[level-membership-for-neurology-category]
Chapter 16 Tics and Tourette syndrome
Introduction
Tourette syndrome (TS), which should be more appropriately called Gilles de la Tourette syndrome, is a neurologic disorder manifested by motor and vocal or phonic tics usually starting during childhood and often accompanied by obsessive-compulsive disorder (OCD), attention-deficit hyperactivity disorder (ADHD), poor impulse control, and other comorbid behavioral problems (Shapiro and Shapiro, 1992; Cohen and Leckman, 1994; Hyde and Weinberger, 1995; Feigin and Clarke, 1998; Leckman and Cohen, 1999; Freeman et al., 2000; Robertson, 2000; Jankovic, 2001a; Leckman et al., 2001; Leckman, 2002; Stein, 2002; Singer, 2005; Albin and Mink, 2006; Leckman et al., 2006; Bloch et al., 2011; Jankovic and Kurlan, 2011). Once considered a rare psychiatric curiosity, TS is now recognized as a relatively common and complex neurobehavioral disorder (Table 16.1). There has been speculation that many notable historical figures, including Dr Samuel Johnson and possibly Wolfgang Amadeus Mozart (Simkin, 1992; Ashoori and Jankovic, 2007), were afflicted with TS.
Table 16.1 Summary of epidemiologic studies in Tourette syndrome (TS)
|
• 6.1% (135/553 children, K–6th grade) had persistent tics (TS); 24% observed to have motor tics during at least one month of the 8-month study
|
One of the earliest reports of TS dates to 1825, when Itard (1825) described a French noblewoman with body tics, barking sounds, and uncontrollable utterances of obscenities. Sixty years later, the French neurologist and a student of Charcot, Georges Gilles de la Tourette (1885) reviewed Itard’s original case and added eight more patients. He noted that all nine patients shared one feature: they all exhibited brief involuntary movements or tics; additionally, six made noises, five shouted obscenities (coprolalia), five repeated the words of others (echolalia), and two mimicked others’ gestures (echopraxia) (Goetz and Klawans, 1982; Kushner, 1999). Although Tourette considered the disorder he described to be hereditary, the etiology was ascribed to psychogenic causes for nearly a century following the original report. The perception of TS began to change in the 1960s, when the beneficial effects of neuroleptic drugs on the symptoms of TS began to be recognized (Shapiro and Shapiro, 1968). This observation helped to refocus attention from psychogenic etiology to central nervous system (CNS) etiology. Despite these advances TS is still often misunderstood and wrongly labeled as a mental or psychiatric disorder. For example, insurance compensation for patients with TS is often compromised because the TS-related diagnostic codes (307.22 = Tic Disorder, 307.23 = Tourette Syndrome) are currently included in the ICD-9 series for mental disorders (290–319). Therefore, the code 333.3 = Tics of Organic Origin or 349.9 = Unspecified Nervous System Disorder may result in more appropriate compensation. If payment is denied, an appeal letter to the insurance company specifying that TS is a neurologic disorder along with some literature and links to a credible website may be required.
The cause of TS is yet unknown, but the disorder appears to be inherited in the majority of patients (Pauls et al., 1988, 1991; Tolosa and Jankovic, 1998; Jankovic, 2001a; Leckman, 2002; Paschou et al., 2004; Singer, 2005). The clinical expression of this genetic defect varies from one individual to another, fluctuations in symptoms are seen within the same individual, and different manifestations occur in various family members (Kurlan, 1994). This variable expression from one individual to another, even within members of the same family, contributes to diagnostic confusion. Without a specific biologic marker, the diagnosis depends on a careful evaluation of the patient’s symptoms and signs by an experienced clinician. Educational efforts directed to physicians, educators, and to the general public have increased awareness about TS. In addition, the media have drawn increasing public attention to this condition. As a result of this improved awareness, the self-referral rate of patients has increased, and the correct diagnosis is made earlier than was the case in the past. Many patients, however, still remain undiagnosed, or their symptoms are wrongly attributed to habits, allergies, asthma, dermatitis, hyperactivity, nervousness, and many other conditions (Jankovic et al., 1998; Hogan and Wilson, 1999).
Phenomenology of tics
Tics, the clinical hallmark of TS, are relatively brief and intermittent movements (motor tics) or sounds (vocal or phonic tics). Recognition of the full spectrum of phenomenology of tics is critical to the diagnosis of TS (Jankovic, 2008). Motor tics typically consist of sudden, abrupt, transient, often repetitive and coordinated (stereotypical) movements that may resemble gestures and mimic fragments of normal behavior, vary in intensity, and are repeated at irregular intervals (Videos 16.1 to 16.8). Currently accepted criteria for the diagnosis of TS require both types of tics to be present (Robertson, 1989; Golden, 1990; Tourette Syndrome Classification Study Group, 1993; Jankovic, 1997; Singer, 2000). This division into motor and vocal/phonic tics, however, is artificial, because vocal/phonic tics are actually motor tics that involve respiratory, laryngeal, pharyngeal, oral, and nasal musculature. Contractions of these muscles may produce sounds by moving air through the nose, mouth, or throat. The term phonic tic is preferable, since not all sounds produced by TS patients involve the vocal cords. 
To better understand the categorization of tics and how they fit in the general schema of movement disorders, it might be helpful to provide a simple classification of movements (Jankovic, 1992). All movements can be categorized into one of four classes:
Recent studies have shown that automatic, learned behaviors appear to be encoded in the sensorimotor portion of the striatum (Jog et al., 1999). Some support for the proposed classification is provided by the findings of Papa and colleagues (1991). They recorded normal premovement (readiness) electroencephalographic, slow, negative potential (the Bereitschaftspotential) 1–1.5 seconds prior to self-induced, internally generated (voluntary) movement in normal individuals but not before externally triggered movement induced by electrical stimulation (Colebatch, 2007). Most tics can be categorized as either semivoluntary (unvoluntary) or involuntary (suppressible). In some cases, learned voluntary motor skills are incorporated into the tic repertoire. This is exemplified by a case of a woman with TS who incorporated sign language into her tic behavior, suggesting that semantics is more important than phonology in the generation of tics (Lang et al., 1993).
Tics may be simple or complex. Simple motor tics involve only one group of muscles, causing a brief, jerk-like movement. They are usually abrupt in onset and rapid (clonic tics), but they may be slower, causing a briefly sustained abnormal posture (dystonic tics) or an isometric contraction (tonic tics) (Jankovic and Fahn, 1986; Jankovic, 1992). Examples of simple clonic motor tics include blinking, nose twitching, and head jerking. Rhythmical clonic tics may rarely resemble tremor or rhythmical myoclonus, such as palatal myoclonus (Schwingenschuh et al., 2007; Adam et al., 2009). Simple dystonic tics include blepharospasm, oculogyric movements, bruxism, sustained mouth opening, torticollis, and shoulder rotation (Videos 16.3 and 16.9). Tensing of abdominal or limb muscles is an example of a tonic tic (Video 16.10). To characterize clonic and dystonic tics further, 156 patients with TS were studied; 89 (57%) exhibited dystonic tics, including oculogyric deviations (28%), blepharospasm (15%), and dystonic neck movements (7%) (Jankovic and Stone, 1991). Since patients with dystonic tics did not differ significantly on any clinical variables from those with only clonic tics, we concluded that despite previous reports (Feinberg et al., 1986), the presence of dystonic tics should not be considered atypical or unusual. In fact, on subsequent observation, the patient in Feinberg and colleagues’ case report was shown to be a case of TS with typical dystonic tics (Fahn, 1987). Dystonic tics should be distinguished from persistent dystonia, which is typically seen in patients with primary dystonia (Jankovic and Fahn, 2002). Dystonic (and tonic) muscle contraction might be responsible for so-called blocking tics (Video 16.11). These blocking tics are due to either prolonged tonic or dystonic tics that interrupt ongoing motor activity such as speech (intrusions) or a sudden inhibition of ongoing motor activity (negative tic). Clonic and dystonic tics may occasionally occur in patients with primary dystonia more frequently than in the general population. In nine patients with coexistent TS and persistent primary dystonia, the onset of tics was at a mean age of 9 years, while dystonia followed the onset of tics by a mean of 22 (10–38) years (Stone and Jankovic, 1991). Other reports have drawn attention to the possible association of tics and dystonia, although the two disorders may coexist by chance alone (Pringsheim et al., 2007). The occasional co-occurrence of tics and dystonia in the same family, however, provides additional evidence for a possible etiologic relationship between the two disorders (Németh et al., 1999; Yaltho et al., 2010). Dopa-responsive dystonia with mutations in the GCH1 gene and TS was found in various members of a large Danish family (Romstad et al., 2003). 
Motor (particularly dystonic) and phonic tics are preceded by premonitory sensations in over 80% of patients (Cohen and Leckman, 1992; Banaschewski et al., 2003; Kwak et al., 2003a; Woods et al., 2005; Prado et al., 2008). This premonitory phenomenon consists of localizable sensations or discomforts, such as a burning feeling in the eye before an eye blink, tension or a crick in the neck that is relieved by stretching of the neck or jerking of the head, a feeling of tightness or constriction that is relieved by arm or leg extension, nasal stuffiness before a sniff, a dry or sore throat before throat clearing or grunting, and itching before a rotatory movement of the scapula. Rarely, these premonitory sensations, termed in one report extracorporeal phantom tics, involve sensations in other people and objects and are temporarily relieved by touching or scratching them (Karp and Hallett, 1996). In one study, premonitory sensations were experienced by 92% of 135 patients with TS, and these were localized chiefly to the shoulder girdle, palms, midline abdominal region, posterior thighs, feet, and eyes (Cohen and Leckman, 1992). We administered a questionnaire regarding various aspects of premonitory sensations associated with their motor tics to 50 TS patients with a mean age of 23.6 ± 16.7 years (Kwak et al., 2003a). Forty-six of 50 (92%) subjects reported some premonitory sensations, the most common of which was an urge to move and an impulse to tic (“had to do it”). Other premonitory sensations included an itch, tingling/burning, numbness, and coldness. Thirty-seven (74%) also reported intensification of premonitory sensations if the patient was prevented from performing a motor tic; 36 (72%) reported relief of premonitory sensations after performing the tic; and 24 (48%) stated that their motor tic would not have occurred if they had had no premonitory sensation. Twenty-seven of 40 patients (68%) described a motor tic as a voluntary motor response to an involuntary sensation rather than as a completely involuntary movement. Besides the local or regional premonitory sensations, this premonitory phenomenon may be a nonlocalizable, less specific, and poorly described feeling, such as an urge, anxiety, anger, and other psychic sensations. The observed movement or sound sometimes occurs in response to these premonitory phenomena, and these movements or sounds have been previously referred to as sensory tics (Kurlan et al., 1989; Chee and Sachdev, 1997). In a study of 60 patients with tic disorders, 41 (68%) thought that all their tics were intentionally produced, and 15 (25%) additional patients had both voluntary and involuntary movements; thus, 93% of the tics were perceived to be “irresistibly but purposefully executed” (Lang, 1991). This “intentional” component of the movement may be a useful feature in differentiating tics from other hyperkinetic movement disorders, such as myoclonus and chorea. The sensations or feelings that often precede motor tics usually occur out of a background of relative normalcy and are clearly involuntary, even though the movements (motor tics) or noises (phonic tics) that occur in response to these premonitory symptoms may be regarded as semivoluntary or unvoluntary. Chee and Sachdev (1997) suggest that sensory tics, which we and others refer to as premonitory sensations, “represent the subjectively experienced component of neural dysfunction below the threshold for motor and phonic tic production.” Many patients report that they have to repeat a particular movement to relieve the uncomfortable urge until “it feels good.” The “just right” feeling has been associated with compulsive behavior, and as such, the “unvoluntary” movement may be regarded as a compulsive tic (Leckman et al., 1994). While many patients describe a gradually increasing inner tension as tic suppression is maintained and a rebound effect of a flurry of tics when the tics are finally expressed, formal objective studies of tic frequency during and after voluntary suppression have failed to detect a rebound increase (Meidinger et al 2005; Himle and Woods 2005) and, when reinforced, tic suppression may last at least 40 consecutive minutes (Woods et al., 2008).
Complex motor tics consist of coordinated, sequenced movements resembling normal motor acts or gestures that are inappropriately intense and timed (Videos 16.6, 16.12, and 16.13). They may be seemingly nonpurposeful, such as head shaking or trunk bending, or they may seem purposeful, such as touching, throwing, hitting, jumping, and kicking. Additional examples of complex motor tics include gesturing “the finger” and grabbing or exposing one’s genitalia (copropraxia) or imitating gestures (echopraxia). Burping, vomiting, and retching have been described as part of the clinical picture of TS, but it is not clear whether this phenomenon represents a complex tic or some other behavioral manifestation of TS (Rickards and Robertson, 1997). Air swallowing is another unusual tic described in TS (Weil et al., 2008). Another unusual tic is ear dyskinesia, which consists of anterior-posterior displacement of the external ear (Cardoso and Faleiro, 1999). Complex motor tics may be difficult to differentiate from compulsions, which frequently accompany tics, particularly in TS. A complex, repetitive movement may be considered a compulsion when it is preceded by, or associated with, a feeling of anxiety or panic, as well as an irresistible urge to perform the movement or sound because of fear that if it is not promptly or properly executed, “something bad” will happen. However, this distinction is not always possible, particularly when the patient is unable to verbalize such feelings. Some coordinated movements resemble complex motor tics but may actually represent pseudovoluntary movements (parakinesias) that are designed to camouflage the tics by incorporating them into seemingly purposeful acts, such as adjusting one’s hair during a head jerk. 
In contrast to other hyperkinetic movement disorders, tics are usually intermittent and may be repetitive and stereotypic (Table 16.2). Tics may occur as short-term bouts or bursting or long-term waxing and waning (Peterson and Leckman, 1998). They vary in frequency and intensity and often change distribution. Typically, tics can be volitionally suppressed, although this might require intense mental effort (Banaschewski et al., 2003). Suppressibility, although characteristic and common in tics, is not unique or specific for tics, and this phenomenon has been well documented in other hyperkinetic movement disorders (Walters et al., 1990). Using functional magnetic resonance imaging (MRI), Peterson and colleagues (1998a) showed decreased neuronal activity during periods of suppression in the ventral globus pallidus, putamen, and thalamus. There was increased activity in the right caudate nucleus, right frontal cortex, and other cortical areas that are normally involved in the inhibition of unwanted impulses (prefrontal, parietal, temporal, and cingulate cortices). Using event-related functional magnetic resonance imaging in 26 children with bipolar disorder, deficits in the ability to engage striatal structures and the right ventral prefrontal cortex were found during unsuccessful inhibition, thus suggesting that deficits in motor inhibition contribute to impulsivity and irritability in children with bipolar disorder and possibly also with TS (Leibenluft et al., 2007).
| A. Primary |
Besides temporary suppressibility, tics are characterized by suggestibility and exacerbation with stress, excitement, boredom, fatigue, and exposure to heat (Lombroso et al., 1991). Emotional stress associated with life events or other stresses have been documented to potentially markedly exacerbate tics, but onset of TS is not necessarily related to stressful life events (Wood et al., 2003; Hoekstra et al., 2004; Horesh et al., 2008). Tics may also increase during relaxation after a period of stress.
In contrast to other hyperkinetic movement disorders that are usually completely suppressed during sleep, motor and phonic tics may persist during all stages of sleep (Cohrs et al., 2001; Jankovic et al., 1984; Silvestri et al., 1990; Fish et al., 1991; Hanna and Jankovic, 2003; Rothenberger et al., 2001). In addition, patients with TS often have disturbances of sleep, such as increased sleep fragmentation, higher frequency of arousals, decreased rapid eye movement (REM) sleep, and enuresis (Hanna and Jankovic, 2003). Many patients note a reduction in their tics when they are distracted while concentrating on mental or physical tasks (such as when playing a video game or during an orgasm). Other patients experience increased frequency and intensity of their tics when distracted, especially when they no longer have the need to suppress the tics. Tics are also typically exacerbated by dopaminergic drugs and by CNS stimulants, including methylphenidate and cocaine (Cardoso and Jankovic, 1993). Finally, it should be noted that there is a broad spectrum of movements that may be present in patients with TS that can be confused with tics, such as akathisia, chorea, dystonia, compulsive movements, and fidgeting as part of hyperactivity associated with ADHD (Jankovic, 1997; Kompoliti and Goetz, 1998; Wilens et al., 2004).
