002], and EE-induced cognitive enhancements are absent [Giza et al., 2005]. These studies of altered developmental plasticity are also supported by the findings of Prins and colleagues using a different model [Prins et al., 2003]. Following a lesion, regrowth of entorhinal cortical axons in a nontraumatically injured brain occurs in a well-characterized fashion. In juvenile rats subjected to experimental TBI, this normal pattern of axonal regrowth was markedly disturbed, with evidence of disruption down to the synaptic level.
Studies of developmental plasticity in traumatically injured children are difficult, as the effects of both age at injury and age at assessment must be considered in the control groups. However, there is growing evidence that pediatric TBI results in severity-dependent altered trajectories of brain development [Babikian and Asarnow, 2009; Catroppa et al., 2008]. First, although children generally have better outcome from TBI than adults, those injured at the earliest ages (i.e., in infancy) actually have worse outcomes [Anderson et al., 2005; Levin et al., 1992; Luerssen et al., 1988]. Some of this may be due to different mechanisms of injury, including nonaccidental trauma, which has a very poor developmental prognosis and is predominantly an etiology seen in infancy. It is also possible that the very young brain is decidedly more vulnerable to injury, as suggested by experimental studies using hypoxia-ischemia [Ikonomidou et al., 1989] and TBI [Bittigau et al., 1999; Pohl et al., 1999]. Second, there are rare anecdotal reports of neonates with head trauma that appear to have resulted in abnormal cortical development that was subsequently confirmed pathologically after epilepsy surgery for intractable seizures [Lombroso, 2000; Marin-Padilla et al., 2002]. Third, studies of severe pediatric TBI show more persistent cognitive deficits when the injury was diffuse rather than focal [Levin et al., 2000]. Lastly, in studies of repeated, mild sports-related TBI sustained during adolescence or early adulthood, subtle but significant cognitive impairments are detected [Collins et al., 1999; Matser et al., 1999].
Examination
The patient’s Glasgow Coma Scale (GCS) score should be determined. The GCS is a quick, reproducible means of rating the severity of the patient’s neurological injury (Table 74-1) [Teasdale and Jennett, 1976]. There are three components to the GCS score:
The total GCS score ranges from 3 (worst) to 15 (normal). There are obvious limitations to this scale in the setting of pediatric trauma, such as how to determine verbal score in infants and young children. Several GCS modifications have been proposed to make it more usable in this age group, and one modified scale is shown in Table 74-1. By convention, mild TBI is defined by a GCS score of 13–15, moderate by 9–12, and severe by 8 or less. A patient with a GCS of 13–15 but having an intracranial lesion would generally be classified as having a moderate TBI [Malec et al., 2007] (see chapter 73).
Sensorimotor examination can also be done quickly, starting with the best motor response elicited on GCS testing. Limb posture and muscle tone should be examined, and particular note should be made of any asymmetry. If the patient is unable to follow commands, then sensorimotor responses may be assessed by application of a peripheral noxious stimulus (e.g., nail bed pressure) in each limb. If no response occurs, then the site of noxious stimuli can be moved centrally (e.g., sternal rub, supraorbital pressure) to distinguish between afferent versus efferent impairment. Application of painful stimulus centrally also allows one to observe more easily whether the child can localize pain (distinction between a 4 and 5 for best motor score). If the child is awake, coordination should be assessed by observation of posture, spontaneous movements, and directed movements when the child reaches for an object or mimics the examiner. Deep tendon reflexes should be checked for asymmetry, hyperreflexia/clonus or areflexia. Flaccid paresis with no reflexes should immediately raise suspicion of a spinal injury. An outline of the rapid trauma examination is provided in Box 74-1.
Box 74-1 Rapid Pediatric Trauma Examination
In the mildly injured child who is conscious at the time of initial evaluation, the neurological examination can be conducted less urgently and more completely. In instances, mental status testing can be more extensive, starting with observation and possibly digit span recall to assess the child’s attention. This can be followed by evaluation of orientation, language, memory (both anterograde – for new items, and retrograde – for past events, including events surrounding the injury), and behavior. Cranial nerve, motor, coordination, and sensory examinations can include voluntary responses to the examiner’s commands, as well as subjective reports of sensory input (visual fields, tactile stimulation). Gait should be observed for signs of unsteadiness or ataxia. As with the more severely injured child, deep tendon reflexes and plantar responses should be checked. Aspects of the neurological examination are reviewed in detail in Chapters 1–4 and 73.
