Toxic Gas, Fume, and Smoke Inhalation
Introduction
Epidemiology
Chronic exposure to inhalation of atmospheric pollution may damage the lung over decades, predisposing to infection, pulmonary fibrosis, or cancer.1 The World Health Organization estimates that more than a billion people, mainly in developing countries, develop airway and pulmonary inflammation resulting from inhaled smoke from indoor cooking fires, forest fires, and burning of crops.2 In the industrial world, chronic inhalation injury may be due to cigarette smoking or occupational exposure (e.g., asbestos). This aspect of inhalation injury will not be further discussed in this textbook, and readers are referred to other resources.3
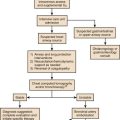
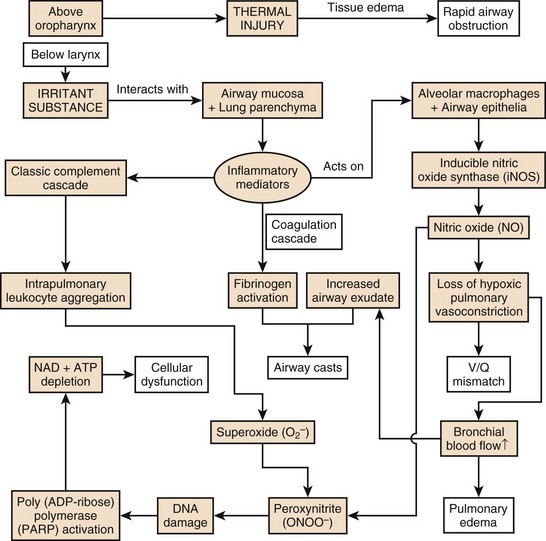
Acute smoke inhalation results in approximately 23,000 injuries and 5000 to 10,000 deaths per year in the United States alone. Among industrial countries, the United States has one of the highest incidences of smoke inhalation injuries.4 The ensuing pulmonary derangements, which follow burn and smoke inhalation injuries, are major contributors to morbidity and mortality in fire victims. The pathophysiology of the injury is multifaceted and induces distant organ dysfunction (Fig. 48.1). The consequences of the profound airway inflammation are heightened by pulmonary shunting and augmented microvascular pressure gradient, resulting in hypoxemic respiratory failure.5 Although survival from burn injury continues to improve, this is not mirrored in inhalation management. The acute lung injury caused by smoke inhalation and pneumonia has a major negative impact on mortality figures in patients with burn injuries. Inhalation injury alone increases mortality in burn victims by approximately 20% and pneumonia increases the rate by approximately 40%, with a maximal increase of approximately 60% when both are present.6 The mainstay of treatment of the smoke inhalation sufferer remains optimal respiratory support with airway toilet, adequate fluid resuscitation, early and aggressive surgical interventions, and precise antimicrobial interventions. Continued research into this systemic process is needed to stop smoke inhalation injury being the major cause of death in fire victims.7
History
Inhalation injuries, both of toxins and smoke, have been recorded in the history books for several thousand years and have been used with enmity from the outset. Thucydides records the Spartans burning pitch, naphtha, and sulfur to produce sulfur dioxide while attacking Athenian cities in 423 BC.8 The fifteenth century brought incendiary devices filled with sulfa and belladonna.9
The history of toxic gases other than smoke tends to run hand in hand with military conflict, and it wasn’t until the First World War that the increased usage sparked off once more, where at least 14 different toxic respiratory agents were used. The years 1914-1915 marked the modern nascence of inhalation warfare when France released chloroacetone and Germany released thousands of liters of chlorine gas in Belgium, at Ypres. More than 1 million casualties were attributed to the use of chemical agents during that war, with sulfur gas being anointed as the “king of battle gases.”10
The 1925 Geneva Protocol signed by many countries pledged never to use gas warfare again.9 Sadly, this has not been adhered to, with Italy being accused of using mustard gas against Abyssinia in Ethiopia. The Chinese suffered gas inhalation at the hands of the Japanese during the Second World War, and the Kurds were victim to similar agents during the attacks by Iraq through the 1980s.11 Sarin gas poisoning was used in Japan in the mid-1990s. In 1994 and 1995, inhaled biochemical weapons have been used for terrorism acts like the sarin gas attacks in Japan.12 Cyanide (CN) may be considered one of the most likely agents of chemical terrorism, as it is capable of causing mass incapacitation and casualties and can cause mass confusion, panic, and social disruption. In addition, cyanide possesses all attributes of an ideal terrorist weapon: it is plentiful, readily available, and easily obtainable because of its widespread use in industry and laboratories.13 Although military interest in biochemical warfare diminished following World War II, there was an understanding of the devastation possible with nuclear weapons; terrorist organizations understand the fear, panic, and collapse of infrastructure that could be realized through the release of such a substance in a busy city. It seems likely that there are many areas of the world where such agents are, or can be, manufactured in great quantities.9
Equally, smoke inhalation has a long record in history, with the first recording by Pliny in the first century AD. He described the execution of prisoners over greenwood smoke, and it seems he may have died through toxic smoke inhalation himself during the eruption of Vesuvius in 79 AD. More recently, two much more industrial occurrences highlighted the problems of smoke inhalation. The Cocoanut Grove fire of 1942 resulted in the deaths of 491 people who were trapped in a burning building. The number of patients who sustained burns was minimal, and it was then that the realization hit that smoke alone could kill as easily, if not more so, than cutaneous burns.14 More than 2000 burn/smoke casualties resulted after the chain of fires and explosions that rippled through refineries and factories in Texas City, Texas, in the 1940s.15 The understanding of smoke and carbon monoxide (CO) inhalation in enclosed spaces was further highlighted in the fire at the MGM Grand Hotel in Las Vegas in 1980 and the Stardust Nightclub fire in Dublin in 1981. Again, the small number of burn injuries was swamped by the deaths resulting from smoke and CO.16 Cyanide’s independent role became clear in the 1980s, particularly after the aircraft fire at Manchester International Airport, Manchester, UK, in 1985 where the majority (87%) of the 54 individuals who died had potentially lethal levels of cyanide in their blood, as opposed to only 21% of these victims having carboxyhemoglobin (COHb) levels exceeding 50%. This event highlighted that different combustants produce different inhalants, and depending on the environment, these may be more lethal than carbon monoxide, which had been regarded as the primary toxic threat.4 Hence, the determinants of inhalants are both environment and material being combusted. It is therefore a mixed toxicology following smoke inhalation.
The terrorism attacks on September 11, 2001, on the World Trade Center in New York were associated with a high incidence of inhalation injuries. Among the 790 injured survivors, 49% suffered from inhalation injury caused by toxic compounds in the smoke and dust.17,18 Industrial catastrophes, biochemical warfare, and terrorism will continue to occur. This chapter’s aim is to help physicians diagnose and then manage patients with inhalation injuries.
