Toxic injury of the CNS


This chapter covers toxins that are known to produce lesions of the CNS. Some of them also cause peripheral neuropathy, but this will be mentioned only briefly (Tables 25.1–25.3).
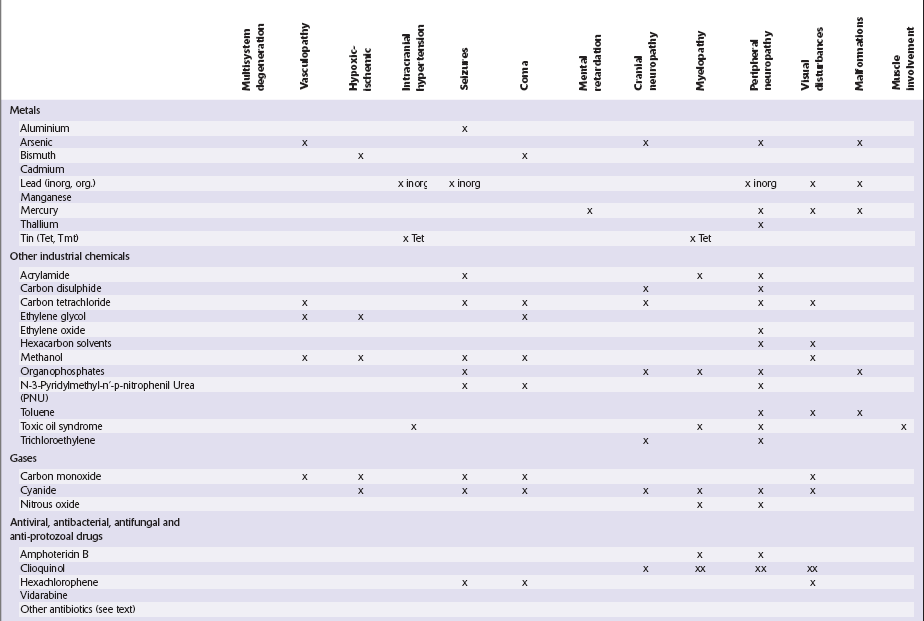
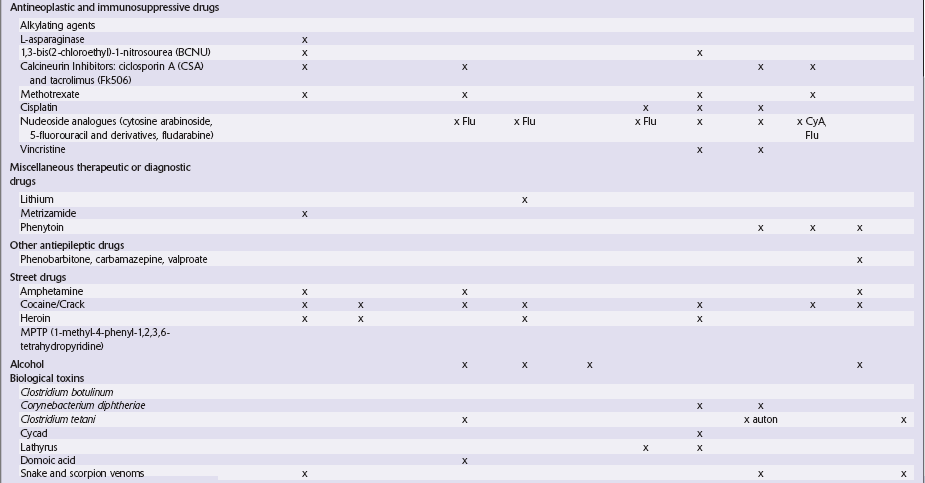
Table 25.1
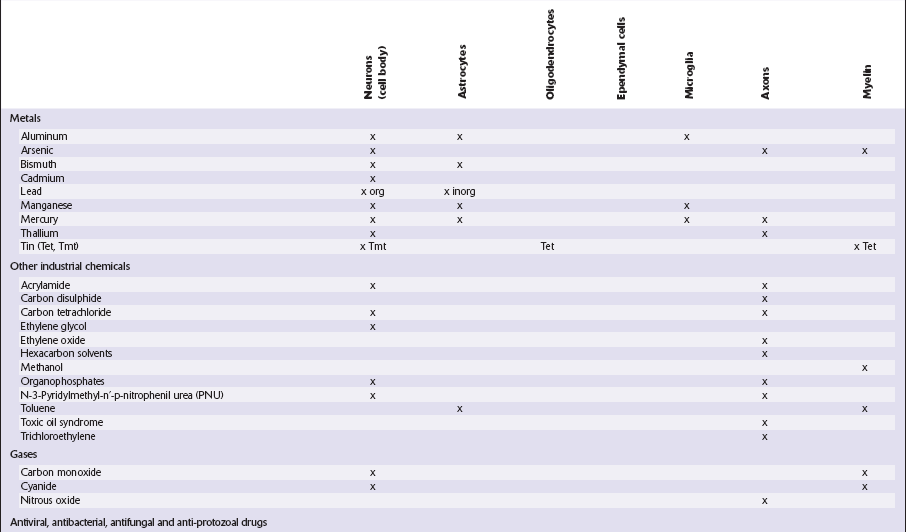
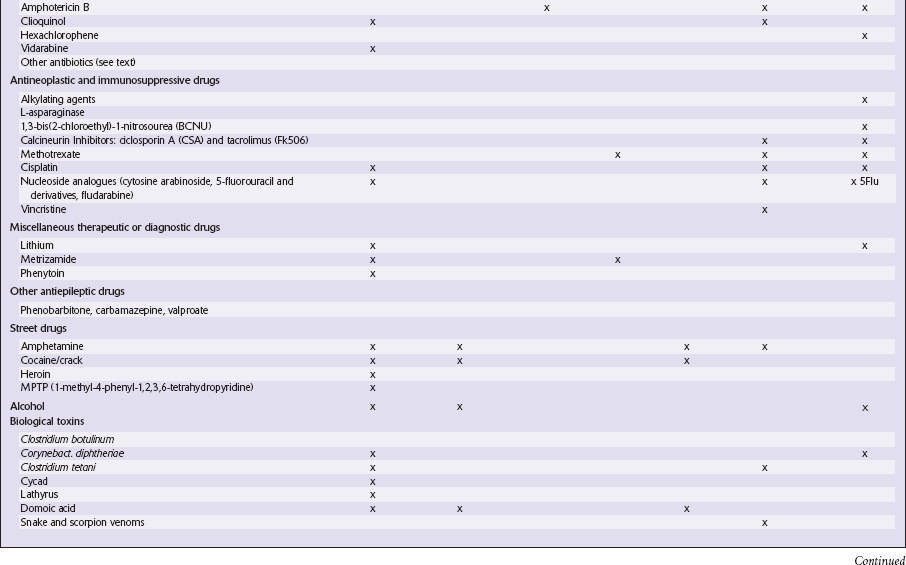
