Chapter 125 Transplantation Frontiers
Introduction
In 1959, Paris Royo and Wilbur Quay reported experiments that were among the earliest steps taken in intraocular retinal cell transplantation.1 Using a paradigm originally developed by Van Dooremaal,2 Royo and Quay transplanted rat fetal retina into the anterior chamber of maternal rat eyes. Prolonged graft survival and a normal rate of graft development and differentiation were noted. Expanding on this work, del Cerro and his colleagues3 transplanted embryonic rat retina into the anterior chamber of adult eyes from various rat strains with similar results. After Gouras and coworkers4 successfully transplanted cultured human retinal pigment epithelium (RPE) cells to monkey Bruch’s membrane from which the retina had been removed, Li and Turner, in 1988, reported prevention of photoreceptor cell degeneration by RPE transplants in the Royal College of Surgeons (RCS) rat, an animal with an inherited defect causing impaired RPE phagocytosis of outer segments and progressive RPE and photoreceptor death.5,6 Li and Turner’s work, which has been replicated by many investigators, was the first successful treatment of retinal degeneration with cellular transplantation.
In principle, successful transplantation requires graft survival and integration with the host. Graft survival depends on a number of factors, both immunological and nonimmunological, that will be discussed in this chapter. Regarding graft–host integration, RPE cell transplants integrate readily with host photoreceptors through the extension of apical villous RPE processes around photoreceptor outer segments. In contrast, synapse formation between retinal grafts and host retina is much more complex. Experiments in the central nervous system, however, have established the possibility that synapse formation between donor and host neural tissue can occur. For example, Lund and others7,8 demonstrated that embryonic retina transplanted into neonatal and adult rat tectum develops the proper layering of the normal retina and extends neuronal projections only to the normal retinorecipient structures, i.e., the superior colliculus, the pretectum, and the dorsal lateral geniculate nucleus. Similarly, intraocular retinal transplants establish with their targets functional connections that elicit light-driven visual responses9 and mediate visual behaviors.10,11 These and other studies12,13 have stimulated interest in transplantation as a treatment for retinal disease. The background and rationale for transplantation, results of transplants in experimental animals and humans, immune response to transplanted tissue, non-immunological causes of graft failure, and future directions for development of transplantation for RPE and photoreceptor grafts will be considered in this chapter.
Background and rationale for RPE transplantation in age-related macular degeneration
Late complications of age-related macular degeneration (AMD) are major causes of irreversible loss of central vision among the elderly.14,15 Severe visual loss in AMD occurs due to growth of abnormal blood vessels (choroidal new vessels, CNVs) under the RPE and retina with secondary exudative retinal detachment, subretinal hemorrhage and lipid exudation, and outer retinal degeneration.16 It can also result from atrophy of RPE with consequent atrophy of overlying photoreceptors and underlying choriocapillaris (geographic atrophy, GA). The prevalence of AMD-associated CNV or GA is approximately 1.47%, increasing dramatically to 10% in people older than 80 years.17 There is no fully effective therapy for either CNVs or GA. Blocking the effects of vascular endothelial growth factor (VEGF) by frequent intravitreal injection of antibodies is the best currently available treatment for CNVs, but only 30–40% of the patients experience a moderate visual improvement.18,19 Cell-based therapy, which involves placing cells in the subretinal space, potentially offers a better option compared to pharmacological monotherapy with anti-VEGF agents. Advantages of such an approach would include long-term therapy without frequent minor surgical injections with medications; since cells like RPE produce numerous factors that help maintain the normal retinal and choroidal anatomy and physiology,18,20–24 transplantation of such cells could provide a richer and more effective therapy that is less susceptible to treatment resistance;25 cell transplantation could potentially restore the subretinal anatomy that is altered by growth (and surgical removal) of CNVs or by atrophy of RPE. Another indication for transplantation of RPE in AMD might be in patients who develop RPE tears,26 while receiving anti-VEGF therapy,27 which have a poor visual prognosis. RPE transplantation in patients with exudative macular degeneration might require prior removal of CNVs, which would result in loss of adjacent native RPE and the subjacent RPE basement membrane,28,29 causing a more complex graft bed than normal. On the other hand, in GA, transplantation of cells would involve resurfacing of the atrophic areas with less surgical manipulation with the goal of restoring the normal structure and function of the photoreceptor–RPE–choriocapillaris complex.
A number of pre-clinical experiments in animal models have demonstrated that transplantation of cells, e.g., RPE or stem cells or cells secreting growth factors, into the subretinal space can prevent photoreceptor degeneration and preserve function.30–36 The RCS rat is used commonly for these studies. A recessive mutation in merTK tyrosine kinase receptor in RPE6 causes a failure of outer segment phagocytosis by the RPE. Consequently, debris accumulates in the subretinal space, and there is eventual loss of photoreceptors causing blindness. Transplantation of RPE into the subretinal space of RCS rats causes a decrease in the amount of subretinal debris,32 prevents loss of photoreceptors,5 maintains synaptic connections of rescued photoreceptors,37 maintains electroretinography responses in areas of rescued retina,38 preserves pupillary light reflexes,39 cortical visual functions,30,33 and visual acuity.33,40,41
Results of macular translocation surgery may mean that establishment of a healthy subfoveal RPE monolayer in AMD eyes can restore macular function. In macular translocation, the retina is moved so that the fovea is relocated to an area away from submacular disease. This maneuver can result in improved visual acuity.42,43 However, after translocation, the fovea rests on previously extrafoveal RPE, Bruch’s membrane, and choriocapillaris. Thus, the procedure is not completely analogous to an RPE transplant per se.
Results of RPE transplants in humans
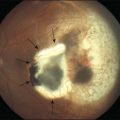
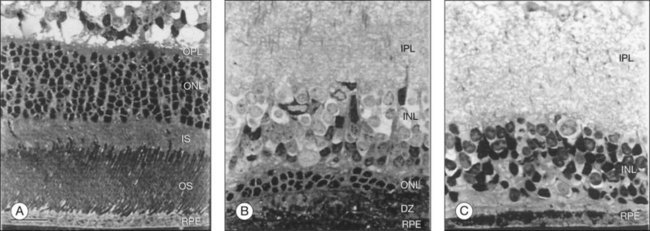
The first human RPE transplant in AMD was reported by Peyman and coworkers in 1991.44 Since then, numerous reports have described the results of allogeneic and autologous RPE transplantation.45–54 These surgeries have involved removal of CNV and placement of suspended allogeneic RPE cells,44 autologous RPE cells,48,54 patches of RPE,46,55 or autografts of RPE-choroid.48,51,52,54 Thus far, RPE transplantation has not been successful in the majority of patients undergoing surgery although a few patients show improvement of two or more lines of visual acuity following autografts.48,50,51,53,54 Allogeneic RPE transplants in AMD patients that have undergone CNV excision have failed with poor visual outcome and, in patients who are not immune-suppressed, subretinal fibrosis and chronic fluid leakage in the dissection bed.45,46,56 In autologous transplants, graft failure has resulted from intra- and postoperative subretinal hemorrhage, failure of graft revascularization, fibrous encapsulation of the graft, and development of epiretinal membrane, proliferative vitreoretinopathy, and retinal detachment.50,51,53 Optical coherence tomography studies indicate the presence of outer retinal atrophy in the majority of patients undergoing autologous RPE transplants, whether as cell suspensions or as sheets of RPE-Bruch’s membrane-choroid.54,57 Immune rejection and graft failure may underlie these results. The allogeneic RPE transplants may not have survived in the subretinal space independent of immune rejection. Tezel and coworkers have shown that if RPE cells cannot adhere to their basement membrane (or comparable surface) within 24 hours, they undergo apoptosis.58 All previous demonstrations of successful RPE transplants in laboratory animals have involved transplantation onto normal Bruch’s membrane (Fig. 125.1) or onto native RPE.4,5,31,32,59–62 In AMD, Bruch’s membrane is itself abnormal.63–66 Uncultured RPE isolated from adult human donor eyes show very limited adherence to aged submacular human Bruch’s membrane in vitro.67 In organ culture experiments, even cultured fetal RPE, which exhibit robust attachment to Bruch’s membrane, cannot survive for more than 1–2 weeks on aged or AMD submacular human Bruch’s membrane,22,68–70 presumably due to age- and AMD-related changes in this substrate. Histopathology of an immune-suppressed patient that underwent CNV excision plus uncultured adult RPE transplantation indicates that the cells were not organized as a monolayer, and there was photoreceptor atrophy over the transplant.55 These results are consistent with the observations of RPE behavior on human submacular Bruch’s membrane in organ culture.
Immune response to RPE transplants
Immune privileged sites and immune privileged tissue
Immune privilege in the eye is the result of multiple anatomic and physiologic factors acting on both the innate and adaptive immune systems.71,72 Streilein et al.72 noted that among the factors and mechanisms responsible for ocular immune privilege are: (1) the blood–ocular barrier, which minimizes contact of allografts with the cells and molecules of the systemic immune system, thus blunting the immune response to alloantigens; (2) deficient lymphatic drainage of the eye, which leads to initial alloantigen presentation in the spleen (versus regional lymph nodes) and, therefore, relatively less inflammatory immune responses; (3) an unusual distribution and functional properties of bone-marrow-derived antigen presenting cells; and (4) an ocular microenvironment rich in soluble or cell membrane-associated immuno-modulatory factors. Examples of such factors include transforming growth factor (TGF)-β2, α-melanocyte stimulating hormone, vasoactive intestinal peptide, calcitonin gene-related peptide, macrophage migration inhibitory factor, interleukin (IL)-1 receptor antagonist, Fas ligand, CD46, CD59, and free cortisol.73–80 These molecules help to create an immune-suppressive microenvironment by affecting the function of immune cells that come in contact with transplanted tissue or inhibiting complement activation. Ocular sympathetic innervation is necessary for maintenance of high ocular levels of TGF-β, and denervation leads to loss of immune privilege.81
Ocular immune privilege is a dynamic state of immune regulation that results in the induction of an antigen-specific “deviant” systemic immune response characterized by: (1) active downregulation of donor-specific delayed hypersensitivity reactions; (2) activation of regulatory cytotoxic CD8+ T-cells that suppress delayed hypersensitivity as well as suppress B-cells producing complement-fixing antibodies; (3) activation of regulatory CD4+ cells that block terminal differentiation of delayed hypersensitivity effector cells; and (4) enhancement of B-cells that produce non-complement-fixing antibodies. Initial descriptions of this response were based on experiments in which alloantigens were placed in the ocular anterior chamber, which led to the acronym, ACAID, anterior chamber-associated immune deviation.73,74,82,83
Immune privilege exists in the subretinal space, but it is not absolute. Subretinal allografts resist immune rejection and induce the development of systemic tolerance in the form of a suppressed antigen-specific delayed hypersensitivity reaction.83,84 RPE cells may create a local immune suppressive microenvironment through the production of immunomodulatory factors such as TGF-β85–87 and through suppression of T-cell activation88,89 and the induction of activated-T-cell apoptosis.90,91 In addition, RPE expresses ligands for T-cell inhibitory receptor programmed cell death-1 (PD-1) receptors on T-cells. These ligands (PDL1 and PDL2), whose expression is upregulated by interferon-γ, bind to PD-1 and suppress T-cells.92,93 RPE also expresses a novel immunosuppressive factor, cathepsin L inhibitor, CTLA-2α, that converts exposed T-cells to regulatory T-cells.94
The RPE can behave as immune privileged tissue. Neonatal RPE sheets, e.g., resist immune rejection at heterotopic sites (i.e., anatomic locations in which the transplanted tissue is not found normally) when compared to non-privileged tissue.95 It is not known whether adult RPE, as a sheet or single cell suspension, is immune privileged. Constitutive expression of Fas ligand on the RPE may contribute to its status as immune privileged tissue.95,96 This status does not mean, however, that the RPE is incapable of sensitizing its host. Allogeneic neonatal RPE grafts can sensitize recipient mice if the grafts are placed at a nonimmune privileged site.95
RPE cells express transplantation antigens. Low levels of major histocompatibility (MHC) class I antigens are present on the surface of fetal RPE cells.97 Also, RPE cells may express minor histocompatibility antigens. They do not seem to express MHC class II antigens normally although they can do so if exposed to interferon (IFN)-γ.98 Culturing RPE cells also can induce MHC antigen expression.99 In addition, RPE cells can process antigen prior to antigen presentation.100 Higher levels of IFN-γ-induced MHC class II antigen expression on RPE cells leads to increased activation of antigen-specific T-cells as manifested by the production of pro-inflammatory tumor necrosis factor-α.101 This activation does not include T-cell proliferation or production of IL-2, however, as is the norm for T-cell activation.
Implantation of neonatal allogeneic RPE cells into ocular immune privileged sites (e.g., the subretinal space) suppresses typical delayed hypersensitivity in the recipients, and the capacity to do so appears to be related to the immune privileged status of the graft site rather than to the immune privileged status of the graft.95 Thus, it may be that if RPE transplantation is done in a setting in which the immune privilege of the recipient site is compromised (e.g., breakdown of the blood–ocular barrier due to removal of native RPE), then allogeneic RPE grafts will be rejected despite their immune privileged status. If native RPE cells are compromised with sodium iodate, for example, tumor cells or ovalbumin injected into the subretinal space do not induce immune deviation.83 Immune deviation appears to be impaired persistently (i.e., after restoration of the blood–retinal barrier is complete) following iodate treatment and may be due to entry of plasma proteins into the subretinal space.83
In patients receiving RPE transplants after CNV excision, the outer blood–retinal barrier will be compromised both by the presence of CNVs preoperatively and by the iatrogenic RPE defect in the transplant bed postoperatively. Experimental studies indicate that the outer blood–retinal barrier is disrupted for approximately 1 week after hydraulic debridement of the native RPE.102,103 Experiments in mice indicate that breakdown of the outer blood–retinal barrier for approximately 14 days (beginning a few days before antigen inoculation) abolished immune privilege and abrogated immune deviation to cell-associated and soluble antigens in the subretinal space.83 It should be noted that placement of alloantigen in the eye in ocular immune privilege experiments involves disruption of the blood–ocular barrier. That immune privilege is extended despite this disruption indicates the importance of the factors in the microenvironment. Persistent loss of the blood–retinal barrier (e.g., due to CNVs) may result in dilution or absence of immunomodulatory factors in the microenvironment in addition to increased access to subretinal space by systemic immune cells. Also, the role of age-related changes in the RPE vis-à-vis subretinal immune privilege is unexplored. Finally, the extent to which immunological abnormalities associated with AMD,104 even in the absence of CNVs, compromise the normal immune suppressive environment of the subretinal space is unknown.
Are RPE transplants rejected?
It is not clear that orthotopic (i.e., transplants placed in an anatomic location in which they are found normally) allogeneic RPE transplants are rejected. Some clinical and experimental evidence indicates that they are rejected.45,46,56,62,105–109 In contrast, other studies indicate that RPE transplants may not be rejected.59,110 In still other studies, it is not clear whether rejection occurred or not.111 Interpretation of most of these studies, however, is complicated by the fact that it was difficult to identify unambiguously the transplanted RPE cells due to lack of a unique histological or clinical marker.
In a pilot study, transplanted allogeneic uncultured adult human RPE did not appear to be rejected in AMD patients who had undergone CNV excision and were treated with systemic azathioprine, prednisone, and cyclosporine, which suggests that immune suppression might prevent RPE transplant rejection in this setting.55 However, these elderly patients were not able to tolerate systemic triple immune suppressive therapy for an extended time. Local immune suppression is somewhat effective in laboratory experiments involving intravitreal injection of cyclosporine,112 but it has not been reported in human RPE transplants.