Pathogenesis of Inhalation Injury
Toxic Smoke Compounds
Smoke is a heterogeneous compound. Each fire produces different toxic features relating to the material combusted and the environment in which the fire occurs, specifically oxygen content. Hence, each patient suffering smoke inhalation may represent a new condition with a possibility of many different inhalants.19 The components of smoke that cause damage are as follows:
• Particulates, deposited in the airways according to their size, with substances smaller than 1 µm in diameter able to reach the alveolar zone suspended in air. At that site, these chemically laden particles increase airway resistance and cause cell lysis and irritation while diminishing pulmonary surfactant production and efficacy.19
• Systemic toxins, such as carbon monoxide and cyanide, which adversely affect oxygen transport by erythrocytes and utilization by mitochondria.20
• Respiratory irritants are implicated in the high mortality rates. Water-soluble gases such as ammonia and hydrogen chloride react with water contained in mucous membranes and produce strong alkalis and acids, which elicit profound inflammatory reactions, which rapidly induce systemic changes via the dense alveolar-capillary interphase. Lipid-soluble irritants (e.g., oxides of nitrogen, phosgene, and aldehydes) exert their effects more slowly as they dissolve into the cellular membrane.21
Heat
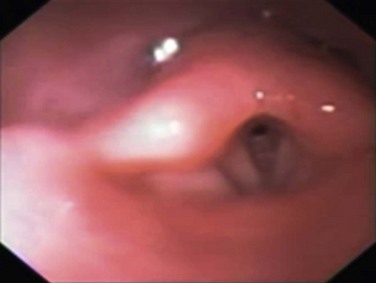
Burns to the nasal and oropharyngeal mucosa are common in fire-exposed victims, but it is rare to encounter thermal injury below the vocal cords. This is because the oropharynx acts as an effective “heat sink,” with the thermal energy of the heated air dissipating into the cells they pass by, causing rapid cell injury, necrosis, and swelling of the upper airway (Fig. 48.2). This can result in upper airway obstruction, which can be fatal before the sequelae of pulmonary burn become apparent. Super-heated steam is an exception, where the oropharynx cannot absorb all the thermal energy, and hence airway burn occurs in this situation.22
Systemic Toxins
Systemic toxins are products of incomplete combustion and include carbon monoxide and hydrogen cyanide. Carbon monoxide intoxication, together with heat incapacitation and a hypoxic environment, is the most common immediate cause of death from fire.20 Carbon monoxide is an odorless, colorless gas that binds to erythrocyte hemoglobin with about 250 times the affinity of oxygen. The resulting COHb molecule is unable to transport oxygen, thus impairing oxygen delivery to the tissue and shifting the oxygen-dissociation curve to the left.23 Furthermore, at the tissue level, carbon monoxide competes with, and inhibits, oxygen binding to the cytochrome oxidase system of enzymes, inhibiting the aerobic metabolism chain and thus incapacitating cellular respiration.21 Hence, CO paralyzes oxygen carriage in the blood and subsequent utilization in the tissue (see Table 48.2, presented later in the chapter). Thermal decomposition of nitrogen-containing polymers in oxygen-poor environments produces smoke containing hydrogen cyanide, inhibiting electron transport and cellular respiration.19
Table 48.2
Carboxyhemoglobin Concentration and Related Symptoms
| COHb [in %] | Symptoms |
| <20 | Slight headache and dilation of peripheral blood vessels |
| 21-40 | Severe headache and pulsating in temporal blood vessels, vertigo, dizziness, nausea and vomiting, circulatory collapse |
| 41-60 | Symptoms as above, syncope, tachycardia, hyperventilation, intermittent seizures, cyanosis, coma, shock, Cheyne-Stokes respiration |
| 61-80 | Coma, intermittent seizures, impaired heart and lung function, weak pulses, slow breathing, death within hours |
| >81 | Death occurs within minutes |
Airway Injury
Although the inhaled gases as described earlier can cause significant and fatal alterations in physiology, the chief mediator of the pathophysiology of smoke inhalation is particulate matter. Carbonaceous particles (soot) impregnated with a multitude of toxins reach the alveolar level suspended in air.24 The chemicals associated with these particles vary depending on the products combusted but commonly include aldehydes from cellulose-based materials such as wood and paper; nitrogen oxides from fabric combustion; halogen acids and sulfur dioxide from rubber; ammonia from wool, silk, and polyurethane; and phosgene from polyvinyl chloride.22 Water-soluble compounds are readily soluble in airway mucus and interact freely with tissue at more proximal levels of the respiratory system. Less water-soluble compounds (such as phosgene) penetrate the airway mucosa deeply and may cause severe delayed damage through late interaction with distal airway tissues, up to 48 hours after exposure. This is an important consideration when treating patients who initially present with apparently mild clinical effects after smoke inhalation.22
Pulmonary Parenchymal Injury
Although thermal injury is mostly adsorbed in the upper airway, the other components of smoke—particulate materials, systemic toxins, and respiratory irritants—descend to the lung and trigger a cascade of events, resulting in pulmonary edema, bronchiolar obstruction, cell death, and ventilation/perfusion (V/Q) mismatch.19
Of paramount importance is the cascade of inflammatory mediators activated by the interaction of irritant substances with the airway mucosa and lung parenchyma. Intrapulmonary leukocyte aggregation following activation of the classic complement cascade releases even more chemokines and cytokines, leading to the production of free radicals of oxygen and nitrogen25 from nitric oxide synthase (NOS)–triggered nitric oxide (NO) and peroxynitrite (ONOO−) production. The vasodilation induced by NO rapidly increases bronchial blood flow and decreases the degree of the protective hypoxic pulmonary vasoconstriction in poorly ventilated areas of the lung, resulting in V/Q mismatch.26 This also intensifies the spread of irritants from the pulmonary to systemic circulation. NO also combines with superoxide (O2−) produced in large quantities by activated neutrophils to form ONOO−. This reactive nitrogen species leads to DNA damage and subsequent activation of poly (Adenosine diphosphate ribose [ADP-ribose]) polymerase, an important enzyme in DNA repair. This activation and subsequent action requires a large amount of chemical energy in the form of adenosine triphosphate (ATP) and Nicotinamide adenine dinucleotide (NAD), the depletion of which causes necrotic cell death of deprived energy-dependent tissues.25 The combination of these effects contributes to tissue injury and increased pulmonary vascular permeability, leading to decreased diffusion, edema, and V/Q mismatch. Furthermore, neutrophils are sequestered from the systemic circulation to the intrapulmonary compartment and are activated, and fibrinogen release by inflammatory mediators causes airway cast formation and widespread plugging. These casts obstruct a number of the smaller airways, and subsequent efforts to mechanically ventilate this inhomogeneous lung can induce ventilator-induced barotrauma as normal lung is overdistended, whereas other regions collapse, and atelectasise. The further tissue injury is heightened with biotrauma of ventilation, and the production of chemokines leads to a potent accumulation of damage.21
Much of the study of smoke inhalation injuries in animal models has focused on aspects of this pathophysiologic sequence. Attempts to manipulate and alter the chain of effects experimentally have reinforced these theories and suggested exciting treatment targets. Nevertheless, experimental treatments have yet to deliver specific therapeutic modalities that improve the course of smoke inhalation injury.5
Diagnostics and Treatment
Initial Prehospital Rescue
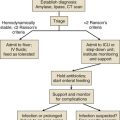
The first priority at the injury scene is rescue of the victim from the source of fire to minimize the exposure time. This is usually the responsibility of firefighters.4 The patient must be assessed as a trauma patient and not merely as the victim of an isolated burn or smoke inhalation injury. Standard early management of severe trauma (EMST) protocols must be observed, including stabilization of the neck. Following the immediate administration of a high flow of O2 to reduce COHb levels, a primary survey must then follow to assess accompanying injuries such as burns or trauma with simultaneous estimation of the extent of smoke inhalation. In addition, it is important to determine whether the victim has been exposed to an explosion and to assess the possibility of blast injury to the lung. If possible, information about comorbidities should be obtained. Standard cardiopulmonary monitoring (electrocardiogram, pulse-oximetry, and noninvasive blood pressure) and intravenous access should be established.27 Carbon monoxide poisoning can result in an erroneously high SaO2 reading due to the light absorption of the classic “cherry red” hemoglobin in smoke inhalation. After these basic measures, the safety of the airway must be assessed. The risk of rapidly developing airway edema has to be taken into account even if no dyspnea is present, but it must be balanced by the real risks faced by endotracheal intubation itself in an unstable, potentially hypoxic patient with possible neck injuries. In the authors’ opinion, endotracheal intubation that is entirely prophylactic is ill advised. Nevertheless, the airway with early edema is likely to worsen, particularly if significant fluid resuscitation is required for burn injury, and hence repeated and thorough assessment of the airway is mandatory. Patients with evidence of stridor or heat and smoke inhalation injury combined with extensive face or neck burns may mandate early intubation. In the case of oral burn without inhalation injury, an airway secured early represents the safest approach. However, victims with smoke inhalation injury but no facial or neck burns can be carefully observed and can be intubated later, if necessary.28 The patient’s head should be elevated to 45 degrees to minimize facial and airway edema. In the field, fluid resuscitation can be minimized to reduce the risk of airway compromise if the necessary skills or equipment are not readily available for intubation. Nebulized adrenaline or corticosteroids may be used in the hope of minimizing upper airway edema, although there is no conclusive evidence for the efficacy of these treatment strategies.29 Bronchospasm is frequently observed, and the nebulized administration of bronchodilators, such as β2-agonists, will reduce this effect, while improving respiratory mechanics by decreasing airflow resistance and peak airway pressures in ventilated patients. This results in improved dynamic compliance. In addition, β2-agonists provide anti-inflammatory properties, represented by a decrease in inflammatory mediators such as histamine, leukotrienes, and TNF-α. Finally, β2-agonists are associated with improved airspace fluid clearance and stimulation of mucosal repair.30–32
After initial stabilization of the patient, information about the type of fire and combustible materials involved, whether the fire occurred in an enclosed space, and the estimated duration of exposure should be sought. In cases of presumed specific intoxication, appropriate therapies should begin.21 Diagnosis of such intoxications is impossible in the field, but a high degree of suspicion must be maintained if the combustion materials and enclosed space lead the treating practitioner to assume risk of CO or CN. All patients should be immediately administered 100% O2 from a high-flow facemask to reduce the CO binding to Hb. Specific therapies exist to treat the toxicity of carbon monoxide and cyanide, which aim to reduce the serum levels of these substances. Depending on the Glasgow Coma Scale, the severity of injury, and the symptoms, the patient’s condition may mandate intubation and mechanical ventilation with an FiO2 of 1.