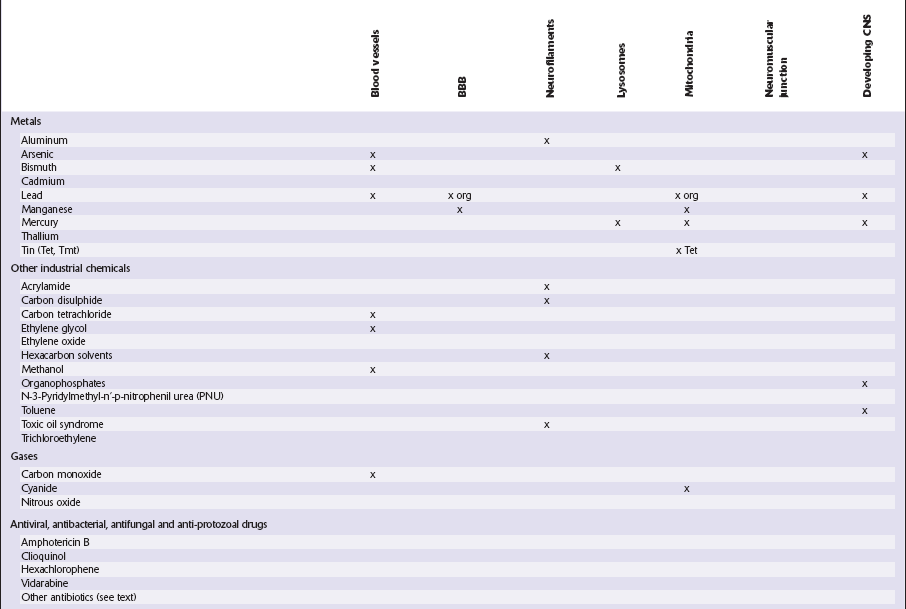
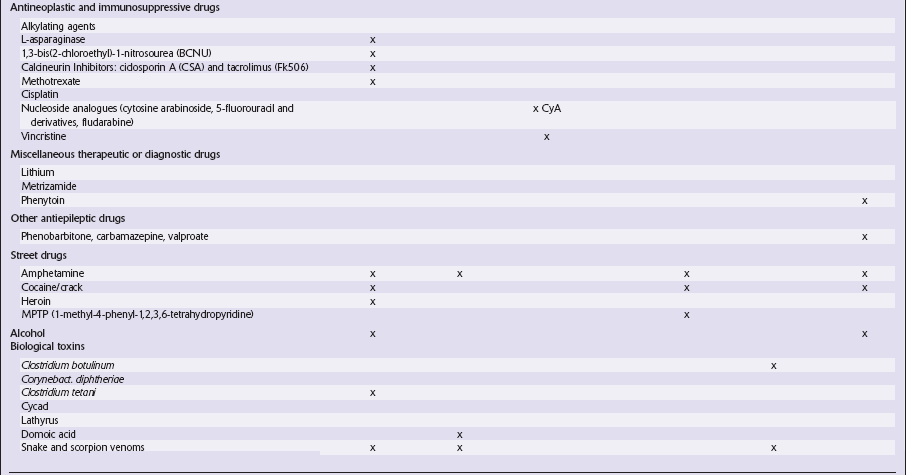
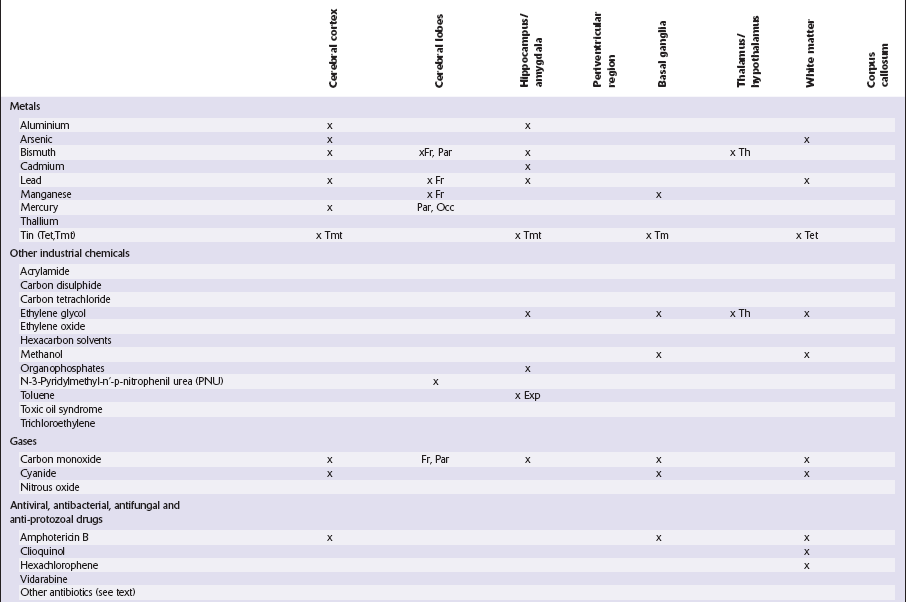
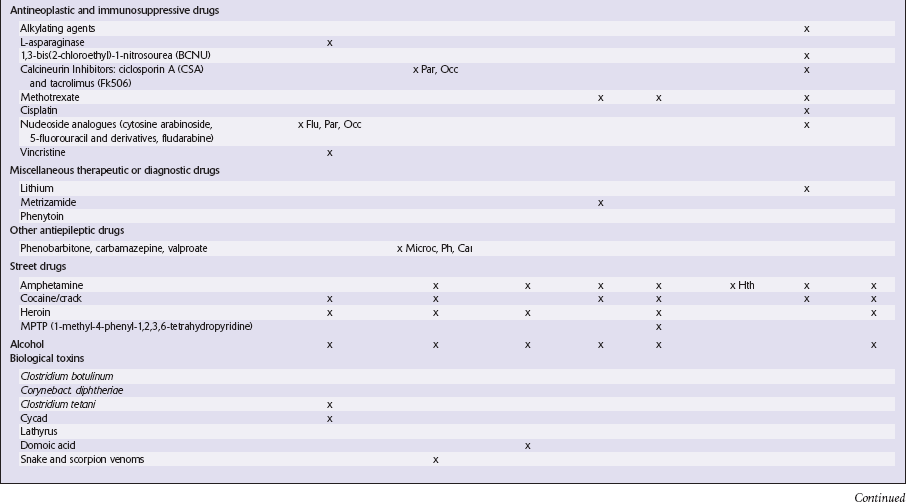
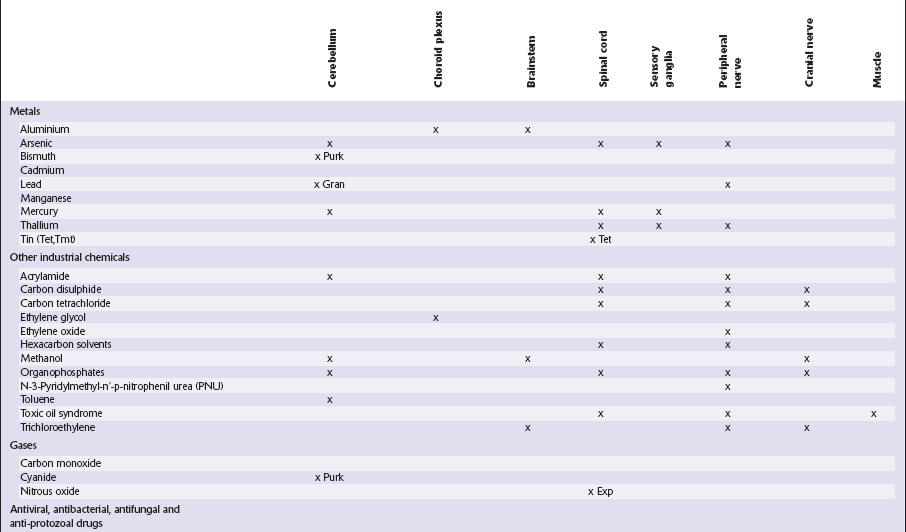
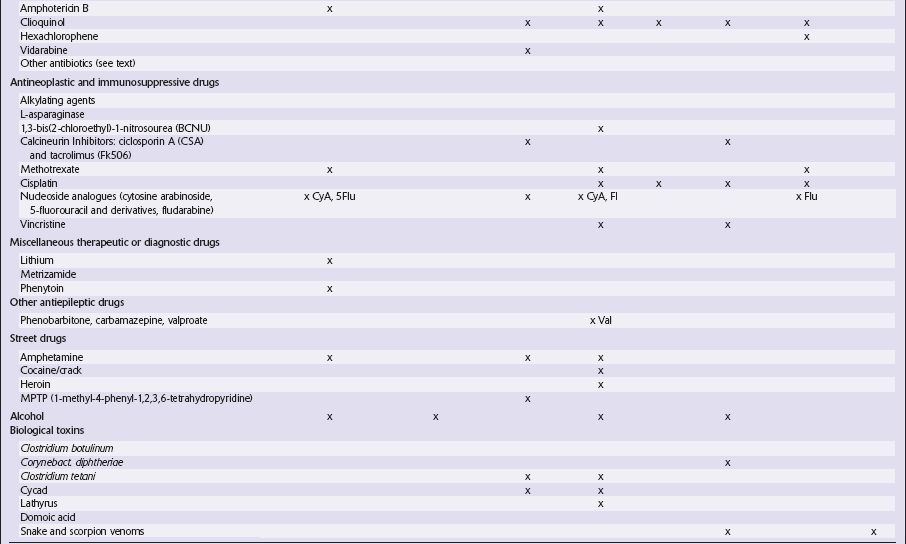
Table 25.3
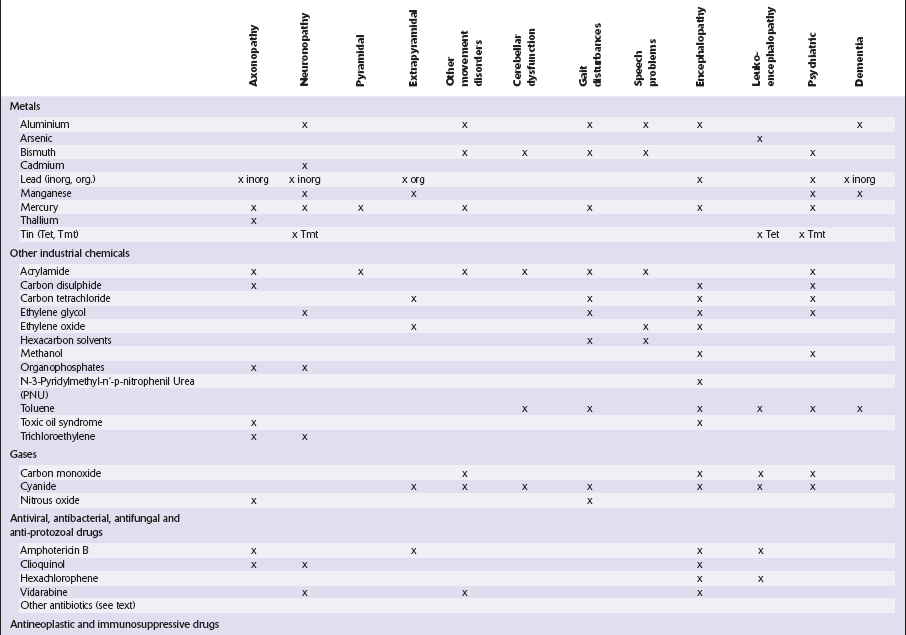
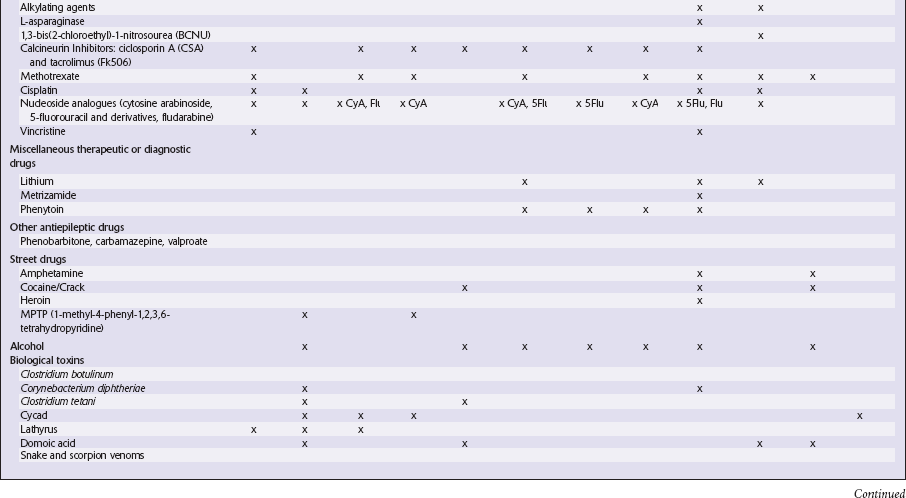
METALS
 ALUMINUM EXPOSURE
ALUMINUM EXPOSURE


