Thyroid Hormone Binding and Variants of Transport Proteins
Characteristics of Hormone Binding
Structure and Binding Characteristics of Normal and Variant Transport Proteins
Function of Iodothyronine-Binding Proteins
Acquired Alterations in the Concentration of Transport Proteins
Competition for Thyroid Hormone Transport Sites
More than 60 years ago, it was shown that circulating thyroid hormones are noncovalently bound to plasma proteins.1 Well over 99% of circulating thyroxine (T4) and triiodothyronine (T3) is protein bound in the circulation, with bound and free moieties in constant rapid equilibrium. An understanding of thyroid hormone binding allows the clinician to better appreciate tissue delivery and interconversion of thyroid hormones, especially when these phenomena change as a result of illness or drug therapy, or when treatment is given to alter thyroid status. This knowledge aids the interpretation of (1) atypical thyroid function tests, in particular unusual relationships between serum thyroid-stimulating hormone (TSH) and circulating thyroid hormones that may indicate hereditary or acquired abnormalities of the three major thyroid hormone–binding proteins; (2) aberrant estimates of serum free T4 that may be due to analytic artifact that results from abnormal tracer binding by a variant protein, and (3) the effects of medications and critical illness on protein binding of the thyroid hormones.
The evolution of thyroid hormone binding to plasma proteins can be traced from fish, which show only albumin binding, through birds, in which T4 binds to both albumin and transthyretin (TTR, previously known as prealbumin). Most larger mammals, with the exception of felines, have in addition a low capacity, high-affinity protein, termed thyroxine-binding globulin (TBG) that carries well over half the total circulating T3 and T4. In humans, only 0.02% to 0.03% (about 1 part in 4000) of T4 and 0.2% to 0.3% (1 part in 400) of T3 circulate in the free or unbound state in undiluted normal human serum or plasma at equilibrium at 37°C. Other iodothyronines, whether synthetic analogues or metabolites, also are generally highly protein bound.2 Numerous other hydrophobic ligands that are unrelated to thyroid hormones can also compete for the various plasma protein–binding sites (see below).
In normal human serum, about 75% of the total circulating T4 concentration of 60 to 140 nmol/L or 4 to 11 µg/dL is carried on TBG, with about 10% to 15% attached to TTR and 10% to 15% bound to albumin. The electrophoretic techniques used to make these estimates allow some dissociation of labeled hormone during separation and thus tend to underestimate proportional carriage on lower-affinity sites. A minor fraction (<5%) of circulating T4 and T3 is associated with lipoprotein.3
Binding to plasma proteins is noncovalent and rapidly reversible; it is important to emphasize that the much larger bound moiety of hormone acts, in effect, as a reservoir. Dissociation of bound hormone almost instantaneously replenishes the free hormone concentration, as this fraction is taken up by tissues or diminished by sample dilution in vitro. A rigid distinction between bound and free hormone moieties may be artificial in light of studies that suggest dissociation and reassociation so rapid that the free and bound moieties interchange several million times per day.4
Marked hereditary differences in the protein binding of iodothyronines lead to wide variations in the total T4 concentration in humans (Fig. 22-1), while the free hormone concentration remains within much narrower limits. This finding supports the free hormone hypothesis (see below), which proposes that the minute free fraction of the total circulating T4 and T3 pool is the major determinant of hormone action, clearance, and negative feedback. Even when different species are compared, the concentration of free hormone varies much less than total concentration.5
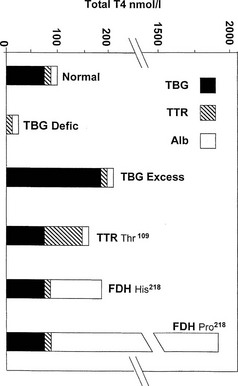
FIGURE 22-1 Estimated proportional carriage of thyroxine (T4) on the three major plasma binding proteins is shown for euthyroid subjects with normal or variant T4 binding proteins. In the face of normal free T4 and thyroid-stimulating hormone (TSH) concentrations, the concentration of total T4 can vary from about 25 nmol/L in total TBG deficiency to about 1800 nmol/L in the Pro218 variant of familial dysalbuminemic hyperthyroxinemia (FDH) described in several Japanese kindreds.
Characteristics of Hormone Binding
The definition of a number of terms assists the understanding of thyroid hormone binding. Capacity expresses the molar concentration of a specific class of ligand-binding site; if one binding site is present per protein molecule, capacity and protein concentration will be identical in molar terms. When about half the binding sites are empty, the free and hormone concentrations will change to about the same extent (i.e., the free fraction will show little change). However, as the total concentration of ligand approaches the binding protein capacity, the free hormone concentration will rise disproportionately, as occurs in thyrotoxicosis as the total T4 serum concentration approaches the capacity of TBG.6
Proportional carriage, the distribution of total hormone between a number of heterogeneous binding proteins, is influenced by the concentration of binding protein, the affinity of binding, and the free hormone concentration, but it is not directly influenced by the total hormone concentration.7 In a heterogeneous mixture of binding proteins, as in serum, proportional carriage can change as the free hormone concentration alters in response to dilution or hormone loading (see below).
Occupancy, the proportion of a particular class of binding site that is filled with hormone, is a direct function of the free hormone concentration and is fundamental to the definition of affinity or kD. TBG is normally about one third occupied by T4; TTR is <1% occupied by T4, and albumin shows negligible occupancy (Table 22-1).
Table 22-1
Properties of the Major Human Thyroid Hormone–Binding Proteins
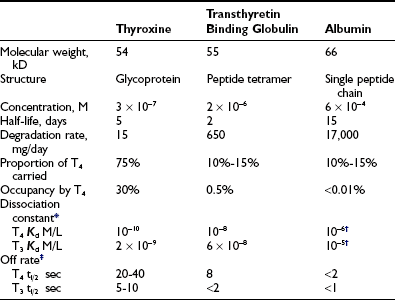
T3, Triiodothyronine; T4, thyroxine.
*Free hormone concentration at half occupancy of binding site at equilibrium.
Dissociation rate (TΩ, sec) or rate constant (sec-1) defines the rate of unidirectional dissociation or delivery of hormone from a binding site.4 The unidirectional maximum rate of hormone delivery is relevant under non–steady state conditions, as, for example, when free hormone is rapidly removed from the circulation during tissue transit. The dissociation rate, as well as kA and kD, are highly temperature dependent. The free T4 fraction is higher at 37°C than at room temperature by a factor of up to two8 and dissociation of ligand is much faster.4
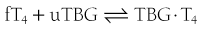
When represented in terms of the association constant, kTBG, this relationship becomes
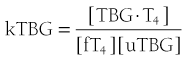
It follows that
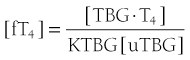
At half occupancy of the binding site, [TBG · T4] will equal [uTBG] and
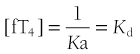
Free Hormone Hypothesis
According to this hypothesis, it is the free or unbound equilibrium concentration of a hormone that determines biological activity. The validity of this hypothesis, which generally is well sustained for the thyroid hormones, has been analyzed in detail.9,10 Earlier liver perfusion experiments that showed the loosely albumin-bound moiety of the total circulating hormone pool to be virtually as readily available as the free hormone11 have now been refuted.9 No conclusive evidence suggests that any particular class of binding protein facilitates tissue uptake of thyroid hormones. When isolated rat liver was perfused with T4 bound to various normal and variant binding proteins,12 tissue uptake of T4 was proportional to the spontaneous dissociation of T4 from each protein.12
Under some circumstances, especially when capillary transit is slow, or when mixing of layers across the diameter of a vessel is incomplete, tissue uptake of hormone may be limited by dissociation of bound hormone.9 Under these circumstances, the local concentration of free hormone at a particular site may be lower than the equilibrium concentration. The albumin-bound moiety with the fastest rate of unidirectional dissociation (see Table 22-1) will then make a large contribution in replenishing the free concentration.9
As formulated by Mendel,9,10 the unmodified free hormone hypothesis will be valid when tissue uptake of hormone is limited by influx or elimination. When flow or dissociation is the limiting condition, for example, when flow is slow and clearance is rapid in a tissue such as the liver, the free hormone hypothesis still holds, with hormone dissociation as an additional critical variable.