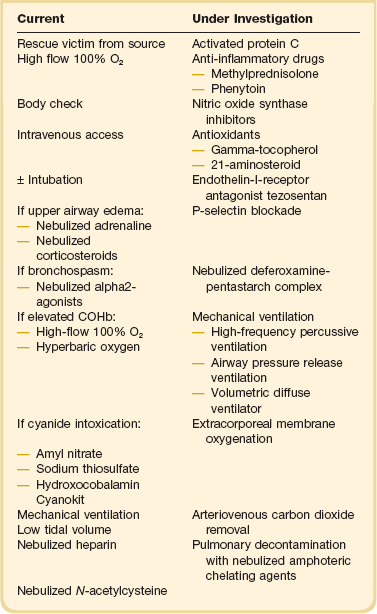
Patients with the possibility of cyanide intoxication require standard supportive care, which may be augmented with specific antidote therapy—the choice of which is the more efficacious remains controversial. Amyl nitrate and sodium thiosulfate are used to oxidize hemoglobin to methemoglobin, which preferentially binds cyanide. In contrast to these antidotes, hydroxycobalamin (vitamin B12a) actively binds CN by forming cyanocobalamin, which is directly excreted via the kidney. Because it does not produce methemoglobin, hydroxocobalamin is safe to use in the preclinical setting. Accordingly, it represents the active compound of the “Cyanokit,” which is used in the prehospital management of smoke inhalation injury in Europe with a reported reduction of mortality33 (Table 48.1).
Airway Management
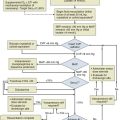
Clinical suspicion of inhalation injury of the upper airway is aroused by the presence of certain risk factors such as history of exposure to fire and smoke in an enclosed space or a period of unconsciousness at the accident scene, burns including the face and neck, singed facial or nasal hair, altered voice, dysphagia, oral or nasal soot deposits, or carbonaceous sputum. The most immediate threat from inhalation injury is upper airway obstruction due to edema (see Fig. 48.2). Early intubation is recommended when this complication threatens and the patient was not intubated on scene.34 However, exposure to smoke does not always lead to severe injury, and in the absence of overt evidence of respiratory distress or failure it may be difficult to identify patients who will experience progressive inflammation and ultimately require intubation of the trachea. When intubating in the field, optimal technique to secure a difficult airway is a contentious issue. Experienced operators may attempt to preserve spontaneous breathing, which allows patients to maintain their own reflexes even when intubation is not possible. Others may elect to perform a rapid sequence induction, which provides better intubating conditions but oblates all of the patient’s own airway reflexes. Attention should be given to gastric residuals during enteric feeding after admission to the burn intensive care unit. In addition, the development of sepsis can slow gastric emptying, which can result in retained fluids in the stomach and risk of aspiration.27
A patient with a compromised airway has evolved to maintain the airway at all costs. This primitive survival instinct is neutered if paralyzing agents or heavy sedation is administered and the safe airway can rapidly become unsalvageable. Intubation of a spontaneously breathing patient, while being more technically challenging, is safer, as the patient will keep breathing at all costs. In terms of anesthetic airway management, the most profound and clinically significant effect of burn injuries on drug response relates to muscle relaxants. Burn injuries influence responses to both succinylcholine and the nondepolarizing muscle relaxants. In burned patients, sensitization to the muscle relaxant effects of succinylcholine can produce exaggerated hyperkalemic responses severe enough to induce cardiac arrest, though this tends to occur 24 to 48 hours post injury, rather than immediately.35 However, recommendations regarding the safe use of succinylcholine after burn injury cannot be given. Various authors recommend avoidance of succinylcholine at intervals ranging from 24 hours to 21 days post burn injury,36 but it seems clear that the hyperkalemic response associated with burn does not occur in the first day and hence the drug can be used with standard precautions at this stage.
An increase in the numbers of acetylcholine receptors and the proliferation of these receptors away from the neuromuscular junction have been suggested as common mechanisms explaining both reduced sensitivity to nondepolarizing relaxants and the exaggerated hyperkalemia that may follow succinylcholine administration in burned patients. Resistance is apparent by 7 days post injury and peaks by approximately 40 days. Sensitivity returns to normal after approximately 70 days. In contrast to other nondepolarizing neuromuscular blockers, mivacurium dosage requirements in pediatric patients appear to be unchanged by burn injury.37
Preoxygenation may be more difficult in the smoke inhalation victim, and relative loss of mandibular mobility may impair airway manipulation, making bag-mask ventilation difficult. The swelling and distortion of the mouth and mandibular aspect of the airway may make preoxygenation and direct laryngoscopy difficult or impossible. Preoxygenation with small aliquots of anxiolysis and analgesics (such as 0.5- to 1-mg increments of midazolam and 25- to 50-µg blouses of fentanyl IV) with sequential direct spraying of lignocaine to the oropharynx and then subsequently under direct vision of the larynx/epiglottic area is technically difficult but safe in experienced hands. Fiberoptic intubation with nebulized or directly sprayed lignocaine spray while maintaining spontaneous ventilation is equally safe but requires a substantial level of skill with the bronchoscope. However, pediatric patients are unable to cooperate and must be adequately sedated. Because deep sedation and full anesthesia cause collapse of pharyngeal tissues and airway obstruction, they are unsuitable for fiberoptic intubation in patients whose airway would be difficult to manage with a mask.38
Agents that may prove useful to facilitate fiberoptic intubation include the ultra-short-acting remifentanil. The novel alpha-2-adrenergic agonist dexmedetomidine, which provides sedation, anxiolysis, and analgesia with much less respiratory depression than other sedatives,39 may be considered, though further data are required until its widespread use is adopted. Ketamine remains a very useful drug, as it provides some analgesia and facilitates a degree of spontaneous breathing while allowing dissociative anesthesia, However, airway secretions may be copious and the clinician must always be aware of this.
The use of a laryngeal mask airway (LMA) has also been successful for airway management during burn surgery for children.40 In the acute phase or in the intensive care unit, the LMA serves as a rescue device when endotracheal intubation fails, but it must be replaced with a definitive airway as soon as is practically possible, as gastroparesis is common in the burn patient.
Monitoring and Investigations
Damages to the airway and lung from inhalation injury often develop with a latency of several hours and are affected by other injuries and degree of fluid resuscitation required. Nevertheless, airway management and the oxygenation status of the patient, regardless of intubation status, need to be frequently reevaluated; such is the dynamic nature of smoke inhalation injury.22 After stabilization of cardiopulmonary hemodynamics and pulmonary gas exchange, the assumed diagnosis of smoke inhalation injury needs to be verified. However, as no uniform criteria are available, diagnosis of smoke inhalation injury has a subjective component based on history, physical examination with supporting imaging, and blood gas assays. Bronchoscopic examination of the airway represents the gold standard to detect a pathognomonic mucosal hyperemia with soot staining below the larynx being diagnosed (Fig. 48.3). Although chest radiographs are mandatory and may reveal injuries consistent with trauma—that is, fractured ribs, pneumothorax, and pulmonary contusion—they provide little information acutely as to the degree of smoke inhalation. Specific changes that may be seen related to the smoke inhalation include signs of diffuse atelectasis, pulmonary edema, or bronchopneumonia.41

Figure 48.3 Soot staining in the hyperemic trachea.
A uniform algorithm for assessing inhalation injury or a reliable indicator of progressive respiratory failure in patients with smoke inhalation injury has not yet been established. This failure is largely explained by the extreme heterogeneity of the clinical presentation. In addition, the delay in the manifestation and development of acute lung injury (ALI) as a consequence of systemic inflammatory response syndrome (SIRS), initiated by accompanying burns or trauma, complicates the evaluation of the isolated effects of smoke inhalation. Frequent blood gas and sputum analyses are useful to monitor patients with smoke inhalation injury.28 In addition, inexplicably high lactate levels, despite adequate fluid resuscitation, will be helpful for the diagnosis of CN poisoning. In a study by Pham and colleagues,42 cyanide poisoning in the canine model showed two phases of injury. The first (compensated) phase had a mechanism consistent with a traditional global oxygen consumption defect. The second (decompensated) phase had a mechanism consistent with heart failure. This heart failure was due to bradycardia. The systolic blood pressure remained relatively constant, whereas diastolic blood pressure decreased by 19%. Cardiac output, heart rate, and DO2 increased to a maximum of 6%, 10%, and 10%, respectively, at 40 minutes, after which they declined to a low of 32%, 28%, and 30% below baseline, respectively. Stroke volume remained constant. Oxygen consumption initially increased by 5% and then decreased to 24% below baseline. The oxygen extraction ratio (OER) initially declined to 35% below baseline and then increased throughout the rest of the study. In clinical practice, serial venous blood gases may unmask cytopathic hypoxia, by insufficient O2 extraction, that may be seen as increased central venous (ScvO2) or mixed venous (SvO2) oxygen saturations.