ARSENIC
 ARSENIC EXPOSURE
ARSENIC EXPOSURE Arsenicals are rapidly absorbed from mucous membranes or through the skin, but do not cross the blood–brain barrier into the CNS to any significant extent.
Arsenicals are rapidly absorbed from mucous membranes or through the skin, but do not cross the blood–brain barrier into the CNS to any significant extent.

 Organic pentavalent arsenicals are present in some drugs used to treat trypanosomiasis, and are used in insecticides and weed killers. Ingestion may produce an acute hemorrhagic leukoencephalopathy, which is thought to be an immune reaction to the organic compound rather than a direct toxic effect of the arsenic. Approximately 2–5% of patients with trypanosomiasis that are treated with melarsoprol develop acute hemorrhagic leukoencephalopathy (see Chapter 20).
Organic pentavalent arsenicals are present in some drugs used to treat trypanosomiasis, and are used in insecticides and weed killers. Ingestion may produce an acute hemorrhagic leukoencephalopathy, which is thought to be an immune reaction to the organic compound rather than a direct toxic effect of the arsenic. Approximately 2–5% of patients with trypanosomiasis that are treated with melarsoprol develop acute hemorrhagic leukoencephalopathy (see Chapter 20).
MACROSCOPIC AND MICROSCOPIC APPEARANCES
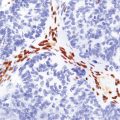
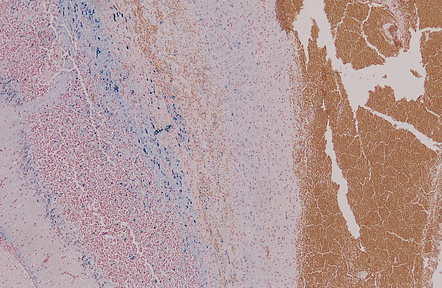
The peripheral neuropathy caused by chronic trivalent arsenical intoxication may be associated with chromatolysis and loss of anterior horn cells. In patients with acute hemorrhagic leukoencephalopathy complicating organic pentavalent arsenical administration, the brain is swollen and contains numerous small and occasional larger foci of hemorrhage (Fig. 25.1). Histology reveals fibrinoid necrosis of many parenchymal blood vessels and hemorrhage into the surrounding tissue (Fig. 25.1). Some of the blood vessels are surrounded by a fibrin exudate containing a mixed inflammatory infiltrate. The findings are described in more detail in Chapter 20.
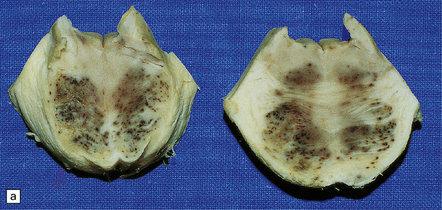
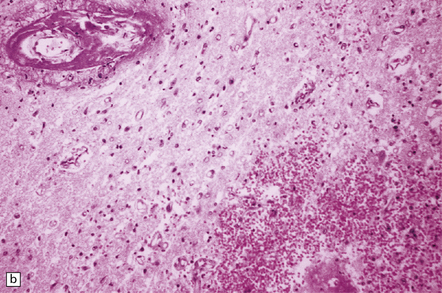
25.1 Acute hemorrhagic encephalopathy complicating melarsoprol treatment.
(a) Petechial hemorrhage and surrounding gray discoloration in the pons. This patient developed acute hemorrhagic leukoencephalopathy as a reaction to melarsoprol treatment. (b) and (c) Histology reveals fibrinoid necrosis of blood vessels and hemorrhage into the surrounding white matter. (Courtesy of Professor Hume Adams, University of Glasgow.)
BISMUTH
 BISMUTH EXPOSURE
BISMUTH EXPOSURE

 BISMUTH TOXICITY
BISMUTH TOXICITY


LEAD
 LEAD EXPOSURE
LEAD EXPOSURE Common sources include lead paint, pottery with lead glazes, herbal and traditional medicines or cosmetics, discarded car batteries, soft water collected in leaded conduits or containers, and alcohol and illicit drugs distilled using lead pipes.
Common sources include lead paint, pottery with lead glazes, herbal and traditional medicines or cosmetics, discarded car batteries, soft water collected in leaded conduits or containers, and alcohol and illicit drugs distilled using lead pipes.
 The lead is absorbed through the gastrointestinal and respiratory tracts.
The lead is absorbed through the gastrointestinal and respiratory tracts.


 Motor vehicle emissions may contribute to chronic low-level toxicity in some communities.
Motor vehicle emissions may contribute to chronic low-level toxicity in some communities.


 LEAD INTOXICATION
LEAD INTOXICATION






MANGANESE
 MANGANESE EXPOSURE
MANGANESE EXPOSURE



MERCURY
 MERCURY EXPOSURE
MERCURY EXPOSURE



 MERCURY TOXICITY
MERCURY TOXICITY Acute toxicity, can produce a range of symptoms including hypersalivation, abdominal cramps, diarrhea, and halitosis which is often described as ‘metallic’. Headache, tiredness, and emotional and psychiatric disturbances may be early manifestations. Other symptoms include tremor (Fig. 25.2), ataxia, vertigo, nystagmus, choreoathetosis, weakness, blurred vision with constricted visual fields, and a sensory neuropathy.
Acute toxicity, can produce a range of symptoms including hypersalivation, abdominal cramps, diarrhea, and halitosis which is often described as ‘metallic’. Headache, tiredness, and emotional and psychiatric disturbances may be early manifestations. Other symptoms include tremor (Fig. 25.2), ataxia, vertigo, nystagmus, choreoathetosis, weakness, blurred vision with constricted visual fields, and a sensory neuropathy.
 Chronic exposure can cause mental retardation, cortical blindness, and quadriplegia.
Chronic exposure can cause mental retardation, cortical blindness, and quadriplegia.

MICROSCOPIC APPEARANCES
There is preferential loss of small neurons, particularly from the granule cell layer of the cerebellum and the primary visual, auditory, and somatosensory regions of the cerebral cortex, in which small neurons predominate. In severe cases, particularly in children, neuronal loss is more extensive and there is spongiform cortical degeneration. Other features include an associated gliosis and, in acute lesions, infiltration by macrophages. Degeneration of dorsal root ganglion cells results in some loss of nerve fibers from the posterior columns of the spinal cord. Sprouting of Purkinje cell dendrites is a prominent feature in long-term survivors, in whom granular deposits of mercury are demonstrable in astrocytes, microglia, and neurons (Fig. 25.2).

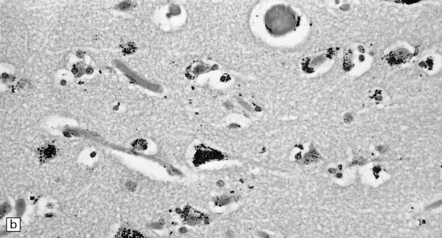
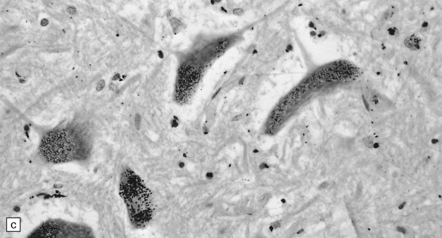
25.2 Mercury poisoning.
(a) Examples of the handwriting and drawing of a patient who 16 years previously had worked for about 18 months filling mercury thermometers and had developed metallic mercury poisoning. (b) Demonstration of intralysosomal mercury in the cerebral cortex. (c) Demonstration of intralysosomal mercury in spinal motor neurons. (b,c) Silver precipitation method of Danscher and Schroeder, see Histochemistry 1979; 60:1–7.
THALLIUM
Most cases of thallium intoxication result from accidental or deliberate ingestion of thallium-containing pesticides or rodenticides. The clinical picture resembles that of trivalent arsenical poisoning. The only consistent abnormalities in the CNS (Fig. 25.3) are those related to the sensorimotor distal axonopathy, which are:


TIN
 ORGANIC TIN EXPOSURE
ORGANIC TIN EXPOSUREInorganic tin is not neurotoxic, but two organotin compounds, triethyltin and trimethyltin, are:



 ORGANOTIN TOXICITY
ORGANOTIN TOXICITY

MACROSCOPIC AND MICROSCOPIC APPEARANCES
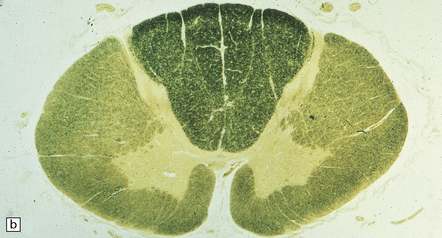
Triethyltin causes striking white matter edema in brain and spinal cord (Fig. 25.4) due to the accumulation of fluid in vacuoles within the myelin sheaths (Fig. 25.4). The vacuoles are formed by separation of myelin along intraperiod lines (Fig. 25.4). Trimethyltin does not cause intramyelinic edema, but is toxic to neurons in the hippocampus, basal ganglia, entorhinal cortex, and amygdala. It elicits specific apoptotic destruction of pyramidal neurons in the CA3 region of the hippocampus and in other limbic structures.
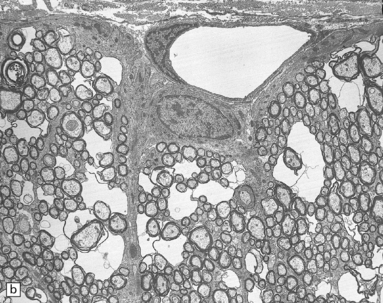
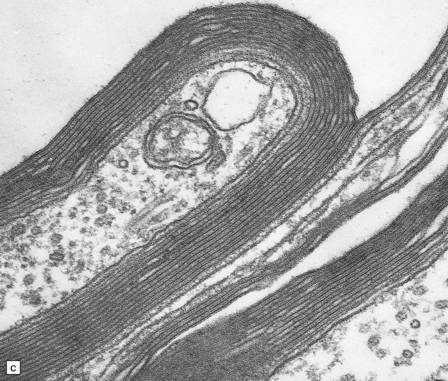
25.4 Triethyltin intoxication.
(a) Widespread vacuolation of the cerebellar white matter in experimental triethyltin intoxication. (b) Electron microscopy reveals accumulations of fluid within the myelin sheaths. (c) At higher magnification, the myelin is seen to have separated along intraperiod lines. (Courtesy of Professor John Cavanagh.)
 CAUSES OF INTRAMYELINIC EDEMA
CAUSES OF INTRAMYELINIC EDEMA









OTHER INDUSTRIAL CHEMICALS
 ACRYLAMIDE EXPOSURE
ACRYLAMIDE EXPOSURE Acrylamide polymers are widely used in industry as flexible sealants, flocculating and grouting agents. They are also used for gel electrophoresis, manufacture of contact lenses and soil conditioning, and form in some foods prepared at high temperatures (e.g. potato and wheat products cooked in oil >120°C).
Acrylamide polymers are widely used in industry as flexible sealants, flocculating and grouting agents. They are also used for gel electrophoresis, manufacture of contact lenses and soil conditioning, and form in some foods prepared at high temperatures (e.g. potato and wheat products cooked in oil >120°C).


