[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 4
Thyroid Hormone Metabolism
Hormone Kinetics and Production
Deiodination and the Iodothyronine Deiodinases
Alternative Routes of Iodothyronine Metabolism
Thyroid Hormone Uptake Into Cells
An Integrated View of Thyroid Hormone Metabolism
The principal secretory product of the thyroid gland, T4, undergoes a complex series of intracellular metabolic alterations in peripheral tissues, as shown in Fig. 4-1. Some of these reactions, such as 5′-deiodination of T4 to form T3 or decarboxylation and deamination of T3 to form the acetic acid analogue termed Triac, result in compounds with considerably greater intrinsic biopotency because of their increased affinity for thyroid hormone receptors.1,2 Other reactions result in the formation of apparently inactive compounds, such as reverse T3 (rT3) formed by 5′-deiodination of T4 or the sulfated and glucuronidated forms of T4 and T3 formed by conjugation of the phenolic ring hydroxyl group.3,4 Progressive deiodination of T4, T3, and rT3 results in the formation of various diiodinated and monoiodinated thyronines and eventually thyronine (T0) itself.5 Most of these compounds are believed to have little or no biological activity. Exceptions to this generalization may be the compounds 3,5-diiodothyronine (3,5-T2) and 3,3′-T2, which have been demonstrated to have effects on mitochondrial function6 and to increase metabolic rate.7–9 In addition, the monoiodinated compound 3-iodothyronamine, formed in vivo by sequential deiodination and decarboxylation, produces bradycardia and dramatic decreases in body temperature and metabolic rate when administered to experimental animals.10,11 This effect, opposite to that expected of a thyroid hormone, is believed to result from a nongenomic mechanism via a G protein–coupled trace amine receptor.10
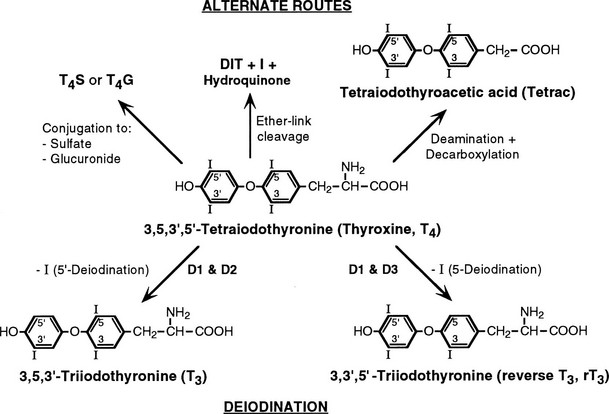
FIGURE 4-1 Graphic representation of the pathways of iodothyronine metabolism. The types 1 and 2 deiodinases (D1, D2) catalyze removal of the 5′ (or chemically equivalent 3′) iodine from T4 and other iodothyronine substrates. Types 1 and 3 enzymes catalyze 5 (or chemically equivalent 3) deiodination. A variety of less common reactions occur via alternative enzymatic pathways. The metabolites shown are subject to further deiodination to form diiodothyronines, monoiodothyronines, and tyrosine. (From St. Germain DL, Galton VA: The deiodinase family of selenoproteins. Thyroid 7:655–658, 1997.)
The enzymatic processes shown in Fig. 4-1 are not mutually exclusive. Indeed, modification of an iodothyronine molecule at one site may markedly alter its susceptibility to other metabolic reactions. For example, Tetrac and Triac are much better substrates for glucuronidation than are T4 and T3.12 The acetic acid analogues and sulfated conjugates also are markedly better substrates for deiodination in the liver and kidney than are the native compounds.13,14 In contrast, sulfation of iodothyronines in certain organs may effectively block further metabolism because these conjugates are poorly reactive with the deiodinase isoforms expressed in other tissues.15,16
Hormone Kinetics and Production
Deiodination at the 5′ or 5 position accounts for approximately 80% of the daily disposal of T4, with the other processes shown in Fig. 4-1 responsible for the remaining metabolism of this compound.5 Such approximations may underestimate the role of both deiodinative and nondeiodinative pathways, however. Many of the products of these reactions, including T3 and rT3, are present primarily in the intracellular compartment and may undergo degradation before they have a chance to exchange with the plasma pool. It thus is difficult in human kinetic studies that use plasma sampling techniques alone to assess the contribution of these processes to overall thyroid hormone metabolism. For example, it has been demonstrated that most of the thyronine (T0) excreted in the urine is in the form of its acetic acid analogue,17 thus suggesting that deamination plays a more prominent role in thyroid hormone metabolism than is apparent from the very low circulating levels of Tetrac and Triac. Such concerns have led to the concept that “hidden pools” of thyroid hormone metabolites may be present in tissues.18
Table 4-119–21 provides estimates of various kinetic parameters of thyroid hormone production and metabolism in humans.5,22,23 The high affinity of T4 for plasma binding proteins, along with its greater production rate, accounts for its relatively high concentration in serum, as well as its long serum half-life. In contrast, T3 and rT3 are present at much lower serum concentrations because of their lower production rates, greater metabolic clearance rates, and lower affinity for thyroxine-binding globulin (TBG). In addition, these two triiodothyronines appear to reside primarily within the intracellular compartment; thus their volumes of distribution are significantly greater than that of T4.
Table 4-1
Normal Thyroid Hormone Kinetics in Humans
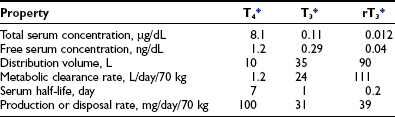
T4, Thyroxine; T3, 3,5,3′-triiodothyronine; rT3, reverse triiodothyronine.
*Conversion factors: T4, 1 mg = 1.3 nmol; T3 and rT3, 1 mg = 1.5 nmol.
The rate of T4 production remains remarkably constant in healthy adults, with alterations noted only during pregnancy and in the aged. Although not well documented, increased T4 secretion probably occurs during pregnancy in response to several factors, including thyroidal stimulation by human chorionic gonadotropin, an increase in the size of the extrathyroidal T4 pool resulting from increased TBG levels, and an increase in the rate of T4 and T3 degradation because of high levels of 5-deiodinase activity in the pregnant uterus and placenta.24,25 One clinical consequence is the need to increase the replacement dose of T4 by 25% to 50% in hypothyroid women during pregnancy.26 Thyroid function appears to be well preserved through age 80 in healthy individuals, although a slight decrease in T4 production and clearance is noted after the age of 60 years, and free T3 levels are lower in centenarians.27,28 That being the case, the decreased T4 requirements of elderly hypothyroid patients probably result more from the presence of chronic illness and the use of concurrent medications than from the aging process per se.29,30
Deiodination and The Iodothyronine Deiodinases
The potential importance of deiodination to thyroid hormone action was first recognized nearly 50 years ago when Gross and Pitt-Rivers demonstrated that although T3 was considerably more potent than T4,31 it was present in the thyroid in much lower amounts.32 This observation suggested that T3 was derived largely from T4 by metabolism in extrathyroidal tissues. This thesis was later proved by Braverman et al., who demonstrated the presence of T3 in the serum of athyreotic subjects injected with T4.33 Research over the past three decades has confirmed the physiologic importance of the 5′- and 5-deiodination processes, has defined the biochemical parameters of these enzymatic reactions, has determined important regulatory factors that influence deiodinase activities, and has identified key structural determinants of the proteins that catalyze these reactions.34,35
Three deiodinase isoforms, termed D1, D2, and D3, are present in vertebrate species. These differ in their catalytic properties, patterns of tissue expression, and mechanisms of regulation.34,36,37 The 5′- and 5-deiodination reactions catalyzed by these enzymes can be considered broadly as activating and inactivating processes, respectively.
Biochemical Characteristics
The biochemical properties of the deiodinases are outlined in Table 4-2.38–45 They are in essence oxidoreductases in that they catalyze the substitution of hydrogen for iodine on the iodothyronine substrate. No other catalytic properties of these enzymes have yet been identified, nor do other known enzymes possess deiodinase activity. The enzymes are remarkable in terms of their substrate specificity and the precise location of the iodine removed.16 D1 is unique in that it can catalyze either 5′- or 5-deiodination, depending on the reacting substrate. Thus rT3 is efficiently deiodinated only at the 5′ position by D1. In contrast, T4 and T3 are poor substrates for this enzyme unless they are first sulfated. This reaction markedly enhances their rate of 5-deiodination and further reduces their susceptibility to 5′-deiodination.46 That D1 is relatively inefficient in converting T4 to T3 presents a considerable paradox, given that a significant proportion of T3 production has traditionally been believed to occur in the liver and other D1-expressing tissues (vide infra).
Table 4-2
Characteristics of Iodothyronine Deiodinases

Data from St. Germain and Galton,38 Leonard and Visser,39 Jakobs et al.,40 Celi et al.,41 Hernández et al.,42 Leonard et al.,43,45 and Curcio-Morelli et al.44
D2 catalyzes only 5′-deiodination and very efficiently converts T4 to T3.16 The T3 thus formed is a poor substrate for 5′-deiodination and is not further metabolized by this enzyme. In contrast, rT3 is a good substrate for D2 as well as D1 and frequently is used in research assays to quantitate 5′-deiodinase activity. D3 exclusively catalyzes 5-deiodination16 and thus serves to convert T4 and T3 to rT3 and 3,3′-T2, respectively—metabolites with little affinity for the nuclear thyroid hormone receptors.
The deiodinases all require the availability of reduced thiol cofactors for efficient catalytic cycling.39 These cofactors presumably function to displace iodine from an enzyme intermediate formed during the reaction and thus to regenerate the active deiodinase.47 In in vitro assay systems using tissue homogenates or cellular subfractions, dithiothreitol typically is added as a cofactor. This small, nonnative four-carbon dithiol efficiently supports deiodination of all three enzymes. Kinetic data derived from broken cell preparations using dithiothreitol as cofactor have demonstrated a Michaelis-Menten constant (Km) value for D1 of approximately 2.3 µmol/L (for T4 5′-deiodination), whereas D2 and D3 manifest much lower Km values in the nanomolar range (see Table 4-2).39 Based on this analysis, D1 sometimes is referred to as a “high Km” enzyme, whereas D2 is said to catalyze a “low Km” 5′-deiodination process. This distinction is somewhat spurious, however, because the kinetic properties of the deiodinases are clearly dependent on the thiol cofactors used in the assay system.48 Thus, when the native thiol cofactor glutathione or thioredoxin is used, Km values for D1 in the nanomolar range are obtained. Unfortunately, the cofactor system supporting deiodination in intact cells remains unknown,49 and evidence suggests that glutathione and thioredoxin may not serve this role.50 Thus the physiologic significance of the kinetic parameters derived in vitro remains uncertain.
Inhibitors
In addition to differences in their catalytic properties, the deiodinases show differential susceptibilities to certain inhibitors39 (see Table 4-2). Most notable is the marked sensitivity of D1 to the antithyroid drug propylthiouracil (PTU). This agent forms an inactive complex with D1 by binding covalently to its active site. Notably, D2 and D3 show little or no susceptibility to inhibition by PTU. The related thioureylene drugs carbimazole and methimazole have no inhibitory effect on deiodination.47
Gold compounds such as aurothioglucose are known to react with the active site selenocysteine residue in glutathione peroxidase and impair its activity.51 This compound also inhibits all three deiodinase isoforms, with D1 again showing a significantly greater sensitivity to this effect.52–54 Other drugs that affect thyroid hormone metabolism include the iodinated radiographic contrast agents iopanoic acid (Telepaque) and sodium ipodate (Oragrafin). These small phenolic compounds act as substrate analogues and inhibit all three deiodinases in a competitive manner. To date, no selective inhibitors of D2 or D3 have been described—a situation that has significantly limited experimental investigations into their physiologic roles.
Tissue Patterns of Expression
An intriguing feature of the deiodinases is their pattern of tissue expression (see Table 4-2). The liver, kidney, thyroid gland, and pituitary gland express high levels of D1, with lesser amounts found in the brain.16,55,56 In contrast, D2 is most abundant in the pituitary gland and brown adipose tissue (in rodents), with significant amounts also noted in the central nervous system.16 Studies using in situ hybridization and immunocytochemistry have demonstrated that D2 expression in the brain appears to be confined to certain subpopulations of astroglial cells, such as the tanycytes lining the third ventricle of the hypothalamus, which suggests that they serve as the principal site of T3 production in this tissue.57–59 In humans, D2 expression appears more widespread; the mRNA for this enzyme has been noted in the heart, skeletal muscle, and thyroid gland,54,60 as well as in coronary artery smooth muscle cells.61
In adult mammals, D3 expression occurs primarily in the brain, with significant amounts also present in the skin.62,63 The brain is thus the only tissue that expresses all three deiodinases, although these are generally found in different subregions of this tissue and in different cell types. Thus, as opposed to the expression of D2 by glial cells, D3 is expressed primarily in neurons.64,65
Finally, as detailed below, deiodinase expression patterns during pregnancy and development are of critical importance, with high levels of D3 activity present in the uterus, the placenta, and several fetal tissues.66,67
Structural Characteristics
Important structural features of the deiodinases have been deduced from the results of molecular cloning experiments.37 All the enzymes have a molecular mass of approximately 29 to 32 kDa, function as homodimers,43–45 and are selenoproteins in that they contain the uncommon amino acid selenocysteine as the reactive residue in the catalytic cleft (Fig. 4-2).68 The importance of this amino acid to enzymatic activity is demonstrated by experiments in which selenocysteine has been replaced by cysteine. Such a substitution decreases the catalytic efficiency of the mutant protein to less than 1% of that of the native enzyme.69 In the case of D3, the cysteine mutant also demonstrates an altered substrate preference.70 It is remarkable that substitution of selenocysteine for a serine residue in a monoclonal antibody raised against T4 confers on the protein deiodinase activity with catalytic properties similar to the D1, including sensitivity to PTU inhibition.71
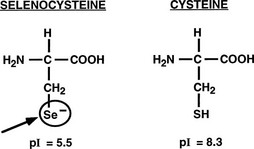
FIGURE 4-2 Comparison of the structures of selenocysteine and cysteine. Selenocysteine is ionized at physiologic pH, which likely contributes to the catalytic efficiencies of the deiodinases.
The importance of selenocysteine to deiodination likely derives from its being ionized at physiologic pH, thus serving as a much more potent nucleophile than cysteine.72 Incorporation of selenocysteine into the deiodinases and other selenoproteins occurs at the time of translation and is directed by a specific stem-loop structure in the 3′-untranslated region of the mRNA termed a selenocysteine insertion sequence.73 A unique tRNA (Sec-tRNA[Sec]), a specific RNA binding protein, and a specialized elongation factor also are required for efficient synthesis of selenoproteins.74,75
In earlier studies, it was suggested that a 29 kDa (p29) nonselenoprotein, almost identical in sequence to a member of the Dickkopf protein family (Dkk3), was a substrate binding subunit of the D2.76 However, Montero-Pedrazuela et al. have demonstrated a marked difference in spatial expression patterns in the brain of the p29 and the D2 selenodeiodinase,77 and as recently reported, mice deficient in the Dkk3 protein do not have any consistent alterations in thyroid hormone status or D2 activity in their tissues.78 These findings strongly suggest that the p29 protein is not relevant to D2 activity or to thyroid hormone homeostasis.