Fluid Resuscitation
Optimal fluid management is critical to the survival of the victim of a major thermal injury, with additional or isolated inhalation injury, and when septic complications occur. Modern fluid resuscitation formulas originate from experimental studies in the pathophysiology of burn shock. Fluid resuscitation in the patient with thermal injury has been recognized as an essential aspect of the care since the first studies were published in 1905.43 The improvement in outcome with cutaneous burns can be related to the development of a protocol based on providing adequate fluid resuscitation to allow optimal organ function while minimizing the physiologic cost associated with over-resuscitation.
Burn shock is both hypovolemic shock and cellular shock and is characterized by specific hemodynamic changes including decreased cardiac output, extracellular fluid, plasma volume, and oliguria. As in the treatment of other shock forms, the primary goal is to restore and preserve tissue perfusion in order to avoid ischemia. However, in burn shock, resuscitation is complicated by obligatory burn edema, and the voluminous transvascular fluid shifts that results from a major burn are unique to thermal trauma.44 Blalock may have been the first to the postulate mechanism, where he induced burn to one side of mongrel dogs and assessed the weight changes in both burned and unburned tissue, demonstrating that burn tissue edema correlated to the drop in blood pressure and was similar in composition to plasma.45 Although the exact pathophysiology of the postburn vascular changes and fluid shifts is still unclear, major components of burn shock are the increase in total body vascular permeability and the changes in microcirculation. Fluid resuscitation is aimed to support the patient throughout the initial 24-hour to 48-hour period of hypovolemia and has existed since the early 1950s.
Cope, in his seminal paper following the Cocoanut Grove disaster, was the first to report the increased fluid requirement seen in patients with smoke inhalation injury.46 This observation has been confirmed by multiple other papers, despite the lack of thermal injury below the larynx; hence fluid loss seems unlikely. One theory, which concurs with much of the known pathophysiology, relates to inflammatory excess seen postsmoke inhalation. Approximately 28% of all neutrophils reside in the human lungs, and this percentage increases postinhalation insult. As described earlier, it is well recognized that the particulate and chemical nature of inhalation injury induces neutrophil activation and the release of numerous cytokines, proteases, and free radicals. This further recruits neutrophils from the systemic circulation, and the local inflammatory state rapidly becomes systemic due to the vast alveolo-capillary interphase transporting these mediators and modulators of vasodilatation to distant organs. The up-regulation is not only in number but also in responsiveness to endotoxin.21,47 Regardless of mechanism, numerous authors have reported that smoke inhalation per se increased the fluid requirement substantially.48–50
Although under-resuscitation in thermal and smoke inhalation injury is known to induce organ failure and death, there are growing concerns with over-resuscitation, where the “fluid creep” can induce increased extravascular water content, resulting in compartment syndrome in extremities or in the abdomen.51 Even after thousands of patients have been saved by fluid resuscitation, resuscitation formulas are still controversially discussed, depending on advantages and disadvantages for the individual patient. Crystalloid solutions, such as lactated Ringer’s solution (sodium concentration 130 Meq/L), are the most popular resuscitation fluids currently utilized. Crystalloid formulas are the “Parkland formula” (which recommends 4 cc/kg/% total body surface area (TBSA) burn in the first 24 hours, with half of the amount administered in the first 8 hours) and the “Modified Brooke formula” (which recommends 2 cc/kg/% TBSA burn). Colloid formulas (Evans, Brooke, Slater), the Dextran formula (Demling), and hypertonic saline formulas (Monafo, Warden) are also in use.44,52–54
Concerns with colloid administration in the resuscitation of early burns began with Goodwin’s work in the 1980s,55 where it was suggested that there was a lack of benefit over crystalloid and potential pulmonary harm, and grew substantially in the late 1990s, but more recent work suggests that concerns of increased capillary leak particularly in the lungs are overplayed, and the pendulum seems to be swinging back toward the judicious use of colloid to minimize the risk of abdominal compartment syndrome (ACS). A randomized controlled trial (RCT) by O’Mara compared plasma to crystalloid in burn resuscitation and showed that colloid resulted in less ACS but no demonstrable improvement in outcome.56
Early studies in fluid resuscitation post smoke inhalation suggest a mean fluid requirement of 5.8 mL/kg/% TBSA burn for optimal outcomes, but more recent studies suggest the figure may be lower than originally expected.49
Hypertonic salt (HTS) solutions have been known for many years for effectiveness in the treatment of burn shock by fluid sparing effects and a reduction of volume load in the early phase of injury.57 Rapid infusion leads to serum hyperosmolarity and hypernatremia, reducing the shift of fluids from intravascular to interstitial areas, which may prevent edema formation and the need for escharotomy.58,59 The use of HTS, however, is controversial because although there may be beneficial effects, other studies have demonstrated an increase in mortality with HTS treatment of major burns.60
Part of the dubiety regarding the exact volume required relates to the lack of consensus of scoring severity of inhalation injury. The extent of cutaneous burn can be readily assessed. Such a scoring system for inhalation injury is lacking, and inhalation can be mild moderate or severe. In a 3-year retrospective review by Gamelli’s group, numerous demographic and injury-related factors, including bronchoscopic scoring, failed to determine which patients with inhalation injury required increased fluid resuscitation. From its 80-patient cohort, the group postulated that a PaO2/FiO2 ratio of <350 may be the best early determinant of which patients would require excess fluid resuscitation.61
In addition to hypovolemia, the risk of infection in smoke inhalation patients is extraordinarily high. Shirani and coworkers6 have shown that inhalation injury alone increased the mortality of burn patients by a maximum of 20%, and pneumonia by a maximum of 40%, with a maximum increase of approximately 60% when both are present. These data indicate that inhalation injury with additional septic complications such as pneumonia has significant, independent, additive effects on burn mortality and that these effects vary with age and burn size. The presence of inhalation injury and sepsis increases the fluid requirements for resuscitation from burn shock after thermal injury.48 Chen and coworkers62 have demonstrated that HTS confers beneficial effects on burn shock, reduces bacterial translocation, and enhances host defenses by several mechanisms in the treatment of burn shock and sepsis. It is of high interest to further investigate HTS in animal models and later on in humans. Future studies with HTS alone and in combination with already established resuscitation formulas are needed to find a risk/benefit ratio for such a treatment. HTS may have the potential to improve resuscitation strategies and outcome in burn care, but it cannot be recommended at the present time due to a lack of evidence.63
Despite the treatment of septic complications following inhalation injury, the gradual shift toward less fluid in both burn and smoke inhalation may relate to the changes in the intensive care unit (ICU) modalities of care—including point-of-care devices to assess oxygenation, less injurious ventilation, and more rapid debridement of burn tissue.64
Treatment of Carbon Monoxide Intoxication
Current treatment recommendations of CO poisoning begin with cessation of exposure, immediate administration of 100% oxygen, and supportive care.65 The rationale of hyperbaric oxygenation therapy (HBOT) is based on its ability to rapidly displace CO from hemoglobin, thus reducing the duration of the hypoxemic state. Administration of 100% oxygen at 3 atmospheres reduces the CO half-life from 250 minutes in room air to 30 minutes.28 Besides this efficiency, its use remains controversial due to the questioned correlation between COHb levels and outcomes. This may also be explained with limited access to the patient during HBOT, which has major impact on the treatment quality of combined burn injuries.28 A Cochrane database review of six randomized controlled trials did not reveal a beneficial effect of HBOT compared to standard treatment in respect to neurologic sequelae.66 However, the results should be interpreted with care, as concerns have been raised regarding design and analyses in all the included trials. In summary, all patients with CO intoxication should be immediately treated with 100% oxygen. Although data do not support its routine use, HBOT may be considered in stable patients with severe neurologic symptoms and high COHb concentrations (>50%), but no major burns and severe pulmonary injury.47
Treatment of Cyanide Intoxication