Autologous transplants (e.g., transplants of uncultured peripheral RPE cells to the submacular space of the same eye) should not undergo graft rejection. Methods for harvesting RPE cells for autologous transplants exist,50,113 but in vitro data indicate that harvested aged human RPE do not adhere well to aged submacular Bruch’s membrane, even in the presence of native RPE basement membrane.67 This result may explain why patients undergoing autologous RPE transplants after CNV excision have generally shown very limited visual improvement.50,54 Also, aged autologous adult RPE may exhibit aging and AMD changes that render them unsuitable for transplantation.114,115 Use of allogeneic cultured RPE could be considered116,117 if immune suppression is either unnecessary or, if necessary, it can be achieved.118 Attempts at xenograft transplantation, even using triple immune suppression, have been unsuccessful thus far in preclinical models.119
RPE graft failure
As noted above, RPE transplants in humans may have failed independent of immune rejection of the graft. Attachment of RPE to Bruch’s membrane after transplantation is crucial as RPE undergo apoptosis otherwise.120 In vitro studies indicate that trypsin-harvested, cultured fetal human RPE can adhere to adult submacular Bruch’s membrane even on surfaces lacking native RPE basement membrane.22,68,69,121 In contrast, uncultured collagenase IV-harvested adult RPE adhere poorly to aged human submacular Bruch’s membrane even in the presence of native RPE basement membrane.67 Del Priore and coworkers have shown higher attachment rates with cultured RPE from older donors when seeded onto peripheral Bruch’s membrane of older persons.122–125 This higher attachment rate might reflect a difference in integrin expression of primary isolated RPE cells versus cultured cells.68,126 It also might reflect differences in extracellular matrix composition of peripheral versus submacular Bruch’s membrane.63,127 Histology of excised CNVs,28,29,128 histopathology of eyes following CNV excision,129,130 and postoperative clinical findings28,131 all suggest that CNV dissection exposes both the superficial and deeper layers of the inner collagenous layer of Bruch’s membrane, which will constitute much of the surface to which transplanted RPE must adhere and on which they must survive if transplants are done after CNV excision. Cultured fetal human RPE can adhere to the superficial and deep inner collagenous layers of aged human submacular Bruch’s membrane, but long-term survival is impaired.22,69,126 An additional age-related change in Bruch’s membrane that could contribute to poor graft survival and function is the increased deposition of cholesterol.132 It is not known if the lipid deposits, which may contribute to the decreased hydraulic conductivity of aged Bruch’s membrane,127 might affect RPE transplant function or survival by masking extracellular matrix ligands. In vitro studies show that fetal RPE can attach to a high degree on the surface of the inner collagenous layer and that the majority of cell death occurs after 7 days in culture, indicating that poor cell survival is a post-attachment event.22 The poor visual results associated with autologous iris pigment epithelium (IPE) transplants might be due to graft failure and, perhaps less likely, from limitations in the ability of IPE to replace RPE.133–135 Stem cell precursors of RPE are an attractive option for transplantation (see below). They are being evaluated for their ability to attach and resurface Bruch’s membrane. One study found that human embryonic stem-cell-derived RPE cells have the potential to resurface aged Bruch’s membrane to a similar extent as cultured fetal RPE, but analysis of protein secretion indicates that these two cells may not behave identically.22
RPE replacement: future directions
Immune rejection
Many different strategies can be pursued to prevent graft rejection, including systemic or local drug therapy with established (e.g., prednisone, azathioprine, cyclosporine) or new (e.g., recombinant cytokines) compounds or via other techniques, e.g., diminishing MHC class-II-positive cells in the transplant.136 Cyclosporine does not appear to interfere with the ability of allogeneic neonatal retinal grafts to induce ACAID, nor does prolonged treatment with systemic cyclosporine interfere with persistence of allospecific suppressor cells for 35 days after transplantation.137 Nonetheless, retina grafts in the anterior chamber do deteriorate in 35-day grafts suggesting that graft destruction is the result either of immune mechanisms not inhibited by cyclosporine and/or non-immunologic factors. Independent of its effectiveness, systemic immune suppression can be complicated by side-effects such as nephrotoxicity, hypertension, risk of malignancy, susceptibility to infection, hepatotoxicity, seizures, and even anaphylaxis, depending on the medications used.138 Thus, local immune suppression has been considered. In one study, local immune suppression of rabbit eyes with cyclosporine prolonged cultured human fetal RPE xenograft survival in the subretinal space, but by 25 weeks, only ~10% of the green fluorescence protein-labeled grafted cells could be identified.112 It may be that the viral vector used to introduce green fluorescence protein induced an inflammatory response. Sustained slow-release cyclosporine delivery was as effective at promoting transplant survival as repeated intravitreal injections of much higher doses. Thus, it is not clear that intraocular immune suppression with cyclosporine alone will prevent allogeneic RPE transplant rejection. High-dose dexamethasone therapy is known to be effective in preventing acute allograft rejection139,140 although lymphocyte resistance to prednisolone can develop.141 Repeated intravitreous injections of dexamethasone can be well tolerated by the retina,142 which leads us to believe that sustained intravitreal dexamethasone delivery may not be complicated by retinal damage. Use of the sustained dexamethasone delivery system has prevented allogeneic corneal transplant rejection in rats.143 Currently, there are several intravitreal sustained-release steroid formulations that could be considered in human transplants.144,145 Thus, if immune suppression of RPE grafts is necessary, this approach may also be effective in preventing RPE transplant rejection.
Transplanted RPE survival and differentiation on aged Bruch’s membrane
There is no fully validated animal model of AMD at present. In vivo and in vitro RPE wound healing and transplantation models exist, but they do not appear to be directly relevant to AMD patients undergoing CNV excision. Previously reported successful in vivo RPE transplants involve attachment to normal Bruch’s membrane or native RPE as noted above. Experiments using aged submacular human Bruch’s membrane in organ culture indicate that aged human RPE do not adhere well to this surface unless they are cultured.67,68 Cultured fetal RPE appear to adhere to aged submacular Bruch’s membrane better than cultured aged RPE, but even in this case the cells do not appear to survive and differentiate well at 7 days in organ culture. It appears that aging changes in Bruch’s membrane are responsible for this deleterious effect on the cells.70,146,147 Thus, one approach to improving RPE transplantation success is to identify and manage these Bruch’s membrane changes. Experiments in vitro have included “cleaning” Bruch’s membrane with Triton X-100 (a detergent) and/or resurfacing the Bruch’s membrane with extracellular matrix molecules, singly or in combination148,149 or resurfacing the Bruch’s membrane with a cell-secreted matrix.70 The latter has shown promise, but translation to clinical application remains a challenge. Another approach has included enhancing the expression of cell-matrix adhesion molecules, i.e., integrins, to improve RPE adhesion and survival.126,150 Finally, an artificial scaffold could be used to transplant RPE. Such a scaffold could provide an environment that would allow easy placement of an RPE graft in the right orientation and/or promote RPE survival and differentiation.151,152 The scaffold might even prevent the age-related changes in Bruch’s membrane from damaging the transplanted RPE cells. An ideal scaffold will be biocompatible, biodegradable or capable of integrating, and should not interfere with membrane transport or permeability. Various polymers like polymethylmethacrylate, polyL-lactide, polyglycolic acid, expanded polytetrafluoroethylene, and others have been used with RPE, stem cells, and Schwann cells.153–155
Native RPE resurfacing of aged Bruch’s membrane
An alternative to RPE transplantation is to stimulate RPE ingrowth from the edge of the dissection bed. Human pathological studies indicate that RPE ingrowth following CNV excision in AMD patients is incomplete and aberrant.28,129,156 Previously described cell culture RPE wound-healing models are associated with complete wound resurfacing, in contrast to the situation in AMD patients undergoing CNV excision.157–159 In vivo RPE wound-healing models (e.g., rabbit, pig) are associated with complete resurfacing of the RPE defect160,161 or involve administration of compounds (e.g., mitomycin C) that complicate further experimental study or have unclear relevance to resurfacing Bruch’s membrane in AMD patients.102,162,163 Experiments from a Bruch’s membrane organ culture model indicate that resurfacing of ~65–85% of a ~3-mm-diameter circular RPE defect in aged submacular human Bruch’s membrane occurs by approximately 10 days if RPE basement membrane or the superficial inner collagenous layer is present. If deeper defects are created, exposing deeper regions of the inner collagenous layer, significantly less resurfacing is observed (~54%) at day 10 indicating that one should expect less resurfacing with greater damage to Bruch’s membrane.164,165 Additional experiments are needed to determine whether one can alter the denuded Bruch’s membrane surface (or the RPE cells at the edge of the defect) so that resurfacing of the RPE defect occurs in situ.
Human clinical studies indicate that periods of macular detachment up to 2 weeks are compatible with recovery of visual acuity of 20/50 or better in a substantial number of patients.166 Monkey and cat experiments indicate that many photoreceptors survive during retinal detachment periods of several weeks duration, although some photoreceptors definitely die.167,168 Approximately 80% of the cat outer nuclear layer survives during 3 days of detachment.169 The numbers of photoreceptor nuclei in detached cat retina do not begin to decline significantly (i.e., >20% decline in density) until detachment periods longer than 13 days.170 Data from cat retinal detachment studies indicate that 14-day detachments followed by 30-day reattachment is associated with rod and cone outer segment length similar to that observed after 5-day detachments.168 The cat retina, however, is rod dominated. Cones are more prone to apoptosis with detachment (versus rods)171 although in one study, approximately 75% of S- and M-cones survived a 1-day detachment (followed by retinal reattachment) in the ground squirrel.172 Thus, at this time, published experimental data do not indicate clearly what the exact survival of cones is after 2-week periods of retinal detachment, but clinical data indicate that it is good enough to warrant developing methods to promote RPE resurfacing. In addition to duration of detachment, the height of detachment influences photoreceptor survival.172,173 The macular detachments that would arise from CNV excision are quite shallow (<1–2 mm height), which also favors photoreceptor survival during a 2-week RPE resurfacing period. Thus, we believe that the RPE resurfacing approach is feasible despite its possible association with photoreceptor death during the resurfacing process.
Alternate source of rpe: stem cells
Stem cells are undifferentiated cells that have the capacity for self-renewal (i.e., can divide indefinitely to produce more karyotypically stable stem cells) and pluripotency (i.e., produce cells that are destined to differentiate into more than one type of cell).174–176 (See Chapter 35, Stem cells and cellular therapy.) During the course of differentiation, stem cells form intermediate populations of increasingly committed progenitor cells that have a decreasing proliferative capacity. Embryonic stem cells (ESCs) are derived from the inner cell mass of the blastocyst and are capable of differentiating into cells of all three germ layers.177,178 Embryonic and fetal tissues (e.g., retina) contain cells that vary in their differentiation status, ranging from progenitor cells, that can proliferate but have a restricted differentiation potential compared to embryonic stem cells, to committed but still immature rods and cones isolated from fetal retina. In adults, multipotent stem cells exist that can differentiate to replace lost or injured cells. Such cells have been isolated from various organs,179,180 including the eye.181,182 Stem cells isolated from a particular tissue can be induced to differentiate into cells of an unrelated tissue. Bone marrow cells, for example, can undergo metaplasia to form skeletal muscle;183 neural stem cells can develop into muscle184 (Table 125.1). It seems that stem cells exhibit plastic behavior depending upon their environment and signals from damaged tissues.185 The central nervous system contains neural progenitor cells. They exist in both embryonic and adult tissues and can differentiate into glial and neuronal lineages. The capacities for self-renewal and pluripotency render stem cells candidates to replace lost or injured cells. In summary, potential sources for replacement of RPE include non-eye-derived embryonic stem cells from the blastocyst, bone-marrow-derived stem cells, neural progenitor cells, and eye-derived progenitor cells.
Table 125.1 Potential sources of cells for retinal cell replacement*
| Cell type | Potential developmental capacity |
|---|---|
| Totipotent stem cell (mammalian zygote, blastomeres) | Can develop into cells derived from all lineages, i.e., ectoderm, mesoderm, and endoderm, as well as supporting tissues like placenta |
| Pluripotent stem cell (e.g., embryonic stem cell from inner cell mass) | Can develop into cells derived from all three germ layers (ectoderm, mesoderm, endoderm) |
| Multipotent stem cell (e.g., retinal progenitor cell) | Can develop into different cell types derived from one lineage |
| Reprogrammed cell (induced pluripotent cell) | Somatic cell reprogrammed by nuclear transfer, limited transfer of transcription factors, or cell fusion to enhance potency |
| Immature post mitotic rod precursor cell | Rod photoreceptors |
| Neural retina and retinal pigment epithelium | Rods, cones, Müller cells, or committed retinal neurons |
* Modified from Jaenisch R, Young R. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell 2008;132:567–82 and Zarbin MA. Retinal pigment epithelium-retina transplantation for retinal degenerative disease. Am J Ophthalmol 2008;146:151–3.
During development, the various retinal cells are derived from a common population of multipotent retinal progenitor cells (RPCs).186 Embryonic retina can be a potential source of such cells. Such cells exist in adults in lower species like fish and amphibians in the ciliary margin zone (CMZ), located in the retinal periphery, surrounded by a pigmented ciliary margin.187–189 These cells can regenerate the neural retina throughout the life of the organism.190 Progenitor cells from chick CMZ can regenerate retina if there is ectopic expression of fibroblastic growth factor and sonic hedgehog.191 More importantly, retinal progenitor cells have been isolated from mouse eyes and from fetal and adult human eyes.182,192–195 Efforts to induce RPCs to differentiate into RPE continue,196,197 but there are no pure, fully characterized RPE cells from RPCs yet.
Ethical considerations preclude the use of stem cells from human embryos although methods have been developed to establish stem cell lines without destruction of the embryo.198,199 It is possible to generate embryonic stem cells by somatic nuclear transfer-DNA transfer from an adult somatic cell into an oocyte, whose nucleus has been removed.200,201 The oocyte would then reprogram the adult DNA and develop in a normal embryonic pattern and form embryonic stem cells. Although the mitochondrial DNA will be derived from the oocyte, the chromosomal DNA would be from the host. Thus, a perfectly matched graft RPE could be prepared. However, ethical considerations are still an issue since it involves human life form. Alternatively, adult fibroblasts can be manipulated by transferring into these cells transcription factors like Oct4, Sox2, Klf4, and c-Myc that can reactivate developmentally regulated genes, thus inducing the cell to become an induced pluripotent stem cell (iPSCs).202,203 Nuclear transfer may be more effective at establishing the ground state of pluripotency than factor-based reprogramming, which can leave an epigenetic memory of the tissue of origin that may influence efforts at directed differentiation for applications in disease modeling or treatment.204 Although iPSCs are pluripotent stem cells, iPSCs and ESCs do differ in some important ways. iPSCs have the theoretical advantage of not being rejected by the patient from whom they are derived (versus ESCs, unless the ESCs were harvested from the patient as an embryo), but abnormal gene expression in some cells differentiated from iPSCs (both via a retroviral and episomal approach) can induce a T-cell-dependent immune response in a syngeneic recipient.205 This response is likely due to the abnormal expression of antigens (e.g., Zg16, Hormad1) not expressed during normal development or differentiation of ESCs, leading to loss of tolerance.205 Expression of these antigens is a reflection of epigenetic differences (e.g., DNA methylation) between iPSCs and ESCs.206,207 Continuous passaging of iPSCs may help attenuate these differences,208 but iPSCs clearly seem to be at greater risk for tumor formation (e.g., due to p53 suppression) than ESCs.
RPE can be derived easily from embryonic stem cells (perhaps because RPE develop early during embryogenesis)40,196,209–211 as well as from iPSCs.151,152,211–214 Transplantation of these human embryonic stem-cell-derived RPE (hES-RPE) into RCS rats also results in improved photoreceptor survival and visual function (although the xenograft does not survive long).36,40 With stem cells, there is a risk of dedifferentiation in the subretinal space and potentially developing into a teratoma. Indeed, in one study, when embryonic stem-cell-derived neural cell precursors were injected into mouse subretinal space, 50% of the eyes developed teratomas within 8 weeks.215 However, in another study, a more stringent selection of 18 different human embryonic stem cell lines did not result in teratoma formation.40 There is a risk of insertional mutagenesis due to the use of viral vectors, and teratoma formation due to the use of oncogenic factors such as c-Myc, in iPSC cell production. Newer techniques of iPSC production without the use of c-Myc216–218 or by using non-integrating episomal vectors219,220 could allay such concerns. Recently, RPE have been generated from human iPSCs (iPS-RPE) and have been shown to express RPE properties such as ion transport, membrane potential, polarized vascular endothelial growth factor secretion, and gene expression profile similar to those of native RPE.221
Bone-marrow-derived mesenchymal stem cells (BM-MSCs) are another potential source of stem cells that have been studied in rats and mice.222 Human BM-MSCs can also be induced to differentiate into retinal cells by co-culturing with RPE cells.223 It is not clear that fully functional RPE cells can be generated with this approach. RPE derived from mouse BM-MSCs have been transplanted into subretinal space of RCS rats and rescue photoreceptors.222 Interestingly, intravenously injected BM-MSCs home into areas of chemically induced RPE loss in mice.224 Autologous cells from umbilical cord tissue can also be a potential source.225
1. Proliferate extensively to generate sufficient quantities of material to serve as a “universal donor”
2. Differentiate into the desired cell type(s). hESC-derived RPE can spontaneously dedifferentiate to non-RPE-like cells and spontaneously redifferentiate into RPE-like cells, indicating phenotypic instability.226 The cultures may not retain a stable phenotype after 5–8 passages. ESCs and iPSCs vary in their tendency to differentiate into cells of a given lineage.204,226 As mentioned above, a number of different criteria should be applied to define a “differentiated” RPE175 and photoreceptor cell.227 The retinal and subretinal microenvironment can influence the differentiation and functionality of transplanted cells, including expression of developmental markers and markers of proliferation.228,229 Abnormalities in Bruch’s membrane may prevent transplanted hESC-derived RPE from surviving and differentiating long term in AMD eyes.22 Since Bruch’s membrane is derived from mesoderm, there is no expectation that hESC- or iPSC-derived RPE will manufacture Bruch’s membrane
3. Survive in the recipient after transplantation. Human iPSC-derived RPE survive ~4 months in RCS rats (xenografts)212
4. Integrate into the surrounding tissue after transplantation
5. Function appropriately for the duration of the recipient’s life.
To be useful for cell-based rescue therapy, stem cells must elaborate needed trophic factors and not proliferate in an uncontrolled manner. In rhodopsin knockout mice, bone-marrow-derived mesenchymal stem cells rescue photoreceptors.230 Subretinal bone-marrow-derived mesenchymal stem cells rescue photoreceptors in RCS rats.231 hESC-derived RPE elaborate neurotrophic substances that have been shown to support photoreceptor survival in preclinical models of retinal degenerative disease.22
RPE cell transplants are an attractive starting point for cell-based combination replacement and rescue therapy in the eye because: (1) hESCs and iPSCs can be induced to differentiate into RPE relatively easily, and, at least in the case of hES-RPE, one can generate large quantities of cells with stable genotype and appropriate phenotype; (2) RPE cells integrate easily with host photoreceptors; (3) RPE cells elaborate trophic substances that support photoreceptors22,33,232; (4) there is abundant evidence for transplant efficacy in pre-clinical models; (5) diseases in which RPE cells appear to be targeted primarily include Best disease233,234 and some forms of retinitis pigmentosa,235,236 and secondarily include Stargardt macular dystrophy237,238 and age-related macular degeneration (AMD).66,104 However, in the case of AMD eyes, RPE survival and proper differentiation on submacular Bruch’s membrane may be problematic.22
Background and rationale for photoreceptor transplantation in retinal dystrophies
Retinitis pigmentosa (RP) is a group of heterogeneous hereditary disorders with an estimated prevalence between 1 : 3000 and 1 : 5000, affecting 50 000 to 100 000 individuals in the USA and approximately 1.5 million people worldwide.239 In the vast majority of cases, the condition results from mutations in photoreceptor cell genes encoding a myriad of structural and phototransduction proteins, and, to a lesser extent, from mutations in genes of the RPE.239 Blindness ultimately results from rod and cone photoreceptor cell death.240 Inner retinal layers, however, remain relatively preserved, especially early in the course of the disease.241,242 As degeneration progresses, however, extensive synaptic rewiring of the inner retina occurs.240,243,244
A variety of approaches to restore vision are under study, including gene therapy,245–249 pharmacotherapy,250,251 visual prostheses,252 endogenous regeneration,253 and transplantation.254–260 The goal of retinal transplantation is to restore and/or maintain visual function by transplanting healthy donor photoreceptors that replace lost photoreceptors by integrating with the host inner retina to reconstruct a functional, neural retinal network. As noted above, this approach to sight restoration is termed “replacement.” Retinal transplantation may also slow down the progression of the disease by means of graft-released trophic substances that rescue residual host photoreceptors,261–264 an approach termed “rescue.” A trophic effect would be most useful, however, only while the critical number of foveal cones required to support useful vision is present.265 In late stages of the disease, when most photoreceptors have degenerated, replacement of lost photoreceptors probably would be more effective.