Structure and Binding Characteristics of Normal and Variant Transport Proteins
The characteristics of the three normal major iodothyronine-binding proteins, thyroxine-binding globulin, transthyretin and albumin, are summarized in Table 22-1. The numerous structural variants of the three major thyroid hormone–binding proteins that have been described (Table 22-2) were initially recognized from the investigation of euthyroid subjects who showed markedly abnormal levels of total serum T4 or T3, but recently, additional variants have been identified from population screening and genetic studies. The total concentrations of serum T4 and T3 range widely in association with variant binding proteins, but none of the multiple hereditary binding alterations has been shown to confer any advantage or disadvantage, or to disturb thyroid hormone action, unless thyroid disease is associated. Plasma TSH concentrations remain normal, as do free T4 and T3 concentrations, provided the method of estimation is free of artifact. In general, TBG abnormalities tend to affect T4 and T3 similarly; by contrast, some albumin variants lead to selective abnormalities of T4 or T3 binding.
Table 22-2
Known Human Variants of Thyroid Hormone–Binding Proteins
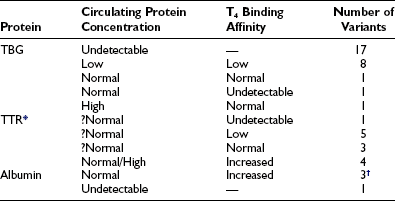
T3, Triiodothyronine; T4, thyroxine; TBG, thyroxine-binding globulin; TTR, transthyretin.
*The T4 binding affinity of more than 30 additional variants remains undefined.
†One variant has selective affinity for T3.
Compiled from references 14, 15, 16, 17, 21, 22, 23, 32, 38, and 53.
• Effect on diagnostic tests. In euthyroid subjects, abnormal total T4 or T3 concentrations occur in association with normal free hormone concentrations. Particularly with the albumin variants, method-dependent artifacts may compromise measurements of free, occasionally total, T4 or T3.
• Modes of inheritance. TBG variants are X-linked, whereas TTR and albumin variants show autosomal dominant inheritance.
• Structure-function relationships. Structure-function relationships of specific hormone-binding sites can be studied by using the large amounts of material available in serum. Albumin variants have shown how structural changes can increase the binding affinity for a particular ligand. In contrast, TBG mutants show either normal or diminished binding affinity, often associated with abnormal heat lability.
• Effects of variant binding proteins. The effects of variant binding proteins on T4 and T3 distribution, clearance, and delivery to tissues can test the hypothesis that it is the free hormone concentration, as determined at equilibrium, that determines hormone clearance and action.
• Changes in binding protein configuration. The recent demonstration that TBG, a member of the serine protease inhibitor (SERPIN) class of proteins, can undergo cleavage or change in configuration at local tissue sites has led to important speculation that changes in binding protein configuration may facilitate tissue-specific hormone delivery (see below).
• Tissue effects of abnormal proteins. Other associated pathology, for example, the familial amyloidosis associated with some TTR variants, can elucidate tissue effects of abnormal proteins.
Thyroxine-Binding Globulin (TBG)
TBG is a single polypeptide chain α globulin, with molecular weight of about 54 kD synthesized as a 415 amino acid protein.13–15 The first 20 amino acid residues of the TBG peptide are hydrophobic in nature and probably represent the signal peptide, which is removed in the endoplasmic reticulum, leaving a mature protein of 395 amino acids in a single chain with a molecular weight of about 44 kD. Multiple glycosylation sites allow an average of 10 terminal sialic acid moieties. The carbohydrate portions of TBG influence protein half-life in blood, stability in vitro, and microheterogeneity on electrophoresis, with only minor effects noted on immunoreactivity or T4 binding. Although TBG is stable in stored serum at 4°C, it gradually loses its binding affinity for T4 at 37°C or above. Differences in the rate of loss of binding affinity at raised temperature have been important in identifying TBG variants. Of particular interest are a variant with markedly increased heat stability (TBG-Chicago)16 and a variant with an extremely heat-labile protein with an abnormally high concentration of denatured TBG and subnormal total T4 (TBG-Gary).17
The amino acid sequence of human TBG shows homology with rat TBG (70%), human cortisol-binding globulin (55%) and members of the serum protease inhibitor family (SERPINS), which includes α-antitrypsin (53% homology) and a 1-antichymotrypsin (58% homology).18 The significance of the structural similarity between human TBG, CBG, and the SERPINS remains unclear, because the hormone-binding proteins do not exhibit antiprotease activity.
Susceptibility to cleavage and change in configuration may modulate hormone delivery from the SERPIN family of binding proteins. The study of Zhou et al.19 confirms that the binding of thyroxine to TBG can alter in response to changes in configuration of the binding protein. Using nonglycosylated recombinant human TBG, investigators reported that thyroxine is carried in a surface pocket and not within the beta barrel of the TBG molecule. With this structural model, conformational changes that result from relocation of a mobile peptide loop within the TBG molecule can favor binding or release of thyroxine. The demonstration of labile interaction between TBG and thyroxine raises the important possibility of modulated or tissue-specific delivery of thyroxine that could vary in response to changes in local pH, temperature, or redox status. The details of how local tissue factors may enhance or limit local hormone release from SERPIN binding proteins remain to be studied (see later section, “Function of Iodothyronine Binding Proteins”).
The normal concentration of human plasma TBG measured by radioimmunoassay is between 10 and 30 mg/L (0.2 to 0.6 µmol/L). TBG is normally 20% to 40% occupied by T4 and <1% occupied by T3. Occupancy may increase markedly in hyperthyroidism owing to increased total T4 and decreased TBG concentrations, leading to a disproportionate rise in free T4 relative to total T4.6 The T4-binding affinity (kD), is about 50 pmol/L at 37°C, consistent with the estimate that TBG is approximately 30% occupied by T4 at the normal free T4 concentration of about 20 pmol/L.20
Hereditary TBG Variants
The single 8 kilobase human TBG gene has been localized to the long arm of the X chromosome at site Xq21-q22.14 Male hemizygotes who express a single mutant allele can show one of three variant phenotypes for T4 binding to TBG: increase, decrease, or absolute deficiency. Despite the presence of two X chromosomes, normal females have TBG levels similar to those of males. Females heterozygous for complete TBG deficiency usually show less than the anticipated 50% reduction in serum TBG, a phenomenon attributed to selective inactivation of the mutant allele.14,15 However, selective inactivation of the normal allele occasionally may result in females showing complete deficiency of TBG.21
Multiple inherited TBG variants, often designated geographically, can result in partial or complete deficiency of immunoreactive TBG in serum. Of at least 24 known X-linked TBG mutants, 15 may cause complete TBG deficiency, and eight other variants are associated with subnormal concentrations of immunoreactive serum TBG, often with reduced affinity for T4.13–15,22 The structural basis of numerous variants is summarized in Fig. 22-2. Several variants of TBG show decreased heat lability in vitro, which generally correlates with accelerated clearance in vivo. TBG deficiency (complete or partial) results from missense or nonsense mutations in the coding exons or in donor or acceptor splice sites.14,15 Thus, single nucleotide deletions or substitutions can lead to a frame shift with premature termination of translation, resulting in a truncated protein that is retained and degraded intracellularly.13–15 In some reports no mutations could be demonstrated in the TBG gene.23
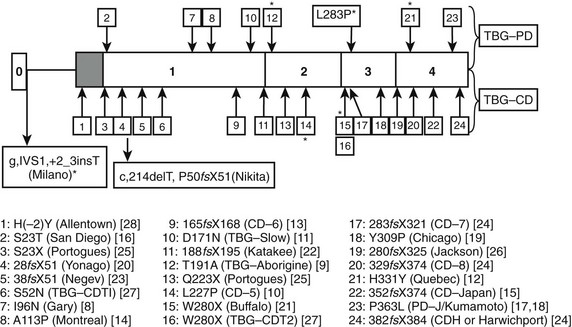
FIGURE 22-2 Summary of reported thyroxine-binding globulin (TBG) deficiency mutations. The numbered panel designates the five exons of TBG. Mutations above that panel refer to partial deficiency of TBG, and those below refer to total TBG deficiency. Polymorphisms are designated by an asterisk. Below the figure, details of each mutation are given, together with geographic designation and literature reference, as in Mannavola et al.14 (From Mannavola et al., 2006.14)
In total TBG deficiency, total T4 is about 25 nmol/L (see Fig. 22-1), associated with normal free T4 and TSH. By contrast, in hemizygous TBG excess, the total T4 concentration is typically over 200 nmol/L, of which over 80% is carried on TBG (see Fig. 22-1).
