[level-membership-for-critical-care-medicine-category]77
Acute Pancreatitis
CLINICAL PRESENTATION AND DIAGNOSIS
EARLY MANAGEMENT OF THE CRITICALLY ILL PATIENT WITH ACUTE PANCREATITIS
MANAGEMENT OF THE LATE COMPLICATIONS OF SEVERE ACUTE PANCREATITIS
Introduction
Acute pancreatitis is a complex disease with a highly variable clinical course. It is responsible for more than 200,000 hospital admissions each year in the United States,1 and it has an annual incidence of 10 to 80 cases in 100,000 in the developed world.2–5 Incidence rates have been increasing.6 For reasons that are unknown, there is seasonal variation in rates of the disease, the incidence being maximal in the spring and fall.7 The crude mortality rate for patients who are hospitalized with acute pancreatitis is less than 2%,8 and the majority of patients with acute pancreatitis experience a benign and self-limited disease, resulting in a hospital stay of only several days and no significant lasting sequelae. In a small percentage of patients, however, acute pancreatitis is sufficiently severe to lead to admission to an intensive care or high dependency unit, and a complicated clinical course with a mortality risk that may exceed 20%.1,9 These latter patients can present the intensivist with formidable challenges during the course of their illness, but if they recover, as most do, they can return to their premorbid state of health with no significant diminution in quality of life.
The prognosis for patients with severe acute pancreatitis has improved considerably since Ranson proposed his widely used severity criteria. In the mid-1970s, the mortality for patients with severe pancreatitis approached 100%10; today the risk is less than one quarter of that figure.11,12 This improved outcome can be ascribed in part to general improvements in the care of critically ill patients and, more important, to fundamental changes in the approach to the medical and surgical care of the patient with acute pancreatitis that, in turn, reflect an evolving understanding of the pathophysiology of the disease.
Definitions and Terminology
Pancreatitis is an acute inflammatory disorder that arises as a consequence of the activation of pancreatic digestive enzymes within the parenchyma of the gland and the surrounding tissues of the peritoneal cavity and retroperitoneum. Its evolution and complications are variable and give rise to terminology that is both confusing and imprecise. The most widely used classification system is that known as the Atlanta classification from 199213 (Table 77.1). Its reproducibility, however, is poor,14 and it is easier to conceptualize the disease as a spectrum of overlapping abnormalities resulting from the leakage of activated pancreatic enzymes, the host response to local tissue injury, and the superimposed complication of infection of what is initially a sterile process (Fig. 77.1).
Table 77.1
The Atlanta Classification System for Acute Pancreatitis
| Term | Definition |
| Acute pancreatitis | Acute inflammation of the pancreas with variable involvement of peripancreatic and remote tissues |
| Mild acute pancreatitis | Edema of pancreas; benign clinical course with minimal organ dysfunction and full recovery |
| Severe acute pancreatitis | Evidence of pancreatic necrosis; complications of infection, pseudocyst; clinical course characterized by organ failure |
| Acute fluid collection | Pancreatic or peripancreatic fluid, evident early in course of disease, and lacking wall |
| Pancreatic pseudocyst | Contained collection of pancreatic juice within a capsule of fibrous tissue and arising following an episode of acute pancreatitis |
| Pancreatic necrosis | Diffuse or focal loss of viability of pancreatic parenchyma or peripancreatic fat, evident as nonenhancing tissue on a contrast-enhanced CT scan |
| Pancreatic abscess | Localized collection of pus within pancreas or peripancreatic region and containing little or no necrotic material |
Adapted from Bradley EL: A clinically based classification system for acute pancreatitis. Arch Surg 1993;128:586-590.
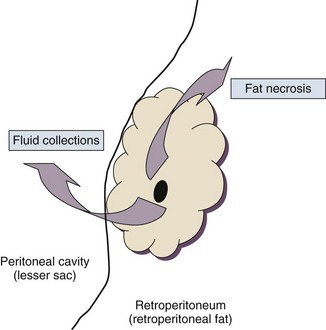
In the mildest form of the disease, leakage of activated enzymes is minimal, and the most prominent manifestation is pancreatic edema secondary to a local inflammatory process. In more severe cases, pancreatic ductal disruption results in leakage of pancreatic enzymes into adjacent tissues. If the leakage occurs anteriorly into the peritoneal cavity, fluid collections will be evident. The local peritoneal inflammatory response triggers coagulation and fibrin deposition, walling the collection off and creating a pseudocyst, so called because it is a fluid collection that lacks an epithelial lining. Drainage of the pancreatic ascites through the diaphragm can create a pleural effusion; typically this is seen on the left. Marked elevation of the amylase level in the pleural fluid establishes the collection as pancreatic juice, rather than as a reaction to an inflammatory process on the abdominal side of the diaphragm. Alternatively, if leakage is into the fatty tissues of the retroperitoneum, necrosis predominates, although small loculated fluid collections are often present. Often in patients with severe acute pancreatitis, computerized tomography shows evidence of both intraperitoneal and retroperitoneal involvement. The older term, hemorrhagic pancreatitis, describes the sequelae of extension of retroperitoneal necrosis into blood vessels of the retroperitoneum. Extraperitoneal tracking of the resulting hematoma gives rise to Grey-Turner’s sign when the ecchymosis is evident in the flanks and Cullen’s sign when it tracks anteriorly through the falciform ligament to present at the umbilicus.15
Pathogenesis
Acute pancreatitis arises in the pancreas through the leakage of activated pancreatic enzymes into pancreatic and peripancreatic tissues. The characteristic clinical syndrome, however, reflects the activation of a massive systemic inflammatory response to that local tissue injury, mediated through the activation of the inflammasome—a multiprotein intracellular complex that leads to the activation of the key inflammatory cytokine, interleukin-1.16
Beyond its role as an endocrine organ, the pancreas plays a fundamental role in the digestion of foodstuffs through the production of enzymes that degrade the major constituents of ingested food: protein (proteases), fat (lipases), starch (amylase), and nucleic acids (nucleases). Pancreatic enzymes are synthesized by the acinar cells lining the pancreatic ductal system and released from the cell as inactive zymogens.17 They pass via the pancreatic duct into the lumen of the duodenum, where they are activated by mucosal enzymes such as enterokinase, and so become capable of degrading their target substrates. Under normal circumstances, activation is vigorously inhibited within the pancreas itself through sequestration of newly synthesized enzymes within zymogen granules, and further through the local action of a specific inhibitor of trypsin activation called SPINK.18 The importance of normal mechanisms that inhibit trypsin activation is underlined by the observation that genetic mutations in the SPINK gene associated with reduced activity have been implicated in the pathogenesis of hereditary pancreatitis.19
Acute pancreatitis arises through the activation of pancreatic enzymes within the pancreas itself, possibly through the activity of lysosomal hydrolases in the acinar cell and initiating autodigestion of the pancreas and surrounding tissues.20,21 Activation of trypsinogen to trypsin appears to be a critical event during the early pathogenesis of the acute process,22,23 and an increase in intracellular calcium concentrations may contribute to activation of trypsinogen.24 Trypsinogen can also be activated by lysosomal cathepsin B within the acinar cell.25 Pancreatic acinar cells can die by either necrosis or apoptosis. Necrotic death is characterized by cell lysis, with the leakage of intracellular constituents into the surrounding microenvironment and the activation of an acute inflammatory response. Neutrophil recruitment in response to the local tissue injury has been implicated in the amplification of cellular damage in acute pancreatitis.26–28 Apoptotic death, in contrast, is noninflammatory. Cells are degraded into membrane-bound vesicles that are taken up by fixed tissue macrophages. Phagocytosis of an apoptotic cell not only prevents the local activation of inflammation but triggers transcriptional programs in the phagocytosing cell that are anti-inflammatory and reparative in nature, with up-regulation of counter-inflammatory cytokines such as interleukin-10 (IL-10).29 The local cellular injury in acute pancreatitis can also be conceptualized as an imbalance between necrotic and apoptotic cell death. Activation of caspases—the intracellular enzymes that mediate apoptosis—attenuate the severity of pancreatitis,30 whereas severe pancreatitis is associated with reduced levels of IL-10.31
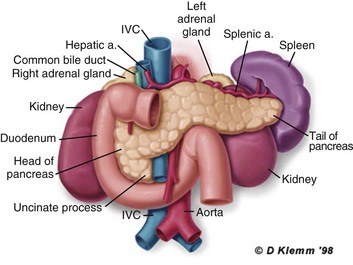
Activated pancreatic enzymes injure not only cells of the pancreas but also those of surrounding tissues. Fat cells appear to be particularly vulnerable, and the disease tends to be more severe in the obese because of the greater amount of peripancreatic fat necrosis.32 However, the degradative effects of pancreatic enzymes, and the secondary tissue injury resulting from the host response, can injure other structures in the vicinity of the pancreas, in particular, the transverse colon, and major vessels such as the splenic artery and vein.33 Pancreatitis rapidly evolves from an inflammatory disease of the pancreas to a chemical burn of the retroperitoneum.