Hydrogen cyanide in fire smoke is an underappreciated threat and one of the most common poisonings in patients who suffer smoke inhalation.67 Its lipophilicity and lack of dissociation allow it to penetrate through mucous membranes with ease, resulting in a myriad of symptoms within minutes, and possible death if the dose is large enough.68 Patients may describe a burning, dry throat with inexplicable feelings of anxiety. Clinical signs range from a patient presenting with tachypnea, confusion (and the classic almond breath fetor) to unconsciousness, cardiovascular collapse, and death, generally from respiratory arrest (see Table 48.3).20 The key for the receiving clinician is to have a high index of suspicion in such a patient, as there is no immediately available lab test. Blood gas analysis may reveal an inexplicably high lactate in the absence of CO poisoning or other trauma. However, CN poisoning frequently coexists with significant CO poisoning, and hence may be overlooked.4,69 For adequate treatment of CN poisoning following smoke inhalation injury, several antidotes are available: The “CN antidote kit” includes amyl nitrite, thiosulfate, and sodium nitrite.70 Because these substances are methemoglobin generators, which may additionally impair oxygen transport, they should be only used in case of proven diagnosis (increased plasma levels of CN) and under continuous monitoring in the intensive care unit, particularly in episodes of poisoning with CO simultaneously.28 Methemoglobin chelates CN to form cyanmethemoglobin. As cyanmethemoglobin dissociates, free CN is converted to thiocyanate by liver mitochondrial enzymes (rhodanese) using thiosulfate as a substrate. Thiocyanate is then excreted into the urine.71 In contrast to these antidotes, hydroxocobalamin, a vitamin B12 derivative, actively binds CN by forming cyanocobalamin, which will be directly excreted by the kidneys.33 Data are growing regarding the safety and efficacy of hydroxocobalamin. Prospective observational studies reveal that empiric administration of hydroxocobalamin was associated with survival among 67% of patients who were confirmed post administration to have had cyanide poisoning. A randomized controlled trial in pigs assessed the efficacy of hydroxocobalamin versus sodium thiosulfate in the treatment of acute cyanide toxicity. The study indicated that in severe poisoning (CN administered to hypotension to 50% of baseline blood pressure), hydroxocobalamin was safe, efficacious, and resulted in much improved survival. Worryingly, sodium thiosulfate failed to reverse cyanide-induced shock. The authors concluded that only hydroxocobalamin was effective.72 In case of intoxication with 1 mg CN, the recommended dose is 50 mg/kg hydroxocobalamin.73 Because of the avoidance of methemoglobin production, hydroxocobalamin can be used safely in the preclinical setting. Accordingly, hydroxocobalamin represents the active compound of the “Cyanokit,” which is used in the prehospital management of smoke inhalation injury in Europe with a reported improvement in mortality.33
Table 48.3
Hydrogen Cyanide Concentration in Air and Related Symptoms
| HCN [ppm] | Symptoms |
| 0.2-5 | Odor threshold |
| 10 | Occupational exposure limit |
| 18-36 | Slight symptoms, headache |
| 45-54 | Will be tolerated for 30-60 minutes |
| 100 | Death within 1 hour |
| 110-135 | Death within 30-60 minutes |
| 181 | Death within 10 minutes |
| 280 | Immediate death |
Aggressive restoration of cardiopulmonary function augments the hepatic clearance of CN via the enzyme rhodanese and has been reported to be successful in severe CN poisoning (blood levels 5.6 to 9 mg/L) as well as after ingestion or smoke inhalation, even without the use of antidotes.74–76 Hydroxocobalamin has been used safely and successfully by emergency personnel in out-of-hospital settings and may represent a new option in cases of suspected or confirmed cyanide poisoning in the field.77 Therefore, the standard care of CN poisoning should combine the aggressive supportive therapy with current data supporting the use of hydroxocobalamin as the optimal specific therapy.47
Bronchoscopy
Within each burn center, diagnostic as well as treatment options are determined by the availability of resources (such as 133Xe scans, fiberoptic bronchoscopy, or 24-hour/day anesthesia staff coverage) and local tradition. A clear diagnosis proving smoke inhalation has occurred allows for the planning and delivery of therapeutic interventions.41 Inhalation injury can be diagnosed with confidence based on clinical presentation and bronchoscopic findings. Changes observed with bronchoscopy include erythema, lesion, erosion, ischemia, or necrosis of the mucous membrane, as well as small to significant edema, blisters, and unidentified damage of the lung parenchyma when the changes in the lower respiratory tract are below the reach of the fiberoptic scope, which can be used for inspection of the upper (oropharynx) and main (trachea and bronchi) respiratory tract78 (see Figs. 48.2 and 48.3).
Early prediction of which patients are vulnerable to resuscitation stresses, increased pulmonary complications, respiratory failure, and mortality is complex and frequently not possible. Many attempts to identify prognostic indicators for patients with smoke inhalation injuries have been made.79,80 It has been difficult to identify reliable indicators of progressive respiratory failure in patients with smoke inhalation injury. Most of these studies have involved small numbers of patients and assessed a small number of clinical features. Prognostic estimations will ultimately rely on a system that allows quantification of the severity of inhalation injury.41 Many observational studies have compared outcomes with various grading systems.61,78,81,82 These grading systems often combine bronchoscopic findings with a small number of other clinical findings. However, it has been recognized that proximal injury observed by bronchoscopy is frequently greater than the peripheral, parenchymal injury. Masanes and colleagues83 found that inhalation injury could be diagnosed by fiberoptic bronchoscopy in some burn patients who were otherwise asymptomatic. Liffner and colleagues 82 found that their scoring system for grading the severity of bronchoscopic evidence of inhalation injury did not correlate with the development of acute respiratory distress syndrome (ARDS). Similarly, although it is generally recognized that inhalation injury increases fluid resuscitation needs in burn patients, Endorf and Gamelli found no correlation of severity of bronchoscopic findings with fluid resuscitation requirements.61 In a review article, Woodson concluded that a consensus may be facilitated when a clinical variable or constellation of variables is identified that is reliably related to the development of respiratory failure or other complications in patients who have inhaled smoke.41 A large multicenter study combining the experience of several institutions is more likely to identify such a correlation. However, consensus in diagnosis and quantification of inhalation injuries may await a theoretic advance with identification of a mediator or marker of cell injury or cell death that reliably and in some concentration-dependent way correlates with pulmonary or systemic complications of inhalation injury. A widely accepted grading system for inhalation injury severity is presently not available. However, a randomized controlled multicenter trial to validate a standardized scoring system for inhalation injury that can be used to both quantify injury severity and predict mortality after inhalation injury is warranted.41
Mechanical Ventilation
Since the advent of positive pressure ventilation, there has been an understanding that this therapeutic maneuver can paradoxically cause harm as well as save lives. Numerous studies have tried to identify optimal ventilatory strategies, positive end-expiratory pressure (PEEP) levels, and modes. An open lung strategy with low-tidal-volume ventilation has been shown to be associated with improved outcome and reduced duration of ventilation.84 In keeping with the ARDSnet data, low tidal volumes are mandated to minimize ventilator-induced lung injury (VILI).85 An open lung strategy is advised due to the decrement of surfactant in this population, but no study has shown an improved outcome with higher levels of PEEP. The frequent casts that occur post smoke inhalation result in areas of collapsed alongside open lung, and application of PEEP may result in tidal hyperinflation in areas of the lung, whereas there is an inability to aerate the collapsed area of lung. Hence, frequent airway toilet with mucolytics is essential to minimize VILI. Newer modalities such as electrical impedance tomography to assess breath-to-breath changes have demonstrated the rapid changes in ventilatory inhomogeneities seen post smoke inhalation.86 A number of specific modalities have been suggested specific to the ventilatory idiosyncrasies associated with smoke inhalation.87
An alternative to the classic ARDSnet low tidal volume ventilation in patients with ARDS may be high-frequency oscillation. A meta-analysis to determine the clinical and physiologic effects of high-frequency oscillation compared with conventional ventilation in patients with ARDS displayed improved survival and it is unlikely to cause harm. As ongoing large multicenter trials will not be completed for several years, these data help clinicians who currently use or are considering this technique for patients with ARDS.87
The volumetric diffusive ventilator (VDR) is a pneumatically powered, pressure-limited ventilator that stacks oscillatory breaths to a selected peak airway pressure by means of a sliding venture called a Phasitron, resulting in low tidal volumes. Exhalation is passive and a level of continuous positive airway pressure (CPAP) can be selected. In addition, VDR reestablishes the physiologic diffusive gas exchange, whereas standard ventilation modes induce a convective gas exchange.88 A prospective clinical analysis revealed an improved gas exchange and a decrease in peak pressures.89 A retrospective study in 330 patients with inhalation injury even reported a lower mortality rate.90 Although these studies compared the VDR to high-volume ventilatory strategies, data regarding a comparison with modern low-tidal volume ventilation are still lacking. This may represent one reason why VDR is not universally accepted. Another factor might be that the VDR differs from other ventilators and, therefore, requires special training. In addition, tidal and minute volumes cannot be monitored, and humidified air as well as nebulized saline are necessary to prevent airway desiccation.47
As in conventional long-term ventilation of patients, common complications must be considered. Mosier and associates91 documented that patients with combined thermal and inhalation injury requiring urgent intubation or prolonged ventilation have a high incidence of bacterial bronchial contamination. Inhalation injury creates a damaged tracheobronchial mucosa, and early intubation provides a portal for bacterial contamination. In patients with smoke inhalation that necessitated urgent intubation, a 50% incidence of ventilator-associated pneumonia (VAP) has been described.92
The use and timing of tracheostomy in burn patients elicit a great deal of passionate discussion but very little solid data. Burn survivors with TBSA >60% are more likely to undergo repeated surgery and have burns to the head and neck region, therefore increasing the requirement for tracheostomy. However, an association has been demonstrated between tracheostomy and high prevalence of chest infection in patients with inhalation injury, greater burn size, and prolonged mechanical ventilation.93 The authors would advise caution in overinterpreting this study, as association does not infer causation, and sicker patients tend to be ventilated for longer periods, resulting in a higher incidence of both tracheostomy and VAP. The tracheostomy allows for less sedation, earlier mobilization, and a more comfortable method of ventilation for a long-term patient.