From newborn screening studies, the prevalence of complete TBG deficiency in males is about 1 : 5000, with 1 : 15,000 showing complete deficiency,15 but marked ethnic differences are noted in the frequency of hereditary TBG deficiency, with complete deficiency being highest in the Japanese.14 Diminished TBG binding of T4 is especially prevalent in Australian aborigines, up to 30% of whom have subnormal serum concentrations of total T4, associated with subnormal serum concentrations of an abnormally heat-labile TBG that shows subnormal affinity for T4.24 Owing to a very high gene frequency in this population, the pattern of inheritance was initially thought to be autosomal dominant.25 Abnormal heat lability at 37°C was found in both male and female subjects from affected families, but the pattern of intermediate heat lability was found exclusively in female subjects,26 demonstrating that inheritance must be X-linked (Fig. 22-3). Hereditary TBG excess, probably due to gene duplication,27 appears to have a prevalence of about 1 : 25,000 in newborn males.15 The binding of T4 to TBG in inherited X-linked TBG excess is indistinguishable from the common type of TBG. In contrast to the albumin variants, no known TBG mutant shows increased T4-binding affinity.
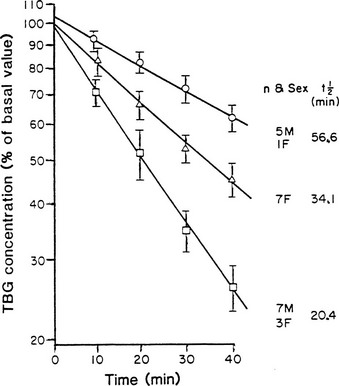
FIGURE 22-3 Heat stability of thyroxine-binding globulin (TBG) at 56°C in sera from Australian aborigines. Both male and female subjects showed either normal (upper line) or markedly reduced stability (lower line). No male subject showed the intermediate affinity (middle line) that demonstrates the heterozygous state, thereby confirming X-linked inheritance. (From Refetoff S, Murata Y: X-chromosome-linked inheritance of the variant thyroxine-binding globulin in Australian Aborigines, J Clin Endocrinol Metab 1985;60:356-360.)
Albumin
Human serum albumin, a highly conserved 66 kD nonglycoprotein,28 has a molar plasma concentration of approximately 600 µmol/L, corresponding to about 40 g/L. As well as being the principal carrier of numerous hydrophobic compounds in serum, albumin binds T4 in its region 2, with an affinity about four orders of magnitude less than that of normal TBG. Albumin normally carries 10% to 15% of circulating T4, but the proportion of albumin occupied by T4 is less than 0.002%.
Hereditary Albumin Variants
Hyperthyroxinemia can result from variant albumins with increased affinity for T4 or T3, the total albumin concentration being normal (Table 22-3). In familial dysalbuminemic hyperthyroxinemia (FDH), the total T4 concentration in affected individuals is about 200 nM,29,30 with over 50% of T4 carried on the variant albumin (see Fig. 22-1). FDH appears to be the most common hereditary T4-binding abnormality, with a prevalence as high as 1 : 1000 in some Latin American populations.31 As with other variants that show enhanced binding affinity or capacity, the increased concentration of total circulating T4 appears to be an appropriate response to maintain a normal free T4 concentration in feedback relationships with TSH.30
Table 22-3
Abnormal Albumin Binding of Iodothyronines

FDH, Familial dysalbuminemic hyperthyroxinemia; T3, triiodothyronine; T4, thyroxine.
In FDH, Scatchard analysis of albumin binding shows two T4-binding sites: a normal site with kD 4.3 mmol/L and an abnormal site with 50- to 100-fold higher affinity, kD 50 nmol/L.32 The capacity of the higher-affinity T4-binding site is approximately 200 µmol/L, suggesting that relative to the molar concentration of albumin, at least one third of albumin molecules have the abnormal binding site.32 As a result, the occupancy of albumin by T4 increases about fivefold to about 0.01%. The common FDH variant is due to Arg-His substitution at position 218 of human albumin,33 with a T4 affinity 65-fold greater than normal,34 similar to the affinity reported for the natural protein more than a decade before.32 Kinetic studies in vivo show altered distribution of T4 in favor of the extracellular compartment and a reduced metabolic clearance rate of T4.35
In FDH, because of a markedly increased affinity of the variant protein for numerous T4-analogue tracers, serum free T4 estimated by early analogue-tracer methods yielded results suggestive of thyrotoxicosis.36 Greater albumin binding of tracer in FDH samples than in normal serum standards made less tracer available for binding to the assay antibody; the spurious decrease in bound counts resulted in a falsely high free T4 estimate.
A related autosomal dominant albumin variant at the same site (Arg 218 Pro), so far reported only in the Japanese,37 shows an even higher selective T4 affinity than the common FDH phenotype found in Caucasians. In euthyroid subjects with normal TSH, total T4 is almost 20-fold elevated at about 1800 nmol/L (see Fig. 22-1), whereas total T3 is only about twofold elevated. Free T4 estimates using analogue tracer also show spurious elevations in this variant.37
To explain the mechanism of increased T4 affinity in both types of FDH, it has been suggested that the guanidine group of arginine 218 normally may give an unfavorable binding interaction with T4. Histidine and proline, which lack the guanidine group, allow higher-affinity binding than is seen with wild-type albumin.37
A recent report described a mutation of the albumin gene at a different site (Leu 66 Pro) with selective affinity for T3 rather than T4.38 Eight euthyroid members of a Thai family showed total T3 levels of 4 to 8 nmol/L (normal, 1.0 to 2.6) when measured by radioimmunoassay with the use of I125 T3. Free T3 and free T4 were normal. This mutant albumin was estimated to have a 40-fold higher T3 affinity than normal albumin, but only a 1.5-fold higher affinity for T4. However, spuriously low total T3 values were found when T3 conjugates were used as tracer in enzyme-linked immunosorbent (ELISA) assays.38 The conjugate tracer, linked to alkaline phosphatase or to peroxidase, showed less binding to the variant albumin than to the natural protein, making more tracer available for binding to antibody in sample than in standard, the opposite artifact to that found with analogue-tracer free T4 estimates in the common type of FDH.
Only a few cases of total hereditary analbuminemia have been described in man, but in one kindred,39 evidence of mild TSH excess was found, consistent with impaired thyroid hormone delivery. In contrast, the Nagase strain of analbuminemic rat showed no evidence of any abnormality of thyroid hormone action or distribution.40
Transthyretin
Transthyretin (TTR, previously known as prealbumin), a protein of approximately 55 kD that circulates in the serum of a wide range of vertebrates, is a tetramer consisting of four identical polypeptide chains held together by noncovalent bonds. Each monomer is a 127 amino acid chain regulated by a single gene on chromosome 18. The tetramer is symmetrical about a central cavity that completely penetrates the molecule and contains two T4-binding sites, one at each end of the central cavity.41
The normal serum concentration of TTR in healthy humans (2 to 8 µmol/L, 100 to 400 mg/L) can decrease rapidly during acute illness or malnutrition as a result of reduced hepatic synthesis.42 At normal concentrations, TTR is <1% occupied by T4, with a T4 affinity (kD ≈ 10 nmol/L) lower than TBG and higher than albumin. TTR has about 10-fold lower affinity for T3 than for T4. The liver is the principal site of synthesis of TTR, but the choroid plexus43 and the pancreatic islets44 are additional sites of TTR synthesis. In evolutionary terms, TTR synthesis at the choroid plexus long preceded the ontogeny of TTR synthesis in the liver.45 The T4-binding domain of TTR appears to have been conserved over the past 350 million years.45
Hereditary Transthyretin Variants
Many autosomal dominant TTR variants characterized by single amino acid substitutions have been described in man, some found in association with familial amyloidosis due to formation of abnormal amyloid fibrils (see reference 46 for review). Complete deficiency of TTR has never been described in humans, suggesting that a deficiency of this protein might be lethal. However, transthyretin-null mice produced by gene knockout show no obvious abnormality of thyroid hormone metabolism or action.47
The Thr109 TTR variant48 can give rise to mild hyperthyroxinemia with a total concentration of T4 of 160 to 200 nmol/L,49 associated with some increase in the total concentration of TTR.49 Kinetic studies with this variant protein show a T4-binding affinity about sevenfold higher than normal TTR.50 In the face of normal TBG and albumin concentrations, Thr109 TTR probably binds about 50% of circulating T4 in euthyroid subjects (see Fig. 22-1). The Val109 and Met119 variants of TTR also have increased affinity for T4, but serum total T4 is outside the reference range only in the former.51,52 TTR variants are not known to cause spurious assay values for total or free T4.