The pathogenesis of the clinical syndrome is further complicated by the presence of the gastrointestinal tract immediately adjacent to the pancreas (Fig. 77.2). The head of the pancreas lies within the curve of the duodenum, immediately behind the stomach and above the transverse colon. Damage to terminal feeding vessels in the colonic fat caused by activated pancreatic enzymes can result in focal perforation of the colon, with leakage of colonic bacteria into the peritoneal cavity.34 Although the stomach and duodenum are only lightly colonized in health, the local ileus induced by the acute pancreatic inflammation promotes proximal gut overgrowth with enteric bacteria.35 In addition, changes in gut mucosal barrier function arising from local inflammation and the absence of enteral nutrients promote the translocation of luminal bacteria and of bacterial products such as endotoxin into the injured or necrotic peripancreatic tissues.36
Ultimately in the more severe cases, the clinical syndrome evolves as a result of a systemically activated inflammatory response, with the same changes in microvascular blood flow, endothelial permeability, and inflammatory mediator release that characterize bacterial sepsis.37
Etiology and Risk Factors
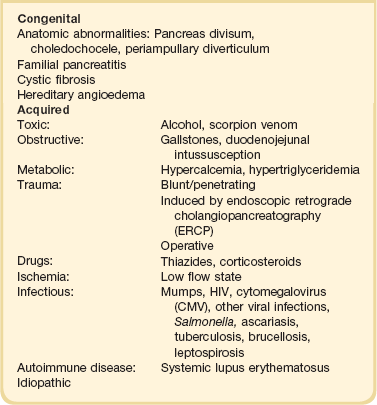
Acute pancreatitis has many causes, although 80% of cases in the developed world result from either alcohol or gallstones (Table 77.2). The mechanism of alcohol-induced pancreatitis is unclear. The acinar cell of the pancreas is capable of metabolizing alcohol and also appears to be the primary target of alcohol-mediated pancreatic injury.38 In vitro studies show that alcohol reduces the sensitivity of isolated acini to zymogen activation by cholecystokinin.39 Gallstone pancreatitis appears to be a consequence of a transient acute increase in pancreatic ductal pressures associated with passage of a small gallstone through the sphincter of Oddi in patients with a common channel for the bile and pancreatic ducts.40 True obstruction of the duct is uncommon, and approximately 90% of patients will be found to have gallstones in the stool, implicating the passage of the stone, rather than an obstructing mechanism, in the etiology of the resulting pancreatic inflammation. The list of drugs implicated in the etiology of acute pancreatitis is long41; some of the more prominent associations are shown in Table 77.2.
Genetic factors have been associated with the development of acute pancreatitis. A polymorphism in the secretory trypsin inhibitor (SPINK1) gene is associated with an increased risk for acute pancreatitis,42 as are mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene,43 and PRSS1.44 Polymorphisms in the genes for tumor necrosis factor α (TNFα) and heat shock protein 70 have also been linked to an increased risk for acute pancreatitis,45 although the association is less clear.
Clinical Presentation and Diagnosis
The causes of shock in the patient with acute pancreatitis are multifactorial. Initially, the acute inflammatory process in the retroperitoneum elicits local inflammation, with an outpouring of fluid into the relatively confined space of the retroperitoneum or into the peritoneal cavity itself. Intra-abdominal inflammation evokes secondary ileus within the gastrointestinal tract, and fluid is sequestered here, increasing the relative intravascular volume deficit. Nausea, vomiting, and a reluctance to take fluids by mouth further exacerbate this fluid deficit. As the process evolves, a systemic inflammatory response to the local abdominal process results in diffuse vasodilatation and capillary leak syndrome. In aggregate, the effective loss of intravascular volume can be enormous, and so the initial resuscitation may require large volumes of intravenous fluid. Acute hypocalcemia may also contribute to the clinical picture of cardiovascular compromise. Indeed the classical prognostic criteria articulated by Ranson (Table 77.3) emphasize the importance of acute inflammation (white cell count), and the secondary hemodynamic (fluid sequestration, base excess, blood urea nitrogen [BUN], and hematocrit) and metabolic (glucose, calcium) sequelae in determining the ultimate prognosis.
Table 77.3
Ranson’s Criteria in Acute Pancreatitis

LDH, Lactate dehydrogenase; AST, aspartate aminotransferase; BUN, blood urea nitrogen.
From Ranson JH, Rifkind KM, Roses DF, et al: Prognostic signs and the role of operative management in acute pancreatitis. Surg Gynecol Obstet 1974;139:69-81.
The diagnosis of pancreatitis is usually straightforward, based on the combination of clinical manifestations and characteristic biochemical findings of elevations in the circulating levels of amylase and lipase, the latter being somewhat more accurate diagnostically.46 The diagnosis can be confirmed, and the severity of the disease evaluated by computerized tomography (Fig. 77.3), using the grading system developed by Balthazar47 (Table 77.4).
Table 77.4
Balthazar Grading of CT Findings in Acute Pancreatitis
| Grade | Findings |
| Grade A | Normal pancreas |
| Grade B | Pancreatic enlargement |
| Grade C | Pancreatic or peripancreatic inflammation |
| Grade D | Single peripancreatic fluid collection |
| Grade E | 2 or more pancreatic collections and/or retroperitoneal air |
From Balthazar EJ: CT diagnosis and staging of acute pancreatitis. Radiol Clin North Am 1989;27:19-37.
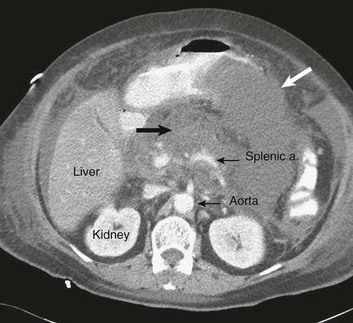
An initial assessment of the severity of the disease is useful primarily for deciding the optimal venue for the early management of the patient, as at least some of the delayed morbidity of acute pancreatitis can be reduced through aggressive initial resuscitation and support. Various approaches have been used to quantify disease severity. Ranson identified 11 variables—5 at initial presentation and 6 over the ensuing 48 hours—that correlated in a graded fashion with the ultimate risk of mortality (see Table 77.3). The Glasgow-Imrie criteria are a modification of Ranson’s scale and represent an alternate model of severity scoring. In head-to-head studies, Acute Physiology, Age, and Chronic Health Evaluation (APACHE) II—a generic severity of illness scale—performs at least as well as the Ranson or Glasgow-Imrie criteria in predicting hospital survival.48 Moreover, even simpler scales appear to provide comparable prognostic information.23,49 Various biochemical measures, including C-reactive protein, procalcitonin, interleukin-6, and trypsinogen activation peptide (TAP), are purported to differentiate mild and severe pancreatitis early during the clinical evolution of the disease50–52; their clinical utility is unclear. Persistence of clinical manifestations of systemic inflammation beyond 48 hours is also associated with subsequent organ dysfunction and a higher mortality.53 Precision in prognostication is far less important than early recognition of the patient for whom close monitoring and aggressive resuscitation can alter the clinical course, and a low threshold for management in a more controlled and monitored setting is an important factor in reducing the complications of pancreatitis.54
Early Management of the Critically Ill Patient with Acute Pancreatitis
Initial Resuscitation
Intravascular fluid deficits early in the course of pancreatitis can be substantial. Early aggressive fluid resuscitation is the cornerstone of initial successful management.55 Volume status should be monitored with a urinary catheter and central venous catheter, and although studies in the specific setting of pancreatitis have not been performed, there is every reason to believe that the principles of goal-directed resuscitation should be followed.56 There are no data to support a preference for colloids or crystalloids during resuscitation; the key, however, is to administer sufficient volumes rapidly enough to restore an adequate circulating volume.57 Typically this comprises many liters of fluid, and not infrequently, the sickest patients will receive more than 10 to 15 L of fluid resuscitation over the first 24 hours. Such aggressive resuscitation is best carried out within the well-monitored environment of the intensive care unit (ICU).
Although aggressive fluid resuscitation can minimize later complications such as pancreatic necrosis and acute renal failure, in the patient with increased vascular permeability it carries a high risk of complications related to interstitial edema. Frequently patients will require endotracheal intubation and mechanical ventilation. Development of the abdominal compartment syndrome is a relatively common and underdiagnosed complication of resuscitated acute pancreatitis.58 Intra-abdominal pressure can be measured by transducing a urinary catheter in the bladder. Normal pressures approximate the central venous pressure, whereas pressures greater than 20 cm H2O indicate intra-abdominal hypertension, and pressures greater than 30 cm H2O indicate a compartment syndrome and carry an increased risk of ischemic injury because of impairment of visceral venous drainage. In severe cases, management requires abdominal decompression by laparotomy. The need for laparotomy in the patient with acute pancreatitis may be avoided by the use of continuous venovenous hemofiltration59 or decompressive anterior abdominal fasciotomy.60
Infection Prophylaxis
Infection is a common complication of acute necrotizing pancreatitis, and its development is associated with an increased risk of morbidity and mortality.61 For the patient with severe acute pancreatitis, these infections may arise within the injured or necrotic peripancreatic tissues or at distant sites as nosocomial infection in a critically ill patient.
The characteristic microbial flora of peripancreatic infection includes organisms normally resident within the gastrointestinal tract62 and organisms that characteristically colonize the proximal gastrointestinal tract of the critically ill patient35 (Box 77.1). Anaerobes may be present but are uncommon, whereas organisms such as Candida, Enterococci, and coagulase-negative Staphylococci are encountered with increasing frequency.63,64 This infecting flora reflects the role of the gastrointestinal tract as the reservoir of organisms inducing superinfection during acute pancreatitis.65
Infection of necrotic tissue in the retroperitoneum can arise by one of several routes. Bacteremic spread from a distant site, retrograde passage up the pancreatic duct, and direct extension through a defect in the adjacent gastrointestinal tract are all plausible mechanisms. However, the most significant mechanism of infection appears to be as a consequence of the translocation of viable microorganisms across an anatomically gastrointestinal (GI) tract, from either the colon or the small intestine. Bacterial translocation is readily demonstrable in animal models of acute pancreatitis,66,67 and indirect evidence suggests that the phenomenon contributes to infectious complications in human pancreatitis.68 Patients with severe pancreatitis, for example, have higher rates of proximal gut colonization with the same enteric organisms that produce infection,69 and intestinal colonization invariably precedes the development of invasive infection.70 Moreover, suppression of pathologic gut colonization reduces the risk of infection.71
Host-microbial interactions within the GI tract are complex,72,73 and the changes that occur in pancreatitis are much more than a state of generalized leakiness. Colectomy, for example, increases rates of small bowel microbial colonization and bacterial translocation.74 Moreover, the anaerobic flora of the gut provides a barrier to intestinal mucosal colonization with pathogenic aerobes and inhibits bacterial translocation.75 Thus, anaerobes are uncommonly found in pancreatic infections, and the isolation of an anaerobic organism from an area of infected pancreatic necrosis is highly suggestive of a physical breach of the gastrointestinal tract.