Nebulization Treatments
As described previously, smoke-inhalation injury causes a destruction of the ciliated epithelium that lines the tracheobronchial tree. Casts produced from these cells, polymorphonuclear leukocytes, and mucus can cause upper-airway obstruction, contributing to pulmonary failure.21 In the early 2000s, it was proposed that a combination of aerosolized heparin and a mucolytic agent, N-acetylcysteine, can ameliorate cast formation and reduce pulmonary failure secondary to smoke inhalation. In a study of 90 consecutive pediatric burn patients at the Shriners Burns Hospital at Galveston, Texas, who had bronchoscopically diagnosed inhalation injury requiring ventilatory support, 5000 units of heparin and 3 mL of a 20% solution of N-acetylcysteine aerosolized every 4 hours the first 7 days after the injury resulted in a significant decrease in reintubation rates, in incidence of atelectasis, and in mortality for patients treated with the regimen when compared with controls, and this practice has ever since been part of the ventilation protocol at this institution.94
Nonventilatory Pulmonary Treatments
Extracorporeal membrane oxygenation (ECMO) is used in specialized centers for neonatal, pediatric, and adult respiratory and cardiac failure. It requires a highly skilled team of intensivists and perfusionists, and echocardiographic support is essential for optimal usage of this modality.95 The goal of ECMO is to support gas exchange, allowing the intensity of mechanical ventilation to be reduced and thus decreasing the potentially injurious effects of ventilator-induced lung injury until recovery. Furthermore, ECMO may be considered the definitive rescue therapy for refractory life-threatening hypoxemia, as pulmonary gas exchange is not required. Our group performed a systematic review of the literature to collect all available clinical data in order to elucidate the role and present evidence of ECMO on severe hypoxemic respiratory failure resulting from burn and smoke inhalation injury.96 Only a small number of clinical trials with a limited number of patients were available. The data suggested a higher ECMO therapy survival than nonsurvival rate of burn patients suffering acute hypoxemic respiratory failure. ECMO run times of less than 200 hours correlate with higher survival compared to 200 hours or more, and scald burns show a tendency of higher survival than flame burns. However, based on the low number of studies and patients, as well as the low grade of evidence of these studies, there are currently inadequate data to support the use of ECMO in burn or smoke inhalation injury. Even though the reports are promising, especially in the pediatric burn population, this review highlighted the lack of evidence for the use of ECMO in this setting. ECMO in adult respiratory failure is controversial, as early randomized trials showed poor outcomes97–99 and use has been limited to highly specialized centers. Nevertheless, ECMO technology and expertise have improved. More recently, the Conventional Ventilation or ECMO for Severe Adult Respiratory Failure trial100 and selected case series have shown improved outcomes, with survival of 75% to 85% in refractory respiratory failure.101,102 Therefore, randomized controlled trials on patients with burn and smoke inhalation injury are warranted to provide definitive recommendations and to further advance this therapeutic option in patients where other ventilatory modalities have failed. Currently, there are large animal studies assessing the efficacy of ECMO in severe smoke inhalation, with promising results.103
Experimental Treatments
Against the background of the current literature, there has been a remarkable increase in our knowledge about the pathogenesis of smoke inhalation injury. There are several promising therapeutic approaches, including the nebulization of β2-agonists, antioxidants, or anticoagulants as well as the use of different ventilation modes.104–112 However, as has been highlighted, smoke inhalation may begin as a single organ injury, but rapidly becomes systemic. Hence, although treatment paradigms may focus on smoke inhalation, it is more frequently a mixed insult of smoke and burn, smoke and pneumonia, smoke and toxicology, and smoke and trauma. Even isolated smoke inhalation generally induces distant organ dysfunction rapidly post injury. Hence, it is unlikely that a single “magic bullet” will be found for this condition, and the clinician is best equipped with a comprehensive understanding of the complex pathophysiology and multitudinous clinical presentations of smoke inhalation injury. The treatment can then be patient and inhalation specific, with a systemic rather than organ-specific approach to these patients (see Table 48.1).
References
1. Vassallo, R, Ryu, JH. Tobacco smoke-related diffuse lung diseases. Semin Respir Crit Care Med. 2008; 29:643–650.
2. Schwela, D. Cooking smoke: A silent killer. People Planet. 1997; 6:24–25.
3. Singh, N, Davis, GS. Review: occupational and environmental lung disease. Curr Opin Pulm Med. 2002; 8:117–125.
4. Alcorta, R. Smoke inhalation & acute cyanide poisoning: Hydrogen cyanide poisoning proves increasingly common in smoke-inhalation victims. JEMS. 2004; 29(Suppl):6–15.
5. Traber, DL, Maybauer, MO, Maybauer, DM, et al. Inhalational and acute lung injury. Shock. 2005; 24(Suppl 1):82–87.
6. Shirani, KZ, Pruitt, BA, Jr., Mason, AD, Jr. The influence of inhalation injury and pneumonia on burn mortality. Ann Surg. 1987; 205:82–87.
7. Maybauer, MO, Maybauer, DM, Herndon, DN. Incidence and outcomes of acute lung injury. N Engl J Med. 2006; 354:416–417.
8. Thucydides, Crawley, RT. The Complete Writings of Thucydides: The Peloponnesian War. New York: Random House; 1951.
9. McManus, J, Huebner, K. Vesicants. Crit Care Clin. 2005; 21:707–718.
10. Haber, LF. The Poisonous Cloud: Chemical Warfare in the First World War. Oxford: Clarendon Press; 1986.
11. Hay, A, Roberts, G. The use of poison gas against the Iraqi Kurds: Analysis of bomb fragments, soil, and wool samples. JAMA. 1990; 263:1065–1066.
12. Yanagisawa, N, Morita, H, Nakajima, T. Sarin experiences in Japan: Acute toxicity and long-term effects. J Neurol Sci. 2006; 249:76–85.
13. Eckstein, M. Cyanide as a chemical terrorism weapon. JEMS. 2004; 29(Suppl):22–31.
14. Saffle, JR. The 1942 fire at Boston’s Cocoanut Grove nightclub. Am J Surg. 1993; 166:581–591.
15. Blocker, V, Blocker, TG, Jr. The Texas City disaster; a survey of 3,000 casualties. Am J Surg. 1949; 78:756–771.
16. Steinberg, JM, Schiller, HJ, Tsvaygenbaum, B, et al. Wood smoke inhalation causes alveolar instability in a dose-dependent fashion. Respir Care. 2005; 50:1062–1070.
17. Yurt, RW, Bessey, PQ, Bauer, GJ, et al. A regional burn center’s response to a disaster: September 11, 2001, and the days beyond. J Burn Care Res. 2005; 26:117–124.
18. Jordan, MH, Hollowed, KA, Turner, DG, et al. The Pentagon attack of September 11, 2001: A burn center’s experience. J Burn Care Res. 2005; 26:109–116.
19. Prien, T, Traber, DL. Toxic smoke compounds and inhalation injury: A review. Burns Incl Therm Inj. 1988; 14:451–460.
20. Baud, FJ, Barriot, P, Toffis, V, et al. Elevated blood cyanide concentrations in victims of smoke inhalation. N Engl J Med. 1991; 325:1761–1766.
21. Maybauer, MO, Rehberg, S, Traber, DL, et al. [Pathophysiology of acute lung injury in severe burn and smoke inhalation injury]. Der Anaesthesist. 2009; 58:805–812.
22. Toon, MH, Maybauer, MO, Greenwood, JE, et al. Management of acute smoke inhalation injury. Crit Care Resusc. 2010; 12:53–61.