Of at least 30 mutations described for TTR, many have now been examined for T4 affinity. A study that compared the interaction of T4 with 10 different naturally occurring human TTR variants showed a wide range of T4 affinities.53 Relative to the wild-type TTR, three show increased affinity for T4—the Thr109 and Val109 variants of sufficient affinity to cause euthyroid hyperthyroxinemia—and three have approximately normal affinity, with five TTR variants showing reduced affinity for T4.54
Function of Iodothyronine-Binding Proteins
The large pool of extracellular thyroxine that is maintained as a result of plasma protein binding is equivalent to 10 to 15 days of hormone secretion from the normal thyroid. Avid protein binding is the basis for the long plasma half-life of about 7 days and the slow metabolic clearance rate of thyroxine, both of which favor sustained thyroid hormone action. A high degree of protein binding also serves to protect against iodine loss from urinary excretion of unbound iodothyronines. It is notable that Ekins had pointed out that protein binding of T4 may have a key role in T4 delivery to the fetus in early pregnancy55 (see below), well before the homology of TBG with the serine antiproteases was demonstrated in 1986.16
Autoradiographic studies suggest that binding proteins facilitate even tissue distribution of iodothyronines56 but indicate that multiple binding protein classes are redundant in this respect. When rat liver lobules were perfused with 125I T4 in the absence of binding protein, almost all of the T4 was taken up by the periportal cells. When 4% human serum albumin or human serum was added to the perfusate, the labeled hormone was taken up uniformly by all cells within the lobule. When a competitor was added to displace T4 from albumin, uptake was again predominantly periportal.56
Interest is increasing in the possibility that selective cleavage or change in the configuration of TBG, as occurs with other SERPIN proteins, may facilitate selective delivery of T4, for example, at sites of inflammation or in the placenta.57,58 Rapid clearance of TBG has been reported in the early postoperative phase after coronary artery surgery,59 and a truncated circulating form of the molecule, with lower affinity for T4, has been reported in sera from patients with sepsis60 and after cardiopulmonary bypass.61 Cleavage of TBG with decreased T4-binding affinity has been demonstrated to result from leukocyte elastase activity.62 These findings have renewed interest in the earlier suggestion63 that high local tissue T4 concentrations might allow release of iodine in concentrations sufficient to enhance leukocyte bactericidal activity. However, it is notable that males who are completely deficient in TBG appear to suffer no disadvantage in terms of their response to infection.
It is speculated that selective placental TBG cleavage with local release of free T4 could facilitate transfer of iodine to the fetus.64 If additional studies confirm these mechanisms, it will become relevant to consider TBG as a selective delivery protein, rather than a molecule that simply prevents rapid T4 clearance and ensures uniform tissue delivery of the hormone. Although TBG does not appear to have a role in targeted hormone delivery via specific cellular receptors for this protein, as shown for corticosteroid-binding globulin,65 a similar effect may be achieved through selective tissue-specific dissociation of thyroid hormone.
Thus, accumulating evidence suggests that the thyroid hormone–binding proteins are more than simply passive reservoirs of bound hormone. Based on cross-species differences and ontogeny, as well as the tissue perfusion study cited above, the trend is to designate the serum carriers of thyroid hormone as “distributor proteins” rather than passive carriers.66 Nevertheless, none of the many human variants in any of the three major binding proteins appears to confer any survival advantage or disadvantage.
Acquired Alterations in the Concentration of Transport Proteins
Numerous drugs can influence the concentrations of TBG and TTR (Table 22-4) through effects that alter protein synthesis or influence degradation; in many instances, the mechanism has not yet been clearly defined. The most common acquired change in TBG is an increase in concentration due to exogenous or endogenous estrogens that result in TBG with a greater proportion of bands with anodal mobility on isoelectric focusing, caused by an increase in sialic acid content of the side chains.67 Reduced TBG degradation due to this oligosaccharide modification appears to be the major mechanism of estrogen-induced TBG excess.68 Transdermal estrogens without predominant effect on the liver do not have this effect.69
Table 22-4
Drug Effects on Serum TBG and TTR Concentrations in Humans
| Increase TBG | Decrease TBG |
| Estrogens | Thyroid hormones |
| Tamoxifen | Androgens, anabolic steroids |
| Heroin | Glucocorticoids |
| Methadone | L-asparaginase |
| 5-Fluorouracil | Interleukin-6 |
| Perphenazine | |
| Clofibrate | |
| Mitotane |
| Increase TTR | Decrease TTR |
| Androgens | Estrogens |
| Glucocorticoids |
Serum concentrations of TBG are reduced in thyrotoxicosis and increased in hypothyroidism in humans70 and in experimental primate studies.71 Serum TBG concentrations are decreased by glucocorticoid excess.72 Unlike human TBG, rat TBG is strongly repressed during adult life but actively expressed during postnatal development, in senescence, in the face of malnutrition, and in hypothyroidism,73 and also after adrenalectomy,74 possibly as part of an adaptive response.
Acquired changes in TTR concentration are common but have relatively minor effects on the total serum concentrations of T4 and T3 because of the low occupancy of this protein. Androgens75 and glucocorticoids72 increase, while estrogens decrease the concentrations of TTR76; these agents have the opposite effect on TBG. Hepatic synthesis of TTR decreases abruptly during any major illness. Some islet cell carcinomas can directly synthesize and release sufficient TTR to cause euthyroid hyperthyroxinemia.77
Competition for Thyroid Hormone Transport Sites
In contrast to binding proteins for corticosteroids, vitamin D, and sex hormones, which are highly specific for a single family of ligands, the iodothyronine-binding proteins show wide cross-reactivity with unrelated hydrophobic ligands, such as nonesterified fatty acids (NEFAs) and numerous drugs (Table 22-5). Each of these substances itself is highly bound to albumin; the unbound rather than the total concentrations of each ligand determine competition.
Table 22-5
Principal Drugs That Displace T4 from TBG Binding in Normal Human Serum
| Drug | Mean Percent Increase in Free T4 Fraction* |
| Salicylates | |
| Acetyl salicylic acid (aspirin) | 62 |
| Salicyl salicylic acid (salsalate) | >100 |
| Furosemide† | 5-30 |
| Fenclofenac | 90 |
| Mefenamic acid | 31 |
| Flufenamic acid | 10 |
| Diclofenac | 7 |
| Diflunisal | 37 |
| Phenytoin | 45 |
| Carbamazepine | 30 |
T4, Thyroxine; TBG, thyroxine-binding globulin.
*Equilibrium dialysis or ultrafiltration of undiluted serum at 37°C in vitro, at appropriate therapeutic concentrations of each drug.
Competition in Serum
When competition is studied by examining displacement of labeled T4 or T3 from an isolated binding protein, using the unlabeled hormone as reference, the affinities of important drug competitors for TBG range from three orders of magnitude less (furosemide) to almost seven orders of magnitude less than T4 itself, in the case of aspirin.78 However, such direct studies do not reflect in vivo competition, because the free serum concentration of a competitor is determined by its binding to sites other than TBG, in particular albumin. Many highly bound drugs circulate in micromolar concentrations and may occupy 5% to 50% of albumin-binding sites; the concentration of albumin, or its occupancy by other ligands, then becomes an important determinant of the free drug concentration. Because of differences in albumin binding, the hierarchy of drug competitor potency at relevant therapeutic concentrations in undiluted serum differs markedly from the affinity of drugs for TBG in isolation.25,26 The data shown in Table 22-5 are influenced by the total concentration of each drug and its free fraction, as well as by its affinity for T4-binding proteins.
Dilution Effects
When working with a highly bound ligand, such as T4, it is technically easier to study binding in diluted serum. In the absence of competitors, such measurements give useful comparisons between samples with high, normal, and low free hormone concentrations. However, it is difficult to establish a system in which the concentrations of free hormone, competitors, and unoccupied binding sites are maintained in the relationships that apply in vivo. Existing literature on competitor effects has become confused because details such as dilution and albumin concentration are often poorly defined. The terms “pre-dilution” and “co-dilution” are useful in defining potential artifacts.7
Pre-dilution occurs when the concentration of binding proteins is progressively decreased before particular concentrations of competitor are added (Fig. 22-4). The lower the concentration of albumin, the higher is the occupancy of available binding sites; this leads to a disproportionate increase in the free competitor concentration, which magnifies apparent competitor potency.79 For example, in the case of a highly albumin-bound competitor, the T4-displacing effect of 5 mM oleic acid added to undiluted serum could be matched almost exactly by 0.5 mM oleic acid in serum diluted 1 : 10.80 In general, pre-dilution of the sample magnifies apparent competition.