Infection of pancreatic necrosis is a relatively late event, occurring maximally during the second or third week after the onset of the acute disease.76 However, circulating bacterial DNA77 or endotoxin from gram-negative bacteria78 can be detected early during the course of the disease.
The role of antibiotic prophylaxis, however, is controversial. An early meta-analysis of randomized controlled trials of prophylactic antibiotics for patients with acute pancreatitis concluded that prophylaxis can improve survival without significantly altering rates of pancreatic infection.79 A consensus conference of critical care organizations, however, has recommended that routine antibiotic prophylaxis should not be used in the absence of more compelling evidence from randomized controlled trials,54 and more recent compilations of data from clinical trials fail to show clear evidence of benefit for antibiotic prophylaxis.80 A push for conservatism is driven by three principal factors: the low methodological quality of trials supporting antibiotic prophylaxis, concern regarding the adverse ecologic consequences of prolonged broad-spectrum antibiotic administration, and the increasing use of an alternate prophylactic strategy, enteral feeding.81 One of the most influential studies of prophylactic antibiotics in pancreatitis, for example, reported a significant survival improvement associated with antibiotic prophylaxis in a cohort of 60 patients82; in that study, however, antibiotic use had no impact on rates of pancreatic infection, and fully three quarters of patients in the control arm received antibiotics early in the course of their disease. The prevention of pathologic gut colonization through the use of selective digestive tract decontamination has been shown to reduce rates of pancreatic infectious complications71 and to reduce mortality associated with gram-negative infections.83 Whether antifungal prophylaxis is efficacious is also controversial.84,85
Nutritional Support
In contrast to the persistent controversy regarding the utility of antibiotic prophylaxis in severe acute pancreatitis, it is now generally accepted that patients do better and, in particular, experience less infectious and inflammatory morbidity if they are fed enterally rather than parenterally. Enteral feeding is associated with maintained epithelial barrier function as well as reduced rates of translocation of endotoxin and viable bacteria in experimental animals.86,87 A meta-analysis of eight trials recruiting 348 patients found a lower rate of infectious complications (relative risk [RR] 0.39; 95% confidence interval [CI] 0.23 to 0.65), a reduced need for operative intervention (RR 0.44; 95% CI 0.29 to 0.67), a reduced risk of organ failure (RR 0.55; 95% CI 0.37 to 0.81), and a reduced risk of death (RR 0.50; 95% CI 0.28 to 0.91).88
Current guidelines recommend the early institution of enteral feeds in patients with severe acute pancreatitis, supplemented, as needed, with parenteral support to meet full nutritional requirements.54,89,90 Although the presence of gastric ileus in acute pancreatitis has resulted in a preference for the nasojejunal rather than the nasogastric route of feeding, there is no evidence of the inferiority of nasogastric feeding in more recent clinical trials.91,92 Similarly, there is no compelling evidence at present to favor any particular nutritional formulation.
Adjuvant Therapy
Despite a substantial body of literature suggesting a benefit for a variety of different adjuvant treatments in preclinical models, there is no evidence for any specific therapy in human disease. A systematic review of the use of cimetidine found no evidence that suppression of gastric acid secretion improved outcome, but rather a trend to a higher risk of complications.93 Pooled data from small studies have suggested that somatostatin or octreotide can improve survival for patients with severe acute pancreatitis,94 but data from larger, more robust trials are lacking. Protease inhibitors have also been suggested to reduce mortality in moderate to severe pancreatitis.95
Platelet-activating factor (PAF) is a potent pro-inflammatory lipid mediator that has been implicated in the local and remote manifestations of acute pancreatitis.96 Inhibition of PAF with either a receptor antagonist97 or through the administration of recombinant PAF acetylhydrolase—the enzyme responsible for degrading PAF98—results in attenuation of injury in animal models. Unfortunately, despite early promise in phase II clinical trials,99,100 a phase III trial failed to show any benefit for adjuvant treatment with the PAF receptor antagonist, lexipafant, in 290 patients with severe acute pancreatitis.101
Other strategies such as inhibition of TNF or blockade of interleukin-1 have shown promise in preclinical models but have not been evaluated in human disease.102
Endoscopic Retrograde Cholangiopancreatography (ERCP) in Acute Gallstone Pancreatitis
Gallstone pancreatitis is caused by a gallstone that has migrated from the gallbladder into the common bile duct producing transient obstruction of the pancreatic duct. This pathogenetic mechanism raises the possibility that measures to relieve the obstruction at the level of the sphincter of Oddi might reduce the severity of the disease. This possibility has been evaluated in at least four clinical trials, the pooled data from which suggest benefit for patients with severe pancreatitis in reducing subsequent complications.103 It is unclear whether the benefit extends to all patients or simply to the subset of patients with persistent common bile duct obstruction and concomitant cholangitis.104,105
Management of the Late Complications of Severe Acute Pancreatitis
It is apparent from the foregoing that the early management of the patient with severe acute pancreatitis is focused primarily on adequate resuscitation and organ support. Early lethal postresuscitation complications are uncommon106 and include intestinal ischemia (typically secondary to a low flow state or mesenteric venous thrombosis following delayed resuscitation)107 and bleeding secondary to erosion into a major vessel. In the absence of these catastrophic and fortunately rare complications, the subsequent management of the critically ill patient with acute pancreatitis involves optimal intensive care,108 close monitoring, and patience.
Pancreatic Pseudocysts
Serial evaluation by computerized tomography shows that the diffuse fluid collections evident in early pancreatitis coalesce to form discrete cystlike collections contained within an organized capsule of fibrin and termed a pseudocyst. Typically these arise in the lesser sac, between the pancreas and the posterior wall of the stomach (Fig. 77.4). Management is expectant in the absence of complications,109 and rarely is intervention required in the ICU setting. The most common symptom precipitating drainage is persistent pain or symptoms of gastric obstruction. Other important complications include infection and hemorrhage into the cyst.
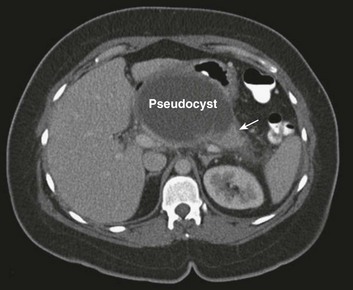
The classic approach to the management of a pancreatic pseudocyst involved waiting until the fibrous capsule of the cyst had matured, then draining the cyst into the back wall of the stomach. Open surgical pseudocyst gastrostomy has largely given way to less invasive approaches, creating a cyst gastrostomy by radiologic image-guided,110 endoscopic,111 or laparoscopic approaches.112 A pseudocyst forms because of injury to the main pancreatic duct, or one of its branches, and persists because of obstruction to the flow of pancreatic juice through the duct. If that obstruction persists, then external drainage will result in a persistent pancreaticocutaneous fistula. Thus, regardless of how it is accomplished, drainage into the stomach avoids this complication by replacing the external fistula with an internal fistula that is without clinical importance. Open operation may be required for massive hemorrhage; angiographic embolization of the bleeding vessel is another alternative.113
Infected Necrosis and Pancreatic Abscess
One of the more significant advances in the management of severe pancreatitis has been the adoption of a policy of surgical conservatism in managing patients with suspected pancreatic or peripancreatic infection. Case series demonstrate improved survival rates when surgery is delayed,114–118 and a single randomized trial showed that delaying surgical intervention for at least 2 weeks in patients with necrotizing pancreatitis improves clinical outcomes.119 This improved outcome can be attributed to a reduced frequency of major bleeding complications associated with the debridement of necrotic, infected retroperitoneal tissue.
Just as serial evaluation reveals localization of pancreatic fluid collections, serial study of areas of pancreatic necrosis shows that they too coalesce with time, becoming more circumscribed and, importantly, developing a wall of granulation tissue between the areas of necrosis and viable surrounding tissues. It is this clear demarcation between viable and nonviable tissue that renders operative intervention safe and obviates the need for repeat operation or open-abdomen approaches. Typically, the process of demarcation takes 3 to 4 weeks or longer to occur; thus, surgical intervention, if contemplated, should be deferred. A strategy of surgical conservatism in the face of a patient with clinical manifestations of sepsis and ongoing organ dysfunction may appear counterintuitive, but it is both well tolerated and safer than the alternative.120
There is general consensus that the indication for operative intervention in a patient with severe pancreatitis is infected necrosis; sterile necrosis should be managed conservatively.115,121,122 The diagnosis of infection of pancreatic necrosis can be challenging to establish, for clinical manifestations of systemic inflammation are common in the acutely ill patient with pancreatitis, even in the absence of infection, as are nosocomial infections in sites other than the necrotic retroperitoneal tissues. A diagnosis of infection may be suggested by new elevations in levels of procalcitonin123,124 or by computed tomography (CT) findings of air in the necrotic tissues (Fig. 77.5). However, definitive diagnosis of infection is best established by CT-guided fine-needle aspiration of the peripancreatic necrotic tissues.125
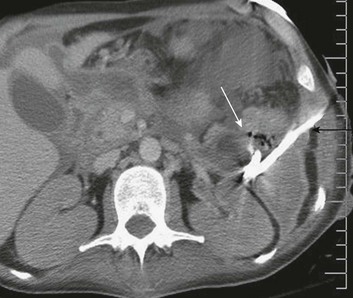
Documentation of infection on a fine-needle aspirate is not an absolute indication for operative intervention. Antibiotics should be administered as guided by the results of culture and sensitivity and then source control options evaluated. Early in the course of the disease, before the demarcation of necrotic and viable tissues, percutaneous drainage of the fluid component of the collection can temporize until operative intervention is safer (see Fig. 77.5). Percutaneous drainage alone may be curative,126,127 and there is even evolving literature indicating that some patients with infected pancreatic necrosis can be successfully managed nonoperatively.128,129
The surgical management of infected pancreatic necrosis entails the debridement of the necrotic retroperitoneal tissues and drainage of the resulting cavity. Approaches span the surgical spectrum from minimal access techniques130–132 to open abdomen approaches.133,134 Improved clinical outcomes have also been seen with the use of less invasive, staged procedures. van Santvoort and colleagues reported a randomized controlled trial of a staged approach to pancreatic necrosectomy, consisting of initial percutaneous drainage of the liquid elements of a pancreatic infection, followed by a minimally invasive necrosectomy, using the drain tract as a route of access.12 The minimally invasive approach resulted in significantly lower rates of major complications and new-onset organ dysfunction, as well as lower rates of delayed complications including diabetes and incisional hernia. This same group has also reported lower rates of new organ dysfunction and pancreatic fistula when patients were managed by transgastric endoscopic necrosectomy when compared with open necrosectomy.135
Laparoscopic techniques have the added advantage of permitting direct visualization of the abscess cavity,132,136,137 but they may be challenging if there is extensive or multiloculated areas of necrosis. Infected necrosis can also be approached via the flank, using a nephroscope or laparoscope to aid in visualization of the abscess contents.138 Further insights into the role of minimally invasive approaches should emerge from several ongoing clinical trials.112,139 The role for open abdomen approaches in the management of infected pancreatic necrosis is diminishing as surgical practice shifts to delayed intervention with more minimally invasive techniques.140
Peritoneal lavage with the objective of evacuating activated pancreatic enzymes enjoyed a period of popularity141 but is now used less frequently. Some authors recommend continuous postoperative lavage following pancreatic necrosectomy,140 though the benefits of this approach are unproven.