The overall amino acid identity of the three deiodinase isoforms is less than 30%. However, a high degree of homology is present in the regions of the selenocysteine and a conserved histidine residue (Fig. 4-3A). These areas show significant structural similarities to the thioredoxin-fold family of proteins and to α-l-iduronidase, respectively.79 Based on these structural homologies, Callebaut et al.79 and others have reported mutagenesis studies that define further the structure-function correlates of the deiodinase isoforms, including the identification of a key serine residue in the D1 active catalytic site that appears to confer sensitivity to PTU.80
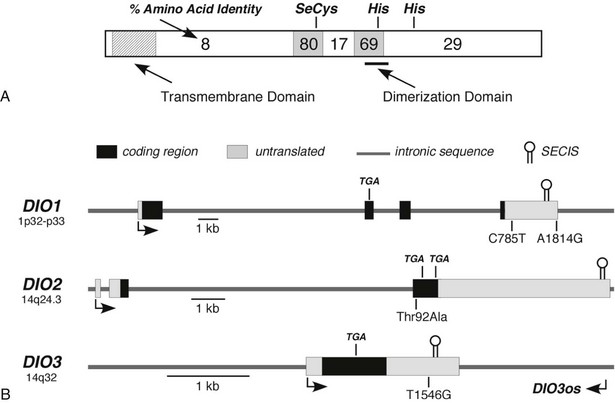
FIGURE 4-3 Structural features of the deiodinase proteins and genes. A, The proteins are composed of 257 to 278 amino acids with predicted molecular masses of 29 to 32 kDa. The hydrophobic transmembrane domain near the amino terminus, the selenocysteine residue (SeC), and two histidines (His) essential for catalytic activity are conserved in all three isoforms. The percentage of amino acid identity between the three isoforms in different portions of the molecule is given. A high degree of homology is noted in the regions surrounding the selenocysteine and one of the histidines. The dimerization domain as defined in the D1 is also shown. B, The structures of the genes, including the locations of the selenocysteine-encoding TGA codons and the regions coding for the selenocysteine insertion sequences (SECIS) located in the 3′-untranslated regions, are shown. Polymorphisms identified in each of the genes are also shown. DIO3os refers to the gene that encodes transcripts from the opposite DNA strand in the DIO3 locus and that remains incompletely characterized.
In addition, all deiodinases contain a hydrophobic region near the N-terminus,81 and subcellular fractionation studies have confirmed that these enzymes are integral membrane proteins.82,83 Such studies, along with fluorescent microscopy of epitope-tagged enzymes, have demonstrated that the D2 is located in the endoplasmic reticulum,82,84 whereas a significant fraction of the D3 has been reported to be in the plasma membrane.85 The location of the D1 may vary in different tissues; in renal epithelial cells, the D1 has been localized to the basolateral plasma membranes,86 whereas in the liver, it appears to reside in the endoplasmic reticulum (Pallud, Croteau, and St. Germain, unpublished observations). Studies by Friesema et al.87 demonstrate that the catalytic effectiveness of all three deiodinases is markedly increased when specific thyroid hormone transporters are coexpressed with the deiodinases in cultured cells. This finding indicates that the active catalytic sites of these enzymes are located in the intracellular compartment.
Genetic Considerations and Knockout Mouse Models
The three deiodinase isoforms are coded in mammals by three different genes located in humans on chromosomes 1 (D1) and 14 (D2 and D3), as indicated in Table 4-2.40–42 Although their genetic structure appears relatively simple in that they contain only four, three, and a single exon, respectively (Fig. 4-3B),40,88,89 surprising complexities that may influence expression patterns have been identified.90,91 For example, the DIO3 gene locus also expresses antisense transcripts from the DIO3os gene coded on the opposite DNA strand, and in the mouse fetus, the D3 is imprinted; transcription preferentially occurs from the paternal allele.91 In humans the DIO3 is also located within an imprinted region of chromosome 14.91 This pattern of organization and expression of the D3 often is observed for genes important in development.92
To enhance our understanding of the physiologic roles of the deiodinases, genetic knockout models of each of the isoforms have been developed and partially characterized (Table 4-3).93–100 D1-deficient mice show no general phenotypic abnormalities, although serum T4 and rT3 levels are modestly elevated, and the excretion patterns of iodothyronines in the feces and of iodine in the urine are significantly altered.93 D2-deficient mice are viable and fertile in a protected laboratory setting. However, observed impairments in hearing,96 thermogenesis,95 and neurocognition97 in these mutants would likely result in lethality in the wild. These animals also manifest elevated T4 and thyroid-stimulating hormone (TSH) levels and show a marked selective resistance to the feedback effects of T4, demonstrating the importance of D2 in pituitary thyrotroph regulation.94 This contrasts with D1-deficient mice, in which TSH levels are normal.93 This implies that the D1 expressed in the pituitary is not involved in TSH regulation.
Table 4-3
Phenotypic Features of Deiodinase Knockout Mice
D1-Deficient Mouse93
D2-Deficient Mouse
A striking finding in the D1- and D2-deficent mouse models is that serum T3 levels are normal.93,94 Indeed, in mice cross-bred to yield a combined D1 and D2 deficiency, the serum T3 level is only minimally decreased (V. A. Galton, unpublished observations).101 This finding is both unexpected and difficult to explain, given that 5′-deiodination in peripheral tissues is supposed to contribute 80% of the T3 production in humans and 45% in rats, with the remainder secreted directly from the thyroid.34,102 Given that the combined D1/D2 knockout mouse has been unequivocally shown to lack all 5′-deiodinating capacity, it is clear that at least in the rodent, thyroidal production and secretion of T3, perhaps combined with a decreased rate of T3 clearance from D1 deficiency, can compensate and maintain serum T3 levels that are essentially normal in the face of an inability to convert T4 to T3 in all tissues. A surprising corollary to this observation is that a D1 deficiency does not appear to significantly compromise T3 secretion from the rodent thyroid, a tissue in which this enzyme is highly expressed and previously was thought to be essential for T3 production.102,103 This suggests that under physiologic conditions, D1 acts primarily as a “scavenger” enzyme that prevents the buildup of lesser iodothyronines (rT3, T2s, T1) and sulfated derivatives in the thyroid gland, blood, and tissues.
The extent to which humans may be able to compensate for at least a partial impairment in 5′-deiodinating capacity has recently been reported. Although to date no mutations in the deiodinase genes in humans have been described, Dumitrescu et al.104 have described two families with partially inactivating mutations in the selenocysteine insertion sequence-binding protein2 (SBP2), which is essential for the synthesis of selenoproteins, including the deiodinases.74 Affected individuals demonstrate short stature and delayed bone age along with modest abnormalities in serum thyroid hormone levels, including increases in total T4 and rT3.104 Notably, total serum T3 levels were only minimally reduced, whereas TSH levels were at the upper part of the reference range and proved relatively resistant to suppression by administered T4, but not T3. These findings are very similar to those observed in knockout mice with a combined deficiency in D1 and D2, as noted previously. Indeed, a deficiency in D2 activity was demonstrated in cultured skin fibroblasts from affected patients.104
Mice deficient in D3 present the most severe phenotype, likely as a result of the importance of this enzyme during the developmental period. Observed abnormalities include significant perinatal mortality, growth retardation, impaired fertility, and altered systemic thyroid hormone levels during the perinatal period (transient hyperthyroidism) and in adulthood (moderate hypothyroidism).99,100 Multiple abnormalities, including impaired response of the thyroid gland to TSH and of the pituitary to TRH, have been observed at all levels of the thyroid axis in the D3-deficient adult mouse. The response to hypothyroidism of TRH mRNA production in the hypothalamus and of pituitary TSH production is also markedly impaired. Many of these abnormalities are similar to those observed in human infants born to mothers with poorly controlled hyperthyroidism during pregnancy105,106 or to rodents treated with large doses of thyroid hormone during the perinatal period.107,108 These findings demonstrate that strict control of thyroid hormone levels during development by D3 is critical for proper development of the thyroid axis.
Regulation
The deiodinases are regulated by multiple hormones, growth factors, and environmental and nutritional factors.34 Foremost among these are the thyroid hormones themselves; alterations in thyroid status induce profound changes in enzyme activity (see Table 4-2). Hypothyroidism is associated with a marked decrease in D1 and D3 levels, whereas D2 activity increases severalfold. Opposite changes occur in hyperthyroidism. These changes result from both pretranslational and posttranslational mechanisms.34 For example, D1 and D3 mRNA levels are increased in the hyperthyroid state,53,109 and D2 mRNA is decreased.54 Hyperthyroidism also results in rapid downregulation of D2 activity by ubiquitination of the D2 protein.110,111 This induces a reversible conformational change in the enzyme’s dimerization structure, which results in reversible loss of activity and eventually may target the enzyme for proteasomal degradation.112 Considerable progress has been made in defining the molecular mechanisms involved in transcriptional control of the deiodinases; this topic has been reviewed recently by Gereben et al.37 Of particular interest has been the observation by Simonides et al.113 that hypoxia induces D3 expression in several cell culture systems, as well as in ischemic myocardium.
Other important regulatory effects on deiodinase activity are noted in the thyroid gland, where TSH and thyroid-stimulating immunoglobulins stimulate both D1 and D2 activity60; in brown adipose tissue, where cold exposure,114 bile acids,115 or intracerebroventricular administration of leptin116 markedly stimulates D2 activity; and in the liver, where nutritional deprivation, diabetes, tumor necrosis factor, and other cytokines decrease D1 activity (see also below).109,117,118 In addition, D2 activity in the brain displays a significant diurnal variation.119 Although the physiologic significance of this remains to be determined, in some seasonal breeding species, rhythmic alterations in D2 and D3 activity in the central nervous system, perhaps mediated by melatonin,120 appear to be integral responses to changes in the photoperiod.121,122
Alternative Routes of Iodothyronine Metabolism
In addition to deiodination, iodothyronines are metabolized by multiple other mechanisms.123 In this regard, conjugation of the phenolic ring hydroxyl group to sulfate or glucuronide probably represents the second most prevalent mechanism of thyroid hormone metabolism.16 Sulfation and glucuronidation are inactivating reactions because the compounds formed are devoid of thyromimetic activity, do not bind to nuclear thyroid hormone receptors, and are rapidly metabolized by D1 or excreted in bile.4,16 However, sulfation is a reversible process that occurs through the action of tissue sulfatases or bacterial sulfatases in the intestine.124 Thus T3 sulfate (T3S) injected into hypothyroid rats demonstrates approximately 20% of the thyromimetic activity of native T3 because of liberation of this active hormone.125
Sulfotransferases in the cytoplasm of many tissues serve to catalyze the formation of iodothyronine sulfate conjugates. In rat tissue homogenates, 3,3′-T2 and T3 appear to be the best substrates for sulfation.46 Although the exact enzymes mediating iodothyronine sulfation are uncertain, the phenol sulfotransferases found in human liver and kidney have been demonstrated in vitro to possess this activity.126 Sensitive and specific radioimmunoassays for various iodothyronine sulfates have been developed and used to demonstrate detectable but very low levels of T3S, rT3S, and T4S in normal adult human serum.46 These low circulating levels probably reflect the rapid metabolism of these compounds by D1. Evidence for this thesis comes from experiments in animals and humans, in which treatment with inhibitors of D1 activity, such as PTU or iopanoic acid, results in marked increases in iodothyronine sulfoconjugate levels in both serum and bile.127,128 In other circumstances where D1 activity is low or impaired, such as during fetal life, in hypothyroidism, or in nonthyroidal illness, serum levels of these compounds also are elevated, either in absolute terms or relative to the native, unconjugated iodothyronines.16 To date, the physiologic role of sulfation in thyroid hormone economy has not been clearly defined, although it appears to represent an important component of the degradative process.
Glucuronidation of iodothyronines followed by their secretion into bile represents another pathway of iodothyronine clearance. Studies in humans, however, suggest that this pathway accounts for less than 1% of the total clearance of T4 by the liver, and only minute amounts of these conjugates are present in plasma.5,129
Two enzymes, l-amino acid oxidase and thyroid hormone aminotransferase, have been implicated in catalyzing the deamination of T4 and T3 to their acetic acid analogues Tetrac and Triac, respectively.130 Current evidence suggests, however, that these reactions occur to only a limited extent in normal humans. Triac has intrinsic biological activity equivalent to that of T3 when tested in in vitro assay systems,130 and it binds with greater avidity than T3 to the β isoform of the thyroid hormone receptor.1 Injection of Triac into humans or animals results in significant physiologic effects; however, relatively large doses are required because of its apparent rapid clearance and degradation via deiodination and glucuronidation.131 Despite its short half-life, Triac has been used successfully in the treatment of thyroid hormone resistance states.1 As in the case of sulfated conjugates, levels of Triac and its conjugates increase in circumstances in which D1 activity is low or impaired, such as during fasting or treatment with PTU or iopanoic acid.130 The physiologic relevance of these observations is uncertain but could be important, given Triac’s significant intrinsic activity. As noted earlier, the novel compound 3-iodothyronamine, formed by sequential deiodination and decarboxylation of thyroid hormones, may have important physiologic effects in some species, although its role in humans has not yet been defined.10,11
A final mechanism of thyroid hormone metabolism involves oxidative cleavage of the ether link of T4 and T3 by phagocytosing leukocytes.132 Current evidence suggests that this pathway is a minor one for iodothyronine degradation, except in the special circumstances of severe bacterial infection.
Thyroid Hormone Uptake Into Cells
Although some nongenomic effects of thyroid hormones may be triggered directly by hormones residing within the extracellular space via binding to cell surface receptors,133 the effects of these compounds on gene transcription, as well as on other cellular events, require their uptake into the cellular compartment. Similarly, the metabolic processes described above likely take place in various intracellular locations.87 Thus, the uptake of thyroid hormones into cells is a prerequisite for their affecting physiologic functions and for their metabolism.
Several membrane proteins capable of transporting thyroid hormones into and perhaps out of cells have been identified recently.134 Members of the organic anion-transporting polypeptide (OATP) family135 and the monocarboxylate transporter (MCT) family136 have been best characterized with regard to their capacity to affect cellular uptake and facilitate metabolism of various thyroid hormones. OATP1C1 has a relatively high affinity for T4 and rT3 and is highly expressed in capillaries in the rodent brain, suggesting a role in the transport of T4 across the blood-brain barrier. It also is widely expressed in the human brain.137 The MCT8 transporter facilitates cellular uptake of T3 and T4 when expressed in Xenopus laevis oocytes and cultured COS1 cells, and results in marked enhancement in the rate of iodothyronine metabolism by the deiodinases.87,138 The transporter is expressed in the choroid plexus and neuronal cells in the brain, as well as in the liver, kidney, heart, and placenta.134 It also appears to facilitate thyroid hormone exit from cells, which is likely important for overall cellular homeostasis.134
Recently, knockout mouse models of MCT8 deficiency have been described that verify the importance of this transporter in thyroid hormone homeostasis.139,140 Key features of these mice include elevations in serum T3 level, decreases in serum T4 and rT3 levels, and decreased brain content of T3. Deiodinase activities are also altered in several tissues secondary to tissue-specific patterns of thyroid hormone uptake. Thus, D1 expression is increased in the liver as the result of thyrotoxicosis in that organ (despite MCT8 deficiency) from high serum T3 levels, whereas the relative brain hypothyroidism results in elevated D2 activity and decreased D3 activity in that tissue. It is surprising to note that the neurologic phenotype of MCT8-deficient mice appears to be mild. This contrasts sharply with the severe phenotype of humans bearing mutations in MCT8, which results in the Allan-Herndon-Dudley syndrome.141 Male patients with this disorder (the MCT8 gene is located on the X chromosome) display poor muscle tone, an inability to speak, and severe mental retardation. Serum thyroid hormone parameters in these patients are analogous to those observed in MCT8-deficient mice. The striking differences in neurological phenotype between the two species is as yet unexplained.