Results of photoreceptor transplants in experimental animals
Animal models of retinal degeneration
Early studies of retinal transplantation utilized normal animals as hosts. Subsequently, investigators turned to animal models of retinal degeneration. Animal models became available either by discovery of naturally occurring genetic mutants such as the retinal degeneration (rd) mouse,266 the retinal degeneration slow (rds) mouse,267 the RCS rat,6 and the rod–cone dysplasia (Rdy) Abyssinian cat,268 or by experimentally induced degenerations such as photoreceptor degeneration induced by phototoxicity269 and by intravitreal injection of a concentrated solution of human hemoglobin.270 In addition, genetically engineered models of homologous human retinitis pigmentosa mutations such as the transgenic rhodopsin (Pro23His) mutant mouse271 and the transgenic rhodopsin (Pro347His) mutant pig272 were developed. The advantages of these models include the rapid and nearly total loss of target cells such as RPE or rod photoreceptor cells as well as their similarity (in some cases, equivalence) to human disease, allowing for the efficient study of retinal transplantation under more clinically relevant conditions. Most work to date has employed rodent models such as the light-damaged rat retina,13,273,274 the rd mouse,256,275–279 the P23H rhodopsin transgenic mouse,280 and the RCS rat.281 The small eye and large lens of the rodent favor transscleral rather than transvitreal graft delivery. Transvitreal delivery is a less traumatic and more pertinent technique to human retinal transplantation than the transscleral approach. Furthermore, the rod to cone ratio in rodent retina282 is different than that of the relatively cone-rich human retina. To overcome these problems, animal models using larger eyes, such as the chemically ablated rabbit retina270 and the Abyssinian Rdy cat,283,284 have been employed as well as normal rabbits,270,285,286 pigs,287 and primates.118 The pig may prove to be a valuable animal model because the anatomy and size of the porcine eye are closer to that of the human,288,289 and the pig retina is well endowed with cones, has an area centralis, and is holangiotic.290 In addition, the transgenic rhodopsin (Pro347His) mutant pig272 carries a mutation similar to that found in a form of human RP and is an animal model closest to human RP, especially with regard to refining surgical procedures.291 However, the pig retina does not have a fovea.
Graft implantation sites and preparations
Retinal transplants have been placed in a surgically induced retinal lesion, in the vitreous cavity (i.e., epiretinally),12,292 and into the host subretinal space.13,293 Grafts from donors of the same species (i.e., allogeneic grafts) and from a different species (i.e., xenogeneic grafts)118,294,295 have been transplanted with or without host immune suppression.
Photoreceptor microaggregates, which are retinal fragments in which tissue integrity is disrupted by gentle trituration with cells remaining attached to each other in small clusters (<0.2 mm2), have been used in retinal transplantation.12 Isolated cell suspensions obtained by mechanical and/or enzymatic dissociation of the retina also have been used.275,277,293,296 Compared to microaggregates, isolated photoreceptors usually are more traumatized, frequently lose their outer segments, display less organization, and often are not properly oriented.255,296 Both preparations often form spherical structures or rosettes, with photoreceptors oriented towards a central lumen similar to those seen in retinoblastomas and retinal dysplasias.12,292,294 Outer segments of misoriented photoreceptors as well as those found in rosettes rapidly degenerate due to their failure to contact RPE cells.297 In addition, both retinal microaggregates and cell suspensions frequently are contaminated with non-photoreceptor retinal cells. Rosette formation prevents the reconstruction of the normal retinal anatomy and interferes with establishing contacts between graft photoreceptor terminals and host second-order neurons.33
To avoid these problems, Silverman and Hughes13 introduced the photoreceptor sheet preparation, created by vibratome-sectioning of adult mammalian retina. Sheet preparations preserve the organization and polarity of grafted photoreceptors and maintain the densely packed arrangement of photoreceptors found in situ and thought to be critical for good visual acuity.298 Furthermore, the inner retina, which could interfere with synaptic interactions between grafted photoreceptors and host second-order neurons, is removed. Sheets can be placed between the host retina and RPE in the proper orientation, and they show a much lower frequency of rosette formation.13,262,270,276 This technique has been modified further to obtain photoreceptor sheets using both vibratome-sectioning and excimer laser ablation of the inner retina.299 A similar approach has been used to transplant sheets of adult full-thickness retina, intact embryonic or fetal retinal sheets,274,281,286 and vibratome-sectioned fetal retina. Aramant and coworkers introduced the approach of co-transplanting fetal neural retina with the adjacent RPE.281 This approach might be advantageous in cases of advanced disease where both retinal cell types have degenerated.258
Age is an important variable in the choice of donor tissue. Studies in neural retinal transplantation have utilized tissue from embryonic, early postnatal, late postnatal, and adult donors as well as transplantation of homo- and heterotypic neural progenitor cells (stem cells, see below). There are several advantages to transplanting embryonic or fetal retinal grafts.254 Immature retinal tissue has a high potential for plasticity,300 lacks immunogenicity,71 and survives and differentiates well in the host.274 However, it is extremely fragile, difficult to handle, and is undifferentiated, which makes obtaining pure photoreceptor sheets difficult or impossible. In one study, attempts to obtain photoreceptor sheets from embryonic retina using vibratome-sectioning resulted in abnormal morphology and poor survival.286 Excimer laser was used to obtain a monolayer of human fetal cones, but the ultrastructure of their synaptic pedicles was disorganized.301 Also, embryonic or fetal retinal grafts often are associated with rosette formation. Finally, obtaining embryonic or fetal tissue for transplantation has many logistical and ethical constraints. Adult retinal grafts seem to show normal morphology and organization in the host retina and are associated with minimal rosette formation, which might allow for better restoration of retinal anatomy,118,270,276 especially if the outer blood–retina barrier remains intact.83 Adult retinal graft preparations have been shown to yield good survival rates.118,270,276 Furthermore, mature retinal tissue is easier to handle and, because it is fully differentiated, reliably yields pure photoreceptor sheets and can be obtained readily from eye banks. Also, it seems that differentiated photoreceptors can integrate with host retina.255
Transplantation aimed at photoreceptor cell rescue
A wide variety of cells, including rod photoreceptors, RPE, IPE, and even nonocular cells such as Schwann cells, as well as progenitor stem cells have been used in transplantation paradigms aimed at rescuing host residual photoreceptors and retarding the progression of retinal degenerative diseases. The strategies employed, including the type of cells used, in these paradigms depend on the primary derangement that causes photoreceptor cell death. For example, Schwann cells, derived from peripheral nerves, have been used as autologous grafts to rescue photoreceptors. Schwann cells are known to produce growth factors such as ciliary neurotrophic factor (CNTF), glial cell line-derived neurotrophic factor (GDNF), and brain-derived neurotrophic factor (BDNF) and are therefore a source for sustained growth factor release. Schwann cells, however, do not phagocytose photoreceptor outer segments. Nevertheless, subretinal Schwann cell transplants have been shown to limit photoreceptor cell loss in RCS rats for as long as 9 months35,302 and for up to 2 months in rhodopsin knockout mice (Fig. 125.2).303 In addition, Schwann cells engineered to secrete GDNF or BDNF promote photoreceptor rescue in RCS rats to an even greater degree than that observed with parent Schwann cells.34
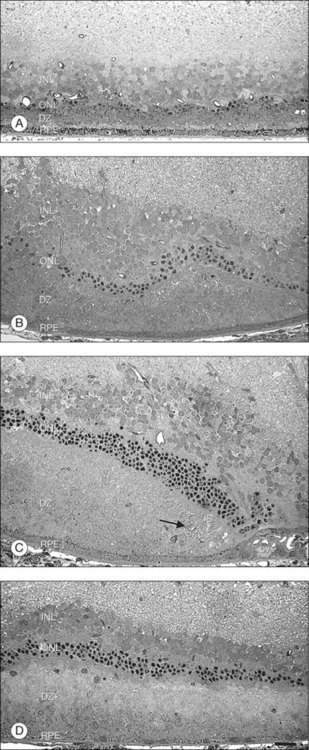
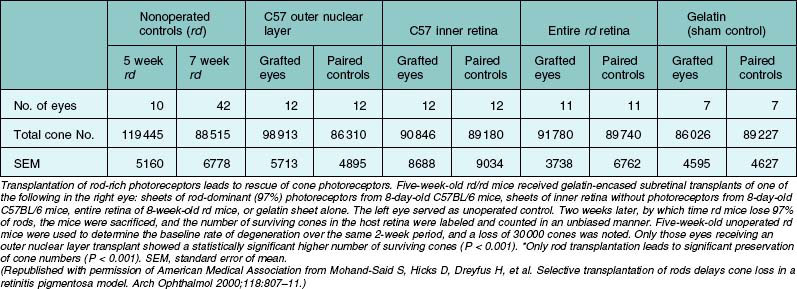
Transplantation of rod photoreceptors with the goal of cone photoreceptor “rescue” has been undertaken because, in the vast majority of RP cases and related animal models, the disease-causing mutations are expressed exclusively in rod cells. Transplanted rods might rescue existing cones that would be lost secondary to rod cell degeneration.304 Subretinal rod photoreceptor transplants have been shown to exert a protective effect on cone photoreceptors in animal models of retinal dystrophies, delaying photoreceptor degeneration and limiting cell death. Mohand-Said and coworkers262 have demonstrated that rod-rich retinal grafts transplanted into the subretinal space of rd mice, a strain with a naturally occurring mutation in the gene encoding the β-subunit of rod cyclic guanosine monophosphate phosphodiesterase (cGMP), preserved cone cells, with cone density 30–40% greater than that of untreated rd retinas (Table 125.2). This rescue effect was observed at some distance from the grafted cells suggesting the existence of diffusible trophic factors released by the transplant. These findings were further substantiated by in vitro co-culture studies where significantly greater numbers of surviving cones were seen in mouse dystrophic retinas cultured with retinas containing normal rods compared to dystrophic retinas cultured in medium alone or with rod-deprived retinas.261 Molecules mediating this paracrine effect have been identified.263,264 Several growth factors, neurotrophic factors, and cytokines such as basic fibroblast growth factor (bFGF), acidic fibroblast growth factor (aFGF), CNTF, GDNF, BDNF, and IL-1β have been shown to exert robust survival-promoting effects in the retina when injected subretinally or intravitreally,250,305 or when delivered by adenovirus gene therapy306 in various animal models of retinal degeneration.307,308 A similar rescue effect has been shown to occur by focal mechanical injury to the retina in light-induced retinal damage309–311 as well as in sham-operated animals during transplantation studies.312,313 This effect is probably mediated by injury-induced upregulation of neurotrophic factors.309,311
Transplantation aimed at photoreceptor cell replacement
Various neural retinal transplantation paradigms have been developed in laboratory animals,117,254,314 some of which have evolved into clinical trials in humans.118,258,315–318 This work has documented the feasibility of retinal transplantation, with long-term survival of the transplant in both animals286,297 and humans.118,258,315–319
While there is evidence that some degree of graft–host integration might occur after retinal transplantation,259 thus far, light and electron microscopy have not demonstrated unambiguous significant direct graft–host synaptic integration.12,13,274,276,277,281,286,320 In many of these studies, a glial limiting membrane, derived from Müller cell processes, formed a barrier between graft and host retinas. In areas where this glial barrier was absent, the transplant and host retinas were in close apposition, and their cell processes intermingled indistinguishably.274 Disruption of the outer limiting membrane seems to improve integration between transplanted photoreceptors and wild-type as well as degenerating retina.321,322
Ultrastructural analysis of full-thickness embryonic retinal sheets transplanted into the subretinal space of normal rabbits has shown bundles of nerve fibers containing mature neuronal processes and growth cones on either side of the graft–host interface in close association with intermingled graft and host Müller cell processes, which form a glial barrier.286 Immunohistochemical analysis of these transplants showed some direct contact between processes of rod bipolar and AII amacrine cells as well as between a few cone bipolar and ganglion cell processes believed to be derived from both transplant and host retinas.286 Identification of graft versus host cells was made based on location, however, not on specific cell markers. Under similar transplant conditions, a sub-population of wide-field amacrine cells expressing neuronal nitric oxide synthase was found to extend processes from the graft into the host inner plexiform layer (IPL), but no direct contacts were observed.323 Synaptic contacts between graft and host retinas are suggested by trans-synaptic tracing studies from the host brain to the transplant. While the previous studies indicate some degree of both graft and host neuronal plasticity, none has provided clear morphological evidence that synaptic connectivity, adequate to relay visual perceptions, was established between the graft and the host following transplantation.
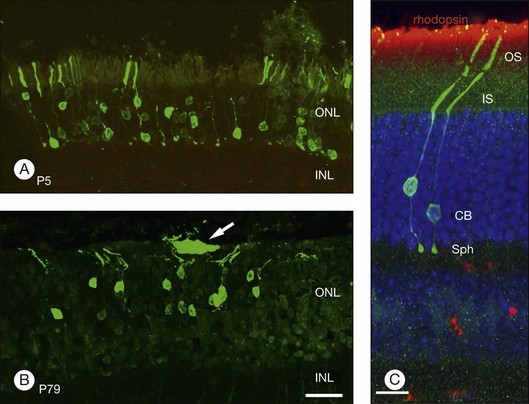
In an important study, MacLaren showed that if donor cells isolated from developing retina at the height of rod development were used for transplantation, integration can occur even if the host has a mature retina.324 Using rds, rd, and rhodopsin knockout mouse models, MacLaren and colleagues demonstrated successful integration of post-mitotic rod precursors by showing the presence of spherule synapse in donor cells (an essential component to communicate with inner retina), ganglion cell recordings in low light intensities that stimulate rods, and by restoration of pupillary light reflex. However, the cells were not morphologically mature even though they expressed photoreceptor markers. Similarly, when donor tissue was used during the development when retina was rich in rod-committed cells, similar integration into the mature retina was seen after transplantation.325 As noted above, this integration is improved when outer limiting membrane is disrupted.321,322 More recently, Gust and Reh have shown that age of the donor tissue is not as critical and that adult photoreceptors were able to integrate into the host retina but had a higher transplant failure rate (Fig. 125.3).255 Whether such integration can occur with human tissues or not remains to be seen.
Although, significant restoration of vision in humans after photoreceptor transplantation has not been documented (see below), there is some indication of improved visual function in laboratory animals such as the RCS rat.326 Recovery of visual function is the goal of retinal transplantation, but it does not constitute evidence of graft–host synaptic connectivity sufficient to reestablish retinal neural circuitry since visual recovery might be due to a “rescue” effect rather than a “replacement” effect. Furthermore, accurate evaluation of visual function is a complex task, particularly in experimental animals. Simple reflexes, electrophysiological testing, and visually guided behaviors have been used to evaluate visual function in laboratory animals. Silverman and coworkers276 reported that visually evoked cortical potentials could be recorded over the retinotopic area that corresponded to the transplant. Light-elicited retinal ganglion cell responses and local light-driven electroretinograms (ERGs) have been recorded from host retina overlying transplants.278 Visual-evoked potentials (VEPs) from areas of the host superior colliculus topographically corresponding to the transplant have also been recorded.259 Recovery of the pupillary light reflex276 as well as behavioral correlates, such as light-flash inhibition of the startle reflex273 and light/dark preference behaviors,277 have also demonstrated some degree of visual function restoration in animal models of retinal degeneration following photoreceptor transplantation. While the results of these tests are promising, the level of elicited responses was often less than that of normal controls.276,278 Moreover, it is not clear that synaptic interactions between the transplant and host underlie the observed improvement.277 A confounding factor in these experiments is the possible trophic effect of the transplant,263,264,279 and, to a lesser extent, of the surgery itself as a form of injury,309,311,312 both of which have been shown to enhance residual host photoreceptor cell survival. A further difficulty is that the validity of some of these tests as accurate measures of visual function remains controversial. For example, Kovalevsky and coworkers327 found no correlation between the intensity of the pupillary light reflex and the number of photoreceptor cells present in the host retina. This result limits the validity of the pupillary light reflex as an accurate tool for evaluating the extent of photoreceptor repopulation or the formation of functional contacts sufficient for the recovery of visual function following retinal transplantation.