Clinical features of Tourette syndrome
Motor symptoms
TS, the most common cause of tics, is manifested by a broad spectrum of motor and behavioral disturbances (Table 16.3). This clinical heterogeneity often causes diagnostic difficulties and presents a major challenge in genetic linkage studies. To aid in the diagnosis of TS, the Tourette Syndrome Classification Study Group (1993) formulated the following criteria for definite TS: (1) Both multiple motor and one or more phonic tics have to be present at some time during the illness, although not necessarily concurrently. (2) Tics must occur many times a day, nearly every day, or intermittently throughout a period of more than 1 year. (3) The anatomic location, number, frequency, type, complexity, or severity of tics must change over time. (4) Onset must be before age 21. (5) Involuntary movements and noises cannot be explained by other medical conditions. (6) Motor and/or phonic tics must be directly witnessed by a reliable examiner at some point during the illness or must be recorded by videotape or cinematography. Probable TS type 1 meets all the criteria except for number 3 and/or number 4, and probable TS type 2 meets all the criteria except for number 1; it includes either a single motor tic with phonic tics or multiple motor tics with possible phonic tics. In contrast to the criteria outlined by the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) (1994), the Tourette Syndrome Classification Study Group criteria do not include a statement about “impairment.” There is considerable controversy about the DSM-IV criteria, which require that “marked distress or significant impairment in social, occupational or other important areas of functioning” be present. Therefore, patients with mild tics that do not produce an impairment would not satisfy the diagnostic criteria for TS, according to DMS-IV. This criterion will be deleted from the fifth edition of the DSM. Kurlan (1997) suggested another set of diagnostic criteria for genetic studies and introduced the term Tourette disorder for patients who have “functional impairment.” This, however, does not take into account the marked fluctuation in symptoms and severity; some patients may be relatively asymptomatic at one time and clearly functionally impaired at another time. The TSA International Genetic Collaboration developed the Diagnostic Confidence Index, which consists of 26 confidence factors, with weightings given to each of them and a total maximum score of 100. The most highly weighted diagnostic confidence factors include history of coprolalia, complex motor or vocal tics, a waxing and waning course, echophenomenon, premonitory sensations, an orchestrated sequence, and age at onset. The Diagnostic Confidence Index was found to be a useful instrument in assessing the lifetime likelihood of TS (Robertson et al., 1999). Several instruments, some based on ratings of videotapes, have been developed to measure and quantitate tics, but they all have some limitations (Goetz and Kompoliti, 2001; Goetz et al., 2001). The most widely used instrument to assess tics is the Yale Global Tic Severity Scale (YGTSS), which consists of two broad domains: Total Tic Severity (with two sub-domains: Motor and Phonic Tics) and Impairment. Within each category there are five dimensions, score 0–5: number of tics, frequency, intensity, complexity, and interference. The total tic score ranges from 0 to 50; the usual ranges in most studies is 15 to 30. A health-related quality of life scale (HR-QOL) has been developed and validated for internal consistency, test–retest reliability and against other clinical scales (Cavanna et al., 2008). Utilization of the HR-QOL scale showed that comorbidities such as ADHD and OCD rather than tic severity are more predictive of the long-term outcome.
Table 16.3 Demographic and clinical features in a large database of patients with TS (Tourette International Consortium) (Freeman et al., 2009)
| Number (80 sites; 60% North America; 66% Psychiatry: 27% Neurology) | 6805 |
| Male : Female ratio | 4.4 : 1 |
| Mean age at onset of tics | 6.4 years |
| Mean age at diagnosis of TS | 13.2 years |
| Delay in diagnosis | 6.4 years |
| Family history | 51.7% |
| TS only, no comorbidity | 14.2% |
| Attention-deficit hyperactivity disorder | 55.6% |
| Obsessive-compulsive disorder/behavior | 54.9% |
| Conduct/oppositional defiant disorder | 12.3% |
| Anger control problems | 27.6% |
| Learning disability | 22.0% |
| Mood disorder | 16.9% |
| Anxiety disorder | 16.8% |
| Pervasive developmental disorder | 4.6% |
The clinical criteria are designed to assist in accurate diagnosis, in genetic linkage studies and in differentiating TS from other tic disorders (Jankovic, 1993b) (Table 16.2). There is a body of evidence to support the notion that many, if not all, patients with other forms of idiopathic tic disorders represent one end of the spectrum in a continuum of TS (Kurlan et al., 1988). The most common and mildest of the idiopathic tic disorders is the transient tic disorder (TTD) of childhood. This disorder is essentially identical to TS except the symptoms last less than 1 year and therefore the diagnosis can be made only in retrospect. Transient tic disorder has been estimated to occur in up to 24% of schoolchildren (Shapiro et al., 1988). Chronic multiple tic disorder is also similar to TS, but the patients have either only motor or, less commonly, only phonic tics lasting at least 1 year. Chronic single tic disorder is the same as chronic multiple tic disorder, but the patients have only a single motor or phonic tic. This separation into transient tic disorder, chronic multiple tic disorder, and chronic single tic disorder seems artificial because all can occur in the same family and probably represent a variable expression of the same genetic defect (Kurlan et al., 1988).
Although the diagnostic criteria require that the onset is present before the age of 21, in 96% of patients the disorder is manifested by age 11 (Robertson, 1989). In 36–48% of patients, the initial symptom is eye blinking, followed by tics involving the face and head. Blink rate in TS is about double of that of normal, age-matched controls (Tulen et al., 1999). During the course of the disease, nearly all patients exhibit tics involving the face or head, two-thirds have tics in the arms, and half have tics involving the trunk or legs. According to one study, the average age at onset of tics is 5.6 years, and the tics usually become most severe at age 10; by 18 years of age, half of the patients are tic-free (Leckman et al., 1998). In a study of 58 adults who had been diagnosed with TS during childhood, Goetz and colleagues (1992) found that tics persisted in all patients but were moderate or severe in only 24%, although 60% had moderate or severe tics during the course of the disease. Tic severity during childhood had no predictive value for the future course, but patients with mild tics during the preadult period had mild tics during adulthood. In a longitudinal study that involved structured interviews of 976 children aged 1–10 years, 776 of whom were reassessed 8, 10, and 15 years later, tics and ADHD symptoms were associated with OCD symptoms in late adolescence and early adulthood (Peterson et al., 2001). Furthermore, ADHD was associated with lower IQ and lower social status, whereas OCD was associated with higher IQ. These findings are similar to those of another study designed to address the long-term prognosis of children with TS as they reach adulthood (Bloch et al., 2006). In this study 46 children with TS underwent a structured interview at a mean age of 11.4 years, and again at 19.0 years. The mean worst-ever tic severity score was 31.6 out of a possible 50 on the YGTSS, and occurred at a mean age of 10.6 years. By the time of the second interview, mean YGTSS score decreased to 10. This first prospective longitudinal study also showed that only 22% continued to experience mild or greater tic symptoms (YGTSS scores, ≥10) at follow-up, while nearly one-third were in complete remission of tic symptoms at follow-up. In contrast to the study by Goetz et al. (1992), the severity of childhood tics was predictive of increased tic severity at follow-up. The peak OCD severity occurred 2 years after peak tic severity. Interestingly, a 10-point increase in baseline IQ increased the risk of OCD symptoms at follow-up by 2.8-fold. The authors point out that the later average onset of OCD symptoms indicates the importance of counseling parents about the possibility of OCD development in children recently diagnosed with tics. Although the long-term prognosis for TS is generally favorable for most patients, a minority of cases may have persistent, severe tic symptoms which may be resistant to medications (Eapen et al., 2002). In another study, the investigators reviewed videotapes of 31 patients, with an average age of 24.2 ± 3.5 years, approximately 12 years after their initial video and found that 90% of the adults still had tics, even though they often considered themselves tic-free (Pappert et al., 2003). There was, however, a significant improvement in tic disability and tic severity. When 40 children and 31 adults with TS were compared no difference in tic phenomenology or severity was found, but children were more frequently managed without medications, and sedation was more common in adults but weight gain was more common in children (Cubo et al., 2008).
Although the vast majority of tics in adults represent recurrences of childhood-onset tics, rare patients may have their first tic occurrence during adulthood (Chouinard and Ford, 2000). In adults with new-onset tics, it is important to search for secondary causes, such as infection, trauma, stroke (Kwak and Jankovic, 2002; Gomis et al., 2008), multiple sclerosis (Nociti et al., 2009), cocaine use, neuroleptic exposure, and peripheral injury (Video 16.14) (Chouinard and Ford, 2000; Jankovic, 2001b; Jankovic and Mejia, 2006). One study of eight patients with adult-onset tics (three of whom had childhood-onset OCD and three of whom had a family history of tics and OCD) found that in comparison to the patients with more typical childhood-onset tics, the former group had more severe symptoms, greater social morbidity, and less favorable response to medications (Eapen et al., 2002). Poor motor control, which can lead to poor penmanship and, at times, almost illegible handwriting, can contribute to the academic difficulties faced by many patients with TS. 
Tics, although rarely disabling, can be quite troublesome for TS patients because they cause embarrassment, interfere with social interactions, and at times can be quite painful or uncomfortable. Rarely, cervical tics may be so forceful and violent, the so-called “whiplash tics,” that they may cause secondary neurologic deficits, such as cervical artery dissection (Norris et al., 2000), and noncompressive (Isaacs et al., 2010) or compressive cervical myelopathy (Krauss and Jankovic, 1996) (Video 16.2). The truncal bending tics, which resemble intermittent, repetitive camptocormia, may cause secondary degenerative changes in the thoracic spine (Azher and Jankovic, 2005) (Video 16.5). These disabling tics and other severe symptoms of TS draw attention to the subset of patients with TS so severe that they may be life-threatening, hence labeled as “malignant” TS (Table 16.4). Of 332 TS patients evaluated at Baylor College of Medicine Movement Disorders Clinic during a 3-year period, 17 (5.1%) met criteria for malignant TS, defined as ≥2 emergency room (ER) visits or ≥1 hospitalizations for TS symptoms or its associated behavioral comorbidities (Cheung et al., 2007). The patients exhibited tic-related injuries, self-injurious behavior (SIB), uncontrollable violence and temper, and suicidal ideation/attempts. Compared to patients with nonmalignant TS, those with malignant TS were significantly more likely to have a personal history of obsessive-compulsive behavior/disorder (OCB/OCD), complex phonic tics, coprolalia, copropraxia, SIB, mood disorder, suicidal ideation, and poor response to medications. Severe or malignant TS, associated with SIB and other disabling features, has been also reported in families, including consanguineous kindreds (Motlagh et al., 2008). 
Table 16.4 Clinical features of severe (“malignant”) TS
Vocalizations have been reported as the initial symptom in 12–37% of patients, throat clearing being the most common (Robertson, 1989). Phonic tics can be quite troublesome for patients and those around them. In addition to involuntary noises, some patients have speech dysfluencies that resemble developmental stuttering, and up to half of all patients with developmental stuttering have been thought to have undiagnosed TS (Abwender et al., 1998). Coprolalia, perhaps the most recognizable and certainly one of the most distressing symptoms of TS, is actually present in only half of patients (Videos 16.8 and 16.15). When describing the distress caused by his severe coprolalia, one of our patients remarked that immediately after shouting an obscenity, he reaches out with his hand in an attempt to “catch the word and bring it back before others can hear it.” Coprolalia appears to be markedly influenced by cultural background. Although in one retrospective analysis of 112 children with TS, only 8% exhibited coprolalia (Goldenberg et al., 1994), the true prevalence of coprolalia in TS children and adults is about 50% in the US population, when mental coprolalia (without actual utterance) is included. In a study of 597 individuals with TS from seven countries, coprolalia occurred at some point in the course of the disease in 19.3% of males and 14.6% of females, and copropraxia in 5.9% of males and 4.9% of females (Freeman et al., 2009). Coprolalia has been reported to occur in only 26% of Danish patients and 4% of Japanese patients (Robertson, 1989). Copropraxia has been found in about 20% of patients, echolalia in 30%, echopraxia in 25%, and palilalia in 15%. Although coprolalia is a characteristic feature of TS and, based on functional MRI studies, attributed to abnormal activation particularly in the left middle frontal gyrus and right precentral gyrus, and possibly the caudate nucleus, cingulate gyrus, cuneus, left angular gyrus, left inferior parietal gyrus, and occipital gyri (Gates et al., 2004), this language abnormality is not universally present or specific for TS. 
Except for tics, the neurologic examination in patients with TS is usually normal. In one case-control study, TS patients were found to have a shorter duration of saccades, but the saccades were performed with a greater mean velocity than in normal controls, and the TS patients had fewer correct antisaccade responses, suggesting a mild oculomotor disturbance in TS (Farber et al., 1999). Although the ability to inhibit reflexive saccades is normal, TS patients make more timing errors, indicating an inability to appropriately inhibit or delay planned motor programs (LeVasseur et al., 2001).
Behavioral symptoms
Patients with TS generally have normal intelligence and may even perform better and faster than age-matched controls on certain tasks that require grammar skills that depend on procedural, rather than declarative memory (Walenski et al., 2007). While TS patients have no cognitive deficits, they often exhibit a variety of behavioral symptoms, particularly ADHD and OCD (Figs 16.1 and 16.2) (Gaze et al., 2006). In the Tourette International Consortium (TIC) database, which includes information on 3500 patients with TS evaluated by neurologists or psychiatrists, only 12% had tics only, without other comorbidities (Freeman et al., 2000). Kurlan and colleagues (2002) interviewed 1596 children, aged 9–17, in schools in Rochester and Monroe Counties, New York, and identified tics in 339 children (21%) after 60–150 minutes of observation. They found the following behavioral problems more frequently (P < 0.05) in children with tics than in those without tics: OCD, ADHD, separation anxiety, overanxious disorder, simple phobia, social phobia, agoraphobia, mania, major depression, and oppositional defiant behavior. Also, children with tics were younger (mean age: 12.5 vs. 13.3 years) and were more likely to require special education services (27% vs. 19.8%).
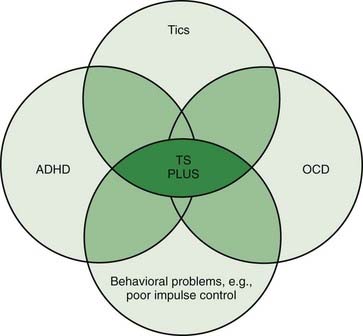
Figure 16.1 An overlap of disorders typically coexisting in patients with TS.
Redrawn from Jankovic J: Tourette’s syndrome. N Engl J Med 2001;345:1184–1192.
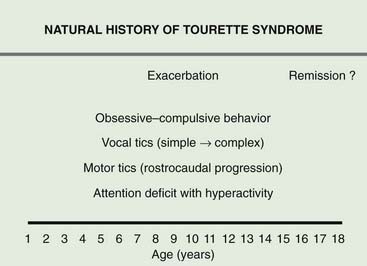
Figure 16.2 Typical progression of symptoms associated with TS.
Redrawn from Jankovic J: Tourette’s syndrome. N Engl J Med 2001;345:1184–1192.
A thorough discussion of the pathogenesis of comorbid disorders and their relationship to TS is beyond the scope of this review, and the reader is referred to some recent reviews on these topics (Tannock, 1998; Stein, 2002). The diagnosis of ADHD and OCD is based on clinical history; there are no laboratory or other tests that reliably diagnose these neurobehavioral disorders (Dulcan and AACAP Works Group on Quality Issues, 1997; Goldman et al., 1998; Swanson et al., 1998). These comorbid behavioral conditions often interfere with learning and with academic and work performance (Singer et al., 1995b). In contrast to tics, ADHD and obsessional symptom severity are significantly associated with impaired social and emotional adjustment (Carter et al., 2000). The clinician should be skilled not only in the recognition and treatment of ADHD but also in documenting the ADHD-related deficits (Richard et al., 1998). Such documentation is essential for the parents and educators in order to provide the optimal educational setting for the affected individual.
Since nearly all studies on the frequency of associated features have been based on a population of TS patients who have been referred to physicians, usually specialists, there is a certain selection bias; therefore, accurate figures on the prevalence of these behavioral disorders in TS patients are not available. It has been estimated, however, that 3–6% of the school-aged population suffers from ADHD (Goldman et al., 1998), and probably a majority of patients with TS have had symptoms of ADHD, OCD, or both at some time during the course of their illness (Coffey and Park, 1997). In a review of 1500 patients with TS, 48% were diagnosed as having ADHD, a figure that is consistent with the results of other studies (Comings and Comings, 1988b). The symptoms of ADHD may be the initial manifestations of TS and may precede the onset of motor and phonic tics by about 3 years. During this time, therapy with stimulant drugs may trigger the onset of tics and may precipitate the emergence of other TS symptoms (Price et al., 1986).