An Integrated View of Thyroid Hormone Metabolism
Altered Thyroid States
Several examples of these principles can be cited, starting first with the response to alterations in thyroid hormone status. Experimental studies, as well as clinical experience, indicate that serum T3 levels tend to be maintained within the normal range in patients with moderate degrees of hypothyroidism despite the attendant hypothyroxinemia.142,143 Several factors, including an increase in the relative proportion of T3 secreted from the thyroid gland and an increase in the proportion of T4 converted to T3 in extrathyroidal tissue, appear to be responsible for this finding144 (Fig. 4-4). Both of these effects result from increased rates of 5′-deiodination. Thus in the thyroid gland, D1 and D2 activities are stimulated by the increase in TSH that accompanies hypothyroidism.60 In peripheral tissues, T4 to T3 conversion mediated by D2, which recently has been postulated to be the major source of plasma T3 in euthyroid humans,145 is also relatively enhanced by increased D2 activity.54 As a result, T4 is used more efficiently for T3 production. In addition, the rate of T3 clearance in extrathyroidal tissues is decreased. This decrease probably results in part from diminished 5-deiodination caused by the decreases in D1 and D3 activity that have been observed in the hypothyroid state. The physiologic importance of these extrathyroidal mechanisms, which in essence act as a form of peripheral autoregulation to help maintain T3 levels, is easily demonstrated by the administration of varying doses of T4 to athyreotic individuals. Under such circumstances, the circulating T3/T4 ratio is highest when low doses of T4 are given, and the ratio progressively declines as full replacement and then supraphysiologic doses are provided.142,143 Alterations in the rates of nondeiodinative pathways may contribute to this response. The net effect of these metabolic adaptations is to minimize the decrease in circulating T3 levels in the face of impaired secretion from the thyroid gland.
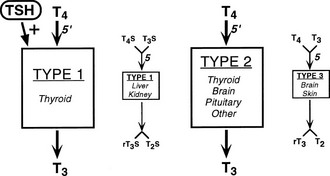
FIGURE 4-4 Autoregulation of thyroid hormone metabolism that accompanies the hypothyroid state, as might occur in iodine deficiency. D1 activity in the thyroid is stimulated by thyroid-stimulating hormone (TSH). Although this enzyme is relatively inefficient in converting T4 to T3, in the high T4 environment of the thyroid gland, this may serve to increase the proportion of T3 formed and secreted by this organ. Concurrently, D1 activity in extrathyroidal tissues is reduced, thereby decreasing the degradation of T4 and T3 by the 5-deiodination of their sulfated analogues. D2 activity is increased in all tissues that express this enzyme, which increases the proportion of T4 to T3 conversion in both the thyroid and peripheral tissues. Finally, D3 activity is decreased, again serving to diminish T4 and T3 degradation by 5-deiodinase processes. The net effect of these changes is to increase the relative rate of T3 production and decrease T3 degradation, thereby preserving the circulating and tissue levels of this active hormone in the presence of a diminished supply of T4.
In the hyperthyroid state that occurs with Graves’ disease, D1 activity is markedly increased both in peripheral tissues (as a result of the thyrotoxic state) and in the thyroid gland (because of thyroid-stimulating immunoglobulins that mimic the D1-stimulating effects of TSH).34 Although relatively inefficient in converting T4 to T3, this enhanced D1 activity has been shown recently to be the major source of circulating T3 in this condition, as well as in toxic multinodular goiter, with much of the D1-mediated T3 production occurring in the hyperactive thyroid gland itself.103 However, given the marked increase in thyroidal T4 and T3 production in the hyperthyroid state, the increase in D1 expression in extrathyroidal tissues appears to be paradoxical in that it would serve to further increase serum T3 levels. But such may not be the case; the administration of high doses of T3 to D1-deficient mice results in both higher serum T3 levels and a greater degree of tissue thyrotoxicosis as compared with thyrotoxic wild-type animals.93 Thus, enhanced expression of D1 in the liver and kidney of hyperthyroid animals may serve to actually mitigate the increase in serum T3, and thus the degree of thyrotoxicosis, presumably as a result of the inactivating 5-deiodinating capability of this enzyme.
The changes in D2 and D3 activity observed in hypothyroidism and hyperthyroidism have important additional effects. In organs that express these enzymes, alterations in activity provide a local mechanism for the maintenance of T3 concentrations at the cellular level. Thus in the hypothyroid rat infused with increasing doses of T4, normalization of the T3 content of D2-expressing tissues, such as the cerebral cortex, cerebellum, and brown adipose tissue, is attained at much lower infusion rates than are required by other tissues.146 This presumably reflects an enhanced rate of T4 to T3 conversion in these tissues by the elevated D2 activity. In contrast, the increased level of D3 expression in the brain that accompanies hyperthyroidism may explain why nuclear T3 levels are observed to be normal in most regions of this organ under this abnormal condition.147 The deiodinases thus appear to provide an additional level of autoregulation in tissues where thyroid hormone effects are especially critical.
Development
Thyroid hormones are of unquestioned importance to the developing fetus and during the neonatal period; hypothyroidism at this time can result in the clinical syndrome of cretinism characterized by severe neurologic impairment.148 However, exposure during development to excessive levels of thyroid hormones promotes premature differentiation of fetal tissues and thus is also detrimental.149,150 Throughout most of gestation, circulating levels of total T4 and total T3 in the mammalian fetus are extremely low,151 although significant amounts of free hormone have been demonstrated in amniotic fluid and fetal serum.152 Thus a key role of the uteroplacental unit is to limit access of the large pool of maternal thyroid hormones to the fetus while at the same time allowing small, but appropriate amounts of hormone to reach the developing embryo.153 This is accomplished by expression in the pregnant uterus25,154 and in the placenta67 of exceedingly high levels of D3 activity. (D2 is also expressed in these tissues, although its role remains undefined.66,155) Uterine D3 and D2 activities are regulated in a complex fashion by estrogens, progesterone, and probably other factors.156,157 However, the marked rise in uterine D3 expression observed during pregnancy is induced immediately and directly by implantation and represents an integral aspect of the uterine decidualization reaction.156 As implied above, high levels of D3 in the pregnant uterus and subsequently in the placenta do not result in a complete barrier to the transfer of maternal thyroid hormones to the fetus, as evidenced by the finding of significant levels of T4 and T3 at term in the serum of athyreotic infants.158 Transporter proteins such as MCT8 likely also play an important role in thyroid hormone homeostasis in the fetal-placental unit.159
Most mammalian fetal tissues, including the human fetal brain,160,161 also express deiodinases, with D3 activity largely predominating in the early and mid stages of development.66 During the later stages of gestation and in the neonatal period, after fetal thyroid function has been initiated, expression of 5′-deiodinases becomes more prevalent. In this regard, expression of D2 in the fetal and neonatal brain appears to be critically important for supplying T3 for the normal development of this tissue.162 This concept has been verified directly with regard to cochlear development; failure of the normal induction of D2 expression in the rodent cochlea shortly after birth results in marked hearing impairment.96,163 However, D2 expression in the developing brain may not be essential, or even important, in all brain regions, as demonstrated by the observation that the neurologic phenotype of the D2KO mouse is relatively mild compared with the rodent with developmental hypothyroidism.97 This observation points to an important role of T3 derived from the serum, which is the sole source of brain T3 in the developing D2KO mouse, in neurologic development.
The ordered and timely expression of the deiodinases during development is also critically important in lower species such as the metamorphosing tadpole.164–167
An important aspect of thyroid hormone metabolism during development relates to the very high levels of sulfated iodothyronines that circulate in the fetus, most likely as a result of the low D1 levels that occur during development and the fact that these metabolites are not efficiently degraded by D3.168 This observation has led to speculation that fetal T3S could serve as a reservoir of T3 that becomes available later in pregnancy through the actions of tissue sulfatases.169
Fasting and Illness
Profound changes occur in thyroid hormone economy during states of fasting and severe systemic illness, as detailed elsewhere in this volume. In humans, as well as in various animal models, the hallmark features induced by these conditions consist of marked decreases in the serum levels of total and free T3, accompanied by an inappropriately normal or reduced serum TSH level.170,171 Serum T4 levels are also reduced in fasted rodents and in critically ill humans. These alterations in circulating thyroid hormone levels are accompanied by reduced tissue levels of T3,172 and the severity of these changes in acute illness correlates closely with patient mortality rates.173 This generalized suppression of the thyroid axis is hypothesized by some observers to be of adaptive benefit in that it is associated with a significant decline in basal metabolic rate (BMR) in both humans and rodents,174,175 and it appears to lessen protein and fat catabolism.176,177
The triggering and homeostatic mechanisms that underlie this response of the thyroid axis are complex and remain poorly understood despite decades of experimental observations. Given the importance of the deiodinases in the maintenance of peripheral and tissue thyroid hormone levels, alterations in the expression and activity of these enzymes have been postulated to play important roles in the development of this syndrome.171 In particular, decreases in serum and tissue T3 levels observed in nutritional deprivation and illness have been attributed, at least in part, to the decreases in D1 activity and/or increases in D3 activity observed experimentally in the liver, muscle, heart, and other tissues of humans and experimental animals.109,113,173,178–180 In fasted or ill rodents, increased activity of D2 and decreased activity of D3 locally in the hypothalamus have been postulated to result in increased T3 levels in this tissue, thereby preventing or blunting the rise in TRH, and subsequently TSH, that otherwise would be expected to result from decreased serum T3 and T4 levels.181–183
However, it is important to note that these concepts are based entirely on indirect evidence. For example, the current concept that peripheral T3 production rates are diminished in fasting and illness is derived largely from in vivo kinetic studies that attempt to define rates of T4 and T3 production and clearance from serum measurements alone.184 Unfortunately, such studies are prone to misinterpretation. Thus, in the only two studies in which the in vivo rate of T4 to T3 conversion was measured directly in whole animals subjected to fasting, by injecting animals with [125]I-T4 and quantifying [125]I-T3 appearance, the fraction of T4 converted to T3 was approximately doubled, and total T3 production was unchanged.185,186 It thus appears that much of the T3 produced during fasting remains in the tissues, where it may be metabolized via other pathways, and is not exchanged with the plasma T3 pool.186 Such results serve to highlight the methodologic shortcomings inherent in kinetic studies of thyroid hormone metabolism in humans or rodents, where sampling is limited to the plasma compartment.
Caution should also be exercised in inferring causality between in vitro–determined deiodination rates in tissue homogenates and observed alterations in systemic thyroid hormone parameters.173 Studies conducted in deiodinase-deficient animals call into question the validity of such correlations. Thus, the changes in serum T4, T3, and TSH observed in fasted D1/D2KO mice are virtually identical to those in fasted wild-type mice (Galton/St. Germain, unpublished observations), and D3KO mice subjected to bacterial infection develop a nonthyroidal illness–type syndrome to the same extent as do infected wild-type mice.187 Notably, however, changes in deiodinase activities could affect T3 availability in selected cells or tissues and therefore could be important in the local response to nutritional deprivation, hypoxia, or inflammation, as suggested by Simonides et al.113
Thermogenesis
T3 and other thyroid hormone derivatives (e.g., 3,5-T2, 3-iodothyronamine) have been implicated in the control of metabolic rate and thermogenesis by both genomic and nongenomic mechanisms11,188 (vide supra). D2 has been demonstrated to be a key modulator of T3 availability and thus thermogenesis in brown adipose tissue (BAT) and other organs.95 The importance of D2 in energetic pathways has been highlighted recently by the report that in mice, the induction of D2 in BAT by bile acids, working through a unique G protein–coupled receptor (TGR5), protects against diet-induced obesity and insulin resistance.115 This suggests that the selective induction of D2 in thermogenic tissues could have therapeutic value in the treatment of obesity and diabetes. In this regard, da Silva et al.189 recently demonstrated that the small polyphenolic molecule kaempferol stimulates D2 activity and energy expenditure in cultured human skeletal muscle myoblasts.
Effects of Selenium
Nutritional selenium deprivation in experimental animals leads to characteristic changes in deiodinase activities and serum thyroid hormone levels.68 The most profound changes are noted in the liver and kidney, where marked decreases in D1 activity are noted secondary to impaired translation of selenoproteins.190 In tissues where selenium levels are better preserved, such as the thyroid gland and brain, deiodinase activities are altered to a lesser extent or not at all.102,191,192 Thus changes in serum thyroid hormone levels in selenium deficiency resemble those observed in other model systems where D1 activity is impaired, namely, T4 is increased and little change occurs in T3 or TSH.193 Similar observations in circulating hormone levels have been noted in human populations susceptible to selenium deficiency.194,195 In these circumstances, dietary selenium supplementation results in small but significant decreases in serum T4 levels and increases in the T3/T4 ratio, thus suggesting restoration of D1 activity. The clinical consequences of these changes have not been determined. It is notable that a recently reported randomized control trial of selenium supplementation in patients in an intensive care unit with severe sepsis found no direct effect of selenium on free and total serum thyroid hormone levels, although patient morbidity seemed to be lessened.196
Of note are reported cases of worsening hypothyroidism developing in individuals with combined selenium and iodine deficiency who were given selenium supplements alone.197 It is postulated that under such circumstances, restoration of D1 activity enhances T4 metabolism and thereby worsens the iodine-induced hypothyroxinemic state. Thus concurrent iodine and selenium supplementation is required to restore thyroid hormone economy in this situation.
Drug Effects
Numerous drugs are known to affect the metabolism of thyroid hormones by directly interfering with enzymatic mechanisms or by altering the regulation of these processes.198 The effect of PTU in inhibiting D1 activity provides it with a theoretical advantage over methimazole for use in the treatment of hyperthyroidism. In this condition, the increased expression of D1, combined with elevations in T4 levels, probably contributes to the overproduction of T3 in the thyroid gland.103 However, this effect of PTU is noted only with relatively high doses of the drug (>1000 mg/day). At the usual doses prescribed for treating hyperthyroidism, methimazole (10 mg three times daily) actually results in more rapid restoration of the euthyroid state than does PTU (100 mg three times daily).199
As noted previously, oral radiographic contrast dyes (e.g., iopanoic acid, sodium ipodate) are potent competitive inhibitors of all three deiodinase isoforms. Although they now are rarely used in clinical medicine as diagnostic agents, the rapidity with which they lower circulating T3 levels makes them extremely useful in the treatment of severe hyperthyroidism. For example, treatment of patients with Graves’ disease with sodium ipodate (1 g daily per os) has been noted to lower serum T3 levels by 58% within 24 hours of initiation of therapy—a decrease that is much greater than that noted with PTU (200 mg three times daily)200 or a saturated solution of potassium iodide (12 drops SSKI daily).201 Of importance, these decreases in serum T3 levels are associated with rapid improvement in cardiovascular complications, with beneficial effects on systemic resistance and cardiac output noted within 3 to 6 hours after treatment initiation.202 However, because of the high iodine content of these drugs, escape from their T3-lowering effects and exacerbation of hyperthyroidism may occur after several days of therapy.201 It thus is important that PTU or methimazole be administered concurrently to impair thyroidal secretion, and that use of the contrast agent be discontinued when thyrotoxicosis has been adequately controlled. It is notable that iopanoic acid (1 g daily per os for 13 days) has been used successfully to rapidly control the hyperthyroid state prior to thyroidectomy in patients with severe amiodarone-induced thyrotoxicosis.203 (Iopanoic acid is currently available only from compounding pharmacies.)