Stem cells in photoreceptor transplantation
Given that the brain and retina are derived from the neurectoderm and that immature neuronal and progenitor cells are intrinsically capable of migrating and differentiating during neural development, it would appear that brain-derived neural progenitor cells could potentially differentiate into photoreceptors in the subretinal space. Several groups have examined this possibility328–331 and noted that there was limited integration of neural progenitor cells into adult host retina,329 but migration of transplanted cells was observed in all layers of a developing immature retina. However, these integrated cells did not express markers of mature retinal cells.328,331,332 Perhaps this result reflects the fact that the lineage is restricted to brain-derived neural cells.333
Alternatively, retinal progenitors isolated from developing retina would not have the above-mentioned lineage restriction. Transplantation of such cells isolated from embryonic retina into young transgenic S334ter rats, an RP model, showed grafted cells forming a multi-layered cellular sheet in the subretinal space and expressing retina-specific neuronal differentiation markers, e.g., recoverin and rhodopsin. Studies by MacLaren and Bartsch mentioned above also demonstrated survival, integration, and expression of mature photoreceptor markers when cells isolated at the peak of rod development were transplanted into the adult retina.325,334 However, isolation of such cells from developing human retina would be unethical.335
Retinal progenitor cells exist in lower vertebrates in the ciliary margin zone and similar cells have been isolated from adult human eyes.193,195 Thus, human donor eyes could be used for isolation of retinal progenitor cells. These cells grow as neurospheres in culture and give rise to both glial and neural cells. Only a small fraction of cells express mature retinal cell types, however, and they do not completely differentiate into functional retinal cells.336,337 These cells can be induced in culture by retroviral transduction of photoreceptor-specific transcription factors to express molecules such as transducin and recoverin, and they differentiate into light-sensitive rod phenotypes.338,339 When transplanted into adult normal or degenerate retina, results have varied with some studies showing no differentiation or expression of mature retinal markers340 and others showing differentiation of the cells with expression of mature retinal markers but without expressing mature retinal cell morphology or integration into host retina.328,341–343 An additional problem with these cells is that they exhibit limited self-renewal in culture.336,344
Müller cells are another potential source of stem cells.345 Müller cells from mammalian retina possess stem-cell-like properties of growing in neurospheres and expression of neural stem cell markers, and they differentiate and express mature retinal markers including peripherin and opsin.346,347 A spontaneously immortalized Müller cell line has been reported by Limb and coworkers.348 Whether these cells can form fully differentiated retinal neurons is not known although, in contrast to retinal progenitor cells, they do not seem to have limited self-renewal. Transplants of Müller-cell-derived stem cells have shown limited integration,346 but treatment of the host retina with chondroitinase (to break down proteoglycans prior to transplantation) resulted in better integration.349
Embryonic stem cells can be cultured indefinitely and can be induced to differentiate into cell lineages of the three germ layers. Culture of these cells requires the use of animal serum or co-culture with animal-derived cells. Approval of use of such cells would be problematic due to potential contamination.335 However, successful culture of human ESCs has been achieved without use of such reagents, and cell lines have been established.350,351 Two groups have developed efficient protocols for producing mature retinal cells from ESCs.213,214,352 Transplants of precursors derived from ESCs via techniques prior to those developed by Lamba or Osakada have been shown to rescue photoreceptors in RCS rats353 or migrate into rabbit retina and express S-opsin and rhodopsin.354 It is not known if the newer techniques of deriving mature human retinal cells create cells that are able to survive, integrate, and function to a greater degree following transplantation. Early experiments in mice have been promising. ESC-derived retinal cells survived and were able to form functional photoreceptors after transplantation in Crx−/− mice,260 a gene knockout animal model of Leber congenital amaurosis that is characterized by reduction or loss of components of phototransduction cycle including rhodopsin, cone opsin, transducin, cone arrestin, and recoverin. Thus, these mice do not have any ERG response. After transplantation, a small ERG response was detected. Human ESCs also replace photoreceptors in mnd mice257 and in rd1 mice.256
Induced pluripotent stem cells (iPSCs) may be a good source of genetically matched stem cells whose isolation may not have the ethical complexity associated with the use of embryonic or fetal tissues. However, the efficiency of production is very poor. Studies have reported on iPSCs being induced to differentiate into photoreceptors as well as the effect of transplantation.355,356 Zhou and coworkers subjected pig iPSCs to a rod differentiation protocol and culture that resulted in rod-like cells that expressed rhodopsin and rod outer-segment-specific membrane protein 1.357 These differentiated cells, when transplanted into the subretinal space of pigs treated with iodoacetic acid to eliminate photoreceptors, were noted in the outer nuclear layer, but only a few of the cells expressed projections resembling outer segments at 3 weeks. Similarly, Lamba and coworkers showed that such cells were able to integrate into mouse retina.355
Finally, following reports of bone-marrow-derived stem cells integrating into retina and differentiating into photoreceptors in rat eyes,358 attempts are being made to induce these cells to develop into retinal cells in vitro.359,360 Injection of bone marrow-derived mesenchymal stem cells into the subretinal space appears to have a rescue effect on photoreceptors.230,231 However, this may be more of a trophic effect than a result of transplanted cells differentiating into photoreceptors. As noted above, intravitreal bone-marrow-derived lineage-negative hematopoietic stem cells rescue photoreceptors (primarily cones) in rd1 and rd10 mice.279
Results of photoreceptor transplants in humans
Neural retinal transplantation has been performed in human volunteers with RP and AMD.118,258,315–318 In these studies pre- and postoperative visual acuity and evaluation of retinal function were done using a variety of psychophysical, electrophysiological, and clinical tests, including macular perimetry, full-field and focal ERGs, fundus examination, fundus photography, and fluorescein angiography. Kaplan and coworkers118 reported the transplantation of vibratome-sectioned adult cadaveric photoreceptor sheets into the subretinal space of two patients with advanced RP and visual acuity of no light perception (NLP). The patients were not immune-suppressed and showed no signs of graft rejection (e.g., focal chorioretinitis, macular edema, vitritis) for up to 12 months after surgery. However, no improvement of visual function was attained. Das and coworkers315 transplanted fetal neuroretinal cells into the subretinal space of one eye in 14 RP patients, temporal or superotemporal to the macula. Patients were followed for as long as 44 months after surgery with no apparent signs of rejection in the absence of immune suppression. Visual improvement was reported in five transplant recipients but was based solely on subjective testing (i.e., improved visual acuity in five patients and a detectable narrow visual field in two patients). Moreover, these patients had some degree of visual perception preoperatively, which may have been underestimated resulting in an apparent increase in visual function after surgery. No improvement of results from more objective tests, such as a full-field ERG or VEPs, was observed. In another study, two patients with advanced RP received subretinal transplants of intact fetal retina.361 Patients were followed for 12 months after surgery with no clinical signs of rejection. Both patients reported subjective visual improvement (new visual sensations), and one patient showed a transient faint positive response during a multifocal ERG that was later undetectable. The same authors co-transplanted sheets of fetal retina together with the adjacent RPE unilaterally into the subretinal space near the fovea in five RP patients with visual acuity of light perception (LP).317 After 6 months follow-up, there was no evidence of rejection, but no visual improvement was observed. This group subsequently reported on ten patients (six RP, four AMD) who received retinal implants in one eye and were followed in a phase II trial conducted in a clinical practice setting.258 Seven patients (three RP, four AMD) showed improved EDTRS visual acuity scores. Three of these patients (one RP, two AMD) showed improvement in both eyes to the same extent. In one RP patient, vision remained the same, but in two RP patients, vision decreased. One RP patient maintained an improvement in vision from 20/800 to 20/320 at the 6-year follow-up, while the non-surgery eye had deteriorated to hand motion vision. This patient also showed a 23% increase in light sensitivity at 5 years compared to microperimetry results at 2 years; the other patients showed no improved sensitivity. Although no HLA match was found between donors and recipients, no rejection of the implanted tissue was observed clinically. Humayun and coworkers316 transplanted fetal retinal microaggregates into the subretinal space of eight patients with advanced RP and visual acuity of LP, as well as a fetal retinal sheet plus microaggregates in one patient (in two separate locations) with advanced neovascular AMD and visual acuity of NLP. Patients were followed for as long as 13 months after surgery with no signs of rejection in the absence of immune suppression. Three patients showed a decline in visual function, and three others showed a transient improvement. The patient with AMD died from an unrelated cause 3 years after receiving the transplant. Ultrastructural and immunocytochemical studies of the eye revealed survival of at least some of the transplanted cells in the subretinal space with no signs of inflammation.319 Although the host retina and transplant were separated in some areas by a fibrocellular membrane composed of Müller cell processes, cellular debris, and collagen, a few GABA-positive and synaptophysin-positive cell processes extended between the transplant and the host retina. However, no synaptic contacts involving these processes were found. In another study, adult human cadaveric photoreceptor sheets harvested with the excimer laser were transplanted into eight patients with advanced RP.318 Subjects were followed for 12 months, but no improvement in visual function was recorded. Siqueir and coworkers362 reported the results of a prospective phase I, nonrandomized open-label study of autologous bone-marrow-derived mononuclear cell transplants in RP patients with best-corrected ETDRS visual acuity worse than 20/200. Patients (three with RP, two with cone–rod dystrophy) received intravitreal injection of 104 autologous bone marrow-derived stem cells/0.1 mL, 3–3.5 mm posterior to the limbus with a 27-gauge needle. No adverse effects occurred (e.g., teratoma, visual loss, intraocular inflammation), but there was no documented benefit (e.g., visual acuity, visual field, ERG, optical coherence tomography) at 10-months follow-up. While these studies have established the feasibility and safety of retinal transplantation in humans as well as the survival of the transplants, no clear improvement in visual function has been demonstrated.
Immune response to photoreceptor transplants
Photoreceptor cells are probably minimally immunogenic due to very low levels of expression of MCH class I molecules.363 Most neural retinal transplantation studies in animal models have employed allografts and, to a lesser extent, xenografts with or without host immunosuppression. In humans, allografts have been used almost exclusively.118,315–318,361 The occurrence of cell-mediated delayed hypersensitivity rejection following retinal transplantation was only reported in a few studies involving xenografts.295 Two distinct aspects of immune privilege may play a role in retinal transplantation. First, immature neural retina is immune privileged tissue,364 and second, the subretinal space is an immune privileged site.73 So far, it is not clear whether adult neural retina also behaves as immune privileged tissue.71 Neural progenitor cells behave like immune privileged tissue as well.365 As noted above, however, neural tissue derived from iPSCs may not be immune privileged because abnormal gene expression in some cells differentiated from iPSCs (both via a retroviral and episomal approach) can induce a T-cell-dependent immune response in a syngeneic recipient.205
Although the majority of neurons, including retinal neurons, do not express MHC molecules, they may express minor histocompatibility antigens.71 These antigens are not displayed in the absence of MHC class I expression, but if cells die, minor histocompatibility antigens can be released, phagocytosed by antigen-presenting cells, and presented to T-lymphocytes.71 Furthermore, retinal glial cells, including Müller cells and astrocytes, constitutively express class I and II MHC molecules.366,367 Neural retinal microglia may also play a major role in the expression of transplantation antigens. Microglia can localize in the lumen of graft rosettes as well as within and surrounding retinal grafts, especially those undergoing rejection.368 Ma and Streilein369 have shown that these cells become activated after transplantation of neural retina into the subretinal space and display upregulated expression of both class I and class II MHC molecules. Donor-derived, activated microglia are thought to act as antigen-presenting cells that initially induce ACAID in their recipients following subretinal transplantation but eventually mediate an atypical delayed hypersensitivity reaction in which slow rejection of the graft takes place instead of the acute graft rejection seen in typical delayed hypersensitivity.
As noted previously, the immune privilege of the subretinal space is not absolute. Whereas gross infiltration by lymphocytes is seen rarely after transplantation into the subretinal space and graft survival is prolonged, tissue rejection may still occur.83,364,370 The underlying mechanism of graft rejection in this immune privileged site is not understood fully. However, it appears to involve an unconventional pattern of immune response that comprises only minimal lymphoreticular cell infiltration with gradual rather than acute graft deterioration.364 Exposure of the subretinal space to systemic immunity, which occurs if the integrity of the blood–retina barrier is compromised due to surgery and/or disease such as RP or AMD, can contribute to graft rejection.83 Breach of the host blood–retina barrier may allow MHC-expressing cells, e.g., dendritic cells from the systemic circulation, to invade the subretinal space and present the transplant antigens to T-lymphocytes. Immune suppression may be needed to prolong the survival of the graft.370
Photoreceptor transplantation: future directions
Studies in experimental animals and humans indicate that absence or paucity of graft–host synaptic interactions remains an unsolved obstacle to successful neural retinal replacement. Reactive glial cells may constitute a barrier between the host retina and the grafted cells.320 Following retinal transplantation, astrocytes and Müller cells become reactive and upregulate expression of the intermediate filament proteins, glial fibrillary acid protein (GFAP), and vimentin.287 Reactive gliosis can create a barrier that separates the area of injury from surrounding healthy tissue,371 which may explain the consistent finding that neuritic processes extend between the graft and the host only in areas of the transplant where the glial barrier is absent274,286 or where the host outer limiting membrane, rich in adherens junctions involving Müller cell processes, is disrupted.320 In an in vitro study, neuronal cell processes extended between two pieces of retina placed adjacent to each other in culture. However, processes did not appear to grow if they encountered glial structures such as the outer or inner limiting membranes.320 In contrast, robust neurite outgrowth was seen in immature isolated retinal cells transplanted into the subretinal space of mutant mice deficient in GFAP and vimentin.372 Disruption of outer limiting membrane has been shown to improve graft integration.321,322 Similarly, breaking down the glial barrier using matrix metalloproteinase delivered through biodegradable polymers also increases graft migration into host retina.373
Alternatively, scarcity of graft–host synaptic integration may involve factors intrinsic to the transplant itself. Retinal neurons, including photoreceptors, display synaptic plasticity during injury or disease243,374–378 and in culture.379,380 This potential for structural plasticity can be geared towards enhancing synaptic interactions between the graft and the host after transplantation. Photoreceptor sheets prepared by vibratome-sectioning undergo significant morphological changes, including rapid retraction of axonal terminals towards their cell bodies in culture.381 This phenomenon has also been seen in studies of experimental retinal detachment170,374 and may be due to separation of photoreceptors from adjacent RPE cells. Retraction of photoreceptor terminals is likely to interfere with synaptic integration following transplantation since physical proximity is required for synaptogenesis to occur between pre- and postsynaptic elements. One can prevent retraction by increasing intracellular cAMP levels in photoreceptor sheets,382 or by inhibiting RhoA,383,384 which may help improve synaptic interactions between pre-treated photoreceptor grafts and host bipolar and horizontal cells.
Identification of the optimal parameters for successful retinal transplantation, such as the source of cells (e.g., human ESCs versus iPSCs versus mature dispersed photoreceptors) or the surgical procedure (e.g., sheets on a biodegradable membrane versus dispersed cells), as well as resolving issues of tissue rejection are important goals, regardless of whether the objective of surgery is “rescue” or “replacement.” We anticipate tremendous progress in generating photoreceptors from stem cells (induced pluripotent or embryonic or bone marrow) and identification of which cell is most capable of integrating and differentiating in a diseased microenvironment during the next decade. Restoration of a functional neural retinal network capable of mediating visual perception, which is a goal unique to “replacement” therapy, depends primarily on establishing graft–host synaptic integration sufficient to rectify the damaged neural circuitry resulting from cell loss. To achieve this goal, additional experimental studies (e.g., identification of molecules that foster synapse formation, development of means to unambiguously identify donor and recipient neurons, identifying methods to manage the synaptic rewiring that accompanies photoreceptor degeneration) and clinical studies must be done. In addition, more rigorous functional tests must be developed to aid in the accurate, objective assessment of incremental changes in vision (particularly for low levels of visual function) following retinal transplantation. Since photoreceptor death is associated with synaptic remodeling in the local retinal circuitry240,243,244,375,385 and may cause transneuronal cell death of inner retinal neurons, relatively early intervention (i.e., before foveal cone cell loss is profound) may play a significant role in the success of retinal transplantation.
1 Royo PE, Quay WB. Retinal transplantation from fetal to maternal mammalian eye. Growth. 1959;23:313–336.
2 Van Dooremaal JC. De onwikkeling van levende weefsels op vreemden bodem geent. Onderz Physiol Lab Rijksuniv Utrechtsche Hoogeschool. 1873;3:277–290.
3 del Cerro M, Gash DM, Rao GN, et al. Intraocular retinal transplants. Invest Ophthalmol Vis Sci. 1985;26:1182–1185.
4 Gouras P, Flood MT, Kjedbye H, et al. Transplantation of cultured human retinal epithelium to Bruch’s membrane of the owl monkey’s eye. Curr Eye Res. 1985;4:253–265.
5 Li LX, Turner JE. Inherited retinal dystrophy in the RCS rat: prevention of photoreceptor degeneration by pigment epithelial cell transplantation. Exp Eye Res. 1988;47:911–917.
6 D’Cruz PM, Yasumura D, Weir J, et al. Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum Mol Genet. 2000;9:645–651.
7 Lund RD, Hauschka SD. Transplanted neural tissue develops connections with host rat brain. Science. 1976;193:582–584.
8 Das GD, Hallas BH. Transplantation of brain tissue in the brain of adult rats. Experientia. 1978;34:1304–1306.
9 Klassen H, Lund RD. Retinal transplants can drive a pupillary reflex in host rat brains. Proc Natl Acad Sci U S A. 1987;84:6958–6960.
10 Coffey PJ, Lund RD, Rawlins JN. Detecting the world through a retinal implant. Progr Brain Res. 1990;82:269–275.
11 Lund RD, Radel JD, Coffey PJ. The impact of intracerebral retinal transplants on types of behavior exhibited by host rats. Trends Neurosci. 1991;14:358–362.
12 Turner JE, Blair JR. Newborn rat retinal cell transplanted into a retinal lesion site in adult host eyes. Dev Brain Res. 1986;26:91–104.
13 Silverman MS, Hughes SE. Transplantation of photoreceptors to light-damaged retina. Invest Ophthalmol Vis Sci. 1989;30:1684–1690.