There are three types of ADHD: predominantly inattentive, predominantly hyperactive-impulsive, and combined (Dulcan AACAP Works Group on Quality Issues, 1997) (Table 16.5). ADHD is one of the most common neurobehavioral disorders, affecting 3–10% of children and 4% of adults (Wilens et al., 2004; Rappley, 2005). Adults with ADHD often have childhood histories of educational and discipline problems; and during adulthood, they usually have lower socioeconomic status, lower rates of professional employment, and higher rates of marital problems, driving violations, and other life failures. In a genome scan of 106 families, including 128 affected sib-pairs with estimated heritability of 60–80%, evidence of a gene locus was found on chromosomes 4, 9, 10, 11, 12, 16, and 17 (Smalley et al., 2001). Although attention deficit is certainly one of the most common and disabling symptoms of TS, in many patients the inability to pay attention is due to not only a coexistent ADHD but also uncontrollable intrusions of thoughts. Some patients are unable to pay attention because of a compulsive fixation of gaze. For example, while they are sitting in a classroom or a theater or during a conversation, their gaze becomes fixated on a particular object, and despite concentrated effort, they are unable to break the fixation. As a result, they miss the teacher’s lesson or a particular action in a play. Another reason for impaired attention in some TS patients is mental concentration exerted in an effort to suppress tics. Yet another cause for inattention is the sedative effect of anti-TS medications. It is therefore important to determine which mechanism or mechanisms are most likely to be responsible for the patient’s attention deficit. This is particularly important in selecting the best therapeutic approach. Despite growing publicity about ADHD, there is little evidence of widespread overdiagnosis or overtreatment of ADHD (Goldman et al., 1998). One study showed that children and adolescents with ADHD as compared to those without ADHD are more likely to have major injuries and asthma, and their 9-year medical costs are double (Leibson et al., 2001). The mechanism of ADHD with or without TS is not well understood, and there are no specific pathologic abnormalities identified. However, by using high-resolution MRI, abnormal morphology was observed in the frontal cortices, reduction in the anterior temporal cortices, and increased gray matter in the posterior and inferior parietal cortices in children and adolescents with ADHD (Sowell et al., 2003).
Table 16.5 Attention-deficit hyperactivity disorder/hyperkinetic disorder (HKD) (ICD-10, DSM-IV)
| Inattention (IN) |
That OCD is a part of the spectrum of neurobehavioral manifestations in TS is now well accepted (Rapoport, 1988; Towbin, 1988; Black, 1992; Stein, 2002). OCD, with an estimated lifetime prevalence of 2–3% (Sasson et al., 1997; Snider and Swedo, 2000) and an incidence of 0.55 per 1000 person-years (Nestadt et al., 1998), is one of the most frequent causes of disability (Jenike, 2004). It may occur alone without other features of TS (Micallef and Blin, 2001). The instrument used most frequently to measure the severity of OCD is the Yale-Brown Obsessive Compulsive Scale (Scahill et al., 1997). A distinction should be made between obsessive-compulsive symptoms or traits, obsessive-compulsive personality disorder, and OCD. Obsessions are characterized by intense, intrusive thoughts, such as concerns about bodily wastes and secretions; unfounded fears; need for exactness, symmetry, evenness, and neatness; excessive religious concerns; perverse sexual thoughts; and intrusions of words, phrases, or music. Compulsions consist of subjective urges to perform meaningless and irrational rituals, such as checking, counting, cleaning, washing, touching, smelling, hoarding, and rearranging. Leckman and colleagues (1994) have drawn attention to the frequent occurrence of the “just right” perception in patients with OCD and TS. While obsessional slowness accounts for some of the school problems experienced by TS patients, cognitive slowing (bradyphrenia) is also a contributing factor (Singer et al., 1995b). Patients with OCD can usually be divided into those with a predominantly cognitive form (in which an idea is followed by a ritual) and those with a sensorimotor type (in which a physical sensation is followed by a movement). In contrast to primary OCD, in which the symptoms relate chiefly to hygiene and cleanliness, the obsessive symptoms that are associated with TS usually involve concerns with symmetry, violent aggressive thoughts, forced touching, fear of harming self or others, and need for saying or doing things “just right” (Eapen et al., 1997b). A principal-components factor analysis of 13 categories that are used to group types of obsessions and compulsions in the Yale-Brown Obsessive Compulsive Scale symptom checklist identified the obsessions and checking and the symmetry and ordering factors as particularly common in patients with tic disorders (Leckman et al., 1997a). Miguel and colleagues (2000) showed that patients who have OCD associated with TS tend to have more bodily sensations occurring either before or during the patient’s performance of the repetitive behaviors as well as mental sensations including urge only, energy release (mental energy that builds up and needs to be discharged), and “just right” perceptions. Cognitive inflexibility manifested by impaired task-switch ability in patients with OCD has been suggested to be due to “imbalance in brain activation between dorsal and ventral striatal circuits (Gu et al., 2008).
In addition to an idiopathic sporadic or familial disorder and TS, OCD has been reported to occur as a result of a variety of lesions in the frontal-limbic-subcortical circuits (Berthier et al., 1996; Kwak and Jankovic, 2002; Voon, 2004). Although both ADHD and OCD are regarded as integral findings of the syndrome, only OCD has been shown to be genetically linked to TS (Alsobrook and Pauls, 1997). A pathogenic link between TS and OCD is also suggested by the finding in one study that 59% of 54 patients with OCD had a lifetime history of tics, and 14% fulfilled the criteria for TS during the 2–7-year follow-up (Leonard, 1992). Alsobrook and Pauls (2002) identified four significant factors: (1) aggressive phenomena (e.g., kicking, temper fits, argumentativeness), (2) purely motor and phonic tic symptoms, (3) compulsive phenomena (e.g., touching of others or objects, repetitive speech, throat clearing), and (4) tapping and absence of grunting, which accounted for 61% of the phenotypic symptom variance in TS probands and their first-degree relatives.
An important link between motor and behavioral manifestations of TS is the loss of impulse control. Many TS patients suffer from poor impulse control, disinhibition of aggression and emotions, and obsessive thoughts that may dictate their actions. Indeed, many behavioral symptoms of TS, including some complex tics, coprolalia, copropraxia, and many behavioral problems, can be explained by loss of normal inhibitory mechanisms (disinhibition) manifested by poor impulse control. It is as though the TS patients have lost their ability to suppress vestiges of primitive behavior. Poor impulse control might also explain the inability to control anger, as a result of which many patients have frequent and sometimes violent, temper outbursts, and rages. Rarely, TS patients exhibit inappropriate sexual aggressiveness and antisocial, oppositional, and even violent, unlawful, or criminal behavior. TS, indeed, serves as a model medical disorder that can predispose one to engage in uncontrollable and offensive behaviors that are misunderstood by the law-abiding community and the legal justice system (Jankovic et al., 2006). The social and legal aspects of TS patients have yet to be investigated, but there is growing concern regarding media misrepresentation that attributes violent criminal behavior in certain individuals to TS. Although TS should not be used as an excuse to justify unlawful or criminal behaviors, studies are needed to determine whether TS-related symptoms and neurobehavioral comorbidities predispose TS patients to engage in such behaviors. Often, the avolitional nature of behaviors in response to involuntary internal thought and emotional patterns is supported by the subsequent remorse and lack of secondary gain. This suggests that the preponderance of unlawful acts committed by TS patients are not premeditated but may result from a variety of TS-related mechanisms such as poor impulse control, OCD associated with addictive behavior (e.g., drugs, alcohol, gambling), attention-deficit disorder (ADD), and distractibility (e.g., motor vehicle accidents). A previous study (Comings and Comings, 1987) compared conduct in 246 TS patients to that of controls for behaviors such as lying, stealing, fighting or inability to stop fighting, violence against animals, physically attacking peers or parents, vandalism, running away from home, starting fires, poor temper control, alcohol and drug abuse, and other misdemeanors. Thirty-five percent of TS patients had a conduct score higher than 13, significantly greater than the 2.1% of controls with high scores (P < 0.0005). All behaviors with the exception of running away from home and trouble with the law occurred at a significantly greater rate in TS patients. Although no difference was found for the law variable, TS patients were significantly more likely to vandalize (P < 0.0005), fight (P < 0.0005), abuse drugs or alcohol (P < 0.003), and steal (P < 0.015). An interesting finding in this study was that certain behaviors, such as starting fires, shouting, and physically attacking, were significantly greater only in TS patients with comorbid ADD, supporting the well-established association of ADD in conduct disorder. It was estimated from this study that 10–30% of conduct disorder cases in non–economically disadvantaged children may be attributed to a possible TS gene. In a more recent study, however, TS accounted for only 2% of all cases that were referred for forensic psychiatric investigation in Stockholm, Sweden, between 1990 and 1995; 15% of the subjects had ADHD, 15% had PDD, and 3% had Asperger syndrome (Siponmaa et al., 2001).
Focal frontal lobe dysfunction, demonstrated in TS by various functional and imaging studies, has been associated with an impulsive subtype of aggressive behavior (Brower and Price, 2001). It has been postulated that impulse disorders stem from exaggerated reward-, pleasure-, or arousal-seeking brain centers, resulting in failure of inhibition. Animal studies of rats with lesions of the nucleus accumbens core, the brain region that is noted for reward and reinforcement, showed that the lesioned rats preferred small, immediate rewards over larger, delayed rewards (Cardinal et al., 2001). In addition to the ventromedial prefrontal cortices, lesions in the amygdala have also been known to result in altered decision-making processes and a disregard for future consequences (Bechara et al., 2000).
One of the most distressing symptoms of TS is a self-injurious behavior (SIB), which has been reported in up to 53% of all patients (Robertson, 1989; Robertson et al., 1990). A common form of SIB is damage of skin by biting, scratching, cutting, engraving, or hitting, particularly in the eye and throat (compulsions), often accompanied by an irresistible urge (obsession) (Jankovic et al., 1998) (Videos 16.16 and 16.17). In some patients SIB can be life-threatening, hence the term “malignant” TS (Cheung et al., 2007). The mechanism of self-injurious behavior is not well understood, but the recently described animal model may shed some light on this very important behavioral disorder that may accompany TS. A mouse with genetic deletion of Sapap3, a gene that codes for postsynaptic scaffolding protein at excitatory striatal synapses, has been proposed as a possible model of OCD as its phenotype is characterized by excessive compulsive grooming resulting in self-injurious behavior, such as facial hair loss and skin lesions (Welch et al., 2007). The similarity of this behavior to OCD is further supported by the observation that the mice markedly improved after treatment with a selective serotonin reuptake inhibitor. Thus, SIB appears to be related to OCD, which has treatment implications. Besides OCD, SIB also correlates with impulsivity and impulse control (Mathews et al., 2004). Conduct disorders and problems with discipline at home and in school are recurrent themes during discussions of behavioral problems with TS families. The inability to suppress or “edit” intentions due to a “dysfunctional intention editor” has been proposed as one of the chief reasons for poor impulse control in patients with TS (Baron-Cohen et al., 1994). 
The TS gene(s) may, in addition to tics, ADHD, and OCD, express itself in a variety of behavioral manifestations, including learning and conduct disorders, schizoid and affective disorders, antisocial behaviors, oppositional defiant disorder, anxiety, depression, conduct disorder, severe temper outbursts, rage attacks, impulse control problems, inappropriate sexual behavior, and other psychiatric problems (Comings, 1987; Robertson, 2000). Personality disorder and depression have been reported in 64% of patients with TS (Robertson et al., 1997). Whether these behavioral problems indeed occur with higher frequency in TS patients and whether they are pathogenically linked to TS are debatable (Comings and Comings, 1988a; Pauls et al., 1988).
Besides comorbid behavioral conditions, TS has been reported to be frequently associated with migraine headaches. In one study, 26.6% of TS patients, with a mean age of 11.9 years, exhibited migraine headaches (Barabas et al., 1984). We found migraine headaches in 25 carefully screened TS patients at a significantly greater rate than the estimated 11–13% in the general adult population (P < 0.0001) and the general pediatric population (P < 0.04) (Kwak et al., 2003b). This compares to 4.0–7.4% in the general population of school-aged children. The Tourette International Consortium Database (Freeman et al., 2000), which at the time of publication included information on 3500 patients with TS collected from 64 centers from around the world, showed that only 12% of patients with TS had no other disorders; ADHD was seen in 60%, symptoms of OCD in 59%, anger control problems in 37%, sleep disorder in 25%, learning disability in 23%, mood disorders in 20%, anxiety disorders in 18%, and SIB in 14%.
Pathogenesis
Neurophysiology
Although the pathogenic mechanisms of TS are still unknown, the weight of evidence supports an organic rather than psychogenic origin, probably involving the basal ganglia circuitry (Leckman et al., 1997b; Palumbo et al., 1997; Jankovic, 2001a; Leckman, 2002; Berardelli et al., 2003; Albin and Mink, 2006) (Fig. 16.3). Despite the observation that some tics may be voluntary, at least in part, physiologic studies suggest that tics are not mediated through normal motor pathways utilized for willed movements (Obeso et al., 1982). Using back-averaging techniques, Obeso and colleagues (1982) observed normal Bereitschaftspotential in six subjects who voluntarily simulated tic-like movements, but no such premovement potential was noted in association with an actual tic. The common absence of premotor potentials in simple motor tics suggests that tics are truly involuntary or that they occur in response to some external cue (Papa et al., 1991). Karp and colleagues (1996), however, documented premotor negativity in two of five patients with simple motor tics. Although the investigators could not correlate the presence of Bereitschaftspotential with the premonitory sensation, the physiology of the premovement phenomenon requires further studies. Transmagnetic stimulation used to study cortical excitability in 11 TS patients found evidence of motor-cortical disinhibition at rest (Heise et al., 2010).
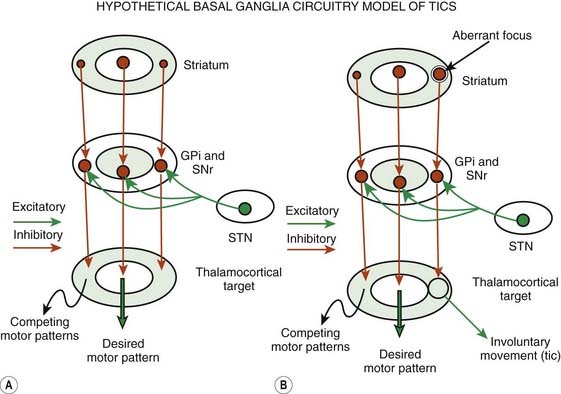
Conventional neurophysiologic investigations have found that TS patients have defective inhibitory mechanisms, as is suggested by the increased duration of the late response of the blink reflex and reduced inhibition at paired pulse testing (Smith and Lees, 1989; Berardelli et al., 2003). TS patients also have exaggerated audiogenic startle response (Gironell et al., 2000). About 20% patients with TS have exaggerated startle responses, which may fail to habituate with repetition (Stell et al., 1995).
As was noted before, functional MRI showed decreased neuronal activity during periods of suppression in the ventral globus pallidus, putamen, and thalamus and increased activity in the right caudate nucleus, right frontal cortex, and other cortical areas that are normally involved in the inhibition of unwanted impulses (prefrontal, parietal, temporal, and cingulate cortices) (Peterson et al., 1998a). In another study of three patients with TS, functional MRI showed marked reduction or absence of activity in secondary motor areas while the patient attempted to maintain a stable grip-load force control (Serrien et al., 2002). The authors interpreted the findings as an ongoing activation of the secondary motor reflecting patients’ involuntary urges to move. In a study of children with ADHD, functional MRI showed increased frontal activation and reduced striatal activation on various tasks and an enhancement of striatal function after treatment with methylphenidate (Vaidya et al., 1998). Functional MRI studies show decreased neuronal activity during periods of suppression in the ventral globus pallidus, putamen, and thalamus and increased activity in the caudate, frontal cortex, and other cortical areas that are normally involved in the inhibition of unwanted impulses (prefrontal, parietal, temporal, and cingulated cortical areas) (Peterson, 2001). By using event-related functional MRI (fMRI) in 10 patients with TS, a brain network of paralimbic areas such as anterior cingulate and insular cortex, supplementary motor area (SMA), and parietal operculum (PO) was identified that predominantly activated before tic onset, whereas at the beginning of tic action, significant fMRI activities were found in sensorimotor areas including superior parietal lobule bilaterally and cerebellum (Bohlhalter et al., 2006). Using resting-state functional connectivity MRI (rs-fcMRI) in 33 adolescents with TS, Church et al. (2009) found anomalous connections primarily in the frontoparietal network, suggesting widespread immature functional connectivity, particularly in regions related to adaptive online control.