Vascular Complications of Necrotizing Pancreatitis
The vascular complications of acute pancreatitis include both thrombosis and hemorrhage.142,143 Thrombosis of the splenic, superior mesenteric, and portal veins develops as a consequence of the pro-thrombotic effects of the adjacent inflamed pancreas and reduced flow prior to full resuscitation. Less commonly, more distant vessels such as the inferior vena cava or renal veins may thrombose. Venous thrombosis may give rise to portal venous gas on computed tomography; in the absence of clinical findings dictating a need for emergent intervention, both the venous thrombosis and the associated CT findings can be managed conservatively.144 Treatment consists of anticoagulation; intestinal infarction secondary to venous thrombosis in patients with acute pancreatitis carries a prohibitive mortality, even with surgery.
Erosion of the retroperitoneal inflammatory process into a major artery can produce bleeding into either the peritoneal cavity or the gut lumen, in which case it presents as a gastrointestinal hemorrhage. Commonly involved vessels include the splenic artery, the gastroduodenal artery, and the pancreaticoduodenal arcade.145 Operative exposure is challenging in the face of acute retroperitoneal inflammation, and whenever feasible, bleeding is best managed by angiographic embolization.113
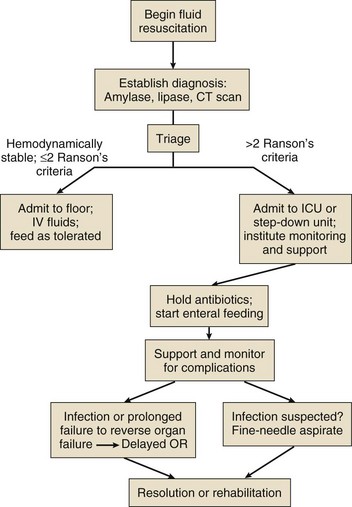
An algorithm summarizing the key elements in the management of the patient with severe acute pancreatitis is provided in Figure 77.6.
Long-Term Outcome and Quality of Life
Acute necrotizing pancreatitis can be an enormously challenging process to treat. The ICU and hospital stay is often prolonged, and in addition to operative procedures undertaken during the acute episode, there is often a need for later intervention to close a stoma, repair an incisional hernia, or excise the gallbladder. Yet there is ample evidence that patients who survive their acute illness return to a health-related quality of life that is no different from age-matched controls.146–149 Abnormalities of both endocrine and exocrine function are commonly evident at follow-up,150 and approximately one third of survivors of severe necrotizing pancreatitis develop diabetes.151
References
1. Whitcomb, DC. Clinical practice: Acute pancreatitis. N Engl J Med. 2006; 354:2142–2150.
2. Shahen, NJ, Hansen, RA, Morgan, DR, et al. The burden of gastrointestinal and liver diseases, 2006. Am J Gastroenterol. 2006; 101:2128–2138.
3. Frey, CF, Zhou, H, Harvey, DJ, White, RH. The incidence and case-fatality rates of acute biliary, alcoholic, and idiopathic pancreatitis in California, 1994-2001. Pancreas. 2006; 33:336–344.
4. Yadav, D, Lowenfels, AB. Trends in the epidemiology of the first attack of acute pancreatitis: A systematic review. Pancreas. 2006; 33:323–330.
5. Andersson, R, Anderrson, B, Haraldsen, P, et al. Incidence, management and recurrence rate of acute pancreatitis. Scand J Gastroenterol. 2004; 39:891–894.
6. Goldacre, MJ, Roberts, SE. Hospital admission for acute pancreatitis in an English population, 1963-98: Database study of incidence and mortality. BMJ. 2004; 328:1466–1469.
7. Gallerani, M, Boari, B, Salmi, R, Manfredini, R. Seasonal variation in the onset of acute pancreatitis. World J Gastroenterol. 2004; 10:3328–3331.
8. Singla, A, Simons, J, Li, Y, et al. Admission volume determines outcome for patients with acute pancreatitis. Gastroenterology. 2009; 137:1995–2001.
9. Swaroop, VS, Chari, ST, Clain, JE. Severe acute pancreatitis. JAMA. 2004; 291:2865–2868.
10. Ranson, JHC, Rifkind, KM, Turner, JW. Prognostic signs and nonoperative peritoneal lavage in acute pancreatitis. Surg Gynecol Obstet. 1976; 143:209.
11. Petrov, MS, Shanbhag, S, Chakraborty, M, et al. Organ failure and infection of pancreatic necrosis as determinants of mortality in patients with acute pancreatitis. Gastroenterology. 2010; 139:813–820.
12. van Santvoort, HC, Besselink, MG, Bakker, OJ, et al. A step-up approach or open necrosectomy for necrotizing pancreatitis. N Engl J Med. 2010; 362:1491–1502.
13. Bradley, EL. A clinically based classification system for acute pancreatitis. Arch Surg. 1993; 128:586–590.
14. Besselink, MG, van Santvoort, HC, Bollen, TL, et al. Describing computed tomography findings in acute necrotizing pancreatitis with the Atlanta classification: An interobserver agreement study. Pancreas. 2007; 33:331–335.
15. Sugimoto, M, Takada, T, Yasuda, H, et al. MPR-hCT imaging of the pancreatic fluid pathway to Grey-Turner’s and Cullen’s sign in acute pancreatitis. Hepatogastroenterology. 2005; 52:1613–1616.
16. Hoque, R, Sohail, M, Malik, A, et al. TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology. 2011; 141:358–369.
17. Pandol, SJ. Acute pancreatitis. Curr Opin Gastroenterol. 2006; 22:481–486.
18. Liddle, RA. Pathophysiology of SPINK mutations in pancreatic development and disease. Endocrinol Metab Clin North Am. 2006; 35:345–356.
19. Halangk, W, Lerch, MM. Early events in acute pancreatitis. Clin Lab Med. 2005; 25:1–15.
20. Gorelick, FS, Otani, T. Mechanisms of intracellular zymogen activation. Bailliere’s Best Pract Res Clin Gastroenterol. 1999; 13:227–240.
21. van Acker, GJ, Perides, G, Steer, ML. Co-localization hypothesis: A mechanism for the intrapancreatic activation of digestive enzymes during the early phases of acute pancreatitis. World J Gastroenterol. 2006; 12:1985–1990.
22. Bhatia, M, Wong, FL, Cao, Y, et al. Pathophysiology of acute pancreatitis. Pancreatology. 2005; 5:132–144.
23. Spitzer, AL, Barcia, AM, Schell, MT, et al. Applying Ockham’s razor to pancreatitis prognostication: A four-variable predictive model. Ann Surg. 2006; 243:380–388.
24. Petersen, OH, Sutton, R. Ca2+ signalling and pancreatitis: Effects of alcohol, bile and coffee. Trends Pharmacol Sci. 2006; 27:113–120.
25. Halangk, W, Lerch, MM, Brandt-Nedelev, B, et al. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J Clin Invest. 2000; 106:773–781.
26. Gukovskaya, AS, Vaquero, E, Zaninovic, V, et al. Neutrophils and NADPH oxidase mediate intrapancreatic trypsin activation in murine experimental acute pancreatitis. Gastroenterology. 2002; 122:974–984.
27. Raraty, MG, Murphy, JA, Mcloughlin, E, et al. Mechanisms of acinar cell injury in acute pancreatitis. Scand J Surg. 2005; 94:89–96.
28. Shields, CJ, Winter, DC, Redmond, HP. Lung injury in acute pancreatitis: Mechanisms, prevention, and therapy. Curr Opin Crit Care. 2002; 8:158–163.
29. Serhan, CN, Savill, J. Resolution of inflammation: The beginning programs the end. Nat Immunol. 2005; 6:1191–1197.
30. Mareninova, OA, Sung, KF, Hong, P, et al. Cell death in pancreatitis: Caspases protect from necrotizing pancreatitis. J Biol Chem. 2006; 281:3370–3381.
31. Laveda, R, Martinez, J, Munoz, C, et al. Different profile of cytokine synthesis according to the severity of acute pancreatitis. World J Gastroenterol. 2005; 11:5309–5313.
32. Martinez, J, Johnson, CD, Sanchez-Paya, J, et al. Obesity is a definitive risk factor of severity and mortality in acute pancreatitis: An updated meta-analysis. Pancreatology. 2006; 6:206–209.
33. Flati, G, Salvatori, F, Porowska, B, et al. Severe hemorrhagic complications in pancreatitis. Ann Ital Chir. 2007; 66:233–237.
34. van Minnen, LP, Besselink, MG, Bosscha, K, et al. Colonic involvement in acute pancreatitis: A retrospective study of 16 patients. Dig Surg. 2004; 21:33–40.