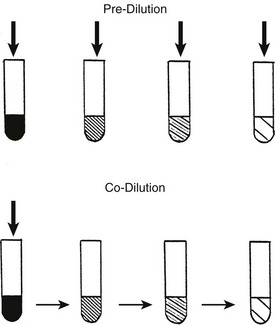
FIGURE 22-4 Pre-dilution: serum is diluted, followed by addition of a particular concentration of competitor. The effect of the competitor will be overestimated as albumin occupancy increases. Co-dilution: The competitor is added to serum, followed by identical simultaneous dilution of binding proteins, hormone, and competitors. If the competitor is less highly bound than the hormone, its effect will be progressively underestimated. (From Stockigt et al: Thyroid hormone transport. In Weetman AP, Grossman A [eds]: Pharmacotherapeutics of the thyroid gland, pp 119–150. Springer, Berlin, 1997.)
A co-dilution effect was the clue that led to the recognition of furosemide as an important inhibitor of T4 binding in serum.81 When T4 binding was studied in serial dilution in critically ill hypothyroxinemic patients, those who had received high-dose furosemide showed a marked increase in free T4 fraction that became less obvious with progressive dilution.81 When the abilities of three commercial free T4 assays to detect the T4-displacing effect of furosemide were compared, the effect was most obvious in the method with least sample dilution82 (Fig. 22-5).
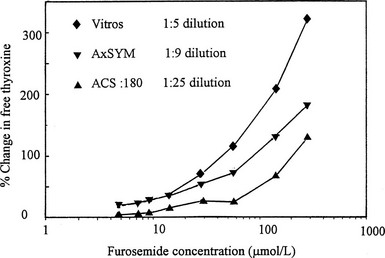
FIGURE 22-5 Effect of addition of furosemide to serum on estimates of free thyroxine (T4) using three commercial free T4 methods that involve varying degrees of sample dilution. The effect of the competitor is progressively obscured with increasing sample dilution. (Data from Hawkins RC. Furosemide interference in newer free thyroxine assays, Clin Chem 1998;44:2550-2551.)
The importance of a pre-dilution effect was demonstrated for therapeutic concentrations of phenytoin and carbamazepine, which increased the free fraction of T4 by 40% to 50% with the use of ultrafiltration of undiluted serum.83 However, no increase in T4 free fraction was seen with a commercial single-step free T4 assay after 1 : 5 serum dilution.83 During continuing drug therapy, total T4 was lowered by 25% to 50%, resulting in calculated free concentrations within the normal range.
Kinetics of The Competitor
The kinetics of the competitor itself will determine how it influences hormone binding in vivo (Fig. 22-6). A competitor of short half-life such as furosemide or salsalate will show fluctuating effects on hormone binding so that free hormone estimates may vary widely, depending on the time between dosage and sampling.84,85 In contrast, a competitor of long half-life will result in a new steady state with a normal free hormone concentration and a lowered total hormone concentration (i.e., an increased free fraction). It is not yet known whether intermittent competitor-induced increases in free hormone concentration can augment hormone action in humans, but it has been shown that a T3– and T4-displacing synthetic flavonoid has a transient thyromimetic effect in rats.86
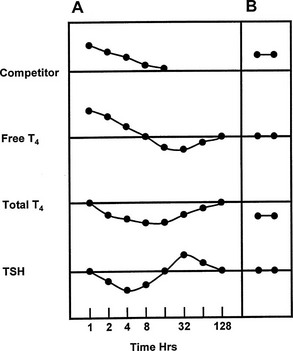
FIGURE 22-6 Representation of the serial changes in serum free thyroxine (T4), total T4, and thyroid-stimulating hormone (TSH) that follow ingestion of a single dose of a potent competitor for T4 binding to thyroxine-binding globulin (TBG) (panel A). An initial increase in free T4 is followed by a decrease in TSH and accelerated T4 clearance, resulting in a decrease in total and free T4, followed by a rebound increase in TSH. Note logarithmic time scale. Panel B shows the effect of a competitor of long half-life, or frequent dosage of a short half-life competitor, with stabilization at a new steady state with normal serum free T4 and TSH concentrations, but a decrease in total T4. (Compiled from data in Newnham et al.84 and Wang et al.85)
Interaction Between Competitors
Increasing concentrations of any substance that shares albumin-binding sites with a competitor can increase the free concentration of that competitor. Two substances that show potential to exert such a “cascade” effect on T4 binding in serum are oleic acid87 and 3-carboxy-4-methyl-5-propyl-2-furanpropanoic acid (CMPF),88 a naturally occurring furanoid acid that accumulates in renal failure. At concentrations that had only a minimal direct effect on the binding of T4 in undiluted normal serum, oleic acid and CMPF can augment the T4-displacing effect in undiluted serum of several drug competitors for T4 binding.88 Through such a mechanism, free hormone concentrations can be influenced by substances that have little direct interaction with hormone-binding sites.
Spurious Competition
The effect of heparin in increasing the apparent free T4 concentration can be misleading owing to another in vitro artifact. As summarized in Fig. 22-7, increases in free T4 that result from heparin treatment may cause in vivo release of lipase, followed by in vitro generation of NEFA, during assay incubation or storage.89 As a result of this artifact, serum NEFA concentrations at the time of assay can be much higher than they were in vivo.90 This effect may account for some reports of apparent increases in free T4 and free T3 fractions in critically ill subjects, although selective degradation of TBG at the site of sepsis or inflammation60,62 would provide an alternative explanation. In cases where heparin has been given, assays of total T4 and T3 are likely to be more informative than assays of free T4 and T3, unless special precautions are taken to avoid in vitro generation of NEFA.90
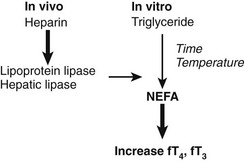
FIGURE 22-7 Summary of the heparin-induced changes that can markedly increase the apparent concentration of serum free thyroxine (T4). Heparin acts in vivo (left) to liberate lipoprotein lipase from vascular endothelium. Lipase acts in vitro (right) to increase the concentration of nonesterified fatty acids (NEFA) to levels >3 mmol/L, resulting in displacement of T4 and triiodothyronine (T3) from thyroxine-binding globulin (TBG). In vitro generation of NEFA is increased by sample storage at room temperature or incubation at 37°C and by high concentration of serum triglyceride. The T4-displacing effect of NEFA is accentuated at low albumin concentrations.80,89
Studies of Specific Binding Sites
If the capacity and affinity of each of the heterogeneous thyroid hormone–binding sites in serum are known, it is possible to manipulate hormone concentration and sample dilution to effectively isolate a single class of binding site.7 In this way, binding and competition can be characterized without isolating the protein. Similar approaches ultimately may be applicable to studies of heterogeneous hormone-binding sites in the intracellular milieu.
Because the T4-binding affinities of TBG and albumin differ by about four orders of magnitude, high-affinity binding can be “dissected” from low-affinity sites by manipulating the free hormone concentration via high sample dilution or hormone loading. For example, if normal serum with a total T4 concentration of 100 nM is diluted 5000 fold, the total T4 concentration will be 20 pM—close to the affinity of TBG. When 125I-T4 of high specific activity is used, the effects of drug competitors on specific T4 binding to TBG can be studied in such a system without the need to isolate the protein. The effects of TTR and albumin are negligible under these conditions.7 Such a method can be used to measure the minute amounts of TBG produced by cultured cells.91 Conversely, low-affinity binding is best examined after hormone loading to achieve a free concentration close to the affinity of that site; at high free hormone concentrations, the low-capacity sites are saturated and are not altered by changes in free hormone concentration. At artificially high hormone concentrations, the abnormally avid binding of T4 to albumin in familial dysalbuminemic hyperthyroxinemia can be readily demonstrated in the presence of 1000-fold relative excess of unlabeled T492 (Fig. 22-8).
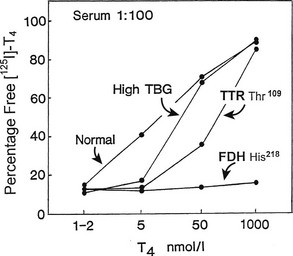
FIGURE 22-8 Effect of progressive increase in the load of unlabeled thyroxine (T4) on dextran-charcoal uptake of 125I T4 at 4°C in sera (diluted in phosphate buffer) from normal, thyroxine-binding globulin (TBG) excess, transthyretin (TTR) Thr,109 and familial dysalbuminemic hyperthyroxinemia (FDH) His218 subjects. In the presence of 1000-fold T4 excess, FDH shows unique persistence of 125I T4 binding at 4°C (low charcoal uptake) characteristic of a high-capacity site of increased affinity.
Data from reference 92. (Modified from Stockigt JR: Serum TSH and thyroid hormone measurements and assessment of thyroid hormone transport. In Braverman LE and Utiger RD, eds: Werner and Ingbar’s The Thyroid, 1996, Seventh Edition, pp 377–396. JB Lippincott, Philadelphia.)