Transcranial magnetic stimulation studies have demonstrated a shortened cortical silent period and defective intracortical inhibition (determined in a conditioning test paired-stimulus paradigm) in patients with TS (Orth et al., 2005) and OCD (Greenberg et al., 1998), thus providing a possible explanation for intrusive phenomena. Subsequent studies utilizing the same technique have demonstrated that patients with tic-related OCD have more abnormal motor cortex excitability than OCD patients without tics (Greenberg et al., 2000). Transcranial magnetic stimulation studies have also demonstrated that TS children have a shorter cortical silent period but that their intracortical inhibition was not different from that of controls, although intracortical inhibition is reduced in children with ADHD (Moll et al., 2001). There is evidence of additive inhibitory deficits, as demonstrated by reduced intracortical inhibition and a shortened cortical silent period in children with TS and comorbid ADHD. In another study, ADHD more than tics were associated with short-interval intracortical inhibition (Gilbert et al., 2004). Both short-interval intracortical inhibition and short-interval afferent inhibition, modulated by nicotinic receptors, were reduced in eight patients with TS (ages 24–38 years) as compared to ten matched healthy controls (Orth et al., 2005). Comprehensive discussion of the neuroscience of ADHD is beyond the scope of this chapter, but the reader is referred to some recent reviews of this topic (Castellanos and Tannock, 2002).
Sleep studies have provided additional evidence that some tics are truly involuntary. Polysomnographic studies in 34 TS patients recorded motor tics in various stages of sleep in 23 patients and phonic tics in 4 patients (Glaze et al., 1983; Jankovic and Rohaidy, 1987). Additional sleep studies have suggested that some patients with TS have alterations of arousal, decreased percentage (up to 30%) of stage 3/4 (slow wave) sleep, decreased percentage of REM sleep, paroxysmal events in stage 4 sleep with sudden intense arousal, disorientation and agitation, restless legs syndrome, and periodic leg movement in sleep (Voderholzer et al., 1997; Chokroverty and Jankovic, 1999; Picchietti et al., 1999). Restless legs syndrome has been reported to be present in about 10% of patients with TS and in 23% of parents (Lesperance et al., 2004). In this regard, it is of interest that in a study in which 14 single-nucleotide polymorphisms were typed spanning the three genomic loci in 298 TS trios, 322 TS cases (including 298 probands from the cohort of TS trios), and 290 control subjects, it was found that the same variant of the gene BTBD9 that increases the risk of restless legs syndrome also increases the risk of TS without obsessive-compulsive disorder or attention-deficit disorder (“pure” TS) (Rivière et al., 2009). Other sleep-related disorders that are associated with TS include sleep apnea, enuresis, sleepwalking and sleep talking, nightmares, myoclonus, bruxism, and other disturbances (Rothenberger et al., 2001; Hanna and Jankovic, 2003).
Neuroimaging
Although standard anatomic neuroimaging studies in TS are unremarkable, by using special volumetric, metabolic, blood flow, ligand, and functional imaging techniques, several interesting findings have been reported that have strong implications for the pathophysiology of TS (Peterson, 2001; Albin and Mink, 2006; Frey and Albin, 2006) (Fig. 16.3). Careful volumetric MRI studies have suggested that the normal asymmetry of the basal ganglia is lost in TS (Peterson et al., 1993; Singer et al., 1993; Peterson, 2001). In one study, the normal left > right asymmetry (Singer et al., 1993) in the volume in the right anterior brain, right caudate, and right pallidum was reversed in TS subjects (Castellanos et al., 1996). Caudate volumes have been reported to correlate significantly and inversely with the severity of tic and OCD in early adulthood (Bloch et al., 2005). The volumetric studies, however, have not always produced consistent results (Moriarty et al., 1997). In another volumetric MRI study, Frederickson and colleagues (2002) found evidence of smaller gray matter volumes in the left frontal lobes of patients with TS, further supporting the findings of loss of normal left > right asymmetry. Quantitative MRI studies have found a subtle, but possibly important, reduction in the volume of caudate nuclei in patients with TS. In 10 pairs of monozygotic twins, the right caudate was smaller in the more severely affected individuals, providing evidence for the role of environmental events in the pathogenesis of TS (Hyde et al., 1995). In contrast, the corpus callosum has been found to be larger in children with TS than in normal controls (Baumgardner et al., 1996). Subsequent study showed that this finding was gender-related and was present only in boys with TS (Mostofsky et al., 1999). An MRI-DTI study of monozygotic twins showed that the mean fractional anisotropy values were significantly lower, particularly in the posterior portion of the corpus callosum, in the twin affected with TS (Cavanna et al., 2010). Using voxel-based morphometry and high-resolution MRI in 31 TS patients, increased gray matter was found in the left mesencephalon compared to 31 controls (Garraux et al., 2006). This finding, however, could not be replicated in a smaller study of 14 boys with TS using the same technique (Ludolph et al., 2006). This latter study found increased gray matter volumes in bilateral ventral putamen and regional decreases in gray matter volumes in the left hippocampus gyrus. The difference was attributed to younger mean age in the latter study compared to the previous study (12.5 vs. 32 years). Diffusion-tensor MRI (DT-MRI) used to investigate the structural integrity of basal ganglia and thalamus in 23 children with TS found increased mean water diffusivity bilaterally in the putamen and decreased anisotropy in the right thalamus, indicating impairment of white matter integrity in the fronto-striatal-thalamic circuit at a microstructural level (Makki et al., 2008). Using tractography of the fronto-striato-thalamic circuit, the same group showed that TS patients had significantly lower probability of connection between caudate nucleus and anterior-dorsolateral-frontal cortex on the left and that OCB was negatively associated with connectivity score of the left caudate and anterior dorsolateral-frontal cortex, and was positively associated with connectivity score for the subcallosal gyrus and for the lentiform nucleus (Makki et al., 2009). Additional imaging studies have identified frontal and parietal cortical thinning, most prominent in ventral portions of the sensory and motor homunculi in patients with TS (Sowell et al., 2008). Dopaminergic hyperfunction (Albin et al., 2003), possibly as a result of overproduction of synapses in mid-childhood (Giedd et al., 1999), might be associated with increased striatal volume in childhood which may be lost in adulthood.
Positron emission tomography (PET) scanning has shown variable rates of glucose utilization in basal ganglia as compared to controls. In one study, [18F]fluorodeoxyglucose PET has shown evidence of increased metabolic activity in the lateral premotor and supplementary motor association cortices and in the midbrain (pattern 1), and decreased metabolic activity in the caudate and thalamic areas (limbic basal ganglia–thalamocortical projection system) (pattern 2) (Eidelberg et al., 1997). Pattern 1 is reportedly associated with tics, and pattern 2 correlates with the overall severity of TS. In a follow-up study, involving 12 TS adult patients (untreated for >2 years) and 12 controls, the investigators found a TS-related metabolic pattern which was characterized by increased premotor cortex and cerebellum activity, and reduced resting activity of the striatum and orbitofrontal cortex (Pourfar et al., 2011). They also provided evidence for an OCD-related pattern which was characterized by reduced activity of the anterior cingulate and dorsolateral prefrontal cortical regions associated with relative increases in primary motor cortex and precuneus. This pattern significantly correlated with the severity of the OCD symptoms.
In contrast to dystonia, which is characterized by lentiform nucleus-thalamic metabolic dissociation, attributed to overactivity of the direct striatopallidal inhibitory pathway, the pattern of TS is characterized by concomitant metabolic reduction in striatal and thalamic function. The authors suggested that this pattern can be explained by a reduction in the indirect pathway, resulting in reduction in subthalamic nucleus (STN) activity. This is in part consistent with another study that found evidence of increased activation in the direct pathway, but the activity in the prefrontal cortex and STN has been found to be increased presumably as a result of compensatory activation (Baym et al., 2008). Using event-related [15O]H2O PET combined with time-synchronized audiotaping and videotaping of six patients with TS, Stern and colleagues (2000) found increased activity in the sensorimotor, language, executive, paralimbic, and frontal-subcortical areas that was temporarily related to the motor and phonic tics and the irresistible urge that precedes these behaviors. Using a similar technique Lerner et al. (2007) found robust activation of cerebellum, insula, thalamus, and putamen during tic release. Rauch and colleagues (1997) showed bilateral medial temporal (hippocampal/parahippocampal) activation on PET in patients with OCD as compared to normal controls and absence of activation of inferior striatum, seen in normal controls. Various neuroimaging studies have also demonstrated moderate reduction in the size of the corpus callosum, basal ganglia (particularly caudate and globus pallidus), and frontal lobes (Filipek et al., 1997) and striatal hypoperfusion (Vaidya et al., 1998) in patients with ADHD.
An autopsy study of three TS brains found consistent increases in dopamine transporter (DAT) and D2 receptor as well as D1 and α-2A density, suggesting that dopaminergic hyperfunction in the frontal lobe may play a role in the pathophysiology of TS (Yoon et al., 2007). Another pathological study showed a marked increase in total number of neurons in the globus pallidus interna (GPi) and decreased number in the globus pallidus externa (GPe) and in the caudate nucleus of brains of patients with TS (Kalanithi et al., 2005). Furthermore, an increased number and proportion of the GPi neurons were positive for the calcium-binding protein parvalbumin in tissue from TS subjects, whereas lower densities of parvalbumin-positive interneurons were observed in both the caudate and putamen of TS subjects. These abnormalities have been interpreted as indicating a developmental defect in the migration of some GABAergic neurons.
Neurochemistry
An alteration in the central neurotransmitters has been suggested chiefly because of relatively consistent responses to modulation of the dopaminergic system (Harris and Singer, 2006). Dopamine antagonists and depletors generally have an ameliorating effect on tics, whereas drugs that enhance central dopaminergic activity exacerbate tics (Jankovic and Rohaidy, 1987; Jankovic and Orman, 1988). Low cerebrospinal fluid homovanillic acid, coupled with a favorable response to dopamine receptor-blocking drugs, has been interpreted as evidence in support of the notion that tics and TS are due to supersensitive dopamine receptors (Singer, 2000). Postmortem binding studies of dopamine receptors, however, have failed to provide support for this hypothesis (Singer, 2000).
Neurochemical studies of TS have been hampered by the lack of available postmortem brain tissue. Biochemical abnormalities in the few postmortem brains that have been studied include low serotonin, low glutamate in the medial globus pallidus, and low cyclic AMP in the cortex (Singer, 2000). Haber and Wolfer (1992) reported that in a blind rating of five TS brains, three had low dynorphin immunoreactivity in the ventral portion of the medial globus pallidus. A defect in tryptophan oxygenase in TS has been proposed by Comings (1990), who analyzed 1400 blood samples from patients with TS or ADHD and their relatives and controls; he found decreased platelet serotonin and low blood tryptophan levels in TS patients and their parents. In support of the “tryptophan hypothesis” is a study that used alpha-[11C]methyl-L-tryptophan (AMT) positron emission tomography (PET) to assess global and focal brain abnormalities of tryptophan metabolism in 26 children with TS and nine controls. The findings indicate that tryptophan uptake is significantly decreased in the dorsolateral prefrontal cortex and increased in the thalamus of TS patients (Behen et al., 2007).
One intriguing hypothesis, supported partly by the increased 3H-mazindol binding to the presynaptic dopamine uptake carrier sites, suggests that TS represents a developmental disorder resulting in dopaminergic hyperinnervation of the ventral striatum and the associated limbic system (nucleus accumbens) (Singer et al., 1991). Using [11C]raclopride PET and amphetamine stimulation, Singer and colleagues (2002) found evidence for increased dopamine release in the putamen of patients with TS. They postulate that in TS, there is increased activity of the dopamine transporter leading to increased dopamine concentration in the dopamine terminals and stimulus-dependent increase in dopaminergic transmission. Support for this hypothesis has been provided by the imaging studies of Albin and colleagues (2003). The authors used PET with the vesicular monoamine transporter type 2 ligand [11C]dihydrotetrabenazine that binds to type 2 vesicular monoamine transporter (VMAT2) to quantify striatal monoaminergic innervation in patients with TS (n = 19) and control subjects (n = 27). With voxel-by-voxel analysis, the investigators found increased [11C]dihydrotetrabenazine binding in the ventral striatum (right > left) in patients with TS as compared to age-matched controls. However, a subsequent PET study involving 33 adults with TS and utilizing not only [11C]dihydrotetrabenazine but also [11C]methylphenidate, a ligand for DAT, binding, found no differences between subjects with TS and controls (Albin et al., 2009). In a study of 8 patients with TS and 8 controls [11C]FLB 457 PET in conjunction with an amphetamine challenge used to evaluate extrastriatal D2/D3 receptor binding and DA release, TS patients showed decreased [11C]FLB 457 binding potentials bilaterally in cortical and subcortical regions outside the striatum, including the cingulate gyrus, middle and superior temporal gyrus, occipital cortex, insula, and thalamus (Steeves et al., 2010). Furthermore, amphetamine challenge induced widespread increased DA release in TS patients, which extended more anteriorly to involve anterior cingulate and medial frontal gyri. The authors suggested that “reductions in D2/D3 receptor binding in both frontal cortex and thalamus are consistent with recently published preliminary data demonstrating similar abnormalities of D2/D3 binding in TS patients using a different PET ligand.” A postmortem examination of two brains of patients with typical childhood-onset TS and one with adult-onset tics showed that the prefrontal cortex rather than the striatum showed most abnormalities, including increased D2 receptor protein, as well as increases in dopamine transporter, VAMP2, and α-2A (Minzer et al., 2004).
Although this idea is highly speculative, it is possible that the genetic defect in TS somehow interferes with the normal regulation of the neuronal progenitor cells during development, thus resulting in the increased innervation of the ventral striatum (Ikonomidou et al., 1999; Itoh et al., 2001; Hanashima et al., 2004). This implies that the genetic defect somehow interferes with the programmed cell suicide that is needed to control cell proliferation in normal development and growth. The ventral striatum is the portion of the basal ganglia that is anatomically and functionally linked to the limbic system. The link between the basal ganglia and the limbic system might explain the frequent association of tics and complex behavioral problems, and a dysfunction in the basal ganglia and the limbic system seems to provide the best explanation for the most fundamental behavioral disturbance in TS, namely, loss of impulse control and a state of apparent “disinhibition.” The notion that deficits in inhibitory functions are at the core of the clinical phenotype associated with TS is supported by studies by Baron-Cohen and colleagues (1994) showing that patients with TS are not able to appropriately edit their intentions and by Swerdlow and colleagues (1996) showing that TS patients demonstrate deficits on the visuospatial priming tasks.
Functional neuroimaging studies have been used to aid in the understanding of neurotransmitter and receptor alterations in TS. Using [123I]β-carboxymethoxy-3β-(4-iodophenyl) tropane (CIT) single photon emission computed tomography scans, Malison and colleagues (1995) demonstrated a mean 37% increase in binding of this dopamine transporter ligand in the striatum in five adult patients with TS as compared to age-matched controls. In contrast, Heinz and colleagues (1998) found no difference in [123I] β-CIT binding in the midbrain, thalamus, or basal ganglia between 10 TS patients and normal control subjects. There was, however, a significant negative correlation between the severity of phonic tics and β-CIT binding in the midbrain and thalamus. In another study involving 12 TS adult patients, β-CIT scans showed evidence of increased dopamine transporter binding (Müller-Vahl et al., 2000). Combining single photon emission computed tomography and MRI, Wolf and colleagues (1996) found 17% greater binding of IBZM, a D2 receptor ligand, in the caudate (but not putamen) nucleus in five of the more affected monozygotic twins who were discordant for TS. It is important to note, however, that two of the five subjects were taking neuroleptics for up to 6 weeks prior to the single photon emission computed tomography studies. These findings, if confirmed by other studies of neuroleptic-naive patients, support the notion that the presynaptic dopamine function is enhanced in TS. This might, in turn, lead to a reduced inhibitory pallidal output to the mediodorsal thalamus. The observation that in patients with Parkinson disease the severity of childhood-onset tics was not influenced by the development of parkinsonism or by its treatment with levodopa, however, argues against the role of dopamine in the pathogenesis of TS symptoms (Kumar and Lang, 1997a). This is supported by the results of PET ligand studies showing normal D2 receptor density (Turjanski et al., 1994). Furthermore, Meyer and colleagues (1999) used PET imaging of (+)-α-[11C]dihydrotetrabenazine to determine the density of vesicular monoamine transporter type 2, a cytoplasm-to-vesicle transporter that is linearly related to monoaminergic nerve terminal density unaffected by medications, in 8 TS patients and 22 controls. This study showed no significant difference in terminal density between patients and controls, thus failing to provide support for the concept of increased striatal innervation. However, these studies do not exclude the possibility of abnormal regulation of dopamine release and uptake. Subsequent study involving 19 adult patients with TS showed that the [11C]dihydrotetrabenazine-binding potential is significantly increased, thus supporting the notion that striatal monoaminergic innervation is increased in the ventral striatum (right > left) of TS patients (Albin et al., 2003). The results of this study contrasted with previous findings of 30–40% increase in dopamine transporter (Müller-Vahl et al., 2000; Singer et al., 2001). Furthermore, in a small sample of TS patients, PET studies, have demonstrated a 25% increase in accumulation of fluorodopa in the left caudate (P = 0.03) and a 53% increase in the right midbrain (P = 0.08) (Ernst et al., 1999). These findings indicate possible dopaminergic dysfunction in the cells of origin and in the dopaminergic terminals, suggesting increased activity of dopa decarboxylase.