35. Marshall, JC, Christou, NV, Meakins, JL. The gastrointestinal tract. The “undrained abscess” of multiple organ failure. Ann Surg. 1993; 218:111–119.
36. Penalva, JC, Martinez, J, Laveda, R, et al. A study of intestinal permeability in relation to the inflammatory response and plasma endocab IgM levels in patients with acute pancreatitis. J Clin Gastroenterol. 2004; 38:512–517.
37. Cuthbertson, CM, Christophi, C. Disturbances of the microcirculation in acute pancreatitis. Br J Surg. 2006; 93:518–530.
38. Apte, MV, Pirola, RC, Wilson, JS. Molecular mechanisms of alcoholic pancreatitis. Dig Dis. 2005; 23:232–240.
39. Gorelick, FS. Alcohol and zymogen activation in the pancreatic acinar cell. Pancreas. 2003; 27:305–310.
40. Acosta, JM, Ledesma, CL. Gallstone migration as a cause of acute pancreatitis. N Engl J Med. 1974; 290:484–487.
41. Trivedi, CD, Pitchumoni, CS. Drug-induced pancreatitis: An update. J Clin Gastroenterol. 2005; 39:709–716.
42. Tukiainen, E, Kylanpaa, ML, Kemppainen, E, et al. Pancreatic secretory trypsin inhibitor (SPINK1) gene mutations in patients with acute pancreatitis. Pancreas. 2005; 30:239–242.
43. Bishop, MD, Freedman, SD, Zielenski, J, et al. The cystic fibrosis transmembrane conductance regulator gene and ion channel function in patients with idiopathic pancreatitis. Hum Genet. 2005; 118:372–381.
44. LaRusch, J, Whitcomb, DC. Genetics of pancreatitis. Curr Opin Gastroenterol. 2011; 27:467–474.
45. Balog, A, Gyulai, Z, Boros, LG, et al. Polymorphism of the TNF-alpha, HSP70-2, and CD14 genes increases susceptibility to severe acute pancreatitis. Pancreas. 2005; 30:46–50.
46. Smith, RC, Southwell-Keely, J, Chesher, D. Should serum pancreatic lipase replace serum amylase as a biomarker of acute pancreatitis? ANZ J Surg. 2005; 75:399–404.
47. Balthazar, EJ. Acute pancreatitis: Assessment of severity with clinical and CT evaluation. Radiology. 2002; 223:603–613.
48. Yeung, YP, Lam, BY, Yip, AW. APACHE system is better than Ranson system in the prediction of severity of acute pancreatitis. Hepatobiliary Pancreat Dis Int. 2006; 5:294–299.
49. Ueda, T, Takeyama, Y, Yasuda, T, et al. Simple scoring system for the prediction of the prognosis of severe acute pancreatitis. Surgery. 2007; 141:51–58.
50. Rau, B, Schilling, MK, Beger, HG. Laboratory markers of severe acute pancreatitis. Dig Dis. 2004; 22:247–257.
51. Papachristou, GI, Whitcomb, DC. Inflammatory markers of disease severity in acute pancreatitis. Clin Lab Med. 2005; 25:17–37.
52. Neoptolemos, JP, Kemppainen, EA, Mayer, JM, et al. Early prediction of severity in acute pancreatitis by urinary trypsinogen activation peptide: A multicentre study. Lancet. 2000; 355:1955–1960.
53. Mofidi, R, Duff, MD, Wigmore, SJ, et al. Association between early systemic inflammatory response, severity of multiorgan dysfunction and death in acute pancreatitis. Br J Surg. 2006; 93:7738–7744.
54. Nathens, AB, Curtis, JR, Beale, RJ, et al. Management of the critically ill patient with severe acute pancreatitis. Crit Care Med. 2004; 32:2524–2536.
55. Tenner, S. Initial management of acute pancreatitis: Critical issues during the first 72 hours. Am J Gastroenterol. 2004; 99:2489–2494.
56. Rivers, E, Nguyen, B, Havstad, S, et al. Early goal-directed therapy in the treatment of severe sepsis and septic shock. N Engl J Med. 2001; 345:1368–1377.
57. Brown, A, Baillargeon, JD, Hughes, MD, Banks, PA. Can fluid resuscitation prevent pancreatic necrosis in severe acute pancreatitis? Pancreatology. 2002; 2:104–107.
58. De Waele, JJ, Hoste, E, Blot, SI, et al. Intra-abdominal hypertension in patients with severe acute pancreatitis. Crit Care. 2005; 9:R452–R457.
59. Sun, ZX, Huang, HR, Zhou, H. Indwelling catheter and conservative measures in the treatment of abdominal compartment syndrome in fulminant acute pancreatitis. World J Gastroenterol. 2006; 12:5068–5070.
60. Leppaniemi, AK, Hienonen, PA, Siren, JE, et al. Treatment of abdominal compartment syndrome with subcutaneous anterior abdominal fasciotomy in severe acute pancreatitis. World J Surg. 2006; 30:1922–1924.
61. Isenmann, R, Rau, B, Beger, HG. Bacterial infection and extent of necrosis are determinants of organ failure in patients with acute necrotizing pancreatitis. Br J Surg. 2007; 86:1020–1024.
62. Schmid, SW, Uhl, W, Friess, H, et al. The role of infection in acute pancreatitis. Gut. 1999; 45:311–316.
63. Shanmugam, N, Isenmann, R, Barkin, JS, Beger, HG. Pancreatic fungal infection. Pancreas. 2007; 27:133–138.
64. Isenmann, R, Schwarz, M, Rau, B, et al. Characteristics of infection with Candida species in patients with necrotizing pancreatitis. World J Surg. 2002; 26:372–376.
65. Ammori, BJ. Role of the gut in the course of severe acute pancreatitis. Pancreas. 2007; 26:122–129.
66. Scwarz, M, Thomsen, J, Meyer, H, et al. Frequency and time course of pancreatic and extrapancreatic bacterial infection in experimental acute pancreatitis in rats. Surgery. 2000; 127:427–432.
67. Kouris, GJ, Liu, Q, Rossi, H, et al. The effect of glucagon-like peptide 2 on intestinal permeability and bacterial translocation in acute necrotizing pancreatitis. Am J Surg. 2001; 181:571–575.
68. Ammori, BJ. Role of the gut in the course of severe acute pancreatitis. Pancreas. 2003; 26:122–129.
69. McNaught, CE, Woodcock, NP, Mitchell, CJ, et al. Gastric colonisation, intestinal permeability and septic morbidity in acute pancreatitis. Pancreatology. 2002; 2:463–468.
70. Luiten, EJ, Hop, WC, Endtz, HP, Bruining, HA. Prognostic importance of gram-negative intestinal colonization preceding pancreatic infection in severe acute pancreatitis: Results of a controlled clinical trial of selective decontamination. Intensive Care Med. 1998; 24:438–445.
71. Luiten, EJ, Hop, WCJ, Lange, JF, Bruining, HA. Controlled clinical trial of selective decontamination for the treatment of severe acute pancreatitis. Ann Surg. 1995; 222:57–65.
72. Hooper, LV, Gordon, JI. Commensal host-bacterial relationships in the gut. Science. 2001; 292:1115–1118.
73. Alverdy, JC, Laughlin, RS, Wu, L. Influence of the critically ill state on host-pathogen interactions within the intestine: Gut-derived sepsis redefined. Crit Care Med. 2003; 31:598–607.
74. van Minnen, LP, Nieuwnhuijs, VB, de Bruijn, MT, et al. Effects of subtotal colectomy on bacterial translocation during experimental acute pancreatitis. Pancreas. 2006; 32:110–114.
75. Marshall, JC. Lipopolysaccharide: An endotoxin or an exogenous hormone? Clin Infect Dis. 2005; 41(Suppl 7):S470–S480.
76. Beger, HG, Bittner, R, Block, S, Buchler, M. Bacterial contamination of pancreatic necrosis: A prospective clinical study. Gastroenterology. 1986; 91:433–438.
77. de Madaria, E, Martinez, J, Lozano, B, et al. Detection and identification of bacterial DNA in serum from patients with acute pancreatitis. Gut. 2005; 54:1293–1297.
78. Buttenschoen, K, Berger, D, Hiki, N, et al. Endotoxin and antiendotoxin antibodies in patients with acute pancreatitis. Eur J Surg. 2000; 166:459–466.
79. Villatoro, E, Bbassi, C, Larvin, M. Antibiotic therapy for prophylaxis against infection of pancreatic necrosis in acute pancreatitis. Cochrane Database Syst Rev. (4):2006.
80. Villatoro, E, Mulla, M, Larvin, M. Antibiotic therapy for prophylaxis against infection of pancreatic necrosis in acute pancreatitis. Cochrane Database Syst Rev. 2010.
81. Targarona Modena, J, Barreda Cevasco, L, Arroyo Basto, C, et al. Total enteral nutrition as prophylactic therapy for pancreatic necrosis infection in severe acute pancreatitis. Pancreatology. 2006; 6:58–64.
82. Sainio, V, Kemppainen, E, Puolakkainen, P, et al. Early antibiotic treatment in acute necrotising pancreatitis. Lancet. 1995; 346:663–667.
83. Luiten, EJ, Hop, WC, Lange, JF, Bruining, HA. Differential prognosis of gram-negative versus gram-positive infected and sterile pancreatic necrosis: Results of a randomized trial in patients with severe acute pancreatitis treated with adjuvant selective decontamination. Clin Infect Dis. 1997; 25:811–816.
84. De Waele, JJ, Vogelaers, D, Blot, S, Colardyn, F. Fungal infections in patients with severe acute pancreatitis and the use of prophylactic therapy. Clin Infect Dis. 2003; 37:208–213.
85. Hoerauf, A, Hammer, S, Muller-Myhsok, B, Rupprecht, H. Intra-abdominal Candida infection during acute necrotizing pancreatitis has a high prevalence and is associated with increased mortality. Crit Care Med. 1998; 26:2010–2015.
86. Kotani, J, Usami, M, Nomura, H, et al. Enteral nutrition prevents bacterial translocation but does not improve survival during acute pancreatitis. Arch Surg. 1999; 134:287–292.