Cell Membrane and Intracellular Transport
Until recently, it has been assumed that thyroid hormones move passively from the extracellular fluid through the cell membrane and cytoplasm to their nuclear receptors, but numerous studies have shown apparently specific binding or transport at both these locations. It is not yet clear whether the apparent competition between T3 or T4 and other ligands is truly specific, or whether apparent saturability and competition merely reflect interaction between intensely lipophilic molecules.93,94 Hennemann and Visser have reviewed the evidence for active cell membrane transport,95 and studies from their group suggest that bilirubin96 and fatty acids97 in sera from critically ill patients may limit the rate of T4 uptake by hepatocytes, thus limiting the amount of substrate available for conversion to T3.
Numerous substances, including calmodulin antagonists and calcium channel blockers, have been shown to inhibit cellular uptake of T3 by mechanisms that appear to be competitive.98 The demonstration in a cultured cell line of a saturable, stereospecific, verapamil-inhibitable mechanism that mediates T3 efflux99 raises the possibility that variable exit of T3 may modify thyroid hormone action. A putative transport protein was identified by photoaffinity labeling.99
Cytoplasmic T3 binding can be competitively inhibited by some anti-inflammatory agents and non–bile acid cholephils,100 but interpretation of these interactions is uncertain because the relevant free concentrations of hormone and competitor at cytoplasmic binding sites are not known. A nicotinamide adenine dinucleotide phosphate (NADPH)-dependent cytosolic T3-binding protein has been reported to limit efflux of T3 from the nucleus101 and has been shown to be expressed mainly in cardiac and neural tissue.102 Numerous possible nongenomic actions of thyroid and steroid hormones have been described,103 but it is not known how novel binding sites or transport proteins might be involved in these effects.
Of the numerous cell membrane proteins that bind thyroid hormone, only monocarboxylate transporter 8 (MCT8) and organic anion-transporting polypeptide 1C1 (OATP-1C1) show sufficient specificity toward thyroid hormone to suggest a physiologic role.104 It is now established that MCT8 is critical to the availability of thyroid hormone for intracellular deiodination and receptor binding, particularly in neural tissues and in placenta.104 The relative importance of these transport systems for inward and outward transit of thyroid hormone remains uncertain.104
Abnormalities of thyroid hormone transport across cell membranes of neural tissues, particularly those related to MCT8 mutations, may be associated with severe disease.105 The demonstration106,107 that various MCT8 mutations are associated with severe X-linked psychomotor retardation defines a new mechanism of disease. In this disorder, known by the eponym Allan-Herndon-Dudley syndrome, affected boys show serum T3 concentrations two- to fourfold in excess of normal, without a clear-cut abnormality of serum T4 or TSH. No deiodinase or thyroid hormone receptor abnormalities have been identified, and affected subjects appear to be euthyroid in tissues other than the brain. This uncommon hereditary disorder demonstrates that active transit of thyroid hormone into cells is crucial for normal brain development in humans.106,107 Thyroid hormone action did not appear to be impaired in tissues such as liver, muscle, or fat. Treatment of this disorder remains unsatisfactory.104–107
References
1. Trevorrow, V. Studies on the nature of the iodine in blood. J Biol Chem. 1939;127:737–750.
2. Robbins, J, Cheng, S-Y, Gershengorn, MC, et al. Thyroxine transport proteins of plasma, molecular properties and biosynthesis. Rec Prog Horm Res. 1978;34:477–519.
3. Benvenga, S, Cahnmann, HJ, Robbins, J. Characterization of thyroid hormone binding to apolipoprotein-E: localization of the binding site in the exon 3-coded domain. Endocrinology. 1993;133:1300–1305.
4. Hillier, AP. Thyroxine dissociation in human plasma: measurement of its rate by a continuous-flow dialysis method. Acta Endocrinologica. 1975;78:32–38.
5. Refetoff, S, Robin, NI, Fang, VS. Parameters of thyroid function in serum from 16 selected vertebrate species. Endocrinology. 1970;86:793–805.
6. Nauman, JA, Nauman, A, Werner, SC. Total and free triiodothyronine in human serum. J Clin Invest. 1967;46:1346–1355.
7. Stockigt, JR, Lim, C-F, Barlow, JW, et al. Thyroid hormone transport. In: Weetman AP, Grossman A, eds. Pharmacotherapeutics of the thyroid gland. Berlin: Springer; 1997:119–150.
8. Korcek, L, Tabachnick, M. Thyroxine-protein interactions, interaction of thyroxine and triiodothyronine with human TBG. J Biol Chem. 1976;251:3558–3562.
9. Mendel, CM. The free hormone hypothesis: a physiologically based mathematical model. Endocr Rev. 1989;10:232–274.
10. Mendel, CM, The free hormone hypothesis and free hormone transport hypothesis: Update 1994 Endocrine Reviews monographs. Clinical and molecular aspects of diseases of the thyroid. Endocr Reviews Monographs. Braverman, LE, Refetoff, S, eds. Clinical and molecular aspects of diseases of the thyroid. Endocr Reviews Monographs, Vol 3. Chevy Chase, Md: The Endocrine Society, 1994:208–209.
11. Pardridge, WM. Transport of protein-bound hormones into tissue in vivo. Endocr Rev. 1981;2:103–123.
12. Mendel, CM, Weisiger, RA. Thyroxine uptake by perfused rat liver: no evidence for facilitation by five different thyroxine-binding proteins. J Clin Invest. 1990;86:1840–1847.
13. Refetoff, S. Inherited thyroxine-binding globulin abnormalities in man. Endocr Rev. 1989;10:275–293.
14. Mannavola, D, Vannucchi, G, Fugazzola, L, et al. TBG deficiency: description of two novel mutations associated with complete TBG deficiency and review of the literature. J Mol Med. 2006;84:864–871.
15. Refetoff, S, Murata, Y, Mori, Y, et al. Thyroxine-binding globulin: organisation of the gene and variants. Horm Res. 1996;45:128–138.
16. Janssen, OE, Chen, B, Büttner, C, et al. Molecular and structural characterization of the heat-resistant thyroxine-binding globulin—Chicago. J Biol Chem. 1995;270:28234–28238.
17. Murata, Y, Takamatsu, J, Refetoff, S. Inherited abnormality of thyroxine-binding globulin with no demonstrable thyroxine-binding activity and high serum levels of denatured thyroxine-binding globulin. N Engl J Med. 1986;314:694–699.
18. Flink, IL, Bailey, TJ, Gustafson, TA, et al. Complete amino acid sequence of human thyroxine-binding globulin deduced from cloned DNA: Close homology to the serine antiproteases. Proc Natl Acad Sci U S A. 1986;83:7708–7712.
19. Zhou, A, Wei, Z, Read, RJ, et al. Structural mechanism for the carriage and release of thyroxine in the blood. Proc Nat Acad Sci U S A. 2006;103:13321–13326.
20. Robbins, J, Bartalena, L. Plasma transport of thyroid hormones. In: Hennemann G, ed. Thyroid Hormone Metabolism. New York: Marcel Dekker; 1986:3–38.
21. Okamoto, H, Mori, Y, Tani, Y, et al. Molecular analysis of females manifesting thyroxine-binding globulin (TBG) deficiency: selective X-chromosome inactivation responsible for the difference between phenotype and genotype in TBG-deficient females. J Clin Endocrinol Metab. 1996;81:2204–2208.
22. Carvalho, GA, Weiss, RE, Refetoff, S. Complete thyroxine-binding globulin (TBG) deficiency produced by a mutation in acceptor splice site causing frameshift and early termination of translation (TBG-Kankakee). J Clin Endocrinol Metab. 1998;83:3604–3608.
23. Retrakul, A, Dimitrescu, A, Macchia, PE, et al. Complete TBG deficiency in two families without mutations in coding or promoter regions of the TBG genes: in vitro demonstration of exon skipping. J Clin Endocrinol Metab. 2002;87:1045–1051.
24. Murata, Y, Refetoff, S, Sarne, DH, et al. Variant thyroxine-binding globulin in serum of Australian aborigines: its physical, chemical and biological properties. J Endocrinol Invest. 1985;8:225–232.
25. Watson, F, Dick, M. Distribution and inheritance of low serum thyroxine-binding globulin levels in Australian Aborigines. Med J Aust. 1980;2:385–387.
26. Refetoff, S, Murata, Y. X-chromosome-linked inheritance of the variant thyroxine-binding globulin in Australian Aborigines. J Clin Endocrinol Metab. 1985;60:356–360.