23. Kirk, MA, Gerace, R, Kulig, KW. Cyanide and methemoglobin kinetics in smoke inhalation victims treated with the cyanide antidote kit. Ann Emerg Med. 1993; 22:1413–1418.
24. Fraser, JF, Venkatesh, B. Recent advances in the management of Burns. Australasian Anaesthesia. 2005; 23–32.
25. Murakami, K, Traber, DL. Pathophysiological basis of smoke inhalation injury. News Physiol Sci. 2003; 18:125–129.
26. Westphal, M, Cox, RA, Traber, LD, et al. Combined burn and smoke inhalation injury impairs ovine hypoxic pulmonary vasoconstriction. Crit Care Med. 2006; 34:1428–1436.
27. Kafka, G, Maybauer, DM, Traber, DL, Maybauer, MO. [Treatment of inhalation injury in preclinical emergency medicine]. Notfall Rettungsmed. 2007; 10:529–540.
28. Maybauer, DM, Traber, DL, Radermacher, P, et al. [Treatment strategies for acute smoke inhalation injury]. Der Anaesthesist. 2006; 55:980–982.
29. Cha, SI, Kim, CH, Lee, JH, et al. Isolated smoke inhalation injuries: Acute respiratory dysfunction, clinical outcomes, and short-term evolution of pulmonary functions with the effects of steroids. Burns. 2007; 33:200–208.
30. Morina, P, Herrera, M, Venegas, J, et al. Effects of nebulized salbutamol on respiratory mechanics in adult respiratory distress syndrome. Intensive Care Med. 1997; 23:58–64.
31. Zhang, H, Kim, YK, Govindarajan, A, et al. Effect of adrenoreceptors on endotoxin-induced cytokines and lipid peroxidation in lung explants. Am J Respir Crit Care Med. 1999; 160:1703–1710.
32. Zhang, Z, Kim, E, Martineau, D. Functional characterization of a piscine retroviral promoter. J Gen Virol. 1999; 80(Pt 12):3065–3072.
33. Fortin, JL, Giocanti, JP, Ruttimann, M, Kowalski, JJ. Prehospital administration of hydroxocobalamin for smoke inhalation-associated cyanide poisoning: 8 years of experience in the Paris Fire Brigade. Clini Toxicol. 2006; 44(Suppl 1):37–44.
34. Woodson, LC, Sherwood, ER, Aarsland, A. Anesthesia for burned patients, 4th ed. Saunders Elservier; 2012.
35. Tolmie, JD, Joyce, TH, Mitchell, GD. Succinylcholine danger in the burned patient. Anesthesiology. 1967; 28:467–470.
36. Yentis, SM. Suxamethonium and hyperkalaemia. Anaesth Intensive Care. 1990; 18:92–101.
37. Martyn, JA, Goudsouzian, NG, Chang, Y, et al. Neuromuscular effects of mivacurium in 2- to 12-yr-old children with burn injury. Anesthesiology. 2000; 92:31–37.
38. Mathru, M, Esch, O, Lang, J, et al. Magnetic resonance imaging of the upper airway: Effects of propofol anesthesia and nasal continuous positive airway pressure in humans. Anesthesiology. 1996; 84:273–279.
39. Walker, J, Maccallum, M, Fischer, C, et al. Sedation using dexmedetomidine in pediatric burn patients. J Burn Care Res. 2006; 27:206–210.
40. McCall, JE, Fischer, CG, Schomaker, E, Young, JM. Laryngeal mask airway use in children with acute burns: Intraoperative airway management. Paediatr Anaesth. 1999; 9:515–520.
41. Woodson, LC. Diagnosis and grading of inhalation injury. J Burn Care Res. 2009; 30:143–145.
42. Pham, JC, Huang, DT, McGeorge, FT, Rivers, EP. Clarification of cyanide’s effect on oxygen transport characteristics in a canine model. Emerg Med J. 2007; 24:152–156.
43. Sneve, H. The treatment of burns and skin grafting. JAMA. 1905; 45:1–8.
44. Warden, GD. Burn shock resuscitation. World J Surg. 1992; 16:16–23.
45. Blalock, A. Experimental shock: The importance of the local loss of fluid in the production of the low blood pressure after burn. Arch Surg. 1931; 22:610–616.
46. Cope, O, Moore, FD. The redistribution of body water and the fluid therapy of the burned patient. Ann Surg. 1947; 126:110–145.
47. Rehberg, S, Maybauer, MO, Enkhbaatar, P, et al. Pathophysiology, management and treatment of smoke inhalation injury. Expert Rev Respir Med. 2009; 3:283–297.
48. Navar, PD, Saffle, JR, Warden, GD. Effect of inhalation injury on fluid resuscitation requirements after thermal injury. Am J Surg. 1985; 150:716–720.
49. Dai, NT, Chen, TM, Cheng, TY, et al. The comparison of early fluid therapy in extensive flame burns between inhalation and noninhalation injuries. Burns. 1998; 24:671–675.
50. Cancio, LC, Chavez, S, Alvarado-Ortega, M, et al. Predicting increased fluid requirements during the resuscitation of thermally injured patients. J Trauma. 2004; 56:404–413.
51. Pruitt, BA, Jr. Protection from excessive resuscitation: “Pushing the pendulum back. ”. J Trauma. 2000; 49:567–568.
52. Baxter, CR, Marvin, JA, Curreri, PW. Early management of thermal burns. Postgrad Med. 1974; 55:131–139.
53. Baxter, CR, Shires, T. Physiological response to crystalloid resuscitation of severe burns. Ann N Y Acad Sci. 1968; 150:874–894.
54. Warden GD: Fluid Resuscitation and early management. Total Burn Care, 4th ed. Chapter 9, pp 115-124.
55. Goodwin, CW, Dorethy, J, Lam, V, Pruitt, BA, Jr. Randomized trial of efficacy of crystalloid and colloid resuscitation on hemodynamic response and lung water following thermal injury. Ann Surg. 1983; 197:520–531.
56. O’Mara, MS, Slater, H, Goldfarb, IW, Caushaj, PF. A prospective, randomized evaluation of intra-abdominal pressures with crystalloid and colloid resuscitation in burn patients. J Trauma. 2005; 58:1011–1018.
57. Guha, SC, Kinsky, MP, Button, B, et al. Burn resuscitation: Crystalloid versus colloid versus hypertonic saline hyperoncotic colloid in sheep. Crit Care Med. 1996; 24:1849–1857.
58. Monafo, WW. The treatment of burn shock by the intravenous and oral administration of hypertonic lactated saline solution. J Trauma. 1970; 10:575–586.
59. Monafo, WW, Halverson, JD, Schechtman, K. The role of concentrated sodium solutions in the resuscitation of patients with severe burns. Surgery. 1984; 95:129–135.
60. Huang, PP, Stucky, FS, Dimick, AR, et al. Hypertonic sodium resuscitation is associated with renal failure and death. Ann Surg. 1995; 221:543–554.
61. Endorf, FW, Gamelli, RL. Inhalation injury, pulmonary perturbations, and fluid resuscitation. J Burn Care Res. 2007; 28:80–83.
62. Chen, LW, Huang, HL, Lee, IT, et al. Hypertonic saline enhances host defense to bacterial challenge by augmenting Toll-like receptors. Crit Care Med. 2006; 34:1758–1768.
63. Maybauer, DM, Maybauer, MO, Traber, DL. Resuscitation with hypertonic saline in burn shock and sepsis. Crit Care Med. 2006; 34:1849–1850.
64. Venkatesh, B, Meacher, R, Muller, MJ, Morgan, TJ, Fraser, J. Monitoring tissue oxygenation during resuscitation of major burns. J Trauma. 2001; 50:485–494.
65. Kealey, GP. Carbon monoxide toxicity. J Burn Care Res. 2009; 30:146–147.
66. Juurlink, DN, Buckley, NA, Stanbrook, MB, et al. Hyperbaric oxygen for carbon monoxide poisoning. Cochrane Database Syst Rev. 2005.
67. Walsh, DW, Eckstein, M. Hydrogen cyanide in fire smoke: An underappreciated threat. Emerg Med Serv. 2004; 33:160–163.
68. Borowitz, JL, Rathinavelu, A, Kanthasamy, A, et al. Accumulation of labeled cyanide in neuronal tissue. Toxicol Appl Pharmacol. 1994; 129:80–85.
69. Eckstein, M, Maniscalco, PM. Focus on smoke inhalation—the most common cause of acute cyanide poisoning. Prehosp Disaster Med. 2006; 21:s49–s55.
70. Barillo, DJ. Diagnosis and treatment of cyanide toxicity. J Burn Care Res. 2009; 30:148–152.
71. Chen, KK, Rose, CL, Clorves, GH. Comparative values of several antidotes in cyanide poisoning. Am J Med Sci. 1934; 188:767–781.