14 Klein R, Klein BE, Linton KL. Prevalence of age-related maculopathy. The Beaver Dam Eye Study. Ophthalmology. 1992;99:933–943.
15 Ferris FL, 3rd., Fine SL, et al. Age-related macular degeneration and blindness due to neovascular maculopathy. Arch Ophthalmol. 1984;102:1640–1642.
16 Klein R, Klein BE, Jensen SC, et al. The five-year incidence and progression of age-related maculopathy: the Beaver Dam Eye Study. Ophthalmology. 1997;104:7–21.
17 Friedman DS, O’Colmain BJ, Munoz B, et al. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004;122:564–572.
18 Rosenfeld PJ, Brown DM, Heier JS, et al. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. 2006;355:1419–1431.
19 Brown DM, Kaiser PK, Michels M, et al. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med. 2006;355:1432–1444.
20 Hackett SF, Schoenfeld CL, Freund J, et al. Neurotrophic factors, cytokines and stress increase expression of basic fibroblast growth factor in retinal pigmented epithelial cells. Exp Eye Res. 1997;64:865–873.
21 Kanuga N, Winton HL, Beauchene L, et al. Characterization of genetically modified human retinal pigment epithelial cells developed for in vitro and transplantation studies. Invest Ophthalmol Vis Sci. 2002;43:546–555.
22 Sugino IK, Sun Q, Wang J, et al. Comparison of FRPE and human embryonic stem cell-derived rpe behavior on aged human Bruch’s membrane. Invest Ophthalmol Vis Sci. 2011;52:4979–4997.
23 Kolomeyer AM, Sugino IK, Zarbin MA. Characterization of conditioned media collected from cultured adult versus fetal retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2011;52:5973–5986.
24 Kolomeyer AM, Sugino IK, Zarbin MA. Characterization of conditioned media collected from aged versus young human eye cups. Invest Ophthalmol Vis Sci. 2011;52:5963–5972.
25 Zarbin MA. Retinal pigment epithelium-retina transplantation for retinal degenerative disease. Am J Ophthalmol. 2008;146:151–153.
26 Maaijwee K, Joussen AM, Kirchhof B, et al. Retinal pigment epithelium (RPE)-choroid graft translocation in the treatment of an RPE tear: preliminary results. Br J Ophthalmol. 2008;92:526–529.
27 Chang LK, Sarraf D. Tears of the retinal pigment epithelium: an old problem in a new era. Retina. 2007;27:523–534.
28 Nasir MA, Sugino I, Zarbin MA. Decreased choriocapillaris perfusion following surgical excision of choroidal neovascular membranes in age-related macular degeneration. Br J Ophthalmol. 1997;81:481–489.
29 Grossniklaus HE, Hutchinson AK, Capone A, et al. Clinicopathologic features of surgically excised choroidal neovascular membranes. Ophthalmology. 1994;101:1099–1111.
30 Coffey PJ, Girman S, Wang SM, et al. Long-term preservation of cortically dependent visual function in RCS rats by transplantation. Nat Neurosci. 2002;5:53–56.
31 Li L, Turner JE. Optimal conditions for long-term photoreceptor cell rescue in RCS rats: the necessity for healthy RPE transplants. Exp Eye Res. 1991;52:669–679.
32 Little CW, Castillo B, DiLoreto DA, et al. Transplantation of human fetal retinal pigment epithelium rescues photoreceptor cells from degeneration in the Royal College of Surgeons rat retina. Invest Ophthalmol Vis Sci. 1996;37:204–211.
33 Lund RD, Adamson P, Sauve Y, et al. Subretinal transplantation of genetically modified human cell lines attenuates loss of visual function in dystrophic rats. Proc Natl Acad Sci U S A. 2001;98:9942–9947.
34 Lawrence JM, Keegan DJ, Muir EM, et al. Transplantation of Schwann cell line clones secreting GDNF or BDNF into the retinas of dystrophic Royal College of Surgeons rats. Invest Ophthalmol Vis Sci. 2004;45:267–274.
35 Lawrence JM, Sauve Y, Keegan DJ, et al. Schwann cell grafting into the retina of the dystrophic RCS rat limits functional deterioration. Invest Ophthalmol Vis Sci. 2000;41:518–528.
36 Lu B, Malcuit C, Wang S, et al. Long-term safety and function of RPE from human embryonic stem cells in preclinical models of macular degeneration. Stem Cells. 2009;27:2126–2135.
37 Sheedlo HJ, Li L, Barnstable CJ, et al. Synaptic and photoreceptor components in retinal pigment epithelial cell transplanted retinas of Royal College of Surgeons dystrophic rats. J Neurosci Res. 1993;36:423–431.
38 Sauve Y, Lu B, Lund RD. The relationship between full field electroretinogram and perimetry-like visual thresholds in RCS rats during photoreceptor degeneration and rescue by cell transplants. Vis Res. 2004;44:9–18.
39 Whiteley SJ, Litchfield TM, Coffey PJ, et al. Improvement of the pupillary light reflex of Royal College of Surgeons rats following RPE cell grafts. Exp Neurol. 1996;140:100–104.
40 Lund RD, Wang S, Klimanskaya I, et al. Human embryonic stem cell-derived cells rescue visual function in dystrophic RCS rats. Cloning Stem Cells. 2006;8:189–199.
41 McGill TJ, Lund RD, Douglas RM, et al. Preservation of vision following cell-based therapies in a model of retinal degenerative disease. Vis Res. 2004;44:2559–2566.
42 Mruthyunjaya P, Stinnett SS, Toth CA. Change in visual function after macular translocation with 360 degrees retinectomy for neovascular age-related macular degeneration. Ophthalmology. 2004;111:1715–1724.
43 Pieramici DJ, De Juan E, Jr., Fujii GY, et al. Limited inferior macular translocation for the treatment of subfoveal choroidal neovascularization secondary to age-related macular degeneration. Am J Ophthalmol. 2000;130:419–428.
44 Peyman GA, Blinder KJ, Paris CL, et al. A technique for retinal pigment epithelium transplantation for age- related macular degeneration secondary to extensive subfoveal scarring. Ophthal Surg. 1991;22:102–108.
45 Algvere PV, Berglin L, Gouras P, et al. Transplantation of fetal retinal pigment epithelium in age-related macular degeneration with subfoveal neovascularization. Graefes Arch Clin Exp Ophthalmol. 1994;232:707–716.
46 Algvere PV, Berglin L, Gouras P, et al. Transplantation of RPE in age-related macular degeneration: observations in disciform lesions and dry RPE atrophy. Graefes Arch Clin Exp Ophthalmol. 1997;235:149–158.
47 Stanga PE, Kychenthal A, Fitzke FW, et al. Retinal pigment epithelium translocation after choroidal neovascular membrane removal in age-related macular degeneration. Ophthalmology. 2002;109:1492–1498.
48 Binder S, Krebs I, Hilgers RD, et al. Outcome of transplantation of autologous retinal pigment epithelium in age-related macular degeneration: a prospective trial. Invest Ophthalmol Vis Sci. 2004;45:4151–4160.
49 Angunawela RI, Williamson TH, Khan MA, et al. Choroidal translocation with a pedicle following excision of a type 1 choroidal neovascular membrane. Br J Ophthalmol. 2005;89:386.
50 van Meurs JC, Van Den Biesen PR. Autologous retinal pigment epithelium and choroid translocation in patients with exudative age-related macular degeneration: short-term follow-up. Am J Ophthalmol. 2003;136:688–695.
51 MacLaren RE, Uppal GS, Balaggan KS, et al. Autologous transplantation of the retinal pigment epithelium and choroid in the treatment of neovascular age-related macular degeneration. Ophthalmology. 2007;114:561–570.
52 Joussen AM, Heussen FM, Joeres S, et al. Autologous translocation of the choroid and retinal pigment epithelium in age-related macular degeneration. Am J Ophthalmol. 2006;142:17–30.
53 Treumer F, Bunse A, Klatt C, et al. Autologous retinal pigment epithelium-choroid sheet transplantation in age related macular degeneration: morphological and functional results. Br J Ophthalmol. 2007;91:349–353.
54 Falkner-Radler CI, Krebs I, Glittenberg C, et al. Human retinal pigment epithelium (RPE) transplantation: outcome after autologous RPE-choroid sheet and RPE cell-suspension in a randomised clinical study. Br J Ophthalmol. 2011;95:370–375.
55 Del Priore LV, Kaplan HJ, Tezel TH, et al. Retinal pigment epithelial cell transplantation after subfoveal membranectomy in age-related macular degeneration: clinicopathologic correlation. Am J Ophthalmol. 2001;131:472–480.
56 Algvere PV, Gouras P, Dafgard Kopp E. Long-term outcome of RPE allografts in non-immunosuppressed patients with AMD. Eur J Ophthalmol. 1999;9:217–230.
57 MacLaren RE, Bird AC, Sathia PJ, et al. Long-term results of submacular surgery combined with macular translocation of the retinal pigment epithelium in neovascular age-related macular degeneration. Ophthalmology. 2005;112:2081–2087.
58 Tezel TH, Del Priore LV, Kaplan HJ. Reattachment of harvested retinal pigment epithelium to a substrate prevents apoptosis. Graefes Arch Clin Exp Ophthalmol. 1997;235:41–47.
59 Sheng Y, Gouras P, Cao H, et al. Patch transplants of human fetal retinal pigment epithelium in rabbit and monkey retina. Invest Ophthalmol Vis Sci. 1995;36:381–390.
60 Li LX, Turner JE. Transplantation of retinal pigment epithelial cells to immature and adult rat hosts: short- and long-term survival characteristics. Exp Eye Res. 1988;47:771–785.
61 Lopez R, Gouras P, Kjeldbye H, et al. Transplanted retinal pigment epithelium modifies the retinal degeneration in the RCS rat. Invest Ophthalmol Vis Sci. 1989;30:586–588.
62 Lai CC, Gouras P, Doi K, et al. Tracking RPE transplants labeled by retroviral gene transfer with green fluorescent protein. Invest Ophthalmol Vis Sci. 1999;40:2141–2146.
63 Karwatowski WS, Jeffries TE, Duance VC, et al. Preparation of Bruch’s membrane and analysis of the age-related changes in the structural collagens. Br J Ophthalmol. 1995;79:944–952.
64 Pauleikhoff D, Harper CA, Marshall J, et al. Aging changes in Bruch’s membrane. A histochemical and morphologic study. Ophthalmology. 1990;97:171–178.
65 Zarbin MA. Age related macular degeneration: review of pathogenesis. Eur J Ophthalmol. 1998;8:133–205.
66 Zarbin MA. Current concepts in the pathogenesis of age-related macular degeneration. Arch Ophthalmol. 2004;122:598–614.
67 Tsukahara I, Ninomiya S, Castellarin A, et al. Early attachment of uncultured retinal pigment epithelium from aged donors onto Bruch’s membrane explants. Exp Eye Res. 2002;74:255–266.
68 Zarbin MA. Analysis of retinal pigment epithelium integrin expression and adhesion to aged submacular human Bruch’s membrane. Trans Am Ophthalmol Soc. 2003;101:499–520.
69 Gullapalli VK, Sugino IK, Van Patten Y, et al. Impaired RPE survival on aged submacular human Bruch’s membrane. Exp Eye Res. 2005;80:235–248.
70 Sugino IK, Gullapalli VK, Sun Q, et al. Cell-deposited matrix improves retinal pigment epithelium survival on aged submacular human Bruch’s membrane. Invest Ophthalmol Vis Sci. 2011;52:1345–1358.
71 Streilein JW. Ocular immune privilege: the eye takes a dim but practical view of immunity and inflammation. J Leukoc Biol. 2003;74:179–185.
72 Streilein JW, Ksander BR, Taylor AW. Immune deviation in relation to ocular immune privilege. J Immunol. 1997;158:3557–3560.
73 Jiang LQ, Jorquera M, Streilein JW. Subretinal space and vitreous cavity as immunologically privileged sites for retinal allografts. Invest Ophthalmol Vis Sci. 1993;34:3347–3354.
74 Ferguson TA. The molecular basis of anterior associated immune deviation (ACAID). Ocul Immunol Inflamm. 1997;5:213–215.
75 Takeuchi M, Alard P, Streilein JW. TGF-beta promotes immune deviation by altering accessory signals of antigen-presenting cells. J Immunol. 1998;160:1589–1597.
76 Taylor AW, Streilein JW, Cousins SW. Identification of alpha-melanocyte stimulating hormone as a potential immunosuppressive factor in aqueous humor. Curr Eye Res. 1992;11:1199–1206.
77 Taylor AW, Streilein JW, Cousins SW. Immunoreactive vasoactive intestinal peptide contributes to the immunosuppressive activity of normal aqueous humor. J Immunol. 1994;153:1080–1086.
78 Apte RS, Sinha D, Mayhew E, et al. Cutting edge: role of macrophage migration inhibitory factor in inhibiting NK cell activity and preserving immune privilege. J Immunol. 1998;160:5693–5696.
79 Griffith TS, Brunner T, Fletcher SM, et al. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science. 1995;270:1189–1192.
80 Bora NS, Gobleman CL, Atkinson JP, et al. Differential expression of the complement regulatory proteins in the human eye. Invest Ophthalmol Vis Sci. 1993;34:3579–3584.
81 Vega JL, Keino H, Masli S. Surgical denervation of ocular sympathetic afferents decreases local transforming growth factor-beta and abolishes immune privilege. Am J Pathol. 2009;175:1218–1225.
82 Streilein JW. Unraveling immune privilege. Science. 1995;270:1158–1159.
83 Wenkel H, Streilein JW. Analysis of immune deviation elicited by antigens injected into the subretinal space. Invest Ophthalmol Vis Sci. 1998;39:1823–1834.
84 Jiang L, Streilein JW. Immunologic privilege evoked by histoincompatible intracameral retinal transplants. Reg Immunol. 1991;3:121–130.
85 Liversidge J, Forrester JV. Regulation of immune responses by the retinal pigment epithelium. In: Marmor MF, Wolfensberger TJ. The retinal pigment epithelium function and disease. New York: Oxford University Press; 1998:511–527.
86 Tanihara H, Yoshida M, Matsumoto M, et al. Identification of transforming growth factor B expressed in cultured human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 1993;34:413–419.
87 Holtkamp GM, de Vos AF, Kijlstra A, et al. Expression of multiple forms of IL-1 receptor antagonist (IL-1ra) by human retinal pigment epithelial cells: identification of a new IL-1ra exon. Eur J Immunol. 1999;29:215–224.
88 Ishida K, Panjwani N, Cao Z, et al. Participation of pigment epithelium in ocular immune privilege. 3. Epithelia cultured from iris, ciliary body, and retina suppress T-cell activation by partially non-overlapping mechanisms. Ocul Immunol Inflamm. 2003;11:91–105.
89 Kaestel CG, Jorgensen A, Nielsen M, et al. Human retinal pigment epithelial cells inhibit proliferation and IL2R expression of activated T cells. Exp Eye Res. 2002;74:627–637.
90 Jorgensen A, Wiencke AK, la Cour M, et al. Human retinal pigment epithelial cell-induced apoptosis in activated T cells. Invest Ophthalmol Vis Sci. 1998;39:1590–1599.
91 Rezai KA, Semnani RT, Farrokh-Siar L, et al. Human fetal retinal pigment epithelial cells induce apoptosis in allogenic T-cells in a fas ligand and PGE2 independent pathway. Curr Eye Res. 1999;18:430–439.
92 Usui Y, Okunuki Y, Hattori T, et al. Functional expression of B7H1 on retinal pigment epithelial cells. Exp Eye Res. 2008;86:52–59.
93 Sugita S, Usui Y, Horie S, et al. T-cell suppression by programmed cell death 1 ligand 1 on retinal pigment epithelium during inflammatory conditions. Invest Ophthalmol Vis Sci. 2009;50:2862–2870.
94 Sugita S, Horie S, Nakamura O, et al. Retinal pigment epithelium-derived CTLA-2alpha induces TGFbeta-producing T regulatory cells. J Immunol. 2008;181:7525–7536.
95 Wenkel H, Streilein JW. Evidence that retinal pigment epithelium functions as an immune- privileged tissue. Invest Ophthalmol Vis Sci. 2000;41:3467–3473.
96 Zamiri P, Zhang Q, Streilein JW. Vulnerability of allogeneic retinal pigment epithelium to immune T-cell-mediated damage in vivo and in vitro. Invest Ophthalmol Vis Sci. 2004;45:177–184.
97 Rezai KA, Semnani RT, Patel SC, et al. The immunogenic potential of human fetal retinal pigment epithelium and its relation to transplantation. Invest Ophthalmol Vis Sci. 1997;38:2662–2671.
98 Liversidge JM, Sewell HF, Forrester JV. Human retinal pigment epithelial cells differentially express MHC class II (HLA, DP, DR and DQ) antigens in response to in vitro stimulation with lymphokine or purified IFN-gamma. Clin Exp Immunol. 1988;73:489–494.
99 Liversidge J, Sewell HF, Thomson AW, et al. Lymphokine-induced MHC class II antigen expression on cultured retinal pigment epithelial cells and the influence of cyclosporin A. Immunology. 1988;63:313–317.
100 Percopo CM, Hooks JJ, Shinohara T, et al. Cytokine-mediated activation of a neuronal retinal resident cell provokes antigen presentation. J Immunol. 1990;145:4101–4107.
101 Sun D, Enzmann V, Lei S, et al. Retinal pigment epithelial cells activate uveitogenic T cells when they express high levels of MHC class II molecules, but inhibit T cell activation when they express restricted levels. J Neuroimmunol. 2003;144:1–8.
102 Leonard DS, Zhang XG, Panozzo G, et al. Clinicopathologic correlation of localized retinal pigment epithelium debridement. Invest Ophthalmol Vis Sci. 1997;38:1094–1109.