 Axonal accumulation of neurofilaments, and impaired neurotransmitter release.
Axonal accumulation of neurofilaments, and impaired neurotransmitter release.
ACRYLAMIDE
 CAUSES OF DISTAL DEGENERATION OF LONG AXONS IN THE CNS
CAUSES OF DISTAL DEGENERATION OF LONG AXONS IN THE CNS













CARBON DISULFIDE
 CARBON DISULFIDE EXPOSURE
CARBON DISULFIDE EXPOSURE

 CARBON DISULFIDE TOXICITY
CARBON DISULFIDE TOXICITY

MACROSCOPIC AND MICROSCOPIC APPEARANCES
The brain and spinal cord appear macroscopically normal, and histologic changes are sparsely documented. In the peripheral nervous system, carbon disulfide intoxication results in axonal swellings (Fig. 25.5), which contain abnormal accumulations of neurofilaments, and distal nerve fiber degeneration. Described CNS abnormalities include distal axonal spinocerebellar degeneration and increased cerebral atherosclerosis. Spinal long tracts contain neurofilamentous axonal swellings (Fig. 25.5). The distal axonopathy is probably secondary to progressive cross-linking and accumulation of neurofilaments during their anterograde transport in long axons.
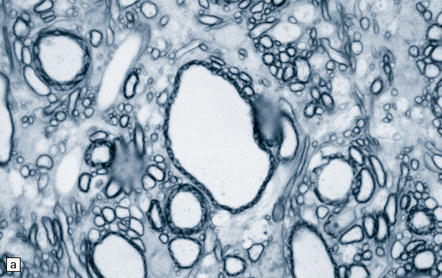
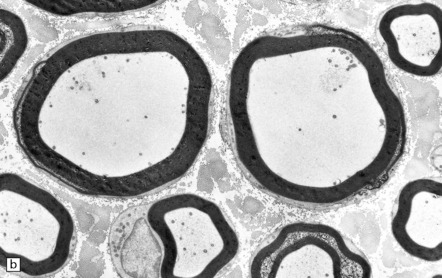
CARBON TETRACHLORIDE
 CARBON TETRACHLORIDE EXPOSURE
CARBON TETRACHLORIDE EXPOSURE

 CARBON TETRACHLORIDE TOXICITY
CARBON TETRACHLORIDE TOXICITY




ETHYLENE GLYCOL
 ETHYLENE GLYCOL EXPOSURE
ETHYLENE GLYCOL EXPOSURE


 ETHYLENE GLYCOL TOXICITY
ETHYLENE GLYCOL TOXICITY



MACROSCOPIC AND MICROSCOPIC APPEARANCES
Acute intoxication produces meningeal congestion and cerebral edema, and, occasionally, petechial hemorrhages. Microscopically, birefringent calcium oxalate deposits can usually be demonstrated by polarized light microscopy (Fig. 25.6) in and around vessels in the meninges, brain parenchyma, and choroid plexus. Neurons may show hypoxic change and there is often white matter edema. Scanty perivascular acute inflammatory infiltrates may be seen, but these are not consistently related to the calcium oxalate deposits and may be a reaction to hypoxic brain injury.
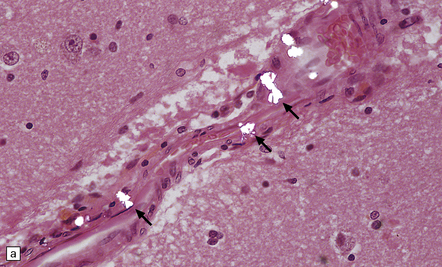
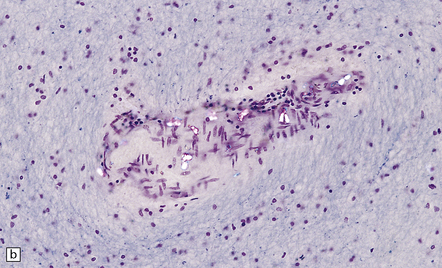
HEXACARBON SOLVENTS (N-HEXANE AND METHYL N-BUTYL KETONE)
MACROSCOPIC AND MICROSCOPIC APPEARANCES
The brain and spinal cord appear macroscopically normal. Histologic changes in the human CNS are sparsely documented, but there are many experimental studies demonstrating the formation of neurofilamentous axonal swellings (Fig. 25.7) and distal degeneration of nerve fibers in long ascending (spinocerebellar and posterior column) and descending (corticospinal) spinal tracts, as occur in the peripheral nerves. As in carbon disulfide intoxication, the changes are probably due to progressive cross-linking and accumulation of neurofilaments during their anterograde transport.
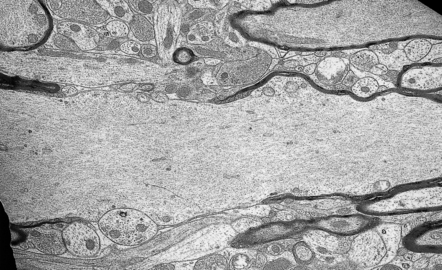
25.7 Experimental 2,5-hexanediol intoxication.
Electron micrograph showing marked neurofilamentous distension of an axon at a node of Ranvier. (Courtesy of Professor John Cavanagh.)
 N-HEXANE AND METHYL N-BUTYL KETONE EXPOSURE
N-HEXANE AND METHYL N-BUTYL KETONE EXPOSURE


 HEXACARBON SOLVENT TOXICITY
HEXACARBON SOLVENT TOXICITY

METHANOL
 METHANOL EXPOSURE
METHANOL EXPOSURE


 METHANOL TOXICITY
METHANOL TOXICITY The manifestations of acute toxicity are delayed for several hours, until after the methanol has been metabolized to form formaldehyde and formic acid.
The manifestations of acute toxicity are delayed for several hours, until after the methanol has been metabolized to form formaldehyde and formic acid.
 Patients develop headache, abdominal pain, nausea, vomiting, and generalized weakness.
Patients develop headache, abdominal pain, nausea, vomiting, and generalized weakness.
 Loss of vision is a common, usually permanent, complication.
Loss of vision is a common, usually permanent, complication.
 Severe intoxication may cause delirium, convulsions, coma, cardiorespiratory failure, and death.
Severe intoxication may cause delirium, convulsions, coma, cardiorespiratory failure, and death.

MACROSCOPIC AND MICROSCOPIC APPEARANCES
In acute methanol intoxication the brain is usually edematous and shows features of global hypoxic injury (see Chapter 8). There may be scattered petechial hemorrhages and larger, symmetric foci of hemorrhagic infarction in the putamen. In severe cases, hemorrhagic necrosis of the putamen is accompanied by more widespread hypoxic damage to the cerebral cortex and extensive white matter necrosis (Figs 25.8–25.10). Survivors of the acute toxicity may show putaminal cavitation, with accumulation of macrophages and reactive astrocytosis (Fig. 25.11). Degeneration of retinal ganglion cells results in optic nerve atrophy and gliosis.
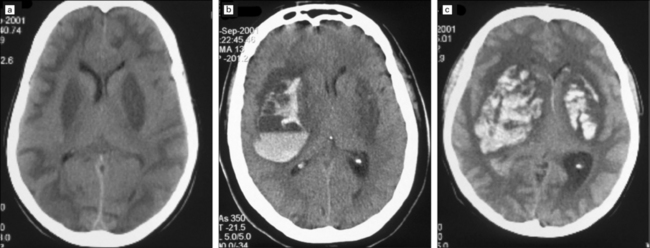
25.8 Acute methanol toxicity – CT head scans.
These CT scans were amongst several obtained 24 hours after ingestion of methanol when approximately 140 people in Pärnu, Estonia, who consumed distilled wood alcohol developed severe metabolic acidosis, visual disturbances and depression of consciousness, progressing to coma. The scans show low attenuation in the putamen, subcortical white matter and parts of the cerebral cortex, most marked in (a) and (c). In (b) and (c) there is also evidence of extensive putaminal hemorrhage, predominantly unilateral in (b), and bilateral in (c). (Images courtesy of Dr Andres Kulla, St Mary’s Hospital, Newport, Isle of Wight.)
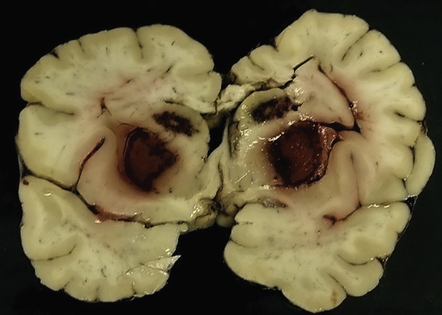
25.9 Acute methanol toxicity – macroscopic findings.
This brain, from one of the Pärnu patients who died from acute methanol intoxication, shows hemorrhage into both the putamina and caudate nuclei. The white matter in the centrum semiovale and corpus callosum was markedly softened. (Courtesy of Dr Andres Kulla.)
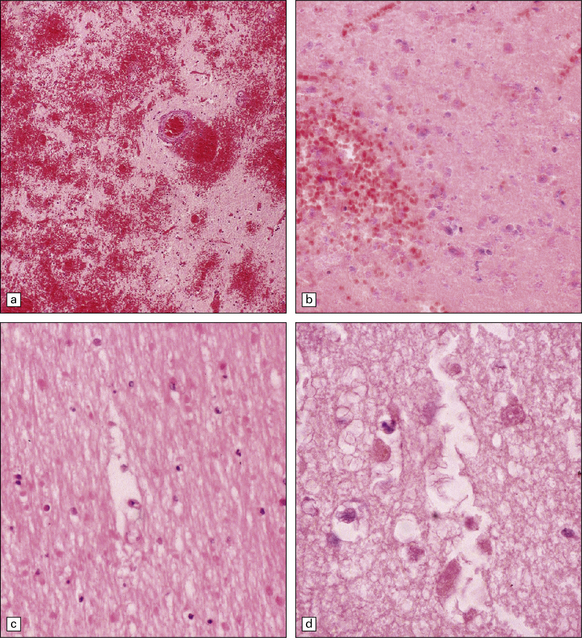
25.10 Acute methanol toxicity.
These sections from one of the Pärnu patients reveal extensive hemorrhagic necrosis in the putamen (a,b). There is also necrosis of subcortical white matter (c) and parts of the cerebral cortex (d). (Courtesy of Dr Andres Kulla.)
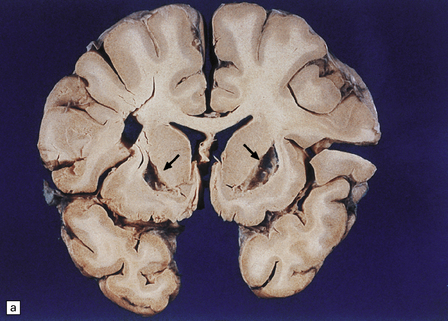
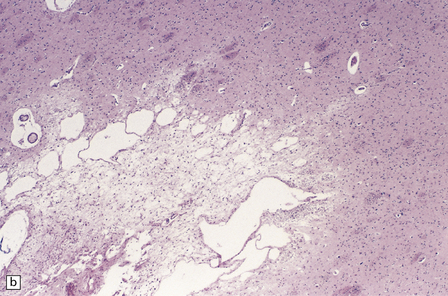
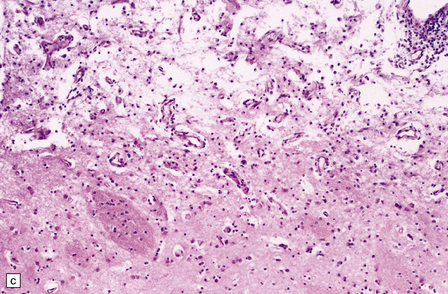
25.11 Methanol toxicity – chronic changes.
(a) Bilateral cavitation (arrows) of medial part of putamen and anterior fibers of internal capsule in methanol poisoning. (The cavitation and splitting of the left centrum semiovale is artefactual.) (Courtesy of Dr J E H Pittella, Department of Pathology, School of Medicine, Federal University of Minas Gerais, Brazil.) (b) Cavitation in the putamen of a long-term survivor of methanol poisoning. (c) Higher magnification view of the cavitated putamen, which contains scattered macrophages and shows some neovascularization.
ORGANOPHOSPHATES
 ORGANOPHOSPHATE EXPOSURE
ORGANOPHOSPHATE EXPOSURE