Functional imaging has provided insights into the mechanisms not only of TS but also of ADHD. In 53 adults with ADHD (without tics) when compared to 44 healthy controls, a reduction in dopamine markers was demonstrated, using 11C-cocaine as a marker for dopamine transporter and 11C-raclopride, a ligand for D2/D3 receptors (Volkow et al., 2009).
Despite some limitations and inconsistencies, the imaging, ligand, and biochemical studies provide support for the hypothesis that the corticostriatal-thalamic-cortical circuit plays an important role in the pathogenesis of TS and related disorders (Witelson, 1993; Peterson, 2001). The dorsolateral prefrontal circuit, which links Brodmann’s area 9 and 10 with the dorsolateral head of the caudate, appears to be involved with executive functions (manipulation of previously learned knowledge, abstract reasoning, organization, verbal fluency, and problem solving; it is closely related to intelligence, education, and social exposure) and motor planning. An abnormality in this circuit has been implicated in ADHD. The lateral orbitofrontal circuit originates in the inferior lateral prefrontal cortex (area 10) and projects to the ventral medial caudate. An abnormality to this circuit is associated with personality changes, mania, disinhibition, and irritability. Last, the anterior cingulate circuit arises in the cingulate gyrus (area 24) and projects to the ventral striatum, which also receives input from the amygdala, hippocampus, medial orbitofrontal cortex, and entorhinal and perirhinal cortex. A variety of behavioral problems, including OCD, may be linked to an abnormality in this circuit.
Immunology
The potential role of immunologic mechanisms and specifically antineuronal antibodies is currently being explored in a variety of neurologic disorders, including TS (Allen, 1997; Hallett and Kiessling, 1997; Hallett et al., 2000; Morshed et al., 2001; Church et al., 2003; Harris and Singer, 2006; Singer et al., 2008; Martino et al., 2009). Several studies have suggested that exacerbations of TS symptoms correlated with an antecedent group A β-hemolytic streptococcus (GABHS) infection (demonstrated by elevated antistreptococcal titers) and the presence of serum antineuronal antibodies (Kiessling et al., 1993). Epitopes of streptococcal M proteins have been found to cross-react with the human brain, particularly the basal ganglia, and may be pathogenetically important in various neurologic disorders, such as Sydenham disease, TS-like syndrome, dystonia, and parkinsonism (Bronze and Dale, 1993; Dale et al., 2001). In 10 patients with poststreptococcal acute disseminated encephalomyelitis (PSADEM) following exposure to GABHS, Dale and colleagues (2001) showed antibasal ganglia antibodies in all with three (60, 67, and 80 kDa) dominant bands. Furthermore, MRI showed hyperintense basal ganglia in 80% of the patients. The B-lymphocyte antigen D8/17 is considered to be a marker for rheumatic fever but is also frequently overexpressed in patients with tics, OCD, and autism (Murphy et al., 1997; Swedo et al., 1997; Hoekstra et al., 2001). In one study, children and adults with TS had significantly higher serum levels of antineuronal antibodies against the putamen, but not the caudate or globus pallidus, as compared to controls (Singer et al., 1998). The potential relevance of this finding has been questioned, however, since there is no relationship between the presence of the antineuronal antibodies and age at onset, severity of tics, or the presence of comorbid disorders. Trifiletti and Packard (1999) have confirmed the presence of a specific brain protein at an apparent molecular weight of 83 kDa that is recognized by antibodies in the serum of 80–90% of patients with TS or OCD (Trifiletti and Packard 1999). They concluded that there may be a subset of patients with TS and OCD, perhaps up to 10% of all cases, in whom a streptococcal infection triggers the onset of symptoms (Trifiletti and Packard, 1999). In a large case-control study of 150 patients with tics compared to 150 controls, Cardona and Orefici (2001) found a correlation between the occurrence of tics and prior exposure to streptococcal antigens and that the severity of tic disorder correlates with the magnitude of the serologic response to streptococcal antigens measured by antistreptolysin O (ASO) titers (38% of the children with tics compared with 2% of the control subjects had ASO titers ≥500 IU, (P < 0.001)). In another study involving 25 adult patients with TS and 25 healthy controls, increased antibody titers against streptococcal M12 and M19 proteins were found in the TS group as compared to the healthy controls (Müller et al., 2001). In yet another study involving 81 patients with TS, 27 with Sydenham disease, 52 with autoimmune disorders, and 67 normal controls, Morshed and colleagues (2001) found elevated titers of IgG antineuronal antibodies in diseased individuals compared to controls, but there was no relationship between the antibody titers and the age, severity of TS, or comorbid disorders. Church and colleagues (2003) studied 100 patients with TS, 50 children with neurologic disease, 40 with recent uncomplicated streptococcal infection, and 50 healthy adults. They found elevated ASO titers in 64% of TS children compared with 15% of pediatric controls (P < 0.0001) and in 68% of adults with TS compared with 12% of adult neurologic controls and 8% of adult healthy controls (P < 0.05). Evidence of basal ganglia antibodies were found in 20–27% of TS patients, compared to 2–4% of controls. Similar to the proposed antigen in Sydenham disease, the most common antigen in TS was 60 kDa protein. Furthermore, 91% of patients with positive basal ganglia antibodies were found to have elevated ASO titers, whereas only 57% of those without such antibodies had high ASO titers. The authors concluded that these findings “suggest a pathogenic similarity between Sydenham disease and some patients with TS.” In another study, 65% of 65 patients with atypical movement disorders, including dystonia and tics, had antibasal ganglia antibodies (Edwards et al., 2004b). Edwards and colleagues (2004a) also reported four adult cases of tic disorder and stereotypies associated with the presence of antibasal ganglia antibodies. There was no difference when clinical features of TS patients (53 children and 75 adults) with and without antinuclear antibodies, determined by Western immunoblot showing bands for at least 1 of 3 reported striatal antigens (40, 45, and 60 kDa), were compared (Martino et al., 2009). When brain MRI T1 and diffusion tensor (DTI) imaging was compared in 9 adults with TS who had antibasal ganglia antibodies with 13 without detectable antibodies, the voxel-based morphometry analysis failed to detect any significant difference in gray matter density between the two groups, again suggesting lack of relevance of the antibasal ganglia antibodies to the pathogenesis of TS. Although there are many inconsistencies in the reported results related to autoantibodies in TS, and increased activation of immune responses in TS has been suggested by some investigators, further studies are needed before it can be concluded that TS is an autoimmune disorder (Martino et al., 2009).
Development of dyskinesias (paw and floor licking, head and paw shaking) and phonic utterances has been reported in rodents after the microinfusion of dilute IgG from TS subjects into their striatum (Hallett et al., 2000). They extended their initial observations by demonstrating that intrastriatal microinfusion of TS sera or gamma immunoglobulins (IgG) in rats produced stereotypies and episodic utterances, analogous to involuntary movements seen in TS, and confirmed the presence of IgG selectively bound to striatal neurons (Hallett et al., 2000). Peterson and colleagues (2000) found that ADHD was associated significantly with titers of two distinct antistreptococcal antibodies, ASO and antideoxyribonuclease B, but no significant association was seen between antibody titers and a diagnosis of either chronic tic disorder or OCD. When basal ganglia volumes were included in these analyses, the relationships between antibody titers and basal ganglia volumes were significantly different in OCD and ADHD subjects compared with other diagnostic groups. Higher antibody titers in these subjects were associated with larger volumes of the putamen and globus pallidus nuclei.
Variably referred to as pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections (PANDAS) or pediatric infection-triggered autoimmune neuropsychiatric disorders (PITANDS), this area is one of the most controversial topics in pediatric neurologic and psychiatric literature (Kurlan, 1998b; Trifiletti and Packard, 1999; Hoekstra et al., 2002; Murphy and Pichichero, 2002; Church et al., 2003; Snider and Swedo, 2003; Kurlan, 2004; Gabbay et al., 2008; Gilbert and Kurlan, 2009; Schrag et al., 2009). The following are the diagnostic criteria for pediatric infection-triggered autoimmune neuropsychiatric disorders, which are similar to those for PANDAS except that they define an episode of illness more specifically:
The PANDAS concept has some other shortcomings: (1) The streptococcal infection may cause stress, which can exacerbate tics; (2) some patients with PANDAS may simply have a variant of Sydenham disease; and (3) there is no established temporal link between antecedent streptococcal infection and the exacerbation of tics. One piece of evidence supporting the notion that PANDAS simply represents one spectrum of Sydenham disease is the frequent occurrence of TS comorbidities in patients with Sydenham disease. One study compared 56 patients with Sydenham disease with 50 rheumatic fever patients and 50 healthy subjects; it found that obsessive-compulsive behavior (OCB) was present in 19%, OCD in 23.2%, and ADHD in 30.4% of Sydenham disease patients, all significantly more frequent than in the rheumatic fever or healthy controls (Maia et al., 2005). Nevertheless, the concept of postinfectious OCD is gradually seeping into the literature, even though definite proof is still lacking (Leonard et al., 1999). One observation, yet to be confirmed, that has been used in support of the autoimmune hypothesis of TS is that infusion of sera from patients with TS who have high titers of antibodies against nuclear or neural protein into ventrolateral striata of rats was associated with a higher rate of oral stereotypies as recorded by “blinded” raters (Taylor et al., 2002). This observation, however, could not be reproduced by other investigators who infused sera from patients with TS who had antiputaminal antibodies (n = 9) and from patients with PANDAS (n = 8) into the striatum of rats (Loiselle et al., 2004). No stereotypic behavior or abnormal movements were observed. Furthermore, there was no difference in antibasal ganglia antibodies between patients with PANDAS and patients with TS as measured by enzyme-linked immunosorbent assay (Singer et al., 2004). In a study of 40 subjects prospectively evaluated with intensive laboratory testing for GABHS for an average of 2 years with additional testing at the time of any clinical exacerbations, Kurlan et al. (2008) found that 5 of 64 exacerbations were temporally associated (within 4 weeks) with a GABHS infection, which was significantly higher than the number that would be expected by chance alone (1.6). In another study no correlation between a variety of immune markers and clinical exacerbations in children with PANDAS was found, arguing against autoimmunity as a pathophysiologic mechanism in this disorder (Singer et al., 2008). In a study that involved consecutive ratings of tics and behavioral symptoms in 45 cases and 41 matched control subjects over a 2-year period, only a small minority of children with TS and early-onset OCD were found to be sensitive to antecedent GABHS infections (Lin et al., 2010). In a case-control study of a large primary care database of 678 862 patients with an average follow-up of 5.08 years, no support was found for a strong relationship between streptococcal infections, neuropsychiatric syndromes such as OCD or TS, or PANDAS (Schrag et al., 2009). Although, in contrast to the Schrag et al. (2009), similar health services database studies in the United States found modest statistical associations between streptococcal infections and tic or OCD diagnoses, “current evidence indicates that for the ordinary TS/OCD phenotype of waxing and waning symptoms, GABHS infection does not seem to be an important etiologic factor and therefore not an appropriate target for assessment or therapy” (Gilbert and Kurlan, 2009).
Untreated GABHS infection is often complicated by rheumatic fever within 10–14 weeks and by Sydenham disease within several months. Several studies have provided evidence for an overlap between TS and Sydenham disease, tics and OCD being manifested in both disorders (Church et al., 2003). It is therefore not clear whether TS and OCD are independent sequelae of GABHS or whether the observed symptoms of TS and OCD are manifestations of Sydenham disease. In a 3-year prospective study of 12 children, 5 to 11 years old, with documented acute GABHS tonsillopharyngitis, Murphy and Pichichero (2002) noted a sudden onset of OCD symptoms in all patients and tics in 3 patients concurrently with the acute infection or within 4 weeks. These symptoms resolved in all patients within 2 weeks of initiation of antibiotic therapy. Although this intriguing hypothesis requires further studies, plasmapheresis, intravenous IgG, and immunosuppressant therapies are currently being investigated in the treatment of TS (Allen, 1997). In a study of 30 children in whom OCD or tics were presumably triggered or exacerbated by GABHS, there were striking improvements in various measures of OCD after intravenous immunoglobulin (IVIg) and in tics after plasma exchange (Perlmutter et al., 1999). Twenty-nine children with PANDAS were randomized in a partially double-blind fashion (no sham plasmapheresis) to an IVIg, IVIg placebo (saline), and plasmapheresis (PEX) group. One month after treatment, the severity of obsessive-compulsive symptoms (OCS) was improved by 58% and 45% in the PEX and IVIg groups, respectively, compared to only 3% in the IVIg control group. In contrast, tic scores were only improved after PEX treatment (i.e., reductions of 49% (PEX), 19% (IVIg), and 12% (IVIg placebo)). Improvements in both tics and OCS were sustained for 1 year. However, there was no control PEX group, and the control comparisons were limited to the 1-month visit. Furthermore, there was no relationship between rate of antibody removal and therapeutic response. Until the results of this study are confirmed, these treatment modalities are not justified in patients with TS. Furthermore, because of uncertainties about the possible cause-and-effect relationship between GABHS and tics and OCD, an antibiotic treatment for acute exacerbations of these symptoms is currently considered unwarranted (Kurlan, 1998b). Some studies have suggested that encephalitis lethargica is also part of the spectrum of poststreptococcal autoimmune diseases (Dale et al., 2004; Vincent, 2004).
Other hypotheses
Although direct evidence is still lacking, TS is currently viewed as a disorder of synaptic transmission involving disinhibition of the corticostriatal-thalamic-cortical circuitry. Several studies have provided evidence in support of the notion that the basal ganglia, particularly the caudate nucleus, and the inferior prefrontal cortex play an important role in the pathogenesis of not only TS but also comorbid disorders, particularly OCD (Baxter et al., 1992; McGuire, 1995; Swoboda and Jenike, 1995). For example, Laplane and colleagues (1989) described eight patients with bilateral basal ganglia lesions (anoxic or toxic encephalopathy) who showed stereotyped activities and OCB; extrapyramidal signs were mild or absent. Several imaging studies (reviewed earlier in the chapter) have implicated the ventral striatum–limbic system complex as playing an important role in the primitive reproductive behavior. A disturbance in sex hormones and certain excitatory neurotransmitters that normally influence the development of these structures may be ultimately expressed as TS (Kurlan, 1992). This hypothesis may explain the remarkable gender difference in TS, with males outnumbering females by 3 to 1; the exacerbation of symptoms at puberty and during the estrogenic phase of the menstrual cycle (Schwabe and Konkol, 1992); the characteristic occurrence of sexually related complex motor and phonic tics; and a variety of behavioral manifestations with sexual content. According to this hypothesis, the gene defect in TS results in an abnormal production of gonadal steroid hormones and increased trophic influence exerted by the excitatory amino acids, causing disordered development and increased innervation of the striatum and the limbic system. This is consistent with the finding of increased presynaptic dopamine uptake sites in brains of patients with TS (Singer et al., 1991).
Although there are no animal models of TS, studies of stereotypies in animals may provide insight into the pathogenesis of habits, rituals, tic-like, and impulsive behaviors in humans (Graybiel, 2008; Cools, 2008). These studies have shown that a broad spectrum of repetitive behaviors and rituals can become habitual and stereotyped through learning, partly as a result of experience-dependent neuroplasticity. Several families of horses with equine self-mutilation syndrome have been described with features that resemble human TS (Dodman et al., 1994). These horses exhibit a variety of stereotypic behaviors, such as glancing, biting at the flank or pectoral areas, bucking, kicking, rubbing, spinning, rolling, and vocalizing (squealing). Similar to TS, this condition is much more common in young males, is typically exacerbated by stress and during restful nonvigilance or boredom, and may be triggered by head trauma and relieved by castration. Genetic factors, however, seem to be most important.