87. Qin, HL, Su, ZD, Hu, LG, et al. Effect of early intrajejunal nutrition on pancreatic pathological features and gut barrier function in dogs with acute pancreatitis. Clin Nutr. 2002; 21:469–473.
88. Al-Omran, M, Albalawi, ZH, Tashkandi, MF, Al-Ansary, LA. Enteral versus parenteral nutrition for acute pancreatitis. Cochrane Database Syst Rev. 2010.
89. Meier, R, Ockenga, J, Pertkiewicz, M, et al. ESPEN Guidelines on Enteral Nutrition: Pancreas. Clin Nutr. 2006; 25:275–284.
90. McClave, SA, Chang, WK, Dhaliwal, R, Heyland, DK. Nutrition support in acute pancreatitis: A systematic review of the literature. JPEN. 2006; 30:143–156.
91. Eatock, FC, Chong, P, Menezes, N, et al. A randomized study of early nasogastric versus nasojejunal feeding in severe acute pancreatitis. Am J Gastroenterol. 2005; 100:432–439.
92. Eckerwall, GE, Axelsson, JB, Andersson, RG. Early nasogastric feeding in predicted severe acute pancreatitis: A clinical, randomized study. Ann Surg. 2006; 244:959–967.
93. Morimoto, T, Noguchi, Y, Sakai, T, et al. Acute pancreatitis and the role of histamine-2 receptor antagonists: A meta-analysis of randomized controlled trials of cimetidine. Eur J Gastroenterol Hepatol. 2002; 14:679–686.
94. Anriulli, A, Leandro, G, Clemente, R, et al. Meta-analysis of somatostatin, octreotide and gabexate mesilate in the therapy of acute pancreatitis. Aliment Pharmacol Ther. 1998; 12:237–245.
95. Seta, T, Noguchi, Y, Shimada, T, et al. Treatment of acute pancreatitis with protease inhibitors: A meta-analysis. Eur J Gastroenterol Hepatol. 2004; 16:1287–1293.
96. Liu, LR, Xia, SH. Role of platelet-activating factor in the pathogenesis of acute pancreatitis. World J Gastroenterol. 2006; 12:539–545.
97. Formela, LJ, Wood, LM, Whittaker, M, Kingsnorth, AN. Amelioration of experimental acute pancreatitis with a potent platelet-activating factor antagonist. Br J Surg. 1994; 81:1783–1785.
98. Hofbauer, B, Saluja, AK, Bhatia, M, et al. Effect of recombinant platelet-activating factor acetylhydrolase on two models of experimental acute pancreatitis. Gastroenterology. 1998; 115:1238–1247.
99. Kingsnorth, AN, Galloway, SW, Formela, LJ. Randomized, double-blind phase II trial of lexipafant, a platelet-activating factor antagonist, in human acute pancreatitis. Brit J Surg. 1995; 82:1414–1420.
100. McKay, CJ, Curran, F, Sharples, C, et al. Prospective placebo-controlled randomized trial of lexipafant in predicted severe acute pancreatitis. Br J Surg. 1997; 84:1239–1243.
101. Johnson, CD, Kingsnorth, AN, Imrie, CW, et al. Double blind, randomised, placebo controlled study of a platelet activating factor antagonist, lexipafant, in the treatment and prevention of organ failure in predicted severe acute pancreatitis. Gut. 2001; 48:62–69.
102. Granger, J, Remick, D. Acute pancreatitis: Models, markers, and mediators. SHOCK. 2005; 24(Suppl 1):45–51.
103. Ayub, K, Imada, R, Slavin, J. Endoscopic retrograde cholangiopancreatography in gallstone-associated acute pancreatitis. Cochrane Database Syst Rev. (4):2004.
104. Folsch, UR, Nitsche, R, Ludtke, R, et al. Early ERCP and papillotomy compared with conservative treatment for acute biliary pancreatitis. N Engl J Med. 1997; 336:237–242.
105. Oria, A, Cimmino, D, Ocampo, C, et al. Early endoscopic intervention versus early conservative management in patients with acute gallstone pancreatitis and biliopancreatic obstruction: A randomized clinical trial. Ann Surg. 2007; 245:10–17.
106. Gloor, B, Muller, CA, Worni, M, et al. Late mortality in patients with severe acute pancreatitis. Br J Surg. 2001; 88:975–979.
107. Hirota, M, Inoue, K, Kimura, Y, et al. Non-occlusive mesenteric ischemia and its associated intestinal gangrene in acute pancreatitis. Pancreatology. 2003; 3:316–322.
108. Dellinger, RP, Carlet, JM, Masur, H, et al. Surviving sepsis campaign guidelines for management of severe sepsis and septic shock. Crit Care Med. 2004; 32:858–873.
109. Pitchumoni, CS, Agarwal, N. Pancreatic pseudocysts: When and how should drainage be performed? Gastroenterol Clin North Am. 1999; 28:615–639.
110. Ferrucci, JT, 3rd., Mueller, PR. Interventional approach to pancreatic fluid collections. Radiol Clin North Am. 2003; 41:1217–1226.
111. Baron, TH. Endoscopic drainage of pancreatic fluid collections and pancreatic necrosis. Gastrointest Endosc Clin N Am. 2003; 13:743–764.
112. Kellogg, TA, Horvath, KD. Minimal-access approaches to complications of acute pancreatitis and benign neoplasms of the pancreas. Surg Endosc. 2003; 17:1692–1704.
113. Beattie, GC, Hardman, JG, Redhead, D, Siriwardena, AK. Evidence for a central role for selective mesenteric angiography in the management of the major vascular complications of pancreatitis. Am J Surg. 2003; 185:96–102.
114. Aultman, DF, Bilton, DB, Zibari, GB, et al. Nonoperative therapy for acute necrotizing pancreatitis. Am Surg. 1997; 63:1114–1117.
115. Ashley, SW, Perez, A, Pierce, EA, et al. Necrotizing pancreatitis: Contemporary analysis of 99 consecutive cases. Ann Surg. 2001; 234:572–580.
116. Hartwig, W, Maksan, SM, Foitzik, T, et al. Reduction in mortality with delayed surgical therapy of severe pancreatitis. J Gastrointest Surg. 2002; 6:481–487.
117. Gotzinger, P, Wamser, P, Exner, R, et al. Surgical treatment of severe acute pancreatitis: Timing of operation is crucial for survival. Surg Infect. 2003; 4:205–211.
118. Hungness, ES, Robb, BW, Seeskin, C, et al. Early debridement for necrotizing pancreatitis: Is it worthwhile? J Am Coll Surg. 2002; 194:740–744.
119. Mier, J, Leon, EL, Castillo, A, et al. Early versus late necrosectomy in severe necrotizing pancreatitis. Am J Surg. 1997; 173:71–75.
120. Bradley, EL, 3rd., Allen, K. A prospective longitudinal study of observation versus surgical intervention in the management of necrotizing pancreatitis. Am J Surg. 1991; 161:19–25.
121. Rau, B, Pralle, U, Uhl, W, et al. Management of sterile necrosis in instances of severe acute pancreatitis. J Am Coll Surg. 1995; 181:279–288.
122. Buchler, MW, Gloor, B, Muller, CA, et al. Acute necrotizing pancreatitis: Treatment strategy according to the status of infection. Ann Surg. 2000; 232:619–626.
123. Olah, A, Belagyi, T, Issekutz, A, et al. Value of procalcitonin quick test in the differentiation between sterile and infected forms of acute pancreatitis. Hepato-gastroenterol. 2005; 52:243–245.
124. Riche, FC, Cholley, BP, Laisne, MJ, et al. Inflammatory cytokines, C reactive protein, and procalcitonin as early predictors of necrosis infection in acute necrotizing pancreatitis. Surgery. 2003; 133:257–262.
125. Rau, B, Pralle, U, Mayer, JM, Beger, HG. Role of ultrasonographically guided fine-needle aspiration cytology in the diagnosis of infected pancreatic necrosis. Br J Surg. 1998; 85:179–184.
126. Freeny, PC, Hauptmann, E, Althaus, SJ, et al. Percutaneous CT-guided catheter drainage of infected acute necrotizing pancreatitis: Techniques and results. AJR Am J Roentgenol. 1998; 170:969–975.
127. Zorger, N, Hamer, OW, Feuerbach, S, Borisch, I. Percutaneous treatment of a patient with infected necrotizing pancreatitis. Nat Clin Pract Gastroenterol Hepatol. 2005; 2:54–57.
128. Olah, A, Belagyi, T, Bartek, P, et al. Alternative treatment modalities of infected pancreatic necrosis. Hepato-gastroenterol. 2007; 53:603–607.
129. Runzi, M, Niebel, W, Goebell, H, et al. Severe acute pancreatitis: Nonsurgical treatment of infected necroses. Pancreas. 2005; 30:195–199.
130. Adamson, GD, Cuschieri, A. Multimedia article: Laparoscopic infracolic necrosectomy for infected pancreatic necrosis. Surg Endosc. 2003; 17:1675.
131. Ammori, BJ. Laparoscopic transgastric pancreatic necrosectomy for infected pancreatic necrosis. Surg Endosc. 2002; 16:1632.
132. Horvath, KD, Kao, LS, Wherry, KL, et al. A technique for laparoscopic-assisted percutaneous drainage of infected pancreatic necrosis and pancreatic abscess. Surg Endosc. 2001; 15:1221–1225.
133. Bradley, EL. A fifteen-year experience with open drainage for infected pancreatic necrosis. Surg Gynecol Obstet. 1993; 177:215–222.
134. Fernandez-del Castillo, C, Rattner, DW, Makary, MA, et al. Debridement and closed packing for the treatment of necrotizing pancreatitis. Ann Surg. 1998; 228:676–684.
135. Bakker, OJ, van Santvoort, HC, van, BS, et al. Endoscopic transgastric vs surgical necrosectomy for infected necrotizing pancreatitis: A randomized trial. JAMA. 2012; 307:1053–1061.