 ORGANOPHOSPHATE TOXICITY
ORGANOPHOSPHATE TOXICITY


TOLUENE
 TOLUENE EXPOSURE
TOLUENE EXPOSURE


 TOLUENE TOXICITY
TOLUENE TOXICITY




TOXIC OIL SYNDROME AND EOSINOPHILIA–MYALGIA SYNDROME
 ETIOLOGY OF TOXIC OIL SYNDROME AND EOSINOPHILIA–MYALGIA SYNDROME
ETIOLOGY OF TOXIC OIL SYNDROME AND EOSINOPHILIA–MYALGIA SYNDROME



 TOXIC OIL SYNDROME AND EOSINOPHILIA–MYALGIA SYNDROME
TOXIC OIL SYNDROME AND EOSINOPHILIA–MYALGIA SYNDROME


GASES
 CARBON MONOXIDE EXPOSURE
CARBON MONOXIDE EXPOSURE Carbon monoxide is a colorless and odorless gas produced by incomplete combustion of carbon fuels. It is a relatively common cause of fatal poisoning, either accidental (e.g. due to inadequate venting of fumes from domestic gas heaters) or suicidal (e.g. due to inhalation of automobile exhaust fumes).
Carbon monoxide is a colorless and odorless gas produced by incomplete combustion of carbon fuels. It is a relatively common cause of fatal poisoning, either accidental (e.g. due to inadequate venting of fumes from domestic gas heaters) or suicidal (e.g. due to inhalation of automobile exhaust fumes).

 CARBON MONOXIDE TOXICITY
CARBON MONOXIDE TOXICITY


MACROSCOPIC APPEARANCES
During the first few hours, the brain is swollen, congested, and cherry-red in color, though this is less striking after prolonged formalin fixation. After 24–48 hours of survival, scattered petechial hemorrhages may be seen in the white matter and larger hemorrhages in the pallidum (Fig. 25.12). These usually involve the dorsal part of the inner segment of the pallidum, but may extend laterally into the outer segment or dorsomedially into the internal capsule. The pallidal lesions are often asymmetric (Fig. 25.12) and may be unilateral or absent. In some cases there are hemorrhages in the hippocampus and cerebral cortex. Following survival of several days or weeks, the lesions in the pallidum appear necrotic or cavitated (Fig. 25.12). Discrete or confluent foci of necrosis may also be evident in the white matter (Fig. 25.12). These tend to spare the arcuate fibers.
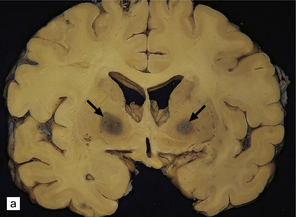
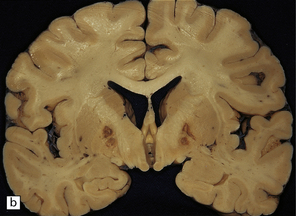
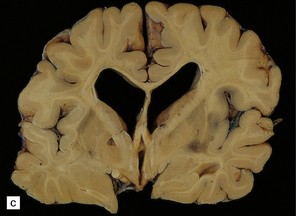
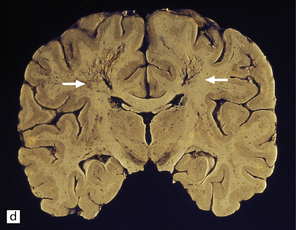
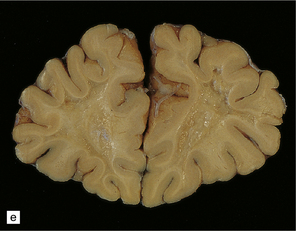
25.12 Carbon monoxide poisoning.
(a) Hemorrhagic discoloration (arrows) of the pallidum in acute carbon monoxide poisoning. The dorsal part of the nucleus is most severely affected. (b) and (c) Examples of symmetric (b) and asymmetric (c) cavitation of the dorsomedial part of the pallidum in survivors of acute carbon monoxide poisoning. Note the ill-defined gray discoloration of the cerebral white matter and thinning of the corpus callosum in (c). (d) Cavitated necrosis (arrows) in the cerebral white matter in a survivor of acute carbon monoxide poisoning. (e) The frontal white matter has a gelatinous gray appearance and is focally cavitated in the brain from a long-term survivor of carbon monoxide poisoning. The arcuate fibers are spared.
MICROSCOPIC APPEARANCES
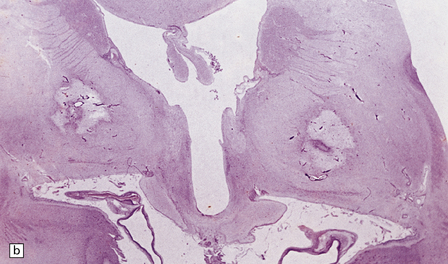
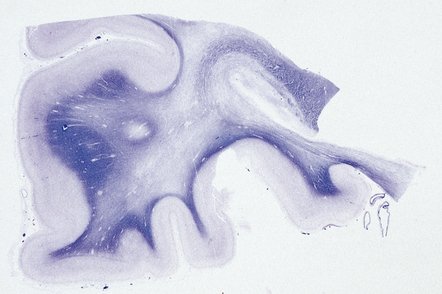
In the acute stage there may be foci of necrosis and/or perivascular hemorrhage in the affected parts of the gray and white matter. Within days, these foci accumulate numerous lipid- or hemosiderin-laden macrophages. Lesions in longer-term survivors are cavitated (Fig. 25.13) or rarefied and gliotic. Typical changes of global brain hypoxia–ischemia of varying severity (see Chapter 8) are usually seen. The white matter lesions may be small and discrete, extensive, or even confluent. In some cases there is a loss of myelin, but relative preservation of axons in the deep white matter. This pattern tends to be associated with a delayed clinical deterioration. Arcuate fibers are usually spared (Fig. 25.14).
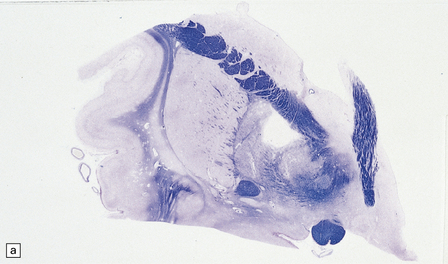
25.13 Histology of the globus pallidus in survivors of acute carbon monoxide poisoning.
(a) Cavitation confined to the dorsomedial part of the globus pallidum. (b) More extensive bilateral pallidal cavitation. (Courtesy of Dr R J Vieira de Mello, Department of Pathology, School of Medicine, Federal University of Pernambuco, Brazil.)
25.14 Extensive loss of myelinated fibers caused by carbon monoxide poisoning.
Note the sparing of the subcortical arcuate fibers.
 CAUSES OF BILATERAL BASAL GANGLIA NECROSIS
CAUSES OF BILATERAL BASAL GANGLIA NECROSIS







CYANIDE
 CYANIDE EXPOSURE
CYANIDE EXPOSURE Cyanide binds strongly to the trivalent iron in cytochrome oxidase, thereby preventing oxidative phosphorylation. Because the cells are therefore unable to use oxygen, this type of toxicity has been termed cytotoxic hypoxia.
Cyanide binds strongly to the trivalent iron in cytochrome oxidase, thereby preventing oxidative phosphorylation. Because the cells are therefore unable to use oxygen, this type of toxicity has been termed cytotoxic hypoxia.

 Industrial exposure can occur during electroplating and gold or silver ore extraction.
Industrial exposure can occur during electroplating and gold or silver ore extraction.

 Hydrogen cyanide is released from many building materials and furnishings during fires.
Hydrogen cyanide is released from many building materials and furnishings during fires.


MACROSCOPIC APPEARANCES
 CYANIDE TOXICITY
CYANIDE TOXICITY



 SAFETY PRECAUTIONS WHEN PERFORMING A NECROPSY IN A CASE OF ACUTE CYANIDE INGESTION
SAFETY PRECAUTIONS WHEN PERFORMING A NECROPSY IN A CASE OF ACUTE CYANIDE INGESTIONNITROUS OXIDE
 NITROUS OXIDE EXPOSURE
NITROUS OXIDE EXPOSURE


 NITROUS OXIDE TOXICITY
NITROUS OXIDE TOXICITY

ANTIVIRAL, ANTIBACTERIAL, ANTIFUNGAL, AND ANTIPROTOZOAL DRUGS
CLIOQUINOL
 CLIOQUINOL EXPOSURE
CLIOQUINOL EXPOSURE

 SUBACUTE MYELO-OPTIC NEUROPATHY (SMON)
SUBACUTE MYELO-OPTIC NEUROPATHY (SMON)
 The full-blown syndrome of SMON is relatively uncommon and consists of impaired vision with optic atrophy, subacute myelopathy producing spasticity and dysesthesiae of the lower limbs and trunk, and peripheral neuropathy.
The full-blown syndrome of SMON is relatively uncommon and consists of impaired vision with optic atrophy, subacute myelopathy producing spasticity and dysesthesiae of the lower limbs and trunk, and peripheral neuropathy.
 Many patients develop isolated optic atrophy or a combination of optic atrophy and myelopathy.
Many patients develop isolated optic atrophy or a combination of optic atrophy and myelopathy.
 High doses of clioquinol can produce an acute encephalopathy with transient global amnesia.
High doses of clioquinol can produce an acute encephalopathy with transient global amnesia.