Propranolol, in relatively modest doses (80 mg/day), also blocks the conversion of T4 to T3 by acting as a competitive inhibitor of 5′-deiodinase activity.204 This drug therefore results in a 20% to 30% decrease in serum T3 levels and a slight elevation in serum T4 in hyperthyroid patients. Other commonly used β-blockers do not have this effect. High doses of glucocorticoids (e.g., 2 mg of dexamethasone four times daily) have been demonstrated to lower T3 levels within 24 hours in hyperthyroid patients with Graves’ disease,205 as well as in euthyroid individuals.23 This activity provides part of the rationale for the use of these agents in severe thyrotoxicosis. Animal studies have demonstrated that this effect is due, at least in part, to diminished total body production of T3, and indeed, decreased D1 activity has been observed in the liver of dexamethasone-treated rats.206 In addition, D2 activity in the brain of experimental animals is altered by several neuropharmacologic agents.207
Finally, the antiarrhythmic agent amiodarone is another iodine-rich compound that competitively inhibits the conversion of T4 to T3 and thus frequently causes a rise in serum T4 levels and a modest decrease in serum T3.208 Although most patients who take this drug remain euthyroid as judged by normal serum TSH levels, the large quantities of iodine liberated during the course of its metabolism can result in hypothyroid or hyperthyroid states, with the latter at times being extremely difficult to treat.209
Emerging Roles of the Deiodinases in Human Disease
Germline mutations in the deiodinase genes that significantly affect their function or levels of expression have not yet been identified in humans. However, in addition to the conditions noted above, several human disease states are associated with alterations in deiodinase activities. Thus, those rare individuals with inactivating mutations in the SBP2 RNA-binding protein manifest a significant decrease in D2 activity104 (vide supra). As a second example, patients with the McCune-Albright syndrome, which stems from activating mutations of the α subunit of the G stimulatory protein, have recently been demonstrated to have increases in thyroidal D1 and D2 activities.210 This provides an explanation for the increase in the serum T3/T4 ratio that is frequently encountered in these patients and the occurrence during childhood of frank T3 toxicosis. The increase in these 5′-deiodinases is believed to be secondary to the elevation in thyroidal cyclic adenosine monophosphate (cAMP) levels that is known to stimulate D2 expression.211
High levels of D1 and/or D2 expression also have been observed in metastatic foci of thyroid follicular carcinoma, again resulting in an increased T3/T4 ratio in patients with this disease.212,213 In cases of widely metastatic disease, T3 toxicosis may develop, depending on the amount of thyroxine supplementation that is provided.213,214 Thus, in patients with this condition, monitoring of the serum T3 level, in addition to serum T4 and TSH, is indicated to avoid a thyrotoxic state. Increased D2215 and D1216 activities also have been observed in toxic adenomas of the thyroid gland, and this likely contributes to the thyrotoxic state.
Perhaps the most dramatic example of deiodinase overexpression is the extraordinarily high level of D3 activity observed in some infantile hepatic hemangiomas.217 The “ectopic” D3 expression in these typically large tumors results in markedly enhanced clearance rates of T4 and T3, which overwhelm the synthetic capability of the thyroid gland and lead to a state of “consumptive hypothyroidism.” Very large doses of thyroid hormone supplements typically need to be administered if these patients are to be maintained in a euthyroid state. Recently, a 3-month-old infant with this condition was treated successfully with liver transplantation.218 Two cases of adults with consumptive hypothyroidism—one with a vascular hepatic tumor219 and another with a large fibrous abdominal tumor—have been described.220 D3 expression has also been reported in TSH-secreting pituitary adenomas, which may contribute to the observed resistance of these tumors to feedback by thyroid hormones.221
Finally, polymorphisms in the three human deiodinase genes have been reported (Fig. 4-3B), and much interest has been focused on possible clinical associations with these genetic variations.222 For example, Mentuccia et al.223 identified a polymorphism in the coding region of the human DIO2 gene that results in a nonconserved amino acid substitution of alanine for threonine at position 92 (Thr92Ala). Evidence has been presented that suggests that patients homozygous for the alanine variant exhibit decreased D2 activity in skeletal muscle.224 In an initial study, the alanine variant was found to be common in Pima Indians and Mexican Americans (allele frequencies of 0.75 and 0.42, respectively), in whom it was strongly associated with insulin resistance, and in patients carrying a second polymorphism in the β3-adrenergic receptor (Trp64Arg), with obesity.223 In small studies that used a different population group, an association with insulin resistance was again noted. However, studies of larger unselected populations have found no association between the alanine variant and the risk for diabetes or obesity.225,226 A similar lack of consistency between study results has been noted with regard to a possible association between this variant and hypertension.227,228 In other studies, polymorphisms in the 3′-untranslated region of the human DIO1 gene have been associated with small (3% to 7%) but significant differences in serum free T4, total T3, and free T3 levels in older subjects, as well as with changes in body composition.229,230
References
1. Takeda, T, Suzuki, S, Liu, RT, et al. Triiodothyroacetic acid has unique potential for therapy of resistance to thyroid hormone. J Clin Endocrinol Metab. 1995;80:2033–2040.
2. Yen, PM, Ando, S, Feng, X, et al. Thyroid hormone action at the cellular, genomic and target gene levels. Mol Cell Endocrinol. 2006;246:121–127.
3. Pittman, HA, Brown, RW, Register, HBJ. Biological activity of 3,3′,5′-triiodo-DL-thyronine. Endocrinology. 1962;70:79–83.
4. Spaulding, SW, Smith, TJ, Hinkle, PM, et al. Studies on the biological activity of triiodothyronine sulfate. J Clin Endocrinol Metab. 1992;74:1062–1067.
5. Engler, D, Burger, AG. The deiodination of the iodothyronines and their derivatives in man. Endo Reviews. 1984;5:151–184.
6. Lombardi, A, Lanni, A, de Lange, P, et al. Acute administration of 3,5-diiodo-L-thyronine to hypothyroid rats affects bioenergetic parameters in rat skeletal muscle mitochondria. FEBS Letters. 2007;581:5911–5916.
7. Moreno, M, Lombardi, A, Beneduce, L, et al. Are the effects of T3 on resting metabolic rate in euthyroid rats entirely caused by T3 itself? Endocrinology. 2002;143:504–510.
8. Grasselli, E, Canesi, L, Voci, A, et al. Effects of 3,5-diiodo-L-thyronine administration on the liver of high fat diet-fed rats. Exp Biol Med. 2008;233:549–557.
9. Lanni, A, Moreno, M, Lombardi, A, et al. 3,5-diiodo-L-thyronine powerfully reduces adiposity in rats by increasing the burning of fats. FASEB J. 2005;19:1552–1554.
10. Scanlan, TS, Suchland, KL, Hart, ME, et al. 3-Iodothyonamine is an endogenous and rapid-acting derivative of thyroid hormone. Nat Med. 2004;10:638–642.
11. Braulke, LJ, Klingenspor, M, DeBarber, A, et al. 3-Iodothyronamine: a novel hormone controlling the balance between glucose and lipid utilization. J Comp Phys B, Biochem, System. Environ Physiol. 2008;178:167–177.
12. Moreno, M, Kaptein, E, Goglia, F, et al. Rapid glucuronidation of tri- and tetraiodothyroacetic acid to ester glucuronides in human liver and to ether glucuronides in rat liver. Endocrinology. 1994;135:1004–1005.
13. Sorimachi, K, Yasumura, Y. High affinity of triiodothyronine (T3) for nonphenolic ring deiodinase and high affinity of tetraiodothyroacetic acid (TETRAC) for phenolic ring deiodinase in cultured monkey hepatocarcinoma cells and in rat liver homogenates. Endocrinol Jap. 1981;28:775–783.
14. Rutgers, M, Heusdens, FA, Visser, TJ. Metabolism of triiodothyroacetic acid (TA3) in rat liver. I. Deiodination of TA3 and TA3 sulfate by microsomes. Endocrinology. 1989;125:424–432.
15. Santini, F, Hurd, RE, Chopra, IJ. A study of metabolism of deaminated and sulfoconjugated iodothyronines by rat placental iodothyronine 5-monodeiodinase. Endocrinology. 1992;131:1689–1694.
16. Visser, TJ. Pathways of thyroid hormone metabolism. Acta Med Aust. 1996;23:10–16.
17. Chopra, IJ, Boado, RJ, Geffner, DL, et al. A radioimmunoassay for measurement of thyronine and its acetic acid analog in urine. J Clin Endocrinol Metab. 1988;67:480–487.
18. LoPresti, JS, Anderson, KP, Nicoloff, JT. Does a hidden pool of reverse triiodothyronine (rT3) production contribute to total thyroxine (T4) disposal in high T4 states in man? J Clin Endocrinol Metab. 1990;70:1479–1484.
19. Chopra, IJ. Nature, sources, and relative biologic significance of circulating thyroid hormones. In: Braverman LE, Utiger RD, eds. The Thyroid. ed 6. New York: Lippincott; 1991:136–143.
20. Chopra, IJ. Simultaneous measurement of free thyroxine and free 3,5,3′-triiodothyronine in undiluted serum by direct equilibrium dialysis/radioimmunoassay: evidence that free triiodothyronine and free thyroxine are normal in many patients with the low triiodothyronine syndrome. Thyroid. 1998;8:249–257.
21. Faber, J, Rogowski, P, Kirkegaard, C, et al. Serum free T4, T3, rT3, 3,3′-diiodothyronine and 3′,5′-diiodothyronine measured by ultrafiltration. Acta Endocrinol. 1984;107:357–365.
22. Nicoloff, JT, Low, JC, Dussault, JH, et al. Simaultaneous measurements of thyroxine and triiodothyronine peripheral turnover kinetics in man. J Clin Invest. 1972;51:473–483.
23. LoPresti, JS, Eigen, A, Kaptein, E, et al. Alterations in 3,3′,5′-triiodothyronine metabolism in response to propylthiouracil, dexamethasone, and thyroxine administration in man. J Clin Invest. 1989;84:1650–1656.
24. Glinoer, D. The regulation of thyroid function in pregnancy: pathways of endocrine adaptation from physiology to pathology. Endo Reviews. 1997;18:404–433.
25. Huang, SA, Dorfman, DM, Genest, DR, et al. Type 3 iodothyronine deiodinase is highly expressed in the human uteroplacental unit and in fetal epithelium. J Clin Endocrinol Metab. 2003;88:1384–1388.
26. Alexander, EK, Marqusee, E, Lawrence, J, et al. Timing and magnitude of increases in levothyroxine requirements during pregnancy in women with hypothyroidism. N Engl J Med. 2004;351:241–249.
27. Mariotti, S, Barbesino, G, Caturegli, P, et al. Complex alteration of thyroid function in healthy centenarians. J Clin Endocrinol Metab. 1993;77:1130–1134.
28. Gregerman, RI, Goffney, GW, Schck, NW. Thyroxine turnover in euthyroid man with special reference to changes with age. J Clin Invest. 1962;41:2065–2074.
29. Kabadi, UM. Variability of L-thyroxine replacement dose in elderly patients with primary hypothyroidism. J Fam Pract. 1987;24:473–477.
30. Peeters, RP. Thyroid hormones and aging. Hormones. 2008;7:28–35.
31. Gross, J, Pitt-Rivers, R. 3:5:3′-triiodothyronine. 2. Physiological activity. Biochem J. 1953;53:652–656.
32. Gross, J, Pitt-Rivers, R. 3:5:3′-triiodothyronine. 1. Isolation from thyroid gland and synthesis. Biochem J. 1953;53:645–652.
33. Braverman, LE, Ingbar, SH, Sterling, K. Conversion of thyroxine to triiodothyronine in athyreotic human subjects. J Clin Invest. 1970;49:855–864.
34. Bianco, AC, Salvatore, D, Gereben, B, et al. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endo Reviews. 2002;23:38–89.
35. Köhrle, J. Iodothyonine deiodinases. Meth Enzymol. 2002;347:125–167.
36. Kohrle, J. Thyroid hormone transporters in health and disease: advances in thyroid hormone deiodination. Best Prac Res Clin Endocrinol Metab. 2007;21:173–191.
37. Gereben, B, Zeold, A, Dentice, M, et al. Activation and inactivation of thyroid hormone by deiodinases: local action with general consequences. Cell Mol Life Sci. 2008;65:570–590.
38. St. Germain, DL, Galton, VA. The deiodinase family of selenoproteins. Thyroid. 1997;7:655–668.
39. Leonard, JL, Visser, TJ. Biochemistry of deiodination. In: Hennemann G, ed. Thyroid Hormone Metabolism. New York: Marcel Dekker; 1986:189–229.
40. Jakobs, TC, Koehler, MR, Schmutzler, C, et al. Structure of the human type I iodothyronine 5′-deiodinase gene and location to chromosome 1p32–p33. Genomics. 1997;42:361–363.
41. Celi, FS, Canettieri, G, Yarnell, DP, et al. Genomic characterization of the coding region of the human type II 5′-deiodinase. Mol Cell Endocrinol. 1998;141:49–52.
42. Hernández, A, Park, J, Lyon, GJ, et al. Localization of the type 3 iodothyronine deiodinase (DIO3) gene to human chromosome 14q32 and mouse chromosome 12F1. Genomics. 1998;53:119–121.
43. Leonard, JL, Visser, TJ, Leonard, DM. Characterization of the subunit structure of the catalytically active type I iodothyronine deiodinase. J Biol Chem. 2001;276:2600–2607.
44. Curcio-Morelli, C, Gereben, B, Zavacki, AM, et al. In vivo dimerization of types 1, 2, and 3 iodothyronine selenodeiodinases. Endocrinology. 2003;144:937–946.
45. Leonard, JL, Simpson, G, Leonard, DM. Characterization of the protein dimerization domain responsible for assembly of functional selenodeiodinases. J Biol Chem. 2005;280:11093–11100.
46. Visser, TJ. Role of sulfation in thyroid hormone metabolism. Chem Biol Interact. 1994;92:293–303.
47. Visser, TJ. Mechanism of inhibition of iodothyronine-5′-deiodinase by thioureylenes and sulfite. Biochim Biophys Acta. 1980;611:371–378.
48. Sharifi, J, St. Germain, DL. The cDNA for the type I iodothyronine 5′-deiodinase encodes an enzyme manifesting both high Km and low Km activity. J Biol Chem. 1992;267:12539–12544.
49. Sarma, BK, Mugesh, G. Thiol cofactors for selenoenzymes and their synthetic mimics. Organ Biomol Chem. 2008;6:965–974.
50. Croteau, W, Bodwell, JE, Richardson, JM, et al. Conserved cysteines in the type 1 deiodinase selenoprotein are not essential for catalytic activity. J Biol Chem. 1998;237:25230–25236.
51. Chaudiere, J, Tappel, AL. Interaction of gold(I) with the active site of selenium-glutathione peroxidase. J Inorg Biochem. 1984;20:313–325.
52. Berry, MJ, Kieffer, JD, Harney, JW, et al. Selenocysteine confers the biochemical properties characteristic of the type I iodothyronine deiodinase. J Biol Chem. 1991;266:14155–14158.
53. Croteau, W, Whittemore, SL, Schneider, MJ, et al. Cloning and expression of a cDNA for a mammalian type III iodothyronine deiodinase. J Biol Chem. 1995;270:16569–16575.
54. Croteau, W, Davey, JC, Galton, VA, et al. Cloning of the mammalian type II iodothyronine deiodinase: a selenoprotein differentially expressed and regulated in the human brain and other tissues. J Clin Invest. 1996;98:405–417.
55. Kohrle, J, Schomburg, L, Drescher, S, et al. Rapid stimulation of type I 5′-deiodinase in rat pituitaries by 3,3′,5′-triiodo-L-thyronine. Mol Cell Endocrinol. 1995;108:17–21.
56. Visser, TJ, Leonard, JL, Kaplan, MM, et al. Kinetic evidence suggesting two mechanisms for iodothyronine 5′-deiodination in rat cerebral cortex. Proc Natl Acad Sci USA. 1982;79:5080–5084.
57. Guadaño-Ferraz, A, Obregón, MJ, St. Germain, DL, et al. The type 2 iodothyronine deiodinase is expressed primarily in glial cells in the neonatal rat brain. Proc Natl Acad Sci USA. 1997;94:10391–10396.
58. Tu, HM, Kim, SW, Salvatore, D, et al. Regional distribution of type 2 thyroxine deiodinase messenger ribonucleic acid in rat hypothalamus and pituitary and its regulation by thyroid hormone. Endocrinology. 1997;138:3359–3368.
59. Diano, S, Leonard, JL, Meli, R, et al. Hypothalamic type II iodothyronine deiodinase: a light and electron microscopic study. Brain Res. 2003;976:130–134.
60. Salvatore, D, Tu, H, Harney, JW, et al. Type 2 iodothyronine deiodinase is highly expressed in human thyroid. J Clin Invest. 1996;98:962–968.
61. Kasahara, T, Tsunekawa, K, Seki, K, et al. Regulation of iodothyronine deiodinase and roles of thyroid hormones in human coronary artery smooth muscle cells. Atherosclerosis. 2006;186:207–214.