Immediate Management
Initial management of the child suffering from moderate or severe TBI centers on implementation of the ABCs and avoidance of secondary insults (see chapter 76). Early management will occur concomitantly with initial evaluation and examination. The airway must be secured and supplemental oxygen provided. In circumstances where the child is unable to maintain his/her airway adequately, then endotracheal intubation should be performed. Appropriate noninvasive monitors should be placed, including cardiac, respiratory, temperature, blood pressure, and those for pulse oximetry. Intubated patients should also have an end tidal CO2 monitor. Intravenous access must be established immediately and fluid resuscitation begun. Blood should be drawn for serum electrolytes, glucose, renal and hepatic function, complete blood count, prothrombin time (PT)/partial thromboplastin time (PTT), type and cross for blood transfusion, and, if appropriate, alcohol or drug screens.
Acute Clinical Syndromes
Herniation Syndromes
Herniation is the displacement of brain tissue from one intracranial compartment to another. This can occur as a result of a space-occupying lesion in one region, diffuse swelling of both cerebral hemispheres, or hydrocephalus (see Figure 74-3 and chapter 73) [Fishman, 1980; Plum and Posner, 1980]. The Cushing response is a paradoxical bradycardia with hypertension and slow irregular respiration that can be seen in the setting of elevated ICP and impending herniation. Definitive treatment for herniation requires surgical removal of the offending mass, if present. In cases where herniation is triggered by edema, positioning the head so it is slightly elevated and in the midline is indicated (to facilitate venous drainage), as well as immediately initiating first-tier measures (see the section on management below) to control elevated ICP: sedation, hyperventilation, and hyperosmolar therapy. Ventriculostomy with CSF drainage should be instituted as soon as possible. Infants can tolerate some elevation of ICP better due to unfused sutures and an open fontanel. These properties also allow for additional physical examination clues to the presence of increased ICP, such as a tense or bulging fontanel or a steadily increasing head circumference (if chronic).
Diffuse Cerebral Swelling
Since trauma, particularly closed head trauma, is by nature a diffuse process, bilateral swelling can occur even in the absence of discrete focal lesions. In fact, diffuse cerebral swelling is more common in children [Aldrich et al., 1992; Lang et al., 1994], while focal cerebral damage, such as contusions, is more common in older adolescents and adults. The etiology of this swelling was originally postulated to be due to hyperemia [Bruce et al., 1981]. However, as previously discussed, original reports of post-traumatic hyperemia in children overestimated the frequency of this phenomenon by comparing CBF values with those of uninjured young adult controls rather than uninjured children [Muizelaar et al., 1989; Zwienenberg and Muizelaar, 1999]. It is more likely that severe degrees of cerebral edema are related to the severity and nature of the injury, and particularly to the degree of cytotoxic and vasogenic edema formation. Cytotoxic edema occurs as a result of injured neurons undergoing energy failure, with loss of membrane ionic gradients and a rapid increase in intracellular water. In contrast, vasogenic edema results from fluid seeping into the extracellular space across a damaged or dysfunctional blood–brain barrier. Cytotoxic edema is felt to predominate after traumatic injury. Diffuse edema can occur as a direct response to trauma, but can also be due to a post-traumatic secondary insult, such as hypotension or hypoxia. At its extreme, diffuse edema can result in central transtentorial herniation and death.
Diffuse Axonal Injury
A clinical correlate to the pathophysiological process of axonal injury and disconnection, DAI is thought to result from shearing and rotational forces on white-matter fiber tracts [Adams et al., 1982]. These forces are usually considerable, and DAI is most commonly seen after a high-impact injury, such as a motor vehicle accident or a high fall.
[/not-level-membership-for-neurology-category]