72. Bebarta, VS, Pitotti, RL, Dixon, P, et al. Hydroxocobalamin versus sodium thiosulfate for the treatment of acute cyanide toxicity in a swine (Sus scrofa) model. Ann Emerg Med. 2012; 59:532–539.
73. Borron, SW, Baud, FJ, Barriot, P, et al. Prospective study of hydroxocobalamin for acute cyanide poisoning in smoke inhalation. Ann Emerg Med. 2007; 49:794–801.
74. Caravati, EM, Litovitz, TL. Pediatric cyanide intoxication and death from an acetonitrile-containing cosmetic. JAMA. 1988; 260:3470–3473.
75. Graham, DL, Laman, D, Theodore, J, Robin, ED. Acute cyanide poisoning complicated by lactic acidosis and pulmonary edema. Arch Intern Med. 1977; 137:1051–1055.
76. Clark, CJ, Campbell, D, Reid, WH. Blood carboxyhaemoglobin and cyanide levels in fire survivors. Lancet. 1981; 1:1332–1335.
77. Hall, AH, Dart, R, Bogdan, G. Sodium thiosulfate or hydroxocobalamin for the empiric treatment of cyanide poisoning? Ann Emerg Med. 2007; 49:806–813.
78. Marek, K, Piotr, W, Stanislaw, S, et al. Fibreoptic bronchoscopy in routine clinical practice in confirming the diagnosis and treatment of inhalation burns. Burns. 2007; 33:554–560.
79. Sellers, BJ, Davis, BL, Larkin, PW, et al. Early prediction of prolonged ventilator dependence in thermally injured patients. J Trauma. 1997; 43:899–903.
80. Edelman, DA, White, MT, Tyburski, JG, Wilson, RF. Factors affecting prognosis of inhalation injury. J Burn Care Res. 2006; 27:848–853.
81. Brown, DL, Archer, SB, Greenhalgh, DG, et al. Inhalation injury severity scoring system: A quantitative method. J Burn Care Rehabil. 1996; 17:552–557.
82. Liffner, G, Bak, Z, Reske, A, Sjoberg, F. Inhalation injury assessed by score does not contribute to the development of acute respiratory distress syndrome in burn victims. Burns. 2005; 31:263–268.
83. Masanes, MJ, Legendre, C, Lioret, N, et al. Fiberoptic bronchoscopy for the early diagnosis of subglottal inhalation injury: Comparative value in the assessment of prognosis. J Trauma. 1994; 36:59–67.
84. Brower, RG, Lanken, PN, MacIntyre, N, et al. Higher versus lower positive end-expiratory pressures in patients with the acute respiratory distress syndrome. N Engl J Med. 2004; 351:327–336.
85. The Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000; 342:1301–1308.
86. Riedel, T, Fraser, JF, Dunster, K, et al. Effect of smoke inhalation on viscoelastic properties and ventilation distribution in sheep. J Appl Physiol. 2006; 101:763–770.
87. Sud, S, Sud, M, Friedrich, JO, et al. High frequency oscillation in patients with acute lung injury and acute respiratory distress syndrome (ARDS): Systematic review and meta-analysis. BMJ. 2010; 340:c2327.
88. Harrington, D. Volumetric diffusive ventilator. J Burn Care Res. 2009; 30:175–176.
89. Carman, B, Cahill, T, Warden, G, McCall, J. A prospective, randomized comparison of the volume diffusive respirator vs conventional ventilation for ventilation of burned children. 2001 ABA paper. J Burn Care Rehabil. 2002; 23:444–448.
90. Rue, LW, 3rd., Cioffi, WG, Mason, AD, et al. Improved survival of burned patients with inhalation injury. Arch Surg. 1993; 128:772–778.
91. Mosier, MJ, Gamelli, RL, Halerz, MM, Silver, G. Microbial contamination in burn patients undergoing urgent intubation as part of their early airway management. J Burn Care Res. 2008; 29:304–310.
92. Eckert, MJ, Wade, TE, Davis, KA, et al. Ventilator-associated pneumonia after combined burn and trauma is caused by associated injuries and not the burn wound. J Burn Care Res. 2006; 27:457–462.
93. Aggarwal, S, Smailes, S, Dziewulski, P. Tracheostomy in burns patients revisited. Burns. 2009; 35:962–966.
94. Desai, MH, Mlcak, R, Richardson, J, et al. Reduction in mortality in pediatric patients with inhalation injury with aerosolized heparin/N-acetylcystine [correction of acetylcystine] therapy. J Burn Care Rehabil. 1998; 19:210–212.
95. Platts, DG, Sedgwick, JF, Burstow, DJ, et al. The role of echocardiography in the management of patients supported by extracorporeal membrane oxygenation. Am Soc Echocardiogr. 2012; 25:131–141.
96. Asmussen, S, Maybauer, DM, Fraser, JF, et al. Extracorporeal membrane oxygenation in burn and smoke inhalation injury. Burns. 2013; 39(3):429–435.
97. Bennett, CC, Johnson, A, Field, DJ, Elbourne, D. UK collaborative randomised trial of neonatal extracorporeal membrane oxygenation: Follow-up to age 4 years. Lancet. 2001; 357:1094–1096.
98. Zapol, WM, Snider, MT, Hill, JD, et al. Extracorporeal membrane oxygenation in severe acute respiratory failure: A randomized prospective study. JAMA. 1979; 242:2193–2196.
99. Morris, AH, Wallace, CJ, Menlove, RL, et al. Randomized clinical trial of pressure-controlled inverse ratio ventilation and extracorporeal CO2 removal for adult respiratory distress syndrome. Am J Respir Crit Care Med. 1994; 149:295–305.
100. Peek, GJ, Mugford, M, Tiruvoipati, R, et al. Efficacy and economic assessment of conventional ventilatory support versus extracorporeal membrane oxygenation for severe adult respiratory failure (CESAR): A multicentre randomised controlled trial. Lancet. 2009; 374:1351–1363.
101. Davies, A, Jones, D, Bailey, M, et al. Extracorporeal membrane oxygenation for 2009 influenza A (H1N1) acute respiratory distress syndrome. JAMA. 2009; 302:1888–1895.
102. Holzgraefe, B, Broome, M, Kalzen, H, et al. Extracorporeal membrane oxygenation for pandemic H1N1 2009 respiratory failure. Minerva Anestesiologica. 2010; 76:1043–1051.
103. Shekar, K, Fung, YL, Diab, S, et al. Development of simulated and ovine models of extracorporeal life support to improve understanding of circuit-host interactions. Crit Care Resusc. 2012; 14:105–111.
104. Maybauer, MO, Maybauer, DM, Fraser, JF, et al. Combined recombinant human activated protein C and ceftazidime prevent the onset of acute respiratory distress syndrome in severe sepsis. Shock. 2012; 37:170–176.
105. Hamahata, A, Enkhbaatar, P, Lange, M, et al. Direct delivery of low-dose 7-nitroindazole into the bronchial artery attenuates pulmonary pathophysiology after smoke inhalation and burn injury in an ovine model. Shock. 2011; 36:575–579.
106. Maybauer, DM, Maybauer, MO, Szabo, C, et al. The peroxynitrite catalyst WW-85 improves microcirculation in ovine smoke inhalation injury and septic shock. Burns. 2011; 37:842–850.
107. Enkhbaatar, P, Wang, J, Saunders, F, et al. Mechanistic aspects of inducible nitric oxide synthase-induced lung injury in burn trauma. Burns. 2011; 37:638–645.
108. Lange, M, Hamahata, A, Traber, DL, et al. Preclinical evaluation of epinephrine nebulization to reduce airway hyperemia and improve oxygenation after smoke inhalation injury. Crit Care Med. 2011; 39:718–724.
109. Maybauer, DM, Maybauer, MO, Szabo, C, et al. The peroxynitrite catalyst WW-85 improves pulmonary function in ovine septic shock. Shock. 2011; 35:148–155.
110. Maybauer, MO, Maybauer, DM, Fraser, JF, et al. Recombinant human activated protein C attenuates cardiovascular and microcirculatory dysfunction in acute lung injury and septic shock. Crit Care. 2010; 14:R217.
111. Traber, DL, Traber, MG, Enkhbaatar, P, Herndon, DN. Tocopherol as treatment for lung injury associated with burn and smoke inhalation. J Burn Care Res. 2009; 30:164–165.
112. Hamahata, A, Enkhbaatar, P, Kraft, ER, et al. Gamma-tocopherol nebulization by a lipid aerosolization device improves pulmonary function in sheep with burn and smoke inhalation injury. Free Radic Biol Med. 2008; 45:425–433.