62. Kaplan, MM, Yaskoski, KA. Phenolic and tyrosyl ring deiodination of iodothyronines in rat brain homogenates. J Clin Invest. 1980;66:551–562.
63. Huang, T, Chopra, IJ, Beredo, A, et al. Skin is an active site of inner ring monodeiodination of thyroxine to 3,3′,5′-triiodothyronine. Endocrinology. 1985;117:2106–2113.
64. Heuer, H, Maier, MK, Iden, S, et al. The monocarboxylate transporter 8 linked to human psychomotor retardation is highly expressed in thyroid hormone-sensitive neuron populations. Endocrinology. 2005;146:1701–1706.
65. Alkemade, A, Friesema, EC, Unmehopa, UA, et al. Neuroanatomical pathways for thyroid hormone feedback in the human hypothalamus. J Clin Endocrinol Metab. 2005;90:4322–4334.
66. Bates, JM, St. Germain, DL, Galton, VA. Expression profiles of the three iodothyronine deiodinases, D1, D2 and D3, in the developing rat. Endocrinology. 1999;140:844–851.
67. Roti, E, Fang, SL, Green, K, et al. Human placenta is an active site of thyroxine and 3,3′,5-triiodothyronine tyrosyl ring deiodination. J Clin Endocrinol Metab. 1981;53:498–501.
68. St. Germain, DL. Selenium, deiodinases, and endocrine function. In: Hatfield DL, ed. Selenium: Its Molecular Biology and Role in Human Health. Boston: Kluwer Academic Publishers; 2001:189–202.
69. Berry, MJ, Maia, AL, Kieffer, JD, et al. Substitution of cysteine for selenocysteine in type I iodothyronine deiodinase reduces the catalytic efficiency of the protein but enhances its translation. Endocrinology. 1992;131:1848–1852.
70. Kuiper, GG, Klootwijk, W, Visser, TJ. Substitution of cysteine for selenocysteine in the catalytic center of type III iodothyronine deiodinase reduces catalytic efficiency and alters substrate preference. Endocrinology. 2003;144:2505–2513.
71. Lian, G, Ding, L, Chen, M, et al. A selenium-containing catalytic antibody with Type I deiodinase activity. Biochem Biophys Res Comm. 2001;283:1007–1012.
72. Stadtman, TC. Selenocysteine. Ann Rev Biochem. 1996;65:83–100.
73. Driscoll, DM, Copeland, PR. Mechanism and regulation of selenoprotein synthesis. Ann Rev Nutr. 2003;23:17–40.
74. Hoffmann, PR, Berry, MJ. Selenoprotein synthesis: a unique translational mechanism used by a diverse family of proteins. Thyroid. 2005;15:769–775.
75. Squires, JE, Berry, MJ. Eukaryotic selenoprotein synthesis: mechanistic insight incorporating new factors and new functions for old factors. IUBMB Life. 2008;60:232–235.
76. Leonard, DM, Stachelek, SJ, Safran, M, et al. Cloning, expression and functional characterization of the substrate binding subunit of rat type II iodothyronine 5′-deiodinase. J Biol Chem. 2000;275:25195–25201.
77. Montero-Pedrazuela, A, Bernal, J, Guadano-Ferraz, A. Divergent expression of type 2 deiodinase and the putative thyroxine-binding protein p29, in rat brain, suggests that they are functionally unrelated proteins. Endocrinology. 2003;144:1045–1052.
78. Barrantes, IdB, Montero-Pedrazuela, A, Guadano-Ferraz, A, et al. Generation and characterization of dickkopf3 mutant mice. Mol Cell Biol. 2006;26:2317–2326.
79. Callebaut, I, Curcio-Morelli, C, Mornon, J-P, et al. The iodothyronine selenodeiodinases are thioredoxin-fold family proteins containing a glycoside hydrolase clan GH-A-like structure. J Biol Chem. 2003;278:36887–36896.
80. Kuiper, GGJM, Klootwijk, W, Morvan Dubois, G, et al. Characterization of recombinant Xenopus laevis type I iodothyronine deiodinase: substitution of a proline residue in the catalytic center by serine (Pro132Ser) restores sensitivity to 6-propyl-2-thiouracil. Endocrinology. 2006;147:3519–3529.
81. Toyoda, N, Berry, MJ, Harney, JW, et al. Topological analysis of the integral membrane protein, type 1 iodothyronine deiodinase (D1). J Biol Chem. 1995;270:12310–12318.
82. Courtin, F, Pelletier, G, Walker, P. Subcellular localization of thyroxine 5′-deiodinase activity in bovine anterior pituitary. Endocrinology. 1985;117:2527–2533.
83. Schoenmakers, CH, Pigmans, IG, Visser, TJ. Investigation of type I and type III iodothyronine deiodinases in rat tissues using N-bromoacetyl-iodothyronine affinity labels. Mol Cell Endocrinol. 1995;107:173–180.
84. Baqui, MMA, Gerebon, B, Harney, JW, et al. Distinct subcellular localization of transiently expressed types 1 and 2 deiodinases as determined by immunofluorescence confocal microscopy. Endocrinology. 2000;141:4309–4312.
85. Baqui, M, Botero, D, Gereben, B, et al. Human type 3 iodothyronine selenodeiodinase is located in the plasma membrane and undergoes rapid internalization to endosomes. J Biol Chem. 2003;278:1206–1211.
86. Leonard, JL, Ekenbarger, DM, Frank, SJ, et al. Localization of type I iodothyronine 5′-deiodinase to the basolateral plasma membrane in renal cortical epithelial cells. J Biol Chem. 1991;266:11262–11269.
87. Friesema, ECH, Kuiper, GGJM, Jansen, J, et al. Thyroid hormone transport by the human monocarboxylate transporter 8 and its rate-limiting role in intracellular metabolism. Mol Endocrinol. 2006;20:2761–2772.
88. Celi, FS, Canettieri, G, Mentuccia, D, et al. Structural organization and chromosomal localization of the human type II deiodinase gene. Eur J Endocrinol. 2000;143:267–271.
89. Hernández, A, Lyon, GJ, Schneider, MJ, et al. Isolation and characterization of the mouse gene for the type 3 iodothyronine deiodinase. Endocrinology. 1999;140:124–130.
90. Gereben, B, Kollar, A, Harney, JW, et al. The mRNA structure has potent regulatory effects on type 2 iodothyronine deiodinase expression. Mol Endocrinol. 2002;16:1667–1679.
91. Hernandez, A, Fiering, S, Martinez, E, et al. The gene locus encoding iodothyronine deiodinase type 3 (Dio3) is imprinted in the fetus and expresses antisense transcripts. Endocrinology. 2002;143:4483–4486.
92. Reik, W, Walter, J. Genomic imprinting: parental influence on the genome. Nat Rev. Genetics. 2001;2:21–32.
93. Schneider, MJ, Fiering, SN, Thai, B, et al. Targeted disruption of the type 1 selenodeiodinase gene (Dio1) results in marked changes in thyroid hormone economy in mice. Endocrinology. 2006;147:580–589.
94. Schneider, MJ, Fiering, SN, Pallud, SE, et al. Targeted disruption of the type 2 selenodeiodinase gene (Dio2) results in a phenotype of pituitary resistance to T4. Mol Endocrinol. 2001;15:2137–2148.
95. de Jesus, LA, Carvalho, SD, Ribeiro, MO, et al. The type 2 iodothyronine deiodinase is essential for adaptive thermogenesis in brown adipose tissue. J Clin Invest. 2001;108:1379–1385.
96. Ng, L, Goodyear, RJ, Woods, CA, et al. Hearing loss and retarded cochlear development in mice lacking type 2 iodothyronine deiodinase. Proc Natl Acad Sci USA. 2004;101:3473–3479.
97. Galton, VA, Wood, ET, St. Germain, EA, et al. Thyroid hormone homeostasis and action in the type 2 deiodinase-deficient brain during development. Endocrinology. 2007;148:3080–3088.
98. Christoffolete, MA, Linardi, CC, de Jesus, L, et al. Mice with targeted disruption of the Dio2 gene have cold-induced overexpression of the uncoupling protein 1 gene but fail to increase brown adipose tissue lipogenesis and adaptive thermogenesis. Diabetes. 2004;53:577–584.
99. Hernandez, A, Martinez, E, Fiering, S, et al. Type 3 deiodinase is critical for the maturation and function of the thyroid axis. J Clin Invest. 2006;116:476–484.
100. Hernandez, A, Martinez, ME, Liao, XH, et al. Type 3 deiodinase deficiency results in functional abnormalities at multiple levels of the thyroid axis. Endocrinology. 2007;148:5680–5687.
101. Christoffolete, MA, Arrojo e Drigo, R, Gazoni, F, et al. Mice with impaired extrathyroidal thyroxine to 3,5,3′-triiodothyronine conversion maintain normal serum 3,5,3′-triiodothyronine concentrations. Endocrinology. 2007;148:954–960.
102. Chanoine, J, Braverman, LE, Farwell, AP, et al. The thyroid gland is a major source of circulating T3 in the rat. J Clin Invest. 1993;91:2709–2713.
103. Laurberg, P, Vestergaard, H, Nielsen, S, et al. Sources of circulating 3,5,3′-triiodothyronine in hyperthyroidism estimated after blocking of type 1 and type 2 iodothyronine deiodinases. [see comment]. J Clin Endocrinol Metab. 2007;92:2149–2156.
104. Dumitrescu, AM, Liao, X-H, Abdullah, MSY, et al. Mutations in SECISBP2 result in abnormal thyroid hormone metabolism. Nat Genetics. 2005;37:1247–1252.
105. Kempers, MJE, van Trotsenburg, ASP, van Tijn, DA, et al. Disturbance of the fetal thyroid hormone state has long-term consequences for treatment of thyroidal and central congenital hypothyroidism. J Clin Endocrinol Metab. 2005;90:4094–4100.
106. Kempers, MJE, van Trotsenburg, ASP, van Rijn, RR, et al. Loss of integrity of thyroid morphology and function in children born to mothers with inadequately treated Graves’ disease. J Clin Endocrinol Metab. 2007:2006–2042.
107. Bakke, JL, Lawrence, NL, Bennett, J, et al. The late effects of neonatal hyperthyroidism upon the feedback regulation of TSH secretion in rats. Endocrinology. 1975;97:659–664.
108. Dussault, JH, Coulombe, P, Walker, P. Effects of neonatal hyperthyroidism on the development of the hypothalamic-pituitary-thyroid axis in the rat. Endocrinology. 1982;110:1037–1042.
109. O’Mara, BA, Dittrich, W, Lauterio, TJ, et al. Pretranslational regulation of type I 5′-deiodinase by thyroid hormones and in fasted and diabetic rats. Endocrinology. 1993;133:1715–1723.
110. Gereben, B, Goncalves, C, Harney, JW, et al. Selective proteolysis of human type 2 deiodinase: a novel ubiquitin-proteasomal mediated mechanism for regulation of hormone activation. Mol Endocrinol. 2000;14:1697–1708.
111. Curcio-Morelli, C, Zavacki, AM, Christofollete, M, et al. Deubiquitination of type 2 iodothyronine deiodinase by von Hippel-Lindau protein-interacting deubiquitinating enzymes regulates thyroid hormone activation. J Clin Invest. 2003;112:189–196.
112. Sagar, GDV, Gereben, B, Callebaut, I, et al. Ubiquitination-induced conformational change within the deiodinase dimer is a switch regulating enzyme activity. Mol Cell Biol. 2007;27:4774–4783.
113. Simonides, WS, Mulcahey, MA, Redout, EM, et al. Hypoxia-inducible factor induces local thyroid hormone inactivation during hypoxic-ischemic disease in rats. J Clin Invest. 2008;118:975–983.
114. Silva, JE, Larsen, PR. Adrenergic activation of triiodothyronine production in brown adipose tissue. Nature. 1983;305:712–713.
115. Watanabe, M, Houten, SM, Mataki, C, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. [see comment]. Nature. 2006;439:484–489.
116. Cettour-Rose, P, Burger, AG, Meier, CA, et al. Central stimulatory effect of leptin on T3 production is mediated by brown adipose tissue type II deiodinase. Am J Physiol. 2002;283:E980–987.
117. Nagaya, T, Fujieda, M, Otsuka, G, et al. A potential role of activated NF-kappa B in the pathogenesis of euthyroid sick syndrome. J Clin Invest. 2000;106:393–402.
118. Yu, J, Koenig, RJ. Regulation of hepatocyte thyroxine 5′-deiodinase by T3 and nuclear receptor coactivators as a model of the sick euthyroid syndrome. J Biol Chem. 2000;275:38296–38301.
119. Campos-Barros, A, Musa, A, Flechner, A, et al. Evidence for circadian variations of thyroid hormone concentrations and type II 5′-iodothyronine deiodinase activity in the rat central nervous system. J Neurochem. 1997;68:795–803.
120. Yasuo, S, Yoshimura, T, Ebihara, S, et al. Temporal dynamics of type 2 deiodinase expression after melatonin injections in Syrian hamsters. Endocrinology. 2007;148:4385–4392.
121. Yasuo, S, Watanabe, M, Nakao, N, et al. The reciprocal switching of two thyroid hormone-activating and -inactivating enzyme genes is involved in the photoperiodic gonadal response of Japanese quail. Endocrinology. 2005;146:2551–2554.
122. Nakao, N, Ono, H, Yamamura, T, et al. Thyrotrophin in the pars tuberalis triggers photoperiodic response. Nature. 2008;452:317–322.
123. Wu, S-Y, Green, WL, Huang, W-S, et al. Alternate pathways of thyroid hormone metabolism. Thyroid. 2005;15:943–958.
124. Kung, MP, Spaulding, SW, Roth, JA. Desulfation of 3,5,3′-triiodothyronine sulfate by microsomes from human and rat tissues. Endocrinology. 1988;122:1195–1200.
125. Santini, F, Hurd, RE, Lee, B, et al. Thyromimetic effects of 3,5,3′-triiodothyronine sulfate in hypothyroid rats. Endocrinology. 1993;133:105–110.
126. Kester, MH, Kaptein, E, Roest, TJ, et al. Characterization of human iodothyronine sulfotransferases. J Clin Endocrinol Metab. 1999;84:1357–1364.
127. Chopra, IJ, Wu, SY, Teco, GN, et al. A radioimmunoassay for measurement of 3,5,3′-triiodothyronine sulfate: studies in thyroidal and nonthyroidal diseases, pregnancy, and neonatal life. J Clin Endocrinol Metab. 1992;75:189–194.
128. Eelkman Rooda, SJ, Kaptein, E, Rutgers, M, et al. Increased plasma 3,5,3′-triiodothyronine sulfate in rats with inhibited type I iodothyronine deiodinase activity, as measured by radioimmunoassay. Endocrinology. 1989;124:740–745.
129. Van Middlesworth, L. Metabolism and excretion of thyroid hormones. In: Greer M, Solomon DH, eds. Handbook of Physiology: Endocrinology. Washington, D.C.: American Physiological Society; 1974:215–231.
130. Siegrist-Kaiser, CA, Burger, AG. Modification of the side chain of thyroid hormones. In: Wu S-Y, Visser TJ, eds. Thyroid Hormone Metabolism. Boca Raton: CRC Press; 1994:175–198.
131. Liang, H, Juge-Aubry, CE, O’Connell, M, et al. Organ-specific effects of 3,5,3′-triiodothyroacetic acid in rats. Eur J Endocrinol. 1997;137:537–544.
132. Green, WL, Ether-link cleavage of iodothyronines. S-Y Wu. TJ Visser. Thyroid hormone metabolism, Boca Raton: CRC Press, 1994:199–221.
133. Davis, PJ, Leonard, JL, Davis, FB. Mechanisms of nongenomic actions of thyroid hormone. Front Neuroendocrinol. 2008;29:211–218.