Genetics
Finding a genetic marker and, ultimately, the gene has been the highest priority in TS research during the past decade (Keen-Kim and Freimer, 2006). Unfortunately, despite concentrated effort by many investigators, the TS gene has thus far eluded this intensive search. A systematic genome scan using 76 affected sib-pair families with a total of 110 sib-pairs showed two regions, 4q and 8p, with a LOD score of 2.38 and 2.09, respectively; four additional regions, on chromosomes 1, 10, 13, and 19, had a LOD score over 1.0 (Tourette Syndrome Association International Consortium on Genetics, 1999). An analysis of 91 Afrikaner nuclear families with one or more affected children provided evidence of linkage to loci on chromosomes 2p11, 8q22, and 11q23–q24, respectively (Simonic et al., 2001). This provides support for the finding in a previous study of a large French Canadian family of a LOD score of 3.24 for association with chromosome 11q23 (Merette et al., 2000). Other gene loci that are of interest in TS include 17q25 (Paschou et al., 2004) and 18q22 (Cuker et al., 2004). Assuming that genetic heterogeneity is not an important factor in TS, over 95% of the genome has been already excluded (Pakstis et al., 1991; Heutink et al., 1992). A linkage of TS with a known gene defect may help in the search for the TS gene. In this regard, a 16-year-old boy with typical TS was found to have a deletion of the terminal portion of the short arm of chromosome 9 (Taylor et al., 1991). A 7;18 translocation was identified in a patient with the sporadic form of TS, and the relevant regions in chromosome 18q22.3 and 7q22–q31 will be searched for possible markers (Boghosian-Sell et al., 1996; Patel, 1996). The identification of balanced reciprocal translocation near 18q22.1 in families with TS and a deletion in the same locus in one patient with tics and OCD (and dysmorphic facial features) led to the tentative assignment of the TS gene in this region (Alsobrook and Pauls, 1997). Two families with translocations involving the 8q13 have been identified (Crawford et al., 2003). Linkage disequilibrium has been demonstrated between a D4 receptor locus (on chromosome 11) and TS (Grice et al., 1996). It is possible that the clue to the TS gene(s) will come from genetic studies of ADHD. In this regard, it is of interest that a genome-wide scan in patients with ADHD identified five “hot spots”: 17p11 (MLS = 2.98) and four nominal regions with MLS values greater than 1.0, including 5p13, 6q14, 11q25, and 20q13 (Ogdie et al., 2003). On the basis of analysis of several genomic regions, 17q25 appears to be of special interest, having the highest LOD score of 2.61 (P = 0.002) (Paschou et al., 2004). Intronic variants in BTBD9, which has been associated with restless legs syndrome and periodic limb movements, have been also associated with TS (Rivière et al., 2009). A genome-wide screening of single nucleotide polymorphism (SNP) genotyping microarray identified five exon-affecting rare variants on 1q21.1, 2p16.3, and 10q21.3 loci, not found in 73 ethnically matched controls (Sundaram et al., 2010). Some of these copy number variants have been implicated previously by other studies in schizophrenia, autism, and ADHD. Although potentially important, this study has many limitations and the data have to be interpreted cautiously (Scharf and Mathews, 2010).
A major advance in the search for the elusive TS gene or genes has been made by the discovery of a frameshift mutation in the Slit and Trk-like 1 (SLITRK1) gene on chromosome 13q31.1 (Abelson et al., 2005). These variants were absent in 172 other TS and in 3600 control chromosomes. SLITRK1 gene mutation, however, is a rare cause of TS (Deng et al., 2006; Fabbrini et al., 2007; Scharf et al., 2008). One parent of a TS patient carried the SLITRK1 mutation but displayed only symptoms of trichotillomania. In a study of 44 families with one or more members who had trichotillomania, two mutations in the SLITRK1 gene were found among some individuals with trichotillomania but not in their unaffected family members (Zuchner et al., 2006). SLITRK1 mutations have been estimated to account for 5% of trichotillomania cases. The SLITRK1 gene has been found to be expressed in brain regions previously implicated in TS, such as the cortex, hippocampus, thalamic, subthalamic, and globus pallidus nuclei, striatum, and cerebellum and it appears to play a role in dendritic growth. Analysis of 988 parents of children with TS estimated the prevalence of SLITRK1 variants as 0.1% (Sharf, personal communication). Another gene mutation implicated in TS is the L-histidine decarboxylase gene (HDC) mutation W317X, located on 15q21.1–15q21.3, inherited in an autosomal dominant fashion in a two-generation family (Ercan-Sencicek et al., 2010). L-histidine decarboxylase is the rate-limiting enzyme that catalyzes the biosynthesis of histamine from histidine. Histaminergic neurons are located chiefly in the posterior hypothalamus but have widespread axonal connections. Although these findings suggest the possibility of using pharmacologic manipulation of histaminergic neurotransmission to treat TS, it is unknown how histamine abnormalities might cause or contribute to TS symptoms. Although potentially an important finding, it is unlikely that the W317X mutation is relevant to the pathogenesis of TS in most cases as the mutation was not found in 720 other patients with TS. Intriguingly, however, chromosomal inv(15) (q13;q22.3), which includes the 15q21.1–15q21.3 region, was identified in one of our patients, a 10-year-old boy with tics (Jankovic and Deng, 2007). Although mutations in HDC and SLITRK1 are only rarely associated with TS, careful studies of monogenic cases may provide insights into the pathogenesis of this complex neurobehavioral disorder. Another potential candidate gene mutation is Gln20X mutation in the 5-hydroxytryptamine receptor 2B gene (HTR2B, MIM 601122), reported to be associated with impulsivity in a Finnish population of violent criminals, but none of the 252 samples of patients with TS showed the Gln20X mutation (Guo et al., 2011).
Current concepts of the genetics of TS support a sex-influenced autosomal dominant mode of inheritance with a nearly complete penetrance for males and 56% penetrance for females when only tics are considered and 70% when OCD is included (Robertson, 1989; Pauls et al., 1991). Comings and Comings (1992), however, have proposed that TS is a semidominant, semirecessive disorder. This model takes into account the common observation that both parents of a TS child often exhibit TS or a forme fruste of TS and that the full and more severe motor-behavioral syndrome in the offspring represents a homozygous state (Fig. 16.4). Indeed, bilineal transmission was noted in 33% (considering tics) and 41% (considering tics or OCB) of TS families (Kurlan et al., 1994). In another study, McMahon and colleagues (1996) examined 175 members of a large, four-generation, TS family as well as 16 spouses who married into this family. Interestingly, they found evidence of TS in 36% of the family members and in 31% of the married-in spouses (some form of a tic was found in 67% and 44%, respectively). Multivariate analysis showed that tics were more severe in the offspring of both parents with tics. This study raises the possibility of assortative mating (like marrying like) in TS, in contrast to random, nonassortative mating, which is presumed in the general population. Thus, bilineal transmission may lead to frequent homozygosity and high density of TS in some families. Walkup and colleagues (1996) failed to find TS in both parents, but when the broader definition was allowed, 19% of families had both parents affected. Using rigorous diagnostic criteria, we found that 25% of our TS cases had both parents with some features of TS: tics, 8%; OCB, 4%; and ADD, 12% (Hanna et al., 1999) (Fig. 16.4). The TS patients were compared with a control population of 1142 students who were observed in second-, fifth-, and eighth-grade classrooms. In contrast to the 5% frequency of ADD in one parent of controls, the occurrence of tics was 31% in at least one parent of TS cases, 45% in at least one parent of ADD cases, and 41% in at least one parent of OCB cases. Among all the parents of TS cases, tics were present in 24%, OCB in 25%, and ADD in 34%, whereas only 3% of parents of controls exhibited ADD. Bilineal transmission violates the standard principle of one-trait-one-locus, and it might explain why a gene marker has not yet been identified in TS despite intense collaborative research effort. These results are similar to those of Lichter and colleagues (1999), who found bilineal transmission in 6% of patients; tics or OCB were represented bilineally in 22% of patients. In a large family study and segregation analysis, Walkup and colleagues (1996) provided evidence for a mixed model of inheritance rather than a simple autosomal mode of inheritance. This complex model of inheritance suggests that the majority of TS patients have two copies of the gene, one from each parent. This is consistent with the observation that many TS patients have both parents affected.
Based on various epidemiological-genetic studies, the rate of TS in the parents of a proband ranges from 6.5% to 16%, with fathers demonstrating a much higher risk (11.9–23.1%) compared to mothers (1–9.6%) (Pauls et al., 1991; Walkup et al., 1996). A Japanese population study, however, found the rate of TS for both parents was 1.9% and the rate of tics for both parents was approximately 11.5% (Kano et al., 2001). Siblings have approximately an 8% risk of TS and a 9.7–22% risk of OCD. The empiric risk to offspring of a proband with TS was reported as 22% for TS and greater than 50% for a tic disorder (McMahon et al., 2003). In bilineal families the risk of TS for offspring increased to 42.8% compared to 15% for unilineal families. Additionally, rates of any tic diagnosis, OCD, and ADHD were increased (McMahon et al., 2003).
Twin studies, showing 89% concordance for TS and 100% concordance for either TS or chronic multiple tic disorder, provide strong support for the genetic etiology of TS (Alsobrook and Pauls, 1997; Singer, 2000). How much influence environmental factors have on the phenotypic expression of this disorder is not known. In searching for nongenetic factors in the pathogenesis of TS, Leckman and colleagues (1990) found that maternal life stress, nausea, and vomiting during the first trimester of pregnancy were some of the perinatal factors that influenced the expression of the TS gene. In a study of 16 pairs of monozygotic twins, 94% of whom were concordant for tics, low birth weight was a strong predictor of tic severity, supporting a relationship between birth weight and phenotypic expression of TS (Hyde et al., 1992). Kurlan (1994) hypothesized that the clinical features associated with TS may occur to a variable degree in the course of normal childhood development and that genetic influences determine the severity of clinical symptoms in a given individual with TS. Eapen and colleagues (1997a) found evidence for earlier age at onset in maternally transmitted cases, suggesting genomic imprinting. The evidence for genomic imprinting in TS is, however, quite weak, and the genetic mechanism of TS remains elusive (Sadovnick and Kurlan, 1997). Because of phenotypic overlap between TS and Rett syndrome and myoclonus–dystonia, we have examined the genes for the latter disorders and found no mutations in our TS population (De Carvalho Aguiar et al., 2004).
One of the major concerns with the genetic linkage studies has been the lack of specificity of the current clinical criteria for diagnosis of TS. The boundaries of neurologic and behavioral manifestations of TS have not been clearly defined. While some investigators believe that the spectrum of behavioral expression of TS gene is quite broad, encompassing, in addition to ADHD and OCD, such diverse symptoms as conduct disorders, stuttering, dyslexia, panic attacks, phobias, depression, mania, and severe anxiety (Comings, 1987; Comings and Comings 1988a), others argue that more restrictive criteria for TS are needed, particularly for genetic linkage studies (Pauls et al., 1991). Pauls and colleagues (1991) studied 338 biological relatives of 86 TS probands and compared them to 21 biologically unrelated relatives of adopted TS probands. They confirmed their earlier observations and concluded that TS and OCD are etiologically related. The debate as to whether TS, OCD, and ADHD are genetically related or whether they merely represent comorbid neurobehavioral disorders will not be resolved until a TS-specific genetic marker is found.
Discovering the gene or genes for OCD would have important implications for TS and would help to clarify the relationship between the two disorders. One study showed that men with OCD are significantly more likely to have both alleles of the gene for catechol-O-methyltransferase in a low activity form (L/L) than in the L/H form (Karayiorgou et al., 1997). As was noted earlier, the B-lymphocyte antigen D8/17 has been suggested to be a possible peripheral marker for childhood-onset OCD and TS (Murphy et al., 1997).
Etiology of tics (secondary tourettism)
While TS is probably the most common cause of tics, there are many other neurologic disorders that are manifested by tics and other symptoms of TS, supporting the notion of a multifactorial genesis of TS (Table 16.2) (Jankovic, 2001a; Jankovic and Mejia, 2006). Of these, tics associated with static encephalopathy, autistic spectrum disorders, neuroacanthocytosis (Spitz et al., 1985; Saiki et al., 2004), Huntington disease (Jankovic and Ashizawa, 1995), pantothenate kinase-associated neurodegeneration (Pellecchia et al., 2005), fragile X syndrome (Schneider et al., 2008), beta-mannosidase deficiency (Sedel et al., 2006) and certain drugs such as dopamine receptor-blocking drugs, cocaine, and antiepileptics (Cardoso and Jankovic, 1993; Bharucha and Sethi, 1995; Kumar and Lang, 1997b; Lombroso, 1999) are particularly important to consider, especially in patients who do not fulfill the clinical criteria of TS. In one report, lamotrigine, a phenyltriazine drug that is structurally unrelated to other antiepileptic medications, was associated with dose-related motor and phonic tics in three children and features of OCD in one (Lombroso, 1999). The author speculated that in these rare cases, lamotrigine in high doses inhibited the presynaptic release of excitatory amino acids and resulted in an abnormal regulation of dopamine uptake in the striatum, leading to the production of tics and related TS symptoms. Rarely, “tourettism” can follow head trauma (Krauss and Jankovic, 1997; Kumar and Lang, 1997b); nonspecific cerebral insults, such as ischemia, hypoxia, and hypothermia (Singer et al., 1997); or striatal encephalitis due to varicella zoster (Dale et al., 2003). The study of the mechanisms of the secondary tourettism could provide insights into the pathogenesis of TS. For example, while TS symptoms have been reported to be exacerbated by various structural lesions involving the basal ganglia and the limbic system (Jankovic, 2001a), one patient with TS had complete resolution of tics after an MRI-proven midbrain lesion due to Wernicke encephalopathy (Pantoni et al., 1997). This suggests that an intact midbrain tegmentum is required for the expression of motor tics.
Autistic spectrum disorders (or pervasive developmental disorders) represent one of the largest groups of pediatric disorders in which features of TS, such as tics, ADHD, and OCD, may be present. In a study of 7288 participants enrolled in the Tourette Syndrome International Database Consortium Registry, 334 (4.6%; 1 of every 22 participants) had a comorbid pervasive developmental disorder, 13 times the expected rate (Burd et al., 2009). Autism is a behaviorally defined syndrome manifested by poor social interaction; disordered language; and atypical responses to people, objects, and events (Volkmar and Pauls, 2003). The syndrome is typically associated with severe disturbances in cognition, language, and behavior that appear before the age of 30 months. Stereotypic movements include body rocking, repeated touching and sniffing of objects, ritualistic ordering, and insistence on precisely following routines. Similar to TS, there is about 3 : 1 male : female preponderance. A study of children 3–10 years old found a 0.34% prevalence of autism with cognitive impairment in 68% (Yeargin-Allsopp et al., 2003). A review of home videos during early development in 17 autistic children found evidence of abnormal movement at the age of 4–6 months and sometimes even at birth, long before the diagnosis of autism (Teitelbaum et al., 1998). The authors suggest that their findings support the view that movement disturbances play an intrinsic part in the phenomenon of autism, that movement disturbances in autistic children are present at birth, and that such movement disturbances can be used to diagnose the presence of autism in the first few months of life. Other studies have suggested that TS symptoms are more common than expected in patients with autism, with an estimated frequency of 6.5% (Baron-Cohen et al., 1999). Asperger syndrome is a form of pervasive developmental disorder in which language and self-help skills are relatively intact. It is often considered a mild form of autism, but its relationship to other autistic disorders has not been well defined. Asperger syndrome patients frequently exhibit repetitive movements (stereotypies) and may have motor and phonic tics in addition to other behavioral abnormalities (Ringman and Jankovic, 2000). Other childhood behavioral disorders that may overlap in symptomatology with autism and TS include Williams syndrome, characterized by remarkable conversational skills and excessive empathy; Prader–Willi syndrome, associated with temper tantrums and obsessive-compulsive behavior; Angelman syndrome, characterized by hyperactivity and a constantly happy disposition (Cassidy and Morris, 2002); and Rett syndrome, an X-linked autistic disorder associated with stereotypies and other movement disorders. However, we excluded mutations in MECP2 gene on chromosome X28, responsible for Rett syndrome (Rosa et al., 2003). We also excluded mutations in the SGCE gene, responsible for the dystonia–myoclonus syndrome, which shares some clinical features with TS, in a population of well-defined TS (De Carvalho Aguiar et al., 2004).
Other related conditions that may have resemblance to tics and TS are the jumping Frenchmen of Maine, “ragin’ Cajuns” of Louisiana, latah of the Malays, and myriachit of Siberia (Lees, 2001). First described in 1878, these culture-bound conditions are characterized by an excessive startle response, sometimes with echolalia, echopraxia, or forced obedience. It is considered by some as an operant conditioned behavior rather than a neurologic or even hysteric disorder (Saint-Hilaire et al., 1986). The rare late-onset startle-induced tics may resemble these culture-bound syndromes and other startle responses (Tijssen et al., 1999). Rarely, psychogenic tics are present in patients with TS, but this very rare occurrence is difficult to confirm clinically (Kurlan et al., 1992).