HEXACHLOROPHENE (USA)/HEXACHLOROPHANE (UK)
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Intoxication causes spongy degeneration of the white matter due to intramyelinic edema (see above), which is most marked in the brain stem (Fig. 25.15). Necrosis of the optic nerves, chiasm, and tracts has also been reported.
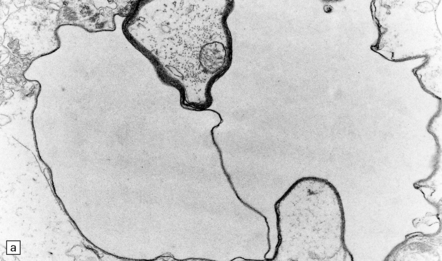
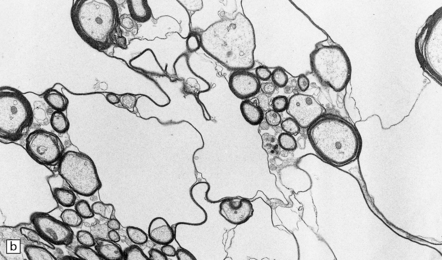
25.15 Hexachlorophene intoxication.
(a) Electron micrograph showing separation of myelin lamellae around an axon in the brain stem of a boy born at 32 weeks’ gestation, who had been washed repeatedly with hexachlorophene. (b) Intramyelinic edema due to experimental hexachlorophene intoxication in the rat. (Courtesy of Dr H Powell, University of California, San Diego.)









ANTINEOPLASTIC AND IMMUNOSUPPRESSIVE AGENTS
CALCINEURIN INHIBITORS: CYCLOSPORINE A (CSA) AND TACROLIMUS (FK506)
 CYCLOSPORINE AND TACROLIMUS TOXICITY
CYCLOSPORINE AND TACROLIMUS TOXICITY
 Patients usually present with posterior reversible encephalopathy syndrome, comprising varying combinations of headache, altered mental functioning, seizures, cortical blindness, and speech or language abnormalities. These abnormalities usually regress after dose reduction or cessation of drug administration, but patients may be left with permanent neurologic deficits.
Patients usually present with posterior reversible encephalopathy syndrome, comprising varying combinations of headache, altered mental functioning, seizures, cortical blindness, and speech or language abnormalities. These abnormalities usually regress after dose reduction or cessation of drug administration, but patients may be left with permanent neurologic deficits.

 Patients may develop hypertension.
Patients may develop hypertension.
 Subarachnoid or parenchymal brain hemorrhage has been reported in a few cases.
Subarachnoid or parenchymal brain hemorrhage has been reported in a few cases.

MACROSCOPIC AND MICROSCOPIC APPEARANCES
There are few reports on the neuropathologic findings in human toxicity. The parieto-occipital white matter may be edematous. Electron microscopy in one case showed detachment of astrocytic foot-processes from blood vessels in the white matter. Vasculitis has also been reported. Rarely, there is also extensive axonal damage and degeneration, with loss of myelin and infiltration by macrophages (Fig. 25.16).
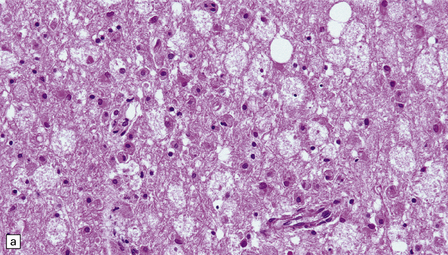
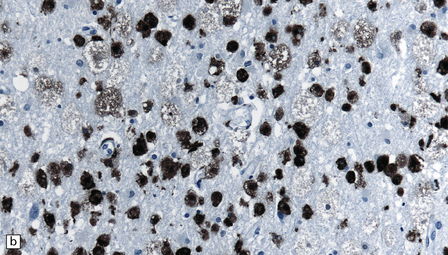

25.16 Cyclosporine neurotoxicity.
(a) Section of occipital white matter from a patient who developed an unusually severe leukoencephalopathy after receiving cyclosporine to prevent rejection of a bone marrow transplant. The tissue is edematous, and infiltrated by foamy cells. (b) Immunohistochemistry for CD68 shows the foamy cells to be macrophages. (c) Immunohistochemistry also reveals abnormal accumulation of β-amyloid precursor protein in swollen, fragmenting axons.
METHOTREXATE
 METHOTREXATE TOXICITY
METHOTREXATE TOXICITY Acute symptoms include headache, confusion, seizures, and, rarely, transient hemiplegia.
Acute symptoms include headache, confusion, seizures, and, rarely, transient hemiplegia.
 Some days or weeks after administration, patients may develop:
Some days or weeks after administration, patients may develop:
l. a subacute encephalopathy characterized by a gradual onset of drowsiness, irritability, mental deterioration, cerebellar dysfunction, parkinsonian features, and seizures.
 These subacute complications are usually irreversible and may be fatal.
These subacute complications are usually irreversible and may be fatal.
 Computerized tomography reveals low-density white matter lesions, calcification, and atrophy.
Computerized tomography reveals low-density white matter lesions, calcification, and atrophy.

MICROSCOPIC APPEARANCES
There is a loss of myelin and swelling and fragmentation of axons within foci of coagulative necrosis in the white matter (Fig. 25.17). Multifocal axonal injury with accumulation of β-amyloid precursor protein may be demonstrable immunohistochemically. Lipid-laden macrophages are plentiful in the early stages. The axonal swellings and other cellular debris tend to calcify. In longer-term survivors the white matter may be reduced to an attenuated layer of gliotic, focally calcified tissue. Vascular abnormalities are present in some cases and include perivascular fibrin exudates, fibrinoid vascular necrosis, and a non-inflammatory mineralizing angiopathy, which is most severe in the lenticular nuclei. Intra-arterial methotrexate administration has caused multiple hemorrhagic infarcts due to fibrinoid necrosis and thrombosis of small blood vessels.
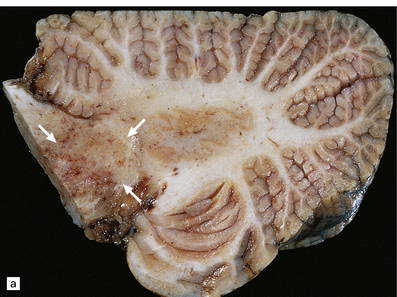
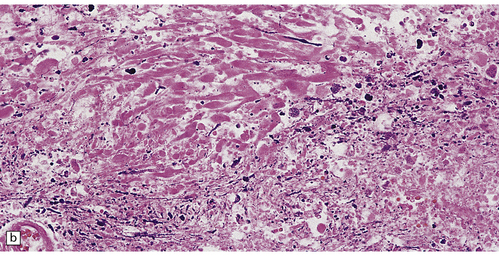
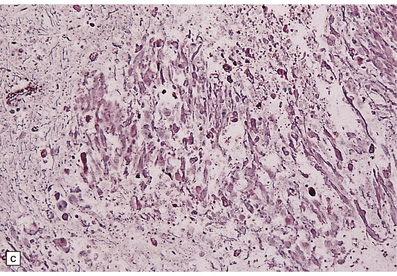
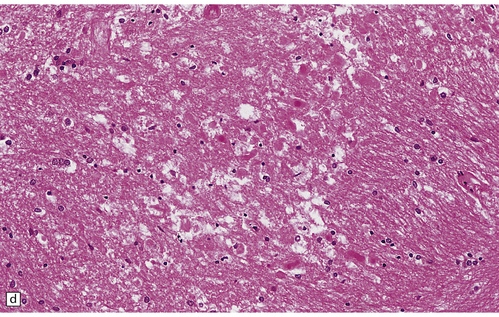
25.17 Methotrexate toxicity.
(a) Large focus of necrosis (arrows) in the middle cerebellar peduncle of a 6-year-old girl who had received repeated intrathecal methotrexate for leukemia with CNS relapses. (b) Histology shows that the lesion contains swollen axons, many of which are mineralized. (c) Bodian silver impregnation of the swollen axons. (Courtesy of Professor E Tessa Hedley-Whyte, Massachusetts General Hospital.) (d) In this case, the changes are much more subtle: swollen and fragmented axons in the cerebral white matter of a child, who died shortly after completing a course of chemotherapy that included high-dose systemic methotrexate for the treatment of lymphoma.
 CAUSES OF MULTIFOCAL NECROTIZING LEUKOENCEPHALOPATHY (MNL)
CAUSES OF MULTIFOCAL NECROTIZING LEUKOENCEPHALOPATHY (MNL)






NUCLEOSIDE ANALOGS
 TOXICITY OF NUCLEOSIDE ANALOGS USED IN CHEMOTHERAPY
TOXICITY OF NUCLEOSIDE ANALOGS USED IN CHEMOTHERAPY









MACROSCOPIC AND MICROSCOPIC APPEARANCES
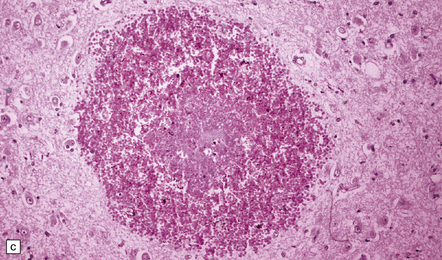
Autopsy examination of the brain in patients with fludarabine-associated neurotoxicity has shown a necrotizing leukoencephalopathy (Fig. 25.18) most severe in the occipital lobes, but also involving the medullary pyramids and posterior spinal columns. The subcortical white matter lesions may extend into the adjacent cerebral cortex.
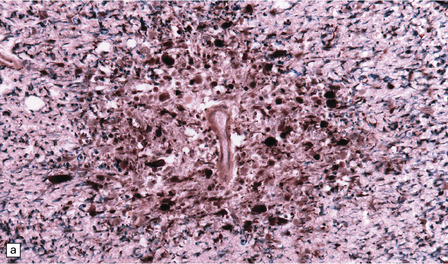
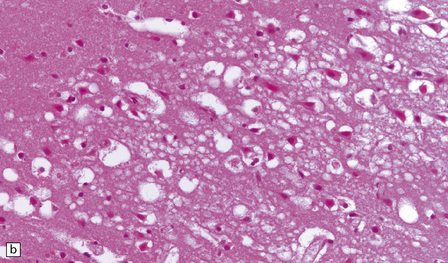
25.18 Fludarabine neurotoxicity.
(a) Unusually severe necrotizing leukoencephalopathy in a leukemic patient who developed an acute fatal encephalopathy after fludarabine administration. This lesion is centered on a blood vessel and shows axonal swelling and fragmentation. Larger lesions in the same patient were not obviously related to blood vessels. (b) Vacuolation and neuronal degeneration in the cerebral cortex overlying a regions of white matter necrosis.
MISCELLANEOUS THERAPEUTIC OR DIAGNOSTIC DRUGS
PHENYTOIN
MACROSCOPIC AND MICROSCOPIC APPEARANCES
There may be obvious atrophy of the cerebellar vermis and hemispheres (Fig. 25.19). Microscopic findings are relatively nonspecific, with severe loss of Purkinje, and usually, granule cells (Fig. 25.19). There is marked proliferation of Bergmann astrocytes, with associated isomorphic gliosis (Fig. 25.19). The cerebellar cortical degeneration is usually diffuse, but may be patchy.
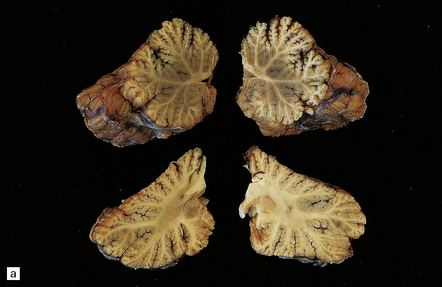
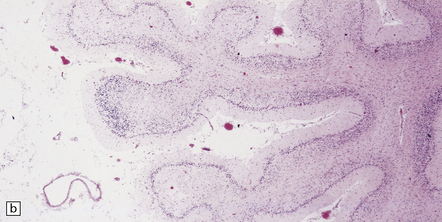
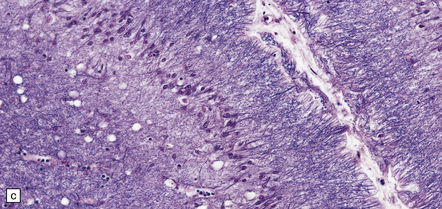
25.19 Phenytoin toxicity.
(a) Atrophy of the cerebellar vermis and hemispheres in a well-controlled epileptic patient who had been taking phenytoin for many years. (b) Severe loss of Purkinje and granule cells. The superficial parts of the folia are most severely affected. (c) Proliferation of Bergmann astrocytes with associated isomorphic gliosis.
 PHENYTOIN EXPOSURE
PHENYTOIN EXPOSURE The principal use of phenytoin (diphenylhydantoin) is as an anticonvulsant, but it has also been used to treat trigeminal neuralgia and ventricular arrhythmias.
The principal use of phenytoin (diphenylhydantoin) is as an anticonvulsant, but it has also been used to treat trigeminal neuralgia and ventricular arrhythmias.