136. Parekh, D. Laparoscopic-assisted pancreatic necrosectomy: A new surgical option for treatment of severe necrotizing pancreatitis. Arch Surg. 2006; 141:895–902.
137. Zhou, ZG, Zheng, YC, Shu, Y, et al. Laparoscopic management of severe acute pancreatitis. Pancreas. 2003; 27:e46–e50.
138. Connor, S, Rraraty, MG, Howes, N, et al. Surgery in the treatment of acute pancreatitis–minimal access pancreatic necrosectomy. Scand J Surg. 2005; 94:135–142.
139. Besselink, MG, van Santvoort, HC, Nieuwenhuijs, VB, et al. Minimally invasive “step-up approach” versus maximal necrosectomy in patients with acute necrotising pancreatitis (PANTER trial): Design and rationale of a randomised controlled multicenter trial [ISRCTN38327949]. BMC Surg. 2006; 6.
140. Besselink, MG, de Bruijn, MT, Rutten, JP, et al. Surgical intervention in patients with necrotizing pancreatitis. Br J Surg. 2006; 93:593–599.
141. Ranson, JHC, Berman, RS. Long peritoneal lavage decreases pancreatic sepsis in acute pancreatitis. Ann Surg. 1990; 211:708–718.
142. Balachandra, S, Siriwardena, AK. Systematic appraisal of the management of the major vascular complications of pancreatitis. Am J Surg. 2005; 190:489–495.
143. Mortele, KJ, Mergo, PJ, Taylor, HM, et al. Peripancreatic vascular abnormalities complicating acute pancreatitis: Contrast-enhanced helical CT findings. Eur J Radiol. 2004; 52:67–72.
144. Iannitti, DA, Gregg, SC, Mayo-Smith, WW, et al. Portal venous gas detected by computed tomography: Is surgery imperative? Dig Surg. 2003; 20:306–315.
145. Testart, J, Boyet, L, Pperrier, G, et al. Arterial erosions in acute pancreatitis. Acta Chir Belg. 2001; 101:232–239.
146. Bosscha, K, Reijnders, K, Jacobs, MH, et al. Quality of life after severe bacterial peritonitis and infected necrotizing pancreatitis treated with open management of the abdomen and planned re-operations. Crit Care Med. 2001; 29:1539–1543.
147. Halonen, KI, Pettila, V, Leppaniemi, AK, et al. Long-term health-related quality of life in survivors of severe acute pancreatitis. Intensive Care Med. 2003; 29:782–786.
148. Soran, A, Chelluri, L, Lee, KK, Tisherman, SA. Outcome and quality of life of patients with acute pancreatitis requiring intensive care. J Surg Res. 2000; 91:89–94.
149. Cinquepalmi, L, Boni, L, Dionigi, G, et al. Long-term results and quality of life of patients undergoing sequential surgical treatment for severe acute pancreatitis complicated by infected pancreatic necrosis. Surg Infect. 2006; 7(Suppl 2):S113–S116.
150. Symersky, T, van Hoorn, B, Masclee, AA. The outcome of a long-term follow-up of pancreatic function after recovery from acute pancreatitis. JOP. 2006; 7:447–453.
[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]77
Acute Pancreatitis
CLINICAL PRESENTATION AND DIAGNOSIS
EARLY MANAGEMENT OF THE CRITICALLY ILL PATIENT WITH ACUTE PANCREATITIS
MANAGEMENT OF THE LATE COMPLICATIONS OF SEVERE ACUTE PANCREATITIS
Introduction
Acute pancreatitis is a complex disease with a highly variable clinical course. It is responsible for more than 200,000 hospital admissions each year in the United States,1 and it has an annual incidence of 10 to 80 cases in 100,000 in the developed world.2–5 Incidence rates have been increasing.6 For reasons that are unknown, there is seasonal variation in rates of the disease, the incidence being maximal in the spring and fall.7 The crude mortality rate for patients who are hospitalized with acute pancreatitis is less than 2%,8 and the majority of patients with acute pancreatitis experience a benign and self-limited disease, resulting in a hospital stay of only several days and no significant lasting sequelae. In a small percentage of patients, however, acute pancreatitis is sufficiently severe to lead to admission to an intensive care or high dependency unit, and a complicated clinical course with a mortality risk that may exceed 20%.1,9 These latter patients can present the intensivist with formidable challenges during the course of their illness, but if they recover, as most do, they can return to their premorbid state of health with no significant diminution in quality of life.
The prognosis for patients with severe acute pancreatitis has improved considerably since Ranson proposed his widely used severity criteria. In the mid-1970s, the mortality for patients with severe pancreatitis approached 100%10; today the risk is less than one quarter of that figure.11,12 This improved outcome can be ascribed in part to general improvements in the care of critically ill patients and, more important, to fundamental changes in the approach to the medical and surgical care of the patient with acute pancreatitis that, in turn, reflect an evolving understanding of the pathophysiology of the disease.
Definitions and Terminology
Pancreatitis is an acute inflammatory disorder that arises as a consequence of the activation of pancreatic digestive enzymes within the parenchyma of the gland and the surrounding tissues of the peritoneal cavity and retroperitoneum. Its evolution and complications are variable and give rise to terminology that is both confusing and imprecise. The most widely used classification system is that known as the Atlanta classification from 199213 (Table 77.1). Its reproducibility, however, is poor,14 and it is easier to conceptualize the disease as a spectrum of overlapping abnormalities resulting from the leakage of activated pancreatic enzymes, the host response to local tissue injury, and the superimposed complication of infection of what is initially a sterile process (Fig. 77.1).
Table 77.1
The Atlanta Classification System for Acute Pancreatitis
| Term | Definition |
| Acute pancreatitis | Acute inflammation of the pancreas with variable involvement of peripancreatic and remote tissues |
| Mild acute pancreatitis | Edema of pancreas; benign clinical course with minimal organ dysfunction and full recovery |
| Severe acute pancreatitis | Evidence of pancreatic necrosis; complications of infection, pseudocyst; clinical course characterized by organ failure |
| Acute fluid collection | Pancreatic or peripancreatic fluid, evident early in course of disease, and lacking wall |
| Pancreatic pseudocyst | Contained collection of pancreatic juice within a capsule of fibrous tissue and arising following an episode of acute pancreatitis |
| Pancreatic necrosis | Diffuse or focal loss of viability of pancreatic parenchyma or peripancreatic fat, evident as nonenhancing tissue on a contrast-enhanced CT scan |
| Pancreatic abscess | Localized collection of pus within pancreas or peripancreatic region and containing little or no necrotic material |
Adapted from Bradley EL: A clinically based classification system for acute pancreatitis. Arch Surg 1993;128:586-590.
In the mildest form of the disease, leakage of activated enzymes is minimal, and the most prominent manifestation is pancreatic edema secondary to a local inflammatory process. In more severe cases, pancreatic ductal disruption results in leakage of pancreatic enzymes into adjacent tissues. If the leakage occurs anteriorly into the peritoneal cavity, fluid collections will be evident. The local peritoneal inflammatory response triggers coagulation and fibrin deposition, walling the collection off and creating a pseudocyst, so called because it is a fluid collection that lacks an epithelial lining. Drainage of the pancreatic ascites through the diaphragm can create a pleural effusion; typically this is seen on the left. Marked elevation of the amylase level in the pleural fluid establishes the collection as pancreatic juice, rather than as a reaction to an inflammatory process on the abdominal side of the diaphragm. Alternatively, if leakage is into the fatty tissues of the retroperitoneum, necrosis predominates, although small loculated fluid collections are often present. Often in patients with severe acute pancreatitis, computerized tomography shows evidence of both intraperitoneal and retroperitoneal involvement. The older term, hemorrhagic pancreatitis, describes the sequelae of extension of retroperitoneal necrosis into blood vessels of the retroperitoneum. Extraperitoneal tracking of the resulting hematoma gives rise to Grey-Turner’s sign when the ecchymosis is evident in the flanks and Cullen’s sign when it tracks anteriorly through the falciform ligament to present at the umbilicus.15
Pathogenesis
Acute pancreatitis arises in the pancreas through the leakage of activated pancreatic enzymes into pancreatic and peripancreatic tissues. The characteristic clinical syndrome, however, reflects the activation of a massive systemic inflammatory response to that local tissue injury, mediated through the activation of the inflammasome—a multiprotein intracellular complex that leads to the activation of the key inflammatory cytokine, interleukin-1.16
Beyond its role as an endocrine organ, the pancreas plays a fundamental role in the digestion of foodstuffs through the production of enzymes that degrade the major constituents of ingested food: protein (proteases), fat (lipases), starch (amylase), and nucleic acids (nucleases). Pancreatic enzymes are synthesized by the acinar cells lining the pancreatic ductal system and released from the cell as inactive zymogens.17 They pass via the pancreatic duct into the lumen of the duodenum, where they are activated by mucosal enzymes such as enterokinase, and so become capable of degrading their target substrates. Under normal circumstances, activation is vigorously inhibited within the pancreas itself through sequestration of newly synthesized enzymes within zymogen granules, and further through the local action of a specific inhibitor of trypsin activation called SPINK.18 The importance of normal mechanisms that inhibit trypsin activation is underlined by the observation that genetic mutations in the SPINK gene associated with reduced activity have been implicated in the pathogenesis of hereditary pancreatitis.19
Acute pancreatitis arises through the activation of pancreatic enzymes within the pancreas itself, possibly through the activity of lysosomal hydrolases in the acinar cell and initiating autodigestion of the pancreas and surrounding tissues.20,21 Activation of trypsinogen to trypsin appears to be a critical event during the early pathogenesis of the acute process,22,23 and an increase in intracellular calcium concentrations may contribute to activation of trypsinogen.24 Trypsinogen can also be activated by lysosomal cathepsin B within the acinar cell.25 Pancreatic acinar cells can die by either necrosis or apoptosis. Necrotic death is characterized by cell lysis, with the leakage of intracellular constituents into the surrounding microenvironment and the activation of an acute inflammatory response. Neutrophil recruitment in response to the local tissue injury has been implicated in the amplification of cellular damage in acute pancreatitis.26–28 Apoptotic death, in contrast, is noninflammatory. Cells are degraded into membrane-bound vesicles that are taken up by fixed tissue macrophages. Phagocytosis of an apoptotic cell not only prevents the local activation of inflammation but triggers transcriptional programs in the phagocytosing cell that are anti-inflammatory and reparative in nature, with up-regulation of counter-inflammatory cytokines such as interleukin-10 (IL-10).29 The local cellular injury in acute pancreatitis can also be conceptualized as an imbalance between necrotic and apoptotic cell death. Activation of caspases—the intracellular enzymes that mediate apoptosis—attenuate the severity of pancreatitis,30 whereas severe pancreatitis is associated with reduced levels of IL-10.31
Activated pancreatic enzymes injure not only cells of the pancreas but also those of surrounding tissues. Fat cells appear to be particularly vulnerable, and the disease tends to be more severe in the obese because of the greater amount of peripancreatic fat necrosis.32 However, the degradative effects of pancreatic enzymes, and the secondary tissue injury resulting from the host response, can injure other structures in the vicinity of the pancreas, in particular, the transverse colon, and major vessels such as the splenic artery and vein.33 Pancreatitis rapidly evolves from an inflammatory disease of the pancreas to a chemical burn of the retroperitoneum.