The discussion of the numerous causes of tics and other manifestations of TS is beyond the scope of this review, and the reader is referred to other reviews of secondary causes of tics (Kumar and Lang, 1997b; Jankovic 2001b; Jankovic and Mejia, 2006).
Epidemiology
Discovery of a disease-specific marker would be helpful not only in improving our understanding of this complex neurobehavioral disorder but also in clarifying the epidemiology of TS (Kadesjo and Gillberg, 2000; Scahill et al., 2001). Up to 24% of children may have tics sometime during their childhood (Robertson, 2008a, 2008b). The prevalence rates have varied markedly and have been estimated to be as high as 4.2% when all types of tic disorders are included (Costello et al., 1996) and 1/83 (1.2%) (Comings and Comings, 1988b) or as low as 28.7/100 000 (0.03%) (Caine et al., 1988) and 1/10 000 (0.01%) (Singer, 2000). There are many reasons for this wide variation, the most important of which are different ascertainment methods, different study populations, and different clinical criteria. Since about one-third of patients with tics do not even recognize their presence, it is difficult to derive more accurate prevalence figures for TS without a well-designed door-to-door survey (Kurlan et al., 1987). In one study, 3034 students in three schools in Los Angeles were monitored over a 2-year period by a school psychologist; the frequency of definite TS was 1 in 95 males and 1 in 759 females (Comings et al., 1990). Because the observed population included special education children, the authors adjusted the final prevalence figure to 0.63%. This figure is similar to that derived from our own observational study of 1142 children in second, fifth, and eighth grades of a general school population, among whom 8 children (0.7%) had some evidence of TS (Hanna et al., 1999). In another school-based study involving 167 randomly selected 13- and 14-year-olds in English high schools, the prevalence of TS based on DSM III-R was estimated at 3%, but 18% screened positive for tics (Mason et al., 1998). A follow-up study with improved methods found a TS prevalence of 0.76–1.85% (Hornsey et al., 2001). Snider and colleagues (2002) observed 553 schoolchildren (kindergarten to sixth grade) monthly over an 8-month period and found tics in 135 (24%); 34 children had persistent tics (6.1%). Those with persistent tics were more likely to be males and had more behavioral problems than those without persistent tics. Kurlan and colleagues (2001) found that 27% of 341 special education students had tics compared to 19.7% of 1255 students in regular classroom programs; the frequency of TS was 7% and 3.8%, respectively. In another study, based on an interview of 1596 children, Kurlan and colleagues (2002) found that 339 (21%) had tics. Children with tics were younger, were more likely to be male and attend special education classes, and had a lower mean IQ. The prevalence of tics was studied in 867 children attending two schools in the Basque country and was estimated to be 6.5% (Linazasoro et al., 2006). Thus, epidemiologic studies have shown that 20–30% of children exhibit tics sometime during childhood and 2–3% of children develop some features of TS, although the worldwide prevalence of TS in children has been reported to range from 0.3% to 0.8% (Scahill et al., 2009).
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 16 Tics and Tourette syndrome
Introduction
Tourette syndrome (TS), which should be more appropriately called Gilles de la Tourette syndrome, is a neurologic disorder manifested by motor and vocal or phonic tics usually starting during childhood and often accompanied by obsessive-compulsive disorder (OCD), attention-deficit hyperactivity disorder (ADHD), poor impulse control, and other comorbid behavioral problems (Shapiro and Shapiro, 1992; Cohen and Leckman, 1994; Hyde and Weinberger, 1995; Feigin and Clarke, 1998; Leckman and Cohen, 1999; Freeman et al., 2000; Robertson, 2000; Jankovic, 2001a; Leckman et al., 2001; Leckman, 2002; Stein, 2002; Singer, 2005; Albin and Mink, 2006; Leckman et al., 2006; Bloch et al., 2011; Jankovic and Kurlan, 2011). Once considered a rare psychiatric curiosity, TS is now recognized as a relatively common and complex neurobehavioral disorder (Table 16.1). There has been speculation that many notable historical figures, including Dr Samuel Johnson and possibly Wolfgang Amadeus Mozart (Simkin, 1992; Ashoori and Jankovic, 2007), were afflicted with TS.
Table 16.1 Summary of epidemiologic studies in Tourette syndrome (TS)
|
• 6.1% (135/553 children, K–6th grade) had persistent tics (TS); 24% observed to have motor tics during at least one month of the 8-month study
|
One of the earliest reports of TS dates to 1825, when Itard (1825) described a French noblewoman with body tics, barking sounds, and uncontrollable utterances of obscenities. Sixty years later, the French neurologist and a student of Charcot, Georges Gilles de la Tourette (1885) reviewed Itard’s original case and added eight more patients. He noted that all nine patients shared one feature: they all exhibited brief involuntary movements or tics; additionally, six made noises, five shouted obscenities (coprolalia), five repeated the words of others (echolalia), and two mimicked others’ gestures (echopraxia) (Goetz and Klawans, 1982; Kushner, 1999). Although Tourette considered the disorder he described to be hereditary, the etiology was ascribed to psychogenic causes for nearly a century following the original report. The perception of TS began to change in the 1960s, when the beneficial effects of neuroleptic drugs on the symptoms of TS began to be recognized (Shapiro and Shapiro, 1968). This observation helped to refocus attention from psychogenic etiology to central nervous system (CNS) etiology. Despite these advances TS is still often misunderstood and wrongly labeled as a mental or psychiatric disorder. For example, insurance compensation for patients with TS is often compromised because the TS-related diagnostic codes (307.22 = Tic Disorder, 307.23 = Tourette Syndrome) are currently included in the ICD-9 series for mental disorders (290–319). Therefore, the code 333.3 = Tics of Organic Origin or 349.9 = Unspecified Nervous System Disorder may result in more appropriate compensation. If payment is denied, an appeal letter to the insurance company specifying that TS is a neurologic disorder along with some literature and links to a credible website may be required.
The cause of TS is yet unknown, but the disorder appears to be inherited in the majority of patients (Pauls et al., 1988, 1991; Tolosa and Jankovic, 1998; Jankovic, 2001a; Leckman, 2002; Paschou et al., 2004; Singer, 2005). The clinical expression of this genetic defect varies from one individual to another, fluctuations in symptoms are seen within the same individual, and different manifestations occur in various family members (Kurlan, 1994). This variable expression from one individual to another, even within members of the same family, contributes to diagnostic confusion. Without a specific biologic marker, the diagnosis depends on a careful evaluation of the patient’s symptoms and signs by an experienced clinician. Educational efforts directed to physicians, educators, and to the general public have increased awareness about TS. In addition, the media have drawn increasing public attention to this condition. As a result of this improved awareness, the self-referral rate of patients has increased, and the correct diagnosis is made earlier than was the case in the past. Many patients, however, still remain undiagnosed, or their symptoms are wrongly attributed to habits, allergies, asthma, dermatitis, hyperactivity, nervousness, and many other conditions (Jankovic et al., 1998; Hogan and Wilson, 1999).
Phenomenology of tics
Tics, the clinical hallmark of TS, are relatively brief and intermittent movements (motor tics) or sounds (vocal or phonic tics). Recognition of the full spectrum of phenomenology of tics is critical to the diagnosis of TS (Jankovic, 2008). Motor tics typically consist of sudden, abrupt, transient, often repetitive and coordinated (stereotypical) movements that may resemble gestures and mimic fragments of normal behavior, vary in intensity, and are repeated at irregular intervals (Videos 16.1 to 16.8). Currently accepted criteria for the diagnosis of TS require both types of tics to be present (Robertson, 1989; Golden, 1990; Tourette Syndrome Classification Study Group, 1993; Jankovic, 1997; Singer, 2000). This division into motor and vocal/phonic tics, however, is artificial, because vocal/phonic tics are actually motor tics that involve respiratory, laryngeal, pharyngeal, oral, and nasal musculature. Contractions of these muscles may produce sounds by moving air through the nose, mouth, or throat. The term phonic tic is preferable, since not all sounds produced by TS patients involve the vocal cords.
To better understand the categorization of tics and how they fit in the general schema of movement disorders, it might be helpful to provide a simple classification of movements (Jankovic, 1992). All movements can be categorized into one of four classes:
Recent studies have shown that automatic, learned behaviors appear to be encoded in the sensorimotor portion of the striatum (Jog et al., 1999). Some support for the proposed classification is provided by the findings of Papa and colleagues (1991). They recorded normal premovement (readiness) electroencephalographic, slow, negative potential (the Bereitschaftspotential) 1–1.5 seconds prior to self-induced, internally generated (voluntary) movement in normal individuals but not before externally triggered movement induced by electrical stimulation (Colebatch, 2007). Most tics can be categorized as either semivoluntary (unvoluntary) or involuntary (suppressible). In some cases, learned voluntary motor skills are incorporated into the tic repertoire. This is exemplified by a case of a woman with TS who incorporated sign language into her tic behavior, suggesting that semantics is more important than phonology in the generation of tics (Lang et al., 1993).
Tics may be simple or complex. Simple motor tics involve only one group of muscles, causing a brief, jerk-like movement. They are usually abrupt in onset and rapid (clonic tics), but they may be slower, causing a briefly sustained abnormal posture (dystonic tics) or an isometric contraction (tonic tics) (Jankovic and Fahn, 1986; Jankovic, 1992). Examples of simple clonic motor tics include blinking, nose twitching, and head jerking. Rhythmical clonic tics may rarely resemble tremor or rhythmical myoclonus, such as palatal myoclonus (Schwingenschuh et al., 2007; Adam et al., 2009). Simple dystonic tics include blepharospasm, oculogyric movements, bruxism, sustained mouth opening, torticollis, and shoulder rotation (Videos 16.3 and 16.9). Tensing of abdominal or limb muscles is an example of a tonic tic (Video 16.10). To characterize clonic and dystonic tics further, 156 patients with TS were studied; 89 (57%) exhibited dystonic tics, including oculogyric deviations (28%), blepharospasm (15%), and dystonic neck movements (7%) (Jankovic and Stone, 1991). Since patients with dystonic tics did not differ significantly on any clinical variables from those with only clonic tics, we concluded that despite previous reports (Feinberg et al., 1986), the presence of dystonic tics should not be considered atypical or unusual. In fact, on subsequent observation, the patient in Feinberg and colleagues’ case report was shown to be a case of TS with typical dystonic tics (Fahn, 1987). Dystonic tics should be distinguished from persistent dystonia, which is typically seen in patients with primary dystonia (Jankovic and Fahn, 2002). Dystonic (and tonic) muscle contraction might be responsible for so-called blocking tics (Video 16.11). These blocking tics are due to either prolonged tonic or dystonic tics that interrupt ongoing motor activity such as speech (intrusions) or a sudden inhibition of ongoing motor activity (negative tic). Clonic and dystonic tics may occasionally occur in patients with primary dystonia more frequently than in the general population. In nine patients with coexistent TS and persistent primary dystonia, the onset of tics was at a mean age of 9 years, while dystonia followed the onset of tics by a mean of 22 (10–38) years (Stone and Jankovic, 1991). Other reports have drawn attention to the possible association of tics and dystonia, although the two disorders may coexist by chance alone (Pringsheim et al., 2007). The occasional co-occurrence of tics and dystonia in the same family, however, provides additional evidence for a possible etiologic relationship between the two disorders (Németh et al., 1999; Yaltho et al., 2010). Dopa-responsive dystonia with mutations in the GCH1 gene and TS was found in various members of a large Danish family (Romstad et al., 2003).
Motor (particularly dystonic) and phonic tics are preceded by premonitory sensations in over 80% of patients (Cohen and Leckman, 1992; Banaschewski et al., 2003; Kwak et al., 2003a; Woods et al., 2005; Prado et al., 2008). This premonitory phenomenon consists of localizable sensations or discomforts, such as a burning feeling in the eye before an eye blink, tension or a crick in the neck that is relieved by stretching of the neck or jerking of the head, a feeling of tightness or constriction that is relieved by arm or leg extension, nasal stuffiness before a sniff, a dry or sore throat before throat clearing or grunting, and itching before a rotatory movement of the scapula. Rarely, these premonitory sensations, termed in one report extracorporeal phantom tics, involve sensations in other people and objects and are temporarily relieved by touching or scratching them (Karp and Hallett, 1996). In one study, premonitory sensations were experienced by 92% of 135 patients with TS, and these were localized chiefly to the shoulder girdle, palms, midline abdominal region, posterior thighs, feet, and eyes (Cohen and Leckman, 1992). We administered a questionnaire regarding various aspects of premonitory sensations associated with their motor tics to 50 TS patients with a mean age of 23.6 ± 16.7 years (Kwak et al., 2003a). Forty-six of 50 (92%) subjects reported some premonitory sensations, the most common of which was an urge to move and an impulse to tic (“had to do it”). Other premonitory sensations included an itch, tingling/burning, numbness, and coldness. Thirty-seven (74%) also reported intensification of premonitory sensations if the patient was prevented from performing a motor tic; 36 (72%) reported relief of premonitory sensations after performing the tic; and 24 (48%) stated that their motor tic would not have occurred if they had had no premonitory sensation. Twenty-seven of 40 patients (68%) described a motor tic as a voluntary motor response to an involuntary sensation rather than as a completely involuntary movement. Besides the local or regional premonitory sensations, this premonitory phenomenon may be a nonlocalizable, less specific, and poorly described feeling, such as an urge, anxiety, anger, and other psychic sensations. The observed movement or sound sometimes occurs in response to these premonitory phenomena, and these movements or sounds have been previously referred to as sensory tics (Kurlan et al., 1989; Chee and Sachdev, 1997). In a study of 60 patients with tic disorders, 41 (68%) thought that all their tics were intentionally produced, and 15 (25%) additional patients had both voluntary and involuntary movements; thus, 93% of the tics were perceived to be “irresistibly but purposefully executed” (Lang, 1991). This “intentional” component of the movement may be a useful feature in differentiating tics from other hyperkinetic movement disorders, such as myoclonus and chorea. The sensations or feelings that often precede motor tics usually occur out of a background of relative normalcy and are clearly involuntary, even though the movements (motor tics) or noises (phonic tics) that occur in response to these premonitory symptoms may be regarded as semivoluntary or unvoluntary. Chee and Sachdev (1997) suggest that sensory tics, which we and others refer to as premonitory sensations, “represent the subjectively experienced component of neural dysfunction below the threshold for motor and phonic tic production.” Many patients report that they have to repeat a particular movement to relieve the uncomfortable urge until “it feels good.” The “just right” feeling has been associated with compulsive behavior, and as such, the “unvoluntary” movement may be regarded as a compulsive tic (Leckman et al., 1994). While many patients describe a gradually increasing inner tension as tic suppression is maintained and a rebound effect of a flurry of tics when the tics are finally expressed, formal objective studies of tic frequency during and after voluntary suppression have failed to detect a rebound increase (Meidinger et al 2005; Himle and Woods 2005) and, when reinforced, tic suppression may last at least 40 consecutive minutes (Woods et al., 2008).
Complex motor tics consist of coordinated, sequenced movements resembling normal motor acts or gestures that are inappropriately intense and timed (Videos 16.6, 16.12, and 16.13). They may be seemingly nonpurposeful, such as head shaking or trunk bending, or they may seem purposeful, such as touching, throwing, hitting, jumping, and kicking. Additional examples of complex motor tics include gesturing “the finger” and grabbing or exposing one’s genitalia (copropraxia) or imitating gestures (echopraxia). Burping, vomiting, and retching have been described as part of the clinical picture of TS, but it is not clear whether this phenomenon represents a complex tic or some other behavioral manifestation of TS (Rickards and Robertson, 1997). Air swallowing is another unusual tic described in TS (Weil et al., 2008). Another unusual tic is ear dyskinesia, which consists of anterior-posterior displacement of the external ear (Cardoso and Faleiro, 1999). Complex motor tics may be difficult to differentiate from compulsions, which frequently accompany tics, particularly in TS. A complex, repetitive movement may be considered a compulsion when it is preceded by, or associated with, a feeling of anxiety or panic, as well as an irresistible urge to perform the movement or sound because of fear that if it is not promptly or properly executed, “something bad” will happen. However, this distinction is not always possible, particularly when the patient is unable to verbalize such feelings. Some coordinated movements resemble complex motor tics but may actually represent pseudovoluntary movements (parakinesias) that are designed to camouflage the tics by incorporating them into seemingly purposeful acts, such as adjusting one’s hair during a head jerk.