103 Del Priore LV, Kaplan HJ, Hornbeck R, et al. Retinal pigment epithelial debridement as a model for the pathogenesis and treatment of macular degeneration. Am J Ophthalmol. 1996;122:629–643.
104 Anderson DH, Radeke MJ, Gallo NB, et al. The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog Retin Eye Res. 2010;29:95–112.
105 Jiang LQ, Streilein JW. Neural retina and retinal pigment epithelium allografts suffer different immunological fates in the eye. Reg Immunol. 1994;6:90–94.
106 Grisanti S, Ishioka M, Kosiewicz M, et al. Immunity and immune privilege elicited by cultured retinal pigment epithelial cell transplants. Invest Ophthalmol Vis Sci. 1997;38:1619–1626.
107 Zhang X, Bok D. Transplantation of retinal pigment epithelial cells and immune response in the subretinal space. Invest Ophthalmol Vis Sci. 1998;39:1021–1027.
108 Weisz JM, Humayun MS, De Juan E, et al. Allogenic fetal retinal pigment epithelial cell transplant in a patient with geographic atrophy. Retina. 1999;19:540–545.
109 Kohen L, Enzmann V, Faude F, et al. Mechanisms of graft rejection in the transplantation of retinal pigment epithelial cells. Ophthal Res. 1997;29:298–304.
110 Berglin L, Gouras P, Sheng Y, et al. Tolerance of human fetal retinal pigment epithelium xenografts in monkey retina. Graefes Arch Clin Exp Ophthalmol. 1997;235:103–110.
111 Crafoord S, Algvere P, et al. Long-term outcome of RPE allografts to the subretinal space of rabbits. Seregard S. Acta Ophthalmol Scand. 1999;77:247–254.
112 Lai CC, Gouras P, Doi K, et al. Local immunosuppression prolongs survival of RPE xenografts labeled by retroviral gene transfer. Invest Ophthalmol Vis Sci. 2000;41:3134–3141.
113 Ishida M, Lui GM, Yamani A, et al. Culture of human retinal pigment epithelial cells from peripheral scleral flap biopsies. Curr Eye Res. 1998;17:392–402.
114 Boulton M, Roanowska M, Wess T. Ageing of the retinal pigment epithelium: implications for transplantation. Graefes Arch Clin Exp Ophthalmol. 2003;242:76–84.
115 Cahill MT, Mruthyunjaya P, Bowes Rickman C, et al. Recurrence of retinal pigment epithelial changes after macular translocation with 360 degrees peripheral retinectomy for geographic atrophy. Arch Ophthalmol. 2005;123:935–938.
116 Ogata N, Nishizawa M, Ando A, et al. Transfection .of basic fibroblast growth factor (bFGF) gene or bFGF antisense gene into human retinal pigment epithelial cells. Graefes Arch Clin Exp Ophthalmol. 1999;237:678–684.
117 Lund RD, Kwan AS, Keegan DJ, et al. Cell transplantation as a treatment for retinal disease. Prog Retin Eye Res. 2001;20:415–449.
118 Kaplan HJ, Tezel TH, Berger AS, et al. Human photoreceptor transplantation in retinitis pigmentosa. Arch Ophthalmol. 1997;115:1168–1172.
119 Del Priore LV, Ishida O, Johnson EW, et al. Triple immune suppression increases short-term survival of porcine fetal retinal pigment epithelium xenografts. Invest Ophthalmol Vis Sci. 2003;44:4044–4053.
120 Tezel TH, Del Priore LV. Reattachment to a substrate prevents apoptosis of human retinal pigment epithelium. Graefes Arch Clin Exp Ophthalmol. 1997;235:41–47.
121 Castellarin AA, Sugino IK, Vargas JA, et al. In vitro transplantation of fetal human retinal pigment epithelial cells onto human cadaver Bruch’s membrane. Exp Eye Res. 1998;66:49–67.
122 Ho TC, Del Priore LV. Reattachment of cultured human retinal pigment epithelium to extracellular matrix and human Bruch’s membrane. Invest Ophthalmol Vis Sci. 1997;38:1110–1118.
123 Tezel TH, Kaplan HJ, Del Priore LV. Fate of human retinal pigment epithelial cells seeded onto layers of human Bruch’s membrane. Invest Ophthalmol Vis Sci. 1999;40:467–476.
124 Tezel TH, Del Priore LV. Repopulation of different layers of host human Bruch’s membrane by retinal pigment epithelial cell grafts. Invest Ophthalmol Vis Sci. 1999;40:767–774.
125 Del Priore LV, Tezel TH. Reattachment rate of human retinal pigment epithelium to layers of human Bruch’s membrane. Arch Ophthalmol. 1998;116:335–341.
126 Gullapalli VK, Sugino IK, Zarbin MA. Culture-induced increase in alpha integrin subunit expression in retinal pigment epithelium is important for improved resurfacing of aged human Bruch’s membrane. Exp Eye Res. 2008;86:189–200.
127 Starita C, Hussain AA, Pagliarini S, et al. Hydrodynamics of ageing Bruch’s membrane: implications for macular disease. Exp Eye Res. 1996;62:565–572.
128 Lopez PF, Grossniklaus HE, Lambert HM, et al. Pathologic features of surgically excised subretinal neovascular membranes in age-related macular degeneration. Am J Ophthalmol. 1991;112:647–656.
129 Hsu JK, Thomas MA, Ibanez H, et al. Clinicopathologic studies of an eye after submacular membranectomy for choroidal neovascularization. Retina. 1995;15:43–52.
130 Castellarin AA, Nasir MA, Sugino IK, et al. Clinicopathological correlation of primary and recurrent choroidal neovascularisation following surgical excision in age related macular degeneration. Br J Ophthalmol. 1998;82:480–487.
131 Ormerod LD, Puklin JE, Frank RN. Long-term outcomes after the surgical removal of advanced subfoveal neovascular membranes in age-related macular degeneration. Ophthalmology. 1994;101:1201–1210.
132 Rudolf M, Curcio CA. Esterified cholesterol is highly localized to Bruch’s membrane, as revealed by lipid histochemistry in wholemounts of human choroid. J Histochem Cytochem. 2009;57:731–739.
133 Thumann G, Bartz-Schmidt KU, El Bakri H, et al. Transplantation of autologous iris pigment epithelium to the subretinal space in rabbits. Transplantation. 1999;68:195–201.
134 Thumann G, Kociok N, Bartz-Schmidt KU, et al. Detection of mRNA for proteins involved in retinol metabolism in iris pigment epithelium. Graefes Arch Clin Exp Ophthalmol. 1999;237:1046–1051.
135 Sugano E, Tomita H, Abe T, et al. Comparative study of cathepsins D and S in rat IPE and RPE cells. Exp Eye Res. 2003;77:203–209.
136 Borlongan CV, Stahl CE, Cameron DF, et al. CNS immunological modulation of neural graft rejection and survival. Neurol Res. 1996;18:297–304.
137 Ishioka M, Okamoto S, Streilein JW, et al. Effect of cyclosporine on anterior chamber-associated immune deviation with retinal transplantation. Invest Ophthalmol Vis Sci. 1997;38:2152–2160.
138 Rao KV, Andersen RC, O’Brien TJ. Successful renal transplantation in a patient with anaphylactic reaction to Solu-Medrol (methylprednisolone sodium succinate). Am J Med. 1982;72:161–163.
139 Mittal R, Agarwal SK, Dash SC, et al. Treatment of acute rejection in live related renal allograft recipients: a comparison of three different protocols. Nephron. 1997;77:186–189.
140 Delmonico FL, Tolkoff RN. Treatment of acute rejection in real transplantation. In: Brenner BM, Stein JH. Contemporary issues in nephrology. Edinburgh: Churchill Livingstone; 1989:129–146.
141 Hirano T, Oka K, Takeuchi H, et al. Clinical significance of glucocorticoid pharmacodynamics assessed by antilymphocyte action in kidney transplantation. Marked difference between prednisolone and methylprednisolone. Transplantation. 1994;57:1341–1348.
142 Yoshizumi MO, Bhavsar AR, Dessouki A, et al. Safety of repeated intravitreous injections of antibiotics and dexamethasone. Retina. 1999;19:437–441.
143 Kagaya F, Usui T, Kamiya K, et al. Intraocular dexamethasone delivery system for corneal transplantation in an animal model. Cornea. 2002;21:200–202.
144 Haller JA, Kuppermann BD, Blumenkranz MS, et al. Randomized controlled trial of an intravitreous dexamethasone drug delivery system in patients with diabetic macular edema. Arch Ophthalmol. 2010;128:289–296.
145 Jaffe GJ, Martin D, Callanan D, et al. Fluocinolone acetonide implant (Retisert) for noninfectious posterior uveitis: thirty-four-week results of a multicenter randomized clinical study. Ophthalmology. 2006;113:1020–1027.
146 Gullapalli VG. Paucity of cellular integrin receptors and aging changes in Bruch’s membrane contribute to poor resurfacing of aged Bruch’s membrane by retinal pigment epithelium. PHD thesis, Graduate School of Biomedical Sciences, UMDNJ; 2005.
147 Gullapalli VK, Sugino IK, Van Patten Y, et al. Retinal pigment epithelium resurfacing of aged submacular human Bruch’s membrane. Trans Am Ophthalmol Soc. 2004;102:123–137. discussion 37–38
148 Tezel TH, Del Priore LV, Kaplan HJ. Reengineering of aged Bruch’s membrane to enhance retinal pigment epithelium repopulation. Invest Ophthalmol Vis Sci. 2004;45:3337–3348.
149 Tezel TH, Oda J, Hornbeck R, et al. RPE repopulation of aged inner collagenous layer (ICL) is inhibited by reversible alterations in Bruch’s membrane. Invest Ophthalmol Vis Sci. 42, 2001. ARVO abstract 4995
150 Fang IM, Yang CH, Yang CM, et al. Overexpression of integrin alpha6 and beta4 enhances adhesion and proliferation of human retinal pigment epithelial cells on layers of porcine Bruch’s membrane. Exp Eye Res. 2009;88:12–21.
151 Lu L, Garcia CA, Mikos AG. Retinal pigment epithelium cell culture on thin biodegradable poly(DL-lactic-co-glycolic acid) films. J Biomater Sci Polym Ed. 1998;9:1187–1205.
152 Tomita M, Lavik E, Klassen H, et al. Biodegradable polymer composite grafts promote the survival and differentiation of retinal progenitor cells. Stem Cells. 2005;23:1579–1588.
153 Treharne AJ, Grossel MC, Lotery AJ, et al. The chemistry of retinal transplantation: the influence of polymer scaffold properties on retinal cell adhesion and control. Br J Ophthalmol. 2011;95:768–773.
154 Krishna Y, Sheridan CM, Kent DL, et al. Polydimethylsiloxane as a substrate for retinal pigment epithelial cell growth. J Biomed Mater Res A. 2007;80:669–678.
155 Thomson HA, Treharne AJ, Walker P, et al. Optimisation of polymer scaffolds for retinal pigment epithelium (RPE) cell transplantation. Br J Ophthalmol. 2011;95:563–568.
156 Castellarin AA, Nasir MA, Sugino IK, et al. Progressive presumed choriocapillaris atrophy after surgery for age-related macular degeneration. Retina. 1998;18:143–149.
157 Hergott GJ, Kalnins VI. Expression of proliferating cell nuclear antigen in migrating retinal pigment epithelial cells during wound healing in organ culture. Exp Cell Res. 1991;195:307–314.
158 Kamei M, Lewis JM, Hayashi A, et al. A new wound healing model of retinal pigment epithelial cells in sheet culture. Curr Eye Res. 1996;15:714–718.
159 MacDonald IM, Peters C, Chen MH. Effect of retinoic acid on wound healing of laser burns to porcine retinal pigment epithelium. Can J Ophthalmol. 1996;31:175–178.
160 Del Priore LV, Hornbeck R, Kaplan HJ, et al. Débridement of the pig retinal pigment epithelium in vivo. Arch Ophthalmol. 1995;113:939–944.
161 Lopez PF, Yan Q, Kohen L, et al. Retinal pigment epithelial wound healing in vivo. Arch Ophthalmol. 1995;113:1437–1446.
162 Valentino TL, Kaplan HJ, Del Priore LV, et al. Retinal pigment epithelial repopulation in monkeys after submacular surgery. Arch Ophthalmol. 1995;113:932–938.
163 Ho TC, Del Priore LV, Hornbeck R. Effect of mitomycin-C on human retinal pigment epithelium in culture. Curr Eye Res. 1997;16:572–576.
164 Wang H, Ninomiya Y, Sugino IK, et al. Retinal pigment epithelium wound healing on human Bruch’s membrane explants. Invest Ophthalmol Vis Sci. 2003;44:2199–2210.
165 Sugino IK, Wang H, Zarbin MA. Age-related macular degeneration and retinal pigment epithelium wound healing. Mol Neurobiol. 2003;28:177–194.
166 Burton TC. Recovery of visual acuity after retinal detachment involving the macula. Trans Am Ophthalmol Soc. 1982;80:475–497.
167 Machemer R. Experimental retinal detachment in the owl monkey. IV. The reattached retina. Am J Ophthalmol. 1968;66:1075–1091.
168 Anderson DH, Guerin CJ, Erickson PA, et al. Morphological recovery in the reattached retina. Invest Ophthalmol Vis Sci. 1986;27:168–183.
169 Mervin K, Valter K, Maslim J, et al. Limiting photoreceptor death and deconstruction during experimental retinal detachment: the value of oxygen supplementation. Am J Ophthalmol. 1999;128:155–164.
170 Erickson PA, Fisher SK, Anderson DH, et al. Retinal detachment in the cat: the outer nuclear and outer plexiform layers. Invest Ophthalmol Vis Sci. 1983;24:927–942.
171 Fisher SK, Stone J, Rex TS, et al. Experimental retinal detachment: a paradigm for understanding the effects of induced photoreceptor degeneration. Prog Brain Res. 2001;131:679–698.
172 Sakai T, Calderone JB, Lewis GP, et al. Cone photoreceptor recovery after experimental detachment and reattachment: an immunocytochemical, morphological, and electrophysiological study. Invest Ophthalmol Vis Sci. 2003;44:416–425.
173 Sakai T, Lewis GP, Linberg KA, et al. The ability of hyperoxia to limit the effects of experimental detachment in cone-dominated retina. Invest Ophthalmol Vis Sci. 2001;42:3264–3273.
174 Wobus AM, Boheler KR. Embryonic stem cells: prospects for developmental biology and cell therapy. Physiol Rev. 2005;85:635–678.
175 Bharti K, Miller SS, Arnheiter H. The new paradigm: retinal pigment epithelium cells generated from embryonic or induced pluripotent stem cells. Pigment Cell Melanoma Res. 2011;24:21–34.
176 Jaenisch R, Young R. Stem cells, the molecular circuitry of pluripotency and nuclear reprogramming. Cell. 2008;132:567–582.
177 Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147.
178 Reubinoff BE, Pera MF, Fong CY, et al. Embryonic stem cell lines from human blastocysts: somatic differentiation in vitro. Nat Biotechnol. 2000;18:399–404.
179 Gage FH. Mammalian neural stem cells. Science. 2000;287:1433–1438.
180 Weissman IL. Translating stem and progenitor cell biology to the clinic: barriers and opportunities. Science. 2000;287:1442–1446.
181 Ahmad I, Tang L, Pham H. Identification of neural progenitors in the adult mammalian eye. Biochem Biophys Res Commun. 2000;270:517–521.
182 Tropepe V, Coles BL, Chiasson BJ, et al. Retinal stem cells in the adult mammalian eye. Science. 2000;287:2032–2036.
183 Ferrari G, Cusella-De Angelis G, Coletta M, et al. Muscle regeneration by bone marrow-derived myogenic progenitors. Science. 1998;279:1528–1530.
184 Galli R, Borello U, Gritti A, et al. Skeletal myogenic potential of human and mouse neural stem cells. Nat Neurosci. 2000;3:986–991.
185 Blau HM, Brazelton TR, Weimann JM. The evolving concept of a stem cell: entity or function? Cell. 2001;105:829–841.
186 Marquardt T. Transcriptional control of neuronal diversification in the retina. Prog Retin Eye Res. 2003;22:567–577.
187 Fischer AJ, Reh TA. Identification of a proliferating marginal zone of retinal progenitors in postnatal chickens. Dev Biol. 2000;220:197–210.
188 Johns PR. Growth of the adult goldfish eye. III. Source of the new retinal cells. J Comp Neurol. 1977;176:343–357.
189 Reh TA, Nagy T. Characterization of Rana germinal neuroepithelial cells in normal and regenerating retina. Neurosci Res Suppl. 1989;10:S151–S161.
190 Raymond PA, Hitchcock PF. Retinal regeneration: common principles but a diversity of mechanisms. Adv Neurol. 1997;72:171–184.
191 Spence JR, Madhavan M, Ewing JD, et al. The hedgehog pathway is a modulator of retina regeneration. Development. 2004;131:4607–4621.
192 Coles BL, Angenieux B, Inoue T, et al. Facile isolation and the characterization of human retinal stem cells. Proc Natl Acad Sci U S A. 2004;101:15772–15777.
193 Mayer EJ, Carter DA, Ren Y, et al. Neural progenitor cells from postmortem adult human retina. Br J Ophthalmol. 2005;89:102–106.
194 Yang P, Seiler MJ, Aramant RB, et al. In vitro isolation and expansion of human retinal progenitor cells. Exp Neurol. 2002;177:326–331.
195 Carter DA, Mayer EJ, Dick AD. The effect of postmortem time, donor age and sex on the generation of neurospheres from adult human retina. Br J Ophthalmol. 2007;91:1216–1218.