The pathogenesis of the clinical syndrome is further complicated by the presence of the gastrointestinal tract immediately adjacent to the pancreas (Fig. 77.2). The head of the pancreas lies within the curve of the duodenum, immediately behind the stomach and above the transverse colon. Damage to terminal feeding vessels in the colonic fat caused by activated pancreatic enzymes can result in focal perforation of the colon, with leakage of colonic bacteria into the peritoneal cavity.34 Although the stomach and duodenum are only lightly colonized in health, the local ileus induced by the acute pancreatic inflammation promotes proximal gut overgrowth with enteric bacteria.35 In addition, changes in gut mucosal barrier function arising from local inflammation and the absence of enteral nutrients promote the translocation of luminal bacteria and of bacterial products such as endotoxin into the injured or necrotic peripancreatic tissues.36
Ultimately in the more severe cases, the clinical syndrome evolves as a result of a systemically activated inflammatory response, with the same changes in microvascular blood flow, endothelial permeability, and inflammatory mediator release that characterize bacterial sepsis.37
Etiology and Risk Factors
Acute pancreatitis has many causes, although 80% of cases in the developed world result from either alcohol or gallstones (Table 77.2). The mechanism of alcohol-induced pancreatitis is unclear. The acinar cell of the pancreas is capable of metabolizing alcohol and also appears to be the primary target of alcohol-mediated pancreatic injury.38 In vitro studies show that alcohol reduces the sensitivity of isolated acini to zymogen activation by cholecystokinin.39 Gallstone pancreatitis appears to be a consequence of a transient acute increase in pancreatic ductal pressures associated with passage of a small gallstone through the sphincter of Oddi in patients with a common channel for the bile and pancreatic ducts.40 True obstruction of the duct is uncommon, and approximately 90% of patients will be found to have gallstones in the stool, implicating the passage of the stone, rather than an obstructing mechanism, in the etiology of the resulting pancreatic inflammation. The list of drugs implicated in the etiology of acute pancreatitis is long41; some of the more prominent associations are shown in Table 77.2.
Genetic factors have been associated with the development of acute pancreatitis. A polymorphism in the secretory trypsin inhibitor (SPINK1) gene is associated with an increased risk for acute pancreatitis,42 as are mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene,43 and PRSS1.44 Polymorphisms in the genes for tumor necrosis factor α (TNFα) and heat shock protein 70 have also been linked to an increased risk for acute pancreatitis,45 although the association is less clear.
Clinical Presentation and Diagnosis
The causes of shock in the patient with acute pancreatitis are multifactorial. Initially, the acute inflammatory process in the retroperitoneum elicits local inflammation, with an outpouring of fluid into the relatively confined space of the retroperitoneum or into the peritoneal cavity itself. Intra-abdominal inflammation evokes secondary ileus within the gastrointestinal tract, and fluid is sequestered here, increasing the relative intravascular volume deficit. Nausea, vomiting, and a reluctance to take fluids by mouth further exacerbate this fluid deficit. As the process evolves, a systemic inflammatory response to the local abdominal process results in diffuse vasodilatation and capillary leak syndrome. In aggregate, the effective loss of intravascular volume can be enormous, and so the initial resuscitation may require large volumes of intravenous fluid. Acute hypocalcemia may also contribute to the clinical picture of cardiovascular compromise. Indeed the classical prognostic criteria articulated by Ranson (Table 77.3) emphasize the importance of acute inflammation (white cell count), and the secondary hemodynamic (fluid sequestration, base excess, blood urea nitrogen [BUN], and hematocrit) and metabolic (glucose, calcium) sequelae in determining the ultimate prognosis.
Table 77.3
Ranson’s Criteria in Acute Pancreatitis
LDH, Lactate dehydrogenase; AST, aspartate aminotransferase; BUN, blood urea nitrogen.
From Ranson JH, Rifkind KM, Roses DF, et al: Prognostic signs and the role of operative management in acute pancreatitis. Surg Gynecol Obstet 1974;139:69-81.
The diagnosis of pancreatitis is usually straightforward, based on the combination of clinical manifestations and characteristic biochemical findings of elevations in the circulating levels of amylase and lipase, the latter being somewhat more accurate diagnostically.46 The diagnosis can be confirmed, and the severity of the disease evaluated by computerized tomography (Fig. 77.3), using the grading system developed by Balthazar47 (Table 77.4).
Table 77.4
Balthazar Grading of CT Findings in Acute Pancreatitis
| Grade | Findings |
| Grade A | Normal pancreas |
| Grade B | Pancreatic enlargement |
| Grade C | Pancreatic or peripancreatic inflammation |
| Grade D | Single peripancreatic fluid collection |
| Grade E | 2 or more pancreatic collections and/or retroperitoneal air |
From Balthazar EJ: CT diagnosis and staging of acute pancreatitis. Radiol Clin North Am 1989;27:19-37.
An initial assessment of the severity of the disease is useful primarily for deciding the optimal venue for the early management of the patient, as at least some of the delayed morbidity of acute pancreatitis can be reduced through aggressive initial resuscitation and support. Various approaches have been used to quantify disease severity. Ranson identified 11 variables—5 at initial presentation and 6 over the ensuing 48 hours—that correlated in a graded fashion with the ultimate risk of mortality (see Table 77.3). The Glasgow-Imrie criteria are a modification of Ranson’s scale and represent an alternate model of severity scoring. In head-to-head studies, Acute Physiology, Age, and Chronic Health Evaluation (APACHE) II—a generic severity of illness scale—performs at least as well as the Ranson or Glasgow-Imrie criteria in predicting hospital survival.48 Moreover, even simpler scales appear to provide comparable prognostic information.23,49 Various biochemical measures, including C-reactive protein, procalcitonin, interleukin-6, and trypsinogen activation peptide (TAP), are purported to differentiate mild and severe pancreatitis early during the clinical evolution of the disease50–52; their clinical utility is unclear. Persistence of clinical manifestations of systemic inflammation beyond 48 hours is also associated with subsequent organ dysfunction and a higher mortality.53 Precision in prognostication is far less important than early recognition of the patient for whom close monitoring and aggressive resuscitation can alter the clinical course, and a low threshold for management in a more controlled and monitored setting is an important factor in reducing the complications of pancreatitis.54
Early Management of the Critically Ill Patient with Acute Pancreatitis
Initial Resuscitation
Intravascular fluid deficits early in the course of pancreatitis can be substantial. Early aggressive fluid resuscitation is the cornerstone of initial successful management.55 Volume status should be monitored with a urinary catheter and central venous catheter, and although studies in the specific setting of pancreatitis have not been performed, there is every reason to believe that the principles of goal-directed resuscitation should be followed.56 There are no data to support a preference for colloids or crystalloids during resuscitation; the key, however, is to administer sufficient volumes rapidly enough to restore an adequate circulating volume.57 Typically this comprises many liters of fluid, and not infrequently, the sickest patients will receive more than 10 to 15 L of fluid resuscitation over the first 24 hours. Such aggressive resuscitation is best carried out within the well-monitored environment of the intensive care unit (ICU).
Although aggressive fluid resuscitation can minimize later complications such as pancreatic necrosis and acute renal failure, in the patient with increased vascular permeability it carries a high risk of complications related to interstitial edema. Frequently patients will require endotracheal intubation and mechanical ventilation. Development of the abdominal compartment syndrome is a relatively common and underdiagnosed complication of resuscitated acute pancreatitis.58 Intra-abdominal pressure can be measured by transducing a urinary catheter in the bladder. Normal pressures approximate the central venous pressure, whereas pressures greater than 20 cm H2O indicate intra-abdominal hypertension, and pressures greater than 30 cm H2O indicate a compartment syndrome and carry an increased risk of ischemic injury because of impairment of visceral venous drainage. In severe cases, management requires abdominal decompression by laparotomy. The need for laparotomy in the patient with acute pancreatitis may be avoided by the use of continuous venovenous hemofiltration59 or decompressive anterior abdominal fasciotomy.60
Infection Prophylaxis
Infection is a common complication of acute necrotizing pancreatitis, and its development is associated with an increased risk of morbidity and mortality.61 For the patient with severe acute pancreatitis, these infections may arise within the injured or necrotic peripancreatic tissues or at distant sites as nosocomial infection in a critically ill patient.
The characteristic microbial flora of peripancreatic infection includes organisms normally resident within the gastrointestinal tract62 and organisms that characteristically colonize the proximal gastrointestinal tract of the critically ill patient35 (Box 77.1). Anaerobes may be present but are uncommon, whereas organisms such as Candida, Enterococci, and coagulase-negative Staphylococci are encountered with increasing frequency.63,64 This infecting flora reflects the role of the gastrointestinal tract as the reservoir of organisms inducing superinfection during acute pancreatitis.65