27. Mori, Y, Miura, Y, Takeuchi, H, et al. Gene amplification as a cause of inherited thyroxine-binding globulin excess in two Japanese families. J Clin Endocrinol Metab. 1995;80:3758–3762.
28. Peters, T, Jr. Serum albumin. Adv Protein Chem. 1985;37:161–245.
29. Hennemann, G, Docter, R, Krenning, EP, et al. Raised total thyroxine and free thyroxine index but normal free thyroxine. Lancet. 1979;1:639–642.
30. Stockigt, JR, Topliss, DJ, Barlow, JW, et al. Familial euthyroid thyroxine excess: an appropriate response to abnormal thyroxine binding associated with albumin. J Clin Endocrinol Metab. 1981;53:353–359.
31. Arevalo, G. Prevalence of familial dysalbuminemic hyperthyroxinemia in serum samples received for thyroid testing. Clin Chem. 1991;37:1430–1431.
32. Barlow, JW, Csicsmann, JM, White, EL, et al. Familial euthyroid thyroxine excess: characterisation of abnormal intermediate-affinity thyroxine binding to albumin. J Clin Endocrinol Metab. 1982;55:244–250.
33. Petersen, CE, Scottolini, AG, Cody, LR, et al. A point mutation in the human serum albumin gene results in familial dysalbuminaemic hyperthyroxinaemia. J Med Genet. 1994;31:355–359.
34. Petersen, CE, Ha, CE, Mandel, M, et al. Expression of a human serum albumin variant with high affinity for thyroxine. Biochem Biophys Res Commun. 1995;214:1121–1129.
35. Mendel, CM, Cavalieri, RR. Thyroxine distribution and metabolism in familial dysalbuminemic hyperthyroxinemia. J Clin Endocrinol Metab. 1984;59:499–504.
36. Stockigt, JR, Stevens, V, White, EL, et al. “Unbound analog” radioimmunoassays for free thyroxin measure the albumin-bound hormone fraction. Clin Chem. 1983;29:1408–1410.
37. Wada, N, Chiba, H, Shimizu, C, et al. A novel missense mutation in codon 218 of the albumin gene in a distinct phenotype of familial dysalbuminemic hyperthyroxinemia in a Japanese kindred. J Clin Endocrinol Metab. 1997;82:3246–3250.
38. Sunthornthepvarakul, T, Likitmaskul, S, Ngowngarmratana, S, et al. Familial dysalbuminemic hypertriiodothyroninemia: a new, dominantly inherited albumin defect. J Clin Endocrinol Metab. 1998;83:1448–1454.
39. Kallee, E. Bennhold’s analbuminemia: a follow-up study of the first two cases (1953–1992). J Lab Clin Med. 1996;127:470–480.
40. Mendel, CM, Cavalieri, RR, Gavin, LA, et al. Thyroxine transport and distribution in Nagase analbuminemic rats. J Clin Invest. 1989;83:143–148.
41. Robbins, J, Cheng, S-Y, Gershengorn, MC, et al. Thyroxine transport proteins of plasma, molecular properties and biosynthesis. Rec Prog Horm Res. 1978;34:477–519.
42. Schreiber, G, Synthesis, processing and secretion of plasma proteins by the liver and other organs and their regulation. The plasma proteins: structure, function and genetic control, ed 2. Putnam, PW, eds. The plasma proteins: structure, function and genetic control, Vol V. Orlando, Fl: Academic Press, 1987:293–363.
43. Schreiber, G, Aldred, AR, Jaworowski, A, et al. Thyroxine transport from blood to brain via transthyretin synthesis in choroid plexus. Am J Physiol. 1990;258:R338–R345.
44. Jacobsson, B, Carlstrom, A, Plotz, A, et al. Transthyretin messenger ribonucleic acid expression in the pancreas and in endocrine tumors of the pancreas and gut. J Clin Endocrinol Metab. 1990;71:875–880.
45. Richardson, SJ, Bradley, AJ, Duan, W, et al. Evolution of marsupial and other vertebrate thyroxine-binding plasma proteins. Am J Physiol. 1994;266:R1359–R1370.
46. Saraiva, MJM. Transthyretin mutations in health and disease. Human Mutation. 1995;5:191–196.
47. Palha, JA, Episkopou, V, Maede, S, et al. Thyroid hormone metabolism in a transthyretin-null mouse strain. J Biol Chem. 1994;269:33135–33139.
48. Moses, AC, Rosen, HN, Moller, DE, et al. A point mutation in transthyretin increases affinity for thyroxine and produces euthyroid hyperthyroxinaemia. J Clin Invest. 1990;86:2025–2033.
49. Moses, AC, Lawlor, J, Haddow, J, et al. Familial euthyroid hyperthyroxinemia resulting from increased thyroxine binding to thyroxine-binding prealbumin. N Engl J Med. 1982;306:966–969.
50. Lalloz, MRA, Byfield, PGH, Himsworth, RL. A prealbumin variant with an increased affinity for T4 and reverse-T3. Clin Endocrinol. 1984;21:331–338.
51. Refetoff, S, Marinov, VSZ, Tunca, H, et al. A new family with hyperthyroxinemia caused by transthyretin Val109 misdiagnosed as thyrotoxicosis and resistance to thyroid hormone—a clinical research centre study. J Clin Endocrinol Metab. 1996;81:3335–3340.
52. Curtis, AJ, Scrimshaw, BJ, Topliss, DJ, et al. Thyroxine binding by human transthyretin variants: mutations at position 119, but not position 54, increase thyroxine binding affinity. J Clin Endocrinol Metab. 1994;78:459–462.
53. Rosen, HN, Moses, AC, Murrell, JR, et al. Thyroxine interactions with transthyretin: a comparison of 10 different naturally occurring human transthyretin variants. J Clin Endocrinol Metab. 1993;77:370–374.
54. Bartalena, L, Thyroid hormone-binding proteins: Update 1994. Clinical and molecular aspects of diseases of the thyroid. Endocrine Review monographs. Braverman, LE, Refetoff, S, eds. Clinical and molecular aspects of diseases of the thyroid. Endocrine Review monographs, vol 3. Chevy Chase, Md: The Endocrine Society, 1994:140–142.
55. Ekins, RP. Roles of serum thyroxine-binding proteins and maternal thyroid hormones in foetal development. Lancet. 1985;1:1129–1132.
56. Mendel, CM, Weisiger, RA, Jones, AL, et al. Thyroid hormone-binding proteins in plasma facilitate uniform distribution of thyroxine within tissues: a perfused rat liver study. Endocrinology. 1987;120:1742–1749.
57. Schussler, GC. Review: the thyroxine-binding proteins. Thyroid. 2000;10:141–149.
58. Robbins, J. Editorial: New ideas in thyroxine-binding globulin biology. J Clin Endocrinol Metab. 2000;85:3994–3995.
59. Afandi, B, Schussler, GC, Arafeh, AH, et al. Selective consumption of thyroxine-binding globulin during cardiac by-pass surgery. Metabolism. 2000;49:270–274.
60. Jirasakuldech, B, Schussler, GC, Yap, MG, et al. A characteristic serpin cleavage product of thyroxine-binding globulin appears in sepsis sera. J Clin Endocrinol Metab. 2000;85:3996–3999.
61. Jirasakuldech, B, Schussler, GC, Yap, MG, et al. Cleavage of thyroxine-binding globulin during cardiac bypass. Metabolism. 2001;50:1113–1116.
62. Janssen, OE, Golcher, HM, Grasberger, H, et al. Characterization of T4-binding globulin cleaved by human elastase. J Clin Endocrinol Metab. 2002;87:1217–1222.
63. Klebanoff, SJ, Green, WL. Degradation of thyroid hormones by phagocytosing human leukocytes. J Clin Invest. 1973;52:60–72.
64. Khan, NS, Schussler, GC, Holden, JB, et al. Thyroxine-binding globulin cleavage in cord blood. J Clin Endocrinol Metab. 2002;87:3321–3323.
65. Rosner, W. The functions of corticosteroid-binding globulin and sex hormone-binding globulin: recent advances. Endocr Rev. 1990;11:80–91.
66. Richardson, SJ, Monk, JA, Shepherdley, CA, et al. Developmentally regulated thyroid hormone distributor proteins in marsupials, a reptile and fish. Am J Physiol Regul Integr Comp Physiol. 2005;288:R1364–R1372.
67. Ain, KB, Mori, Y, Refetoff, S. Reduced clearance rate of thyroxine-binding globulin (TBG) with increased sialylation: a mechanism for estrogen-induced elevation of serum TBG concentration. J Clin Endocrinol Metab. 1987;65:689–696.