196 Gong J, Sagiv O, Cai H, et al. Effects of extracellular matrix and neighboring cells on induction of human embryonic stem cells into retinal or retinal pigment epithelial progenitors. Exp Eye Res. 2008;86:957–965.
197 Vossmerbaeumer U, Kuehl S, Kern S, et al. Induction of retinal pigment epithelium properties in ciliary margin progenitor cells. Clin Exp Ophthalmol. 2008;36:358–366.
198 Klimanskaya I, Chung Y, Becker S, et al. Human embryonic stem cell lines derived from single blastomeres. Nature. 2006.
199 Chung Y, Klimanskaya I, Becker S, et al. Human embryonic stem cell lines generated without embryo destruction. Cell Stem Cell. 2008;2:113–117.
200 Wilmut I, Schnieke AE, McWhir J, et al. Viable offspring derived from fetal and adult mammalian cells. Nature. 1997;385:810–813.
201 Byrne JA, Pedersen DA, Clepper LL, et al. Producing primate embryonic stem cells by somatic cell nuclear transfer. Nature. 2007;450:497–502.
202 Park IH, Zhao R, West JA, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141–146.
203 Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872.
204 Kim K, Doi A, Wen B, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:285–290.
205 Zhao T, Zhang ZN, Rong Z, et al. Immunogenicity of induced pluripotent stem cells. Nature. 2011;474:212–215.
206 Chin MH, Mason MJ, Xie W, et al. Induced pluripotent stem cells and embryonic stem cells are distinguished by gene expression signatures. Cell Stem Cell. 2009;5:111–123.
207 Doi A, Park IH, Wen B, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nature Gen. 2009;41:1350–1353.
208 Polo JM, Liu S, Figueroa ME, et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat Biotechnol. 2010;28:848–855.
209 Haruta M, Sasai Y, Kawasaki H, et al. In vitro and in vivo characterization of pigment epithelial cells differentiated from primate embryonic stem cells. Invest Ophthalmol Vis Sci. 2004;45:1020–1025.
210 Klimanskaya I, Hipp J, Rezai KA, et al. Derivation and comparative assessment of retinal pigment epithelium from human embryonic stem cells using transcriptomics. Cloning Stem Cells. 2004;6:217–245.
211 Idelson M, Alper R, Obolensky A, et al. Directed differentiation of human embryonic stem cells into functional retinal pigment epithelium cells. Cell Stem Cell. 2009;5:396–408.
212 Buchholz DE, Hikita ST, Rowland TJ, et al. Derivation of functional retinal pigmented epithelium from induced pluripotent stem cells. Stem Cells. 2009;27:2427–2434.
213 Osakada F, Ikeda H, Sasai Y, et al. Stepwise differentiation of pluripotent stem cells into retinal cells. Nat Protoc. 2009;4:811–824.
214 Osakada F, Ikeda H, Mandai M, et al. Toward the generation of rod and cone photoreceptors from mouse, monkey and human embryonic stem cells. Nat Biotechnol. 2008;26:215–224.
215 Arnhold S, Klein H, Semkova I, et al. Neurally selected embryonic stem cells induce tumor formation after long-term survival following engraftment into the subretinal space. Invest Ophthalmol Vis Sci. 2004;45:4251–4255.
216 Nakagawa M, Koyanagi M, Tanabe K, et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 2008;26:101–106.
217 Huangfu D, Osafune K, Maehr R, et al. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat Biotechnol. 2008;26:1269–1275.
218 Shi Y, Desponts C, Do JT, et al. Induction of pluripotent stem cells from mouse embryonic fibroblasts by Oct4 and Klf4 with small-molecule compounds. Cell Stem Cell. 2008;3:568–574.
219 Yu J, Hu K, Smuga-Otto K, et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324:797–801.
220 Kaji K, Norrby K, Paca A, et al. Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature. 2009;458:771–775.
221 Kokkinaki M, Sahibzada N, Golestaneh N. Human induced pluripotent stem-derived retinal pigment epithelium (RPE) cells exhibit ion transport, membrane potential, polarized vascular endothelial growth factor secretion, and gene expression pattern similar to native RPE. Stem Cells. 2011;29:825–835.
222 Arnhold S, Heiduschka P, Klein H, et al. Adenovirally transduced bone marrow stromal cells differentiate into pigment epithelial cells and induce rescue effects in RCS rats. Invest Ophthalmol Vis Sci. 2006;47:4121–4129.
223 Chiou SH, Kao CL, Peng CH, et al. A novel in vitro retinal differentiation model by co-culturing adult human bone marrow stem cells with retinal pigmented epithelium cells. Biochem Biophys Res Commun. 2005;326:578–585.
224 Atmaca-Sonmez P, Li Y, Yamauchi Y, et al. Systemically transferred hematopoietic stem cells home to the subretinal space and express RPE-65 in a mouse model of retinal pigment epithelium damage. Exp Eye Res. 2006;83:1295–1302.
225 Lund RD, Wang S, Lu B, et al. Cells isolated from umbilical cord tissue rescue photoreceptors and visual functions in a rodent model of retinal disease. Stem Cells. 2007;25:602–611.
226 Feng Q, Lu SJ, Klimanskaya I, et al. Hemangioblastic derivatives from human induced pluripotent stem cells exhibit limited expansion and early senescence. Stem Cells. 2010;28:704–712.
227 Lamba DA, Reh TA. Microarray characterization of human embryonic stem cell-derived retinal cultures. Invest Ophthalmol Vis Sci. 2011;52:4897–4906.
228 Banin E, Obolensky A, Idelson M, et al. Retinal incorporation and differentiation of neural precursors derived from human embryonic stem cells. Stem Cells. 2006;24:246–257.
229 Vugler A, Carr AJ, Lawrence J, et al. Elucidating the phenomenon of HESC-derived RPE: anatomy of cell genesis, expansion and retinal transplantation. Exp Neurol. 2008;214:347–361.
230 Arnhold S, Absenger Y, Klein H, et al. Transplantation of bone marrow-derived mesenchymal stem cells rescue photoreceptor cells in the dystrophic retina of the rhodopsin knockout mouse. Graefes Arch Clin Exp Ophthalmol. 2007;245:414–422.
231 Inoue Y, Iriyama A, Ueno S, et al. Subretinal transplantation of bone marrow mesenchymal stem cells delays retinal degeneration in the RCS rat model of retinal degeneration. Exp Eye Res. 2007;85:234–241.
232 Kolomeyer A, Sugino I, Zarbin M. Characterization of conditioned media collected from cultured adult versus fetal retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2011;52:5973–5986.
233 Marmorstein AD, Marmorstein LY, Rayborn M, et al. Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium. Proc Natl Acad Sci U S A. 2000;97:12758–12763.
234 Bakall B, Radu RA, Stanton JB, et al. Enhanced accumulation of A2E in individuals homozygous or heterozygous for mutations in BEST1 (VMD2). Exp Eye Res. 2007;85:34–43.
235 Morimura H, Fishman GA, Grover SA, et al. Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or leber congenital amaurosis. Proc Natl Acad Sci U S A. 1998;95:3088–3093.
236 Maw MA, Kennedy B, Knight A, et al. Mutation of the gene encoding cellular retinaldehyde-binding protein in autosomal recessive retinitis pigmentosa. Nat Genet. 1997;17:198–200.
237 Mata NL, Weng J, Travis GH. Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc Natl Acad Sci U S A. 2000;97:7154–7159.
238 Allikmets R, Singh N, Sun H, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15:236–246.
239 Berson EL. Retinitis pigmentosa: unfolding its mystery. Proc Natl Acad Sci U S A. 1996;93:4526–4528.
240 Milam AH, Li ZY, Fariss RN. Histopathology of the human retina in retinitis pigmentosa. Prog Retin Eye Res. 1998;17:175–205.
241 Santos A, Humayun MS, de Juan E, et al. Preservation of the inner retina in retinitis pigmentosa. A morphometric analysis. Arch Ophthalmol. 1997;115:511–515.
242 Humayun MS, Prince M, de Juan E, et al. Morphometric analysis of the extramacular retina from postmortem eyes with retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1999;40:143–148.
243 Li ZY, Kljavin IJ, Milam AH. Rod photoreceptor neurite sprouting in retinitis pigmentosa. J Neurosci. 1995;15:5429–5438.
244 Jones BW, Watt CB, Frederick JM, et al. Retinal remodeling triggered by photoreceptor degenerations. J Comp Neurol. 2003;464:1–16.
245 Ali RR, Sarra GM, Stephens C, et al. Restoration of photoreceptor ultrastructure and function in retinal degeneration slow mice by gene therapy. Nat Genet. 2000;25:306–310.
246 Bennett J, Tanabe T, Sun D, et al. Photoreceptor cell rescue in retinal degeneration (rd) mice by in vivo gene therapy. Nat Med. 1996;2:649–654.
247 Vollrath D, Feng W, Duncan JL, et al. Correction of the retinal dystrophy phenotype of the RCS rat by viral gene transfer of Mertk. Proc Natl Acad Sci U S A. 2001;98:12584–12589.
248 Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med. 2008;358:2240–2248.
249 Bainbridge JW, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. 2008;358:2231–2239.
250 LaVail MM, Yasumura D, Matthes MT, et al. Protection of mouse photoreceptors by survival factors in retinal degenerations. Invest Ophthalmol Vis Sci. 1998;39:592–602.
251 Sieving PA, Caruso RC, Tao W, et al. Ciliary neurotrophic factor (CNTF) for human retinal degeneration: phase I trial of CNTF delivered by encapsulated cell intraocular implants. Proc Natl Acad Sci U S A. 2006;103:3896–3901.
252 Lakhanpal RR, Yanai D, Weiland JD, et al. Advances in the development of visual prostheses. Curr Opin Ophthalmol. 2003;14:122–127.
253 Karl MO, Reh TA. Regenerative medicine for retinal diseases: activating endogenous repair mechanisms. Trends Mol Med. 2010;16:193–202.
254 del Cerro M, Lazar ES, Diloreto D, Jr. The first decade of continuous progress in retinal transplantation. Microsc Res Tech. 1997;36:130–141.
255 Gust J, Reh TA. Adult donor rod photoreceptors integrate into the mature mouse retina. Invest Ophthalmol Vis Sci. 2011;52:5266–5272.
256 Meyer JS, Katz ML, Maruniak JA, et al. Neural differentiation of mouse embryonic stem cells in vitro and after transplantation into eyes of mutant mice with rapid retinal degeneration. Brain Res. 2004;1014:131–144.
257 Meyer JS, Katz ML, Maruniak JA, et al. Embryonic stem cell-derived neural progenitors incorporate into degenerating retina and enhance survival of host photoreceptors. Stem Cells. 2006;24:274–283.
258 Radtke ND, Aramant RB, Petry HM, et al. Vision improvement in retinal degeneration patients by implantation of retina together with retinal pigment epithelium. Am J Ophthalmol. 2008;146:172–182.
259 Seiler MJ, Aramant RB, Thomas BB, et al. Visual restoration and transplant connectivity in degenerate rats implanted with retinal progenitor sheets. Eur J Neurosci. 2010;31:508–520.
260 Lamba DA, Gust J, Reh TA. Transplantation of human embryonic stem cell-derived photoreceptors restores some visual function in Crx-deficient mice. Cell Stem Cell. 2009;4:73–79.
261 Mohand-Said S, Deudon-Combe A, Hicks D, et al. Normal retina releases a diffusible factor stimulating cone survival in the retinal degeneration mouse. Proc Natl Acad Sci U S A. 1998;95:8357–8362.
262 Mohand-Said S, Hicks D, Dreyfus H, et al. Selective transplantation of rods delays cone loss in a retinitis pigmentosa model. Arch Ophthalmol. 2000;118:807–811.
263 Leveillard T, Mohand-Said S, Lorentz O, et al. Identification and characterization of rod-derived cone viability factor. Nat Genet. 2004;36:755–759.
264 Chalmel F, Leveillard T, Jaillard C, et al. Rod-derived Cone Viability Factor-2 is a novel bifunctional-thioredoxin-like protein with therapeutic potential. BMC Mol Biol. 2007;8:74.
265 Geller AM, Sieving PA. How many cones are required to “see?”: lessons from Stargardt’s macular dystrophy and from modeling with degenerate photoreceptor arrays. In: Hollyfield JG, Anderson RE, La Vail MM. Retinal degeneration. New York: Plenum Press; 1993:25–34.
266 Lavail M. Analysis of neurological mutants with inherited retinal degeneration. Invest Ophthalmol Vis Sci. 1981;21:638–657.
267 Sanyal S, De Ruiter A, Hawkins RK. Development and degeneration of retina in rds mutant mice: light microscopy. J Comp Neurol. 1980;194:193–207.
268 Narfstrom K, Nilsson SE. Hereditary rod-cone degeneration in a strain of Abyssinian cats. Prog Clin Biol Res. 1987;247:349–368.
269 Noell WK, Walker VS, Kang BS, et al. Retinal damage by light in rats. Invest Ophthalmol. 1966;5:450–473.
270 Schuschereba ST, Silverman MS. Retinal cell and photoreceptor transplantation between adult New Zealand red rabbit retinas. Exp Neurol. 1992;115:95–99.
271 Olsson JE, Gordon JW, Pawlyk BS, et al. Transgenic mice with a rhodopsin mutation (Pro23His): a mouse model of autosomal dominant retinitis pigmentosa. Neuron. 1992;9:815–830.
272 Petters RM, Alexander CA, Wells KD, et al. Genetically engineered large animal model for studying cone photoreceptor survival and degeneration in retinitis pigmentosa. Nat Biotechnol. 1997;15:965–970.
273 del Cerro M, Ison JR, Bowen GP, et al. Intraretinal grafting restores visual function in light-blinded rats. Neuroreport. 1991;2:529–532.
274 Seiler MJ, Aramant RB. Intact sheets of fetal retina transplanted to restore damaged rat retinas. Invest Ophthalmol Vis Sci. 1998;39:2121–2131.
275 Gouras P, Du J, Kjeldbye H, et al. Reconstruction of degenerate rd mouse retina by transplantation of transgenic photoreceptors. Invest Ophthal Vis Sci. 1992;33:2579–2586.
276 Silverman MS, Hughes SE, Valentino TL, et al. Photoreceptor transplantation: Anatomic, electrophysiologic, and behavioral evidence for the functional reconstruction of retinas lacking photoreceptors. Exp Neurol. 1992;115:87–94.
277 Kwan AS, Wang S, Lund RD. Photoreceptor layer reconstruction in a rodent model of retinal degeneration. Exp Neurol. 1999;159:21–33.
278 Radner W, Sadda SR, Humayun MS, et al. Light-driven retinal ganglion cell responses in blind rd mice after neural retinal transplantation. Invest Ophthalmol Vis Sci. 2001;42:1057–1065.
279 Otani A, Dorrell MI, Kinder K, et al. Rescue of retinal degeneration by intravitreally injected adult bone marrow-derived lineage-negative hematopoietic stem cells. J Clin Invest. 2004;114:765–774.
280 Yang Y, Mohand-Said S, Leveillard T, et al. Transplantation of photoreceptor and total neural retina preserves cone function in P23H rhodopsin transgenic rat. PloS ONE. 2010;5:e13469.
281 Aramant RB, Seiler MJ, Ball SL. Successful cotransplantation of intact sheets of fetal retina with retinal pigment epithelium. Invest Ophthalmol Vis Sci. 1999;40:1557–1564.
282 Carter-Dawson LD, LaVail MM. Rods and cones in the mouse retina: Structural analysis using light and electron microscopy. J Comp Neurol. 1979;188:245–262.
283 Ivert L, Gouras P, Naeser P, et al. Photoreceptor allografts in a feline model of retinal degeneration. Graefes Arch Clin Exp Ophthalmol. 1998;236:844–852.
284 Bragadottir R, Narfstrom K. Lens sparing pars plana vitrectomy and retinal transplantation in cats. Vet Ophthalmol. 2003;6:135–139.
285 Bergstrom A, Ehinger B, Wilke K, et al. Transplantation of embryonic retina to the subretinal space in rabbits. Exp Eye Res. 1992;55:29–37.
286 Ghosh F, Bruun A, Ehinger B. Immunohistochemical markers in full thickness embryonic rabbit retinal transplants. Ophthalmic Res. 1999;31:5–15.
287 Ghosh F, Arner K. Transplantation of full-thickness retina in the normal porcine eye: surgical and morphologic aspects. Retina. 2002;22:478–486.
288 Prince JH Diesem CD, Eglitis I, et al. Anatomy and histology of the eye and orbit in domestic animals. Springfield IL: Charles C. Thomas; 1960. p. 260–97
289 Beauchemin ML. The fine structure of the pig’s retina. Graefes Arch Clin Exp Ophthalmol. 1974;190:27–45.
290 Simoens P, De Schaepdrijver L, Lauwers H. Morphologic and clinical study of the retinal circulation in the miniature pig. A: Morphology of the retinal microvasculature. Exp Eye Res. 1992;54:965–973.
291 Ghosh F, Engelsberg K, English RV, et al. Long-term neuroretinal full-thickness transplants in a large animal model of severe retinitis pigmentosa. Graefes Arch Clin Exp Ophthalmol. 2007;245:835–846.
292 Seiler M, Aramant RB, Ehinger B, et al. Transplantation of embryonic retina to adult retina in rabbits. Exp Eye Res. 1990;51:225–228.
293 Lazar E, del Cerro M. A new procedure for multiple intraretinal transplantation into mammalian eyes. J Neurosci Methods. 1992;43:157–169.
294 del Cerro M, Kordower JH, Lazar E, et al. Photoreceptor differentiation in retinal xenografts of fetal monkey retina. Brain Res. 1992;574:1–8.
295 Rauer O, Ghosh F. Survival of full-thickness retinal xenotransplants without immunosuppression. Graefes Arch Clin Exp Ophthalmol. 2001;239:145–151.