134. Visser, WE, Friesema, ECH, Jansen, J, et al. Thyroid hormone transport in and out of cells. Trends Endocrinol Metab. 2008;19:50–56.
135. Hagenbuch, B. Cellular entry of thyroid hormones by organic anion transporting polypeptides. Best Pract Res Clin Endocrin Metab. 2007;21:209–221.
136. Visser, WE, Friesema, ECH, Jansen, J, et al. Thyroid hormone transport by monocarboxylate transporters. Best Pract Res Clin Endocrin Metab. 2007;21:223–236.
137. Pizzagalli, F, Hagenbuch, B, Stieger, B, et al. Identification of a novel human organic anion transporting polypeptide as a high affinity thyroxine transporter. Mol Endocrinol. 2002;16:2283–2296.
138. Friesema, EC, Ganguly, S, Abdalla, A, et al. Identification of monocaraboxylate transporter 8 as a specific thyroid hormone transporter. J Biol Chem. 2003;278:40128–40135.
139. Dumitrescu, AM, Liao, XH, Weiss, RE, et al. Tissue-specific thyroid hormone deprivation and excess in monocarboxylate transporter (mct) 8-deficient mice. Endocrinology. 2006:4036–4043.
140. Trajkovic, M, Visser, TJ, Mittag, J, et al. Abnormal thyroid hormone metabolism in mice lacking the momcarboxylate transporter 8. J Clin Invest. 2007;117:627–635.
141. Schwartz, CE, Stevenson, RE. The MCT8 thyroid hormone transporter and Allan-Herndon-Dudley syndrome. Best Pract Res Clin Endocrin Metab. 2007;21:307–321.
142. Lum, SM, Nicoloff, JT, Spencer, CA, et al. Peripheral tissue mechanism for maintenance of serum triiodothyronine values in a thyroxine-deficient state in man. J Clin Invest. 1984;73:570–575.
143. Keck, FS, Loos, U, Duntas, L, et al, Evidence for peripheral autoregulation of thyroxine conversion. G. Medeiros-Neto, E. Gaitan. New York: Plenum Medical Book Co., 1986:525–530.
144. Nicoloff, JT, LoPresti, JS. Alternate pathways of thyroid hormone metabolism. In: Wu S-Y, ed. Thyroid Hormone Metabolism. Boston: Blackwell Scientific Publications; 1991:55–64.
145. Maia, AL, Kim, BW, Huang, SA, et al. Type 2 deiodinase is the major source of plasma T3 in euthyroid humans. J Clin Invest. 2005;115:2524–2533.
146. Escobar-Morreale, H, Obregón, MJ, Escobar del Rey, F, et al. Replacement therapy for hypothyroidism with thyroxine alone does not ensure euthyroidism in all tissues, as studied in thyroidectomized rats. J Clin Invest. 1995;96:2828–2838.
147. Broedel, O, Eravci, M, Fuxius, S, et al. Effects of hyper- and hypothyroidism on thyroid hormone concentrations in regions of the rat brain. Am J Physiol. 2003;285:E470–480.
148. Porterfield, SP, Hendrich, CE. The role of thyroid hormones in prenatal and neonatal neurological development—current perspectives. Endo Reviews. 1993;14:94–106.
149. Daneman, D, Howard, NJ. Neonatal thyrotoxicosis: intellectual impairment and craniosynostosis in later years. J Pediatr. 1980;97:257–259.
150. Figueiredo, BC, Almazan, G, Ma, Y, et al. Gene expression in the developing cerebellum during perinatal hypo- and hyperthyroidism. Mol Brain Res. 1993;17:258–268.
151. Burrow, GN, Fisher, DA, Larsen, PR. Maternal and fetal thyroid function. N Engl J Med. 1994;331:1072–1078.
152. Calvo, RM, Jauniaux, E, Gulbis, B, et al. Fetal tissues are exposed to biologically relevant free thyroxine concentrations during early phases of development. J Clin Endocrinol Metab. 2002;87:1768–1777.
153. Morreale de Escobar, G, Obregon, MJ, Escobar del Rey, F. Is neuropsychological development related to maternal hypothyroidism or to maternal hypothyroxinemia? J Clin Endocrinol Metab. 2000;85:3975–3987.
154. Galton, VA, Martinez, E, Hernandez, A, et al. Pregnant rat uterus expresses high levels of the type 3 iodothyronine deiodinase. J Clin Invest. 1999;103:979–987.
155. Galton, VA, Martinez, E, Hernandez, A, et al. The type 2 iodothyronine deiodinase is expressed in the rat uterus and induced during pregnancy. Endocrinology. 2001;142:2123–2128.
156. Wasco, EC, Martinez, E, Grant, KS, et al. Determinants of iodothyronine deiodinase activities in rodent uterus. Endocrinology. 2003;144:4253–4261.
157. Kester, MHA, Kuiper, GGJM, Versteeg, R, et al. Regulation of type III iodothyronine deiodinase expression in human cell lines. Endocrinology. 2006;147:5845–5854.
158. Vulsma, T, Gons, MH, de Vijlder, JJM. Maternal-fetal transfer of thyroxine in congenital hypothyroidism due to a total organification defect or thyroid agenesis. N Engl J Med. 1989;321:13–16.
159. James, SR, Franklyn, JA, Kilby, MD. Placental transport of thyroid hormone. Best Pract Res Clin Endo Metab. 2007;21:253–264.
160. Chan, S, Kachilele, S, McCabe, CJ, et al. Early expression of thyroid hormone deiodinases and receptors in human fetal cerebral cortex. Develop Brain Res. 2002;138:109–116.
161. Kester, HA, Martinez de Mena, R, Obregon, MJ, et al. Iodothyronine levels in the human developing brain: major regulatory roles of iodothyronine deiodinases in different areas. J Clin Endocrinol Metab. 2004;89:3117–3128.
162. Calvo, R, Obregón, MJ, Ruiz de Ona, C, et al. Congenital hypothyroidism, as studied in rats: crucial role of maternal thyroxine but not 3,5,3′-triiodothyronine in the protection of the fetal brain. J Clin Invest. 1990;86:889–899.
163. Campos-Barros, A, Amma, LL, Faris, JS, et al. Type 2 iodothyronine deiodinase expression in the cochlea before the onset of hearing. Proc Natl Acad Sci USA. 2000;97:1287–1292.
164. Becker, KB, Stephens, KC, Davey, JC, et al. The type 2 and type 3 iodothyronine deiodinases play important roles in coordinating development in Rana catesbeiana tadpoles. Endocrinology. 1997;138:2989–2997.
165. Marsh-Armstrong, N, Huang, H, Remo, BF, et al. Asymmetric growth and development of the Xenopus laevis retina during metamorphosis is controlled by type III deiodinase. Neuron. 1999;24:871–878.
166. Huang, H, Cai, L, Remo, BF, et al. Timing of metamorphosis and the onset of the negative feedback loop between the thyroid gland and the pituitary is controlled by type II iodothyronine deiodinase in Xenopus laevis. Proc Natl Acad Sci USA. 2001;98:7348–7353.
167. Cai, L, Brown, DD. Expression of type II iodothyronine deiodinase marks the time that a tissue responds to thyroid hormone-induced metamorphosis in Xenopus laevis. Devel Biol. 2004;266:87–95.
168. Fisher, DA, Polk, DH, Wu, SY. Fetal thyroid hormone metabolism. Thyroid. 1994;4:367–371.
169. Santini, F, Chopra, IJ, Wu, S-Y, et al. Metabolism of 3,5,3′-triiodothyronine sulfate by tissues of the fetal rat: a consideration of the role of desulfation of 3,5,3′-triiodothyronine sulfate as a source of T3. Pediatr Res. 1992;31:541–544.
170. Wartofsky, L, Burman, K. Alterations in thyroid function in patients with systemic illness: the “euthyroid sick syndrome. Endo Reviews. 1982;3:164–217.
171. Peeters, RP, Debaveye, Y, Fliers, E, et al. Changes within the thyroid axis during critical illness. Crit Care Clinics. 2006;22:41–55.
172. Peeters, RP, Van der Geyten, S, Wouters, PJ, et al. Tissue thyroid hormone levels in critical illness. J Clin Endocrinol Metab. 2005;90:6498–6507.
173. Peeters, RP, Wouters, PJ, van Toor, H, et al. Serum 3,3′,5′-triiodothyronine (rT3) and 3,5,3′-triiodothyronine/rT3 are prognostic markers in critically ill patients and are associated with postmortem tissue deiodinase activities. J Clin Endocrinol Metab. 2005;90:4559–4565.
174. Wells, S, Campbell, R. Decrease in resting metabolic rate during rapid weight loss is reversed by low dose thyroid hormone treatment. Metabolism. 1986;35:289–291.
175. Forsum, E, Hillman, PE, Nesheim, MC. Effect of energy restriction on total heat production, basal metabolic rate, and specific dynamic action of food in rats. J Nutr. 1981;111:1691–1697.
176. Carter, WJ, Shakir, KM, Hodges, S, et al. Effect of thyroid hormone on metabolic adaptation to fasting. Metabolism. 1975;24:1177–1183.
177. Gardner, DF, Kaplan, MM, Stanley, CA, et al. Effect of tri-iodothyronine replacement on the metabolic and pituitary responses to starvation. N Engl J Med. 1979;300:579–584.
178. Boelen, A, Kwakkel, J, Alkemade, A, et al. Induction of type 3 deiodinase in inflammatory cells of mice with chronic local inflammation. Endocrinology. 2005;146:5128–5134.
179. Peeters, RP, Wouters, PJ, Kaptein, E, et al. Reduced activation and increased inactivation of thyroid hormone in tissues of critically ill patients. J Clin Endocrinol Metab. 2003;88:3202–3211.
180. Olivares, EL, Marassi, MP, Fortunato, RS, et al. Thyroid function disturbance and type 3 iodothyronine deiodinase induction after myocardial infarction in rats—a time course study. Endocrinology. 2007;148:4786–4792.
181. Chang, M, Reddy, CC. Active transcription of the selenium-dependent glutathione peroxidase gene in selenium-deficient rats. Biochem Biophys Res Comm. 1991;181:1431–1436.
182. Coppola, A, Hughes, J, Esposito, E, et al. Suppression of hypothalamic deiodinase type II activity blunts TRH mRNA decline during fasting. FEBS Letters. 2005;579:4654–4658.
183. Sanchez, E, Singru, PS, Fekete, C, et al. Induction of type 2 iodothyronine deiodinase in the mediobasal hypothalamus by bacterial lipopolysaccharide: role of corticosterone. Endocrinology. 2008;149:2484–2493.
184. Lankford, SP, Cosson, P, Bonifacino, JS, et al. Transmembrane domain length affects charge-mediated retention and degradation of proteins within the endoplasmic reticulum. J Biol Chem. 1993;268:4814–4820.
185. Kinlaw, WB, Schwartz, HL, Oppenheimer, JH. Decreased serum triiodothyronine in starving rats is due primarily to diminished thyroidal secretion of thyroxine. J Clin Invest. 1984;75:1238–1241.
186. Yen, YM, DiStefano, JJ, III., Yamada, H, et al. Direct measurement of whole body thyroid hormone pool sizes and interconversion rates in fasted rats: hormone regulation implications. Endocrinology. 1994;134:1700–1709.
187. Boelen A, Kwakkel J, Wieland CW, et al: Impaired bacterial clearance in type 3 deiodinase deficient mice infected with Streptococcal pneumoniae, Endocrinology 150:1984–1989, 2009.
188. Kim, B. Thyroid hormone as a determinant of energy expenditure and the basal metabolic rate. Thyroid. 2008;18:141–144.
189. da Silva, WS, Harney, JW, Kim, BW, et al. The small polyphenolic molecule kaempferol increases cellular energy expenditure and thyroid hormone activation. Diabetes. 2007;56:767–776.
190. DePalo, D, Kinlaw, WB, Zhao, C, et al. Effect of selenium deficiency on type I 5′-deiodinase. J Biol Chem. 1994;269:16223–16228.
191. Meinhold, H, Campos-Barros, A, Walzog, B, et al. Effects of selenium and iodine deficiency on type I, type II, and type III iodothyronine deiodinases and circulating thyroid hormones in the rat. Exp Clin Endocrinol. 1993;101:87–93.
192. Bates, JM, Spate, VL, Morris, JS, et al. Effects of selenium deficiency on tissue selenium content, deiodinase activity, and thyroid hormone economy in the rat during development. Endocrinology. 2000;141:2490–2500.
193. Chanoine, J, Safran, M, Farwell, AP, et al. Effects of selenium deficiency on thyroid hormone economy in rats. Endocrinology. 1992;131:1787–1792.
194. Olivieri, O, Girelli, D, Azzini, M, et al. Low selenium status in the elderly influences thyroid hormones. Clin Sci. 1995;89:637–642.
195. Kauf, E, Dawczynski, H, Jahreis, G, et al. Sodium selenite therapy and thyroid-hormone status in cystic fibrosis and congenital hypothyroidism. Biol Trace Elem Res. 1994;40:247–253.
196. Angstwurm, MWA, Schopohl, J, Gaertner, R. Selenium substitution has no direct effect on thyroid hormone metabolism in critically ill patients. Eur J Endo. 2004;151:47–54.
197. Contempré, B, Duale, NL, Dumont, JE, et al. Effect of selenium supplementation on thyroid hormone metabolism in an iodine and selenium deficient population. Clin Endocrinol. 1992;36:579–583.
198. Cavalieri, RR. Effects of drugs on human thyroid hormone metabolism. In: Hennemann G, ed. Thyroid Hormone Metabolism. New York: Marcel Dekker, Inc; 1986:359–379.
199. Okamura, K, Ikenoue, H, Shiroozu, A, et al. Reevaluation of the effects of methylmercaptoimidazole and propylthiouracil in patients with Graves’ hyperthyroidism. J Clin Endocrinol Metab. 1987;65:719–723.
200. Wu, S, Shyh, T, Chopra, IJ, et al. Comparison of sodium ipodate (Oragrafin) and propylthiouracil in early treatment of hyperthyroidism. J Clin Endocrinol Metab. 1982;54:630–634.
201. Roti, E, Robuschi, G, Manfredi, A, et al. Comparative effects of sodium ipodate and iodide on serum thyroid hormone concentrations in patients with Graves’ disease. Clin Endocrinol. 1985;22:489–496.
202. Seclen, SN, Pretell, EA, Tapia, FA, et al. Rapid amelioriation of severe cardiovascular complications of thyrotoxic patients with sodium ipodate. In: Medeiros-Neto G, Gaitan E, eds. Frontiers in Thyroidology. New York: Plenum Medical Book Company; 1986:1101–1105.
203. Bogazzi, F, Miccoli, P, Berti, P, et al. Preparation with iopanoic acid rapidly controls thyrotoxicosis in patients with amiodarone-induced thyrotoxicosis before thyroidectomy. Surgery. 2002;132:1114–1117.
204. Wiersinga, WM. Propranolol and thyroid hormone metabolism. Thyroid. 1991;1:273–277.
205. Chopra, IJ, Williams, DE, Orgiazzi, J, et al. Opposite effects of dexamethasone on serum concentrations of 3,3′,5′-triiodothyronine (reverse T3) and 3,3′5-triiodothyronine (T3). J Clin Endocrinol Metab. 1975;41:911–920.
206. Cavaleri, RH, Castle, JN, McMahon, FA. Effects of dexamethasone on kinetics and distribution of triiodothyronine in the rat. Endocrinology. 1984;114:215–221.
207. Eravci, M, Pinna, G, Meinhold, H, et al. Effects of pharmacological and nonpharmacological treatments on thyroid hormone metabolism and concentrations in rat brain. Endocrinology. 2000;141:1027–1040.
208. Newman, CM, Price, A, Davies, DW, et al. Amiodarone and the thyroid: a practical guide to the management of thyroid dysfunction induced by amiodarone therapy. Heart. 1998;79:121–127.