68. Ain, KB, Refetoff, S. Relationship of oligosaccharide modification to the cause of serum thyroxine-binding globulin excess. J Clin Endocrinol Metab. 1988;66:1037–1043.
69. Chetkowski, RJ, Meldrum, DR, Steingold, KA, et al. Biologic effects of transdermal estradiol. N Engl J Med. 1986;314:1615–1620.
70. Konno, N, Kakinoki, K, Hagiwara, K. Serum concentrations of unsaturated thyroxine-binding globulin in hyper- and hypothyroidism. Clin Endocrinol. 1985;22:249–255.
71. Glinoer, D, McGuire, R, Dubois, A, et al. Thyroxine-binding globulin metabolism in Rhesus monkeys: effects of hyper- and hypothyroidism. Endocrinology. 1979;104:175–183.
72. Oppenheimer, J, Werner, S. Effect of prednisone on thyroxine-binding proteins. J Clin Endocrinol Metab. 1966;26:715–721.
73. Rouaze-Romet, M, Savu, L, Vranckx, R, et al. Re-expression of thyroxine-binding globulin in post-weaning rats during protein or energy malnutrition. Acta Endocrinologica. 1992;127:441–448.
74. Emerson, CH, Seiler, CM, Alex, S, et al. Gene expression and serum thyroxine-binding globulin are regulated by adrenal status and corticosterone in the rat. Endocrinology. 1993;133:1192–1196.
75. Arafah, BM. Decreased levothyroxine requirement in women with hypothyroidism during androgen therapy for breast cancer. Ann Intern Med. 1994;121:247.
76. Man, EB, Reid, WA, Hellegers, AE. Thyroid function in human pregnancy. III. Serum thyroxine binding prealbumin (TBPA) and thyroxine-binding globulin (TBG) of pregnant women. Am J Obstet Gynecol. 1969;103:338–347.
77. Maye, P, Bisetti, A, Burger, A, et al. Hyperprealbuminemia, euthyroid hyperthyroxinemia, Zollinger-Ellison-like syndrome and hypercorticism in a pancreatic endocrine tumour. Acta Endocrinol. 1989;120:87–91.
78. Munro, SL, Lim, C-F, Hall, JG, et al. Drug competition for thyroxine binding to transthyretin (prealbumin): comparison with effects on thyroxine-binding globulin. J Clin Endocrinol Metab. 1989;68:1141–1147.
79. Mendel, CM, Frost, PH, Cavalieri, RR. Effect of free fatty acids on the concentration of free thyroxine in human serum: the role of albumin. J Clin Endocrinol Metab. 1986;63:1394–1399.
80. Lim, C-F, Bai, Y, Topliss, DJ, et al. Drug and fatty acid effects on serum thyroid hormone binding. J Clin Endocrinol Metab. 1988;67:682–688.
81. Stockigt, JR, Lim, C-F, Barlow, JW, et al. High concentrations of furosemide inhibit plasma binding of thyroxine. J Clin Endocrinol Metab. 1984;59:62–66.
82. Hawkins, RC. Furosemide interference in newer free thyroxine assays. Clin Chem. 1998;44:2550–2551.
83. Surks, MI, Defesi, CR. Normal serum free thyroid hormone concentrations in patients treated with phenytoin or carbamazepine: a paradox resolved. JAMA. 1996;275:1495–1498.
84. Newnham, HH, Hamblin, PS, Long, F, et al. Effect of oral furosemide on diagnostic indices of thyroid function. Clin Endocrinol. 1987;26:423–431.
85. Wang, R, Nelson, JC, Wilcox, RB. Salsalate administration in a potential pharmacological model of the sick euthyroid syndrome. J Clin Endocrinol Metab. 1998;83:3095–3099.
86. Lueprasitsakul, W, Alex, S, Fang, SL, et al. Flavonoid administration immediately displaces thyroxine (T4) from serum transthyretin, increases serum free T4 and decreases serum thyrotropin in the rat. Endocrinology. 1990;126:2890–2895.
87. Lim, C-F, Curtis, AJ, Barlow, JW, et al. Interactions between oleic acid and drug competitors influence specific binding of thyroxine in serum. J Clin Endocrinol Metab. 1991;73:1106–1110.
88. Lim, C-F, Stockigt, JR, Curtis, AJ, et al. Influence of a naturally-occurring furanoid acid on the potency of drug competitors for specific thyroxine binding in serum. Metabolism. 1993;42:1468–1474.
89. Mendel, CM, Frost, PH, Kunitake, ST, et al. Mechanism of the heparin-induced increase in the concentration of free thyroxine in plasma. J Clin Endocrinol Metab. 1987;65:1259–1264.
90. Zambon, A, Hashimoto, SI, Brunzell, JD. Analysis of techniques to obtain plasma for measurement of levels of free fatty acids. J Lipid Res. 1993;34:1021–1028.
91. Crowe, TC, Cowen, NL, Loidl, NM, et al. Down-regulation of thyroxine-binding globulin messenger ribonucleic acid by 3,5,3′-triiodothyronine in human hepatoblastoma cells. J Clin Endocrinol Metab. 1995;80:2233–2237.
92. Stockigt, JR, Dyer, SA, Mohr, VS, et al. Specific methods to identify plasma binding abnormalities in euthyroid hyperthyroxinemia. J Clin Endocrinol Metab. 1986;62:230–233.
93. Hulbert, AJ. Thyroid hormones and their effects: a new perspective. Biological Reviews of the Cambridge Philosophical Society. 2000;75:519–631.
94. Scholz, GH, Vieweg, S, Uhlig, M, et al. Inhibition of thyroid hormone uptake by calcium antagonists of the dihydropyridine class. J Med Chem. 1997;40:1530–1538.
95. Hennemann, G, Visser, TJ. Thyroid hormone synthesis, plasma membrane transport and metabolism. In: Weetman AP, Grossman A, eds. Pharmacotherapeutics of the thyroid gland. Berlin: Springer; 1997:75–117.
96. Lim, C-F, Docter, E, Visser, TJ, et al. Inhibition of thyroxine transport into cultured rat hepatocytes by serum of nonuremic critically ill patients: effects of bilirubin and nonesterified fatty acids. J Clin Endocrinol Metab. 1993;76:1165–1172.
97. Lim, C-F, Bernard, BF, de Jong, M, et al. A furan fatty acid and indoxyl sulfate are the putative inhibitors of thyroxine hepatocyte transport in uremia. J Clin Endocrinol Metab. 1993;76:318–324.
98. Topliss, DJ, Kolliniatis, E, Barlow, JW, et al. Uptake of T3 cultured rat hepatoma cells is inhibitable by non-bile acid cholephils, diphenylhydantoin and non-steroidal anti-inflammatory drugs. Endocrinology. 1989;124:980–986.
99. Cavalieri, RR, Simeoni, LA, Park, SW, et al. Thyroid hormone export in rat FRTL-5 thyroid cells and mouse NIH-3T3 cells is carrier mediated, verapamil sensitive and stereospecific. Endocrinology. 1994;130:4948–4954.
100. Barlow, JW, Curtis, AJ, Raggatt, LE, et al. Drug competition for intracellular triiodothyronine-binding sites. Eur J Endocrinol. 1994;130:417–421.
101. Mori, J, Suzuki, S, Kobayashi, M, et al. Nicotine adenine nucleotide phosphate-dependent cytosolic T3 binding protein as a regulator for T3 mediated transactivation. Endocrinology. 2002;143:1538–1544.
102. Suzuki, S, Mori, J, Kobayashi, M, et al. Cell-specific expression of NADPH-dependent cytosolic 3,5,3′-triiodothyronine-L-thyroxine binding protein (p38CTBP). Eur J Endocrinol. 2003;148:259–268.
103. Davis, PJ, Tillmann, HC, Davis, FB, et al. Comparison of the mechanisms of nongenomic actions of thyroid hormones and steroid hormones. J Endocrinol Invest. 2002;25:377–388.
104. Visser, WE, Friesema, ECH, Jansen, J, et al. Thyroid hormone transport in and out of cells. Trends in Endocrinology and Metabolism. 2007;19:50–56.
105. Friesema, EC, Ganguly, S, Abdalla, A, et al. Identification of monodecarboxylate transporter 8 as a specific thyroid hormone transporter. J Biol Chem. 2003;278:40128–40135.
106. Dumitrescu, AM, Liao, XH, Best, TB, et al. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet. 2004;74:168–175.
107. Friesema, EC, Grueters, A, Biebermann, H, et al. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet. 2004;364:1435–1437.