In contrast to other hyperkinetic movement disorders, tics are usually intermittent and may be repetitive and stereotypic (Table 16.2). Tics may occur as short-term bouts or bursting or long-term waxing and waning (Peterson and Leckman, 1998). They vary in frequency and intensity and often change distribution. Typically, tics can be volitionally suppressed, although this might require intense mental effort (Banaschewski et al., 2003). Suppressibility, although characteristic and common in tics, is not unique or specific for tics, and this phenomenon has been well documented in other hyperkinetic movement disorders (Walters et al., 1990). Using functional magnetic resonance imaging (MRI), Peterson and colleagues (1998a) showed decreased neuronal activity during periods of suppression in the ventral globus pallidus, putamen, and thalamus. There was increased activity in the right caudate nucleus, right frontal cortex, and other cortical areas that are normally involved in the inhibition of unwanted impulses (prefrontal, parietal, temporal, and cingulate cortices). Using event-related functional magnetic resonance imaging in 26 children with bipolar disorder, deficits in the ability to engage striatal structures and the right ventral prefrontal cortex were found during unsuccessful inhibition, thus suggesting that deficits in motor inhibition contribute to impulsivity and irritability in children with bipolar disorder and possibly also with TS (Leibenluft et al., 2007).
| A. Primary |
Besides temporary suppressibility, tics are characterized by suggestibility and exacerbation with stress, excitement, boredom, fatigue, and exposure to heat (Lombroso et al., 1991). Emotional stress associated with life events or other stresses have been documented to potentially markedly exacerbate tics, but onset of TS is not necessarily related to stressful life events (Wood et al., 2003; Hoekstra et al., 2004; Horesh et al., 2008). Tics may also increase during relaxation after a period of stress.
In contrast to other hyperkinetic movement disorders that are usually completely suppressed during sleep, motor and phonic tics may persist during all stages of sleep (Cohrs et al., 2001; Jankovic et al., 1984; Silvestri et al., 1990; Fish et al., 1991; Hanna and Jankovic, 2003; Rothenberger et al., 2001). In addition, patients with TS often have disturbances of sleep, such as increased sleep fragmentation, higher frequency of arousals, decreased rapid eye movement (REM) sleep, and enuresis (Hanna and Jankovic, 2003). Many patients note a reduction in their tics when they are distracted while concentrating on mental or physical tasks (such as when playing a video game or during an orgasm). Other patients experience increased frequency and intensity of their tics when distracted, especially when they no longer have the need to suppress the tics. Tics are also typically exacerbated by dopaminergic drugs and by CNS stimulants, including methylphenidate and cocaine (Cardoso and Jankovic, 1993). Finally, it should be noted that there is a broad spectrum of movements that may be present in patients with TS that can be confused with tics, such as akathisia, chorea, dystonia, compulsive movements, and fidgeting as part of hyperactivity associated with ADHD (Jankovic, 1997; Kompoliti and Goetz, 1998; Wilens et al., 2004).
Clinical features of Tourette syndrome
Motor symptoms
TS, the most common cause of tics, is manifested by a broad spectrum of motor and behavioral disturbances (Table 16.3). This clinical heterogeneity often causes diagnostic difficulties and presents a major challenge in genetic linkage studies. To aid in the diagnosis of TS, the Tourette Syndrome Classification Study Group (1993) formulated the following criteria for definite TS: (1) Both multiple motor and one or more phonic tics have to be present at some time during the illness, although not necessarily concurrently. (2) Tics must occur many times a day, nearly every day, or intermittently throughout a period of more than 1 year. (3) The anatomic location, number, frequency, type, complexity, or severity of tics must change over time. (4) Onset must be before age 21. (5) Involuntary movements and noises cannot be explained by other medical conditions. (6) Motor and/or phonic tics must be directly witnessed by a reliable examiner at some point during the illness or must be recorded by videotape or cinematography. Probable TS type 1 meets all the criteria except for number 3 and/or number 4, and probable TS type 2 meets all the criteria except for number 1; it includes either a single motor tic with phonic tics or multiple motor tics with possible phonic tics. In contrast to the criteria outlined by the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) (1994), the Tourette Syndrome Classification Study Group criteria do not include a statement about “impairment.” There is considerable controversy about the DSM-IV criteria, which require that “marked distress or significant impairment in social, occupational or other important areas of functioning” be present. Therefore, patients with mild tics that do not produce an impairment would not satisfy the diagnostic criteria for TS, according to DMS-IV. This criterion will be deleted from the fifth edition of the DSM. Kurlan (1997) suggested another set of diagnostic criteria for genetic studies and introduced the term Tourette disorder for patients who have “functional impairment.” This, however, does not take into account the marked fluctuation in symptoms and severity; some patients may be relatively asymptomatic at one time and clearly functionally impaired at another time. The TSA International Genetic Collaboration developed the Diagnostic Confidence Index, which consists of 26 confidence factors, with weightings given to each of them and a total maximum score of 100. The most highly weighted diagnostic confidence factors include history of coprolalia, complex motor or vocal tics, a waxing and waning course, echophenomenon, premonitory sensations, an orchestrated sequence, and age at onset. The Diagnostic Confidence Index was found to be a useful instrument in assessing the lifetime likelihood of TS (Robertson et al., 1999). Several instruments, some based on ratings of videotapes, have been developed to measure and quantitate tics, but they all have some limitations (Goetz and Kompoliti, 2001; Goetz et al., 2001). The most widely used instrument to assess tics is the Yale Global Tic Severity Scale (YGTSS), which consists of two broad domains: Total Tic Severity (with two sub-domains: Motor and Phonic Tics) and Impairment. Within each category there are five dimensions, score 0–5: number of tics, frequency, intensity, complexity, and interference. The total tic score ranges from 0 to 50; the usual ranges in most studies is 15 to 30. A health-related quality of life scale (HR-QOL) has been developed and validated for internal consistency, test–retest reliability and against other clinical scales (Cavanna et al., 2008). Utilization of the HR-QOL scale showed that comorbidities such as ADHD and OCD rather than tic severity are more predictive of the long-term outcome.
Table 16.3 Demographic and clinical features in a large database of patients with TS (Tourette International Consortium) (Freeman et al., 2009)
| Number (80 sites; 60% North America; 66% Psychiatry: 27% Neurology) | 6805 |
| Male : Female ratio | 4.4 : 1 |
| Mean age at onset of tics | 6.4 years |
| Mean age at diagnosis of TS | 13.2 years |
| Delay in diagnosis | 6.4 years |
| Family history | 51.7% |
| TS only, no comorbidity | 14.2% |
| Attention-deficit hyperactivity disorder | 55.6% |
| Obsessive-compulsive disorder/behavior | 54.9% |
| Conduct/oppositional defiant disorder | 12.3% |
| Anger control problems | 27.6% |
| Learning disability | 22.0% |
| Mood disorder | 16.9% |
| Anxiety disorder | 16.8% |
| Pervasive developmental disorder | 4.6% |
The clinical criteria are designed to assist in accurate diagnosis, in genetic linkage studies and in differentiating TS from other tic disorders (Jankovic, 1993b) (Table 16.2). There is a body of evidence to support the notion that many, if not all, patients with other forms of idiopathic tic disorders represent one end of the spectrum in a continuum of TS (Kurlan et al., 1988). The most common and mildest of the idiopathic tic disorders is the transient tic disorder (TTD) of childhood. This disorder is essentially identical to TS except the symptoms last less than 1 year and therefore the diagnosis can be made only in retrospect. Transient tic disorder has been estimated to occur in up to 24% of schoolchildren (Shapiro et al., 1988). Chronic multiple tic disorder is also similar to TS, but the patients have either only motor or, less commonly, only phonic tics lasting at least 1 year. Chronic single tic disorder is the same as chronic multiple tic disorder, but the patients have only a single motor or phonic tic. This separation into transient tic disorder, chronic multiple tic disorder, and chronic single tic disorder seems artificial because all can occur in the same family and probably represent a variable expression of the same genetic defect (Kurlan et al., 1988).
Although the diagnostic criteria require that the onset is present before the age of 21, in 96% of patients the disorder is manifested by age 11 (Robertson, 1989). In 36–48% of patients, the initial symptom is eye blinking, followed by tics involving the face and head. Blink rate in TS is about double of that of normal, age-matched controls (Tulen et al., 1999). During the course of the disease, nearly all patients exhibit tics involving the face or head, two-thirds have tics in the arms, and half have tics involving the trunk or legs. According to one study, the average age at onset of tics is 5.6 years, and the tics usually become most severe at age 10; by 18 years of age, half of the patients are tic-free (Leckman et al., 1998). In a study of 58 adults who had been diagnosed with TS during childhood, Goetz and colleagues (1992) found that tics persisted in all patients but were moderate or severe in only 24%, although 60% had moderate or severe tics during the course of the disease. Tic severity during childhood had no predictive value for the future course, but patients with mild tics during the preadult period had mild tics during adulthood. In a longitudinal study that involved structured interviews of 976 children aged 1–10 years, 776 of whom were reassessed 8, 10, and 15 years later, tics and ADHD symptoms were associated with OCD symptoms in late adolescence and early adulthood (Peterson et al., 2001). Furthermore, ADHD was associated with lower IQ and lower social status, whereas OCD was associated with higher IQ. These findings are similar to those of another study designed to address the long-term prognosis of children with TS as they reach adulthood (Bloch et al., 2006). In this study 46 children with TS underwent a structured interview at a mean age of 11.4 years, and again at 19.0 years. The mean worst-ever tic severity score was 31.6 out of a possible 50 on the YGTSS, and occurred at a mean age of 10.6 years. By the time of the second interview, mean YGTSS score decreased to 10. This first prospective longitudinal study also showed that only 22% continued to experience mild or greater tic symptoms (YGTSS scores, ≥10) at follow-up, while nearly one-third were in complete remission of tic symptoms at follow-up. In contrast to the study by Goetz et al. (1992), the severity of childhood tics was predictive of increased tic severity at follow-up. The peak OCD severity occurred 2 years after peak tic severity. Interestingly, a 10-point increase in baseline IQ increased the risk of OCD symptoms at follow-up by 2.8-fold. The authors point out that the later average onset of OCD symptoms indicates the importance of counseling parents about the possibility of OCD development in children recently diagnosed with tics. Although the long-term prognosis for TS is generally favorable for most patients, a minority of cases may have persistent, severe tic symptoms which may be resistant to medications (Eapen et al., 2002). In another study, the investigators reviewed videotapes of 31 patients, with an average age of 24.2 ± 3.5 years, approximately 12 years after their initial video and found that 90% of the adults still had tics, even though they often considered themselves tic-free (Pappert et al., 2003). There was, however, a significant improvement in tic disability and tic severity. When 40 children and 31 adults with TS were compared no difference in tic phenomenology or severity was found, but children were more frequently managed without medications, and sedation was more common in adults but weight gain was more common in children (Cubo et al., 2008).
Although the vast majority of tics in adults represent recurrences of childhood-onset tics, rare patients may have their first tic occurrence during adulthood (Chouinard and Ford, 2000). In adults with new-onset tics, it is important to search for secondary causes, such as infection, trauma, stroke (Kwak and Jankovic, 2002; Gomis et al., 2008), multiple sclerosis (Nociti et al., 2009), cocaine use, neuroleptic exposure, and peripheral injury (Video 16.14) (Chouinard and Ford, 2000; Jankovic, 2001b; Jankovic and Mejia, 2006). One study of eight patients with adult-onset tics (three of whom had childhood-onset OCD and three of whom had a family history of tics and OCD) found that in comparison to the patients with more typical childhood-onset tics, the former group had more severe symptoms, greater social morbidity, and less favorable response to medications (Eapen et al., 2002). Poor motor control, which can lead to poor penmanship and, at times, almost illegible handwriting, can contribute to the academic difficulties faced by many patients with TS.
Tics, although rarely disabling, can be quite troublesome for TS patients because they cause embarrassment, interfere with social interactions, and at times can be quite painful or uncomfortable. Rarely, cervical tics may be so forceful and violent, the so-called “whiplash tics,” that they may cause secondary neurologic deficits, such as cervical artery dissection (Norris et al., 2000), and noncompressive (Isaacs et al., 2010) or compressive cervical myelopathy (Krauss and Jankovic, 1996) (Video 16.2). The truncal bending tics, which resemble intermittent, repetitive camptocormia, may cause secondary degenerative changes in the thoracic spine (Azher and Jankovic, 2005) (Video 16.5). These disabling tics and other severe symptoms of TS draw attention to the subset of patients with TS so severe that they may be life-threatening, hence labeled as “malignant” TS (Table 16.4). Of 332 TS patients evaluated at Baylor College of Medicine Movement Disorders Clinic during a 3-year period, 17 (5.1%) met criteria for malignant TS, defined as ≥2 emergency room (ER) visits or ≥1 hospitalizations for TS symptoms or its associated behavioral comorbidities (Cheung et al., 2007). The patients exhibited tic-related injuries, self-injurious behavior (SIB), uncontrollable violence and temper, and suicidal ideation/attempts. Compared to patients with nonmalignant TS, those with malignant TS were significantly more likely to have a personal history of obsessive-compulsive behavior/disorder (OCB/OCD), complex phonic tics, coprolalia, copropraxia, SIB, mood disorder, suicidal ideation, and poor response to medications. Severe or malignant TS, associated with SIB and other disabling features, has been also reported in families, including consanguineous kindreds (Motlagh et al., 2008).
Table 16.4 Clinical features of severe (“malignant”) TS
Vocalizations have been reported as the initial symptom in 12–37% of patients, throat clearing being the most common (Robertson, 1989). Phonic tics can be quite troublesome for patients and those around them. In addition to involuntary noises, some patients have speech dysfluencies that resemble developmental stuttering, and up to half of all patients with developmental stuttering have been thought to have undiagnosed TS (Abwender et al., 1998). Coprolalia, perhaps the most recognizable and certainly one of the most distressing symptoms of TS, is actually present in only half of patients (Videos 16.8 and 16.15). When describing the distress caused by his severe coprolalia, one of our patients remarked that immediately after shouting an obscenity, he reaches out with his hand in an attempt to “catch the word and bring it back before others can hear it.” Coprolalia appears to be markedly influenced by cultural background. Although in one retrospective analysis of 112 children with TS, only 8% exhibited coprolalia (Goldenberg et al., 1994), the true prevalence of coprolalia in TS children and adults is about 50% in the US population, when mental coprolalia (without actual utterance) is included. In a study of 597 individuals with TS from seven countries, coprolalia occurred at some point in the course of the disease in 19.3% of males and 14.6% of females, and copropraxia in 5.9% of males and 4.9% of females (Freeman et al., 2009). Coprolalia has been reported to occur in only 26% of Danish patients and 4% of Japanese patients (Robertson, 1989). Copropraxia has been found in about 20% of patients, echolalia in 30%, echopraxia in 25%, and palilalia in 15%. Although coprolalia is a characteristic feature of TS and, based on functional MRI studies, attributed to abnormal activation particularly in the left middle frontal gyrus and right precentral gyrus, and possibly the caudate nucleus, cingulate gyrus, cuneus, left angular gyrus, left inferior parietal gyrus, and occipital gyri (Gates et al., 2004), this language abnormality is not universally present or specific for TS.
Except for tics, the neurologic examination in patients with TS is usually normal. In one case-control study, TS patients were found to have a shorter duration of saccades, but the saccades were performed with a greater mean velocity than in normal controls, and the TS patients had fewer correct antisaccade responses, suggesting a mild oculomotor disturbance in TS (Farber et al., 1999). Although the ability to inhibit reflexive saccades is normal, TS patients make more timing errors, indicating an inability to appropriately inhibit or delay planned motor programs (LeVasseur et al., 2001).
Behavioral symptoms
Patients with TS generally have normal intelligence and may even perform better and faster than age-matched controls on certain tasks that require grammar skills that depend on procedural, rather than declarative memory (Walenski et al., 2007). While TS patients have no cognitive deficits, they often exhibit a variety of behavioral symptoms, particularly ADHD and OCD (Figs 16.1 and 16.2) (Gaze et al., 2006). In the Tourette International Consortium (TIC) database, which includes information on 3500 patients with TS evaluated by neurologists or psychiatrists, only 12% had tics only, without other comorbidities (Freeman et al., 2000). Kurlan and colleagues (2002) interviewed 1596 children, aged 9–17, in schools in Rochester and Monroe Counties, New York, and identified tics in 339 children (21%) after 60–150 minutes of observation. They found the following behavioral problems more frequently (P < 0.05) in children with tics than in those without tics: OCD, ADHD, separation anxiety, overanxious disorder, simple phobia, social phobia, agoraphobia, mania, major depression, and oppositional defiant behavior. Also, children with tics were younger (mean age: 12.5 vs. 13.3 years) and were more likely to require special education services (27% vs. 19.8%).
Figure 16.1 An overlap of disorders typically coexisting in patients with TS.
Redrawn from Jankovic J: Tourette’s syndrome. N Engl J Med 2001;345:1184–1192.
[/not-level-membership-for-neurology-category]