209. Ursella, S, Testa, A, Mazzone, M, et al. Amiodarone-induced thyroid dysfunction in clinical practice. Eur Rev Med Pharm Sci. 2006;10:269–278.
210. Celi, FS, Coppotelli, G, Chidakel, A, et al. The role of type 1 and type 2 5′-deiodinase in the pathophysiology of the 3,5,3′-triiodothyronine toxicosis of McCune-Albright syndrome. J Clin Endocrinol Metab. 2008;93:2383–2389.
211. Bartha, T, Kim, SW, Salvatore, D, et al. Characterization of the 5′-flanking and 5′-untranslated regions of the cyclic adenosine 3′,5′-monophosphate-responsive human type 2 iodothyronine deiodinase gene. Endocrinology. 2000;141:229–237.
212. Kim, BW, Daniels, GH, Harrison, BJ, et al. Overexpression of type 2 iodothyronine deiodinase in follicular carcinoma as a cause of low circulating free thyroxine levels. J Clin Endocrinol Metab. 2003;88:594–598.
213. Miyauchi, A, Takamura, Y, Ito, Y, et al. 3,5,3′-Triiodothyronine thyrotoxicosis due to increased conversion of administered levothyroxine in patients with massive metastatic follicular thyroid carcinoma. J Clin Endocrinol Metab. 2008;93:2239–2242.
214. Takano, T, Miyauchi, A, Ito, Y, et al. Thyroxine to triiodothyronine hyperconversion thyrotoxicosis in patients with large metastases of follicular thyroid carcinoma. Thyroid. 2006;16:615–618.
215. Murakami, M, Araki, O, Hosoi, Y, et al. Expression and regulation of type II iodothyronine deiodinase in human thyroid gland. Endocrinology. 2001;142:2961–2967.
216. Brtko, J, Bobalova, J, Podoba, J, et al. Thyroid hormone receptors and type I iodothyronine 5′-deiodinase activity of human thyroid toxic adenomas and benign cold nodules. Exp Clin Endo Diab. 2002;110:166–170.
217. Huang, SA, Tu, HM, Harney, JW, et al. Severe hypothyroidism caused by type 3 iodothyronine deiodinase in infantile hemangiomas. N Engl J Med. 2000;343:185–189.
218. Balazs, AE, Athanassaki, I, Gunn, SK, et al. Rapid resolution of consumptive hypothyroidism in a child with hepatic hemangioendothelioma following liver transplantation. Ann Clin Lab Sci. 2007;37:280–284.
219. Huang, SA, Fish, SA, Dorfman, DM, et al. A 21-year-old woman with consumptive hypothyroidism due to a vascular tumor expressing type 3 iodothyronine deiodinase. J Clin Endocrinol Metab. 2002;87:4457–4461.
220. Ruppe, MD, Huang, SA, Jan de Beur, SM. Consumptive hypothyroidism caused by paraneoplastic production of type 3 iodothyronine deiodinase. Thyroid. 2005;15:1369–1372.
221. Tannahill, LA, Visser, TJ, McCabe, CJ, et al. Dysregulation of iodothyronine deiodinase enzyme expression and function in human pituitary tumours. Clin Endocrinol. 2002;56:735–743.
222. Peeters, RP, van Toor, H, Klootwijk, W, et al. Polymorphisms in thyroid hormone pathway genes are associated with plasma TSH and iodothyronine levels in healthy subjects. J Clin Endocrinol Metab. 2003;88:2880–2888.
223. Mentuccia, D, Proietti-Pannunzi, L, Tanner, K, et al. Association between a novel variant of the human type 2 deiodinase gene Thr92Ala and insulin resistance: evidence of interaction with the Trp64Arg variant of the beta-3-adrenergic receptor. Diabetes. 2002;51:880–883.
224. Canani, LH, Capp, C, Dora, JM, et al. The type 2 deiodinase A/G (Thr92Ala) polymorphism is associated with decreased enzyme velocity and increased insulin resistance in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2005;90:3472–3478.
225. Maia, AL, Dupuis, J, Manning, A, et al. The type 2 deiodinase (DIO2) A/G polymorphism is not associated with glycemic traits: the Framingham Heart Study. Thyroid. 2007;17:199–202.
226. Grarup, N, Andersen, MK, Andreasen, CH, et al. Studies of the common DIO2 Thr92Ala polymorphism and metabolic phenotypes in 7342 Danish white subjects. J Clin Endocrinol Metab. 2007;92:363–366.
227. Gumieniak, O, Perlstein, TS, Williams, JS, et al. Ala92 type 2 deiodinase allele increases risk for the development of hypertension. [see comment]. Hypertension. 2007;49:461–466.
228. Maia, AL, Hwang, S-J, Levy, D, et al. Lack of association between the type 2 deiodinase A/G polymorphism and hypertensive traits: the Framingham Heart Study. Hypertension. 2008;51:e22–23.
229. de Jong, FJ, Peeters, RP, den Heijer, T, et al. The association of polymorphisms in the type 1 and 2 deiodinase genes with circulating thyroid hormone parameters and atrophy of the medial temporal lobe. J Clin Endocrinol Metab. 2007;92:636–640.
230. Peeters, RP, van den Beld, AW, van Toor, H, et al. A polymorphism in type I deiodinase is associated with circulating free insulin-like growth factor I levels and body composition in humans. J Clin Endocrinol Metab. 2005;90:256–263.
231. Galton, V, Wood, E, St. Germain, EA, et al. Thyroid hormone homeostasis and action in the type 2 deiodinase-deficient rodent brain during development. Endocrinology. 2007;148:3080–3088.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 4
Thyroid Hormone Metabolism
Hormone Kinetics and Production
Deiodination and the Iodothyronine Deiodinases
Alternative Routes of Iodothyronine Metabolism
Thyroid Hormone Uptake Into Cells
An Integrated View of Thyroid Hormone Metabolism
The principal secretory product of the thyroid gland, T4, undergoes a complex series of intracellular metabolic alterations in peripheral tissues, as shown in Fig. 4-1. Some of these reactions, such as 5′-deiodination of T4 to form T3 or decarboxylation and deamination of T3 to form the acetic acid analogue termed Triac, result in compounds with considerably greater intrinsic biopotency because of their increased affinity for thyroid hormone receptors.1,2 Other reactions result in the formation of apparently inactive compounds, such as reverse T3 (rT3) formed by 5′-deiodination of T4 or the sulfated and glucuronidated forms of T4 and T3 formed by conjugation of the phenolic ring hydroxyl group.3,4 Progressive deiodination of T4, T3, and rT3 results in the formation of various diiodinated and monoiodinated thyronines and eventually thyronine (T0) itself.5 Most of these compounds are believed to have little or no biological activity. Exceptions to this generalization may be the compounds 3,5-diiodothyronine (3,5-T2) and 3,3′-T2, which have been demonstrated to have effects on mitochondrial function6 and to increase metabolic rate.7–9 In addition, the monoiodinated compound 3-iodothyronamine, formed in vivo by sequential deiodination and decarboxylation, produces bradycardia and dramatic decreases in body temperature and metabolic rate when administered to experimental animals.10,11 This effect, opposite to that expected of a thyroid hormone, is believed to result from a nongenomic mechanism via a G protein–coupled trace amine receptor.10
FIGURE 4-1 Graphic representation of the pathways of iodothyronine metabolism. The types 1 and 2 deiodinases (D1, D2) catalyze removal of the 5′ (or chemically equivalent 3′) iodine from T4 and other iodothyronine substrates. Types 1 and 3 enzymes catalyze 5 (or chemically equivalent 3) deiodination. A variety of less common reactions occur via alternative enzymatic pathways. The metabolites shown are subject to further deiodination to form diiodothyronines, monoiodothyronines, and tyrosine. (From St. Germain DL, Galton VA: The deiodinase family of selenoproteins. Thyroid 7:655–658, 1997.)
The enzymatic processes shown in Fig. 4-1 are not mutually exclusive. Indeed, modification of an iodothyronine molecule at one site may markedly alter its susceptibility to other metabolic reactions. For example, Tetrac and Triac are much better substrates for glucuronidation than are T4 and T3.12 The acetic acid analogues and sulfated conjugates also are markedly better substrates for deiodination in the liver and kidney than are the native compounds.13,14 In contrast, sulfation of iodothyronines in certain organs may effectively block further metabolism because these conjugates are poorly reactive with the deiodinase isoforms expressed in other tissues.15,16
Hormone Kinetics and Production
Deiodination at the 5′ or 5 position accounts for approximately 80% of the daily disposal of T4, with the other processes shown in Fig. 4-1 responsible for the remaining metabolism of this compound.5 Such approximations may underestimate the role of both deiodinative and nondeiodinative pathways, however. Many of the products of these reactions, including T3 and rT3, are present primarily in the intracellular compartment and may undergo degradation before they have a chance to exchange with the plasma pool. It thus is difficult in human kinetic studies that use plasma sampling techniques alone to assess the contribution of these processes to overall thyroid hormone metabolism. For example, it has been demonstrated that most of the thyronine (T0) excreted in the urine is in the form of its acetic acid analogue,17 thus suggesting that deamination plays a more prominent role in thyroid hormone metabolism than is apparent from the very low circulating levels of Tetrac and Triac. Such concerns have led to the concept that “hidden pools” of thyroid hormone metabolites may be present in tissues.18
Table 4-119–21 provides estimates of various kinetic parameters of thyroid hormone production and metabolism in humans.5,22,23 The high affinity of T4 for plasma binding proteins, along with its greater production rate, accounts for its relatively high concentration in serum, as well as its long serum half-life. In contrast, T3 and rT3 are present at much lower serum concentrations because of their lower production rates, greater metabolic clearance rates, and lower affinity for thyroxine-binding globulin (TBG). In addition, these two triiodothyronines appear to reside primarily within the intracellular compartment; thus their volumes of distribution are significantly greater than that of T4.
Table 4-1
Normal Thyroid Hormone Kinetics in Humans
T4, Thyroxine; T3, 3,5,3′-triiodothyronine; rT3, reverse triiodothyronine.
*Conversion factors: T4, 1 mg = 1.3 nmol; T3 and rT3, 1 mg = 1.5 nmol.
The rate of T4 production remains remarkably constant in healthy adults, with alterations noted only during pregnancy and in the aged. Although not well documented, increased T4 secretion probably occurs during pregnancy in response to several factors, including thyroidal stimulation by human chorionic gonadotropin, an increase in the size of the extrathyroidal T4 pool resulting from increased TBG levels, and an increase in the rate of T4 and T3 degradation because of high levels of 5-deiodinase activity in the pregnant uterus and placenta.24,25 One clinical consequence is the need to increase the replacement dose of T4 by 25% to 50% in hypothyroid women during pregnancy.26 Thyroid function appears to be well preserved through age 80 in healthy individuals, although a slight decrease in T4 production and clearance is noted after the age of 60 years, and free T3 levels are lower in centenarians.27,28 That being the case, the decreased T4 requirements of elderly hypothyroid patients probably result more from the presence of chronic illness and the use of concurrent medications than from the aging process per se.29,30
Deiodination and The Iodothyronine Deiodinases
The potential importance of deiodination to thyroid hormone action was first recognized nearly 50 years ago when Gross and Pitt-Rivers demonstrated that although T3 was considerably more potent than T4,31 it was present in the thyroid in much lower amounts.32 This observation suggested that T3 was derived largely from T4 by metabolism in extrathyroidal tissues. This thesis was later proved by Braverman et al., who demonstrated the presence of T3 in the serum of athyreotic subjects injected with T4.33 Research over the past three decades has confirmed the physiologic importance of the 5′- and 5-deiodination processes, has defined the biochemical parameters of these enzymatic reactions, has determined important regulatory factors that influence deiodinase activities, and has identified key structural determinants of the proteins that catalyze these reactions.34,35
Three deiodinase isoforms, termed D1, D2, and D3, are present in vertebrate species. These differ in their catalytic properties, patterns of tissue expression, and mechanisms of regulation.34,36,37 The 5′- and 5-deiodination reactions catalyzed by these enzymes can be considered broadly as activating and inactivating processes, respectively.
Biochemical Characteristics
The biochemical properties of the deiodinases are outlined in Table 4-2.38–45 They are in essence oxidoreductases in that they catalyze the substitution of hydrogen for iodine on the iodothyronine substrate. No other catalytic properties of these enzymes have yet been identified, nor do other known enzymes possess deiodinase activity. The enzymes are remarkable in terms of their substrate specificity and the precise location of the iodine removed.16 D1 is unique in that it can catalyze either 5′- or 5-deiodination, depending on the reacting substrate. Thus rT3 is efficiently deiodinated only at the 5′ position by D1. In contrast, T4 and T3 are poor substrates for this enzyme unless they are first sulfated. This reaction markedly enhances their rate of 5-deiodination and further reduces their susceptibility to 5′-deiodination.46 That D1 is relatively inefficient in converting T4 to T3 presents a considerable paradox, given that a significant proportion of T3 production has traditionally been believed to occur in the liver and other D1-expressing tissues (vide infra).
Table 4-2
Characteristics of Iodothyronine Deiodinases
Data from St. Germain and Galton,38 Leonard and Visser,39 Jakobs et al.,40 Celi et al.,41 Hernández et al.,42 Leonard et al.,43,45 and Curcio-Morelli et al.44
D2 catalyzes only 5′-deiodination and very efficiently converts T4 to T3.16 The T3 thus formed is a poor substrate for 5′-deiodination and is not further metabolized by this enzyme. In contrast, rT3 is a good substrate for D2 as well as D1 and frequently is used in research assays to quantitate 5′-deiodinase activity. D3 exclusively catalyzes 5-deiodination16 and thus serves to convert T4 and T3 to rT3 and 3,3′-T2, respectively—metabolites with little affinity for the nuclear thyroid hormone receptors.
The deiodinases all require the availability of reduced thiol cofactors for efficient catalytic cycling.39 These cofactors presumably function to displace iodine from an enzyme intermediate formed during the reaction and thus to regenerate the active deiodinase.47 In in vitro assay systems using tissue homogenates or cellular subfractions, dithiothreitol typically is added as a cofactor. This small, nonnative four-carbon dithiol efficiently supports deiodination of all three enzymes. Kinetic data derived from broken cell preparations using dithiothreitol as cofactor have demonstrated a Michaelis-Menten constant (Km) value for D1 of approximately 2.3 µmol/L (for T4 5′-deiodination), whereas D2 and D3 manifest much lower Km values in the nanomolar range (see Table 4-2).39 Based on this analysis, D1 sometimes is referred to as a “high Km” enzyme, whereas D2 is said to catalyze a “low Km” 5′-deiodination process. This distinction is somewhat spurious, however, because the kinetic properties of the deiodinases are clearly dependent on the thiol cofactors used in the assay system.48 Thus, when the native thiol cofactor glutathione or thioredoxin is used, Km values for D1 in the nanomolar range are obtained. Unfortunately, the cofactor system supporting deiodination in intact cells remains unknown,49 and evidence suggests that glutathione and thioredoxin may not serve this role.50 Thus the physiologic significance of the kinetic parameters derived in vitro remains uncertain.
Inhibitors
In addition to differences in their catalytic properties, the deiodinases show differential susceptibilities to certain inhibitors39 (see Table 4-2). Most notable is the marked sensitivity of D1 to the antithyroid drug propylthiouracil (PTU). This agent forms an inactive complex with D1 by binding covalently to its active site. Notably, D2 and D3 show little or no susceptibility to inhibition by PTU. The related thioureylene drugs carbimazole and methimazole have no inhibitory effect on deiodination.47
Gold compounds such as aurothioglucose are known to react with the active site selenocysteine residue in glutathione peroxidase and impair its activity.51 This compound also inhibits all three deiodinase isoforms, with D1 again showing a significantly greater sensitivity to this effect.52–54 Other drugs that affect thyroid hormone metabolism include the iodinated radiographic contrast agents iopanoic acid (Telepaque) and sodium ipodate (Oragrafin). These small phenolic compounds act as substrate analogues and inhibit all three deiodinases in a competitive manner. To date, no selective inhibitors of D2 or D3 have been described—a situation that has significantly limited experimental investigations into their physiologic roles.