l. has occurred in patients who have never had seizures, to whom phenytoin was given for cardiac arrhythmias or prophylactically after uncomplicated subarachnoid hemorrhage
l. is not related to the frequency or total number of seizures
l. can occur in patients treated for partial seizures and not just in those subject to generalized tonic–clonic convulsions.
 PHENYTOIN TOXICITY
PHENYTOIN TOXICITY

 ANTIEPILEPTIC DRUGS AND FETAL DEVELOPMENT
ANTIEPILEPTIC DRUGS AND FETAL DEVELOPMENT
 Most of the commonly used antiepileptic drugs are teratogenic, especially in high doses, if used in combination, and if administered during the first trimester. Overall, the risk of major fetal malformations is increased by about 3–5%.
Most of the commonly used antiepileptic drugs are teratogenic, especially in high doses, if used in combination, and if administered during the first trimester. Overall, the risk of major fetal malformations is increased by about 3–5%.

 Valproate administration carries an approximately 2% risk of spina bifida.
Valproate administration carries an approximately 2% risk of spina bifida.
STREET DRUGS
COCAINE
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Neuropathologic complications include arterial infarcts, and subarachnoid or parenchymal hemorrhages (Fig. 25.20):



The spinal cord may be affected.
Rupture of saccular (berry) aneurysms (see Chapter 10) is a well-documented complication of cocaine abuse in young adults and is thought to be due to the transient, but occasionally marked, hypertension.
HEROIN (DIAMORPHINE)
Heroin is usually injected intravenously or snorted. Neurologic manifestations of toxicity range from anorexia, nausea, and vomiting, to cardiorespiratory depression, coma, and death. Ischemic CNS complications include cerebral infarcts, ischemic myelopathy, and global hypoxic–ischemic brain injury (Fig. 25.21).
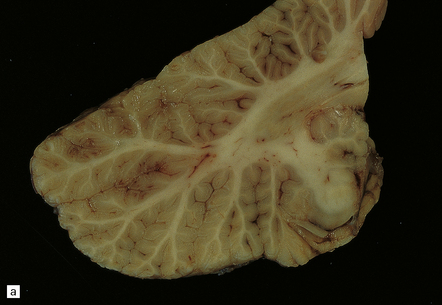
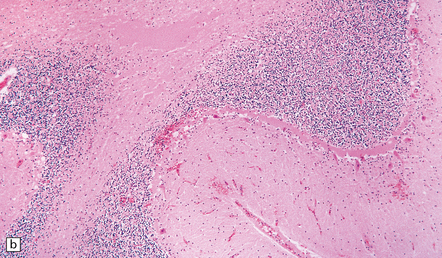
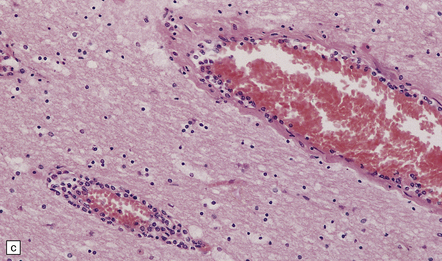
25.21 Acute cerebellar infarct in a heroin addict.
(a) Folia in the superior part of the cerebellar hemisphere show dusky discoloration and loss of definition of the white matter. (b) Histology confirms acute infarction, with congestion and edema. (c) Blood vessels in the adjacent white matter show margination and scanty perivascular cuffing by mononuclear inflammatory cells, but no vasculitis.
MACROSCOPIC AND MICROSCOPIC APPEARANCES
 CEREBROVASCULAR COMPLICATIONS OF ‘RECREATIONAL’ DRUGS
CEREBROVASCULAR COMPLICATIONS OF ‘RECREATIONAL’ DRUGS


 ABUSE OF VASOACTIVE DRUGS DURING PREGNANCY
ABUSE OF VASOACTIVE DRUGS DURING PREGNANCY

Angiographic findings have been suggestive of a small vessel angiitis in some patients. However, the predominant findings have been watershed infarcts, laminar sclerosis, and bilateral infarcts of the globus pallidum, all presumably due to a combination of reduced cerebral perfusion and global hypoxia. The spinal cord may be affected. Refractile embolic particles of foreign material have been observed in the skin, and rarely in the spinal cord of heroin users, but not in the brain.
MPTP (1-METHYL-4-PHENYL-1,2,3,6-TETRAHYDROPYRIDINE)
 MPTP
MPTP


MACROSCOPIC AND MICROSCOPIC APPEARANCES
Neuropathologic examination reveals nigrostriatal neuronal degeneration in a pattern closely resembling that of idiopathic Parkinson’s disease (see Chapter 29). Typical Lewy bodies are, however, not seen. In experimental studies:
 inclusion bodies have been noted within distorted mitochondria in the substantia nigra and locus ceruleus after acute intoxication.
inclusion bodies have been noted within distorted mitochondria in the substantia nigra and locus ceruleus after acute intoxication.

 CAUSES OF IRREVERSIBLE, DRUG-INDUCED PARKINSONISM
CAUSES OF IRREVERSIBLE, DRUG-INDUCED PARKINSONISM








ETHANOL (‘ALCOHOL’)
Ethanol affects the nervous system through a variety of direct and indirect mechanisms, the latter including metabolic disturbances produced by ethanol-induced organ (especially liver) damage and various nutritional deficiencies that tend to complicate chronic alcohol abuse. In addition, alcoholism predisposes to infections, is associated with an increased likelihood of sustaining traumatic injuries, and increases the risk of hemorrhagic strokes. Alcoholism and its associated nutritional deficiencies are important causes of peripheral neuropathy. Some of the direct and indirect toxic effects of ethanol on the CNS are discussed below, while Wernicke–Korsakoff syndrome and pellagra are discussed in the context of nutritional deficiencies (see Chapter 21). Central pontine myelinolysis is considered with other metabolic disorders in Chapter 22.
CNS EFFECTS OF CHRONIC ALCOHOLISM
CNS effects include cerebral atrophy, cerebellar degeneration, Marchiafava–Bignami disease, Morel’s laminar sclerosis, and fetal alcohol syndrome. Chronic alcoholism also predisposes to a range of other disorders including Wernicke’s encephalopathy, Wernicke– Korsakoff syndrome, and pellagra (see Chapter 21), and central pontine myelinolysis (see Chapter 22).
CEREBELLAR DEGENERATION
MACROSCOPIC AND MICROSCOPIC APPEARANCES
Neuropathologic examination reveals atrophy of the superior part of the vermis and adjacent regions of the cerebellar hemispheres, with atrophy of the folia and widening of the sulci (Fig. 25.22). In contrast to the pattern of degeneration resulting from hypoxia, the crests of the folia tend to be more severely affected than the depths of the sulci. There is a loss of Purkinje cells, a patchy loss of granule cells with resulting atrophy of the molecular layer, and proliferation of Bergmann glia (Fig. 25.22). Neuronal loss and gliosis are usually evident in the dorsal layer of the inferior olivary nuclei (Fig. 25.22). The vestibular, fastigial, globose, and emboliform nuclei may show mild degenerative changes.
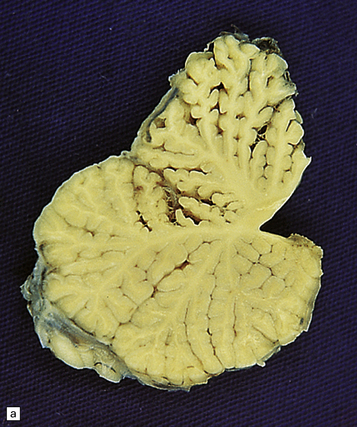
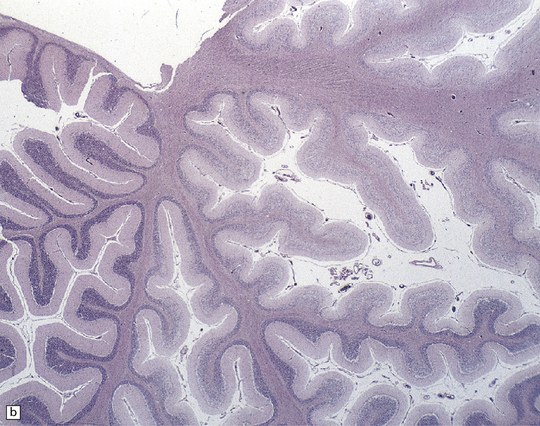
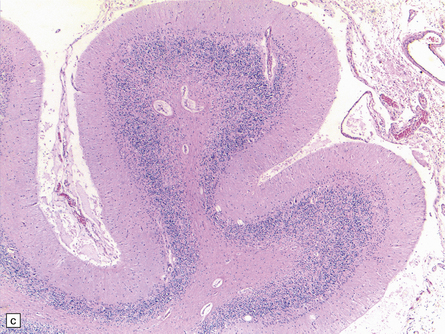
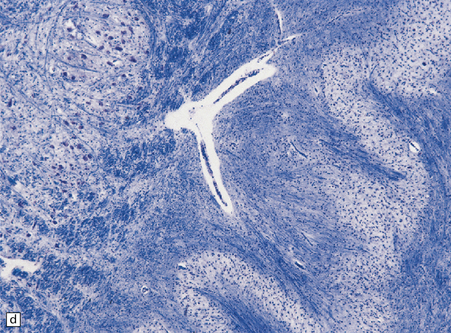
25.22 Alcoholic cerebellar degeneration.
(a) Atrophy of the superior part of the vermis complicating chronic alcoholism. (b) Histology shows the crests of the folia usually to be more severely affected than the depths of the sulci in chronic alcoholism, but the distribution of degeneration may be rather patchy. (c) Higher magnification reveals loss of Purkinje cells with thinning of the molecular layer and proliferation of Bergmann glia. (d) Neuronal loss and gliosis in the dorsal layer of the inferior olivary nucleus. In contrast, the dorsal accessory olivary nucleus (towards the left of the figure) is well preserved.
MARCHIAFAVA–BIGNAMI DISEASE
 MARCHIAFAVA–BIGNAMI DISEASE
MARCHIAFAVA–BIGNAMI DISEASE Although left-sided apraxia, hemidyslexia, and other callosal disconnection syndromes have been reported, the clinical features are variable and often nonspecific. They include tremor, unsteadiness of gait, and confusion, and in acute cases, convulsions progressing to coma.
Although left-sided apraxia, hemidyslexia, and other callosal disconnection syndromes have been reported, the clinical features are variable and often nonspecific. They include tremor, unsteadiness of gait, and confusion, and in acute cases, convulsions progressing to coma.
 The disease is often fatal, though spontaneous recovery may occur.
The disease is often fatal, though spontaneous recovery may occur.