296 Juliusson B, Bergström A, van Veen T, et al. Cellular organization in retinal transplants using cell suspensions or fragments of embryonic retinal tissue. Cell Transplant. 1993;2:411–418.
297 Sharma RK. Dissection and cotransplantation of large pieces of RPE and neural retina; effect of protease K on the development. Acta Ophthalmol Scand. 2000;78:3–8.
298 Geller AM, Sieving PA. Assessment of foveal cone photoreceptors in Stargardt’s macular dystrophy using a small dot detection task. Vis Res. 1993;33:1509–1524.
299 Tezel TH, Kaplan HJ. Harvest and storage of adult human photoreceptor cells: the vibratome compared to the excimer laser. Curr Eye Res. 1998;17:748–756.
300 Aramant R, Seiler M, Turner JE. Donor age influences on the success of retinal grafts to adult rat retina. Invest Ophthalmol Vis Sci. 1988;29:498–503.
301 Salchow DJ, Trokel SL, Kjeldbye H, et al. Isolation of human fetal cones. Curr Eye Res. 2001;22:85–89.
302 Pinilla I, Cuenca N, Martinez-Navarrete G, et al. Intraretinal processing following photoreceptor rescue by non-retinal cells. Vis Res. 2009;49:2067–2077.
303 Keegan DJ, Kenna P, Humphries MM, et al. Transplantation of syngeneic Schwann cells to the retina of the rhodopsin knockout (rho(−/−)) mouse. Invest Ophthalmol Vis Sci. 2003;44:3526–3532.
304 Hicks D, Sahel J. The implications of rod-dependent cone survival for basic and clinical research. Invest Ophthalmol Vis Sci. 1999;40:3071–3074.
305 LaVail MM, Unoki K, Yasumura D, et al. Multiple growth factors, cytokines, and neurotrophins rescue photoreceptors from the damaging effects of constant light. Proc Natl Acad Sci U S A. 1992;89:11249–11253.
306 Cayouette M, Gravel C. Adenovirus-mediated gene transfer of ciliary neurotrophic factor can prevent photoreceptor degeneration in the retinal degeneration (rd) mouse. Hum Gene Ther. 1997;8:423–430.
307 Chaum E. Retinal neuroprotection by growth factors: a mechanistic perspective. J Cell Biochem. 2003;88:57–75.
308 Wenzel A, Grimm C, Samardzija M, et al. Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Prog Retin Eye Res. 2005;24:275–306.
309 Wen R, Song Y, Cheng T, et al. Injury-induced upregulation of bFGF and CNTF mRNAS in the rat retina. J Neurosci. 1995;15:7377–7385.
310 Faktorovich EG, Steinberg RH, Yasumura D, et al. Photoreceptor degeneration in inherited retinal dystrophy delayed by basic fibroblast growth factor. Nature. 1990;347:83–86.
311 Cao W, Wen R, Li F, et al. Mechanical injury increases bFGF and CNTF mRNA expression in the mouse retina. Exp Eye Res. 1997;65:241–248.
312 Silverman MS, Hughes SE. Photoreceptor rescue in the RCS rat without pigment epithelium transplantation. Curr Eye Res. 1990;9:183–191.
313 Sharma RK, Ehinger B. Retinal cell transplants: How close to clinical application? Acta Ophthalmol Scand. 1997;75:355–363.
314 Coffey PJ, Whiteley SJ, Lund RD. Preservation and restoration of vision following transplantation. Prog Brain Res. 2000;127:489–499.
315 Das T, del Cerro M, Jalali S, et al. The transplantation of human fetal neuroretinal cells in advanced retinitis pigmentosa patients: results of a long-term safety study. Exp Neurol. 1999;157:58–68.
316 Humayun MS, de Juan E, Jr., del Cerro M, et al. Human neural retinal transplantation. Invest Ophthalmol Vis Sci. 2000;41:3100–3106.
317 Radtke ND, Seiler MJ, Aramant RB, et al. Transplantation of intact sheets of fetal neural retina with its retinal pigment epithelium in retinitis pigmentosa patients. Am J Ophthalmol. 2002;133:544–550.
318 Berger AS, Tezel TH, Del Priore LV, et al. Photoreceptor transplantation in retinitis pigmentosa: short-term follow-up. Ophthalmology. 2003;110:383–391.
319 del Cerro M, Humayun MS, Sadda SR, et al. Histologic correlation of human neural retinal transplantation. Invest Ophthalmol Vis Sci. 2000;41:3142–3148.
320 Zhang Y, Arner K, Ehinger B, et al. Limitation of anatomical integration between subretinal transplants and the host retina. Invest Ophthalmol Vis Sci. 2003;44:324–331.
321 West EL, Pearson RA, Tschernutter M, et al. Pharmacological disruption of the outer limiting membrane leads to increased retinal integration of transplanted photoreceptor precursors. Exp Eye Res. 2008;86:601–611.
322 Pearson RA, Barber AC, West EL, et al. Targeted disruption of outer limiting membrane junctional proteins (Crb1 and ZO-1) increases integration of transplanted photoreceptor precursors into the adult wild-type and degenerating retina. Cell Transplant. 2010;19:487–503.
323 Zhang Y, Sharma RK, Ehinger B, et al. Nitric oxide-producing cells project from retinal grafts to the inner plexiform layer of the host retina. Invest Ophthalmol Vis Sci. 1999;40:3062–3066.
324 MacLaren RE, Pearson RA, MacNeil A, et al. Retinal repair by transplantation of photoreceptor precursors. Nature. 2006;444:203–207.
325 Bartsch U, Oriyakhel W, Kenna PF, et al. Retinal cells integrate into the outer nuclear layer and differentiate into mature photoreceptors after subretinal transplantation into adult mice. Exp Eye Res. 2008;86:691–700.
326 Gamm DM, Wang S, Lu B, et al. Protection of visual functions by human neural progenitors in a rat model of retinal disease. PloS One. 2007;2:e338.
327 Kovalevsky G, DiLoreto D, Jr., Wyatt J, et al. The intensity of the pupillary light reflex does not correlate with the number of retinal photoreceptor cells. Exp Neurol. 1995;133:43–49.
328 Chacko DM, Rogers JA, Turner JE, et al. Survival and differentiation of cultured retinal progenitors transplanted in the subretinal space of the rat. Biochem Biophys Res Commun. 2000;268:842–846.
329 Sakaguchi DS, Van Hoffelen SJ, Grozdanic SD, et al. Neural progenitor cell transplants into the developing and mature central nervous system. Ann N Y Acad Sci. 2005;1049:118–134.
330 Klassen H, Schwartz PH, Ziaeian B, et al. Neural precursors isolated from the developing cat brain show retinal integration following transplantation to the retina of the dystrophic cat. Vet Ophthal. 2007;10:245–253.
331 Van Hoffelen SJ, Young MJ, Shatos MA, et al. Incorporation of murine brain progenitor cells into the developing mammalian retina. Invest Ophthalmol Vis Sci. 2003;44:426–434.
332 Sam TN, Xiao J, Roehrich H, et al. Engrafted neural progenitor cells express a tissue-restricted reporter gene associated with differentiated retinal photoreceptor cells. Cell Transplant. 2006;15:147–160.
333 Klassen H, Sakaguchi DS, Young MJ. Stem cells and retinal repair. Prog Retin Eye Res. 2004;23:149–181.
334 Maclaren RE, Pearson RA, Macneil A, et al. Retinal repair by transplantation of photoreceptor precursors. Nature. 2006.
335 Gruen L, Grabel L. Concise review: scientific and ethical roadblocks to human embryonic stem cell therapy. Stem Cells. 2006;24:2162–2169.
336 MacNeil A, Pearson RA, MacLaren RE, et al. Comparative analysis of progenitor cells isolated from the iris, pars plana, and ciliary body of the adult porcine eye. Stem Cells. 2007;25:2430–2438.
337 Kokkinopoulos I, Pearson RA, Macneil A, et al. Isolation and characterisation of neural progenitor cells from the adult Chx10(orJ/orJ) central neural retina. Mol Cell Neurosci. 2008;38:359–373.
338 Akagi T, Mandai M, Ooto S, et al. Otx2 homeobox gene induces photoreceptor-specific phenotypes in cells derived from adult iris and ciliary tissue. Invest Ophthalmol Vis Sci. 2004;45:4570–4575.
339 Jomary C, Jones SE. Induction of functional photoreceptor phenotype by exogenous Crx expression in mouse retinal stem cells. Invest Ophthalmol Vis Sci. 2008;49:429–437.
340 Yang P, Seiler MJ, Aramant RB, et al. Differential lineage restriction of rat retinal progenitor cells in vitro and in vivo. J Neurosci Res. 2002;69:466–476.
341 Canola K, Angenieux B, Tekaya M, et al. Retinal stem cells transplanted into models of late stages of retinitis pigmentosa preferentially adopt a glial or a retinal ganglion cell fate. Invest Ophthalmol Vis Sci. 2007;48:446–454.
342 Klassen H, Kiilgaard JF, Zahir T, et al. Progenitor cells from the porcine neural retina express photoreceptor markers after transplantation to the subretinal space of allorecipients. Stem Cells. 2007;25:1222–1230.
343 Klassen HJ, Ng TF, Kurimoto Y, et al. Multipotent retinal progenitors express developmental markers, differentiate into retinal neurons, and preserve light-mediated behavior. Invest Ophthalmol Vis Sci. 2004;45:4167–4173.
344 Liu IH, Chen SJ, Ku HH, et al. Comparison of the proliferation and differentiation ability between adult rat retinal stem cells and cerebral cortex-derived neural stem cells. Ophthalmologica. 2005;219:171–176.
345 Del Debbio CB, Balasubramanian S, Parameswaran S, et al. Notch and Wnt signaling mediated rod photoreceptor regeneration by Muller cells in adult mammalian retina. PloS One. 2010;5:e12425.
346 Lawrence JM, Singhal S, Bhatia B, et al. MIO-M1 cells and similar Muller glial cell lines derived from adult human retina exhibit neural stem cell characteristics. Stem Cells. 2007;25:2033–2043.
347 Das AV, Mallya KB, Zhao X, et al. Neural stem cell properties of Muller glia in the mammalian retina: regulation by Notch and Wnt signaling. Dev Biol. 2006;299:283–302.
348 Limb GA, Salt TE, Munro PM, et al. In vitro characterization of a spontaneously immortalized human Muller cell line (MIO-M1). Invest Ophthalmol Vis Sci. 2002;43:864–869.
349 Singhal S, Lawrence JM, Bhatia B, et al. Chondroitin sulfate proteoglycans and microglia prevent migration and integration of grafted Muller stem cells into degenerating retina. Stem Cells. 2008;26:1074–1082.
350 Xu C, Inokuma MS, Denham J, et al. Feeder-free growth of undifferentiated human embryonic stem cells. Nat Biotechnol. 2001;19:971–974.
351 Ludwig TE, Bergendahl V, Levenstein ME, et al. Feeder-independent culture of human embryonic stem cells. Nat Methods. 2006;3:637–646.
352 Lamba DA, Karl MO, Ware CB, et al. Efficient generation of retinal progenitor cells from human embryonic stem cells. Proc Natl Acad Sci U S A. 2006;103:12769–12774.
353 Schraermeyer U, Thumann G, Luther T, et al. Subretinally transplanted embryonic stem cells rescue photoreceptor cells from degeneration in the RCS rats. Cell Transplant. 2001;10:673–680.
354 Amirpour N, Karamali F, Rabiee F, et al. Differentiation of human embryonic stem cell-derived retinal progenitors into retinal cells by sonic hedgehog and/or retinal pigmented epithelium and transplantation into the subretinal space of sodium iodate-injected rabbits. Stem Cells Dev. 2011;21:42–53.
355 Lamba DA, McUsic A, Hirata RK, et al. Generation, purification and transplantation of photoreceptors derived from human induced pluripotent stem cells. PloS One. 2010;5:e8763.
356 Tucker BA, Park IH, Qi SD, et al. Transplantation of adult mouse iPS cell-derived photoreceptor precursors restores retinal structure and function in degenerative mice. PloS One. 2011;6:e18992.
357 Zhou L, Wang W, Liu Y, et al. Differentiation of induced pluripotent stem cells of Swine into rod photoreceptors and their integration into the retina. Stem Cells. 2011;29:972–980.
358 Kicic A, Shen WY, Wilson AS, et al. Differentiation of marrow stromal cells into photoreceptors in the rat eye. J Neurosci. 2003;23:7742–7749.
359 Sun X, Chen M, Li J, et al. E13.5 retinal progenitors induce mouse bone marrow mesenchymal stromal cells to differentiate into retinal progenitor-like cells. Cytotherapy. 2011;13:294–303.
360 Tao YX, Xu HW, Zheng QY, et al. Noggin induces human bone marrow-derived mesenchymal stem cells to differentiate into neural and photoreceptor cells. Indian J Exp Biol. 2010;48:444–452.
361 Radtke ND, Aramant RB, Seiler M, et al. Preliminary report: indications of improved visual function after retinal sheet transplantation in retinitis pigmentosa patients. Am J Ophthalmol. 1999;128:384–387.
362 Siqueira RC, Messias A, Voltarelli JC, et al. Intravitreal injection of autologous bone marrow-derived mononuclear cells for hereditary retinal dystrophy: a phase I trial. Retina. 2011;31:1207–1214.
363 Wang HM, Kaplan HJ, Chan WC, et al. The distribution and ontogeny of MHC antigens in murine ocular tissue. Invest Ophthalmol Vis Sci. 1987;28:1383–1389.
364 Jiang LQ, Jorquera M, Streilein JW, et al. Unconventional rejection of neural retinal allografts implanted into the immunologically privileged site of the eye. Transplantation. 1995;59:1201–1207.
365 Hori J, Ng TF, Shatos M, et al. Neural progenitor cells lack immunogenicity and resist destruction as allografts. Stem Cells. 2003;21:405–416.
366 Yang P, Chen L, Zwart R, et al. Immune cells in the porcine retina: distribution, characterization and morphological features. Invest Ophthalmol Vis Sci. 2002;43:1488–1492.
367 Provis JM, Penfold PL, Edwards AJ, et al. Human retinal microglia: expression of immune markers and relationship to the glia limitans. Glia. 1995;14:243–256.
368 Aramant RB, Seiler MJ. Human embryonic retinal cell transplants in athymic immunodeficient rat hosts. Cell Transplant. 1994;3:461–474.
369 Ma J, Xu L, Othersen DK, et al. Cloning and localization of RPE65 mRNA in salamander cone photoreceptor cells1. Biochim Biophys Acta. 1998;1443:255–261.
370 West EL, Pearson RA, Barker SE, et al. Long-term survival of photoreceptors transplanted into the adult murine neural retina requires immune modulation. Stem Cells. 2010;28:1997–2007.
371 Fawcett JW, Asher RA. The glial scar and central nervous system repair. Brain Res Bull. 1999;49:377–391.
372 Kinouchi R, Takeda M, Yang L, et al. Robust neural integration from retinal transplants in mice deficient in GFAP and vimentin. Nat Neurosci. 2003;6:863–868.
373 Tucker BA, Redenti SM, Jiang C, et al. The use of progenitor cell/biodegradable MMP2-PLGA polymer constructs to enhance cellular integration and retinal repopulation. Biomaterials. 2010;31:9–19.
374 Lewis GP, Linberg KA, Fisher SK. Neurite outgrowth from bipolar and horizontal cells after experimental retinal detachment. Invest Ophthalmol Vis Sci. 1998;39:424–434.
375 Jansen HG, Hawkins RK, Sanyal S. Synaptic growth in the rod terminals of mice after partial photoreceptor cell loss: a three-dimensional ultrastructural study. Microsc Res Tech. 1997;36:96–105.
376 Fei Y. Cone neurite sprouting: an early onset abnormality of the cone photoreceptors in the retinal degeneration mouse. Mol Vis. 2002;8:306–314.
377 Fariss RN, Li ZY, Milam AH. Abnormalities in rod photoreceptors, amacrine cells, and horizontal cells in human retinas with retinitis pigmentosa. Am J Ophthalmol. 2000;129:215–223.
378 Fisher SK, Lewis GP. Muller cell and neuronal remodeling in retinal detachment and reattachment and their potential consequences for visual recovery: a review and reconsideration of recent data. Vis Res. 2003;43:887–897.
379 Nachman-Clewner M, Townes-Anderson E. Injury-induced remodelling and regeneration of the ribbon presynaptic terminal in vitro. J Neurocytol. 1996;25:597–613.
380 Mandell JW, MacLeish PR, Townes-Anderson E. Process outgrowth and synaptic varicosity formation by adult photoreceptors in vitro. J Neurosci. 1993;13:3533–3548.
381 Khodair MA, Zarbin MA, Townes-Anderson E. Synaptic plasticity in mammalian photoreceptors prepared as sheets for retinal transplantation. Invest Ophthalmol Vis Sci. 2003;44:4976–4988.
382 Khodair MA, Zarbin MA, Townes-Anderson E. Cyclic AMP prevents retraction of axon terminals in photoreceptors prepared for transplantation: an in vitro study. Invest Ophthalmol Vis Sci. 2005;46:967–973.
383 Fontainhas AM, Townes-Anderson E. RhoA inactivation prevents photoreceptor axon retraction in an in vitro model of acute retinal detachment. Invest Ophthalmol Vis Sci. 2011;52:579–587.
384 Fontainhas AM, Townes-Anderson E. RhoA and its role in synaptic structural plasticity of isolated salamander photoreceptors. Invest Ophthalmol Vis Sci. 2008;49:4177–4187.
385 Marc RE, Jones BW, Watt CB, et al. Neural remodeling in retinal degeneration. Prog Retin Eye Res. 2003;22:607–655.