Tissue Patterns of Expression
An intriguing feature of the deiodinases is their pattern of tissue expression (see Table 4-2). The liver, kidney, thyroid gland, and pituitary gland express high levels of D1, with lesser amounts found in the brain.16,55,56 In contrast, D2 is most abundant in the pituitary gland and brown adipose tissue (in rodents), with significant amounts also noted in the central nervous system.16 Studies using in situ hybridization and immunocytochemistry have demonstrated that D2 expression in the brain appears to be confined to certain subpopulations of astroglial cells, such as the tanycytes lining the third ventricle of the hypothalamus, which suggests that they serve as the principal site of T3 production in this tissue.57–59 In humans, D2 expression appears more widespread; the mRNA for this enzyme has been noted in the heart, skeletal muscle, and thyroid gland,54,60 as well as in coronary artery smooth muscle cells.61
In adult mammals, D3 expression occurs primarily in the brain, with significant amounts also present in the skin.62,63 The brain is thus the only tissue that expresses all three deiodinases, although these are generally found in different subregions of this tissue and in different cell types. Thus, as opposed to the expression of D2 by glial cells, D3 is expressed primarily in neurons.64,65
Finally, as detailed below, deiodinase expression patterns during pregnancy and development are of critical importance, with high levels of D3 activity present in the uterus, the placenta, and several fetal tissues.66,67
Structural Characteristics
Important structural features of the deiodinases have been deduced from the results of molecular cloning experiments.37 All the enzymes have a molecular mass of approximately 29 to 32 kDa, function as homodimers,43–45 and are selenoproteins in that they contain the uncommon amino acid selenocysteine as the reactive residue in the catalytic cleft (Fig. 4-2).68 The importance of this amino acid to enzymatic activity is demonstrated by experiments in which selenocysteine has been replaced by cysteine. Such a substitution decreases the catalytic efficiency of the mutant protein to less than 1% of that of the native enzyme.69 In the case of D3, the cysteine mutant also demonstrates an altered substrate preference.70 It is remarkable that substitution of selenocysteine for a serine residue in a monoclonal antibody raised against T4 confers on the protein deiodinase activity with catalytic properties similar to the D1, including sensitivity to PTU inhibition.71
FIGURE 4-2 Comparison of the structures of selenocysteine and cysteine. Selenocysteine is ionized at physiologic pH, which likely contributes to the catalytic efficiencies of the deiodinases.
The importance of selenocysteine to deiodination likely derives from its being ionized at physiologic pH, thus serving as a much more potent nucleophile than cysteine.72 Incorporation of selenocysteine into the deiodinases and other selenoproteins occurs at the time of translation and is directed by a specific stem-loop structure in the 3′-untranslated region of the mRNA termed a selenocysteine insertion sequence.73 A unique tRNA (Sec-tRNA[Sec]), a specific RNA binding protein, and a specialized elongation factor also are required for efficient synthesis of selenoproteins.74,75
In earlier studies, it was suggested that a 29 kDa (p29) nonselenoprotein, almost identical in sequence to a member of the Dickkopf protein family (Dkk3), was a substrate binding subunit of the D2.76 However, Montero-Pedrazuela et al. have demonstrated a marked difference in spatial expression patterns in the brain of the p29 and the D2 selenodeiodinase,77 and as recently reported, mice deficient in the Dkk3 protein do not have any consistent alterations in thyroid hormone status or D2 activity in their tissues.78 These findings strongly suggest that the p29 protein is not relevant to D2 activity or to thyroid hormone homeostasis.
The overall amino acid identity of the three deiodinase isoforms is less than 30%. However, a high degree of homology is present in the regions of the selenocysteine and a conserved histidine residue (Fig. 4-3A). These areas show significant structural similarities to the thioredoxin-fold family of proteins and to α-l-iduronidase, respectively.79 Based on these structural homologies, Callebaut et al.79 and others have reported mutagenesis studies that define further the structure-function correlates of the deiodinase isoforms, including the identification of a key serine residue in the D1 active catalytic site that appears to confer sensitivity to PTU.80
FIGURE 4-3 Structural features of the deiodinase proteins and genes. A, The proteins are composed of 257 to 278 amino acids with predicted molecular masses of 29 to 32 kDa. The hydrophobic transmembrane domain near the amino terminus, the selenocysteine residue (SeC), and two histidines (His) essential for catalytic activity are conserved in all three isoforms. The percentage of amino acid identity between the three isoforms in different portions of the molecule is given. A high degree of homology is noted in the regions surrounding the selenocysteine and one of the histidines. The dimerization domain as defined in the D1 is also shown. B, The structures of the genes, including the locations of the selenocysteine-encoding TGA codons and the regions coding for the selenocysteine insertion sequences (SECIS) located in the 3′-untranslated regions, are shown. Polymorphisms identified in each of the genes are also shown. DIO3os refers to the gene that encodes transcripts from the opposite DNA strand in the DIO3 locus and that remains incompletely characterized.
In addition, all deiodinases contain a hydrophobic region near the N-terminus,81 and subcellular fractionation studies have confirmed that these enzymes are integral membrane proteins.82,83 Such studies, along with fluorescent microscopy of epitope-tagged enzymes, have demonstrated that the D2 is located in the endoplasmic reticulum,82,84 whereas a significant fraction of the D3 has been reported to be in the plasma membrane.85 The location of the D1 may vary in different tissues; in renal epithelial cells, the D1 has been localized to the basolateral plasma membranes,86 whereas in the liver, it appears to reside in the endoplasmic reticulum (Pallud, Croteau, and St. Germain, unpublished observations). Studies by Friesema et al.87 demonstrate that the catalytic effectiveness of all three deiodinases is markedly increased when specific thyroid hormone transporters are coexpressed with the deiodinases in cultured cells. This finding indicates that the active catalytic sites of these enzymes are located in the intracellular compartment.
Genetic Considerations and Knockout Mouse Models
The three deiodinase isoforms are coded in mammals by three different genes located in humans on chromosomes 1 (D1) and 14 (D2 and D3), as indicated in Table 4-2.40–42 Although their genetic structure appears relatively simple in that they contain only four, three, and a single exon, respectively (Fig. 4-3B),40,88,89 surprising complexities that may influence expression patterns have been identified.90,91 For example, the DIO3 gene locus also expresses antisense transcripts from the DIO3os gene coded on the opposite DNA strand, and in the mouse fetus, the D3 is imprinted; transcription preferentially occurs from the paternal allele.91 In humans the DIO3 is also located within an imprinted region of chromosome 14.91 This pattern of organization and expression of the D3 often is observed for genes important in development.92
To enhance our understanding of the physiologic roles of the deiodinases, genetic knockout models of each of the isoforms have been developed and partially characterized (Table 4-3).93–100 D1-deficient mice show no general phenotypic abnormalities, although serum T4 and rT3 levels are modestly elevated, and the excretion patterns of iodothyronines in the feces and of iodine in the urine are significantly altered.93 D2-deficient mice are viable and fertile in a protected laboratory setting. However, observed impairments in hearing,96 thermogenesis,95 and neurocognition97 in these mutants would likely result in lethality in the wild. These animals also manifest elevated T4 and thyroid-stimulating hormone (TSH) levels and show a marked selective resistance to the feedback effects of T4, demonstrating the importance of D2 in pituitary thyrotroph regulation.94 This contrasts with D1-deficient mice, in which TSH levels are normal.93 This implies that the D1 expressed in the pituitary is not involved in TSH regulation.
Table 4-3
Phenotypic Features of Deiodinase Knockout Mice
D1-Deficient Mouse93
D2-Deficient Mouse
A striking finding in the D1- and D2-deficent mouse models is that serum T3 levels are normal.93,94 Indeed, in mice cross-bred to yield a combined D1 and D2 deficiency, the serum T3 level is only minimally decreased (V. A. Galton, unpublished observations).101 This finding is both unexpected and difficult to explain, given that 5′-deiodination in peripheral tissues is supposed to contribute 80% of the T3 production in humans and 45% in rats, with the remainder secreted directly from the thyroid.34,102 Given that the combined D1/D2 knockout mouse has been unequivocally shown to lack all 5′-deiodinating capacity, it is clear that at least in the rodent, thyroidal production and secretion of T3, perhaps combined with a decreased rate of T3 clearance from D1 deficiency, can compensate and maintain serum T3 levels that are essentially normal in the face of an inability to convert T4 to T3 in all tissues. A surprising corollary to this observation is that a D1 deficiency does not appear to significantly compromise T3 secretion from the rodent thyroid, a tissue in which this enzyme is highly expressed and previously was thought to be essential for T3 production.102,103 This suggests that under physiologic conditions, D1 acts primarily as a “scavenger” enzyme that prevents the buildup of lesser iodothyronines (rT3, T2s, T1) and sulfated derivatives in the thyroid gland, blood, and tissues.
The extent to which humans may be able to compensate for at least a partial impairment in 5′-deiodinating capacity has recently been reported. Although to date no mutations in the deiodinase genes in humans have been described, Dumitrescu et al.104 have described two families with partially inactivating mutations in the selenocysteine insertion sequence-binding protein2 (SBP2), which is essential for the synthesis of selenoproteins, including the deiodinases.74 Affected individuals demonstrate short stature and delayed bone age along with modest abnormalities in serum thyroid hormone levels, including increases in total T4 and rT3.104 Notably, total serum T3 levels were only minimally reduced, whereas TSH levels were at the upper part of the reference range and proved relatively resistant to suppression by administered T4, but not T3. These findings are very similar to those observed in knockout mice with a combined deficiency in D1 and D2, as noted previously. Indeed, a deficiency in D2 activity was demonstrated in cultured skin fibroblasts from affected patients.104
Mice deficient in D3 present the most severe phenotype, likely as a result of the importance of this enzyme during the developmental period. Observed abnormalities include significant perinatal mortality, growth retardation, impaired fertility, and altered systemic thyroid hormone levels during the perinatal period (transient hyperthyroidism) and in adulthood (moderate hypothyroidism).99,100 Multiple abnormalities, including impaired response of the thyroid gland to TSH and of the pituitary to TRH, have been observed at all levels of the thyroid axis in the D3-deficient adult mouse. The response to hypothyroidism of TRH mRNA production in the hypothalamus and of pituitary TSH production is also markedly impaired. Many of these abnormalities are similar to those observed in human infants born to mothers with poorly controlled hyperthyroidism during pregnancy105,106 or to rodents treated with large doses of thyroid hormone during the perinatal period.107,108 These findings demonstrate that strict control of thyroid hormone levels during development by D3 is critical for proper development of the thyroid axis.
Regulation
The deiodinases are regulated by multiple hormones, growth factors, and environmental and nutritional factors.34 Foremost among these are the thyroid hormones themselves; alterations in thyroid status induce profound changes in enzyme activity (see Table 4-2). Hypothyroidism is associated with a marked decrease in D1 and D3 levels, whereas D2 activity increases severalfold. Opposite changes occur in hyperthyroidism. These changes result from both pretranslational and posttranslational mechanisms.34 For example, D1 and D3 mRNA levels are increased in the hyperthyroid state,53,109 and D2 mRNA is decreased.54 Hyperthyroidism also results in rapid downregulation of D2 activity by ubiquitination of the D2 protein.110,111 This induces a reversible conformational change in the enzyme’s dimerization structure, which results in reversible loss of activity and eventually may target the enzyme for proteasomal degradation.112 Considerable progress has been made in defining the molecular mechanisms involved in transcriptional control of the deiodinases; this topic has been reviewed recently by Gereben et al.37 Of particular interest has been the observation by Simonides et al.113 that hypoxia induces D3 expression in several cell culture systems, as well as in ischemic myocardium.
Other important regulatory effects on deiodinase activity are noted in the thyroid gland, where TSH and thyroid-stimulating immunoglobulins stimulate both D1 and D2 activity60; in brown adipose tissue, where cold exposure,114 bile acids,115 or intracerebroventricular administration of leptin116 markedly stimulates D2 activity; and in the liver, where nutritional deprivation, diabetes, tumor necrosis factor, and other cytokines decrease D1 activity (see also below).109,117,118 In addition, D2 activity in the brain displays a significant diurnal variation.119 Although the physiologic significance of this remains to be determined, in some seasonal breeding species, rhythmic alterations in D2 and D3 activity in the central nervous system, perhaps mediated by melatonin,120 appear to be integral responses to changes in the photoperiod.121,122
Alternative Routes of Iodothyronine Metabolism
In addition to deiodination, iodothyronines are metabolized by multiple other mechanisms.123 In this regard, conjugation of the phenolic ring hydroxyl group to sulfate or glucuronide probably represents the second most prevalent mechanism of thyroid hormone metabolism.16 Sulfation and glucuronidation are inactivating reactions because the compounds formed are devoid of thyromimetic activity, do not bind to nuclear thyroid hormone receptors, and are rapidly metabolized by D1 or excreted in bile.4,16 However, sulfation is a reversible process that occurs through the action of tissue sulfatases or bacterial sulfatases in the intestine.124 Thus T3 sulfate (T3S) injected into hypothyroid rats demonstrates approximately 20% of the thyromimetic activity of native T3 because of liberation of this active hormone.125
Sulfotransferases in the cytoplasm of many tissues serve to catalyze the formation of iodothyronine sulfate conjugates. In rat tissue homogenates, 3,3′-T2 and T3 appear to be the best substrates for sulfation.46 Although the exact enzymes mediating iodothyronine sulfation are uncertain, the phenol sulfotransferases found in human liver and kidney have been demonstrated in vitro to possess this activity.126 Sensitive and specific radioimmunoassays for various iodothyronine sulfates have been developed and used to demonstrate detectable but very low levels of T3S, rT3S, and T4S in normal adult human serum.46 These low circulating levels probably reflect the rapid metabolism of these compounds by D1. Evidence for this thesis comes from experiments in animals and humans, in which treatment with inhibitors of D1 activity, such as PTU or iopanoic acid, results in marked increases in iodothyronine sulfoconjugate levels in both serum and bile.127,128 In other circumstances where D1 activity is low or impaired, such as during fetal life, in hypothyroidism, or in nonthyroidal illness, serum levels of these compounds also are elevated, either in absolute terms or relative to the native, unconjugated iodothyronines.16 To date, the physiologic role of sulfation in thyroid hormone economy has not been clearly defined, although it appears to represent an important component of the degradative process.
Glucuronidation of iodothyronines followed by their secretion into bile represents another pathway of iodothyronine clearance. Studies in humans, however, suggest that this pathway accounts for less than 1% of the total clearance of T4 by the liver, and only minute amounts of these conjugates are present in plasma.5,129
Two enzymes, l-amino acid oxidase and thyroid hormone aminotransferase, have been implicated in catalyzing the deamination of T4 and T3 to their acetic acid analogues Tetrac and Triac, respectively.130 Current evidence suggests, however, that these reactions occur to only a limited extent in normal humans. Triac has intrinsic biological activity equivalent to that of T3 when tested in in vitro assay systems,130 and it binds with greater avidity than T3 to the β isoform of the thyroid hormone receptor.1 Injection of Triac into humans or animals results in significant physiologic effects; however, relatively large doses are required because of its apparent rapid clearance and degradation via deiodination and glucuronidation.131 Despite its short half-life, Triac has been used successfully in the treatment of thyroid hormone resistance states.1 As in the case of sulfated conjugates, levels of Triac and its conjugates increase in circumstances in which D1 activity is low or impaired, such as during fasting or treatment with PTU or iopanoic acid.130 The physiologic relevance of these observations is uncertain but could be important, given Triac’s significant intrinsic activity. As noted earlier, the novel compound 3-iodothyronamine, formed by sequential deiodination and decarboxylation of thyroid hormones, may have important physiologic effects in some species, although its role in humans has not yet been defined.10,11
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