MACROSCOPIC APPEARANCES
Macroscopically, the central part of the corpus callosum appears shrunken, and dusky gray or yellow, depending on the age of the lesion (Fig. 25.23), and there may be cavitation. Similar lesions are occasionally seen in the optic chiasm, anterior and posterior commissures, and middle cerebellar peduncles. Striatal necrosis is an inconstant finding.
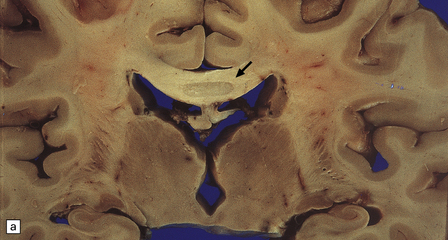

MICROSCOPIC APPEARANCES
Histologically, the disorder is characterized by variable combinations of necrosis and demyelination. These most consistently involve the genu and body of the corpus callosum and spare a thin layer of myelinated fibers along the dorsal and ventral surfaces (Fig. 25.23). Other white matter structures may be involved, as noted above. Macrophages are abundant in the early stages, but lymphocytic inflammation is not a feature. Oligodendrocytes are markedly reduced in number, and astrocytes generally show only modest reactive changes. A striking feature is the proliferation and hyaline thickening of small blood vessels.
MOREL’S LAMINAR SCLEROSIS
This is characterized by a band of spongiosis and gliosis involving the third layer (and, in some cases, deeper layers also) of the frontal and temporal cortex (Fig. 25.24). It is usually associated with, and may be secondary to, the callosal lesions of Marchiafava–Bignami disease, but has also been reported as an isolated finding in chronic alcoholics.
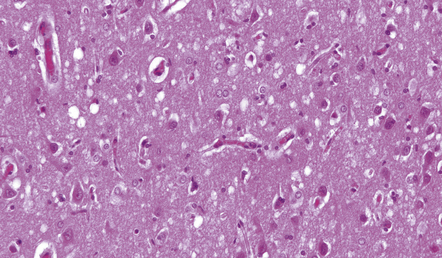
25.24 Morel’s laminar sclerosis.
This patient developed hepatic encephalopathy due to alcoholic cirrhosis. Autopsy examination of the brain revealed laminar spongiosis in the deeper parts of the frontal and temporal cortex. The abnormal cortex contains scattered reactive and Alzheimer type II astrocytes, and shows vacuolation resembling that of Creutzfeldt–Jakob disease, with which these histologic changes may be confused.
BIOLOGIC TOXINS
CYCAD TOXIN
It has been suggested that environmental factors (possibly the local use of seeds of the indigenous cycad, Cycas circinalis, in food and medicine) are involved in the development of the distinct variants of amyotrophic lateral sclerosis (ALS) and parkinsonism–dementia syndrome that are particularly prevalent in Guam and other Western Pacific islands (see Chapter 28). The cycad’s seeds contain the amino acid L-β-methylaminoalanine, which has been reported to produce a disorder resembling motor neuron disease in rhesus monkeys.
DOMOIC ACID
 DOMOIC ACID (DA) EXPOSURE
DOMOIC ACID (DA) EXPOSURE

 AMNESIC SHELLFISH POISONING
AMNESIC SHELLFISH POISONING

REFERENCES
Bolon, B., Garman, R., Jensen, K., Ad Hoc Working Group of the STP Scientific and Regulatory Policy Committee. A ‘best practices’ approach to neuropathologic assessment in developmental neurotoxicity testing – for today. Toxicol Pathol. 2006;34:296–313.
Calabrese, E.J. Astrocytes: adaptive responses to low doses of neurotoxins. Crit Rev Toxicol.. 2008;38:463–471.
Eicher, T., Avery, E. Toxic encephalopathies. Neurol Clin.. 2005;23:353–376.
Finkelstein, Y., Milatovic, D., Lazarovici, P., et al. Peaceful use of disastrous neurotoxicants. Neurotoxicology.. 2010;31:608–620.
Genuis, S.J. Toxic causes of mental illness are overlooked. Neurotoxicology.. 2008;29:1147–1149.
Guidotti, M., Chiveri, L., Mauri, M. Acute encephalopathies. Neurol Sci.. 2006;27:S55–S56.
Philbert, M.A., Billingsley, M.L., Reuhl, K.R. Mechanisms of injury in the central nervous system. Toxicol Pathol.. 2000;28:43–53.
Segura Aguilar, J., Kostrzewa, R.M. Neurotoxins and neurotoxic species implicated in neurodegeneration. Neurotox Res. 2004;6:615–630.
Wallace, D.R. Overview of molecular, cellular, and genetic neurotoxicology. Neurol Clin.. 2005;23:307–320.
Bondy, S.C. The neurotoxicity of environmental aluminum is still an issue. Neurotoxicology.. 2010;31:575–581.
Burton, N.C., Guilarte, T.R. Manganese neurotoxicity: lessons learned from longitudinal studies in nonhuman primates. Environ Health Perspect.. 2009;117:325–332.
Ceccatelli, S., Daré, E., Moors, M. Methylmercury-induced neurotoxicity and apoptosis. Chem Biol Interact.. 2010;188:301–308.
Crossgrove, J., Zheng, W. Manganese toxicity upon overexposure. NMR Biomed.. 2004;17:544–553.
Dobson, A.W., Erikson, K.M., Aschner, M. Manganese neurotoxicity. Ann N Y Acad Sci.. 2004;1012:115–128.
Fitsanakis, V.A., Aschner, M. The importance of glutamate, glycine, and gamma-aminobutyric acid transport and regulation in manganese, mercury and lead neurotoxicity. Toxicol Appl Pharmacol.. 2005;204:343–354.
Jenkins, S.M., Barone, S. The neurotoxicant trimethyltin induces apoptosis via caspase activation, p38 protein kinase, and oxidative stress in PC12 cells. Toxicol Lett.. 2004;147:63–72.
Lidsky, T.I., Schneider, J.S. Lead neurotoxicity in children: basic mechanisms and clinical correlates. Brain.. 2003;126:5–19.
Murata, K., Iwata, T., Dakeishi, M., et al. Lead toxicity: does the critical level of lead resulting in adverse effects differ between adults and children? J Occup Health.. 2009;51:1–12.
Sanders, T., Liu, Y., Buchner, V., et al. Neurotoxic effects and biomarkers of lead exposure: a review. Rev Environ Health.. 2009;24:15–45.
White, L.D., Cory-Slechta, D.A., Gilbert, M.E., et al. New and evolving concepts in the neurotoxicology of lead. Toxicol Appl Pharmacol.. 2007;225:1–27.
Wright, R.O., Baccarelli, A. Metals and neurotoxicology. J Nutr.. 2007;137:2809–2813.
Exon, J.H. A review of the toxicology of acrylamide. J Toxicol Environ Health B Crit Rev.. 2006;9:397–412.
LoPachin, R.M. Acrylamide neurotoxicity: neurological, morphological and molecular endpoints in animal models. Adv Exp Med Biol.. 2005;561:21–37.
van Valen, E., Wekking, E., van der Laan, G., et al. The course of chronic solvent induced encephalopathy: a systematic review. Neurotoxicology.. 2009;30:1172–1186.
Hu, L.F., Lu, M., Wong, P.T., et al. Hydrogen sulfide: neurophysiology and neuropathology. Antioxid Redox Signal.. 2011;15:405–419.
Nishiwaki, Y., Takebayashi, T., O’Uchi, T., et al. Six year observational cohort study of the effect of carbon disulphide on brain MRI in rayon manufacturing workers. Occup Environ Med.. 2004;61:225–232.
Antiviral, antibacterial, antifungal, and antiprotozoal drugs
Preziosi, P. Isoniazid: metabolic aspects and toxicological correlates. Curr Drug Metab.. 2007;8:839–851.
Antineoplastic and immunosuppressive agents
Choi, S.M., Lee, S.H., Yang, Y.S., et al. 5-fluorouracil-induced leukoencephalopathy in patients with breast cancer. J Korean Med Sci.. 2001;16:328–334.
Gijtenbeek, J.M., van den Bent, M.J., Vecht, C.J. Cyclosporine neurotoxicity: a review. J Neurol.. 1999;246:339–346.
Pirzada, N.A., Ali, I.I., Dafer, R.M. Fluorouracil-induced neurotoxicity. Ann Pharmacother.. 2000;34:35–38.
Cavaletti, G., Nicolini, G., Marmiroli, P. Neurotoxic effects of antineoplastic drugs: the lesson of pre-clinical studies. Front Biosci.. 2008;13:3506–3524.
Windebank, A.J., Grisold, W. Chemotherapy-induced neuropathy. J Peripher Nerv Syst.. 2008;13:27–46.